

Abstract
Background
Brain edema is a common complication of brain metastases (BM) and associated treatment. The extent to which cytotoxic edema, the first step in the sequence that leads to ionic edema, vasogenic edema, and brain swelling, contributes to radiation-induced brain edema during BM remains unknown. This study aimed to determine whether radiation-associated treatment of BM induces cytotoxic edema and the consequences of blocking the edema in preclinical models of breast-cancer brain metastases (BCBM).
Methods
Using in vitro and in vivo models, we measured astrocytic swelling, trans-electric resistance (TEER), and aquaporin 4 (AQP4) expression following radiation. Genetic and pharmacological inhibition of AQP4 in astrocytes and cancer cells was used to assess the role of AQP4 in astrocytic swelling and brain water intake. An anti-epileptic drug that blocks AQP4 function (topiramate) was used to prevent cytotoxic edema in models of BM.
Results
Radiation-induced astrocytic swelling and transient upregulation of AQP4 occurred within the first 24 hours following radiation. Topiramate decreased radiation-induced astrocytic swelling and loss of TEER in astrocytes in vitro, and acute short-term treatment (but not continuous administration), prevented radiation-induced increase in brain water content without pro-tumorigenic effects in multiple preclinical models of BCBM. AQP4 was expressed in clinical BM and breast-cancer cell lines, but AQP4 targeting had limited direct pro-tumorigenic or radioprotective effects in cancer cells that could impact its clinical translation.
Conclusions
Patients with BM could find additional benefits from acute and temporary preventive treatment of radiation-induced cytotoxic edema using anti-epileptic drugs able to block AQP4 function.
Keywords: Aquaporin 4, Brain metastasis, Brain radiation, Brain Edema, Cytotoxic Edema, Topiramate
Graphical Abstract
Graphical Abstract.
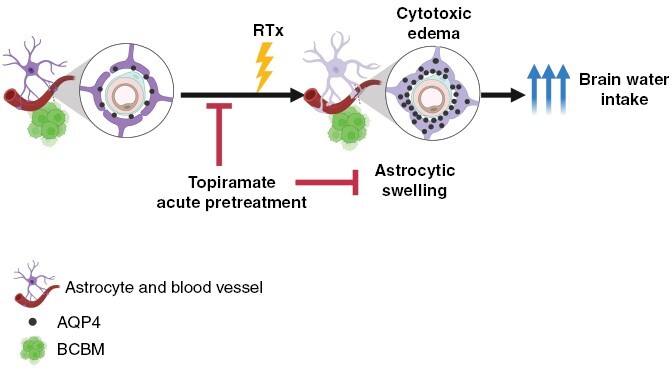
Key Points.
Radiation induces cytotoxic edema via acute dysregulation of AQP4 in astrocytes in preclinical models of brain metastases.
Pharmacologic blockage of AQP4 function prevents water intake, astrocytic swelling, and restores TEER in vitro.
pretreatment with single-dose topiramate prevents brain radiation-induced brain edema without direct tumor effects in preclinical models of breast-cancer brain metastases.
Importance of the Study.
In this study, we describe a novel role for astrocytic swelling and cytotoxic edema in the progression of stereotactic-radio-surgery (SRS)-induced brain edema during BM treatment. While radiation-induced edema has been fully attributed to the disruption of the blood-brain barrier (BBB) and ensuing vasogenic effects, our results suggest that cytotoxic edema affecting astrocytes in the acute setting plays an important role in the progression of brain edema during BM standard of care. Current standard of care for brain edema involves pretreatment with steroids and the use of bevacizumab only after clinically significant edema develops. Both interventions are presumed to target vasogenic edema. This study suggests that patients with BM could find additional benefits from acute and temporary preventive treatment of radiation-induced cytotoxic edema using an already FDA-approved anti-epileptic drug. Such early prevention strategy can be easily clinically implemented with the goal of minimizing treatment-related toxicities.
Brain metastases (BM) represent the most common intracranial malignancy in adults, and breast cancer is a frequent primary site leading to BM. Standard of care for BM includes stereotactic radiosurgery (SRS) alone or in combination with surgery, systemic chemotherapy, or targeted therapies.1 A common complication of BM and SRS is brain edema, a pathological state in which brain volume increases due to abnormal accumulation of fluid and ions within the cerebral parenchyma.2 Unlike edema in other organs, brain edema can be lethal or significantly impair quality of life due to intracranial hypertension.
Brain edema associated with BM and SRS is attributed mainly to vasogenic edema, a disruption of the BBB resulting in the extravasation of serum components and entry of fluids to the interstitial space from the bloodstream.3 However, a first step in the sequence that leads to ionic edema, vasogenic edema, and ultimate brain swelling and necrosis,4,5 is cytotoxic edema, a premorbid cellular process, whereby extracellular Na+ and other cations enter into neurons and astrocytes and accumulate intracellularly, resulting in osmotic expansion of the cells and leading to necrotic cell death.3,4,6 While cytotoxic edema has been described in spinal cord injury, traumatic brain injury, and stroke, the extent to which cytotoxic edema contributes to the pathophysiology of BM and SRS remains unknown.
Cytotoxic edema is characterized by astrocytic swelling and dysregulation of AQP4 levels, the main bilateral water channel in the central nervous system (CNS).7–10 AQP4 is expressed abundantly in astrocytes and plays different roles in development and resolution of CNS edema, with water flow through AQP4 driving the development of cytotoxic edema in the early post-injury stage but later clearing vasogenic edema.11,12 Therefore, reversible inhibition of AQP4 function during the acute phase of CNS trauma has been proposed as a viable strategy to prevent CNS edema.13,14 We have shown that radiation induces AQP4 dysregulation in immortalized astrocytes in vitro and that combination of radiation with cytotoxic drugs (i.e T-DM1 used to treat Her2+ BM)15 further increases AQP4 dysregulation and astrocytic cell death, suggesting AQP4 and cytotoxic edema play a role in radiation-induced edema in BM. Herein, we demonstrate that radiation induces AQP4 dysregulation and cytotoxic edema in human primary astrocytes in vitro and in vivo, and show that an FDA-approved anti-epileptic drug topiramate, reported blocking AQP4 function16 can be used to prevent radiation-induced cytotoxic edema in preclinical models of breast-cancer brain metastases (BCBM).
Methods
Cells
HuAST (primary human astrocytes from ScienceCell #1800-10), HAL (immortalized human astrocytes from Dr. Patricia S. Steeg, NCI, USA), THV (immortalized human astrocytes, from Dr. Paul B. Fisher). Human breast-cancer cells were: BT474 (from Dr. Dihua Yu, MD Anderson Cancer Center, TX, USA); MCF-7 (RRID: CVCL_0031 https://scicrunch.org/resources/data/source/SCR_013869-1/search?q=CVCL_0031&l=CVCL_0031), JmT1BR3 and 231BR (from Dr. Patricia Steeg, NIH, USA); and F2-7cells.17 Murine mammary gland tumor cells were: 4T1BR5 (derived from 4T1 from Dr. Suyun Huang, MD Anderson Cancer Center, TX, USA) and E0771BR (derived from E0771 RRID: CVCL_GR23 https://scicrunch.org/resources/data/source/SCR_013869-1/search?q=CVCL_%20GR23&l=CVCL_%20GR23). Details in Supplementary Methods.
Human-Derived BM
De-identified paraffin-embedded archived BM samples from female breast-cancer patients were obtained from consenting donors under approved IRB protocol 20-1669 at the University of Colorado.
Drugs
Topiramate (TPM, Selleckchem S7998) was used in vitro at 10-100 μM and in vivo at 50 mg/Kg/day in 10% DMSO. N-(1,3,4-thiadiazol-2-yl) pyridine-3-carboxamide dihydrochloride (TGN-020, Tocris Cat No 5425) was used in vitro at 10-100 μM. T-DM1 (Genetech) was used in vitro at 1 μg/mL.
Cell Proliferation
Of 1000–3000 cells were plated in 96 well plates, pretreated for 2 hours before radiation when indicated, and imaged over time using Live Cell Incucyte Imaging (Essen Bioscience). Cell confluence was calculated in 6 fields per well, in at least 5 replicates per treatment, in 3 independent experiments.
Cell-by-Cell Analysis for Detection of Apoptosis
Apoptotic HAL and HuAST cells were detected using NucLight Rapid Red Reagent (Cat 4717) and Incucyte® Caspase-3/7 Green Dye (Cat 4440). Cells were treated with Vehicle (DMSO), T-DM1(1 µg/mL), TPM (100 µM), or T-DM1+TPM 2 hours before radiation (± 8 Gy), using the RS2000 X-Ray irradiator. Plates were imaged for 3 days in a cell-by-cell module of Incucyte S3 (Essen BioScience). Analysis of apoptotic cells (green cells with red nuclei) was performed using the Incucyte software. n = 6 replicates in at least 2 independent experiments. Details in Supplementary Methods.
Western Blot
When indicated, cells for western blot were irradiated with 0–8 Gy using the RS2000 X-Ray irradiator. PVDF membrane was then incubated overnight at 4°C with primary antibodies (Supplementary table 1). Membranes were imaged using Li-COR Odissey CLx and analyzed using Image Studio™ Software V5.2. Details in Supplementary Methods.
Animal Experiments
All animal experiments were approved by the University of Colorado Institutional Animal Care and Use Committee and the DoD-ACURO. Whole brain radiation therapy (WBRT) in female mice was performed using the Precision X-Ray X-Rad 225Cx Micro IGRT and SmART Systems, using 4.8 to 5.8 Gy/minutes in a single dose. To model radiation-induced brain edema, immune-compromised NOD-SCID IL2Rγnull (NSG) female mice received 10 Gy, and immunocompetent C57black/6J (Jackson Labs) female received 10 Gy or 35 Gy18,19 depending on the experiment. Experimental BM were developed by intracardial injection (ic) of 250000 JmT1BR3-GFP-luciferase cells or 50000 E0771BR1-GFP-luciferase cells in the left ventricle in 14–18 weeks old female mice (n = 8 to 14 per group) and metastatic burden measured via IVIS. All animal interventions were performed by investigators blinded to the experimental groups.
Brain Water Content
Freshly collected right brain hemispheres were put on a pre-weighed aluminum foil sheet, weighted (Melter Toledo, ME54E accuracy of 0.0001 g), dried at 99°C, and weighed after 24 hours. The percentage of brain water content was determined as: (%) water content = 100 × (wet weight-dry weight)/wet weight.20
Electron Microscopy
For astrocytic end feet analysis, mice were anesthetized with isoflurane and intracardially perfused with PBS, followed by 2.5% glutaraldehyde, 2% PFA in 0.1 M Na-cacodylate pH 7.4. Brains were kept at 4°C in fixative solution and processed at the Electron Microscopy Center SOM (Details in Supplementary Methods). Images were acquired on a Tecnai T12 transmission electron microscope (ThermoFisher) equipped with a LaB6 source at 80 kV using an XR80 (8Mpix) camera (AMT). Ten vessels/samples were imaged, and tight junctions from each vessel were imagined separately at higher magnification. Details in Supplementary Methods.
Image Acquisition and Analysis
Microvessel images were collected using a 3i Marianas Spinning Disk Confocal microscope attached to a Zeiss Axio Observer Z1 with Yokogawa CSU-X1 microscope, with a 63x Oil objective. Maximum Z projections were made using SlideBook 6 software. For immunofluorescence (IF) analysis, images were collected using a Nikon Eclipse Ti-S inverted microscope using a Nikon CFI Plan Apo 20X/0.75 DIC M/N2/0.17 WD 1.0 Objective MRD00205. Image resolution was 1280 × 1024 px. Pixel size 0.32 µm/px. Region of interest were manually delineated using the drawing tool in Fiji software and mean Intensity and area of interest were calculated using the measure tool in Fiji software.21 Details in Supplementary Methods.
Transepithelial Electrical Resistance (TEER)
TEER in monolayer of HAL and HuAST plated on cell culture inserts was measured using a volt ohmmeter (EVOM2, World Precision Instruments) with a chopstick electrode (STX3, World Precision Instruments). Measures were made before radiation and pretreatments, and at the indicated times following radiation. The unit area resistance (Ωcm2) was obtained by multiplying the Ω reading by the surface area of the filter membrane (0.3 cm2). The TEER from empty inserts with media alone was subtracted from the TEER of cultures (values between ~160–200 Ω). Details in Supplementary Methods.
Statistical Analysis
Specific statistical tests are listed in each figure legend, along with the number of samples assessed. Data statistical analysis and graphs were completed using GraphPad Prism software version 9.5.0. Randomization was used for all mice studies. All in vitro experiments were repeated at least 3 times unless indicated otherwise. Data were analyzed using unpaired t-test, Mann–Whitney test, and two-way analysis of variance (ANOVA) with multiple comparison tests as appropriate. A P value ≤0.05 was considered significant.
Results
Radiation-Induced Astrocytic Swelling Via AQP4 Upregulation is an Early Step in Brain Edema
Consistent with our previous report15; immortalized human astrocytes (HAL and THV) and primary human astrocytes (HuAST) express a baseline level of AQP4 (Figure 1A) which transiently increases within the first 24 hours following radiation (8 Gy, P = .0325) (Figure 1B, Supplementary Figure 1A). To determine whether radiation-induced AQP4 upregulation resulted in water intake and astrocyte swelling, we measured the cell area of HuAST 24 hours post-radiation. A dose of 8 Gy (previously shown to induce AQP4 upregulation and astrocytic swelling in immortalized astrocytes15) increased the astrocyte area by 4.8 fold compared with non-irradiated cells (P < .0001) (Figure 1C). Since radiation induces senescence in astrocytes and senescent cells are hypertrophic,22 we assessed whether radiation-induced astrocytic swelling was due to senescence. Radiation increased the percentage of senescent HuAST (SA-β-Gal+) 48 hours after a single-8 Gy dose (P = .002) (Figure 1D, 1E-left). However, swollen astrocytes following 8 Gy radiation were non-senescent (SA-β-Galneg) (P < .0001) (Figure 1E-right). These results suggest that radiation-induced astrocytic swelling is not due to radiation-induced senescence.
Figure 1.
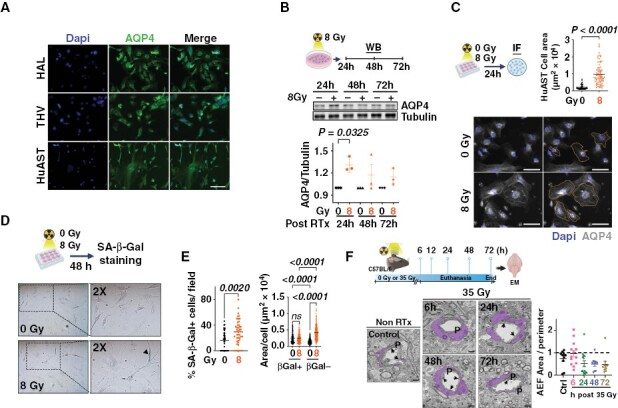
Radiation induces astrocytic swelling in vitro and in vivo. (A) AQP4 (green) expression in human astrocytes. Nuclei (Dapi-blue). Scale 100 µm. (B) WB shows AQP4 levels in HuAST ± 8 Gy at the indicated times. Graph shows fold change AQP4/tubulin levels relative to non-irradiated cells (n = 3). Paired T-test. (C) Astrocytic area in AQP4-stained HuAST (gray) ±8 Gy 24 hours post-radiation. Orange line shows astrocyte area delimitation. Scale 100 µm. Graph shows area of single cells (n = 86). (D) SA-β-gal staining in HuAST treated ± 8 Gy. Blackhead arrow shows SA-b-gal+ cells. (E) Left: Percentage of SA-β-Gal+ HuAST ± 8 Gy at 48 hours, quantified in 45 fields/condition on 3 independent slides. Right: Individual cell area of βGal+ or βGal- HuAST ± 8 Gy at 48 hours, quantified in bright field images from 3 coverslips per condition using imageJ, analyzed by Kruskal–Wallis test followed by Dunn’s post hoc test. (F) C57Bl6 mice were irradiated with 35 Gy whole brain radiation therapy and euthanized at the indicated times. Representative microphotographs of cortex brain microvessels showing perivascular end feet astrocytes (purple), pericytes (P), and tight junction (black arrows) at the indicated times. Scale 1 µm. Graph shows astrocytic end feet area normalized to the vessel perimeter.
To determine if radiation-induced astrocytic swelling occurs in vivo, female C57Bl6 mice were treated with a single 35Gy-WBRT and the astrocytic end feet (AEF) surrounding microvessels in the brain cortex was analyzed using electron microscopy (Figure 1F). A larger fraction of AEF with an enlarged area (>1 AEF/vessel perimeter) was observed 6 hours following radiation compared to non-irradiated mice (46.5% vs. 20.0% enlarged AEF, respectively) (Figure 1F), without significant changes in tight junction BBB integrity (Supplementary Figure 1B), suggesting that radiation induces astrocytic swelling and transient upregulation of AQP4 without leading to acute disruption of the BBB within the first 24 hours following radiation.
Topiramate Protects Human Astrocytes From Radiation-Induced Cell Swelling In Vitro
AQP4 is critical for survival of astrocytes in vitro.23 Consistently, stable knockdown of AQP4 using shRNAs prevented radiation-induced astrocytic swelling in HuAST but resulted in cell death (Supplementary Figure 2A–C). Thus, we used a pharmacologic approach to block AQP4 function and hypothesized that this will prevent radiation-induced cytotoxic swelling. We used a selective AQP4 inhibitor, TGN-020,24 or an FDA-approved anti-epileptic drug topiramate (TPM),25 previously reported to function as an AQP4 inhibitor.16 Neither TGN-020 nor TPM induced cell death in THV astrocytes at 10–100 µM (Supplementary Figure 2D). Pretreatment with TGN-020 slightly reduced the fraction of radiation-induced swollen astrocytes, but this effect was not statistically significant at the doses tested. By contrast, a 2 hours pretreatment with 100 µM of TPM was effective in protecting THV from radiation-induced swelling (P < .0001) (Figure 2A). Therefore, we sought to determine if TPM could prevent astrocytic swelling in HuAST alone or in combination with cytotoxic therapies known to promote radiation-induced edema.
Figure 2.
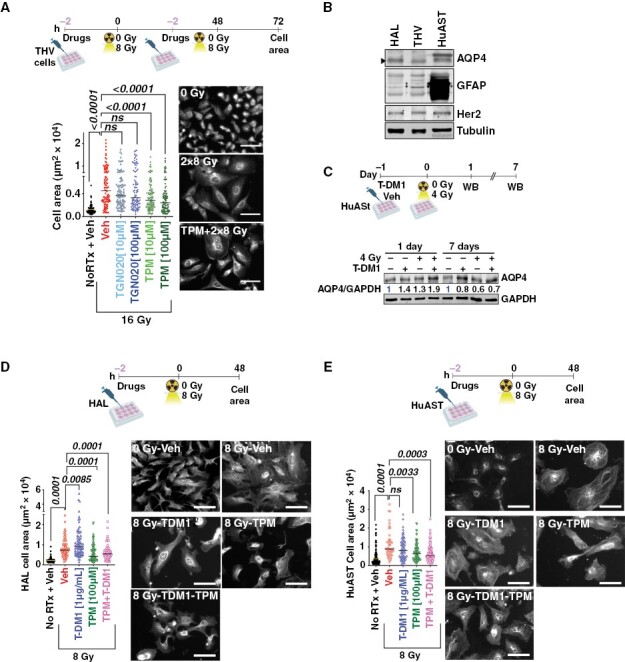
Topiramate prevents radiation-induced cell swelling in human astrocytes in vitro. (A) THV cells were pretreated with Veh (DMSO), TGN-020 (10–100 µM), or TPM (10–100 µM) 2 hours prior to ± 8 Gy. Graph shows THV cell area 24 hours after 2 doses of 8 Gy. IF images of THV stained with AQP4 (gray). (B) WB shows AQP4, GFAP, and Her2 expression in human astrocytes. Tubulin is loading control. (C) HuAST were pretreated with vehicle or T-DM1 [1 µg/mL] 2 hours before ± 4 Gy. WB shows AQP4 levels 1 and 7 days post-radiation. GAPDH is loading control. Numbers are AQP4/GAPDH relative to untreated cells. (D) HAL cells were pretreated with Veh (DMSO), T-DM1 (1 µg/mL), TPM (100 µM), or TPM+T-DM1, 2 hours prior to 8 Gy. Graphs show cell area in µm2 48 hours post-radiation. Representative images of AQP4 stained HAL cells (gray). (E) Cell size 48 hours post-irradiation in HuAST treated as in D. For all images, scale bar 100 µm. Data were analyzed with Kruskal–Wallis followed by post hoc corrections (A) or Mann–Whitney test for paired comparisons (D, E).
We previously showed that the Her2-targeting agent T-DM1 increased radiation-induced astrocytic swelling in immortalized human astrocytes, which express normal levels of Her2 and are an unintended target of T-DM1.15 Accordingly, T-DM1 increased AQP4 within 24 hours post-radiation in Her2+ HAL and HuAST, returning to normal levels 7 days post-radiation (Figure 2B-C). To test whether TPM could prevent this transient dysfunction of AQP4, HAL and HuAST were pretreated with 100 μM of TPM for 2 hours before radiation and T-DM1 treatments (Figure 2D-E). TPM prevented astrocytic swelling induced by radiation alone or in combination with T-DM1 in both HAL and HuAST cells, P < .0001 in HAL (Figure 2D), and P = .0033 in HuAST (Figure 2E).
To determine if preventing astrocytic swelling could also preserve astrocytic function in the BBB alone or in combination with T-DM1, we measured TEER of normal and irradiated astrocytic monolayers in vitro (Figure 3A). Radiation changed TEER in a biphasic fashion; increasing TEER in the first 20 minutes (P = .0313 in HAL and P < .0001 in HuAST) followed by a significant decrease after 24 hours (P = .0152) (Figure 3A). Therefore, we tested whether pretreatment with TPM could prevent the loss of TEER 24 hours following radiation alone or in combination with T-DM1 (Figure 3B). T-DM1 and radiation significantly decreased TEER compared to non-irradiated cells (HAL, P = .0054) and (HuAST, P < .0001) (Figure 3B), and TPM pretreatment restored TEER in T-DM1/Radiation-treated cells by 73% in HAL cells (P = .0459) and 48% in HuAST (P = .0122) as compared to T-DM1/Radiation-treated cells (Figure 3B). Since radiation and T-DM1 can induce astrocytic cell death by mechanisms other than AQP4 dysregulation including direct DNA damage (Supplementary Figure 3A), we measured the impact of TPM in radiation and T-DM1-induced cell death. Using a live-imaging cell-by-cell detection of apoptosis T-DM1 and radiation alone and in combination significantly induced apoptosis by 72 hours compared to untreated cells in both HAL and HuAST (Figure 3C). TPM showed a modest reduction in apoptosis in HAL cells and no effects in T-DM1/Radiation-induced apoptosis of HuAST (Figure 3C). Consistent with these results, TPM did not affect radiation-induced double-strand DNA damage as measured by pH2A.X activation, (Supplementary Figure 3A), caspase-3 activation (Supplementary Figure 3B) or activation of radiation-induced survival pathways (Supplementary Figure 3C). Thus, TPM can prevent astrocytic swelling and partially protects the physiological properties of astrocytes but cannot reverse radiation-induced cell death.
Figure 3.
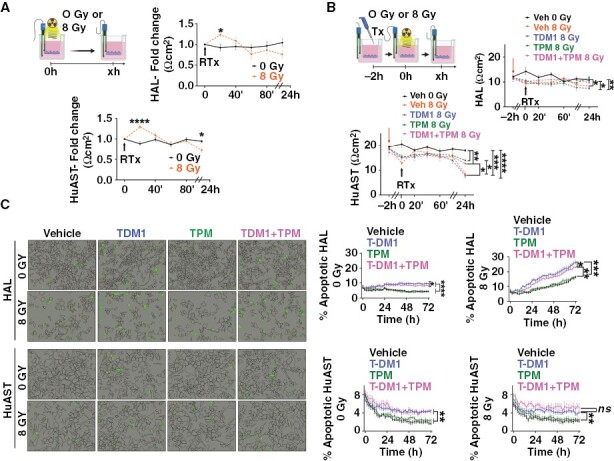
Topiramate restores radiation and T-DM1-induced loss of TEER but does not prevent astrocytic cell death in vitro. (A) Dynamics of TEER changes in monolayer of astrocytes grown on transwell inserts and irradiated with 8 Gy. Graphs show TEER expressed as a fold change of Ωcm2 relative to time zero in HAL and HuAST. (B) TEER in a monolayer of astrocytes pretreated with Veh (DMSO), T-DM1 (1 µg/mL), TPM (100 µM), or (TPM+T-DM1) 2 hours prior to 8 Gy. Graphs show mean TEER in Ωcm2 (n = 9 transwell/treatment). Lines are mean± SEM. (C) HAL or HuAST were treated as B and Apoptotic and Caspase 3/7+ cells (green mask) measured over time. Left: representative images 72 hours after treatment. Right: Graphs shows percentage of apoptotic cells over time. Mean ± SEM (n = 6 wells). *P < .05, **P < .01 ***P < .001, ****P < .0001. (two-way ANOVA-Geisser-Greenhouse correction followed by post hoc test).
Short-Term pretreatment With Topiramate Prevents Radiation-Induced Brain Edema In Vivo
Changes in brain water content are widely used to measure water channel dysfunction and brain edema in preclinical mouse models.11,26 To determine whether TPM could prevent radiation-induced astrocytic swelling and brain edema in vivo, we first measured water content in brains of untreated C57BL6 mice, or mice that had been exposed 24 hours before, to a single 35 Gy dose of WBRT, pretreated or not with TPM (Figure 4A). While radiation increased AQP4 in AEF in brain microvessels (Figure 4B), there were no changes in brain water content 24 hours following irradiation in vehicle or TPM-treated mice as compared to untreated mice (Figure 4C), suggesting that radiation-induced early astrocytic swelling does not result in acute changes in brain water content in the absence of BM.
Figure 4.
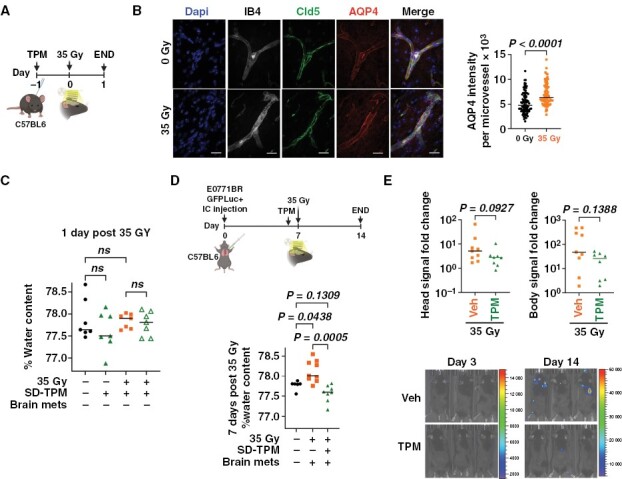
Pretreatment with topiramate prevents radiation-induced brain water accumulation in a syngeneic model of breast-cancer brain metastases. (A) C57BL6 mice were pretreated with a single dose of vehicle or TPM [50 mg/Kg] 24 hours prior to receiving or not 35 Gy Whole brain radiation therapy (WBRT), and euthanized 24 hours later (n = 10). (B) Confocal z-stack maximum projection of end-feet astrocytes AQP4+ (red) in IB4+ (Isolectin B4, gray) and Cld5+ (Claudin-5, green) microvessels. Dapi stains nuclei (Blue). Scale bar 20 µm. Graph shows mean AQP4 intensity per brain microvessel (n = 100 from 3 mice/treatment). Two-tail Mann–Whitney test. (C) Percentage brain water content per mouse (n = 6 per treatment) in mice from A. One-way ANOVA followed by Sidak’s post hoc test. (D) C57BL6 female mice were injected intracardially with E0771BR-GFP-Luciferase cells and metastasis was allowed to grow for 7 days. Mice were then randomized to receive a single dose of TPM [50 mg/Kg] or vehicle 4 hours prior to receiving 35 Gy WBRT and euthanized 7 days later. Graph shows the percentage of brain water content per mouse (n = 8,9 mice/group). Non-irradiated naïve C57BL6 mice (n = 6) were included as baseline water content. Data were analyzed using Kruskal–Wallis followed by 2-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli multiple comparisons test. (E) Fold change IVIS signal per mouse in head (left) and body (right). Mann–Whitney test. Representative images of IVIS signal 3 and 14 days after cell injection.
We next determined whether TPM could prevent radiation-induced edema in a preclinical model of BM. For this, breast-cancer cells syngeneic to C57Bl6 mice (E0771-GFP-Luciferase) were injected intracardially in 8–12-week-old female mice, and BM were allowed to form for 7 days. Mice were then randomized to receive vehicle or TPM 4 hours before a single 35 Gy WBRT, and brain water content was quantified 7 days later (Figure 4D). In this model of radiation-induced edema in BM-carrying mice, TPM significantly reduced brain water content compared to vehicle-treated mice (Figure 4D). Importantly, TPM prior to radiation did not affect brain metastatic burden (Figure 4E), suggesting that short-term TPM is sufficient to prevent radiation-induced brain edema and does not increase BM burden in this model.
Topiramate Does Not Alter the Response of Cancer Cells to Radiation or T-DM1 Treatment In Vitro
While E0771 cells were not responsive to short-term TPM in vivo, AQP4 has been shown to play various roles in breast-cancer cells,27,28 and interventions targeting AQP4 have the potential to impact tumor burden. IHC staining in a cohort of breast-cancer BM showed that AQP4 is heterogeneously expressed in BM percentage positive tumor area ranging from 1.6% to 91.1%, median of 10.5%, (Figure 5A, B). AQP4 was also expressed in 5 of the 7 metastatic breast-cancer cell lines from all breast-cancer subtypes, as well as in murine mammary cancer models (Figure 5C). Moreover, radiation modified AQP4 expression in a cell line-dependent manner (ie, radiation increased AQP4 expression in MCF-7 and E0771 cells but reduced its expression in 4T1BR5 cells (Figure 5C).
Figure 5.
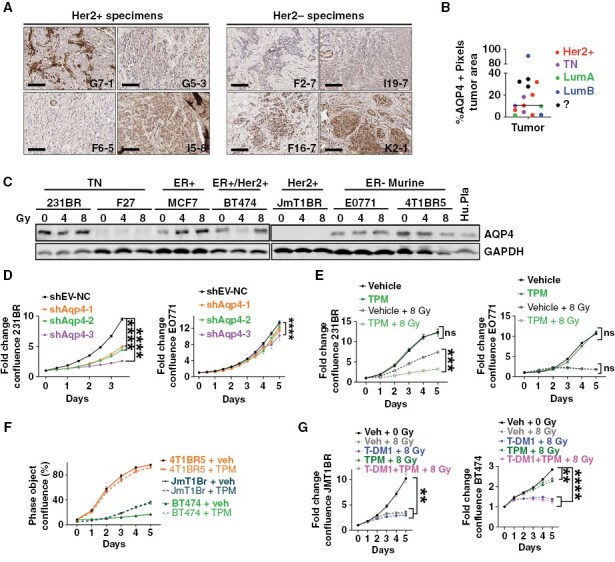
AQP4 is expressed in breast-cancer brain metastasis. (A) AQP4 IHC staining in Her2+ and Her2- breast-cancer brain metastases (BCBM) clinical samples. (B) Percentage of positive pixels in tumor area in a cohort of 14 BCBM patients from the indicated subtypes. (C) AQP4 WB in human and murine breast-cancer cells treated with 0, 4, and 8 Gy. (D) Fold change confluence of 231BR or E0771 cells infected with shRNA shEV or 3 shRNAs targeting human (231 BR) or murine (E0771) AQP4, respectively. (E) Fold change in confluence of 231BR and E0771 cells treated with vehicle (DMSO) or TPM [100 µMl] 2 hours prior ± 8 Gy. (F) Percentage confluence of indicated breast-cancer cells treated with Vehicle (DMSO) or TPM [100 µM]. (G) Her2+ JmT1BR or BT474 cells treated with vehicle (DMSO), T-DM1 [1 µg/mL], TPM [100 µM], or TPM+T-DM1, 2 hours prior to 8 Gy. For all graphs, line is mean ± SEM. Data were analyzed using Two-way ANOVA with the Geisser-Greenhouse correction followed by Tukey’s multiple comparisons tests.
To test whether targeting AQP4 could directly influence cancer cell proliferation, AQP4 was knocked down in 231BR and E0771BR cells, using 3 different lentiviral shRNAs targeting human or murine AQP4, respectively. AQP4 knockdown in the human 231BR cells decreased their proliferation significantly compared with control cells (P < .0001), while AQP4 knockdown in the murine E0771 cells had negligible effects on cell proliferation (Figure 5D). Consistently, TPM increased the sensitivity to radiation in 231BR, but had no effect on E0771 cells (Figure 5E). Moreover, TPM treatment did not impact proliferation of other breast-cancer cell lines (Figure 5F), nor did it impact sensitivity to T-DM1 and radiation in 2 Her2+ cell lines (Figure 5G). Taken together, these results suggest that TPM does not have direct pro-tumorigenic effects, nor does it have a radioprotective effects in cancer cells that could impact its clinical translation for preventing radiation-induced brain edema.
Long-term topiramate decreased brain edema but increased tumor burden in syngeneic mouse models but not in immunocompromised models.
Since TPM is an FDA-approved anti-epileptic drug used daily, we sought to determine whether a daily TPM administration could improve long-term control of radiation-induced brain edema in preclinical models of BM. We chose models that were not sensitive to TPM in vitro (JmT1Br3 and E0771) to be able to measure the effects of TPM on radiation-induced edema independent of tumor burden. First, we injected AQP4neg human JmT1Br3 cells in severely immunocompromised NSG mice, allowed metastases to form for 7 days, and then randomized mice to receiving, or not, a single 10 Gy dose (maximum permissible dose in NSG mice) and a daily dose of TPM or vehicle for the following 14 days (Figure 6A). Consistent with the results found using short-term use of TPM in the E0771/C57BL6 model, sustained TPM treatment reduced brain water content following irradiation (Figure 6A) without changes in tumor burden in the brain or in systemic metastases (Figure 6B). Unexpectedly, sustained TPM treatment in C57Bl6 mice carrying E0771 BM reduced brain water content in irradiated mice (Figure 6C) but led to a significant increase in metastatic tumor burden (Figure 6D). Since TPM did not directly alter the growth of E0771-GFP-Luciferase cells in vitro (Figure 5F), and this pro-metastatic effect was not observed in the JmT1BR3/NSG model, we proposed that the immune environment was responsible for this pro-tumorigenic effect of long-term TPM. Consistent with this hypothesis, sustained TPM in NSG mice injected with E0771-GFP-Luciferase (Figure 6E) resulted in decreased water content (Figure 6E) but did not lead to increases in metastatic burden (Figure 6F). Thus, long-term use of TPM in the context of metastatic breast cancer may impact metastatic burden through modulation of the immune microenvironment.
Figure 6.
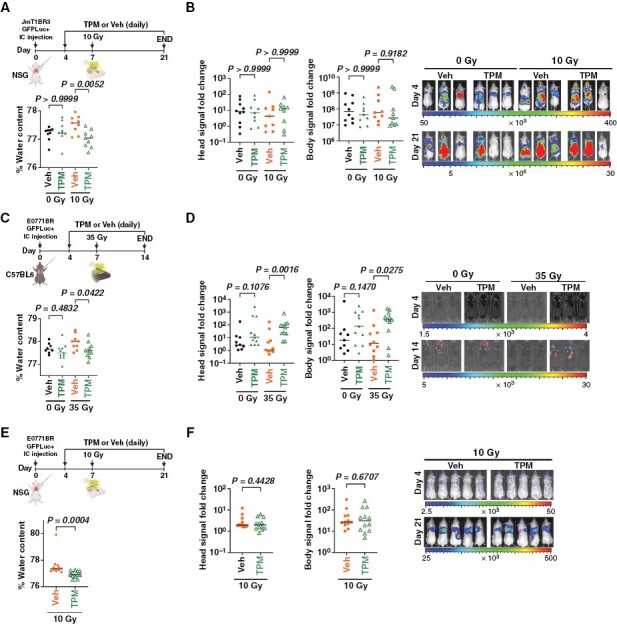
Long-term topiramate decreased brain edema but increased tumor burden in syngeneic mouse models but not in immunocompromised models. (A) NSG mice were injected intracardially with JmT1BR3-GFP-Luciferase cells and 4 days later randomized to receive daily doses of vehicle or TPM [50 mg/Kg]. Mice received or not 10 Gy Whole brain radiation therapy (WBRT) 7 days post-injection and were euthanized 14 days later. Graph shows percentage of brain water content per mouse (n = 9 per group). (B) Fold change metastatic burden in the head (left) and body (right) measured via IVIS in mice from A. Panel shows representative IVIS images 4 and 21 days after cancer cell injections. (C) C57BL6 mice were injected intracardially with E0771BR-GFP-Luciferase cells and treated as in A, except that received 35 Gy WBRT on day 7 and were euthanized 7 days later. Graph shows percentage of brain water content per mouse (n = 9 to 11/group). (D) Fold change metastatic burden in the head (left) and body (right) measured via IVIS in mice from C. Panel shows representative IVIS images 4 and 14 days after cell injections. (E) NSG mice were injected intracardially with E0771BR-GFP-Luciferase cells and treated as in A, except that all mice received 10 Gy WBRT. Graph shows percentage of brain water content per mouse (n = 12/group). (F) Fold change of metastatic burden in the head (left) and body (right) measured via IVIS in mice from E. Panel shows representative IVIS images 4 and 21 days after cancer cell injections. (A–D) Data were analyzed using Kruskal–Wallis followed by Dunn’s post hoc test. (E–F) was analyzed using Mann–Whitney test.
Discussion
In this study, we describe a novel role for AQP4 and cytotoxic edema in BM and radiation-induced brain edema. While edema in the context of BM has been fully attributed to tumor-induced disruption of the BBB leading to vasogenic edema, our results suggest that cytotoxic edema affecting astrocytes also plays an important role in the progression of brain edema during BM following SRS.
Using in vitro models of human astrocytes and in vivo models of WBRT, we show that radiation induces astrocytic swelling and transient upregulation of AQP4, hallmarks of cytotoxic edema in other brain injuries.10,29–32 In vitro radiation doses used in this study correspond to previously reported doses able to induce astrocytic swelling in immortalized astrocytes,15 without causing acute cell death. We show that radiation-induced astrocytic swelling is not occurring in senescent cells; thus, increased astrocytic cell size can be attributed to disrupted water intake and not hypertrophy occurring during senescence.22,33 Moreover, the fact that AQP4 blockage can decrease astrocytic swelling and restore TEER function in vitro but does not protect from cell death induced by radiation or cytotoxic drugs such as T-DM1, further suggests that acute cytotoxic edema and AQP4 dysfunction are not mechanistically linked to DNA damage or microtubule disruption, the main cytotoxic drivers of radiation and T-DM1, respectively.
In contrast to prior reports showing that 35 Gy of WBRT on C57BL/6J mice induced brain water accumulation after 1 day of radiation,19 we observed that a 35 Gy dose lead to AQP4 upregulation in vivo, without leading to acute disruption of the BBB or changes in brain water content within the first 24 hours following radiation. While we recognize that this single WBRT high dose is not comparable to focal-fractionated SRS used commonly in humans, modeling of BCBM requires hematogenous dissemination which produces multiple, diffuse lesions throughout the whole brain and precludes the use of mouse SRS. Despite this limitation, WBRT doses between 10 and 35 Gy induced a significant increase in brain water content in BM-carrying mice, and a single pretreatment dose of TPM administered within 4 hours from radiation were able to block this effect, further supporting the hypothesis that acute cytotoxic edema plays a role in the pathobiology of radiation-induced edema and can be targeted in the context of BM.
Acute inhibition of AQP4 has been shown to decrease edema and infarct lesion volume in preclinical models of stroke34; however, the existing AQP4-specific inhibitors are far from clinical application. In this study, we use TPM due to its previously reported role in blocking AQP4-dependent water flux,16 and the fact that is an FDA-approved anti-epileptic drug with rapid translational potential. However, we recognize that TPM has additional functions and may decrease cytotoxic edema through other mechanisms. Recent studies have shown that AQP4 subcellular re-localization to the plasma membrane is required to drive cytotoxic edema in models of hypoxia-induced cell swelling.11 AQP4 translocation to the membrane was shown to require an influx of Ca2+ ions to activate calmodulin, which in turn activates adenylyl cyclases leading to PKA activation and subsequent phosphorylation of AQP4, ultimately responsible for AQP4 translocation to the plasma membrane.11 Since TPM targets calcium channels and TPM has been reported to decrease Ca2+ influx in neurons,35 it is possible that TPM alters the intracellular concentration of Ca2+ also in astrocytes, thus, reducing AQP4 function at the plasma membrane. This potential mechanism for TPM in blocking AQP4 function could explain why TPM but not the AQP4 specific inhibitor TGN-020 which targets AQP4 channel blocker function, decreased radiation-induced astrocytic swelling in vitro.
Patients with BM are at significant risk for developing seizures,36 and anti-epileptic drugs are prescribed to control this side effect. Thus, we tested whether daily TPM administration could improve long-term control of radiation-induced brain edema in preclinical models of BM. Unexpectedly, our results showed that long-term use of TPM in the context of BCBM increased systemic metastatic burden, raising concerns about the potential side effects of TPM and other anti-epileptic drugs targeting AQP4 in patients with systemic metastasis. While E0771 cells express AQP4, neither AQP4 KD or TPM altered E0711 cell proliferation in vitro, and TPM did not alter E0711 growth in NK and T-cell depleted NSG mice, suggesting that long-term TPM treatment promotes metastatic progression through modulation of immune cells. It has been shown that AQP4 is expressed on murine CD4+ and CD8+ T cells, and that blockage of AQP4 using a small molecule (AER-270) inhibits T cell proliferation in vitro and alters T-cell trafficking in vivo in a model of cardiac transplantation.37 Thus, we propose long-term TPM or AQP4 inhibitors may suppress antitumoral T-cell function and thus indirectly facilitate metastatic tumor growth. Further studies are warranted to decipher the mechanisms whereby TPM and other anti-epileptic drugs targeting AQP4 could influence immune cells and tumor progression.
Expression of aquaporins in brain tumors and other malignancies has been shown to promote migration and proliferation38 and can alter the response of conventional anticancer therapies27,39; however, little is known about AQP4 function in BCBM. While AQP4 was expressed in several breast-cancer cell lines, the majority were not susceptible to AQP4 blockage (either genetic or via TPM), suggesting that TPM or AQP4 blockage can be used to prevent cytotoxic edema without direct pro or antitumoral effects.
Finally, our studies have important clinical implications. First, current standard of care for brain edema involves post-edema use of bevacizumab and steroids targeting vasogenic edema. Our results suggest that patients with BM could find additional benefits from acute and temporary preventive treatment of SRS-induced cytotoxic edema using an already FDA-approved anti-epileptic drug. The proposed short use of TPM is similar to current clinical use of steroids in this setting. Finally, our studies highlight the need for better understanding of the interactions between drugs commonly used in the care of patients with BM and their cancer-specific treatments.
Supplementary Material
Acknowledgments
We thank Dr. A. Van Bokhoven at the Biorepository Core Facility, and personnel at the University of Colorado Neurosurgery Nervous System Biorepository for providing de-identified human tissues. Dr. D. Yu for providing breast-cancer cells BT474, and Dr. P. Steeg for providing 231BR and JmT1BR3 cells. Special thanks to Benjamin Van Court and Brooke Neupert in the Small-Animal Irradiator Shared Resource, Dr. Jennifer Bourne (†) and Dr. Anza Darehshouri in the Electron Microscopy Center at University of Colorado Anschutz Medical Campus.
Contributor Information
Maria J Contreras-Zárate, Department of Pathology, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA.
Karen L F Alvarez-Eraso, Department of Pathology, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA.
Jenny A Jaramillo-Gómez, Department of Pathology, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA.
Zachary Littrell, Department of Pathology, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA.
Nikki Tsuji, Office of Laboratory Animal Resources, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA.
D Ryan Ormond, Department of Neurosurgery, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA.
Sana D Karam, Department of Radiation Oncology, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA.
Peter Kabos, Department of Medicine, Division of Medical Oncology, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA.
Diana M Cittelly, Department of Pathology, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA.
Funding
This work was supported by the Department of Defense (DOD) Breast Cancer Research Program (BCRP) DOD BCRP W81XWH-19-1-0033 (MJC). DMC is supported by National Cancer Institute (NCI) R37CA227984 and DOD BCRP W81XWH-22-1-0042. PK is supported by National Institutes of Health NIH R01-CA20544 R01 CA258766. SDK is supported by the National Institute of Dental and Craniofacial Research - National Cancer Institute NIDCR/NCI R01 DE028528-01; R01 DE028282-01; P50 CA261605-01. The University of Colorado Comprehensive Cancer Center shared resources supported by the National Cancer Institute NCI P30CA046934 and the Clinical and Translational Science Awards (CTSA) National Institutes of Health (NIH) CTSA UL1TR001082 Center grants. The Advanced Light Microscopy Core at University of Colorado Anschutz Medical Campus supported by Rocky Mountain Neurological Disorders Core National Institutes of Health (NIH) P30 NS048154 and by Diabetes Research Center P30 DK116073. The Animal Imaging Shared Resource funded by the National Institutes of Health (NIH) S10 Shared Instrumentations grants (S10 OD023485 and S10 OD027023), and P30 CA046934.
Conflict of Interest
MJC, DMC, KA, JAJ, ZL, and NT: no conflicts. DRO: Clinical trial research funding from Integra Biosciences, Servier Pharmaceuticals. SDK: Clinical trial, preclinical research funding from AstraZeneca, Genentech, Ionis and Roche Pharmaceuticals are all unrelated to this work. PK: Clinical research grant support from Genentech, Radius Health, and Eli Lilly all unrelated to this work.
Author Contribution
Study supervision: DMC; Conception and design: DMC, MJC; Data acquisition, analysis, resources: DMC, MJC, KA, JAJ, ZL, NT, DO, SK, PK. All authors contributed to writing and approved manuscript.
References
- 1. Arvold ND, Lee EQ, Mehta MP, et al. Updates in the management of brain metastases. Neuro Oncol. 2016;18(8):1043–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stokum JA, Gerzanich V, Simard JM.. Molecular pathophysiology of cerebral edema. J Cereb Blood Flow Metab. 2016;36(3):513–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Klatzo I. Evolution of brain edema concepts. In: Brain Edema IX, et al. Acta Neurochirurgica, vol 60; 1994. Springer, Vienna. [DOI] [PubMed] [Google Scholar]
- 4. Liang D, Bhatta S, Gerzanich V, Simard JM.. Cytotoxic edema: Mechanisms of pathological cell swelling. Neurosurg Focus. 2007;22(5):E2–E2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bardutzky J, Schwab S.. Antiedema therapy in ischemic stroke. Stroke. 2007;38(11):3084–3094. [DOI] [PubMed] [Google Scholar]
- 6. Jha RM, Raikwar SP, Mihaljevic S, et al. Emerging therapeutic targets for cerebral edema. Expert Opin Ther Targets. 2021;25(11):917–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stokum JA, Kurland DB, Gerzanich V, Simard JM.. Mechanisms of astrocyte-mediated cerebral edema. Neurochem Res. 2015;40(2):317–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thrane AS, Rappold PM, Fujita T, et al. Critical role of aquaporin-4 (AQP4) in astrocytic Ca2+ signaling events elicited by cerebral edema. Proc Natl Acad Sci USA. 2011;108(2):846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shi Z, Zhang W, Lu Y, et al. Aquaporin 4-mediated glutamate-induced astrocyte swelling is partially mediated through metabotropic glutamate receptor 5 activation. Front Cell Neurosci. 2017;11(116). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vella J, Zammit C, Di Giovanni G, Muscat R, Valentino M.. The central role of aquaporins in the pathophysiology of ischemic stroke. Front Cell Neurosci. 2015;9:108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kitchen P, Salman MM, Halsey AM, et al. Targeting aquaporin-4 subcellular localization to treat central nervous system edema. Cell. 2020;181(4):784–799.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang B, Zador Z, Verkman AS.. Glial cell aquaporin-4 overexpression in transgenic mice accelerates cytotoxic brain swelling. J Biol Chem. 2008;283(22):15280–15286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Verkman AS, Smith AJ, Phuan PW, Tradtrantip L, Anderson MO.. The aquaporin-4 water channel as a potential drug target in neurological disorders. Expert Opin Ther Targets. 2017;21(12):1161–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Papadopoulos MC, Verkman AS.. Aquaporin water channels in the nervous system. Nat Rev Neurosci. 2013;14(4):265–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stumpf PK, Cittelly DM, Robin TP, et al. Combination of trastuzumab emtansine and stereotactic radiosurgery results in high rates of clinically significant radionecrosis and dysregulation of aquaporin-4. Clin Cancer Res. 2019;25(13):3946–3953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huber VJ, Tsujita M, Kwee IL, Nakada T.. Inhibition of aquaporin 4 by antiepileptic drugs. Bioorg Med Chem. 2009;17(1):418–424. [DOI] [PubMed] [Google Scholar]
- 17. Contreras-Zárate MJ, Ormond DR, Gillen AE, et al. Development of novel patient-derived xenografts from breast cancer brain metastases. Front Oncol. 2017;7(252). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boria AJ, Perez-Torres CJ.. Impact of mouse strain and sex when modeling radiation necrosis. Radiat Oncol. 2020;15(1):141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moore AH, Olschowka JA, Williams JP, Paige SL, O’Banion MK.. Radiation-induced edema is dependent on cyclooxygenase 2 activity in mouse brain. Radiat Res. 2004;161(2):153–160. [DOI] [PubMed] [Google Scholar]
- 20. Keep RF, Hua Y, Xi G.. Brain water content. A misunderstood measurement? Transl Stroke Res. 2012;3(2):263–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: An open-source platform for biological-image analysis. Nat Methods. 2012;9(7):676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Turnquist C, Beck JA, Horikawa I, et al. Radiation-induced astrocyte senescence is rescued by Δ133p53. Neuro Oncol. 2019;21(4):474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nicchia GP, Frigeri A, Liuzzi GM, Svelto M.. Inhibition of aquaporin-4 expression in astrocytes by RNAi determines alteration in cell morphology, growth, and water transport and induces changes in ischemia-related genes. FASEB J. 2003;17(11):1508–1510. [DOI] [PubMed] [Google Scholar]
- 24. Igarashi H, Huber VJ, Tsujita M, Nakada T.. Pretreatment with a novel aquaporin 4 inhibitor, TGN-020, significantly reduces ischemic cerebral edema. Neurol Sci. 2011;32(1):113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Topiramate (Topamax) FDA label. 2012; https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/020844s041lbl.pdf. Accessed December 27, 2021.
- 26. Kimbler DE, Shields J, Yanasak N, Vender JR, Dhandapani KM.. Activation of P2X7 promotes cerebral edema and neurological injury after traumatic brain injury in mice. PLoS One. 2012;7(7):e41229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Charlestin V, Fulkerson D, Arias Matus CE, et al. Aquaporins: New players in breast cancer progression and treatment response. Front Oncol. 2022;12:988119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moon CS, Moon D, Kang SK.. Aquaporins in cancer biology. Front Oncol. 2022;12:782829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fukuda AM, Adami A, Pop V, et al. Posttraumatic reduction of edema with aquaporin-4 RNA interference improves acute and chronic functional recovery. J. Cereb. Blood Flow Metab. 2013;33(10):1621–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hirt L, Fukuda AM, Ambadipudi K, et al. Improved long-term outcome after transient cerebral ischemia in aquaporin-4 knockout mice. J. Cereb. Blood Flow Metab. 2017;37(1):277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sucha P, Hermanova Z, Chmelova M, et al. The absence of AQP4/TRPV4 complex substantially reduces acute cytotoxic edema following ischemic injury. Front Cell Neurosci. 2022;16:1054919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Manley GT, Fujimura M, Ma T, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med. 2000;6(2):159–163. [DOI] [PubMed] [Google Scholar]
- 33. Blagosklonny MV. Cell senescence: Hypertrophic arrest beyond the restriction point. J Cell Physiol. 2006;209(3):592–597. [DOI] [PubMed] [Google Scholar]
- 34. Sylvain NJ, Salman MM, Pushie MJ, et al. The effects of trifluoperazine on brain edema, aquaporin-4 expression and metabolic markers during the acute phase of stroke using photothrombotic mouse model. Biochim Biophys Acta Biomembr. 2021;1863(5):183573. [DOI] [PubMed] [Google Scholar]
- 35. Poulsen CF, Simeone TA, Maar TE, et al. Modulation by topiramate of AMPA and kainate mediated calcium influx in cultured cerebral cortical, hippocampal and cerebellar neurons. Neurochem Res. 2004;29(1):275–282. [DOI] [PubMed] [Google Scholar]
- 36. Singh G, Rees JH, Sander JW.. Seizures and epilepsy in oncological practice: Causes, course, mechanisms and treatment. J Neurol Neurosurg Psychiatry. 2007;78(4):342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nicosia M, Miyairi S, Beavers A, et al. Aquaporin 4 inhibition alters chemokine receptor expression and T cell trafficking. Sci Rep. 2019;9(1):7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Papadopoulos MC, Saadoun S.. Key roles of aquaporins in tumor biology. Biochim Biophys Acta. 2015;1848(10 Pt B):2576–2583. [DOI] [PubMed] [Google Scholar]
- 39. Edamana S, Pedersen SF, Nejsum LN.. Aquaporin water channels affect the response of conventional anticancer therapies of 3D grown breast cancer cells. Biochem Biophys Res Commun. 2023;639:126–133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.