

Abstract
Background
Ependymoma (EPN) posterior fossa group A (PFA) has the highest rate of recurrence and the worst prognosis of all EPN molecular groups. At relapse, it is typically incurable even with re-resection and re-irradiation. The biology of recurrent PFA remains largely unknown; however, the increasing use of surgery at first recurrence has now provided access to clinical samples to facilitate a better understanding of this.
Methods
In this large longitudinal international multicenter study, we examined matched samples of primary and recurrent disease from PFA patients to investigate the biology of recurrence.
Results
DNA methylome derived copy number variants (CNVs) revealed large-scale chromosome gains and losses at recurrence in PFA. CNV changes were dominated by chromosome 1q gain and/or 6q loss, both previously identified as high-risk factors in PFA, which were present in 23% at presentation but increased to 61% at first recurrence. Multivariate survival analyses of this cohort showed that cases with 1q gain or 6q loss at first recurrence were significantly more likely to recur again. Predisposition to 1q+/6q− CNV changes at recurrence correlated with hypomethylation of heterochromatin-associated DNA at presentation. Cellular and molecular analyses revealed that 1q+/6q− PFA had significantly higher proportions of proliferative neuroepithelial undifferentiated progenitors and decreased differentiated neoplastic subpopulations.
Conclusions
This study provides clinically and preclinically actionable insights into the biology of PFA recurrence. The hypomethylation predisposition signature in PFA is a potential risk-classifier for trial stratification. We show that the cellular heterogeneity of PFAs evolves largely because of genetic evolution of neoplastic cells.
Keywords: chromosomal instability, ependymoma, prognosis, recurrence
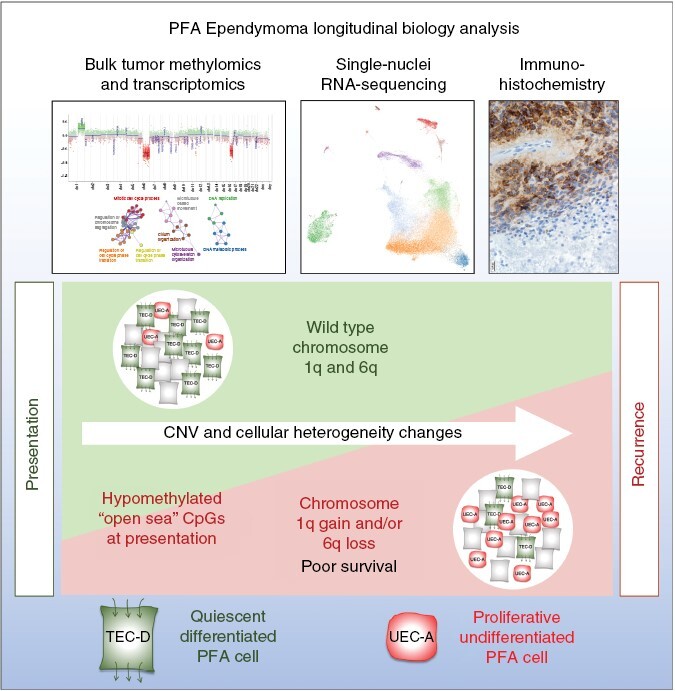
Graphical Abstract
Graphical Abstract.
Key Points.
High-risk CNVs are acquired at a significant rate at recurrence in PFA.
High-risk CNVs are associated with a proliferative, undifferentiated cellular phenotype in PFA.
Importance of the Study.
We present a large multicenter study of matched presentation and recurrent samples of ependymoma (EPN) posterior fossa group A (PFA). This study provides new information on the development of 1q gain and 6q loss at recurrence and the undifferentiated tumor biology associated with these CNVs. Data are presented indicating that DNA methylation status of PFA at presentation may predict those tumors which will develop 1q+ and/or 6q− at 1st recurrence, a potentially impactful finding that requires validation first in independent cohort methylation datasets and then in prospective trials. The dominance of high-risk 1q+/6q− CNVs at recurrence in PFA mandate that both these abnormalities be routinely tested for in clinical care and used for clinical trial stratification. To better inform such trials, future preclinical testing should be performed using available in vitro and in vivo 1q+/6q− PFA models.
Childhood ependymoma is characterized by a chemotherapy resistant phenotype, with approximately 50% of patients progressing after the current standard treatment of surgery and radiation therapy.1 Bulk genomic, transcriptomic, and methylomic analyses have been used to thoroughly investigate primary cases of ependymoma, identifying prognostic copy number variants (CNVs) and molecular subgroups.2 The commonest and most deadly childhood ependymoma type, posterior fossa group A (PFA), has been further characterized by single cell and spatial transcriptomic analyses, identifying a high level of cellular heterogeneity.3,4,5 Such heterogeneity includes diverse neuroepithelial and mesenchymal neoplastic subpopulations and a variety of infiltrating immune cell types.5,6 The most established prognostic biological factors in ependymoma are currently group-specific CNV variants, the foremost of these being gain of chromosome 1q (1q+) in PFA.7,8 Chromosome 6q loss (6q−) has not been as extensively studied as 1q+ but has recently been identified as an ultra-high-risk factor in PFA, that often co-occurs with 1q gain.9
An understanding of the evolutionary factors that drive progression is needed to improve the treatment strategies for PFA. Although the impact of CNVs and biology on outcome in PFA at initial presentation has been extensively studied, large studies on changes in CNVs and other biological characteristics at recurrence using matched presentation and recurrent tumor pairs have been lacking, often consisting of smaller patient cohorts due to the rarity of these specimens.4,6,10–12 However, over recent years, surgery targeting gross total resection is increasingly recognized as an important prognostic factor at relapse. This surgical approach, alongside first radiation or reirradiation is now standard of care at first recurrence.1,13–16 To provide a clearer understanding of the biology of recurrence in PFA, we charted genomic and cellular trajectories between presentation and subsequent recurrent tumors using a large international multi-institutional cohort.
Methods
Cohort Details
Matched primary and recurrence cohort tumor samples were obtained from multiple international institutions (Supplementary Table 1). Colorado and St Jude PFA samples used for methylation, bulk RNAseq, single nuclei RNAseq or immunohistochemistry were collected and snap-frozen in liquid nitrogen or formalin fixed paraffin embedded (FFPE) at the time of surgery with institutional consent (see Supplementary Material). Heidelberg methylation data were collected as part of a previous study.2 Samples from Nottingham, UK, were collected as part of two clinical trials (SIOP ependymoma I and SIOP 99-02) alongside ethically approved collection of retrospective ependymoma cases. CNS WHO grades were retrieved from diagnostic pathology reports that were compiled by each of the 4 collaborating institutions in this study, and no centralized retrospective CNS WHO grading was performed. Clinical variables were collected where available.
Methylomic Analysis
Methylomic analysis of DNA was performed using the Illumina 450K or 850K methylation array using DNA extracted from either snap frozen or FFPE samples. PFA molecular subgroups/subtypes and chromosomal copy number variants (CNVs) were obtained by uploading methylation data to the MolecularNeuropathology.org classifier. CpG methylation beta values were determined using RnBeads (http://rnbeads.mpi-inf.mpg.de/). Raw intensity data from Illumina 450K and 850K arrays were extracted using the ChAMP package (version 2.18.0) in R (version 4.1.0) for further processing. Quality control was performed on each sample using the ChAMP pipeline, and samples were excluded if they had a detection p-value > 0.05 or if they showed evidence of technical artifacts such as low signal intensity or high background noise. The raw data were then normalized and preprocessed using the functional normalization method implemented in ChAMP, which corrects for technical variation between arrays by adjusting for probe-dependent biases in the data. The resulting beta values were then adjusted for batch effects using ComBat to account for any systematic variation introduced by processing the arrays in different batches. The final beta values were then obtained by calculating the ratio of the methylated probe intensity to the overall intensity, with values ranging from 0 (completely unmethylated) to 1 (completely methylated).
Shared 450K and 850K CpG probes were compiled for downstream analyses and CpG loci were annotated for location within open sea, shelf, shore (subdivided into north and south shelf and shore) or island regions. Differentially methylated CpGs in 1q+ and/or 6q− predisposed samples (n = 24) versus those samples that were 1q/6q wildtype at presentation and 1st recurrence (n = 29) were analyzed to measure enrichment or reduction of CpG methylation in open sea, shelf, shore or island regions.
Survival Analysis
Univariate and multivariate modeling was used to assess the effect of age at diagnosis (<5 years), gender, WHO grade at presentation and 1st recurrence, extent of resection at presentation and 1st recurrence, 1q gain at presentation and 1st recurrence, 6q loss at presentation and 1st recurrence, and combined 1q gain and/or 6q loss on progression free survival and overall survival from presentation and 1st recurrence. For overall survival from presentation, we treated 1q gain/6q loss as time-dependent variables to model the effect the effect of CNV changes at recurrence.
Cytogenetics
Karyotype analysis was performed at the Colorado Genetics Laboratory (Aurora, CO) as described previously.17 Monolayer cell cultures in log-phase growth were harvested by standard cytogenetics methods after mitotic arrest with colcemid (0.05 mg/mL) for 4 hours and enzymatic dispersal with trypsin/EDTA. Cell suspensions were spotted onto microscope glass slides, and G-banding metaphase spreads images were acquired and analyzed. At least 20 metaphases were analyzed for each cell line, and karyotyping designation followed the International System for Human Cytogenetic Nomenclature.18
Single-Nuclei RNA-Sequencing
Single nuclei were isolated from 14 (7 matched primary/recurrent pairs) PFA frozen samples using CHAPS-based buffer protocol as previously described by Slyper et al.19 Single-nuclei RNA-sequencing (snRNAseq) was performed using the 10xGenomics Chromium platform as described previously.3 Processing and filtering of sequencing data resulted in 52,020 nuclei across all samples. After normalization, these nuclei were clustered using the Seurat workflow. We applied Harmony alignment (Theta = 2) to correct for inter-sample variation due to experimental or sequencing batch effects.20 Coarse nucleus types (tumor, immune, myeloid, and stroma cells) were assigned based on marker gene expression and single-nuclei CNV profiling. Single nuclei CNVs were inferred from snRNAseq data based on average relative expression in variable genomic windows containing 101 genes using inferCNV. Nuclei classified as non-neoplastic based on gene expression were used to define a baseline of normal karyotype such that their average copy number value was subtracted from all nuclei. Differential expression and marker gene identification was performed using Presto.21
Neoplastic subpopulations were characterized by direct examination of subpopulation gene lists and by comparison with previously defined PFA neoplastic subpopulations.5 Overlap between single-nuclei clusters and existing neoplastic subpopulation marker genelists was measured by hypergeometric enrichment analysis.
Bulk RNAseq
RNA was isolated from snap frozen PFA samples and sequenced using the Illumina NovaSeq 6000. Sequencing data were aligned using STAR and quantification of RNA expression, as fragments per kilobase per million (FPKM), was derived by Cufflinks as described previously.5 Differential gene expression analysis was performed between samples that were grouped according to CNV status then subjected to gene ontology biological process GOterm enrichment analyses using Metascape and Cytoscape.22,23 Single sample geneset enrichment analysis (ssGSEA) was performed on individual sample RNAseq FPKM data using the GenePattern portal.24,25
Immunohistochemistry
Standard immunohistochemistry was performed for H3K27me3 (Cell Signaling Technology; clone C36B11; 1:200), Ki-67 (Neomarkers/Thermo Scientific; clone SP6; 1:500), and COL9A2 (Thermo PA5-63286; polyclonal; 1:50) and counterstained with hematoxylin as described previously.5,33 COL9A2 antibody was previously selected and validated as a UEC-A IHC marker based on differential expression analysis of scRNAseq and spatial transcriptomic data, and correlation of staining in FFPE sections with transcript levels in a cohort of PFA patient samples.3,5 Blinded immunohistochemical scoring of H3K27me3 (R.J.C and T.R.R), Ki-67 and COL9A2 (N.W.) were performed using positive/negative staining, positive cell count of the highest high-powered field and a subjective scoring scale (range 0–4), respectively.
Statistics
Statistical analyses were performed using R, Prism (GraphPad), and Excel (Microsoft) software.
Results
Incomplete Resection and WHO Grade 3 at 1st Recurrence are Significant Predictors of Shorter Survival in Recurrent PFA
Ninety-five children with recurrent posterior fossa group A ependymoma (PFA) were included in the recurrence cohort. All samples were defined as PFA based on DNA methylation array classification (molecularneuropathology.org). A subset of presentation/1st recurrence pairs were also assessed for absence of H3K27me3 by immunohistochemistry (IHC), a diagnostic marker of PFA,27 revealing that the H3K27 trimethyl mark remained absent at recurrence in the majority of pairs (11/12). Median overall survival (OS) from presentation (n = 88) was 69 months (Supplementary Table 2). Median progression free survival (PFS) from presentation was 24 months (n = 87) and from first recurrence was 16.6 months (n = 61).
The prognostic association of clinical variables (Supplementary Table 2) was assessed using multivariate analysis that included modeling for the variable effect of factors such as CNV status and tumor grades which often changed at recurrence. Consistent with previous findings,16 extent of resection at 1st recurrence was significantly associated with PFS and OS (Supplementary Table 3). Patients with subtotally resected tumors had a 3.1-fold (95% CI: 1.2–7.7) higher risk of progression and 4.1-fold (95% CI: 1.4–11.6) higher risk of death than patients with gross total resection (GTR). GTR at presentation was paradoxically associated with a shorter PFS and OS from 1st presentation compared to those patients that received an initial subtotal resection, suggesting that those patients that relapse after GTR harbor more clinically aggressive disease than those that likely recurred due to incomplete resection. PFAs were classified as WHO grade 3 in 51% at presentation, which significantly increased to 69% at 1st recurrence (Fisher’s exact test p = 0.0005). CNS WHO grade 3 tumors at 1st recurrence were significantly associated with shorter OS, having a 5.7-fold (95% CI: 1.8–18.2) risk of death from 1st recurrence.
Chromosome 1q Gain and/or 6q Loss in PFA Show Increased Prevalence from Diagnosis to First Recurrence and is Associated with Excess Mortality
DNA methylation was used to measure individual chromosomal copy number variants (CNVs), which were then averaged for each occurrence: presentation (n = 76), 1st recurrence (n = 72), second recurrence (n = 15), and combined subsequent recurrences including autopsy material (n = 13) (Supplementary Table 4). At presentation the study samples had comparable CNV profiles to a larger published cohort of PFA from presentation (n = 240)2 with no significant difference in number of CNVs per sample (Student’s t-test p = 0.36) (Figure 1A). Progressively increasing proportions of chromosome gains and losses were observed in successive recurrences, increasing from an average of 2.0 CNVs per sample at presentation to 4.2 CNVs per sample at 1st recurrence (p = 0.0042) and 6.7 CNVs per sample at 2nd recurrence (p = 0.00035). Consequently, samples with balanced chromosomal copy numbers reduced from 58% at presentation to 24% at 1st recurrence and <10% at later recurrences (Figure 1A). The dominant CNVs in the Pajtler cohort were gain of 1q (1q+) and loss of 6q (6q−), which matched the CNV profile of presentation samples in the study cohort.
Figure 1.
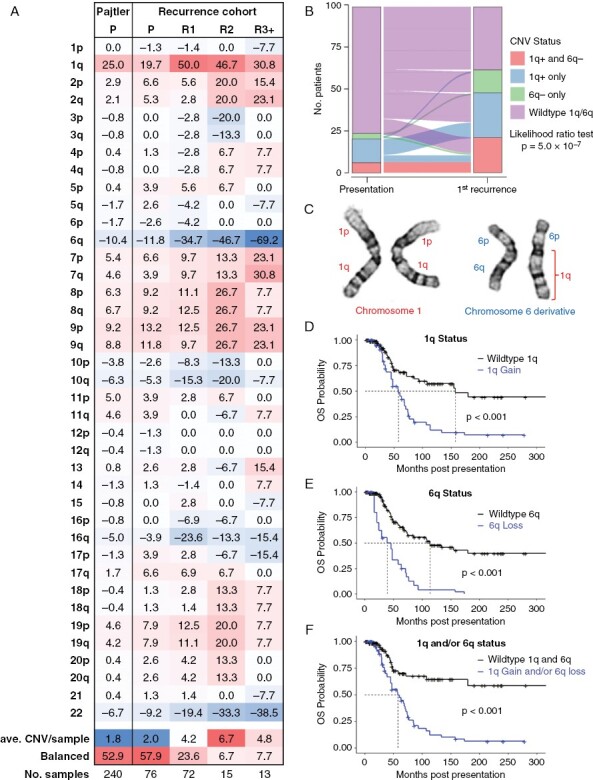
Chromosome 1q gain and/or 6q loss in PFA show increased prevalence at recurrence and are associated with a high rate of mortality. (A) Average copy numbers for all chromosome arms at presentation (P), 1st recurrence (R1), 2nd recurrence (R2) and subsequent recurrences and autopsy (R3+) and a publicly available cohort of presentation PFA samples published by Pajtler et al.2 (Pajtler P). The average CNV per sample and percentage of patients with a balanced genome are included. (B) Sankey plot showing changes in (i) 1q+ and 6q− co-occurrence (1q+ and 6q−), (ii) 1q+ with wildtype 6q (1q+), (iii) 6q− with wildtype 1q (6q−) and (iv) wildtype 1q/6q, between presentation and 1st recurrence. (C) example of translocation of chromosome 1q and 6q in UPN-811-R3, resulting in derivative chromosome 6 with one copy of 6q replaced with 1q. Overall survival from presentation for (D) 1q+,(E) 6q−, and (F) combined 1q+ and/or 6q− in the recurrence cohort (n = 88) with CNVs considered as time dependent variables to incorporate CNV changes at recurrence.
Strikingly, both 1q+ and 6q− proportions were particularly altered in matched recurrent samples, with 1q+ increasing from 20% at presentation to 50% at 1st recurrence (McNemar’s chi-squared test with continuity correction p = 1.1 × 10−5) and 6q− from 12% to 35% (p = 3.8 × 10−8) (Figure 1A,B). Co-occurrence of 1q+ and 6q− in individual samples was a common event, particularly at recurrence (Figure 1B) for each of the four institutional cohorts (Supplementary Figure 1), which included cases that had 1q+ at presentation losing 6q at recurrence (Figure 1B). The cumulative proportion of 1q+ and/or 6q− and vice versa was 23% at presentation, increasing to 61% at 1st recurrence (likelihood ratio test p = 0.0001 with a mixed multinomial logic model). Consistent with the general increase in CNVs at recurrence in the entire cohort, additional CNVs at 1st recurrence were observed in a subset (11/16) of those cases with 1q+ and/or 6q− at presentation. Of these 10q loss was the most frequent CNV (5/11), implicating tumor suppressor PTEN loss as an important factor in PFA tumorigenesis. The impact of upfront radiation on development of 1q and 6q CNVs was assessed, showing no significant difference in the frequency of these CNVs at recurrence in those that received radiation versus those that did not. Karyotype analysis of 1q+/6q− co-occurrence, in 2 of 2 assessable cases, was a result of translocation between the long arms of chromosomes 1 and 6, resulting in a net gain of 1q and loss of 6q (Figure 1C).17
Both 1q+ and 6q− have been established as high-risk markers in PFA ependymoma at presentation.7–9 As previously reported by Baroni et al.9 1q+ at presentation was associated with a significantly higher rate of metastatic recurrence (54%) compared to 1q wildtype (15%) (Fishers exact test (two-sided) p = 0.0056), whereas no significantly different rate was seen between 6q− and 6q wildtype. Neither 1q+ nor 6q− at recurrence was associated with metastasis at recurrence. Multivariate survival analysis was used to determine the prognostic impact of 1q+ and 6q− at presentation and 1st recurrence in the recurrence cohort using a temporal variable effect model that considers CNV changes at recurrence. In some instances, outcome associations for 1q+ and 6q− did not show an impact on outcome (Supplementary Table 3; Supplementary Figure 2) possibly due to (i) the poor baseline outcome of the recurrence cohort, and (ii) individualization of 1q+ and 6q− outcome associations creating comparisons where the 1q wildtype includes high risk 6q− samples and vice versa, thus comparing high-risk factors. Despite this, consistent associations of 1q+ and 6q− with overall survival were observed, both individually, and more significantly in poor combination. By multivariate analysis 1q+ and 6q− were associated with a 4.3-fold (95% CI: 1.8–10.4) and 4.2-fold (2.0–9.0) increased risk of death, respectively (Figure 1D,E; Supplementary Table 3; Supplementary Figure 2). Combined 1q+ and/or 6q− showed a particularly high impact on outcome with a 6.1-fold (2.6–14.0) greater risk of death (Figure 1F, Supplementary Table 3, Supplementary Figure 3) emphasizing the impact of these high-risk CNVs at presentation and in the increased instances at recurrence.
Presentation PFA Samples Predisposed to 1q+ and/or 6q− are Hypomethylated in Heterochromatin-associated Open Sea CpG Regions
PFA have been subclassified into two major molecular groups, PFA1 and PFA2, and subtypes therein based on methylation profiling,28 where subtype PFA1c was shown to harbor the highest frequency of 1q+ and 6q− at presentation. We examined changes in PFA subtypes at 1st recurrence, which revealed classification changes within, but never between PFA1 and PFA2 methylation group cases (Figure 2A), suggesting subtypes within PFA1 and PFA2 are impacted by biological changes including CNV changes associated with recurrence. While 1q+ and 6q− CNVs are largely restricted to PFA1c at presentation, these CNVs became broadly distributed across subtypes at recurrence (Figure 2B), with 1q+ being significantly increased from 9% at presentation to 52% at 1st recurrence in PFA2 subtypes (Fisher’s exact test p = 0.0031).
Figure 2.
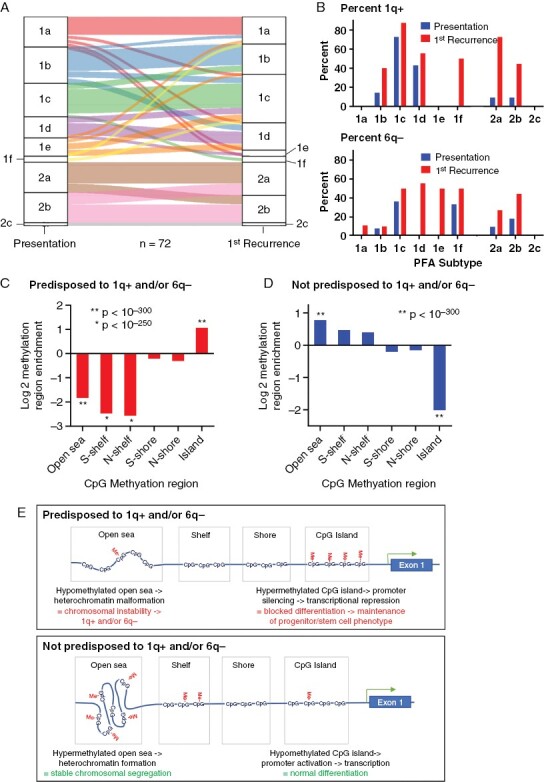
PFA methylomic subtype changes at recurrence and 1q+ and/or 6q- predisposition methylation characteristics. (A) Sankey plot showing changes in PFA methylation subtype between presentation and 1st recurrence. (B) DNA proportion of 1q+ (top panel) and 6q− cases (bottom panel) per subtype at presentation and 1st recurrence. (C) Enrichment (hypergeometric analysis) of methylated CpGs in open sea, shelf, shore (subdivided into north and south shelf and shore) and islands in CpGs hypermethylated in presentation samples predisposed to 1q+ and/or 6q− samples versus not predisposed to 1q+ and/or 6q− (with wildtype 1q and 6q at presentation and recurrence) and (D) CpG hypermethylated in presentation samples not predisposed to 1q/6q changes versus 1q+ and/or 6q− predisposed. (E) Schema depicting hypothetical consequences of differential CpG methylation (Me) patterns in open sea, shelf, shore and islands in presentation samples not predisposed to 1q/6q CNVs versus 1q+ and/or 6q− predisposed samples.
Given the high proportion (~40%) of samples that gained 1q and/or lost 6q at recurrence, we sought to identify biological markers that predisposed tumors for the chromosomal instability observed in this high-risk group. Chromosomal instability has been correlated with aberrant DNA methylation, specifically hypomethylation of “open sea” CpGs regions that are associated with heterochromatin.29–31 Open sea CpGs are thus named due to their location in the genome outside of CpG islands, shores and shelves. The latter CpG regions are defined based on their relationship to genes, where CpG islands correspond to gene promotor regions, CpG shores are ~2 kb from CpG islands, and CpG shelves are ~4 kb from CpG islands. We compared CpG methylation in presentation samples that were predisposed to 1q gain and/or 6q loss at 1st recurrence (n = 24) to those specimens with wildtype 1q and 6q at both presentation and recurrence (n = 29). Hypergeometric enrichment analysis of CpGs with higher methylation beta values (p < 0.05) in 1q+ and/or 6q− predisposed versus the unchanged samples revealed a significantly reduced proportion of methylated CpGs located in open sea regions (p < 10−300) and enrichment of CpGs island site methylation (p < 10−300) and (Figure 2C). The converse analysis showed that CpGs with increased methylation in those specimens with no 1q/6q abnormalities at presentation or recurrence were enriched for open sea CpG methylation and decreased for CpG island methylation (Figure 2D). These findings suggest that open sea CpG hypomethylation predisposes PFAs to 1q+ and 6q− CNVs (Figure 2E).
Single Cell Analysis of CNVs in Matched Primary and Recurrent PFA Show No Evidence of Rare Recurrence-Associated CNV Subclones at Presentation
The increase in copy number aberrations in PFA recurrences suggest that rare subclones containing recurrence associated CNVs may exist at presentation. We addressed this question by performing single-nuclei RNAseq CNV analysis of 52,020 cells from 14 matched patient PFA samples from presentation and 1st recurrence, each with CNV changes at recurrence. Cell gene-expression matrices were projected as 2D uniform manifold approximation and projection (UMAP) plots, revealing multiple clusters of cells that were shared between samples (Figure 3A). Cluster gene expression profiles (Supplementary Data 1) were used to define the cellular identity of clusters, identifying subpopulations of neoplastic and non-neoplastic cells by comparison with a prior scRNAseq study by our laboratory3 (Figure 3A; data from Supplementary Figure 4).
Figure 3.
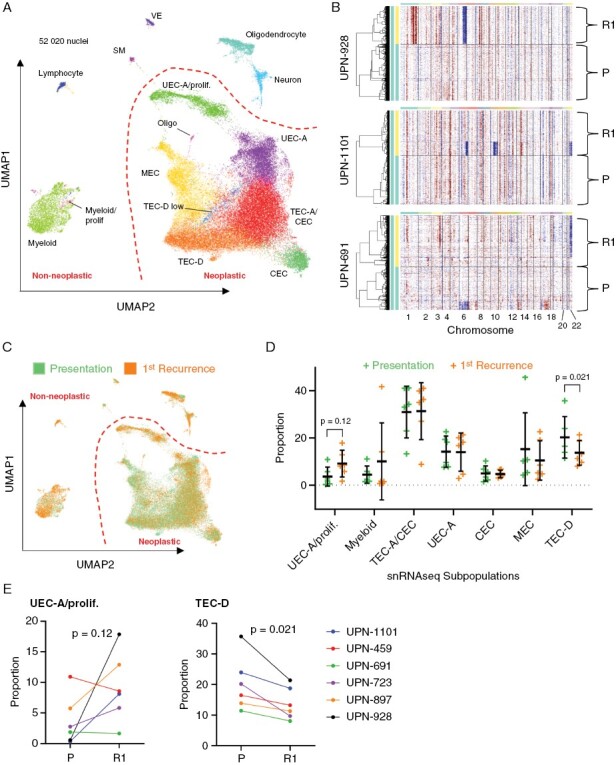
Single-nuclei RNAseq analysis of subclone and neoplastic subpopulation trajectories from presentation to recurrence in PFA. (A) Aligned UMAP projection of single-nuclei expression data of 7 PFA patient samples pairs from presentation and 1st recurrence reveals neoplastic and non-neoplastic cell type clusters (Abbreviations: CEC, ciliated EPN cell; MEC, mesenchymal EPN cell; SM; smooth muscle; TEC, transportive EPN cell; UEC, undifferentiated EPN cell; VE, vascular endothelium). (B) Inference of CNVs (inferCNV) in neoplastic single nuclei in three representative PFA presentation (P)/1st recurrence (R1) pairs, exemplifying gain of 1q and loss of 6q at recurrence (top panel), subclonal loss of 6q, 10q, and 22 at recurrence (middle panel), and loss of 22 at recurrence (bottom panel). (C) tumor stage overlaid on to aligned UMAP projection of PFA single nuclei. (D) Major neoplastic and non-neoplastic subpopulation proportions presentation and 1st recurrence samples. (E) ladder plot depicting individual sample changes in the UEC-A/proliferative (left panel) and TEC-D ependymally differentiated (right panel) neoplastic cluster proportion between presentation and 1st recurrence.
Single cell CNV inference analysis (inferCNV) identified copy number gain and loss signatures in individual neoplastic cells from 12 of the 14 samples that had an evaluable neoplastic component. Single cell CNV profiles were concordant with those measured by CNV analysis of methylation data from corresponding bulk tumor samples (Supplementary Figure 5). Each of the 6 PFA presentation/1st recurrence pairs revealed changes in chromosome copy number, with the commonest change being loss 6q at recurrence (5/6), two of which also gained 1q (Figure 3B; Supplementary Figure 5). A single sample lost chromosome 22 at recurrence. Evidence of CNV altered subclones were identified at presentation (1/6) and recurrence (2/6) (Figure 3B). However, no evidence of minor subclones of recurrence-associated CNV signatures were identified in any of the matched presentation samples, either by visual examination or by clustering analysis.
Neoplastic Cellular Subpopulations Evolve to a Less Differentiated Phenotype at Recurrence
Single nuclei RNAseq analysis provides a direct measure of the evolution of neoplastic and non-neoplastic cellular subpopulations between presentation and recurrence. We annotated neoplastic and non-neoplastic clusters based on recent scRNAseq studies performed by our laboratory and others3,4 and gene set enrichment analysis (Supplementary Data 2). Neoplastic clusters harbored gene-expression profiles that correlated with each of the previously defined major neoplastic PFA subpopulations that correspond to undifferentiated ependymoma cells (UEC); ependymally differentiated transportive (TEC) and ciliated (CEC) ependymoma cells; and mesenchymal differentiated (MEC) ependymoma cells (Figure 3A; Supplementary Figure 4). More recent spatial transcriptomic analysis identified further insights into neoplastic subpopulations, showing heterogeneity within the PFA tumor microenvironment that was broadly divided into neuroepithelial and mesenchymally differentiated zones.5 Neuroepithelial zones contained subpopulations with differing levels of ependymal differentiation: including a proliferative, undifferentiated neuroepithelial progenitor-like cell (UEC-A) that was predictive of recurrence in PFA, and a highly ependymally differentiated subpopulation (TEC-D) that was more abundant in non-recurrent PFAs.5 Accordingly, Single-nuclei RNAseq identified subpopulations corresponding to both of UEC-A and TEC-D (Figure 3A; Supplementary Figure 4). The most abundant non-neoplastic subpopulation was myeloid cells (CD33, ITGAX(CD11c), TYROBP) that included a small proliferating subpopulation. Rarer non-neoplastic subpopulations included lymphocytes (CD3D, CD3E, GZMH), oligodendrocytes (MAG, OLIG1, OLIG2), neurons (GABRA1, GABRA2, NEUROD1), vascular endothelium (VWF, ERG, PECAM1) and smooth muscle (CALD1, TPM1).
We observed that neoplastic and non-neoplastic subpopulation proportions were altered between presentation and 1st recurrence (Figure 3C,D). The largest change in proportion was in the neoplastic UEC-A/proliferative subpopulation that was on average 2.48-fold higher at 1st recurrence than presentation, increasing in 4/6 cases (Figure 3E) with a marginal p-value of 0.12. Conversely, the ependymally differentiated TEC-D subpopulation was significantly decreased at recurrence in all cases (1.48-fold, p = 0.021) (Fig 3D,E). Collectively, these subpopulation changes suggest that PFA undergo a process of de-differentiation between presentation and recurrence.
Co-occurrence of Chromosome 1q Gain and 6q Loss is Associated with a Particularly Undifferentiated Proliferative Cellular Phenotype
The increased frequency of 1q+/6q− co-occurrence in recurrent PFA and associated mortality warrant development of therapy to target this specific molecular group. We therefore sought to characterize the biology of 1q+/6q− PFAs using bulk RNAseq transcriptomic analysis of a cohort PFA surgical samples (n = 72). This clinically and molecularly annotated cohort was obtained from presentation samples and subsequent recurrences with documented CNVs obtained from matched methylomic data, identifying samples with 1q+ and/or 6q− (n = 26) and with wild type 1q and 6q (n = 46) (Supplementary Table 4). Genes differentially expressed between these two groups were examined for enrichment of Gene Ontology (GOterm) biological process genesets, revealing a dominance of cell cycle-related gene functions in 1q+ and/or 6q− samples (Figure 4A; Supplementary Data 3), that included CENPF, NEK2, and ASPM. To examine the impact of 1q+ and 6q− co-occurrence versus the impact of each CNV individually we performed single-sample gene set enrichment analysis (ssGSEA) for the most enriched GOterm geneset “mitotic cell cycle process.” This showed that 1q+/6q− co-occurrence (n = 12) had a significantly higher enrichment of cell division GOterm genes than 1q/6q wildtype samples (n = 46), whereas 1q+ (n = 10) and 6q− (n = 4) individually were not significantly higher than wild type (Figure 4B). Conversely GOterm biological process genesets enriched in wildtype versus 1q+ and/or 6q− samples were associated with gliogenesis (NTRK2, OLIG1, OLIG2) and cell adhesion (AQP4, NCAM2), suggesting a more differentiated phenotype in 1q/6q wild type tumors (Figure 4C). SsGSEA showed that 1q+/6q− co-occurrence, but not 1q+ or 6q− individually, had a significantly lower enrichment of GOterm “gliogenesis” genes (Figure 4D; Supplementary Data 3).
Figure 4.
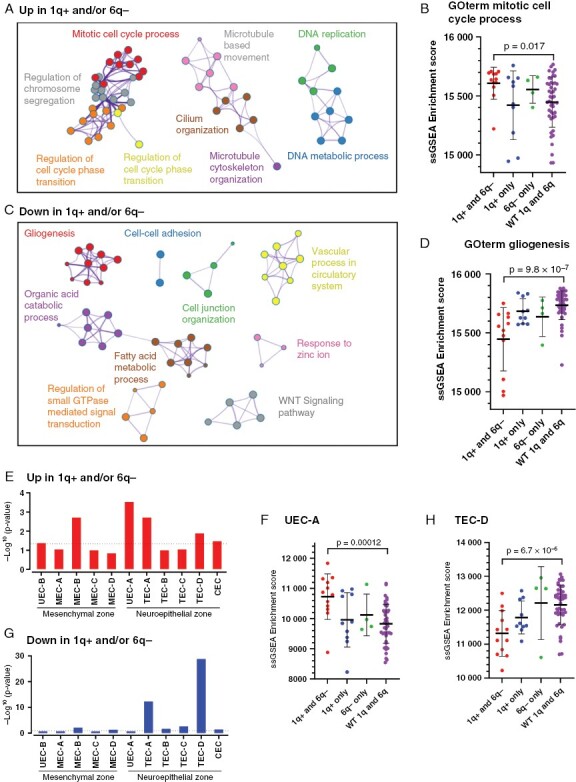
Co-occurrence of chromosome 1q gain and 6q loss is associated with an undifferentiated proliferative cellular phenotype in PFA. (A) Cytoscape clustering of gene ontologies enriched in genes overexpressed in PFA with 1q+ and/or 6q− (n = 26) versus those with wildtype 1q/6q (n = 46). (B), single sample gene set enrichment analysis (ssGSEA) of top 1q+ and/or 6q− PFA associated GOterm mitotic cell cycle process in individual patient samples grouped according to (i) 1q+ and 6q− co-occurrence (1q+ and 6q−; n = 12), (ii) 1q+ with wildtype 6q (1q+ only; n = 10), (iii) 6q− with wildtype 1q (6q− only; n = 4), and (iv) wildtype 1q and 6q (WT 1q and 6q; n = 46). C, Ontology clustering of genes underexpressed in 1q+ and/or 6q−, and (D) individual sample enrichment of GOterm gliogenesis grouped according to CNV status. (E) Overexpressed genes in 1q+ and/or 6q− PFA were compared to previously identified PFA neoplastic cluster marker genesets5 (top 50) by hypergeometric analysis. (F) Deconvolution of individual patient samples for UEC-A (undifferentiated EPN cell-A) neuroepithelial progenitor subpopulation using ssGSEA. (G) Corresponding neoplastic cluster gene enrichment analysis for genes underexpressed in 1q+ and/or 6q− PFA. (H) Deconvolution of individual patient samples for differentiated TEC-D (transportive EPN cell-D) subpopulation using ssGSEA.
We next estimated the relative proportion of neoplastic subpopulations in bulk RNAseq data by deconvolution with subpopulation-specific genesets previously defined by single cell and spatial transcriptomic analyses.3,5 Consistent with snRNAseq findings, deconvolution showed that proliferative neuroepithelial progenitor UEC-A was the most enriched subpopulation 1q+ and/or 6q− samples (Figure 4E), and by ssGSEA was highest in 1q+/6q− co-occurrence samples (Figure 4F). Wild type 1q/6q showed a significant enrichment of TEC-D subpopulation signature genes (Figure 4G), that was particularly low in 1q+/6q− co-occurrence samples (Figure 4H).
To confirm the association of cell cycle-related gene enrichment with 1q+ and/or 6q− we performed IHC for proliferation marker Ki-67 in a cohort of PFA cases from presentation and recurrences for which FFPE material was available (n = 70). Ki-67 expression was typically higher in hypercellular areas commonly seen in samples with 1q+ and/or 6q− compared to hypocellular areas seen in those samples with wildtype 1q/6q (Figure 5A) and those cases with 1q+ and 6q− co-occurrence showed significantly higher Ki-67+ cell numbers (per highest high-powered field) than wildtype 1q/6q (Figure 5B). Consistent with this, we observed a significant association of 1q+ and/or 6q− with WHO grade 3 tumor histology in samples from all tumor stages in the recurrence cohort (n = 152) (Figure 5C). COL9A2 was previously identified as an IHC marker of UEC-A that was shown to be coexpressed with Ki-67 on a cellular level.5 We used COL9A2 in the present study to confirm the increased proportion of UEC-A in 1q+ and/or 6q− cases inferred by bulk transcriptomic analyses. We demonstrated increased expression of COL9A2 in hypercellular areas as observed for Ki-67 (Figure 5D) and a significantly higher expression of COL9A2 in samples with 1q+/6q− co-occurrence and 1q+ alone when compared to samples with wildtype 1q and 6q (Figure 5E). These immunohistochemical findings are consistent with bulk transcriptomic deconvolution data that together reveal enrichment of high-risk proliferative progenitor UEC-A in 1q+/6q− PFAs.
Figure 5.
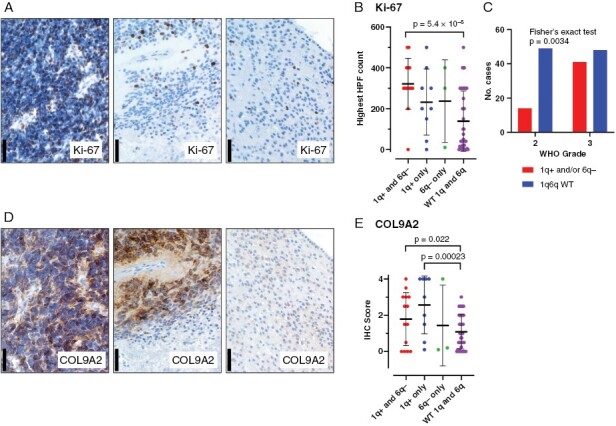
Histological characteristics of 1q+ and/or 6q− samples with respect to proliferative marker Ki-67 and UEC-A progenitor subpopulation marker COL9A2. (A) Representative IHC staining of proliferative marker Ki-67 in 1q+/6q− co-occurrence (left panel), 1q+ only (middle panel) and 1q/6q wildtype PFA (scale bars = 50 µm). (B) Ki-67 positive cell count per highest high power field (HPF) in individual patient samples grouped according to (i) 1q+ and 6q− co-occurrence (1q+ and 6q−; n = 14), (ii) 1q+ with wildtype 6q (1q+ only; n = 9), (iii) 6q− with wildtype 1q (6q− only; n = 3), and (iv) wildtype 1q and 6q (WT 1q and 6q; n = 27). (C) Contingency analysis of CNVs status versus WHO grade (n = 152) (Fisher’s exact test). (D) Representative IHC staining of proliferative neuroepithelial UEC-A subpopulation marker COL9A2 in 1q+/6q− co-occurrence (left panel), 1q+ only (middle panel) and 1q/6q wildtype PFA. (E) COL9A2 IHC scores in individual patient samples grouped according to CNV status as in panel B.
Discussion
This is currently the largest study of matched presentation and relapse samples to examine molecular changes at recurrence in PFA. The ability to perform this study was facilitated by surgery at first recurrence becoming a standard of care intervention for PFA ependymoma.14 We have identified striking increases in CNVs in PFA patients that fail initial standard of care treatment. Foremost amongst these chromosomal changes were gains of 1q and loss of 6q, often co-occurring, and these conferred a worse survival in this cohort of patients that failed standard therapy.
Hypomethylation of heterochromatin-associated open sea regions was observed in those PFA that subsequently acquired 1q/6q CNVs at recurrence. Heterochromatin-associated open sea CpGs are hypermethylated in normal cells, assisting in formation of heterochromatin by DNA condensation.30 Open sea hypomethylation, and associated heterochromatin malformation, has previously been identified as a driver of chromosomal instability in cancer29,31,32 and potentially explains the chromosomal instability seen in PFAs. Of potential relevance to PFA, hypomethylation of pericentromeric heterochromatin regions in 1q and 16q has been previously correlated with in 1q/16q translocations resulting in 1q+ and 16q− in childhood Wilms tumors.31 Conversely, CpG island hypermethylation (CIMP), as seen in the 1q+ and/or 6q− predisposed samples, is a common event in cancer and was previously implicated in the biology of posterior fossa ependymoma where it was shown to block differentiation via silencing of the PRC2 complex.26 A subsequent epigenetic study revealed DNA hypomethylation outside of GpG islands in all PFA compared to PFB,27 matching the methylation profile seen in 1q+ and/or 6q− predisposed PFA in the present study, and hinting that PFA as a whole may be predisposed to development of CNVs. Identification of open sea CpG hypomethylation in PFA as a potential marker of predisposition for subsequent 1q/6q translocations in PFA further supports the potentiating impact of DNA methylation aberrations in chromosomal instability, and adds a further dimension to the role of aberrant DNA methylation in the pathobiology of PFA (Figure 2E).
We used single nuclei RNAseq analysis to measure CNV clonality at presentation and recurrence which showed that in all cases assessed, rare recurrence-associated CNVs were not identified at presentation. Collectively, these findings support a hypothesis that recurrence CNV clones arise at some time after initial resection and radiation therapy, in those samples that have a predisposition to chromosomal instability. Given current treatment protocols in both North America and Europe, it is also likely that all, or almost all, of the patients in our study also received first radiation or reirradiation as part of their recurrence treatment, in addition to surgery. However, the retrospective nature of our cohort precludes determination of the impact of particular types of therapy on recurrence changes and this remains an important question for future research.
Given the known abysmal survival associated with 1q+/6q− PFAs highlighted by this study and others,7,9 it is crucial that patients with these copy number changes are treated with experimental therapy as part of clinical trials. The frequency of high-risk 1q+ and/or 6q− CNVs acquired at recurrence provides an important biological factor for stratification of these trials. The high prevalence of these specific CNVs at recurrence had previously been underappreciated, largely due to infrequent testing, particularly for 6q. In our opinion, these CNVs should now be routinely assessed by DNA methylation analysis, which, through the European BIOMECA study, has been shown to be an appropriate approach for copy number analysis in a prospective ependymoma clinical trial setting.33 Although methylomic analysis is not part of routine procedure for the majority of treating centers, this diagnostic modality is becoming more widely used, and centralized methylomic workup is currently offered by a number of larger institutions in the US and Europe. Future, ideally prospective studies, are needed to establish heterochromatin-associated DNA methylation analysis as an upfront predictor of PFAs predisposed to chromosomal instability, that could be either stratified with 1q+ and/or 6q− PFAs or to an additional high-risk study arm. Preclinical studies in PFA would also benefit by testing in appropriate 1q+/6q− high-risk models. To this end, our group has recently established both in vitro and in vivo models of PFA with both 1q+ and 6q− (MAF-811 and MAF-928) that are freely available to the research community.17,34 These models have already be utilized for preclinical in vitro screening of FDA-approved oncology drugs for PFA selective efficacy35 and for in vivo validation of select compounds such as metformin.36
The present study advances preclinical efforts by providing insights into the specific biological characteristics of PFA with 1q+ and/or 6q− and how this relates to the biology of recurrence in PFA. We utilized single-nuclei and bulk transcriptomic datasets to identify a proliferative phenotype in cases with 1q+ and/or 6q− that was confirmed by protein analysis in a cohort of FFPE samples. Consistent with this finding, we found that 1q+ and/or 6q− was associated with WHO grade 3 PFA, and a diagnosis of WHO grade 3 tumor PFA at 1st recurrence was associated with a shorter overall survival, likely reflecting the underlying impact of 1q+ and/or 6q− on outcome. Accompanying the more proliferative phenotype in 1q+ and/or 6q− PFA was an increased proportion of the undifferentiated UEC-A neoplastic subpopulation that was recently identified using single-cell and spatial transcriptomic studies.3,5 UEC-A were previously shown to be more abundant in presentation PFA samples that later progressed versus those that were cured,5 and in the present study we show that UEC-A is often further increased at recurrence. The higher proportion of UEC-A at presentation in those tumors that subsequently recur relates to the impact of minimal residual disease as a driver of recurrence in PFA. We also speculate that UEC-A are radio- and chemotherapy resistant due to interplay between this specific cellular phenotype and an underlying loss of tumor suppressor gene(s) located on chromosome 6q. The association of UEC-A with 1q+ and/or 6q− was identified by both snRNAseq and deconvolution of bulk transcriptomic data, which was then confirmed by IHC scoring of COL9A2 expression in FFPE material, a test with potential clinical utility in the identification of high-risk PFA. Consistent with the increased proportion of cells with an undifferentiated phenotype, 1q+ and/or 6q− PFA had reduced proportions of the differentiated and relatively quiescent TEC-D neoplastic subpopulation that has been associated with a favorable clinical outcome.5 These studies reveal that the cellular heterogeneity of PFAs evolves largely because of genetic evolution of neoplastic cells, where 1q+ and/or 6q− skew the cellular phenotype of PFA to a more treatment-resistant, undifferentiated proliferative phenotype.
The high prevalence of 1q+/6q− at recurrence and the associated shortened survival mandates that preclinical therapeutics should be tested using 1q+/6q− PFA models, that these chromosomal abnormalities should be routinely tested for and used for trial stratification. Patients with 1q+/6q− at recurrence need experimental therapies, delivered through clinical trials, targeted at the underlying biology of this particularly aggressive type of PFA ependymoma.
Supplementary Material
Acknowledgements
The authors appreciate the contribution to immunohistochemical studies made by E. Erin Smith, HTL (ASCP) CMQIHC of the University of Colorado Denver Histology Shared Resource.
Contributor Information
Andrew M Donson, Department of Pediatrics, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA; Morgan Adams Foundation Pediatric Brain Tumor Research Program, Children’s Hospital Colorado, Aurora, Colorado, USA.
Kelsey C Bertrand, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Kent A Riemondy, RNA Biosciences Initiative, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA.
Dexiang Gao, Department of Pediatrics, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA; University of Colorado Cancer Center Biostatistics and Bioinformatics Shared Resource, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA.
Yonghua Zhuang, Department of Pediatrics, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA; University of Colorado Cancer Center Biostatistics and Bioinformatics Shared Resource, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA.
Bridget Sanford, Department of Pediatrics, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA.
Gregory A Norris, Department of Pediatrics, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA; Morgan Adams Foundation Pediatric Brain Tumor Research Program, Children’s Hospital Colorado, Aurora, Colorado, USA.
Rebecca J Chapman, Children’s Brain Tumor Research Centre, University of Nottingham, Nottingham, UK.
Rui Fu, Computational Biology, New York Genome Center, New York, New York, USA.
Nicholas Willard, Department of Pathology, University of Colorado Denver, Aurora, Colorado, USA.
Andrea M Griesinger, Department of Pediatrics, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA; Morgan Adams Foundation Pediatric Brain Tumor Research Program, Children’s Hospital Colorado, Aurora, Colorado, USA.
Graziella Ribeiro de Sousa, Department of Pediatrics, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA; Morgan Adams Foundation Pediatric Brain Tumor Research Program, Children’s Hospital Colorado, Aurora, Colorado, USA.
Vladimir Amani, Department of Pediatrics, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA; Morgan Adams Foundation Pediatric Brain Tumor Research Program, Children’s Hospital Colorado, Aurora, Colorado, USA.
Enrique Grimaldo, Department of Pediatrics, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA; Morgan Adams Foundation Pediatric Brain Tumor Research Program, Children’s Hospital Colorado, Aurora, Colorado, USA.
Todd C Hankinson, Morgan Adams Foundation Pediatric Brain Tumor Research Program, Children’s Hospital Colorado, Aurora, Colorado, USA; Department of Neurosurgery, University of Colorado Denver, Aurora, Colorado, USA.
Ffyona Booker, Children’s Brain Tumor Research Centre, University of Nottingham, Nottingham, UK.
Martin Sill, Hopp-Children’s Cancer Center Heidelberg (KiTZ), Heidelberg, Germany; Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany.
Richard G Grundy, Children’s Brain Tumor Research Centre, University of Nottingham, Nottingham, UK.
Kristian W Pajtler, Hopp-Children’s Cancer Center Heidelberg (KiTZ), Heidelberg, Germany; Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany; Department of Pediatric Oncology, Hematology, and Immunology and Pulmonology, Heidelberg University Hospital, Heidelberg, Germany.
David W Ellison, St. Jude Children’s Research Hospital, Memphis, TN, USA.
Nicholas K Foreman, Department of Pediatrics, University of Colorado Anschutz Medical Campus, Aurora, Colorado, USA; Morgan Adams Foundation Pediatric Brain Tumor Research Program, Children’s Hospital Colorado, Aurora, Colorado, USA.
Timothy A Ritzmann, Children’s Brain Tumor Research Centre, University of Nottingham, Nottingham, UK.
Funding
This study was supported by NIH grant R01CA237608 (R.F., K.A.R., A.G., N.K.F., and A.M.D.). K.A.R. and R.F were supported as informatics fellows of the RNA Bioscience Initiative, University of Colorado School of Medicine. The University of Colorado Denver Genomics and Microarray, and Histology Shared Resources are supported by the University of Colorado NIH/NCI Cancer Center (P30CA046934). Additional support was provided by the Tanner Seebaum Foundation and the Morgan Adams Foundation. Funding for collection, profiling and annotation of the UK specimens was provided by Fighting Ependymoma and the James Tudor Foundation.
Data Availability
Methylation, snRNAseq and bulk RNAseq data are available through GEO SuperSeries accession number GSE226961 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE226961). SnRNAseq data are available at the Pediatric Neuro-Oncology Cell Atlas browser (pneuroonccellatlas.org).
Conflict of interest statement
The authors have no conflicts of interest to disclose.
Authorship statement
All experiments were performed with assistance by A.M.D., G.A.N., R.J.C., A.M.G., G.R.S., V.A., E.G., and T.C.H. K.A.R. R.F, D.G., Y.Z and B.S. performed bioinformatic and biostatistical analyses. N.W. assisted with neuropathology concerns. A.M.D, K.C.B., G.A.N., F.B., M.S., N.K.F., R.G.G, K.W.P., D.W.E., N.K.F. and T.A.R. provided data and were responsible for the design of the study. All authors assisted with manuscript preparation.
References
- 1. Ritzmann TA, Rogers HA, Paine SML, et al. A retrospective analysis of recurrent pediatric ependymoma reveals extremely poor survival and ineffectiveness of current treatments across central nervous system locations and molecular subgroups. Pediatr Blood Cancer. 2020;67(9):e28426. [DOI] [PubMed] [Google Scholar]
- 2. Pajtler KW, Witt H, Sill M, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell. 2015;27(5):728–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gillen AE, Riemondy KA, Amani V, et al. Single-Cell RNA sequencing of childhood ependymoma reveals neoplastic cell subpopulations that impact molecular classification and etiology. Cell Rep. 2020;32(6):108023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gojo J, Englinger B, Jiang L, et al. Single-cell RNA-Seq reveals cellular hierarchies and impaired developmental trajectories in pediatric ependymoma. Cancer Cell. 2020;38(1):44–59.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fu R, Norris GA, Willard N, et al. Spatial transcriptomic analysis delineates epithelial and mesenchymal subpopulations and transition stages in childhood ependymoma. Neuro Oncol. 2022;25(4):786–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aubin RG, Troisi EC, Montelongo J, et al. Pro-inflammatory cytokines mediate the epithelial-to-mesenchymal-like transition of pediatric posterior fossa ependymoma. Nat Commun. 2022;13(1):3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Merchant TE, Bendel AE, Sabin ND, et al. Conformal radiation therapy for pediatric ependymoma, chemotherapy for incompletely resected ependymoma, and observation for completely resected, supratentorial ependymoma. J Clin Oncol. 2019;37(12):974–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dyer S, Prebble E, Davison V, et al. Genomic imbalances in pediatric intracranial ependymomas define clinically relevant groups. Am J Pathol. 2002;161(6):2133–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baroni LV, Sundaresan L, Heled A, et al. Ultra high-risk PFA ependymoma is characterized by loss of chromosome 6q. Neuro Oncol. 2021;23(8):1360–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhao S, Li J, Zhang H, et al. Epigenetic alterations of repeated relapses in patient-matched childhood ependymomas. Nat Commun. 2022;13(1):6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hoffman LM, Donson AM, Nakachi I, et al. Molecular sub-group-specific immunophenotypic changes are associated with outcome in recurrent posterior fossa ependymoma. Acta Neuropathol. 2014;127(5):731–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang D, Holsten T, Bornigen D, et al. Ependymoma relapse goes along with a relatively stable epigenome, but a severely altered tumor morphology. Brain Pathol. 2021;31(1):33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mak DY, Laperriere N, Ramaswamy V, et al. Reevaluating surgery and re-irradiation for locally recurrent pediatric ependymoma-a multi-institutional study. Neurooncol Adv. 2021;3(1):vdab158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ruda R, Bruno F, Pellerino A, Soffietti RE.. Evaluation and management updates. Curr Oncol Rep. 2022;24(8):985–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ruda R, Reifenberger G, Frappaz D, et al. EANO guidelines for the diagnosis and treatment of ependymal tumors. Neuro Oncol. 2018;20(4):445–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Adolph JE, Fleischhack G, Mikasch R, et al. Local and systemic therapy of recurrent ependymoma in children and adolescents: short- and long-term results of the E-HIT-REZ 2005 study. Neuro Oncol. 2021;23(6):1012–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Amani V, Donson AM, Lummus SC, et al. Characterization of 2 novel ependymoma cell lines with chromosome 1q gain derived from posterior fossa tumors of childhood. J Neuropathol Exp Neurol. 2017;76(7):595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shaffer LG, McGowan-Jordan J, Schmid M.. ISCN 2013: an international system for human cytogenetic nomenclature. Karger. 2013. [Google Scholar]
- 19. Slyper M, Porter CBM, Ashenberg O, et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat Med. 2020;26(5):792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Korsunsky I, Millard N, Fan J, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16(12):1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Korsunsky I, Nathan A, Millard N, Raychaudhuri S.. Presto scales Wilcoxon and auROC analyses to millions of observations. bioRxiv. 2019;653253. [Google Scholar]
- 22. Childhood Ependymoma Treatment (PDQ(R)): Health Professional Version. PDQ Cancer Information Summaries. Bethesda (MD); 2002. [Google Scholar]
- 23. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barbie DA, Tamayo P, Boehm JS, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462(7269):108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102(43):15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mack SC, Witt H, Piro RM, et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature. 2014;506(7489):445–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bayliss J, Mukherjee P, Lu C, et al. Lowered H3K27me3 and DNA hypomethylation define poorly prognostic pediatric posterior fossa ependymomas. Sci Transl Med. 2016;8(366):366ra161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pajtler KW, Wen J, Sill M, et al. Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathol. 2018;136(2):211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Visone R, Bacalini MG, Di Franco S, et al. DNA methylation of shelf, shore and open sea CpG positions distinguish high microsatellite instability from low or stable microsatellite status colon cancer stem cells. Epigenomics. 2019;11(6):587–604. [DOI] [PubMed] [Google Scholar]
- 30. Richards EJ, Elgin SC.. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell. 2002;108(4):489–500. [DOI] [PubMed] [Google Scholar]
- 31. Qu GZ, Grundy PE, Narayan A, Ehrlich M.. Frequent hypomethylation in Wilms tumors of pericentromeric DNA in chromosomes 1 and 16. Cancer Genet Cytogenet. 1999;109(1):34–39. [DOI] [PubMed] [Google Scholar]
- 32. Ehrlich M. DNA methylation in cancer: too much, but also too little. Oncogene. 2002;21(35):5400–5413. [DOI] [PubMed] [Google Scholar]
- 33. Chapman RJ, Ghasemi DR, Andreiuolo F, et al. Optimising biomarkers for accurate ependymoma diagnosis, prognostication and stratification within International Clinical Trials: A BIOMECA study. Neuro-Oncology. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pierce AM, Witt DA, Donson AM, et al. Establishment of patient-derived orthotopic xenograft model of 1q+ posterior fossa group A ependymoma. Neuro Oncol. 2019;21(12):1540–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Donson AM, Amani V, Warner EA, et al. Identification of FDA-approved oncology drugs with selective potency in high-risk childhood ependymoma. Mol Cancer Ther. 2018;17(9):1984–1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Panwalkar P, Tamrazi B, Dang D, et al. Targeting integrated epigenetic and metabolic pathways in lethal childhood PFA ependymomas. Sci Transl Med. 2021;13(614):eabc0497. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Methylation, snRNAseq and bulk RNAseq data are available through GEO SuperSeries accession number GSE226961 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE226961). SnRNAseq data are available at the Pediatric Neuro-Oncology Cell Atlas browser (pneuroonccellatlas.org).