

Abstract
Background
Distinguishing the cellular origins of childhood brain tumors is key for understanding tumor initiation and identifying lineage-restricted, tumor-specific therapeutic targets. Previous strategies to map the cell-of-origin typically involved comparing human tumors to murine embryonal tissues, which is potentially limited due to species-specific differences. The aim of this study was to unravel the cellular origins of the 3 most common pediatric brain tumors, ependymoma, pilocytic astrocytoma, and medulloblastoma, using a developing human cerebellar atlas.
Methods
We used a single-nucleus atlas of the normal developing human cerebellum consisting of 176 645 cells as a reference for an in-depth comparison to 4416 bulk and single-cell transcriptome tumor datasets, using gene set variation analysis, correlation, and single-cell matching techniques.
Results
We find that the astroglial cerebellar lineage is potentially the origin for posterior fossa ependymomas. We propose that infratentorial pilocytic astrocytomas originate from the oligodendrocyte lineage and MHC II genes are specifically enriched in these tumors. We confirm that SHH and Group 3/4 medulloblastomas originate from the granule cell and unipolar brush cell lineages. Radiation-induced gliomas stem from cerebellar glial lineages and demonstrate distinct origins from the primary medulloblastoma. We identify tumor genes that are expressed in the cerebellar lineage of origin, and genes that are tumor specific; both gene sets represent promising therapeutic targets for future study.
Conclusion
Based on our results, individual cells within a tumor may resemble different cell types along a restricted developmental lineage. Therefore, we suggest that tumors can arise from multiple cellular states along the cerebellar “lineage of origin.”
Keywords: cerebellum, ependymoma, medulloblastoma, pilocytic astrocytoma, radiation-induced glioma, tumor origin
Key Points.
Common pediatric brain cancers arise from distinct lineages of origin in the developing cerebellum.
Radiation-induced gliomas arise as de novo secondary tumors.
Developmental genes represent possible opportunities to target tumor vulnerabilities.
Importance of the Study.
We compile the largest collection of transcriptome datasets for cerebellar-localized pediatric brain tumors: posterior fossa ependymoma, pilocytic astrocytoma, and medulloblastoma. We compare this large dataset to a unique and in-depth atlas of human cerebellum development. With these 2 datasets, we uncover the potential lineage-of-origins for the most common cerebellar brain tumors in children; we also find that radiation-induced gliomas, secondary tumors arising after cranial irradiation, have cellular origins independent of the primary tumor. We identify genes active during development and still expressed in the tumor, which may represent promising therapeutic targets for targeted interventions.
Pediatric central nervous system (CNS) tumors represent one of the most fatal disease entities in children.1 Current radiation and chemotherapy approaches frequently result in neurocognitive disorders and life-long side effects, including secondary malignancies in patients surviving their primary disease.2 Future therapeutic approaches should target tumor-specific vulnerabilities,3 ideally without affecting normal tissue architecture—a difficult task due to the functional and cellular complexity of the brain.4 One such approach is first to identify the developmental lineage from which tumors arose and then to identify selective lineage vulnerabilities of the tumor that are no longer needed by normal cells once embryonic development is complete.
The cerebellum is the most frequent anatomic location of pediatric brain tumors,5,6 including posterior fossa subgroup A ependymoma7 (PFA-ependymoma), pilocytic astrocytoma,8 and medulloblastoma.9 Based on comparison with murine cerebellar atlases,6 PFA-ependymomas may originate from radial glial cell subtypes,10 including “roof-plate-like stem cells” and “gliogenic progenitors”; however, the exact cellular origins are still unclear.11 While the origin of pilocytic astrocytomas is unknown, these tumors are hypothesized to arise from developing oligodendrocytes.12 Different subgroups of medulloblastoma are predicted to have distinct cellular origins. While WNT-medulloblastoma is hypothesized to arise outside of the cerebellum,13 SHH-medulloblastomas originate from granule cell precursors.14,15 Groups 3 and 4 medulloblastomas are predicted to arise from the subventricular zone of the cerebellar rhombic lip,16,17 a progenitor zone for glutamatergic unipolar brush cells. Except for these recently published studies for medulloblastoma,16,17 the cellular origins of pediatric tumors have been deduced from murine cell atlases.6,18 As exemplified by Smith et al. and Hendrikse et al.,16,17 murine-to-human comparisons may miss key aspects of human development relevant for tumor formation, including human-specific developmental gene expression.19,20 Thus, we re-examined the cellular origins of pediatric tumors based on a comprehensive human cell atlas.
We compared the transcriptomes of cell types, subtypes, and differentiation states from the developing human cerebellum21 to gene expression profiles from pediatric CNS tumors (Supplementary Figure S1A and B). Because of the rarity of tumor samples, we compiled published (N = 2892) and unpublished (N = 1425) bulk transcriptome datasets (total = 4317) and published single-cell transcriptome datasets (N = 99), which together represent the largest collection of CNS tumor transcriptome samples. Our integrated bioinformatic analyses of these datasets revealed the most closely matching cellular lineages of origin for ependymoma, pilocytic astrocytoma, and medulloblastoma, as well as putative lineage-specific and tumor-specific target genes. Our results can be further explored at brain-match.org.
Materials and Methods
Ethics Statement
The tumor molecular bulk and single data materials were collected from a group of published studies listed in Supplementary Table S2 under full regulation of corresponding ethics standards.
Human Cerebellum Single-Nuclei RNA Sequencing Data
The single-nucleus RNA sequencing (snRNA-seq) data generation and annotation are described in detail in the corresponding paper.21 Briefly, 38 libraries from 31 independent human cerebellum samples were produced using Chromium (10x Genomics) version 2 or 3 reagents. The dataset includes 176 645 high-quality cells from 10 developmental stages ranging from postconceptional week 7 to adult age. Louvain algorithm was used to cluster the merged dataset into 68 clusters and 611 subclusters. Hierarchical annotation of the dataset classified the cells into 21 cell types and 37 cell differentiation states. Cells from 12 cell states were further divided into 37 subtypes. For the purpose of the current study, we combined the cell state and subtype level annotations to divide the dataset into 65 subgroups. For simplicity, we refer to these subgroups as “subtypes” throughout the study.
Bulk Tumor Transcriptome Data Integration
The main microarray tumor cohort used for our analyses was generated from the combination of three mixed CNS tumor cohorts resulting in 2923 unique samples, classified via tumor random forest model application with reference created from tumors with available methylation-based profiles via mnp v12.3 system (molecularneuropathology.org/mnp). Bulk RNA-seq profiles (Reads Per Kilobase of transcript, per Million mapped reads (RPKM), normalized gene expression counts) were obtained for 1394 samples from 4 independent cohorts and processed as described in the Supplementary Methods.
Comparison of Bulk Tumor Profiles to the Normal Cerebellum Single Nuclei Data
We compared the transcriptomic signatures of tumor samples to those of normal cerebellum cell states or subtypes (as reference) based on a combination of correlation estimates and Gene Set Variance Analysis (GSVA22 package v1.28). Expression profiles of normal cerebellar cells were aggregated into pseudobulk across cell state and subtypes and normalized into RPKM values. The details on verification of the computational methods are provided in the Supplementary Methods.
Differential Gene Expression Analysis Among Cerebellum Cell Types and Tumor Tissues
The differentially expressed genes among assigned cerebellum cell types were identified via methods from Seurat v.3.2.2 (Wilcoxon Rank Sum and distance-based assignment). The genes differentially expressed among tumor classes were adjusted following selection of limma-derived group-specific comparison (target tumor class vs all other samples in cohort), using minimum adjusted P-value .05 filtering limit. Afterward only the top 2000 genes were selected from P-value sorted result gene list.
Single-Cell Tumor Transcriptome Data Integration
Single-cell datasets were collated for medulloblastoma, ependymoma and pilocytic astrocytoma as described in Supplementary Table S2, representing 10x Genomics (v2 and v3) and SmartSeq2 platforms. Detailed data processing is described in the Supplementary Methods.
Comparison of Single-Cell Tumor Profiles to Normal Cerebellum Single-Nuclei Data
The support vector machine (SVM) based multi-class predication model was used to assign each tumor cell to its closest matching identity in the reference cell state annotation. To study the trajectory of tumor cells with respect to the cerebellum cell type trajectory, we integrated each tumor sample to the best matching lineage of that sample’s tumor type (pilocytic astrocytoma: oligodendrocytes, ependymoma: astrocytes, medulloblastoma: GC/UBC). We used a batch correction method to integrate tumor samples to the matching normal lineage, with the assumption that the tumorigenic effect on cell transcriptome is independent (and hence orthogonal) to normal developmental programs and can be corrected. The specific details are provided in Supplementary Methods.
Available Resources
The results of all main comparisons of normal cell types to tumors (both bulk and single cell) and common differentially expressed genes can be browsed using the ShinyApp R application at brain-match.org.
Results
Specific Origins for Distinct Brain Tumor Classes
We recently generated an extensive snRNA-seq dataset that covers the development of the human cerebellum from the beginning of neurogenesis to adulthood.21 In this reference atlas, 176 645 cells (Figure 1A) are grouped into 21 cell types (eg, astroglia) and further divided into 37 cell differentiation states (eg, progenitor), hereafter referred to as cell states (Supplementary Figure S1A; Figure 1B). In addition, subtypes are specified for cell types and states, resulting in 65 transcriptomically defined subtypes of cells (eg, progenitors divided into 9 spatio-temporal subtypes including rhombic lip progenitors). Notably, cell states within a cell type form a continuum along the differentiation trajectory, which we refer to as “cellular lineage” in this study. For instance, the astroglial lineage includes progenitor (neuroepithelial and radial glial cells), glioblast, and astrocyte cell states.
Figure 1.
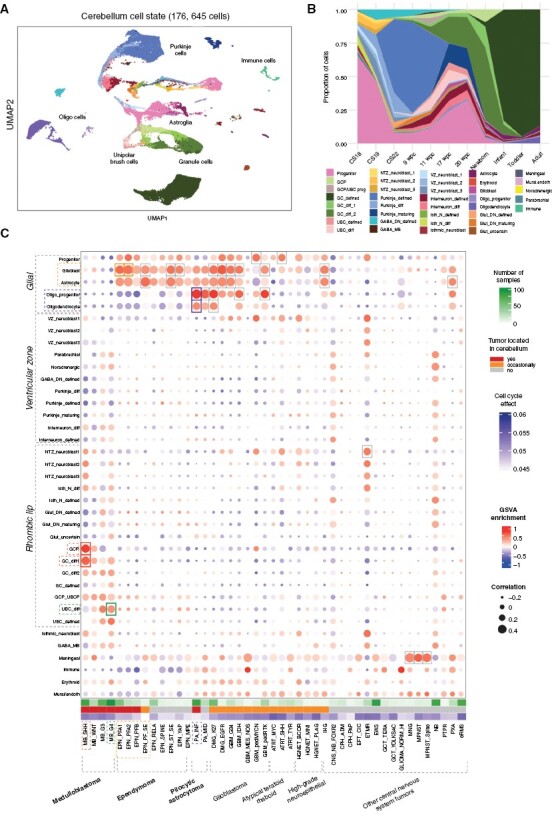
Human single-nuclei developmental cerebellum cell atlas comparisons reveal putative lineages-of-origin of cerebellar tumors. (A) UMAP of human developmental cerebellum single-nuclei data used as a reference. Color codes represent cell state annotation and labels denote the main cell types. (B) Proportions of different states across developmental time. (C) Heatmap of comparisons of bulk central nervous system tumor transcriptome profiles (in columns) to cerebellar cell state (in rows), based on gene signature enrichment score (via Gene Set Variance Analysis (GSVA, indicated by color) and Pearson correlation score (indicated by size). Highest associations marked by rectangles, based on cutoff limits of min GSVA enrichment = 0.4 and min correlation = 0.4.
We collected published and unpublished bulk microarray expression data to use as our main tumor transcriptome discovery cohort (Supplementary Figure S1C). We annotated tumor type and subtype using methylation-based classification as previously described23 (Supplementary Figure S1D,E). This discovery cohort represents 2923 samples annotated into 45 defined molecular classes (eg, medulloblastoma Group 3 (G3)) and 68 methylation subclasses (eg, medulloblastoma Group 3/4, subtype II (G34_II)) (Supplementary Table S1). We also compared our developmental reference atlas to an independent collection of 4 published bulk RNA-seq datasets (N = 1394 samples) as our validation cohort (Supplementary Table S2).
After exploration of existing computational approaches,6,18,24 we selected 2 main measures to determine the similarity of tumors to normal cell types: correlation of expression patterns of shared highly variable genes and gene set variance analysis (GSVA).22,24 These measures were validated using SHH-medulloblastoma as a positive control, given that this tumor class has been experimentally shown to arise from the granule cell progenitor (GCP) lineage14,15 (ICGC cohort,25N = 160, Supplementary Figure S2A). Additionally, we estimated the required filtering thresholds for these computed values (Supplementary Figure S2B–E, see Supplementary Methods). To independently verify both methods, we applied deconvolution on the control cohort using the CIBERSORT26 toolkit (Supplementary Figure S2F).
We next compared the large global transcriptome tumor cohort to normal cerebellar cells (Figure 1C). We included non-cerebellar tumor classes, such as meningioma (MNG), or glioma profiles enriched with immune cells (GLIOMA_NORM_HI) as external controls for our global comparison.
From this global comparison, we found that the 2 classes of posterior fossa A (PFA) ependymoma, PFA1, and PFA2, were most similar to cells in the astroglial lineage. This lineage includes progenitors, glioblasts, and astrocytes; however, ependymal glial cells are missing in our human cerebellum dataset, even though this cell type also belongs to astroglial lineage. Other posterior fossa ependymoma tumor classes, including posterior fossa B (PFB) and posterior fossa subependymoma (PF_SE) that are much rarer in children, showed similar associations to the astroglial lineage.
Infratentorial pilocytic astrocytoma (PA_INF), a tumor class nearly exclusively occurring in the cerebellum, showed the closest association with the oligodendrocyte lineage, including oligodendrocyte progenitor cells (OPCs) and oligodendrocytes. These results increase the evidence that pilocytic astrocytomas indeed arise from the oligodendrocyte lineage, as previously suggested.12 Several glioblastoma (GBM) molecular classes that are only occasionally located in the cerebellum27,28 demonstrated similarities to various glial cell states, including astrocytes and oligodendrocytes, and their respective precursors.
As expected, we confirmed the closest association of SHH-medulloblastoma to the granule cell lineage (GCP and differentiating granule cells, GC_diff1). We found that Group 4 medulloblastoma most closely resembles the unipolar brush cell lineage (differentiating unipolar brush cells, UBC_diff), further supporting previous results.6,16–18 Group 3 medulloblastoma did not match any specific lineage within filtering thresholds, but was most closely associated to the GCP and UBC lineages. WNT-medulloblastoma also did not match any cerebellar cell state, which is in line with its proposed origins from the developing brainstem, that is, outside of the cerebellum.13
The results we obtained using an external RNA-seq cohort (CBTN) as a validation dataset agreed with the results from our discovery cohort (Supplementary Figure S3A), thereby verifying our approach using independent gene expression datasets. Using deconvolution on this validation dataset, the tumor lineage-of-origin was consistent in the majority of cases (Supplementary Figure S3B). As a further control, we used an independent human cerebellum atlas as an alternative reference,29 and all major findings listed were confirmed as well (Supplementary Figure S3C).
Posterior Fossa Ependymomas Likely Arise From the Astroglial Lineage
We next investigated the cellular origins of the main pediatric brain tumor types arising in the cerebellum in greater detail and at single-cell resolution. The 2 major classes of posterior fossa ependymoma (PFA and PFB) were previously proposed to originate from radial glial cells.10 This result was partly confirmed by a study that used a mouse cerebellum-based reference map and found an association with “roof-plate like stem cells” and “gliogenic progenitors.”6 Using our human cerebellum single-cell dataset, we found that the transcriptional profiles of posterior fossa ependymomas most closely resembled various states of differentiation within the astroglial lineage of the cerebellum, starting from radial glial and neuroepithelial progenitors, and extending to glioblasts and astrocytes (Figure 1C, Supplementary Figure S4A). These human cell states in-turn demonstrated similarities to murine “roof-plate-like stem cells” and “gliogenic progenitors”6 (Supplementary Figure S4B), thus confirming the result and demonstrating the potential benefit of using human data for a more precise comparison.
To increase the resolution of cell type associations, we further focused on cell subtypes within the astroglial lineage (Figure 2A). Two spatially segregated gliogenic paths in the cerebellum start from gliogenic progenitors (producing Bergmann glia and parenchymal astrocytes) and bipotent progenitors (producing GABAergic interneurons and parenchymal astrocytes), respectively.21 They progress via their corresponding glioblast subtypes toward mature astrocytes.30 Applying the same methods above for bulk transcriptome data on this restricted lineage, we found that PFA and PFB ependymoma transcriptomes matched bipotent progenitors, prospective white matter glioblast (glioblast_PWM), astroblast, and mature parenchymal astrocyte subtype signatures (Figure 2B).
Figure 2.
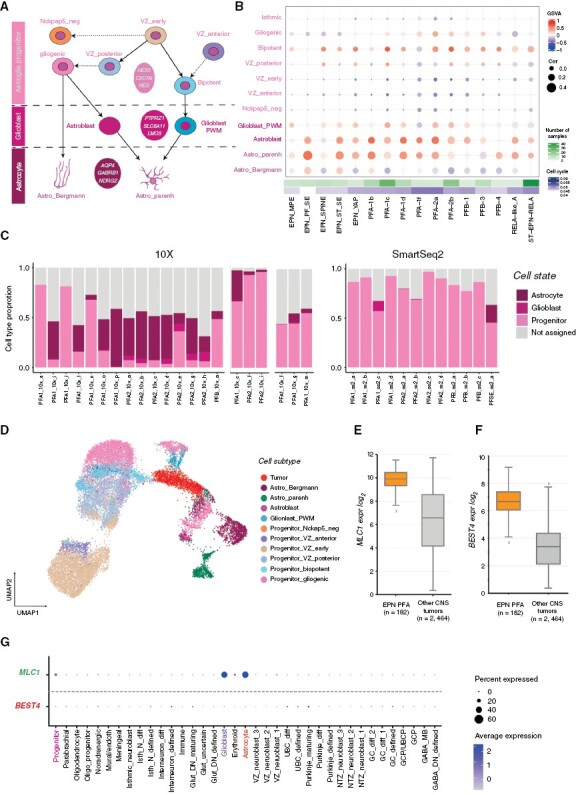
Ependymoma tumors arise from the astroglial lineage. (A) Astroglial cell lineage derived from early ventricular zone (VZ) progenitors. (B) Comparison of bulk ependymoma tumor gene expression profiles to cell subtypes of the astroglial lineage, based on GSVA enrichment and correlation measures. (C) Ependymoma single cells assigned to the closest normal cerebellum cell state; cells with <50% similar are classified as “not assigned.” (D) Projection of an individual single-cell tumor data (PFA1_10x_g, 10x v2) onto astroglial cell subtypes as visualized via UMAP. (E) Median expression boxplot of MLC1, a PFA-ependymoma enriched gene expressed within the astroglial lineage (limma adjusted P-value: 5.67E-31). (F) Median expression boxplot of BEST4, a PFA-ependymoma-specific gene (limma adjusted P-value: 1.75E−61). (G) Specificity of MLC1 and BEST4 gene expression across cerebellum cell states.
Based on these observations, we hypothesized that either the tumor develops from an earlier node within the astroglial lineage and differentiates along the specified trajectory after malignant transformation, or that the transcriptome of posterior fossa ependymoma tumor cells is comprised of a mix of progenitor and more differentiated cell signatures. To investigate these possibilities further, we used published posterior fossa ependymoma single-cell data from three independent studies.6,31,32 We assigned each tumor cell within a sample to its closest matching cell state identity, using SVM multi-class prediction model (see Supplementary Methods for details, Supplementary Figure S4C). Using this assignment, we also found nonmalignant immune cell clusters in PF ependymoma samples analyzed using 10x chemistry (Supplementary Figure S4D). These cell clusters had normal DNA copy number profiles when compared to tumor cell clusters, confirming their nonmalignant identity (Supplementary Figure S4E); hence, we excluded them from further analysis to prevent obscuring the tumor signature. Similarly, we also filtered out cells with assigned identities that did not arise from the astroglial lineage (for example, meningeal, GCP, or oligo-progenitors). These cells do not form independent cell clusters and may represent rare, nonimmune normal cells from the microenvironment. Subsequently, we found that each tumor sample had cells resembling progenitor, glioblast, and mature astrocyte-like cells, but with different proportional distributions within tumors of the same class (Figure 2C). This result suggests that ependymoma PF tumors can comprise cells along a differentiation trajectory within the lineage of origin.
We tested this hypothesis by overlaying tumor cells onto the normal astroglial lineage and found that the tumor cells integrated along the entire lineage, suggesting a gradient of cell differentiation arising originally from bipotent and gliogenic progenitors (Figure 2D). Surprisingly, the transcriptomic signatures of progenitor-like and astrocyte-like tumor cells had mixed marker gene expression, expressing genes normally present exclusively in progenitor cells together with genes normally present exclusively in astrocyte cells (eg, VIM, PTN, CST3 expressed together in a single-tumor cell); however, relative enrichment of marker genes (eg, progenitor: TENM3, glioblast: SMOC1, astrocyte: AQP4) supported the assigned resemblance of these tumor cells to the corresponding cell types (Supplementary Figure S4F). We thus predict that even “mature” tumor cells maintain proliferative capacity, as evidenced by the expression of progenitor marker genes (eg, VIM, PTN). We also observed a similar gradient of differentiation when merging tumor cells with the full cerebellum lineage (Supplementary Figure S4G). Altogether, our findings suggest that PF tumors are composed of malignant cells differentiating along the astroglial lineage.
We next identified the genes shared between the astroglial lineage and PFA-ependymomas (Supplementary Table S3). We focused on genes that are specifically expressed in the brain19 and/or cerebellum, and are either plasma membrane localized or possible drug targets, based on the human protein atlas.33 We identified the potentially druggable MLC1 gene expressed in ependymomas (Figure 2E), which was also expressed specifically in the astroglial lineage, but not in any other cell lineage in the cerebellum (Figure 2G) nor expressed outside of the brain (Supplementary Figure S4H). Similarly, we identified and filtered tumor-specific gene candidates, such as the gene BEST4 (Figure 2F). BEST4 encodes a membrane protein and was not detected in any cerebellar cell lineage (Supplementary Table S4, Figure 2G). It is also absent or lowly expressed in other normal tissues (Supplementary Figure S4I). These 2 membrane proteins could be explored as CAR-T targets to specifically recognize PFA-ependymoma cells.34
Pilocytic Astrocytomas Transcriptomically Resemble Postnatal Oligodendrocyte Precursor Cells (OPCs)
Our global comparisons showed that 2 pilocytic astrocytoma molecular classes, PA_INF (infratentorial or posterior fossa, predominantly cerebellum) and PA_MID (supratentorial midline) most closely resembled oligodendrocyte progenitor cells (OPCs, Figure 1C). To investigate this result further, we focused on a bulk RNA-seq cohort35 comprised of additional pediatric low-grade gliomas classes (Supplementary Figure S5A). We compared pilocytic astrocytomas to the OPC lineage (Figure 3A), using supratentorial gangliogliomas (GG_ST) and H3K27 altered diffuse midline gliomas (DMG_K27) as controls. We found that pilocytic astrocytomas best match late oligodendrocyte progenitor cells (OPC_late) present in the postnatal cerebellum (Figure 3B; Supplementary Figure S5A,B). Infratentorial pilocytic astrocytomas matched better than midline pilocytic astrocytomas to cerebellar OPC_late cells (Figure 3B, GSVA/correlation difference: 0.34/0.04), hinting that the anatomical location of the tumor also influences cell type matching. Using single-cell data,6,12 we found that pilocytic astrocytomas are comprised of tumor cells resembling oligodendrocyte progenitors (Figure 3C); normal cells were filtered out for this comparison. We then merged single-cell tumor data with the normal oligodendrocyte lineage and found that most tumor cells integrated with the OPC_late cluster, with some cells overlapping with early OPCs and mature oligodendrocytes (Figure 3D). This pattern was also observed when merging tumor cells with the full cerebellum atlas (Supplementary Figure S5C).
Figure 3.
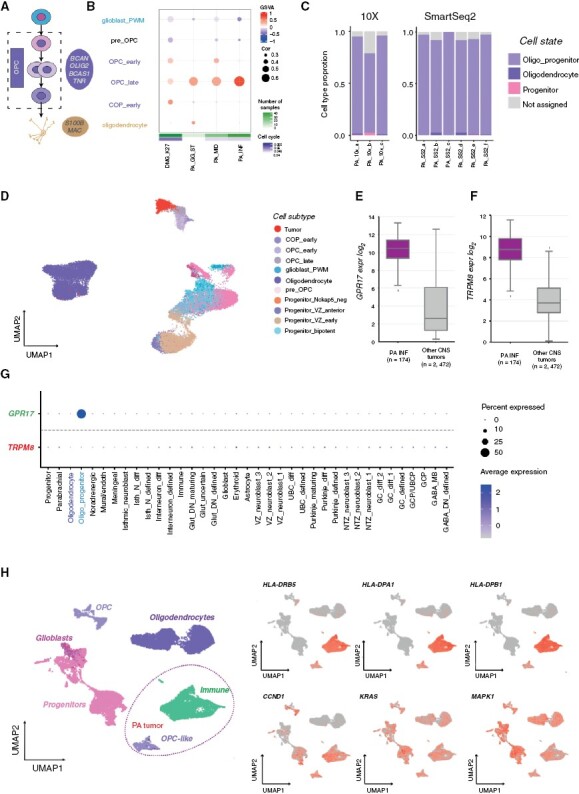
Pilocytic astrocytomas arise from the oligodendrocyte lineage. (A) Oligodendrocyte cerebellar lineage. (B) Comparison of bulk pilocytic astrocytoma gene expression profiles to cell subtypes from the oligodendrocyte lineage based on GSVA enrichment and correlation measures. (C) Pilocytic astrocytoma single-cell data SVM-based comparison to cerebellum cell state. (D) Projection of a single-cell tumor sample (ID: PA_10X_a, 10x v2) onto astroglia and oligodendrocyte cell subtypes as visualized via UMAP. (E) Median expression boxplot of GPR17, a pilocytic astrocytoma enriched gene expressed within the oligodendrocyte lineage (limma adjusted P-value: 1.25E−105). (F) Median expression boxplot of TRPM8, a pilocytic astrocytoma unique gene (limma adjusted P-value: 2.14E−72). (G) Specificity of GPR17 and TRPM8 gene expression across cerebellum cell states. (H) Comparison of a single-cell sample (ID: PA_10X_a, nontumorous immune cells included) with the oligodendrocyte lineage, and relative gene expression of MHC- and MAPK-associated genes.
We assessed shared and unique genes between pilocytic astrocytomas and OPCs (Supplementary Tables S3, S4), and identified possible candidate cell surface genes, including GPR17 and TRPM8 (Figure 3E–G; Supplementary Figure S5D,E). Using gene-set enrichment analysis (GSEA), we found that OPC and pilocytic astrocytomas both express genes involved in oligodendrocyte development (Supplementary Figure S5F). Surprisingly, tumor-specific genes were enriched in major histocompability complex class II (MHC II) and immune-associated genes (Supplementary Figure S5F). Because this immune signature could result from immune cells within the tumor micro-environment, we excluded non-malignant cells. Even after filtering, we still detected expression of MHC class II genes (eg, CD74, HLA-DRB5) in the remaining tumor cells (Supplementary Figure S5G). These same tumor cells also express downstream targets of MAPK signaling (Figure 3H), a tumor-promoting pathway constitutively activated in pilocytic astrocytoma.36 The immunological role of this pattern and its contribution to tumorigenesis and senescence commonly seen in pilocytic astrocytoma remains to be investigated.
Medulloblastomas Demonstrate the Closest Similarity to the GC/UBC Lineage in Transcriptome Profiles
From our global comparison, SHH, Group 3, and Group 4 medulloblastoma are associated with one or both branches of the GC/UBC lineage (Figure 1A); therefore, we focused on a high-resolution lineage map to delineate the potential origins of different medulloblastoma groups (Figure 4A). We found that the majority of SHH-medulloblastoma subgroups corresponded to GCPs and nascent postmitotic GCs (GC_diff1, Figure 4B). SHH-medulloblastoma subgroups I, II, and IV showed high similarity to early GCPs and to the late subpopulation of differentiating GCs that emerge postnatally (GC_diff1_late, Figure 4B). SHH-II (beta) subgroup, which is associated with infant onset and metastasis,37 instead resembled later stages along the differentiation trajectory, including mature GCs (GC_defined, Figure 4B).
Figure 4.
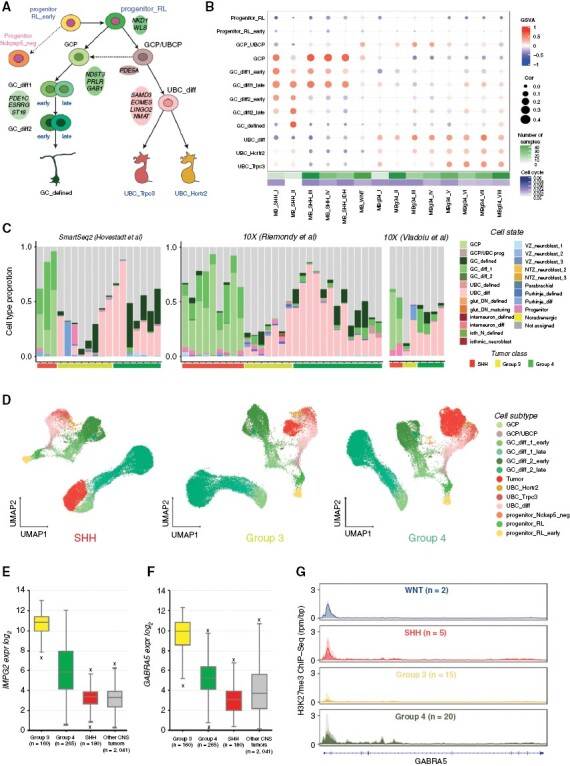
Medulloblastomas arise from the granule cell and unipolar brush cell lineages. (A) Granule/unipolar brush cell (GC/UBC) lineage arising from rhombic lip progenitors. (B) Comparison of bulk medulloblastoma gene expression profiles to the GC/UBC lineage, based on GSVA enrichment and correlation measures. (C) Medulloblastoma single-cell data SVM-based comparison to cerebellum cell state. (D) 10X genomics medulloblastoma samples (IDs: SHH_10X_b, G3_10x_b, G4_10x_c) projection onto the GC/UBC lineage as visualized via UMAP. (E) Median expression boxplot of IMPG2, a medulloblastoma enriched gene expressed within the GC/UBC lineage (limma adjusted p-value: 5.72E−148). (F) Median expression boxplot GABRA5, a medulloblastoma-specific gene (limma adjusted P-value: 2.89E−77). (G) H3K27me3 ChIP-seq profiles (reads per million mapped reads per bp, rpm/bp) of GABRA5 loci in the four medulloblastoma groups.
Groups 3 and 4 medulloblastoma subgroups range from I to VIII, with subgroups II–IV restricted to Group 3 and subgroups VI/VIII restricted to Group 4.9,38 Subgroups V–VIII (mostly Group 4 medulloblastoma) best matched to UBCs (differentiating and defined, Figure 4B). Subgroups I–IV (mostly Group 3 medulloblastoma) displayed low transcriptome correlations with subtypes in the GC/UBC lineage; nevertheless, GSVA enrichment scores suggested some similarity with GC/UBC progenitors and early differentiating UBCs (Figure 4B).
To examine the cell types comprising the tumors, we analyzed published medulloblastoma single-cell data.6,18,39 Cell-label transfer analysis confirmed the similarity of SHH medulloblastoma to GCPs and demonstrated that the majority of Group 3/4 cells match to differentiating UBCs (Figure 4C, Supplementary Figure S6A). This similarity was also observed after merging with normal cells in the GC/UBC lineage (Figure 4D, Supplementary Figure S6B). As suggested in previous studies,18 we also found that tumor cells expressed GABAergic genes(eg, GABRA5), Purkinje cell genes (eg, BCL11A, FOXP2), and photoreceptor markers (eg, CRX, NRL), genes which are not normally expressed in the glutamatergic GC/UBC lineage (Supplementary Figure S6C). Therefore, this noncerebellar gene expression appears to drive the identity of tumor cells away from the GC/UBC lineage. We further examined differentiated GC-like cells in Group 4 tumors, and found these cells express markers of the UBC lineage, including LMX1A and EOMES (Supplementary Figure S6C). This result suggests that these tumor cells likely originate from bi-potent GC/UBC cells, thus keeping their dual identity intact. Further resolution of the cellular origin of Groups 3 and 4 medulloblastoma will require a greater breadth and depth of single-cell tumor data.
We hypothesized that somatic MYC/MYCN changes may deviate tumor cells away from the original lineage. Indeed, the lowest similarity to normal cell types was observed for Group 3/4 subgroup II (Figure 4B,C), which is known to be associated with MYC amplifications.38 In addition, when examining MYC- (Group 3) and MYCN- (SHH and Group 4) amplified tumors in isolation, these specific tumors did not show the same degree of similarity to the GC/UBC signature (Supplementary Figure S6D). We next examined whether expression of normal marker genes of the GC/UBC lineage correlated with patient outcome. While there was no effect on survival in SHH or Group 4, we found that Group 3 patients with low expression of UBC lineage genes had worse overall and progression-free survival compared to patients with high expression of GC/UBC lineage genes (Supplementary Figure S6E,F). This result suggests that as a Group 3 tumor deviates from the normal GC/UBC lineage, patient outcome worsens.
To identify potentially therapeutically relevant candidate genes, we focused on 2 gene sets: those genes that were shared between the medulloblastoma groups and the GC/UBC lineage, with little or no expression in other tissues, and those genes that were specific to medulloblastoma (Supplementary Table S3,4). Among possible tumor unique candidates, we identified the surface protein coding gene39IMPG2, another photoreceptor-associated gene specific for Group 3 tumors and lowly expressed in normal tissue (Figure 4E; Supplementary Figure S7A). In addition, Group 3/4 tumors express the somatostatin receptor SSTR2 (Supplementary Figure S7B), which is involved in driving rod photoreceptor differentiation in the retina,40 suggesting receptor expressing tumor cells may respond to Somatostatin therapy.41
We next identified the gene ontology pathways associated with these gene groups (Supplementary Figure S7C–E). Group 3 tumor-specific genes were highly enriched for neural progenitor cell (NPC) genes that are usually suppressed by H3K27me3 signal. (Supplementary Figure S7D).42 This group of genes is normally off in normal NPCs, but they are aberrantly expressed in Group 3. Some of these genes, for example, GABRA5 (Figure 4F), were tumor-specific. Interestingly, we found loss of H3K27me3 histone marks in the promoters of these genes specifically in Group 3 medulloblastoma (Figure 4G, Supplementary Figure S7F,G), confirming prior reports that H3K27me3 mis-regulation could play important role in Group 3 medulloblastoma formation or maintenance.43
Radiation-Induced Gliomas Arise de novo as Secondary Tumors
Radiation-induced gliomas (RIGs) sometimes occur as a consequence of cranial radiotherapy commonly used to treat CNS tumors,44 including medulloblastoma.45 Based on their glioblastoma (GBM)-specific genetic landscape, lack of overlapping mutations with the initial medulloblastoma,46,47 and on functional studies in nonhuman primates,48 it has been assumed that these tumors arise de novo from healthy cells after radiation rather than by trans-differentiation of residual medulloblastoma cells; however, this has never been formally demonstrated. To investigate this hypothesis, we examined secondary radiation-induced gliomas (N = 11) and their respective primary medulloblastoma samples (N = 11), and compared them to cerebellum cell types (Supplementary Table S5, Supplementary Figure S8A). Copy number variation analysis showed distinct, nonoverlapping profiles for primary and secondary tumors, as previously described46 (Supplementary Figure S8B). This finding was confirmed using methylation classification. Unsupervised hierarchical clustering of gene expression profiles further distinguished primary medulloblastomas from secondary RIGs (Figure 5A). For a subset of formalin-fixed paraffin-embedded (FFPE) primary-relapse matched samples, we compared transcriptome profiles with an additional FFPE bulk RNA-seq dataset (N = 410), representing sporadic medulloblastoma and glioblastoma samples (Supplementary Table S2). UMAP visualization of the tumor pairs with control data resulted in grouping of the primary and secondary tumors with medulloblastoma and GBM clusters, respectively (Figure 5B).
Figure 5.
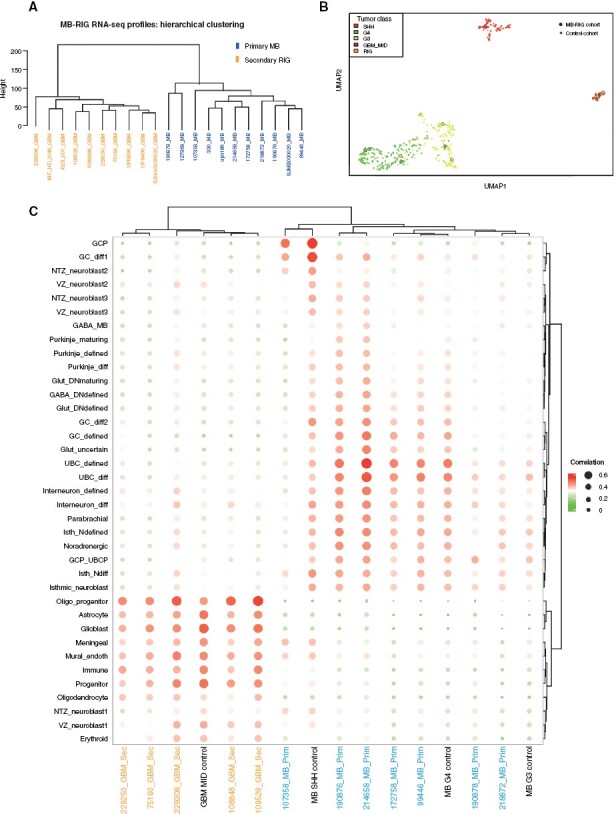
Secondary radiation-induced gliomas (RIGs) originate de novo from the glial cell lineage and not from the original primary tumor. (A) Unsupervised hierarchical clustering of batch-effect adjusted RNA-seq profiles from primary medulloblastoma and secondary RIG tumors (based on top 500 HVGs). (B) UMAP visualization of medulloblastoma and glioblastomas. Primary medulloblastoma and corresponding secondary RIGs are marked with circles. (C) Comparison of gene expression profiles of primary medulloblastoma and secondary RIG tumors combined with corresponding FFPE bulk control to cerebellum cell state, based on correlation measures.
We next compared each medulloblastoma-RIG pair to cerebellar cell states, using correlation analyses only; we could not use GSVA-derived methods because of limited sample size. RIGs, along with glioblastoma controls, were strongly associated with glioblast, astrocyte, and oligodendrocyte cells within the glial lineage, whereas medulloblastoma primary tumors and their controls were most similar to cells from the GC/UBC lineage (Figure 5C). We also merged all RIG samples into a single group and computed both correlation and GSVA scores; these samples demonstrated mapping to oligodendrocyte progenitors and glioblasts (Supplementary Figure S8C). We verified this result using deconvolution analysis for each sample (Supplementary Figure S8D). From this data, we conclude that the lineage-of-origin is different between primary medulloblastoma and secondary RIG tumors and that secondary RIGs arise de novo from a glial lineage.
Discussion
Using an unprecedented cell atlas of the developing human cerebellum, we performed a comprehensive comparison of childhood brain tumor cohorts to normal developmental cells. We determined the most probable cellular lineages of origin for the most common tumor types arising in the cerebellum (Table 1). One of the remarkable observations from our exhaustive analysis was that rarely any tumor class exhibited associations with only one state of a cell type. Instead, tumors at the time of sampling typically contained a gradient of differentiating cells along a cellular lineage. Furthermore, tumors exhibit compositional heterogeneity of cell states along the lineage of origin. Based on our results, we hypothesize that tumor expansion often follows a pre-specified trajectory, which closely resembles the developmental trajectory of the lineage of origin. Importantly, our findings also indicate that tumors exploit developmental and functional characteristics of the lineage of origin to grow and develop into their complex form, similar to a tissue containing cells of different functions.
Table 1.
Summary of the cerebellum-specific CNS tumor associations to normal cell lineages and potential candidate target genes for further investigation
| Tumor class | Closest lineage association to the normal cerebellum | Common tumor-normal genes (full | surfaceome | druggable) |
Tumor unique genes (full | surfaceome | druggable) |
|---|---|---|---|
| EPN PFA | Astroglial | 140 | 33 | 20 eg, MLC1, LRP2, GABRG1 |
389 | 59 | 55 eg, BEST4, PPARG, CYP4F12 |
| PA INT | Oligo | 145 | 30 | 16 eg, GPR17, TNR, GRM5 |
206 | 53 | 41 eg, TRPM8, ANKRD55, TLR7 |
| MB SHH | Granule cells | 135 | 33 | 22 eg, PRLR, BOC, CDK6 |
136 | 21 | 46 eg, GPR68, NRIP2, KCNA3 |
| MB G3 | Unipolar brush cells | 88 | 13 | 14 eg, GLRA1, EPS8, SLC8A1 |
167 | 34 | 42 eg, IMPG2, EPHA8, GABRA5 |
| MB G4 | Unipolar brush cells | 91 | 14 | 13 eg, GRIK1, GRM8, UNC5D |
179 | 28 | 48 eg, SSTR2, PTPN5, HTR2C |
We propose that the astroglial lineage could be the lineage of origin for posterior fossa ependymomas. The astroglial progenitors in the human cerebellar atlas are similar to murine “roof-plate-like stem cells” (possibly choroid plexus or ependymal precursors) and gliogenic progenitors.6 In the future, expanding our analyses to regions outside of the cerebellum, including posterior hindbrain regions, will help refine this observation and uncover cell types that were inadvertently missed, including ependymal cells.
In our analysis, pilocytic astrocytomas are associated with postnatal OPCs, with a best fit to late OPC. This result suggests that pilocytic astrocytoma cells are “stuck” in this cell state, and are unable to proceed in differentiation toward mature oligodendrocytes. Importantly, we also found that MHC class II members are abundantly expressed in tumor cells, similar to previous observations in pilocytic astrocytoma for some MHC class I members,12 which suggests these tumor cells may be better recognized by the immune system, in contrast to other CNS tumors. The favorable prognosis of diagnosed patients could be the result of slow tumor growth, MAPK-induced senescence, increased immunogenicity, or a combination thereof.
For SHH, Group 3, and Group 4 medulloblastoma, we found the 2 branches of the GC/UBC lineage as the putative lineage of origin. While these tumors are associated with the same cerebellar glutamatergic lineage, the relative proportions of GC and UBC cells within a tumor varied depending on the tumor class/subclass.
We found that while Group 4 tumors are enriched with differentiating UBCs, both Group 3 and 4 tumor cells express markers of UBC lineage including EOMES, OTX2, and LMX1A. The presence of GC-like cells suggests that both Group 3/4 tumors arise from an earlier bi-potent precursor cell that is capable to generate both UBC and GC, a hypothesis that still needs to be experimentally verified. This result aligns with recently published studies suggesting that Group 4 tumors arise from the subventricular zone of the rhombic lip.16,17 Group 3 medulloblastomas show similarity to both GC and UBC cells; however, tumor cells were not significantly enriched with either signature. Of note, many cells within Group 3 tumors were “not-assigned” after filtering, or co-expressed a non-GC/UBC lineage signature (eg, differentiating Purkinje cells) with GC/UBC marker genes. The inability to assign a specific identity to Group 3 tumor cells could occur if the cell type of origin was not captured in our cerebellar atlas if oncogene expression drives the tumor cell away from all normal lineages, or if cells stuck in a normal developmental trajectory amplify pathways that were previously only lowly expressed. This hypothesis remains to be experimentally tested.
We identified specific genes that are expressed in both tumors and their lineage of origin, as well as genes that are tumor-specific (Table 1). The combination of tumor-specific and lineage-restricted genes overexpressed in tumors can help improve the specificity of multi-factor therapeutic approaches after experimental validation and correlating with patient outcomes.49 In addition, some lineage-specific genes remain active only in progenitors within a lineage (eg, GPR17 in pilocytic astrocytomas are expressed only in OPCs), and could represent developmental dependencies based on their persistent expression in resulting tumors. Importantly, if these tumor-specific genes are not detected or are only minimally expressed in postnatal non-brain tissue, and if the gene expresses a protein localizes to the plasma membrane, this target could be used in next generation CAR-T therapies.
One limitation of our study is that malignant transformation of tumor cells naturally changes their transcriptional profiles, making it difficult to definitively prove cell-of-origin by transcriptional comparisons alone. In the future, rigorous functional validation and integrating other data types including epigenetic states is required.
Current limitations in the understanding of oncogenic vulnerabilities of normal cell types and transformation to malignancy are major roadblocks in identifying new therapies in pediatric neuro-oncology. In our work, we focused on uncovering the potential lineage of origin by integrating a human single-cell cerebellum atlas to pediatric CNS tumors via global comparisons. We have made the results freely available via an interactive graphical interface at brain-match.org for further hypothesis generation and investigation.
Supplementary Material
Contributor Information
Konstantin Okonechnikov, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany.
Piyush Joshi, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany; Developmental Origins of Pediatric Cancer Junior Research Group, German Cancer Research Center (DKFZ), Heidelberg, Germany.
Mari Sepp, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany; Center for Molecular Biology of Heidelberg University (ZMBH), DKFZ-ZMBH Alliance, Heidelberg, Germany.
Kevin Leiss, Center for Molecular Biology of Heidelberg University (ZMBH), DKFZ-ZMBH Alliance, Heidelberg, Germany.
Ioannis Sarropoulos, Center for Molecular Biology of Heidelberg University (ZMBH), DKFZ-ZMBH Alliance, Heidelberg, Germany.
Florent Murat, Center for Molecular Biology of Heidelberg University (ZMBH), DKFZ-ZMBH Alliance, Heidelberg, Germany; INRAE, LPGP, Rennes, France.
Pengbo Beck, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany.
Kenneth Chun-Ho Chan, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany.
Andrey Korshunov, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Clinical Cooperation Unit Neuropathology, German Cancer Research Center (DKFZ), Heidelberg, Germany; Department of Neuropathology, Institute of Pathology, Heidelberg University Hospital, Heidelberg, Germany.
Felix Sah, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Clinical Cooperation Unit Neuropathology, German Cancer Research Center (DKFZ), Heidelberg, Germany; Department of Neuropathology, Institute of Pathology, Heidelberg University Hospital, Heidelberg, Germany.
Maximilian Y Deng, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Division of Pediatric Glioma Research, German Cancer Research Center (DKFZ), Heidelberg, Germany.
Dominik Sturm, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Division of Pediatric Glioma Research, German Cancer Research Center (DKFZ), Heidelberg, Germany; Department of Pediatric Hematology and Oncology, Heidelberg University Hospital, Heidelberg, Germany.
John DeSisto, Morgan Adams Foundation Pediatric Brain Tumor Research Program, University of Colorado School of Medicine, Aurora, CO, USA.
Andrew M Donson, Morgan Adams Foundation Pediatric Brain Tumor Research Program, University of Colorado School of Medicine, Aurora, CO, USA.
Nicholas K Foreman, Morgan Adams Foundation Pediatric Brain Tumor Research Program, University of Colorado School of Medicine, Aurora, CO, USA; Children’s Hospital Colorado, Aurora, CO, USA.
Adam L Green, Morgan Adams Foundation Pediatric Brain Tumor Research Program, University of Colorado School of Medicine, Aurora, CO, USA; Children’s Hospital Colorado, Aurora, CO, USA.
Giles Robinson, Department of Oncology, St Jude Children’s Research Hospital, Memphis, TN, USA.
Brent A Orr, Department of Pathology, St Jude Children’s Research Hospital, Memphis, TN, USA.
Qingsong Gao, Department of Pathology, St Jude Children’s Research Hospital, Memphis, TN, USA; Department of Developmental Neurobiology, St Jude Children’s Research Hospital, Memphis, TN, USA.
Emily Darrow, Department of Developmental Neurobiology, St Jude Children’s Research Hospital, Memphis, TN, USA.
Jennifer L Hadley, Department of Developmental Neurobiology, St Jude Children’s Research Hospital, Memphis, TN, USA.
Paul A Northcott, Department of Developmental Neurobiology, St Jude Children’s Research Hospital, Memphis, TN, USA.
Johannes Gojo, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany; Department of Pediatrics and Adolescent Medicine, Comprehensive Center for Pediatrics and Comprehensive Cancer Center, Medical University of Vienna, 1090 Vienna, Austria; Department of Neuropathology, NN Burdenko Neurosurgical Institute, Moscow, Russia.
Daisuke Kawauchi, Department of Biochemistry and Cellular Biology, National Institute of Neuroscience, NCNP, Tokyo, Japan.
Volker Hovestadt, Department of Pediatric Oncology, Dana-Farber Boston Children’s Cancer and Blood Disorders Center, Boston, USA; Broad Institute of Harvard and MIT, Cambridge, USA.
Mariella G Filbin, Department of Pediatric Oncology, Dana-Farber Boston Children’s Cancer and Blood Disorders Center, Boston, USA; Broad Institute of Harvard and MIT, Cambridge, USA.
Andreas von Deimling, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Clinical Cooperation Unit Neuropathology, German Cancer Research Center (DKFZ), Heidelberg, Germany; Department of Neuropathology, Institute of Pathology, Heidelberg University Hospital, Heidelberg, Germany.
Marc Zuckermann, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany.
Kristian W Pajtler, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany; Clinical Cooperation Unit Neuropathology, German Cancer Research Center (DKFZ), Heidelberg, Germany.
Marcel Kool, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany; Princess Máxima Center for Pediatric Oncology, 3584 CS Utrecht, the Netherlands.
David T W Jones, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Division of Pediatric Glioma Research, German Cancer Research Center (DKFZ), Heidelberg, Germany.
Natalie Jäger, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany.
Lena M Kutscher, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Developmental Origins of Pediatric Cancer Junior Research Group, German Cancer Research Center (DKFZ), Heidelberg, Germany.
Henrik Kaessmann, Center for Molecular Biology of Heidelberg University (ZMBH), DKFZ-ZMBH Alliance, Heidelberg, Germany.
Stefan M Pfister, Hopp Children’s Cancer Center (KiTZ), Heidelberg, Germany; Division of Pediatric Neurooncology, German Cancer Research Center (DKFZ) and German Cancer Consortium (DKTK), Heidelberg, Germany; Division of Pediatric Glioma Research, German Cancer Research Center (DKFZ), Heidelberg, Germany.
Conflict of interest statement
The authors declare no competing financial interests.
Funding
This project was supported by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 819894) and Seventh Framework Programme (FP7-2007-2013) (grant agreement no. 615253).
Authorship
K.O. and P.J. performed bioinformatics analysis related to the comparison of tumors to normal cerebellum cell types. M.Se. and K.S. processed the data and provided detailed annotations for the single-nucleus RNA-sequencing atlas of human cerebellum development. M.Se., K.S., I.S., F.M. and H.K. contributed to discussions about the design and interpretation of analyses. M.Si., P.B., and N.J provided additional tumor bulk data analysis materials. K.C.C., D.K., and M.Z. contributed to the gene selection control. A.K., F.S., M.Y.D., D.S., J.D. N.K.F., A.M.D., A.L.G., J.G., G.R., B.A.O., Q.G., E.D., and J.L.H. provided tumor tissue samples along with processed data when possible and contributed to the design of the study. P.N., J.G., D.K., V.H., M.G.F., A.v.D., M.Z., K.W.P., M.K., D.T.W.J., and N.J. provided expert advice and helped to design the study. K.O., P.J., L.M.K., and S.M.P. prepared the figures and wrote the manuscript based on feedback from all authors. L.M.K, H.K., and S.M.P. conceived and co-led the study.
Data Availability
The human cerebellum snRNA-seq dataset is available in the heiDATA repository QDOC4E. Access information for all tumor bulk and single-cell transcriptome data sets is provided via the interactive online interface at www.brain-match.org in the “Data description” sections as well as in Supplementary Table S2.
References
- 1. Udaka YT, Packer RJ.. Pediatric brain tumors. Neurol Clin. 2018;36(3):533–556. [DOI] [PubMed] [Google Scholar]
- 2. Chemaitilly W, Armstrong GT, Gajjar A, Hudson MM.. Hypothalamic-pituitary axis dysfunction in survivors of childhood CNS tumors: importance of systematic follow-up and early endocrine consultation. J Clin Oncol. 2016;34(36):4315–4319. [DOI] [PubMed] [Google Scholar]
- 3. Jones DT, Banito A, Grünewald TG, et al. Molecular characteristics and therapeutic vulnerabilities across paediatric solid tumours. Nat Rev Cancer. 2019;19(8):420–438. [DOI] [PubMed] [Google Scholar]
- 4. Baslan T, Hicks J.. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat Rev Cancer. 2017;17(9):557–569. [DOI] [PubMed] [Google Scholar]
- 5. Kaatsch P, Rickert CH, Kühl J, Schüz J, Michaelis J.. Population-based epidemiologic data on brain tumors in German children. Cancer: Interdisc Int J Am Cancer Soc. 2001;92(12):3155–3164. [DOI] [PubMed] [Google Scholar]
- 6. Vladoiu MC, El-Hamamy I, Donovan LK, et al. Childhood cerebellar tumours mirror conserved fetal transcriptional programs. Nature. 2019;572(7767):67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pajtler KW, Witt H, Sill M, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell. 2015;27(5):728–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Colin C, Padovani L, Chappe C, et al. Outcome analysis of childhood pilocytic astrocytomas: a retrospective study of 148 cases at a single institution. Neuropathol Appl Neurobiol. 2013;39(6):693–705. [DOI] [PubMed] [Google Scholar]
- 9. Northcott PA, Robinson GW, Kratz CP, et al. Medulloblastoma. Nat Rev Dis Primers. 2019;5(1):1–20. [DOI] [PubMed] [Google Scholar]
- 10. Taylor MD, Poppleton H, Fuller C, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8(4):323–335. [DOI] [PubMed] [Google Scholar]
- 11. Jenseit A, Camgöz A, Pfister SM, Kool ME.. EZHIP: a new piece of the puzzle towards understanding pediatric posterior fossa ependymoma. Acta Neuropathol. 2021;143(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Reitman ZJ, Paolella BR, Bergthold G, et al. Mitogenic and progenitor gene programmes in single pilocytic astrocytoma cells. Nat Commun. 2019;10(1):1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gibson P, Tong Y, Robinson G, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468(7327):1095–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kim JY, Nelson AL, Algon SA, et al. Medulloblastoma tumorigenesis diverges from cerebellar granule cell differentiation in patched heterozygous mice. Dev Biol. 2003;263(1):50–66. [DOI] [PubMed] [Google Scholar]
- 15. Wallace VA. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol. 1999;9(8):445–448. [DOI] [PubMed] [Google Scholar]
- 16. Smith KS, Bihannic L, Gudenas BL, et al. Unified rhombic lip origins of group 3 and group 4 medulloblastoma. Nature. 2022;609(7929):1012–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hendrikse LD, Haldipur P, Saulnier O, et al. Failure of human rhombic lip differentiation underlies medulloblastoma formation. Nature. 2022;609(7929):1021–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hovestadt V, Smith KS, Bihannic L, et al. Resolving medulloblastoma cellular architecture by single-cell genomics. Nature. 2019;572(7767):74–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cardoso-Moreira M, Halbert J, Valloton D, et al. Gene expression across mammalian organ development. Nature. 2019;571(7766):505–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hodge RD, Bakken TE, Miller JA, et al. Conserved cell types with divergent features in human versus mouse cortex. Nature. 2019;573(7772):61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sepp M, Leiss K, Sarropoulos I, et al. Cellular development and evolution of the mammalian cerebellum. bioRxiv. 2012;2021(2021):2020–.473443. [Google Scholar]
- 22. Hänzelmann S, Castelo R, Guinney JG.. gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 2013;14(1):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Capper D, Jones DT, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Diaz-Mejia JJ, Meng EC, Pico AR, et al. Evaluation of methods to assign cell type labels to cell clusters from single-cell RNA-sequencing data. F1000Research. 20198(296):ISCB Comm J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Northcott PA, Buchhalter I, Morrissy AS, et al. The whole-genome landscape of medulloblastoma subtypes. Nature. 2017;547(7663):311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen B, Khodadoust MS, Liu CL, Newman AM, Alizadeh AA.. Profiling tumor infiltrating immune cells with CIBERSORT. Cancer Systems Biology: Methods and Protocols. 2018;1711(1):243–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Venteicher AS, Tirosh I, He bert C, et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science (New York, N.Y.). 2017;355(6332):eaai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Filbin MG, Tirosh I, Hovestadt V, et al. Developmental and oncogenic programs in H3K27M gliomas dissected by single-cell RNA-seq. Science (New York, N.Y.). 2018;360(6386):331–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aldinger KA, Thomson Z, Phelps IG, et al. Spatial and cell type transcriptional landscape of human cerebellar development. Nat Neurosci. 2021;24(8):1163–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cerrato V, Parmigiani E, Figueres-Oñate M, et al. Multiple origins and modularity in the spatiotemporal emergence of cerebellar astrocyte heterogeneity. PLoS Biol. 2018;16(9):e2005513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gojo J, Englinger B, Jiang L, et al. Single-cell RNA-seq reveals cellular hierarchies and impaired developmental trajectories in pediatric ependymoma. Cancer Cell. 2020;38(1):44–59.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gillen AE, Riemondy KA, Amani V, et al. Single-cell RNA sequencing of childhood ependymoma reveals neoplastic cell subpopulations that impact molecular classification and etiology. Cell Rep. 2020;32(6):108023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thul PJ, Åkesson L, Wiking M, et al. A subcellular map of the human proteome. Science. 2017;356(6340):eaal3321. [DOI] [PubMed] [Google Scholar]
- 34. June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC.. CAR T cell immunotherapy for human cancer. Science. 2018;359(6382):1361–1365. [DOI] [PubMed] [Google Scholar]
- 35. van Tilburg CM, Pfaff E, Pajtler KW, et al. The pediatric precision oncology study INFORM: Clinical outcome and benefit for molecular subgroups. J Clin Oncol. 2020 2020;38(18_suppl):LBA10503 [Google Scholar]
- 36. Jones DT, Gronych J, Lichter P, Witt O, Pfister SM.. MAPK pathway activation in pilocytic astrocytoma. Cell Mol Life Sci. 2012;69(11):1799–1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cavalli FM, Remke M, Rampasek L, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. 2017;31(6):737–754.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sharma T, Schwalbe EC, Williamson D, et al. Second-generation molecular subgrouping of medulloblastoma: an international meta-analysis of Group 3 and Group 4 subtypes. Acta Neuropathol. 2019;138(2):309–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Riemondy KA, Venkataraman S, Willard N, et al. Neoplastic and immune single-cell transcriptomics define subgroup-specific intra-tumoral heterogeneity of childhood medulloblastoma. Neuro-Oncology. 2021;24.(2):273–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen M, Mao X, Huang D, et al. Somatostatin signalling promotes the differentiation of rod photoreceptors in human pluripotent stem cell-derived retinal organoid. Cell Prolif. 2022;55(7):e13254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Galvis L, Gonzalez D, Bonilla C.. Relapsed high-risk medulloblastoma: stable disease after two years of treatment with somatostatin analog-case report. Cureus. 2016;8(1):e446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mikkelsen TS, Ku M, Jaffe DB, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448(7153):553–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dubuc AM, Remke M, Korshunov A, et al. Aberrant patterns of H3K4 and H3K27 histone lysine methylation occur across subgroups in medulloblastoma. Acta Neuropathol. 2013;125(3):373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Relling MV, Rubnitz JE, Rivera GK, et al. High incidence of secondary brain tumours after radiotherapy and antimetabolites. The Lancet. 1999;354(9172):34–39. [DOI] [PubMed] [Google Scholar]
- 45. Yamanaka R, Hayano A, Kanayama T.. Radiation-induced gliomas: a comprehensive review and meta-analysis. Neurosurg Rev. 2018;41(3):719–731. [DOI] [PubMed] [Google Scholar]
- 46. Deng MY, Sturm D, Pfaff E, et al. Radiation-induced gliomas represent H3-/IDH-wild type pediatric gliomas with recurrent PDGFRA amplification and loss of CDKN2A/B. Nat Commun. 2021;12(1):5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. DeSisto J, Lucas JT, Xu K, et al. Comprehensive molecular characterization of pediatric radiation-induced high-grade glioma. Nat Commun. 2021;12(1):1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lonser RR, Walbridge S, Vortmeyer AO, et al. Induction of glioblastoma multiforme in nonhuman primates after therapeutic doses of fractionated whole-brain radiation therapy. J Neurosurg. 2002;97(6):1378–1389. [DOI] [PubMed] [Google Scholar]
- 49. Land CA, Musich PR, Haydar D, Krenciute G, Xie Q.. Chimeric antigen receptor T-cell therapy in glioblastoma: charging the T cells to fight. J Transl Med. 2020;18(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The human cerebellum snRNA-seq dataset is available in the heiDATA repository QDOC4E. Access information for all tumor bulk and single-cell transcriptome data sets is provided via the interactive online interface at www.brain-match.org in the “Data description” sections as well as in Supplementary Table S2.