

Abstract
Background
Glioma stem cells (GSCs) are a subpopulation of tumor cells with self-renewal and tumorigenic capabilities in glioblastomas (GBMs). Diffuse infiltration of GSCs facilitates tumor progression and frustrates efforts at effective treatment. Further compounding this situation is the currently limited understanding of what drives GSC invasion. Here we comprehensively evaluated the significance of a novel invasion-related protein, Family with Sequence Similarity 129 Member A (FAM129A), in infiltrative GSCs.
Methods
Western blotting, immunohistochemistry, and gene expression analysis were used to quantify FAM129A in glioma specimens and cancer datasets. Overexpression and knockdown of FAM129A in GSCs were used to investigate its effects on tumor growth and invasion. RNA-seq, qRT-PCR, western blotting, and co-precipitation assays were used to investigate FAM129A signaling mechanisms.
Results
FAM129A is preferentially expressed in invasive frontiers. Targeting FAM129A impairs GSC invasion and self-renewal. Mechanistically, FAM129A acted as a positive regulator of Notch signaling by binding with the Notch1 intracellular domain (NICD1) and preventing its degradation.
Conclusions
FAM129A and NICD1 provide a precise indicator for identifying tumor margins and aiding prognosis. Targeting them may provide a significantly therapeutic strategy for GSCs.
Keywords: FAM129A, glioma stem cell, NICD, Notch pathway, tumor invasion
Graphical Abstract
Graphical Abstract.

Key Points.
FAM129A and NICD1 are highly elevated in areas of GSC invasion and correlate with poor prognosis.
Diminishing FAM129A reduces the self-renewal capacity and tumorigenic potential of GSCs.
FAM129A contributes to GBM progression and GSC invasion via binding to the NICD1 and activating Notch signaling.
Importance of the Study.
Invasive GSCs exhibit diverse features of diffuse infiltration, such as perivascular adhesion, infiltration along myelin sheaths, and collective migration. However, the molecular features of GSCs in above invasive status are poorly understood. Here, we demonstrate that the FAM129A-NICD1 complex is preferentially expressed in areas of GSC invasion, revealing a new feature for intratumoral heterogeneity. Notch signaling-dependent cellular communication in infiltrative GSCs requires the involvement of FAM129A. Our work will serve the purpose of recognizing the margins and the degree of tumor invasion and provide a potential drug target for both Notch pathway antagonism and anti-GSCs therapy.
Malignant gliomas, especially glioblastomas, are predominantly lethal tumors of the central nervous system.1 The heterogeneity and plasticity of GBM are critical factors contributing to the resistance to chemoradiotherapy and postoperative recurrence.2,3 These malignant features are thought to originated from GSCs, a subpopulation of tumor cells with self-renewing and tumorigenic capabilities.4,5 The main distinction between GBM and other solid tumors is that GBM (and GSCs) extensively infiltrates the surrounding brain tissue, but rarely metastasizes to other organs.6 The infiltrating potential derived from residual or distantly migrated GSCs impedes the efficacy of surgical resection and contributes to recurrence.7 However, the molecular mechanisms that drive the formation of invasion potential in GSCs remain unknown.
In this study, we analyzed the molecular characteristics of GSCs located in the infiltrating area. FAM129A was one of the key candidate proteins involved in GSC invasion. FAM129A, aliases C1orf24, GIG39 or Niban, is an endoplasmic reticulum stress related protein that regulates the phosphorylation of eIF2a and S6K1/4E-BP1 to modulate cell death.8 FAM129A is upregulated in several cancers, including renal cancers,9,10 prostate cancers,11,12 thyroid carcinomas,13 and hepatocellular carcinoma.14 In hepatocellular carcinoma and prostate cancer, loss of FAM129A affects the focal adhesion kinase signaling pathway and unfolded protein response.11,14 FAM129A may also inhibit autophagy by activation of the AKT/mTOR/p70S6K axis in thyroid carcinoma cell lines.13 However, how FAM129A regulates GSC behaviors remains unknown, and the mechanism by which it accelerates GBM progression is poorly understood.
We observed elevated expression of the FAM129A in the invasive region of GBM. Downregulation of FAM129A compromised the self-renewal ability and tumorigenic potential of GSCs and further prevented its infiltration into normal brain parenchyma. Thus, we propose that FAM129A is crucial to the maintenance of infiltrative potential in GSC and represents a therapeutic target for the treatment of GBM.
Materials and Methods
Patients and Clinical Samples
All glioma samples were surgically obtained from January 2016 to March 2022 at Tongji Hospital of Huazhong University of Science and Technology (Wuhan, China), and all patients had signed informed written consent before surgery in accordance with a protocol approved by the Research Ethics Committee of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China (Serial no. TJ-IBR20181111). No patients received radiation or chemotherapy prior to surgery. The clinical information was detailed in Supplementary Tables 1 and 2.
Cells and Cell Culture
All GSC lines (T3359, T456, T3691 and T387) and neural progenitor cells (NPCs) were gifted by professor Shideng Bao and Jeremy N. Rich. All cells were cultured in a humidified 37°C, 5% CO2 incubator. Moreover, all cultured cell lines were confirmed to be free from mycoplasma infection. Details were described in Supplementary Materials and Methods.
Animals and Intracranial Tumor Assay
The experimental BALB/C nude mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd (Beijing, China). And the animal experiments were authorized by the Institutional Animal Care and Use Committee (IACUC) of Huazhong University of Science and Technology. Ketamine and xylazine cocktail were used for anesthesia by intraperitoneal injection before all surgical procedures. GSCs (2 × 104 cells/mouse) after anticipatory treatment were implanted into the right frontal lobes of 4-week-old mice. And the manifestation of neurological signs, severe weight loss, and physical impairment were considered as clinical endpoints for the survival experiments.
Immunohistochemistry (IHC)
The protocol of IHC was implemented as described previously.15 The antibodies used and quantification of protein expression levels located in specific regions of the tumor were described in Supplementary Materials and Methods. The histochemical scoring (H-SCORE) was utilized to measure the expression level of FAM129A and NICD1 in the tissue microarray of the glioma cohort (Figure 6). Detailed scoring data is summarized in Supplementary Table 2.
Figure 6.
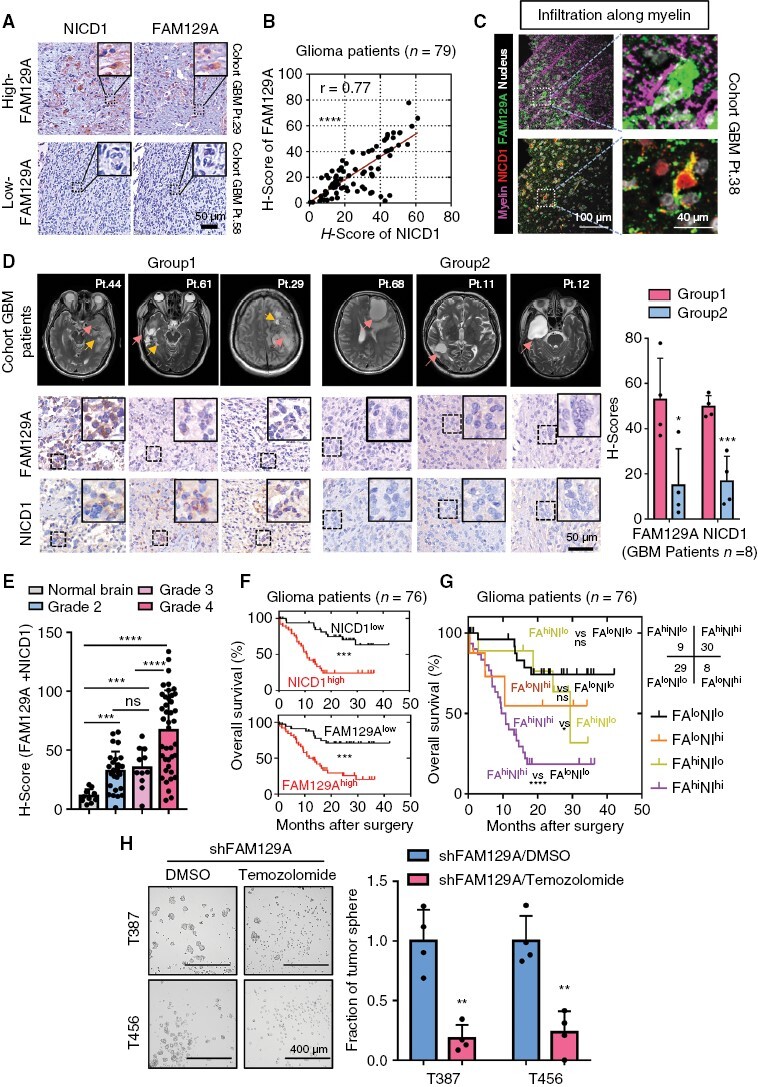
The clinicopathological features and diagnostic value for prognosis of NICD1 and FAM129A complex in glioma cohort. (A) Immunohistochemical staining of FAM129A and NICD1 in same human GBM tissues. Scale bars: 50 μm. (B) Scatter plot indicating the correlation between FAM129A and NICD1 in 79 gliomas tissues. The clinical information and H-scores of 79 patients with glioma were detailed in Supplementary Table 2, which also corresponding to (B, D–G). (C) IF staining of FAM129A and NICD1 in infiltrated area along the myelin sheath. Scale bars: 100 μm; enlarged image: 40 μm. The thickness of tumor sections was 10 μm. (D) MRI T2 images of GBM patients and the corresponding histochemical staining of FAM129A and NICD1. All MRI images were obtained from the patients’ preoperative examination, at which time they did not receive any treatment. Pink arrows: main tumor body; Yellow arrows: the infiltrating front. Scale bars of IHC image: 50 μm. (E) Histogram describing the distribution of H-scores (FAM129A + NICD1) in different histopathological subgroups. Normal brain, n = 10; Grade 2, n = 26; Grade 3, n = 11; Grade 4, n = 41. (F) Overall survival curves of 76 glioma subjects based on immunohistochemical staining data of NICD1 (above) and FAM129A (bottom). (G) Kaplan–Meier survival curves of 76 glioma subjects based on the combination of FAM129A and NICD1 immunohistochemical staining data. (H) Bright-field microscopy indicating the representative images of tumor-sphere formation (scale bars: 400 μm). And histogram showing the quantification of tumor-spheres formation (n = 4). Data are presented as the mean ± SD. ns, no significance; *P < .05; **P < .01; ***P < .001; ****P < .0001.
Western Blotting, Co-immunoprecipitation Assay (Co-IP) and Immunofluorescent Staining (IF)
The protocol of western blotting, co-IP and IF was implemented as described previously.16 The thickness of all tumor sections used in IF was 10 μm. Supplementary Materials and Methods listed the antibodies used.
RNA Isolation and Real-time PCR (qRT-PCR)
The protocol of RNA isolation and real-time PCR was implemented as described previously.16 All primer sequences are listed in Supplementary Table 3.
GSCs Self-renewal Assays
Cell Viability Assay, Sphere Formation, Limiting Dilution Assay, and EdU incorporation assay were employed to measure cellular self-renewal capacity as described previously.16 Detailed experimental steps were described in Supplementary Materials and Methods.
GSCs Movement and Invasion Assay
Transwell invasion assay and cell spheroid seeding assay were employed to measure the invasion and movement ability of GSC. Detailed experimental steps were described in Supplementary Materials and Methods.
Plasmid Generation, Lentiviral Transfection, and Reagent Treatment
In this study, the sequence of shRNAs for inhibiting FAM129A or nontargeting NT shRNA was acquired from Sigma-Aldrich. We screened two shRNA sequences (shFAM129A#1, TRCN0000122151; shFAM129A#2, TRCN0000140457) for all subsequently related experiments that were performed at least in triplicate. Cell lines stably expressing either FAM129A or NICD1 were constructed using lentivirus.
For the application of cell reagent, the following is in detail: (1), lysosome inhibitor chloroquine (MCE, #54-05-7), 5 μM, incubated with GSCs for 6 h; (2), proteasome inhibitor MG132 (MCE, #133407-82-6), 5 μM, incubated with GSCs for 6 hours; (3), γ-secretase inhibitor DAPT (MCE, #HY-13027), 40 μM, incubated with GSCs for 24 hours; (4) caspase-3 inhibitor Z-DEVD-FMK (MCE, # HY-12466), 20 μM. Detailed experimental steps and the application of cell reagent were described in Supplementary Materials and Methods.
Single Sample Gene Set Enrichment Analysis (ssGSEA), Bioinformatics Databases and RNA-seq Analysis
Detailed bioinformatics screening methods and data processing were described in Supplementary Materials and Methods.
Quantification and Statistical Analysis
All statistical processing was conducted by utilizing R software (version 4.0.2) or GraphPad Prism (version 7.0), and all data were presented as mean ± SD. The unpaired Student’s t-test was used for comparison between two groups and one-way ANOVA for multiple group comparison. Moreover, the generation of Kaplan–Meier curves was validated by using the Log-rank test. P < .05 was considered statistically significant.
Results
Identification of the Key Characteristic Molecules in Invasive GSCs
To elucidate the typical histopathological features of infiltrative GSC in GBM tissues, we labeled GSCs with SOX2 or CD133. From this, we identified at least three patterns of GSC invasion: including perivascular invasion, collective invasion and infiltration along myelin sheaths (Figure 1A). Based on these observations, we speculated that the infiltrative GSC has a distinctive spatiotemporal gene expression signature. Thus, we employed three independent transcriptome databases (TCGA, CGGA, and GSE119834) for screening the distinctive molecular patterns in infiltrative GSCs. We first mapped out the specific gene clusters of GSCs (Figure 1B). We then utilized the ssGSEA algorithm to calculate the invasion and migration gene signature scores of the GBM tissues in the CGGA-GBM cohort and divided 249 GBM patients into two clusters, with cluster 2 having a more aggressive profile (Figure 1C). Our results showed nine genes (TNC, FAM129A, METTL7B, FCGR2A, CHI3L1, SHOX2, HSPA6, HOXD10, and TNFAIP6) met all the following screening criteria: (1) enriched in cluster 2, (2) high expression in the GBM tissue versus normal brain tissue, and (3) elevated expression in GSCs (Figure 1D and E). Prognostic analysis of mesenchymal GBM further identified FAM129A as the candidate of our subsequent study (Figure 1E and Supplementary Figure S1).
Figure 1.
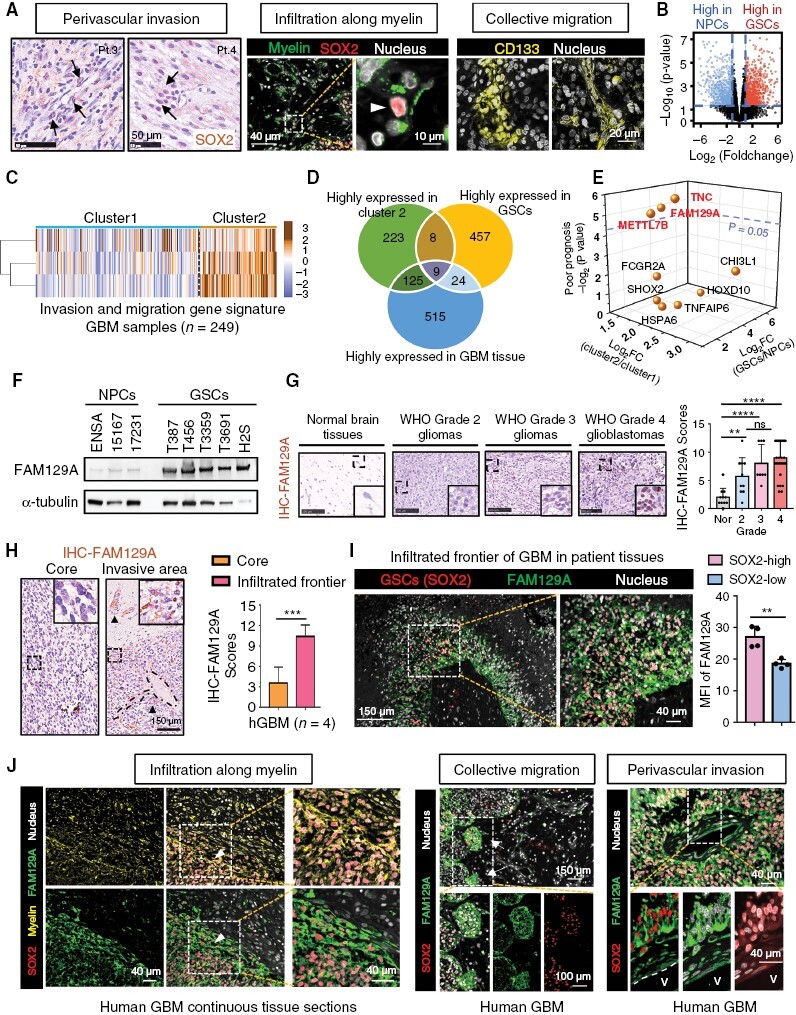
Identification of essential molecules in invasive GSCs. (A) Three manifestations of GSCs invasion. Left: immunohistochemical staining of SOX2 (marker of GSC) in perivascular areas of GBM, scale bars: 50 μm; Middle and right: Immunofluorescence staining in human GBM, MBP (marker of myelin sheath) is shown in green, SOX2 in red, CD133 (marker of GSC) in yellow and DAPI in gray. Scale bars: middle, 10 μm; right, 20 μm. (B) Volcano plot showing the distribution of differential genes in 44 GSCs and 10 NPCs (GSE119834). High-expressed genes in GSCs is shown in red, and that in NPCs is blue. (Cutoff: fold change >2, adjusted P value < .01). (C) Heatmap reflecting cluster grouping according to the ssGSEA scores of invasive and migrative gene signatures. Dataset for running ssGSEA: CGGA-GBM cohort (n = 249; cluster 1: low invasion and migration scores, n = 171; cluster 2: high invasion and migration scores, n = 78). (D) Venn diagram showing genes that were highly expressed in cluster 2 GBM patients (compared to cluster 1), GSC (compared to NPC) and GBM tissues (compared to normal brain tissues). (E) Scatter plot showing the distribution of priority of candidate genes. (F) Western blotting of FAM129A protein in GSCs and NPCs. (G) Immunohistochemical staining of FAM129A in normal brain tissues and gliomas. Scale bars: 100 μm. And the right histogram describing the distribution of FAM129A IHC-scores (ns, no significance; **P < .01; ****P < .0001, one-way ANOVA followed by Tukey’s test). (H) Immunohistochemical staining of FAM129A in core and invasive region. Dotted line: blood vessels; Arrows: perivascular FAM129A positive cells. Scale bars: 100 μm. And right histogram depicting the IHC quantification of FAM129A in center and invasive frontier (n = 4). See the detailed pictures in Supplementary Figure S3. (I) Immunofluorescence staining of FAM129A (green) and SOX2 (red) in human GBM tissues. DAPI is shown in gray. Also shown is quantification of the mean fluorescence intensity (MFI) of FAM129A (right, n = 4). Scale bars: 150 μm; enlarged image: 40 μm. (J) Immunofluorescence staining in human GBM, MBP is shown in yellow, SOX2 in red, FAM129A in green and DAPI in gray. Scale bars: 40 μm; middle, 100 μm; right: 40 μm.
High Correlation of FAM129A with Invasive GSC
To further distinguish between FAM129A expression in myeloid, vascular, and GSC cell populations, we re-analyzed the single cell sequencing datasets (GSE138794) and found that FAM129A is predominantly expressed in glioma tumor cells, although also expressed in endothelial cells (Supplementary Figure S2A). IF co-staining of CD31, CD68, Nestin and Iba1 with FAM129A further confirmed that the predominate localization of FAM129A was within tumor cells in GBM tissues (Supplementary Figure S2B). Next, we found that FAM129A was preferentially expressed in GSCs but not in bulk tumor, matched differentiated cells, or NPCs (Figure 1F and Supplementary Figure S2C–G).
To verify the aforementioned screening criteria, we subsequently detected FAM129A expression in normal brain tissues (n = 11) and glioma tissues with different WHO grades (grade 2, n = 10; grade 3, n = 8; grade 4, n = 32). The results confirmed that FAM129A expression in glioma tissues, especially in GBM, is unambiguously higher than normal brain tissues (Figure 1G). Moreover, IHC staining confirmed that FAM129A+ cells were mainly located in the infiltrated areas instead of the central tumor area (Figure 1H and Supplementary Figure S3). Confocal imaging affirmed that the majority of these FAM129A+ cells were GSCs in the infiltrating region (Figure 1I). And we confirmed that FAM129A was enriched in the infiltrating areas of GSC, coinciding with our screening results (Figure 1J). Ultimately, we ascertained the feasibility of FAM129A as a signature molecule in the infiltrative GSCs.
FAM129A Maintains GSC Self-renewal in an Apoptosis—Independent Manner
We next investigated the potential dependency of GSCs on FAM129A by inhibiting or overexpressing FAM129A. As shown by cell viability assay, the proliferation of GSCs was suppressed by FAM129A silencing and accelerated after overexpression (Figure 2A and B). Tumor-sphere formation assay revealed that disruption or enrichment of FAM129A caused the impairment or elevation of GSC spheres formation (Figure 2C, D and Supplementary Figure S4A–C). As assessed by extreme limiting dilution assay, FAM129A elimination or supplementation resulted in the restraint or acceleration of self-renewal, respectively (Figure 2E, F and Supplementary Figure S4D). Next, we employed the EdU incorporation assay and found that downregulation of FAM129A decreased DNA replication of GSCs (Figure 2G and Supplementary Figure S4E, F). Unexpectedly, SOX2, not OLIG2, appeared to decrease after interfering with the expression of FAM129A, although both SOX2 and OLIG2 are key transcription factors indispensable to GSC self-renewal (Supplementary Figure S5A). Similarly, we also observed that upregulation of apoptosis-related proteins (cleaved PARP and caspase-3) occurred after the knockdown of FAM129A (Supplementary Figure S5B). To clarify whether the decrease in “stemness” of GSCs was due to apoptosis caused by FAM129A knockdown, we used inhibitors of caspase-3 and demonstrated that the reduced self-renewal ability did not ameliorate after inhibition of apoptosis, and that SOX2 remained at a depressed level (Figure 2H and Supplementary Figure S5C). This indicated that FAM129A exerts its control on SOX2 expression through an apoptosis-independent pathway.
Figure 2.
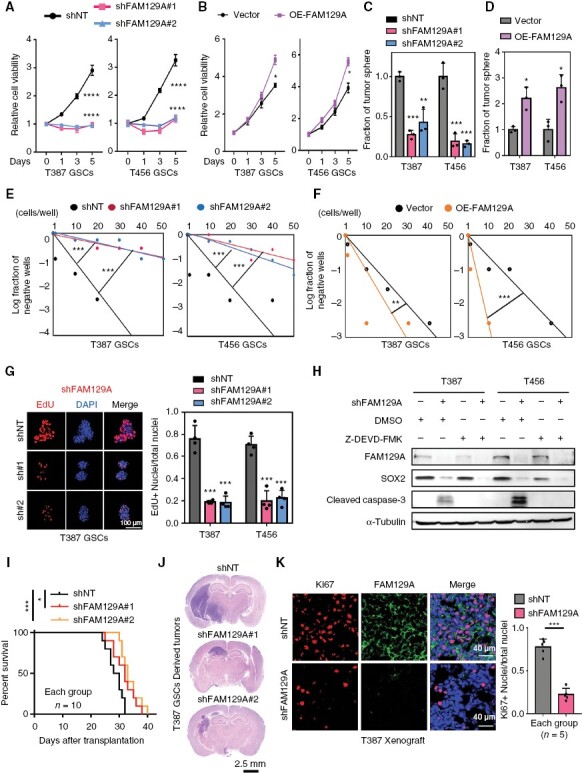
FAM129A maintains the self-renewal and tumorigenic potential of GSCs. (A, B) CellTiter-Glo assays displaying the measurement of GSC proliferation (n = 6). (C, D) Histogram showing the quantification of tumor-spheres formation (n = 3). (E, F) Limiting dilution assays showing the spheroidization efficiency in the indicated cells. (G) Confocal imaging (left) of EdU incorporation in GSC tumor-spheres (n = 4). And EDU is shown in red, DAPI in blue. Also shown is quantification of the fraction of EdU+ cells (right). Scale bars: 100 μm. (H) Western blot analysis of cleaved caspase-3 and SOX2 proteins in the indicated cells. Z-DEVD-FMK (20 μM), a caspase-3 inhibitor. (I) Kaplan–Meier survival curve of immunocompromised mice bearing T387 GSCs expressing shNT or shFAM129A (n = 10 for each group). (J) H&E staining of brain sections from immunocompromised mice 24 days after transplantation. Scale bars: 2.5 mm. (K) IF image (left) in xenografts. Ki67 is in red, FAM129A in green and DAPI in blue. Scale bars: 40 μm. Also shown is the quantification of the Ki67+ cell in GBM xenografts (n=5 for each group). Data are presented as the mean ± SD. *P < .05; **P < .01; ***P < .001.
In vivo, our experiments showed that shFAM129A mice survived significantly longer than shNT mice (Figure 2I). Hematoxylin and eosin (H&E) staining revealed larger tumor masses in shNT mice than in shFAM129A mice (Figure 2J). Finally, we compared the apoptotic level and proliferation ability of tumors from shNT and shFAM129A mice by IF staining of cleaved caspase-3 and Ki67. Consistent with the in vitro results, tumors from shFAM129A mice exhibited high levels of cleaved caspase-3 and lower Ki67 expression (Figure 2K and Supplementary Figure S5D, E).
FAM129A Affects the Invasion and Migration of GSCs
When we conducted the H&E staining of xenograft tumors, we observed that shFAM129A mice showed smoother tumor borders (Supplementary Figure S6A). We also observed typical features of invasive GSC in xenograft tumors that mimic those in human GBM tissue (Figure 3A). Fluorescent labeling of N-cadherin revealed that collective invasion was not isolated and disorganized, but rather accompanied by well-defined migratory bands (Figure 3A). Moreover, the elevated expression of FAM129A in the infiltrating margin was similar to that of human samples (Figure 3B). Subsequently, we found that coincident with the attenuation of FAM129A, the group invasion was curbed, and its associated molecules MMP9 and N-cadherin were also downregulated (Figure 3C, D, and Supplementary Figure S6B).
Figure 3.
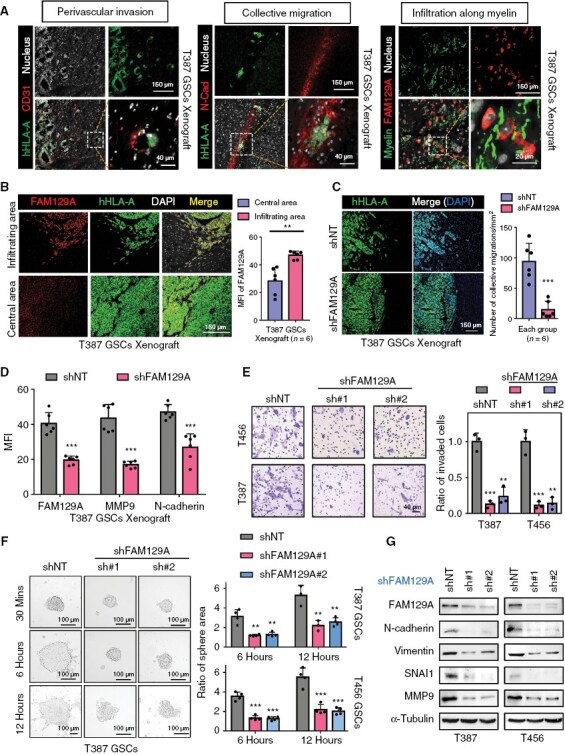
FAM129A is essential for invasion of GSCs. (A) Three manifestations of GSCs invasion in xenograft. hHLA-A: specific labeling of human-derived tumor cells; CD31: marker of vessels; N-cad: N-cadherin, a cell adhesion molecule; MBP: marker of myelin; Scale bars: 150 μm; enlarged image: 20 μm. (B) IF staining of FAM129A and hHLA-A in tumor periphery or center of xenograft. Also shown is quantification of the mean fluorescence intensity (MFI) of FAM129A (right, n = 6). Scale bars: 150 μm. (C) IF staining of hHLA-A in xenograft. Also shown is quantification of number of collective migrations in tumor periphery (right, n = 6). Scale bars: 150 μm. (D) MFI quantification of FAM129A, MMP9 and N-cadherin in T387 GSCs xenograft transduced with shNT or shFAM129A (n = 6). See the detailed pictures in Supplementary Figure S6B. (E) Transwell assay showing the effect of FAM129A on GSCs invasion. Also shown is quantification of number of invading cells (right, n = 3). Scale bars: 40 μm. (F) Bright-field microscopy (left) showing the tumor-sphere extension of T387 GSCs transduced with shNT or shFAM129A. And the quantification of relative extension is shown (right, n = 4). Scale bars: 100 μm. (G) Western blot analysis of FAM129A and EMT-related proteins in the indicated cells. Data are presented as the mean ± SD. *P < .05; **P < .01; ***P < .001.
By interfering with the expression of FAM129A in vitro, transwell assay demonstrated that the invasive ability of GSCs was diminished. Conversely, GSC invasion was enhanced after overexpression of FAM129A (Figure 3E and Supplementary Figure S6C). FAM129A silencing suppressed the movement and cytoskeletal extensions of GSCs (Figure 3F and Supplementary Figure S6D). We speculated that the above phenomenon is associated with altered expression patterns of epidermal mesenchymal transformation (EMT)-related molecules. Congruent with this, we discovered a high correlation between FAM129A and EMT-related representative molecules in GBM patients based on transcriptomic data of CGGA and TCGA (Supplementary Figure S6E, F). Western blotting further verified that protein levels of EMT-related molecules (N-cadherin, vimentin, and SNAI1) were attenuated after knockdown of FAM129A under in vitro culture conditions (Figure 3G). We conclude that FAM129A knockdown effectively interrupted the maintenance of invasive properties of GSCs in vivo and in vitro.
FAM129A is a Novel Notch-related Molecule of GSCs
To map the assumed signaling mechanisms by which FAM129A may support GSCs’ self-renewal and invasion, we performed next-generation RNA sequencing on the T387 GSCs infected with either shNT or shFAM129A (Figure 4A). Gene ontology analysis of downregulated gene sets suggested that loss of FAM129A caused perturbation of nervous system development, cell proliferation, cell adhesion, and cell migration, highly consistent with our previous observation (Figure 4B). Furthermore, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis identified the impairment of Notch signaling after FAM129A deficiency in T387 GSCs (Figure 4C). We found FAM129A had a highly positive correlation with Notch-related molecules, such as Notch1, Notch3, Hey1, Hes1 and MYC (Supplementary Figure S7A–C).
Figure 4.
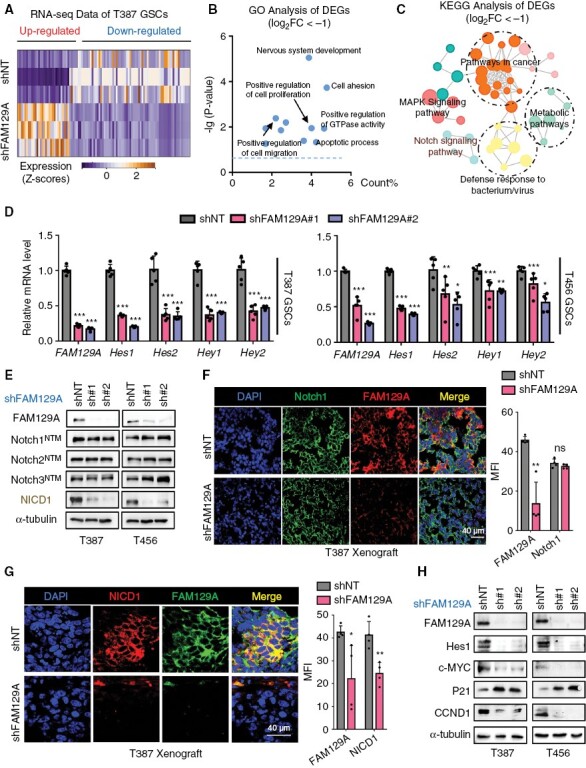
FAM129A knockdown inhibits Notch signaling in GSCs. (A) Heatmap reflecting the results of RNA-seq upon FAM129A knockdown. (B, C) Scatter plot reflecting the results of GO (B) and KEGG analysis (C) using RNA-seq data. (D) qRT-PCR analysis of mRNA expression of FAM129A, Hes1, Hes2, Hey1 and Hey2 in T387 and T456 GSCs transduced with shNT or shFAM129A (n = 5; one-way ANOVA with Tukey’s multiple comparisons). (E) Western blotting of FAM129A, Notch1NTM, Notch2NTM, Notch3NTM and NICD1 protein expression in the indicated cells. (F) IF co-staining of FAM129A and Notch1 in xenografts. And the quantification of MFI is shown (right, n = 4). Scale bars: 40 μm. (G) IF co-staining of FAM129A and NICD1 in xenografts. And the quantification of MFI is shown (right, n = 4). Scale bars: 40 μm. (H) Western blotting of FAM129A and NICD1 targets (CCND1, Hes1, c-MYC, P21 and MMP9) in indicated cells. Data are presented as the mean ± SD. ns, no statistical difference; *P < .05; **P < .01; ***P < .001.
To further explore the functional relevance of FAM129A to the modulation of Notch signaling, we performed qRT-PCR analyses after knockdown of FAM129A in T387 and T456 GSCs lines. Our data showed that the mRNA levels of Notch target genes (Hes1, Hes2, Hey1, Hey2, MYC) were downregulated after silencing FAM129A (Figure 4D and Supplementary Figure S7D). Subsequently, we performed western blotting to detect the attenuation of the Notch pathway and the result showed that deletion of FAM129A mainly led to the reduction of NICD1 both in T387 and T456 GSCs lines (Figure 4E). No variation in Notch1–3 was perceptible, however (Figure 4E). IF also showed that FAM129A disruption predominantly downregulated the levels of NICD1, not Notch1, to interfere with Notch signal activation in vivo and in vitro (Figure 4F, G and Supplementary Figure S8A, B). Moreover, our results showed that FAM129A deficiency led to an increase of P21 and a decrease of c-MYC, CCND1, Hey1, and Hes1 (Figure 4H and Supplementary Figure S8C). Thus, FAM129A facilitates the activation of the Notch pathway via regulating NICD1 in GSCs.
FAM129A Prevents Ubiquitylation of NICD to Control its Turnover
Given the importance and complexity of the Notch pathway in controlling stem cell niches, the level of NICD is highly regulated.17,18 We used DAPT to inhibit its generation, but FAM129A did not show significant changes in transcript levels (Supplementary Figure S9A), and its protein levels fluctuated with increasing concentrations (Supplementary Figure S9B). We speculated that this trend may be due to adaptive changes caused by downregulation of NICD1. Besides, we were unable to discern any variations in Notch ligands or in γ-secretase expression (Supplementary Figure S9C).
It has been demonstrated that ubiquitination functionally involves the turnover of NICD1 in mammals.19,20 IF staining evidenced the presence of significant co-localization of FAM129A with NICD1 (Figure 5A and Supplementary Figure S9D). Thus, we hypothesized that FAM129A binds to NICD1 to control its turnover. We verified the interaction between NICD1 and FAM129A by using co-immunoprecipitation (Figure 5B). Therefore, we treated GSCs with MG132 (a proteasomal inhibitor) or chloroquine (a lysosomal acidification inhibitor). After FAM129A knockdown, DMSO-treated cells (as a control group) exhibited reduced NICD1 levels (Figure 5CD and Supplementary Figure S9E). However, this reduction in NICD1 level was ablated by treatment with MG132 (Figure 5C, D). NICD1 levels were not rescued by chloroquine treatment (Supplementary Figure S9E). Next, we sought to determine whether increased ubiquitylation of endogenous NICD1 occurs when FAM129A is silenced. Co-IP of NICD1 and ubiquitin indicated that NICD1 was highly ubiquitinated in MG132-treated GSCs after FAM129A knockdown (Figure 5E and Supplementary Figure S9F).
Figure 5.
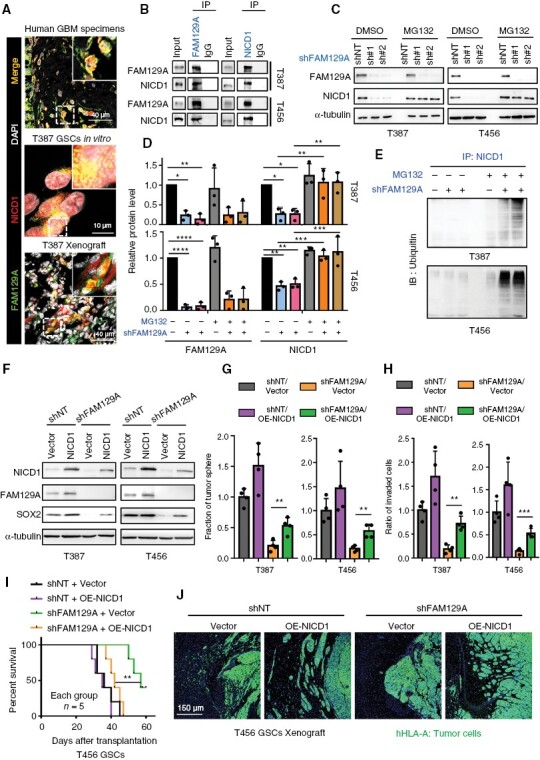
Depletion of FAM129A caused proteasome-mediated degradation of NICD1. (A) IF co-staining of FAM129A and NICD1 in human GBM (scale bars, 40 μm), GSC (scale bars, 10 μm) and xenograft (scale bars, 40 μm). The thickness of tumor sections was 10 μm. (B) Co-immunoprecipitation of FAM129A (left) or NICD1 (right) in T456 and T387 GSC lines (IgG as a control antibody). (C, D) Western blotting of FAM129A and NICD1 in indicated cells. GSC was treated with MG132 6 h for harvest. And the quantification of protein is shown in (D). (E) Co-IP of NICD1 and then western blotted with anti-ubiquitin in T387 and T456 GSCs. Input is shown in Supplementary Figure S9F. (F) Western blotting of FAM129A, NICD1 and SOX2 in indicated cells. (G, H) Histogram showing the quantification of tumor-spheres formation (G) and number of invading cells (H). See the detailed pictures in Supplementary Figure S10B, C. (I) Kaplan–Meier survival curve of immunocompromised mice bearing T456 GSCs in indicated groups (n = 5 for each group). (J) IF staining of hHLA-A in xenograft. Scale bars: 150 μm. Data are presented as the mean ± SD. ns, no statistical difference; *P < .05; **P < .01; ***P < .001.
To verify whether the Notch pathway is involved in the maintenance of the infiltrative GSC by FAM129A, we overexpressed NICD1. The loss of invasive capacity and self-renewal ability of GSCs that we had observed following FAM129A downregulation was rescued by the overexpression of NICD1 (Figure 5FH, and Supplementary Figure S10A–C). The intracranial transplantation model further illustrated that the remediation of Notch signaling accelerates the tumor progression and invasion (Figure 5I, J). Collectively, these results suggest that NICD1 is degraded through the proteasome and that FAM129A binds it to restrain its degradation in GSCs.
The Interaction of FAM129A and NICD1 is a Diagnostic or Therapeutic Target
We assigned a cohort containing 79 glioma subjects (Grade 1, n = 1; Grade 2, n = 26; Grade 3, n = 11; Grade 4, n = 41) from Tongji Hospital and examined whether FAM129A could be exploited diagnostically in GBM.
First, we employed IHC staining of the same area in continuous tissue sections, which showed a positive correlation between the expression of FAM129A and NICD1 (Figure 6A, B). We confirmed the spatial consistency correlation between FAM129A and NICD1 in characteristic infiltrated areas of glioma (Figure 6C and Supplementary Figure S11A–C). We then collected T2 MRI images of patients with clinically significant GBM and divided the patients into two groups based on the characteristics of the tumor body margins, where group 2 possessed smoother margins. Group 1 possessed invasive anterior segment or distant lesions, and the histological scores of such patients reflected consistently high expression of FAM129A and NICD1 (Figure 6D).
Next, we evaluated the clinicopathological relevance of these two proteins against its H-scores. FAM129AhighNICD1high subjects manifested a strikingly higher histopathological grade (Figure 6E). Ultimately, Kaplan–Meier and univariate Cox proportional hazard regression analyses of 76 glioma patients (41 GBM patients) with prognostic follow-up information revealed that raised FAM129A and NICD1 levels were associated with an unfavorable prognosis (Figure 6F). The combination of FAM129A and NICD1 expression exhibited that the cases with FAM129AhighNICD1high had a distinctly poorer prognosis (OS, overall survival, 10.1425 months) compared with the other groups (FAM129AlowNICD1low, OS: 29.3589 months; FAM129AlowNICD1high, OS: 13.4877 months; FAM129AhighNICD1low, OS: 22.7689 months) (Figure 6G).
Considering the treatment-related implications of FAM129A, we reviewed public databases and collected tissues from recurrent GBM treated with temozolomide after surgery, and there was no significant difference in FAM129A levels compared to primary GBM, as well as in vitro cultured GSC compared to treated with temozolomide (Supplementary Figure S12A–E). Then we found knockdown of FAM129A in combination with temozolomide led to the extinction of GSC, suggesting a possible involvement of FAM129A in temozolomide resistance (Figure 6H and Supplementary Figure S12F).
Taken together, evidence from the human glioma cohort highlighted that the combination of FAM129A and NICD1 provides a precise indicator of identifying tumor margins and targeting them could be a significant therapeutic strategy for GBM.
Discussion
Invasive GSCs exhibit diverse features of diffuse infiltration of brain parenchyma, such as perivascular invasion, infiltration along myelin sheaths, and collective migration, and retain intercellular signaling and communication.21,22 Notch signaling is considered one of the key factors involved in maintaining these invasive behaviors.23,24 The Notch1-SOX2 positive feedback loop has been shown to be a driving component of GSC migration along white matter tracts, and GSCs in the invasive frontier exhibit excessive activation of Notch signaling.25 In this study, FAM129A silencing caused the reduction of SOX2 but not OLIG2, and the decrease of Notch signal targets (MMP9, c-MYC, and Hes1) also can be observed, suggesting that FAM129A may act as an influential contributor to the Notch1-SOX2 pathways that drives GSC invasion. Importantly, we observed that FAM129A and NICD1 were more visible in tumor-infiltrating regions compared to central regions, and we revealed that elevated FAM129A inhibits proteasome-mediated degradation of NICD by binding to it in GSCs. Our work reveals that FAM129A is indispensable for GSC growth and diffuse brain infiltration and provides a new viewpoint for the hyperactivation of Notch signaling in invasive GSCs.
Based on previous study,8,12 we speculated that the increase of FAM129A is induced by cellular stress due to the transformations that occurred in the tumor microenvironment, accompanying the migration of GSCs. Migrating GSCs often exhibit morphological extensions and frequent interaction with nearby normal structures, such as white matter tracts, axons, and blood vessels.26–28 GSCs themselves, differentiated tumor cells as well as normal cells located in the peritumoral brain parenchyma, such as vascular endothelial cells and neurons, all can furnish Notch ligands to activate Notch receptors of adjacent GSCs, further increasing the formation of NICD, which functionally associated with the preservation of a dedifferentiation state of GSCs in invasive niches.25,29,30 The exact role of FAM129A in GSC cellular interactions needs to be further explored. Overall, elevated FAM129A is used to anchor accumulated NICD in GSCs, maintaining their self-renewal characteristics and invasive potential in the infiltration area.
Knockdown of FAM129A caused a reduction in the self-renewal capacity of the GSC and a decrease in proliferation ability. And we demonstrate that the FAM129A post-knockdown state is not a drug-resistant state, but instead promotes susceptibility to temozolomide. FAM129A may be involved in the maintenance of temozolomide resistance in recurrent tumors, but its potential role and specific mechanisms need to be further explored. In summary, our work details a novel adaptative feature of GSCs in the migratory state, that is, FAM129A inhibits the proteasomal degradation of NICD to stabilize overactivated Notch signaling of GSCs, thereby accelerating GBM progression and recurrence. Therefore, the FAM129A/NICD complex is critical for the maintenance of GSCs infiltrating niches. Targeting FAM129A may represent a novel strategy to conquer diffuse brain infiltration and clinically fatal outcomes in GBM.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Contributor Information
Guohao Liu, Department of Neurosurgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Po Zhang, Department of Neurosurgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Sui Chen, Department of Neurosurgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Zirong Chen, Department of Neurosurgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Yanmei Qiu, Department of Neurology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Peng Peng, Department of Neurosurgery, Xiangyang Central Hospital, Affiliated Hospital to Hubei University of Arts and Science, Xiangyang, China.
Wenda Huang, Department of Neurosurgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Fangling Cheng, Hepatic Surgery Centre, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Yang Zhang, Department of Histology and Embryology, School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Huan Li, Department of Histology and Embryology, School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Qungen Xiao, Department of Neurosurgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Feng Mao, Department of Neurosurgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Baofeng Wang, Department of Neurosurgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Xiaobing Jiang, Department of Neurosurgery, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Feng Wan, Department of Neurosurgery, Guangdong Provincial People’s Hospital, Guangdong Academy of Medical Sciences, Guangzhou, China.
Dongsheng Guo, Department of Neurosurgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Xingjiang Yu, Department of Histology and Embryology, School of Basic Medicine, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China.
Conflict of Interest Statement
The authors declare no potential conflicts of interest. The research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
Supported by (1) HUST Academic Frontier Youth Team, (2) Huazhong University of Science and Technology Independent Innovation Research Fund Project (NO.2019kfyXJJS187), and (3) National Natural Science Foundation of China (NO. 81874086, NO.81974452 and NO. 82072797).
Author Contributions
Conception and design: DG and XY; conceived the study and verified the data: PZ and GL; implementation of the project, preparation of the manuscript and analysis of data: GL, PZ, SC and ZC; writing, review, and/or revision of the manuscript: GL, SC, PP, WH, YZ, HL, YQ, FM, BW, XJ and FW; collection of glioma tissues and clinical information: FC, QX.
Data Availability
The data of this study are available within the Supplementary materials. Further inquiries can be directed to contact the corresponding authors.
References
- 1. Weller M, Wick W, Aldape K, et al. Glioma. Nat Rev Dis Primers. 2015;1:15017. [DOI] [PubMed] [Google Scholar]
- 2. Iwadate Y. Plasticity in glioma stem cell phenotype and its therapeutic implication. Neurol Med Chir (Tokyo). 2018;58(2):61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nicholson JG, Fine HA.. Diffuse glioma heterogeneity and its therapeutic implications. Cancer Discov. 2021;11(3):575–590. [DOI] [PubMed] [Google Scholar]
- 4. Justin DL, Mack SC, Mulkearns-Hubert EE, Valentim CLL, Rich JN.. Cancer stem cells in glioblastoma. Genes Dev. 2015;29(12):1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444(7120):756–760. [DOI] [PubMed] [Google Scholar]
- 6. Uyar R. Glioblastoma microenvironment: the stromal interactions. Pathol Res Pract. 2022;232:153813. [DOI] [PubMed] [Google Scholar]
- 7. Cheng L, Huang Z, Zhou W, et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153(1):139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun GD, Kobayashi T, Abe M, et al. The endoplasmic reticulum stress-inducible protein Niban regulates eIF2alpha and S6K1/4E-BP1 phosphorylation. Biochem Biophys Res Commun. 2007;360(1):181–187. [DOI] [PubMed] [Google Scholar]
- 9. Adachi H, Majima S, Kon S, et al. Niban gene is commonly expressed in the renal tumors: a new candidate marker for renal carcinogenesis. Oncogene. 2004;23(19):3495–3500. [DOI] [PubMed] [Google Scholar]
- 10. Feng X, Yan N, Sun W, et al. miR-4521-FAM129A axial regulation on ccRCC progression through TIMP-1/MMP2/MMP9 and MDM2/p53/Bcl2/Bax pathways. Cell Death Discov. 2019;5:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shaw GL, Whitaker H, Corcoran M, et al. The early effects of rapid androgen deprivation on human prostate cancer. Eur Urol. 2016;70(2):214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pallmann N, Livgard M, Tesikova M, et al. Regulation of the unfolded protein response through ATF4 and FAM129A in prostate cancer. Oncogene. 2019;38(35):6301–6318. [DOI] [PubMed] [Google Scholar]
- 13. Nozima BH, Mendes TB, Pereira G, et al. FAM129A regulates autophagy in thyroid carcinomas in an oncogene-dependent manner. Endocr Relat Cancer. 2019;26(1):227–238. [DOI] [PubMed] [Google Scholar]
- 14. Ayesha M, Majid A, Zhao D, et al. MiR-4521 plays a tumor repressive role in growth and metastasis of hepatocarcinoma cells by suppressing phosphorylation of FAK/AKT pathway via targeting FAM129A. J Adv Res. 2022;36:147–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang P, Liu G, Hu J, et al. Tenascin-C can serve as an indicator for the immunosuppressive microenvironment of diffuse low-grade gliomas. Front Immunol. 2022;13:824586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Man J, Yu X, Huang H, et al. Hypoxic induction of vasorin regulates notch1 turnover to maintain glioma stem-like cells. Cell Stem Cell. 2018;22(1):104–118.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li L, Guturi KKN, Gautreau B, et al. Ubiquitin ligase RNF8 suppresses Notch signaling to regulate mammary development and tumorigenesis. J Clin Invest. 2018;128(10):4525–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Plaks V, Kong N, Werb Z.. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16(3):225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu G, Lyapina S, Das I, et al. SEL-10 is an inhibitor of notch signaling that targets notch for ubiquitin-mediated protein degradation. Mol Cell Biol. 2001;21(21):7403–7415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kar R, Jha SK, Ojha S, et al. The FBXW7-NOTCH interactome: a ubiquitin proteasomal system-induced crosstalk modulating oncogenic transformation in human tissues. Cancer Rep (Hoboken). 2021;4(4):e1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gritsenko PG, Atlasy N, Dieteren CEJ, et al. p120-catenin-dependent collective brain infiltration by glioma cell networks. Nat Cell Biol. 2020;22(1):97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Friedl P, Locker J, Sahai E, Segall JE.. Classifying collective cancer cell invasion. Nat Cell Biol. 2012;14(8):777–783. [DOI] [PubMed] [Google Scholar]
- 23. Zhu TS, Costello MA, Talsma CE, et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011;71(18):6061–6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang H, Zhou C, Zhang Z, et al. Jagged1-Notch1-deployed tumor perivascular niche promotes breast cancer stem cell phenotype through Zeb1. Nat Commun. 2020;11(1):5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang J, Xu SL, Duan JJ, et al. Invasion of white matter tracts by glioma stem cells is regulated by a NOTCH1-SOX2 positive-feedback loop. Nat Neurosci. 2019;22(1):91–105. [DOI] [PubMed] [Google Scholar]
- 26. Sahm F, Capper D, Jeibmann A, et al. Addressing diffuse glioma as a systemic brain disease with single-cell analysis. Arch Neurol. 2012;69(4):523–526. [DOI] [PubMed] [Google Scholar]
- 27. Winkler F, Kienast Y, Fuhrmann M, et al. Imaging glioma cell invasion in vivo reveals mechanisms of dissemination and peritumoral angiogenesis. Glia. 2009;57(12):1306–1315. [DOI] [PubMed] [Google Scholar]
- 28. Tsai HH, Niu J, Munji R, et al. Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science. 2016;351(6271):379–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Osswald M, Jung E, Sahm F, et al. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015;528(7580):93–98. [DOI] [PubMed] [Google Scholar]
- 30. Jung E, Alfonso J, Osswald M, et al. Emerging intersections between neuroscience and glioma biology. Nat Neurosci. 2019;22(12):1951–1960. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data of this study are available within the Supplementary materials. Further inquiries can be directed to contact the corresponding authors.