

Abstract
In the 5th edition of the WHO CNS tumor classification (CNS5, 2021), multiple molecular characteristics became essential diagnostic criteria for many additional CNS tumor types. For those tumors, an integrated, “histomolecular” diagnosis is required. A variety of approaches exists for determining the status of the underlying molecular markers. The present guideline focuses on the methods that can be used for assessment of the currently most informative diagnostic and prognostic molecular markers for the diagnosis of gliomas, glioneuronal and neuronal tumors. The main characteristics of the molecular methods are systematically discussed, followed by recommendations and information on available evidence levels for diagnostic measures. The recommendations cover DNA and RNA next-generation-sequencing, methylome profiling, and select assays for single/limited target analyses, including immunohistochemistry. Additionally, because of its importance as a predictive marker in IDH-wildtype glioblastomas, tools for the analysis of MGMT promoter methylation status are covered. A structured overview of the different assays with their characteristics, especially their advantages and limitations, is provided, and requirements for input material and reporting of results are clarified. General aspects of molecular diagnostic testing regarding clinical relevance, accessibility, cost, implementation, regulatory, and ethical aspects are discussed as well. Finally, we provide an outlook on new developments in the landscape of molecular testing technologies in neuro-oncology.
Keywords: glioma, glioneuronal tumors, molecular classification, molecular diagnostics, neuronal tumors, WHO classification
Gliomas, glioneuronal, and neuronal tumors represent the most frequent and diverse category of tumors originating from the central nervous system (CNS) parenchyma. Gliomas are traditionally divided into “diffuse gliomas” (characterized by very extensive migration of tumor cells within the CNS parenchyma), and tumors demonstrating less invasive growth properties (eg, more circumscribed astrocytic tumors and ependymal neoplasms). Diffuse gliomas represent the majority of tumors diagnosed in adult neuro-oncology practice.1 Up to the revised 4th edition of the World Health Organization (WHO) Classification of CNS Tumors (published in 2016), definitions of CNS tumors were mainly based on histological and immunohistochemical characteristics and their resemblance to a supposed cell type of origin.
However, microscopy-based classification of CNS tumors suffered from considerable inter-observer variation. In addition, it became increasingly clear that recognition of clinically relevant subgroups of gliomas and glioneuronal tumors solely based on their morphological characteristics was challenging and inevitably imprecise.2–5 During the last 2 decades, the rapidly increasing knowledge of molecular characteristics of these tumors allowed for the introduction of more robust diagnostic markers into clinical practice.
In the revised 4th edition of the WHO classification of CNS tumors (2016),6 molecular alterations were for the first time included in the definition of diffuse gliomas and certain types of ependymal and embryonal tumors. In the 5th edition of the WHO classification (2021),7,8 multiple molecular characteristics became essential diagnostic criteria for many more CNS tumor types. For these tumors, an integrated, that is, “histomolecular” diagnosis is required. Furthermore, in the 2021 classification, diffuse gliomas are for the first time separated into adult-type and pediatric-type diffuse gliomas. Adult-type diffuse gliomas include astrocytoma, isocitrate dehydrogenase (IDH)-mutant, oligodendroglioma, IDH-mutant and 1p/19q-codeleted, and glioblastoma, IDH-wildtype. CNS WHO grades are now assigned within these types using Arabic (instead of Roman) numerals. In addition to histological features, particular genetic parameters are now being used for assigning the highest malignancy grade to adult-type IDH-wildtype and IDH-mutant diffuse astrocytic gliomas.8
Since the release of the WHO classification of CNS tumors in 2021, several comprehensive reviews and guidelines were published on testing strategies and diagnostic algorithms.9,10 The present guideline highlights the methods that can be used for assessment of the currently most informative diagnostic and prognostic molecular markers. Some of these markers may also have predictive value, that is, predict the likelihood of a response to a particular therapy, and assays hence may yield information on a combination of diagnostic, prognostic, and predictive markers. Since many of these assays are comparatively novel, and have both unique and redundant features, each method is comprehensively reviewed to provide a common ground and understanding before subsequently providing recommendations. Covering the entire realm of available methods in one guideline was considered to be of importance, especially with the new diagnostic requirements. Likewise, the different technologies may not be equally available at different sites or may be subject to different regulatory and reimbursement schemes. This is also a recurring topic of debate after the release of the current WHO classification. Thus, we also include considerations on implementation and socioeconomic factors of diagnostic marker testing. For a recent review on the most relevant predictive molecular markers in gliomas and glioneuronal tumors in adults, the reader is referred to the recent companion European Association of Neuro-Oncology (EANO) guideline on testing for therapeutic targets beyond the WHO 2021 diagnosis.11
Table 1 presents the glioma, glioneuronal and neuronal tumor types listed in the WHO 2021 classification, together with their most relevant diagnostic and prognostic molecular markers. Of note, pediatric-type tumors occasionally may occur in adults and adult-type tumors may rarely be diagnosed in children or adolescents. Also, we included MGMT promoter methylation as a predictive marker for the success of alkylating chemotherapy in patients with IDH-wildtype glioblastoma because of its importance in clinical diagnostic practice. The main characteristics of the molecular methods that can be used for the assessment of the markers listed in Table 1 are systematically discussed in separate paragraphs, followed by recommendations and where possible information on evidence.
Table 1.
Molecular Characteristics of Gliomas, Glioneuronal Tumors and Neuronal Tumors as Listed in the Tables with Essential and Desirable Diagnostic Criteria of the WHO 2021 Classification
| TUMOR FAMILY/tumor type | Most relevant diagnostic and prognostic molecular alterations |
|---|---|
| ADULT-TYPE DIFFUSE GLIOMAS | |
| Astrocytoma, IDH-mutant | IDH1 p.R132 1 # or IDH2 p.R1721#; no chr. 1p/19q5; ATRX2,3; CDKN2A/B4@; TP532,3; MP |
| Oligodendroglioma, IDH-mutant and 1p/19q-codeleted | IDH1 p.R132 1 # or IDH2 p.R1721#; chr. 1p/19q5; TERT promoter2; MP; IHC: retained nuclear ATRX expression |
| Glioblastoma, IDH-wildtype | IDH-wt and H3-wt; TERT promoter2; EGFR6; chr. +7/−107; MP; predictive: MGMT promoter methylation§ |
| PEDIATRIC-TYPE DIFFUSE LOW-GRADE GLIOMAS | |
| Diffuse astrocytoma, MYB- or MYBL1-altered | MYB 8, MYBL18; IDH1-wt and H3-wt; MP |
| Angiocentric glioma | MYB 8; MP |
| Polymorphous low-grade neuroepithelial tumor of the young (PLNTY) | MAPK alteration such as BRAF p.V600 1 , FGFR2 8 , FGFR3 8 , or other; IDH-wt; no 1p/19q5 |
| Diffuse low-grade glioma, MAPK pathway-altered | MAPK alteration; IDH-wt and H3-wt; no CDKN2A/B4; MP: absence of profile of other FGFR- or BRAF-altered tumor |
| PEDIATRIC-TYPE DIFFUSE HIGH-GRADE GLIOMAS | |
| Diffuse midline glioma, H3K27-altered | IHC: loss of H3 p.K28me3 (K27me3) in tumor cell nuclei; p.K28M (K27M) or pK28I (K27I) mutation in H3.3, H3.1, or H3.2 for H3 K27-mutant subtypes1^; EGFR2,6; EZHIP10; MP |
| Diffuse hemispheric glioma, H3G34-mutant | H3.3 p.G35 (G34) 1^ ; MP; loss of ATRX expression, diffuse p53 immunopositivity |
| Diffuse pediatric-type high-grade glioma, H3- and IDH-wildtype | PDGFRA 2,6, EGFR2,6 or MYCN6; IDH-wt and H3-wt; MP |
| Infant-type hemispheric glioma | RTK alteration such as NTRK family gene 8 , ROS1 8, MET18, ALK8; MP |
| CIRCUMSCRIBED ASTROCYTIC GLIOMAS | |
| Pilocytic astrocytoma | MAPK alteration, such as in BRAF (mostly KIAA1549::BRAF) |
| High-grade astrocytoma with piloid features | MP; MAPK alteration; CDKN2A/B2,4; CDK46; ATRX2 |
| Pleomorphic xanthoastrocytoma | BRAF 1 or other MAPK alteration; CDKN2A/B4; MP |
| Subependymal giant cell astrocytoma | TSC1 2 or TSC22; MP |
| Chordoid glioma | PRKCA p.D463H1; MP |
| Astroblastoma, MN1-altered | MN1 8; MP |
| GLIONEURONAL AND NEURONAL TUMORS | |
| Ganglioglioma | BRAF 1 or other MAPK alteration; MP |
| Gangliocytoma | – |
| Desmoplastic infantile ganglioglioma/desmoplastic infantile astrocytoma | MP; BRAF1,2,8 or RAF12,8; no CDKN2A/B4 |
| Dysembryoplastic neuroepithelial tumor | FGFR1 9,8,2; MP |
| Diffuse glioneuronal tumor with oligodendroglioma-like features and nuclear clusters* | MP; chr. 143 |
| Papillary glioneuronal tumor | PRKCA 8 (mostly SLC44A1::PRKCA); MP |
| Rosette-forming glioneuronal tumor | FGFR1 2 , with co-occurring PIK3CA2 and/or NF12; MP |
| Myxoid glioneuronal tumor | PDGFRA p.K3851; PDGFRA2; MP |
| Diffuse leptomeningeal glioneuronal tumor | 1p 3 ; MAPK alteration, most frequent BRAF 8 /KIAA1549::BRAF; MP |
| Multinodular and vacuolating neuronal tumor | MAPK alteration |
| Dysplastic cerebellar gangliocytoma (Lhermitte-Duclos disease) | PTEN 2,3 |
| Central neurocytoma | MP |
| Extraventricular neurocytoma | IDH-wt; MP; FGFR18 (mostly FGFR1::TACC1) |
| Cerebellar liponeurocytoma | MP |
| EPENDYMAL TUMORS | |
| Supratentorial ependymoma, ZFTA fusion positive | ZFTA (= C11orf95) 8 (mostly ZFTA::RELA); MP |
| Supratentorial ependymoma, YAP1 fusion positive | YAP1 8; MP |
| Posterior fossa ependymoma, group A (PFA) | MP; IHC: H3 p.K28me3 (K27me3) in tumor cell nuclei reduced |
| Posterior fossa ependymoma, group B (PFB) | MP; IHC: H3 p.K28me3 (K27me3) in tumor cell nuclei retained; chromosomal instability/aneuploidy |
| Spinal ependymoma | 22q3; MP; no MYCN6 |
| Spinal ependymoma, MYCN-amplified | MYCN 6; MP |
| Myxopapillary ependyoma | MP |
| Subependymoma | MP |
Of note, the list of molecular alterations in this table is not exhaustive; for example, MAPK alteration includes alterations in BRAF, NF1, MAP2K1, MET, or a member of the FGFR or NTRK family. Also, some of the alterations are generally mutually exclusive (eg, IDH1 versus IDH2 mutation), while others can occur in combination in the same tumor (eg, TERT promoter mutation, EGFR amplification, and + 7/−10). Last but not least, demonstration of the lack of particular alterations (such as absence of complete 1p/19q co-deletion, or IDH-wt and H3-wt status of a tumor) can be essential for establishing the correct diagnosis as well. The reader should refer to the 2021 WHO CNS tumor classification for more information on how apply these molecular markers and/or IHC for surrogate markers for the clinical diagnosis of these tumors.
Abbreviations: IHC, immunohistochemistry; MAPK alteration, MAPK pathway driving genetic alteration; wt, wildtype.
Essential diagnostic criteria are written in bold (except those that are listed as “for unresolved lesions” only). Meaning of numbers in superscript and of symbols: 1—hotspot mutation; 2—mutation; 3—loss; 4—homozygous loss; 5—combined whole-arm deletion; 6—amplification; 7—combined whole chromosome gain (+) and whole chromosome loss (−); 8—gene fusion; 9—tyrosine kinase duplication (TKD)/internal tandem duplication (ITD); 10—overexpression.
#Rarely, IDH1 p.R100 and IDH2 p.R140 variant are described.
@Listed in a separate table as criterion for grading.
§Due to its role in neuropathology testing as predictive marker, inclusion of MGMT promoter testing in this guideline was decided.
^According to the present Human Genome Organization (HUGO) Gene Nomenclature Committee (HGNC; see https://www.genenames.org) H3.3 corresponds to H3-3A (also known as (a.k.a.) H3F3A), H3.1 to H3C2 (a.k.a. HIST1H3B), and H3.2 to H3C14 (a.k.a. HIST2H3C); rarely, other H3 genes are involved in the oncogenesis of H3K27-mutant gliomas.
*Provisional tumor type.
The value of immunohistochemistry as an alternative approach for some of these tests is briefly covered as well (partly in the Supplementary information). In addition, an overview of the different assays with their characteristics, especially advantages and limitations are provided in Table 2 and Figure 1. In the last part of this guideline some more general aspects of molecular diagnostic testing with respect to clinical relevance, test availability, costs, implementation, emerging technologies and ethical aspects are discussed.
Table 2.
Characteristics of Assays Discussed in this Guideline
| Method | SNVs/InDels | CNV | Rearrangements | Methyl. class | MGMT | Comment | Throughput samples | Throughput markers | Duration** | Cost |
|---|---|---|---|---|---|---|---|---|---|---|
| NGS DNA | Yes | Yes | Yes | No | No | Sole method for efficient comprehensive variant detection, up to 10-fold lower LOD than Sanger seq, larger target size and higher multiplexing than ddPCR | High and flexible, potentially scalable > 96 samples | High (select targeted genes as panel up to exome/genome as target region); CNV calling depending on target region and unbiased library generation | Depending on exact method and target size 2–5 d from extracted analyte to raw result; pooling of samples for cost-efficiency must be considered | CapEx $$$ Reagents $$ Marker $ Interpr $$$ |
| NGS RNA | (Yes) | No | Yes | No | No | Most efficient method for detection of gene fusions with unknown partners or screening for variety of possible fusions; may allow for detection of activating SNVs and InDels | High and flexible, potentially scalable > 96 samples | High (select gene fusions up to complete coding RNA or transcriptome) | Depending on exact method and target size 2–5 d from extracted analyte to raw result; pooling of samples for cost-efficiency must be considered | CapEx $$$ Reagents $$ Marker $ Interpr $$$ |
| Methylome profiling (“850k”) | No* | Yes | (Yes) | Yes | Yes | Sole method for DNA methylation-based classification, efficient genome-wide CNV analysis (besides DNA-based NGS) | High, array design allows only multiples of 8 samples | High: Coverage of methylation classes depends on subsequent algorithm (classifier), not on assay setup; covers genome-wide CNVs with pre-selection; suggests (but not proves or excludes) several rearrangements depending on their exact configuration (BRAF, FGFR1, FGFR3, ZFTA, YAP1 and others) | 3 d from analyte to raw result; pooling of samples for cost-efficiency must be considered due to chip design | CapEx $$$ Reagents $$ Marker $ Interpr. $$ |
| FISH | No | Yes | Yes | No | No | Efficient assay for interrogating suspected single genomic event like specific copy-number alteration, specific single rearrangements; more prone to artificial false results in CNV analysis than 850k | Typically, one marker (may consist of two loci, eg, break apart FISH) per test | within 1 d | CapEx $ Reagents $$ Marker $$$ Interpr $ |
|
| Sanger | Yes | No | No | No | No | Typically, one marker (hotspot variant) or small region with few markers (eg, H3-3A K28 and G35) per test | Within 1 d | CapEx $ Reagents $ Marker $$ Interpr $ |
||
| Pyro-Seq | Yes | No | No | No | Yes | Typically, one marker (eg, hotspot variant) or small region (MGMT CpG sites) with one test | Within 1 d | CapEx $$ Reagents $$ Marker $$ Interpr $ |
||
| (MS-) MLPA |
Yes | Yes | No | No | Yes | Typically, one or two combined markers per test (eg, 1p/19q and IDH1/IDH2, or IDH1/IDH2 and MGMT promoter) | Within 1 d | CapEx $ Reagents $$ Marker $$ Interpr $ |
||
| qPCR | No | No | Yes | No | No | Typically, one marker per test | Within 1 d | CapEx $ Reagents $ Marker $ Interpr $ |
||
| RT-PCR | No | No | Yes | No | No | Typically, one marker per test | Within 1 d | CapEx $ Reagents $$ Marker $$ Interpr $ |
||
| MSP | No | No | No | No | Yes | Typically, one marker per test | Within 1 d | CapEx $ Reagents $$ Marker $$ Interpr $ |
||
| MSA (LOH) | No | Yes | No | No | No | Within 1 d | CapEx $ Reagents $$ Marker $$ Interpr $ |
|||
| ddPCR | Yes | No | (Yes) | No | No | Requires low amount of DNA; Very high sensitivity and allows quantitative analysis |
Typically, one or few markers per test (eg, FGFR1 mutation and duplication or BRAFV600E mutation and duplication) | Within 1 d | CapEx $$ Reagents $ Marker $ Interpr $ |
Some methods could potentially be employed for other information as indicated here (eg, RNA-seq could be screened for indication of SNVs), but these are use cases not typically employed and validated in diagnostic settings.
Bold: essential criterion.
Abbreviations: NGS, next-generation-sequencing; FISH, fluorescence in-situ hybridization; (MS-)MLPA, (methylation-specific) multiplex ligation-dependent probe amplification; qPCR, quatitative polymerase chain reaction; RT, real time; MSP, methylation-specific PCR; MSA, microsatellite analysis; LOH, loss of heterozygosity; dd, droplet digital; $–$$$ rough categorization of cost for the items “CapEx”, “Reagents”, “Marker”, "Interpr'; CapEx, Capital Expense for equipment required to run assay; Reagents, reagents for assay; Marker, cost per marker covered per run; Interpr., interpretation cost, from $ (low, marker can be readily evaluated) to $$$ (needs extensive bionformatics pipeline and expertise); LOD, limit of detection.
*850k analysis does not identify SNVs or small insertions/deletions, but the methylation prediction can suggest presence of a certain genomic alteration, for example, if an astrocytoma, IDH-mutant is predicted. Still, it does not discriminate between the different variants of oncogenic IDH alterations.
**Duration applies to mere lab workflow from already extracted analyte to raw results; some assays require pooling samples over several days or even weeks depending on laboratory size, multiplying the duration. In turn, pooling samples in order to streamline lab processes applies for most assays. However, this aspect is only specifically mentioned for those assays which cannot run efficiently for single/low number of samples.
Figure 1.
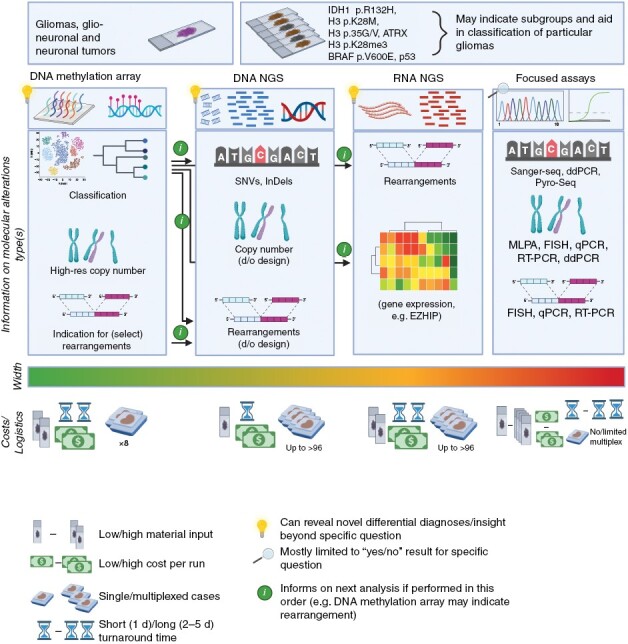
Overview of major molecular diagnostic technologies. The technologies discussed in this guideline are depicted with focus on the high-throughput, more recently established methods. For simplicity, turnaround times represent a scenario in which the protocol is instantly initiated for a given sample, not accounting for other timeframes in typical routine workflows, for example, once-weekly methylation runs, or other platforms with the need to collect samples over several days for generating reasonable batch sizes. Exact cost, material input, timelines, but also width of analysis results depend on the local setting: While DNA methylation arrays are highly standardized, DNA and RNA NGS platforms can be designed at different scale, from very narrow target regions to a whole exome/genome or transcriptome, respectively. Figure generated with BioRender.
Methods
The review and recommendations were conducted by a task force as nominated by the EANO Guidelines Committee. The topic and lead author selection was approved by the EANO Executive Board. The task force was compiled with the aim to represent experts from both diagnostic and clinical disciplines with a demonstrated record in development and/or diagnostic application and/or clinical interpretation of molecular tools in neuro-oncology. The guideline was then composed in an iterative process of virtual meetings and circulated drafts. Subgroups of authors were responsible for drafting paragraphs of their specific expertise, with all other authors subsequently reviewing the text.
Due to the wide range of methodology covered, it was decided not to survey the complete literature with specific search terms, but leave reference selection to the authors. After completion of paragraphs, the respective author subgroups proposed “expert recommendations” summarizing key messages from the text, based on literature but also their experience and common practice, assigned levels of recommendation and, where applicable, evidence. These were then again reviewed and discussed by the entire author group, resulting in “consensus recommendations”. In the paragraphs on “Clinical Relevance” and “Availability, Cost and Implementation”, no recommendations but consensus statements are provided.
Classification of recommendations into classes (C I–IV) and levels (L A–C) for diagnostic tests followed the guidelines published by the European Federation of Neurological Societies,12 for reference provided in Supplementary Table 1. Of note, common practices that are mandated by several national guidelines and/or followed in multiple studies, even if these practices themselves were not the parameters tested for, are attributed to “C I, L A” (eg, stating the genes covered in panel sequencing). Consensus statements on socio-political aspects do not have class or level annotations.
Molecular Diagnostic Techniques
Requirements for Input Material
Not only for biopsies, but also for neurosurgical resections, a limited tissue sample is often submitted for pathological analysis, as resection of these tumors is usually performed using ultrasound aspiration rather than en-bloc resection, which may limit tissue availability and typically impedes spatial annotation of the submitted sample. In this context, comprehensive tissue annotation, supplemented by imaging and intraoperative information, can be helpful for optimal interpretation of the results of tissue analysis. It is also recommended that no tissue is discarded, that is, even ultrasound aspirates should be collected and can be used for histological and molecular analyses.13
To obtain adequate molecular diagnostic results, a selection of representative tissue with sufficient tumor cell content, and extraction of high-quality DNA/RNA are essential. In particular in diffuse gliomas, this can be challenging, for example, due to a high fraction of non-neoplastic cells admixed with tumor cells, or a low amount of viable neoplastic tissue in largely necrotic areas. For cases where the tumor cells can be unequivocally labeled (eg, by a mutation-specific antibody), a visual estimate of tumor cell fraction can be more reliably established. For other scenarios, the tumor cell estimate should be made by taking into account the fraction of non-neoplastic components, such as stromal cells, immune cells, and by estimating the tumor cell fraction based on morphological characteristics such as cellular and nuclear atypia. However, the limitations of a morphological approach should be considered throughout the diagnostic process, and the output from the tumor cell purity estimate derived from molecular analysis (eg, methylation array) may need to be taken into account when formulating an integrated diagnosis. A higher variant allele frequency (VAF) of a driver mutation than expected from histology may indicate a higher fraction of tumor cells. Other possible events affecting VAF include copy-number alterations or presence of the variant in the germline/non-neoplastic tissue. Implications of low tumor cell content or low DNA/RNA quality are discussed in the relevant sections below.
Ideally, both snap-frozen and formalin-fixed and paraffin-embedded (FFPE) tissue samples should be collected for diagnostic purposes. Also, the use of tissue for clinical diagnosis should have priority over biobanking for research. Frozen tissue has higher analytical quality for certain molecular procedures, such as for high molecular weight RNA extraction and increasingly relevant long-read DNA technology (eg, for Nanopore assays), while formalin-fixation and paraffin embedding remains the gold standard for optimal preservation of tissue morphology and is also suitable for most immunohistochemistry and DNA-based assays.
-
Consensus recommendations
Diagnostic work-up should have priority over biobanking for research. (C IV; L C)
Information on tumor location and origin of tissue samples should be provided by the surgeon (or at least be available from hospital patient information systems) for optimal diagnostic interpretation, incl. the assessment of representativeness. (C IV; L C)
Selection of tissue area for DNA/RNA extraction should be done (or supervised) by an experienced (neuro)pathologist. Enrichment of tumor cell fraction by tissue microdissection should be performed in cases with apparently low tumor cell content to increase analytical sensitivity. (C I; L A)
Whenever feasible, simultaneous submission of snap-frozen and FFPE tissue, or of fresh tissue that can be processed into FFPE and the remainder stored frozen, should be attempted. (C III; L B)
If tissue volume is limited, the potential value and input requirements of high-throughput analyses should be considered early in the diagnostic decision-making process, taking into account the strengths and weaknesses of analytical methods as outlined below, especially in cases that do not qualify for targeted analyses (eg, no specific differential diagnoses). (C IV; L C)
DNA Sequencing for Mutational Analysis
Among the variety of molecular alterations required for integrated diagnosis of CNS tumors, single nucleotide variants (SNVs), small insertions/deletions (InDels, both widely and herein often still referred to as “mutation”, especially when of established relevance), and gain or loss of complete chromosomes/chromosome-arms are the most common. They are typically interrogated by DNA sequencing unless mutation-specific antibodies are available. While DNA methylation analysis can be considered as a reliable surrogate test for certain mutations (eg, for IDH-mutant glioma), sequencing may still be required to identify the exact type of mutation, especially when a specific mutation is used as predictor for targeted therapy or when different mutations are associated with distinct outcomes, for example, H3.3 (H3-3A) vs H3.1 (H3C2) or H3.2 (H3C14) p.K28M (p.K27M) mutations in H3 K27-altered diffuse midline gliomas (DMGs).
Next-generation-sequencing (NGS) can cover a series of genes (a “gene panel”) in a single analysis, and “molecular barcoding” allows for multiplexing and thus processing of several samples in parallel. Coding sequences and other genomic regions (eg, TERT promoter) included are referred to as the “target” or “target regions”. Several specific gene sets have been proposed for neuro-oncology panels.14–16 Technically, regions of interest can either be amplified by primers (amplicon-based) or enriched by hybridization to probes designed to bind these regions (hybrid-capture). Typically, amplicon-based panels are used for smaller panels, for example, interrogating 20–50 genes, while hybrid-capture panels can cover several mega-bases and are also employed for whole exome sequencing (WES). Sensitivity for the detection of mutations is generally similar for both methods. The amplicon-based approach may be superior for mutations at very low abundance. However, the amplification steps may obscure subtle copy-number differences. For both approaches, sensitivity depends not only on tumor cell content and DNA quality, but also on the read depth of subsequent sequencing, that is, the unique sequence coverage.
Apart from the high-throughput, the ability to quantitatively determine the variant allele frequency (VAF) is an advantage over the limited estimation of this parameter by Sanger sequencing. VAFs must be interpreted in the context of histology and other information on tumor purity (eg, derived from VAFs of driver mutations in IDH1/IDH2, H3-3A or the TERT promoter, or from genome-wide copy-number-profiles from methylation analysis or larger NGS panels). This may allow to distinguish between clonal and subclonal alterations, and may also provide hints towards possible germline alterations. Quantification is also possible with pyrosequencing and droplet digital polymerase chain reaction (ddPCR). However, these latter methods allow only for sequencing of small target regions.
DNA sequencing can also inform about gene fusions in case the relevant potential genomic breakpoints are covered in the target region. However, most fusion breakpoints are in regions typically not covered by NGS panels (introns). Also, the sensitivity of the existing bioinformatics tools is so far limited, and interpretation typically requires additional information like genomic copy-number variations (CNVs), or confirmation by additional RNA sequencing, fluorescent in situ hybridization (FISH) or reverse transcriptase (RT)-PCR analysis when specific fusion partners are suspected.
CNVs can also be inferred from DNA sequencing data obtained by NGS, either by inferring large variation from the “average” baseline, presuming that most signals result from two chromosomal copies, or by assessing the VAF of SNVs. The latter allows for higher resolution and can also identify copy-neutral loss of heterozygosity (LOH). However, a genome-wide CNV profile that reliably informs on the chromosomal arms and single loci relevant in neuro-oncology requires either a specific design for that purpose, that is, beyond the relevant genes for mutational analysis with a wide distribution of targets across the genome, or (low coverage) WES or whole genome sequencing (WGS). New approaches such as nanopore-based long-read NGS can also provide diagnostic copy-number and mutational profiles, however, these so far require high molecular weight DNA extracted from frozen tissue.17
With decreasing sequencing costs, WES or WGS will be increasingly used. However, high costs are currently still prohibitive for routine use, especially for samples requiring high read depth because of limited tumor cell content. Also, the number of SNVs and InDels detected with these methods results in considerable workload for variant interpretation. Interpretation of both WES/WGS, but also of large panels can benefit from concurrent analysis of constitutional DNA of the patients. This supports the detection of somatic variants and also may identify clinically relevant germline variants. However, informed patient consent must be obtained for large-scale sequencing of constitutional DNA because of the possible detection of germline variants, and national laws regarding genetic counseling and management of unsolicited findings must be considered (see below). Caution is warranted not to setup a data analysis pipeline that standardly subtracts all SNVs/InDels in the constitutional DNA from those in the tumor DNA, since this may obscure tumor-relevant (germline) alterations. In adults, sequencing of non-neoplastic tissue is not mandatory for the identification of the most relevant somatic changes since subtraction of general population SNVs/InDels is often sufficient, at least in target sizes < 1 Mb. This population data can be derived from databases like gnomAD (https://gnomad.broadinstitute.org).
Reporting of NGS data should include the list of the genes interrogated by the test, or a reference to them, and to provide unambiguous details of the identified alterations according to international standards as released by the Human Genome Variation Society (https://varnomen.hgvs.org/), including transcript identification or genomic location with reference genome version, nucleotide and amino acid exchange, read depth at the respective position, and VAF. Biological interpretation of the relevance of identified variants should follow established guidelines, also with respect to the diagnosis. A consensus across various guidelines on the reporting of somatic variants is currently lacking and subject to dynamic development. The Clinical Genome resource (ClinGen), the Cancer Genomics Consortium (CGC) and the Variant Interpretation in Cancer Consortium (VICC) have recently published a guideline specific for the classification of somatic variants in cancer.18 This intends to overcome the limited applicability of classification guidelines that were first applied for germline data.
-
Consensus recommendations
The advantages of NGS in covering a variety of alterations, including those of low abundance, within a single assay and with small input amounts should be considered when selecting the testing methodology; this is particularly relevant when the diagnosis is challenging and thus the spectrum of potentially relevant genetic variants being broad. (C IV; L B)
The target regions covered in the NGS analysis should be clearly stated in the neuropathology report. (C I; L A).
Reporting of specific NGS findings should include the transcript identification and/or genomic location with reference genome version, nucleotide and amino acid exchange, read depth at the respective position, and VAF. (C I; L A).
Biological interpretation of the findings should be provided and follow standards like the ClinGen/CGC/VICC guidelines. (C I; L:A)
Reports putting the NGS results in the context of a histomolecular diagnosis should be made by an experienced (neuro)pathologist, including plausibility of the molecular findings regarding diagnosis and tumor cell content; the latter is especially of relevance when morphologically estimated tumor cell content and VAFs are highly divergent. (C III; L B).
RNA Sequencing
RNA sequencing has become a valuable tool for the detection of gene fusions.19 Gene fusions can result in a hybrid protein that is constitutively active (eg, when a dimerization domain is fused to a receptor tyrosine kinase), or with altered expression (eg, by promoter rearrangement or enhancer hijacking). Genes that have been identified as component of driver fusion-genes in glial, neuronal and glioneuronal tumors include BRAF, ROS1, NTRK1/2/3, FGFR1/3, EGFR, PDGFRA, and others. Many of these genes can bind to one of several partners, though some have a clearly preferred partner (eg, KIAA1549::BRAF in pilocytic astrocytomas)20–22 (www.tumorfusions.org)
The two main approaches for detection of gene fusions by RNA sequencing are targeted and whole transcriptome RNA sequencing.23 Targeted RNA sequencing can achieve a higher sensitivity than whole transcriptome sequencing even at lower sequencing depth. Targeted RNA sequencing also requires less input material and benefits from simpler bioinformatic analyses, making it particularly suitable for routine diagnostics. Whole transcriptome sequencing allows for the identification and detailed characterization of fusion partners of both pre-defined and novel fusion-genes; however, interpretation of the data requires more bioinformatic skills.
RNA degradation, especially in FFPE tissue samples, presents an important limitation for RNA sequencing in routine diagnostic practice.24,25 Also, as RNA sequencing requires presence of at least one exon of either fusion partner, it is not suitable to detect genomic rearrangements that involve enhancer hijacking or promoter rearrangement. Such events may be rare in CNS tumors but have been described.26
Bioinformatics tools have been developed to detect fusion-genes but with varied detection sensitivities which are due to intrinsic difficulties (eg, because fusion transcripts cannot be mapped to a standard transcriptome) and/or sequencing artifacts. Multiple unique sequence reads directly spanning the breakpoint are indicative but are not always detected. Novel tools are continuously being developed and their efficacy can vary depending on the specific analytical workflow, making it difficult to define the most effective one. However, STAR-Fusion, Arriba and STAR-SEQR appeared to be the most accurate in a comparison of 23 tools,27 and pipelines based on a combination of fusion gene callers may be used for added confidence.28 In some instances, it is possible to identify the genomic breakpoint using reads derived from unspliced (pre-)RNA species.29 Multiple studies have reported the efficacy of RNA sequencing to detect gene fusions in CNS tumors.30–33
RNA sequencing is also well suited to identify intragenic rearrangements and deletions such as those in EGFRvIII (characterized by an in-frame deletion of exons 2–7).34 Moreover, if a gene is expressed at sufficiently high levels, SNVs and InDels may also be detected by RNA sequencing. However, for detection of SNVs and InDels, DNA sequencing is preferable. Strong overexpression of a proto-oncogene may point to gene amplification, but unequivocal criteria for detecting gene amplification and overexpression by RNA-seq are still lacking, as are the criteria for overexpression in absence of gene amplification.35 Transcriptome analysis can also be used for classifying CNS tumors (eg, subtyping of medulloblastomas),36 but so far this has been mainly used for research purposes, especially after DNA methylation has proven to be superior in this task.37,38
-
Consensus recommendations
In case of technical failure due to the extent of RNA degradation, repeating the assay using a snap-frozen sample if available should be considered. (C IV; L C)
The report should include the assay type (whole transcriptome versus targeted), covered genes/regions, the applied bioinformatic pipeline and the number of fusion reads. (C I; L A)
Significance and functional plausibility (eg, retention of the kinase domain in a tyrosine kinase receptor) should be checked before reporting the presence of a gene fusion (C IV; L C)
DNA sequencing or methylation array should be regarded as superior over RNA sequencing to detect SNVs and gene amplifications, respectively.
Methylome Profiling (850k Array), Including CNV Profile and MGMT Promoter Methylation Status Analysis
Genome-wide methylation profiling is a tool to establish, confirm and/or fine-tune the diagnosis of CNS tumors, and to help avoiding potential misdiagnoses in up to 25% of cases.39–42 This diagnostic tool is distinct from other assays discussed here, as it does not detect a particular molecular alteration as such, but provides a classification of the specimen based on its DNA methylome “fingerprint” and can be considered as surrogate marker for some (eg, IDH, histone) mutations. A dataset derived from over 2800 CNS tumors was used to build an initial version of a publicly accessible “Brain Tumor Classifier” (www.molecularneuropathology.org). In a rapidly increasing number of centers, this tool is now applied for assigning a specific methylation class for the diagnosis of CNS tumors and for sub-classifying tumor types into clinically relevant subtypes.42–55 Data are currently (as of early 2023) generated by the Methylation BeadChip (EPIC) 850k array, assessing presence/absence of a methyl group on 850 000 CpG sites across the entire human genome. The IDAT file with the raw data can then be uploaded via the webpage (see above) and will be matched with a reference cohort consisting of thousands of CNS tumors. New versions of the array or entirely different technologies to obtain methylation data thus require updates of this classifier. Subsequently, a report is issued, containing information to what extent the methylation profile of the test sample matches the pre-defined methylation classes in the Brain Tumor Classifier. In this report, a “calibrated score” is added, indicating the likelihood for the given sample to match to a specific reference class. A score ≥ 0.9 represents a “match” and usually indicates a reliable indication of the diagnosis.
Subsequently, it has been reported that using the older v11b4 version of the Brain Tumor Classifier a threshold of 0.84 was non-inferior to the 0.9 threshold.40 However, in more recent versions of the classifier this may well be different, also because more, and new tumor types and methylation classes are recognized (eg, in the recently launched version v12.5 there are 184 classes, including meningioma and medulloblastoma subclasses). A calibrated score of < 0.9 (or 0.84 in v11b4) should be interpreted with great caution; such a result may indicate suboptimal quality and/or quantity of DNA in the sample, a tumor type that is not yet well represented in the classifier, and/or a tumor in a patient with a genetic tumor predisposition syndrome which has a somewhat different molecular oncogenesis. Only in a small percentage of cases methylome profiling analysis results with a high calibrated score in an erroneous/misleading suggestion for the diagnosis. It is therefore of critical importance that the interpretation of the results is done in the context of histopathological +/− other molecular information by a (neuro)pathologist familiar with the potential pitfalls of this diagnostic tool. Reanalyzing data against another or a new classifier may help in cases of discrepancy between different layers of molecular and/or clinical/histological information. Some cases may also be resolved by applying other classifiers, for example, the sarcoma classifier could identify mesenchymal tumors that are not covered in the Brain Tumor classifier.56
The genome-wide coverage of the EPIC bead chip also allows for obtaining a CNV profile across all chromosomes, issued with the report, and thus is useful for assessing chromosomal alterations that are potentially relevant for the diagnosis and/or assessment of the WHO grade of CNS tumors. However, no specific thresholds for the detection of presence of a homozygous versus hemizygous deletion (eg, of CDKN2A/B) can currently be given. Furthermore, the presence of fusions like KIAA1549::BRAF (which is associated with a tandem duplication on 7q34), TACC1::FGFR1 and others can often also be inferred from the CNV plot,57 but these indications are neither ultimate proof for their presence, nor does their absence exclude a fusion. Of note, only the most relevant genes are annotated in the report file of the Brain Tumor Classifier, but other focal alterations of relevance may be visible in the CNV profile as well. To inspect these in detail, a file compatible with the integrated genome viewer (IGV) is provided in the downloaded package from the website. As a third “layer” of information, this diagnostic tool provides information on the MGMT promoter status including the confidence interval.58,59 However, the optimal methods and respective cut-offs for assessment of this molecular marker are debated (see section on MGMT testing below).
DNA methylome profiling can be performed from DNA extracted from FFPE tissue, as well as from snap-frozen or fresh tissue (and, currently in experimental settings, also from liquid biopsies60). Selection of suitable tissue areas is of paramount importance to achieve an optimal result. Furthermore, rather than providing information on single reads or other allele-specific information, methylation profiling represents analysis of compound signals and is thus less sensitive for tissue samples with a low tumor cell percentage. Bioinformatic approaches for deconvolution of cellular composition are being developed but have so far not entered diagnostic routine.61 For reliable analysis, 200 ng of high-quality DNA from a sample with > 50% tumor cells is generally sufficient, but even 20 ng of representative DNA from very small biopsies and lower tumor cell fractions may work.43
A potential risk of the methylation array technology for clinical purposes, specifically the diagnosis of tumors, is the dependence on the products and services of a single manufacturer. Also, the costs for its implementation and use can be substantial, and the need to pool samples in batches of currently eight may pose challenges to institutions with lower throughput. At the same time, application of this tool may be relatively cost-effective in a situation when it replaces the necessity to perform multiple other analyses, for example, for assessment of clinically relevant CNVs and/or of the MGMT promoter status, and if no diagnosis can be established otherwise, or only with a wider range of other markers, for example, larger panels of immunohistochemical stains. Of note, none of the methylome-based classifiers are currently certified as in vitro diagnostic device (ie, they have no CE-IVD label), and their use for diagnostic purposes is at present not endorsed by the institution hosting the classifier (DKFZ, Heidelberg, Germany). Users must thus take care for approval and, if required, accreditation of this method at the own institution for diagnostic purposes.
As the workflow of the analysis itself (from DNA extraction to reading the chip) already takes about five working days, clinicians should anticipate that results will generally become available at least two weeks after the surgery has been performed. The pathology report should contain information on (1) estimated tumor cell fraction of the extracted DNA, (2) amount of DNA input, (3) quality of bisulfite conversion, (4) classifier version(s) used, (5) highest scoring methylation category with the respective calibrated score(s), (6) sub-classification with score(s) if applicable and (7) the MGMT promoter methylation status, especially for adult-type diffuse gliomas. For cases without a sufficient calibrated score (<0.9 or, alternatively, <0.84 in v11b4), the reporting of lower scores depends on multiple factors such as tumor cell content, plausibility of the diagnosis in the context of other molecular and morphological findings, and quality of the methylation data (eg, low conversion rate may reduce or falsely alter scores, and low amplitudes of CNVs may indicate low tumor cell content or clonal heterogeneity). CNVs relevant for the specific case should be stated in the pathology report as well, taking the overall quality of the CNV profile and tumor cell content into consideration.
-
Consensus recommendations
Access to DNA methylation analysis, on-site or via referral, should be made available at any institution involved in the diagnostics of CNS tumors. (C II; L A).
The report of methylome profiling analysis should contain information on the estimated tumor cell content/fraction of the extracted DNA, amount of DNA input, quality of bisulfite conversion, classifier version(s) used, the highest scoring methylation category with the respective calibrated score(s), and sub-classification with score(s) if applicable. (C III; L A).
DNA methylome profiling with array-based methods can identify specific genomic alterations; however, in particular for tumors with indication of gene fusions and/or for which therapeutic approaches are considered, ultimate proof must be provided by orthogonal methodology (eg, sequencing) (C II; L A).
The classifier score or any other use of methylation data provides an adjunct in diagnostics, not a diagnosis itself; thus, it should be incorporated into the integrated diagnosis by an experience (neuro)pathologist. (C I; L A).
Single/Limited Target Analysis
In this section several more commonly used methods for single/limited analysis of molecular characteristics are briefly described. See Supplementary information for other methods that can be used in this context, including multiplex ligation-dependent probe amplification (MLPA), reverse transcription PCR (RT-PCR), quantitative PCR (qPCR), microsatellite analysis for detection of LOH. These assays may have advantages in terms of lower cost and in part also lower input requirements (Table 2). For example, if only few slides are available that do not allow for sufficient DNA extraction, a FISH analysis for the appropriate target may be highly informative. Yet, it does not provide comprehensive information and may waste tissue if that specific marker turns out to be uninformative. Hence, for a single marker, one of these assays may be the most expeditious method for assessment. Yet, limitations on other information that can be retrieved may outweigh this advantage.
FISH allows in situ detection of defined chromosomal aberrations in tissue sections with fluorescently labeled probes that hybridize to their complementary nuclear DNA target sequences. FISH can detect diagnostically relevant low-level copy-number variations, such as gain of chromosome 7 and loss of chromosome 10 (+7/−10), 1p/19q co-deletion, homozygous deletions (eg, CDKN2A/B) or gene-amplifications (eg, EGFR, MYC, MYCN, C19MC). FISH can also detect gene fusions/translocations with dual-color FISH probes for distinct fusion partners, such as KIAA1549 and BRAF, or by using break-apart probes for individual target genes. FISH works on high-quality frozen and FFPE tissue sections, even of tiny biopsies. Standardized evaluation by an experienced assessor is key for optimal interpretation.62–65 Importantly, the use of FISH probes that are not representative for the entire chromosome or chromosomal arm can lead to overcalling chromosomal loss or gain (eg, partial losses on 1p or 19q, which do not suffice as diagnostic marker like complete 1p/19q co-deletion).
Sanger sequencing is one of the oldest and still most widely available methods for DNA sequencing. It relies on chain termination using dideoxynucleotide diphosphates, often labeled with fluorescent dyes specific for each deoxynucleotide triphosphate (dNTP). The sequencing reaction is then size-separated (eg, using capillary electrophoresis). Sanger sequencing is ideal for the analysis of relatively short DNA segments (100–1000 bp). The sensitivity of mutation detection by Sanger sequencing is lower compared to other sequencing methods.66
Pyrosequencing is a technique of direct sequencing but, unlike Sanger sequencing, the dNTPs are added sequentially, that is, after integration of a dNTP into the newly synthetized sequence a pyrophosphate is released that activates luciferase and the resulting light signal is detected by the sequencer. This technique can detect point mutations and also be used for quantitative detection of MGMT promoter methylation by targeted sequencing of selected CpG sites following bisulfite conversion of DNA. The method works well with DNA extracted from frozen and FFPE samples. The advantages over Sanger sequencing are a higher analytical sensitivity and the quantitative determination of mutant/methylated allele frequencies.67
Droplet digital PCR (ddPCR) is a rapid, cost-effective and highly sensitive method to detect and quantify genetic alterations by using very small amounts of template DNA. The template DNA molecules are distributed across multiple replicate reactions for quantitative measurement without the need of an external standard. This technique is suitable for DNA extracted from snap-frozen or FFPE tissue specimens as well as from liquid biopsies (CSF, plasma). It is highly accurate to detect point mutations, such as the hotspot mutations in IDH1, IDH2, H3-3A, BRAF, PIK3CA, FGFR1 and the TERT promoter, but can also reliably detect gene deletions, duplications (such as FGFR1, BRAF duplication associated with KIAA1549::BRAF fusion) and amplifications. The analytical sensitivity of ddPCR is higher compared to Sanger sequencing. In comparison to NGS approaches, ddPCR is limited in the simultaneous detection of several genetic abnormalities, although multiplex ddPCR techniques have been developed to detect recurrent genetic alterations in diffuse gliomas and glioneuronal tumors.68–72
-
Consensus recommendations
Pitfalls in the assessment of chromosomal copy-number variations should be recognized, such as detection of partial losses on 1p or 19q when analyzing only single loci on each chromosome arm, for example, by FISH analysis. (C III; L C)
Mutation detection by pyrosequencing or ddPCR may be more sensitive than Sanger sequencing in samples with low tumor cell content. (C II; L C)
MGMT Promoter Methylation Analysis
The methylation status of the promoter of the MGMT gene is a predictive factor for benefit from alkylating agent therapy such as temozolomide (TMZ) in IDH-wildtype glioblastoma and prognostic in glioblastoma patients treated with TMZ.73 Hypermethylation of the CpG island in the promoter region leads to transcriptional silencing of the MGMT gene and thereby inactivates the repair capability of the cells to remove the most toxic adduct induced by alkylating agents, namely O6-methylguanine. The 5ʹ CpG island of MGMT comprises 98 CpGs, whereof 2 regions, DMR1 and DMR2, are highly correlated with loss of expression when methylated.74 Of note, this predictive value of the MGMT promoter methylation status has only been established for IDH-wildtype glioblastoma that due to frequent loss of chromosome 10 only needs to inactivate the remaining MGMT allele at 10q26 by promoter methylation. In a randomized phase 3 study on newly diagnosed anaplastic gliomas without 1p/19q co-deletion (CATNON/EORTC study 26053-22054), MGMT promoter methylation determined by the MGMT-STP27 algorithm was not predictive for outcome to TMZ in IDH-mutant anaplastic astrocytomas.75 The clinical value of assessment of the MGMT promoter methylation status for other glial, glioneuronal and neuronal tumor types remains to be determined.
The most commonly used methods for assessment of MGMT promoter methylation are based on variations of quantitative methylation-specific PCR (qMSP) or methylation-specific sequencing-based technology such as MS-pyrosequencing (PSQ), and more recently, BeadChip based technology. Different technologies have distinct pattern recognition features and do not necessarily interrogate the same sets of CpGs. Even with the same technology, different sets of CpGs are analyzed, and various definitions for cut-offs are used to discriminate methylated from unmethylated [eg, based on the confidence interval versus based on survival (optimal risk cut-off)].76,77 In case of discrepant results of different validated tests on the same DNA sample, it is currently not known which test or cut-off most precisely predicts response to TMZ. Some assays define a “gray zone” referring to cases that cannot be unequivocally classified as either methylated or unmethylated.59,76,77 Such an equivocal test result may necessitate further investigation of the “true” MGMT promoter status, for example, when the question arises whether to treat with TMZ only, omitting RT, or in trials omitting TMZ in the test arm where only “truly” unmethylated patients should be enrolled.76 Thus, the gray zone must be clearly communicated and put into context of histology and other pathology information (eg, tumor cell content) and clinical decisions must be taken considering the individual patient situation. See Figure 2 and Supplementary information for some more information on differences in CpGs interrogated by the various assays. Immunohistochemistry is not suitable for determination of the MGMT status for clinical purposes.78 This may be due to problems with discriminating staining of non-neoplastic versus neoplastic cells and differences in MGMT protein levels before and after challenging the tumor with alkylating agents.
Figure 2.
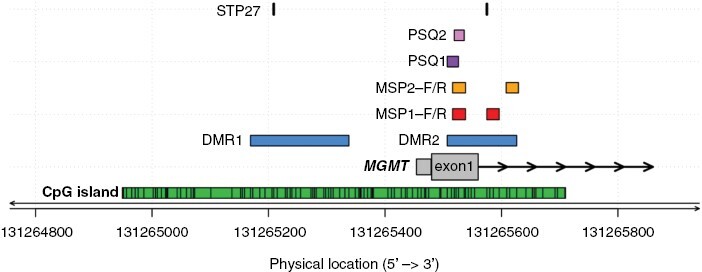
CpGs interrogated in commonly used MGMT promoter methylation assays. The physical location of sites in the CpG island (green) interrogated by commonly used MGMT methylation assays are shown (CpGs, numbered 1–98, genome build GRCh37/hg19). The differentially methylated regions 1 and 2 (DMR1 and 2) are marked in blue.74 The CpGs from the original MSP assay commonly examined by qualitative and quantitative MSP assays are marked in red (MSP1, CpGs 76–80 and 84–87);73,77,79–81 CpGs analyzed in a quantitative MSP assay (MSP2, CpGs 76–80 and 88–90) used in large clinical trials are marked in yellow.76 Two sets of CpGs used commonly for pyrosequencing, PSQ1 (CpGs 74–78) and PSQ2 (CpGs 76–79), are marked in purple and pink, respectively.77,80 The locations of the CpGs (31, 84) selected from the BeadChip array analyzed by the MGMT-STP27 procedure, are marked in black.58 Figure by Pierre Bady.
-
Consensus recommendations
The establishment of an MGMT assay for clinical application requires intra-laboratory technical validation and a quality controlled (QC) setup, with a defined cut-off proper to the assay; for bisulfite-based assays, the completeness of the bisulfite conversion needs to be controlled; the performance of some assays, including the limit of detection, may be affected by the type of input material (frozen vs FFPE) and may require some technical adaptations. (C IV; L C)
For adequate interpretation of the test results (also at a referral center) it is crucial to cite the assay and CpGs assessed, report the value of the test, indicate cut-offs and possible gray zone results, provide information of corresponding relevance for clinical decision-making. (C IV; L C)
Immunohistochemistry is discouraged for determination of MGMT status as basis for clinical decision-making. (C III; L C)
Immunohistochemistry as an Alternative Approach
The number of diagnostically relevant antibodies that may substitute for nuclear acid-based molecular analyses is steadily increasing and represents a relatively easy, fast and less expensive method for facilitating integrated histomolecular classification of CNS tumors according to WHO 2021 criteria. The most important molecular markers amenable to immunohistochemical analysis in this context (IDH1 p.R132H, H3 p.K28M and p.G35R/V, BRAF p.V600E, ATRX, p53 and H3 p.K28me3) are described in the Supplementary material.
-
Consensus recommendations
To avoid misinterpretation of immunohistochemical stainings it is critical to choose antibodies that work well on FFPE material, to optimize and validate tissue pretreatment and staining protocols stringently, and to perform tests alongside appropriate negative and positive controls. For example, ATRX and H3 p.K28me3 immunohistochemistry are sensitive to hypoxia and crush artifacts; in this setting nuclear staining in non-neoplastic (eg, endothelial, inflammatory) cells can serve as a positive internal control. (C III; L C)
Since internal positive controls are lacking for “mutation-specific” antibodies, use of a separate positive control, optimally on-slide, is recommended. (C IV; L C)
Alternative alterations in case of negative immunohistochemistry must be considered and interrogated (eg, non-canonical IDH1 variants by sequencing). (C III; L C)
Practical implications
Clinical Relevance
Molecular diagnostics is an essential part for neuropathological diagnostic practice and for clinical trials since molecular parameters became part of the WHO CNS tumor classification in 2016, and in the 2021 edition even more diagnoses require integration of molecular information. Integrated histomolecular classification allows for more accurate counseling and a superior prognostication of patients in a potentially critical medical situation and for informing on the choice of optimal post-operative treatment, with IDH-mutations being the most prominent examples. These mutations separate adult-type diffuse gliomas into biologically and prognostically distinct groups and allow for—yet trial-based—mutation-specific treatments.82,83 Less common and sometimes puzzling tumor entities are often resolved when more advanced diagnostic methods are used, and incorrect diagnoses can be rectified in up to 25% of cases using DNA methylome profiling,39–42 resulting in decreased burden of treatment, initiation of the appropriate therapy, and better stratification into clinical (trial) cohorts to evaluate novel therapeutic approaches.
Advanced molecular diagnostics by NGS panels, WES/WGS or methylation arrays as opposed to single assays assessing individual makers will be cost-effective long-term as they enable the assessment of multiple individual molecular parameters that are required for classification according to WHO in a single assay, reduce turnaround times and reveal information that may otherwise not have been possible with a stepwise single-assay approach. In addition, these high-throughput analyses will provide knowledge supporting further scientific development in this oncology subdiscipline.84,85 With the discovery of sometimes rare and diverse driving molecular alterations in CNS tumors, mainly in children but also in adults, and with molecularly guided treatments for some entities becoming available, high-throughput analyses have become a requirement to identify patients candidable to specific treatment options, for example, directed at BRAF, NTRK, MAPK or PDGFR pathway alterations.86–88 Gene fusions appear to be particularly attractive targets, though thus far with limited efficacy for CNS tumors.11,89 Panel diagnostics require less tumor material and can at times provide a diagnosis in the absence of a clear histopathological tumor diagnosis, avoiding the need for a repeat biopsy.90 This is very similar to other areas in oncology where the role of advanced molecular diagnostics is increasing rapidly: optimal oncological treatment starts with the right diagnosis in all cases.
Of note, re-analysis of tissue after recurrence may be indicated in some settings, for example in cases in which molecular markers inform about grading, or to distinguish recurrence from secondary neoplasms. Presence of treatment targets may also change upon recurrence, potentially even induced by earlier therapy. However, this is outside the scope of this guideline.
With the pivotal role of molecular diagnostics for both the routine classification of CNS tumors and to establish adequate treatment plans for the patients, reimbursement for standard of care pathology including molecular diagnosis is essential. The weight is most prominent in patients with no or only first-line therapy guideline supported medical treatment options. For these patients, in addition to the eminent role in proper diagnostics, the mentioned molecular analyses are regarded critical to develop novel options. Similar to surgery, chemotherapy and MR imaging being part of the reimbursed day-to-day care of CNS tumor patients, proper tumor diagnoses built on molecular analysis is part of standard of care. This requires adequate reimbursement of pathology including molecular tests to meet the standard of care as detailed in current international and national guidelines.91–93 As a next step, the relevance of treatment related diagnostics should be proven and care providers should reimburse also deducted therapies.
-
Consensus statements
Molecular diagnostics is an essential part of care for patients with glial, glioneuronal and neuronal tumors.
Standard of technical practice tests as mentioned in this guideline need to be reimbursed in addition to more traditional (ie, morphological) analysis.
Tests should be done as effectively and with the least burden for patients as possible.
Availability, Cost and Implementation
Availability and access to modern molecular diagnostic techniques can vary substantially within and between countries. Advanced molecular techniques and/or bioinformatic expertise, tools and algorithms for correct interpretation are available mostly in specialized academic and tertiary care institutions and are usually not available in the primary and secondary health care setting. Costs for integrated histomolecular diagnostic work-up may be significant and in many countries currently not or only partially reimbursed by health care providers. Guaranteed and equal access of patients to state-of-the-art diagnostic assessment according to the WHO classification 2021 is a challenge for health care professionals in the field of neuro-oncology world-wide and its implementation needs to be recognized as a priority to political decision-makers.
Mutation-specific antibodies, for which costs are comparatively low, have been implemented in many institutions worldwide, enabling pathologists to distinguish between, for example, IDH-mutant and -wildtype gliomas. The high feasibility and robustness of NGS panels have also led to widespread use of commercial panels in routine diagnostics and have increased the use of precision diagnostics in the clinical pathological setting. Yet, not all markers relevant for neuro-oncology diagnostics are covered by commercial panels, thus requiring customization, and as a result some academic centers have established glioma-tailored panels which cover also rare molecular alterations. DNA methylation-based tumor classification of CNS tumors has been implemented in the clinical diagnostic practice in many academic centers, and their networking efforts have facilitated implementation of the technique and reduced turnaround times.44 Implementation of NGS and methylation profiling requires continued efforts to keep up with molecular neuropathology expertise, for example, methylation classes of novel subtypes, or variant interpretation in NGS. Apart from the cost for equipment and reagents, recruitment and training of qualified personnel remains a challenge.
Importantly, these comprehensive methods have compelling advantages also from economical perspectives: Assessing a series of markers in a single assay is faster than consecutive single-assay steps, consumes less tissue, and may also yield information that the morphological impression would not have been triggered to test for, thus reducing the risk of testing bias and missing important information. Over the course of further treatment, an inaccurate or incomplete initial information of the molecular characteristics of a neoplasm may eventually result in substantially higher cost through inadequate treatment than an initial comprehensive work-up of the tissue.
-
Consensus statements
Equal access to state-of-the-art integrated histomorphological and molecular diagnostic assessment of CNS tumors according to the most current version of the WHO classification should be pursued.
Every center should participate in quality assurance programs, either according to local regulations or international norms (DIN EN ISO 17020 or 15189) or setup own inter-institutional quality circles.
Larger centers are called to assist others in implementing novel technology and provide samples for validation.
Regulations in Europe and Ethical Aspects (Including Germline Testing)
In the light of all these technological advances it is important to realize that the use of in vitro diagnostics has become more strict in the European Union (EU) under the new in vitro diagnostics regulation (IVDR, EU directive 2017/746)94,95 also depicted in Supplementary Figure 1. Most innovative tests are introduced as lab-developed tests (LDTs). Although LDTs can be exempt from the IVDR, there are several conditions that need to be met that are mentioned in chapter 2, article 5.5 of the regulation.96,97 The two most important thereof are that the laboratory is compliant with EN ISO 15189 standards or, where applicable, national provisions (article 5.5c) and provides a justification for the use of the LDT, that is, why the specific needs for that test cannot be met by an equivalent (IVD) device on the market (article 5.5d). Still, even then the relevant general safety and performance requirements set out in Annex I of the IVDR need to be documented for the LDT. These procedures typically also result in several validation analyses, that may vary considerably between national legislation and international standards/norms. Typically, intra- and inter-assay validation runs are performed, employing samples that representatively cover the targets assessed with this method. This setup easily amounts to dozens of analyses for only a single assay validation. Laboratories should be aware of these conditions to make sure technological advances, as described above, can make it into routine patient care.
With all these innovations there is also an increased chance of unsolicited findings, that is, findings that are unrelated to the initial clinical question the test was performed for, but that may still be of medical relevance to the health of the patient or the family. Prime example are indications for hereditary disorders (germline findings). Obviously, when normal DNA of the patient is used as a reference in the sequencing of tumor DNA there is a high chance of identifying germline pathogenic variants in patients carrying such a variant,98 but also tumor DNA-only sequencing may provide indications for germline involvement. The same goes for methylation profiling that may identify the tumor as a tumor entity having a high chance of having a germline pathogenic variant (eg, in the DICER1 or SMARCB1 genes). The interpretation and communication of these findings need to be handled with caution and may be subject to specific regulations in individual countries. Depending on the chances of identifying unsolicited findings these could be discussed upfront with the patient and may require an informed consent before initiation of molecular testing.99,100
Future Perspectives and Conclusions
Emerging Technologies
Technologies are rapidly advancing and are likely to soon further impact on tumor classification. Implementation of such novel sequencing capacities and other technologies in clinical practice is so far hampered by the costs and by the fact that many (neuro)pathology departments only store FFPE tissues, thus severely limiting the use of WES and long-read sequencing technologies. Also, single cell or single nucleus sequencing technologies at present generally still require either fresh tissue (single cell) or snap-frozen (single nucleus) samples as starting material, but recent advances now make those methods applicable on FFPE tissues. Obviously, a change of current pathology practice would facilitate the implementation of techniques that (so far) require fresh or snap-frozen tissue samples.
Spatially resolved transcriptomics has been selected as method of the year 2020 by Nature Methods.101 Such spatial techniques are of interest as they may identify resistant or malignant clones within a single tumor even in very small biopsies. There are several methods that can be used to perform spatial transcriptomics including (but not limited to) hybridization-based techniques, cleavable oligonucleotides and multiplexed in situ hybridization. Some of these techniques are now also suitable for use on FFPE tissues. The level of detail provided by these techniques is impressive, but there is still significant development required until they can be implemented into clinical diagnostic settings.
One drawback of sequencing technologies is that they detect nucleotide entities (DNA/RNA) where proteins are considered to comprise most active components driving oncogenesis (miRNAs, long non-coding RNAs, ncRNAs, or ribosomes are at present not exploited for diagnostic purposes, and targeting non-coding RNAs is challenging). Over the past decades several interesting advances have been made in proteomics and the technique can now also be used on FFPE tissues and provide spatial information.102 However, an additional increase in detection sensitivity is likely required for use in a clinical diagnostic setting. Especially in specialized centers, metabolomics or MR spectroscopy can be employed to detect D-2HG as marker for non-invasive assessment of the presence of IDH-mutations.103
Many efforts are made to evaluate the use of liquid biopsies, which hold the promise to bypass surgery purely for diagnostic purposes, and to provide a minimally invasive approach for active tumor monitoring. At present however, the detection of CNS tumors using liquid biopsies/blood seems feasible but with quite limited sensitivity.60,104 Cerebrospinal fluid may be more useful as it appears to hold more circulating CNS tumor DNA.105 Further (prospective) clinical studies are needed to assess the role of such liquid biopsy procedures.
We can also expect to see an advance in the use of artificial intelligence and artificial neural networks for image recognition. A prime example is ResNet50 (see https://viso.ai/deep-learning/resnet-residual-neural-network/). Such analysis will most likely find its way into image analysis of histological sections and MRI images to aid diagnosis and guide treatment decisions. All these developments are also expected to reduce the turnaround-time until information about molecular markers is available, overcoming a critical limitation of current high-throughput analyses.
Conclusions
Comprehensive molecular diagnostics emerged as an essential part of clinical practice, and is not limited to clinical trials, since molecular markers became part of the WHO CNS tumor classification in 2016. This development has been accelerated with the 2021 edition of the WHO classification, in which even more diagnoses are based on integration of molecular information. Thereby, these novel markers assist in rendering a more precise diagnosis and reducing the inter-diagnostician variation, which is a known risk of histopathological assessment. For the clinical setting, this allows improved prognostication and, consequently, more accurate counseling of patients with critical clinical conditions on the optimal post-operative treatment. The availability of a wide spectrum of markers and the variety of assays poses a challenge to diagnostic laboratories. Finding the best compromise between cost of infrastructure, cost per sample, turnaround times and typical sample submission numbers is crucial for the sustainable provision of comprehensive diagnostics. With the advances in next- and now third-generation sequencing, wider analyses like exome- or even genome-wide analyses, and genome-wide methylation profiling become more feasible from an operational and economical perspective. Most recent developments in the market for sequencing devices and reagents, triggered by the expiry of some of the patents held by the de facto monopolist, may serve as catalysator of this transformation. Carefully curated data of this comprehensive type in turn is the basis for further scientific insight into these tumor types and the discovery of novel diagnostic, and potentially also therapeutic markers.
Regardless, the power of molecular data is only leveraged when contextualized with the corresponding tissue. Thus, reporting of molecular findings should include characteristics of histopathological features, the method applied for the analyses, and unequivocal description of the molecular findings, to derive an integrated diagnosis. When rendering the integrated diagnosis, a comment should be provided as to how the data were weighted and integrated, unless all layers are entirely consistent. With the increasing granularity and extent of neuropathology specimen work-up, mandated by national and international guidelines, any inadequate reflection of reimbursement of this decisive step in clinical care needs to be overcome by capturing cost as precisely as possible and to incorporate them into reimbursement schemes.
In conclusion, thanks to the translation of scientific findings on molecular characteristics of gliomas, glioneuronal and neuronal tumors into diagnostic practice, major improvements have been achieved in rendering a more precise, tissue-based diagnosis of these tumors. Yet, much work remains to be done to improve the availability of the diagnostic tools that allow for optimal assessment of the relevant molecular alterations, and to translate the improved, tissue-based diagnoses of gliomas, glioneuronal and neuronal tumors into a better outcome for the patients diagnosed with such tumors.
Supplementary Material
Contributor Information
Felix Sahm, Department of Neuropathology, University Hospital Heidelberg, Heidelberg, Germany; CCU Neuropathology, German Concortium for Translational Cancer Research (DKTK), German Cancer Research Center (DKFZ), Heidelberg, Germany.
Sebastian Brandner, Department of Neurodegenerative Disease, UCL Queen Square Institute of Neurology and Division of Neuropathology, The National Hospital for Neurology and Neurosurgery, University College London Hospitals NHS Foundation Trust, London, UK.
Luca Bertero, Pathology Unit, Department of Medical Sciences, University of Turin, Turin, Italy.
David Capper, Department of Neuropathology, Charité, Universitätsmedizin Berlin, Freie Universität Berlin and Humboldt-Universität zu Berlin, Berlin, Germany; German Cancer Consortium (DKTK), Partner Site Berlin, German Cancer Research Center (DKFZ), Heidelberg, Germany.
Pim J French, Department of Neurology, Brain Tumor Center at Erasmus MC Cancer Center, 3015 GD Rotterdam, The Netherlands.
Dominique Figarella-Branger, Aix-Marseille University, APHM, CNRS, INP, Institute Neurophysiopathol, CHU Timone, Service d’Anatomie Pathologique et de Neuropathologie, Marseille, France.
Felice Giangaspero, Department of Radiological, Oncological and Anatomo-Pathological Sciences, University Sapienza of Rome, Rome, Italy.
Christine Haberler, Division of Neuropathology and Neurochemistry, Department of Neurology, Medical University of Vienna, Austria.
Monika E Hegi, Neuroscience Research Center and Neurosurgery, Lausanne University Hospital and University of Lausanne, Switzerland.
Bjarne W Kristensen, Department of Clinical Medicine and Biotech Research and Innovation Center (BRIC), University of Copenhagen, Denmark; Department of Pathology, The Bartholin Institute, Rigshospitalet, Copenhagen University Hospital, Copenhagen, Denmark.
Kathreena M Kurian, Brain Tumour Research Centre, University of Bristol, UK.
Matthias Preusser, Division of Oncology, Department of Medicine I, Medical University of Vienna, Austria.
Bastiaan B J Tops, Princess Máxima Center for Pediatric Oncology, Utrecht, The Netherlands.
Martin van den Bent, The Brain Tumor Center at Erasmus MC Cancer Institute, Rotterdam, The Netherlands.
Wolfgang Wick, Department of Neurology and Neurooncology Program, National Center for Tumor Diseases, Heidelberg University Hospital; Clinical Cooperation Unit Neurooncology, German Cancer Consortium (DKTK), German Cancer Research Center (DKFZ), Heidelberg, Germany.
Guido Reifenberger, Institute of Neuropathology, Heinrich Heine University, Medical Faculty, and University Hospital Düsseldorf, and German Cancer Consortium (DKTK), Partner Site Essen/Düsseldorf, Düsseldorf, Germany.
Pieter Wesseling, Department of Pathology, Amsterdam University Medical Centers, Amsterdam, The Netherlands; Princess Máxima Center for Pediatric Oncology, Utrecht, The Netherlands (P.W.).
Conflict of interest statement: FS: Co-inventor on patents filed related to methylation profiling, co-founder of Heidelberg Epignostix. The paragraph on DNA methylation analysis has hence been in the responsibility of SB and PW. Honoraria from Illumina and Bayer. DC: Co-inventor on patents filed related to methylation profiling and mutation-specific antibodies, co-founder of Heidelberg Epignostix. The paragraph on DNA methylation analysis and mutation-specific immunohistochemistry has hence not been in his responsibility. MP: received honoraria for lectures, consultation or advisory board participation from the following for-profit companies: Bayer, Bristol-Myers Squibb, Novartis, Gerson Lehrman Group (GLG), CMC Contrast, GlaxoSmithKline, Mundipharma, Roche, BMJ Journals, MedMedia, Astra Zeneca, AbbVie, Lilly, Medahead, Daiichi Sankyo, Sanofi, Merck Sharp & Dome, Tocagen, Adastra, Gan & Lee Pharmaceuticals, Servier; MvdB: received honoraria for consultancy from Genenta, Servier, Astra Zeneca, Boehringer-Ingelheim, Carthera, Nerviano, Chimerix, Roche, Fore Biotherapeutics, Menarini-Stemline, Incyte and Sumitomo Pharma Oncology; WW: reports honoraria for consultation or non-financial clinical trial support from Apogenix, Bayer, Merck Sharp & Dome, AstraZeneca, Merck Serono, Novartis, Roche and Mundipharma, with compensation paid to the University Clinic. SB, LB, PF, DFB, FG, CH, MH, BWK, KK, BBJT, GR, PW: none.
Dedication
This work is dedicated to co-author Prof. Felice Giangaspero, who unfortunately passed away at the time this manuscript was submitted for publication to Neuro-Oncology. We and the EANO community honor Felice for his major and continuous contributions to improved classification of pediatric and other CNS tumors, and for his role in mentoring many younger colleagues in the field. We will remember Felice as a very kind, knowledgeable and wise colleague and as a true gentleman; he will be dearly missed.
References
- 1. Wesseling P, Capper D.. WHO 2016 Classification of gliomas. Neuropathol Appl Neurobiol. 2018;44(2):139–150. [DOI] [PubMed] [Google Scholar]
- 2. Coons SW, Johnson PC, Scheithauer BW, Yates AJ, Pearl DK.. Improving diagnostic accuracy and interobserver concordance in the classification and grading of primary gliomas. Cancer. 1997;79(7):1381–1393. [DOI] [PubMed] [Google Scholar]
- 3. Kros JM, Gorlia T, Kouwenhoven MC, et al. Panel review of anaplastic oligodendroglioma from European Organization For Research and Treatment of Cancer Trial 26951: assessment of consensus in diagnosis, influence of 1p/19q loss, and correlations with outcome. J Neuropathol Exp Neurol. 2007;66(6):545–551. [DOI] [PubMed] [Google Scholar]
- 4. Kros JM. From expert opinion to evidence-based: changes in the gold standard of primary brain tumour diagnosis. J Pathol. 2007;213(1):1–3. [DOI] [PubMed] [Google Scholar]
- 5. van den Bent MJ. Interobserver variation of the histopathological diagnosis in clinical trials on glioma: a clinician’s perspective. Acta Neuropathol. 2010;120(3):297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Louis DN, et al. . WHO Classification of Tumours of the Central Nervous System. revised. 4th ed. Lyon; 2016. [Google Scholar]
- 7. Board, W.C.o.T.E. Central Nervous System Tumours. 5th ed. Lyon; 2016. [Google Scholar]
- 8. Louis DN, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Brat DJ, Aldape K, Bridge JA, et al. Molecular biomarker testing for the diagnosis of diffuse gliomas. Arch Pathol Lab Med. 2022;146(5):547–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Horbinski C, Berger T, Packer RJ, Wen PY.. Clinical implications of the 2021 edition of the WHO classification of central nervous system tumours. Nat Rev Neurol. 2022;18(9):515–529. [DOI] [PubMed] [Google Scholar]
- 11. Capper D, et al. EANO guideline on rational molecular testing of gliomas, glioneuronal and neuronal tumors in adults for targeted therapy selection. Neuro Oncol. 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Brainin M, Barnes M, Baron J-C, et al. . Guidance for the preparation of neurological management guidelines by EFNS scientific task forces - revised recommendations 2004. Eur J Neurol. 2004;11(9):577–581. [DOI] [PubMed] [Google Scholar]
- 13. Zadeh G, Salehi F, An S, et al. Diagnostic implications of histological analysis of neurosurgical aspirate in addition to routine resections. Neuropathology. 2012;32(1):44–50. [DOI] [PubMed] [Google Scholar]
- 14. Zacher A, Kaulich K, Stepanow S, et al. Molecular diagnostics of gliomas using next generation sequencing of a glioma-tailored gene panel. Brain Pathol. 2017;27(2):146–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sahm F, Schrimpf D, Jones DTW, et al. Next-generation sequencing in routine brain tumor diagnostics enables an integrated diagnosis and identifies actionable targets. Acta Neuropathol. 2016;131(6):903–910. [DOI] [PubMed] [Google Scholar]
- 16. Nikiforova MN, Wald AI, Melan MA, et al. Targeted next-generation sequencing panel (GlioSeq) provides comprehensive genetic profiling of central nervous system tumors. Neuro Oncol. 2016;18(3):379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patel A, Dogan H, Payne A, et al. Rapid-CNS(2): rapid comprehensive adaptive nanopore-sequencing of CNS tumors, a proof-of-concept study. Acta Neuropathol. 2022;143(5):609–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Horak P, Griffith M, Danos AM, et al. Standards for the classification of pathogenicity of somatic variants in cancer (oncogenicity): Joint recommendations of Clinical Genome Resource (ClinGen), Cancer Genomics Consortium (CGC), and Variant Interpretation for Cancer Consortium (VICC). Genet Med. 2022;24(5):986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schram AM, Chang MT, Jonsson P, Drilon A.. Fusions in solid tumours: diagnostic strategies, targeted therapy, and acquired resistance. Nat Rev Clin Oncol. 2017;14(12):735–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. You G, Fan X, Hu H, Jiang T, Chen CC.. Fusion genes altered in adult malignant gliomas. Front Neurol. 2021;12:715206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jones DT, Hutter B, Jäger N, et al. ; International Cancer Genome Consortium PedBrain Tumor Project. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45(8):927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Singh D, Chan JM, Zoppoli P, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337(6099):1231–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heyer EE, Deveson IW, Wooi D, et al. Diagnosis of fusion genes using targeted RNA sequencing. Nat Commun. 2019;10(1):1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fujii T, Uchiyama T, Matsuoka M, et al. Evaluation of DNA and RNA quality from archival formalin-fixed paraffin-embedded tissue for next-generation sequencing—retrospective study in Japanese single institution. Pathol Int. 2020;70(9):602–611. [DOI] [PubMed] [Google Scholar]
- 25. Davila JI, Fadra NM, Wang X, et al. Impact of RNA degradation on fusion detection by RNA-seq. BMC Genom. 2016;17(1):814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Barthel FP, Wei W, Tang M, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet. 2017;49(3):349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haas BJ, Dobin A, Li B, et al. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019;20(1):213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. LaHaye S, Fitch JR, Voytovich KJ, et al. Discovery of clinically relevant fusions in pediatric cancer. BMC Genom. 2021;22(1):872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hoogstrate Y, Komor MA, Böttcher R, et al. Fusion transcripts and their genomic breakpoints in polyadenylated and ribosomal RNA-minus RNA sequencing data. GigaScience. 2021;10(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hehir-Kwa JY, Koudijs MJ, Verwiel ETP, et al. Improved gene fusion detection in childhood cancer diagnostics using RNA sequencing. JCO Precis Oncol. 2022;6:e2000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Na K, Kim H-S, Shim HS, et al. Targeted next-generation sequencing panel (TruSight Tumor 170) in diffuse glioma: a single institutional experience of 135 cases. J Neurooncol. 2019;142(3):445–454. [DOI] [PubMed] [Google Scholar]
- 32. van Tilburg CM, Pfaff E, Pajtler KW, et al. The Pediatric Precision Oncology INFORM Registry: clinical outcome and benefit for patients with very high-evidence targets. Cancer Discov. 2021;11(11):2764–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stichel D, Schrimpf D, Casalini B, et al. Routine RNA sequencing of formalin-fixed paraffin-embedded specimens in neuropathology diagnostics identifies diagnostically and therapeutically relevant gene fusions. Acta Neuropathol. 2019;138(5):827–835. [DOI] [PubMed] [Google Scholar]
- 34. Hoogstrate Y, Vallentgoed W, Kros JM, et al. EGFR mutations are associated with response to depatux-m in combination with temozolomide and result in a receptor that is hypersensitive to ligand. Neurooncol Adv. 2020;2(1):vdz051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. French PJ, et al. Defining EGFR amplification status for clinical trial inclusion. Neuro Oncol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Juraschka K, Taylor MD.. Medulloblastoma in the age of molecular subgroups: a review. J Neurosurg Pediatr. 2019;24(4):353–363. [DOI] [PubMed] [Google Scholar]
- 37. Korshunov A, Chavez L, Northcott PA, et al. DNA-methylation profiling discloses significant advantages over NanoString method for molecular classification of medulloblastoma. Acta Neuropathol. 2017;134(6):965–967. [DOI] [PubMed] [Google Scholar]
- 38. Korshunov A, Sahm F, Zheludkova O, et al. DNA methylation profiling is a method of choice for molecular verification of pediatric WNT-activated medulloblastomas. Neuro Oncol. 2019;21(2):214–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Capper D, Jones DTW, Sill M, et al. DNA methylation-based classification of central nervous system tumours. Nature. 2018;555(7697):469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Capper D, Stichel D, Sahm F, et al. Practical implementation of DNA methylation and copy-number-based CNS tumor diagnostics: the Heidelberg experience. Acta Neuropathol. 2018;136(2):181–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wu Z, Abdullaev Z, Pratt D, et al. Impact of the methylation classifier and ancillary methods on CNS tumor diagnostics. Neuro Oncol. 2022;24(4):571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pickles JC, Fairchild AR, Stone TJ, et al. DNA methylation-based profiling for paediatric CNS tumour diagnosis and treatment: a population-based study. Lancet Child Adolesc Health. 2020;4(2):121–130. [DOI] [PubMed] [Google Scholar]
- 43. Jaunmuktane Z, Capper D, Jones DTW, et al. Methylation array profiling of adult brain tumours: diagnostic outcomes in a large, single centre. Acta Neuropathol Commun. 2019;7(1):24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Priesterbach-Ackley LP, Boldt HB, Petersen JK, et al. Brain tumour diagnostics using a DNA methylation-based classifier as a diagnostic support tool. Neuropathol Appl Neurobiol. 2020;46(5):478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Pickles JC, Stone TJ, Jacques TS.. Methylation-based algorithms for diagnosis: experience from neuro-oncology. J Pathol. 2020;250(5):510–517. [DOI] [PubMed] [Google Scholar]
- 46. Pajtler KW, Witt H, Sill M, et al. Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell. 2015;27(5):728–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ghasemi DR, Sill M, Okonechnikov K, et al. MYCN amplification drives an aggressive form of spinal ependymoma. Acta Neuropathol. 2019;138(6):1075–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pajtler KW, Mack SC, Ramaswamy V, et al. The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Acta Neuropathol. 2017;133(1):5–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Korshunov A, et al. DNA-methylation profiling is a method of choice for molecular verification of pediatric WNT activated medulloblastomas. Neuro Oncol. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schwalbe EC, Lindsey JC, Nakjang S, et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. Lancet Oncol. 2017;18(7):958–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cavalli FMG, Remke M, Rampasek L, et al. Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell. 2017;31(6):737–754.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ramaswamy V, Remke M, Bouffet E, et al. Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol. 2016;131(6):821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sturm D, Orr BA, Toprak UH, et al. New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell. 2016;164(5):1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Suppiah S, Nassiri F, Bi WL, et al. ; International Consortium on Meningiomas. Molecular and translational advances in meningiomas. Neuro Oncol. 2019;21(Suppl 1):i4–i17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Maas SLN, Stichel D, Hielscher T, et al. ; German Consortium on Aggressive Meningiomas (KAM). Integrated molecular-morphologic meningioma classification: a multicenter retrospective analysis, retrospectively and prospectively validated. J Clin Oncol. 2021;39(34):3839–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Koelsche C, Schrimpf D, Stichel D, et al. Sarcoma classification by DNA methylation profiling. Nat Commun. 2021;12(1):498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stichel D, Schrimpf D, Sievers P, et al. Accurate calling of KIAA1549-BRAF fusions from DNA of human brain tumours using methylation array-based copy number and gene panel sequencing data. Neuropathol Appl Neurobiol. 2021;47(3):406–414. [DOI] [PubMed] [Google Scholar]
- 58. Bady P, Sciuscio D, Diserens A-C, et al. MGMT methylation analysis of glioblastoma on the Infinium methylation BeadChip identifies two distinct CpG regions associated with gene silencing and outcome, yielding a prediction model for comparisons across datasets, tumor grades, and CIMP-status. Acta Neuropathol. 2012;124(4):547–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bady P, Delorenzi M, Hegi ME.. Sensitivity analysis of the MGMT-STP27 model and impact of genetic and epigenetic context to predict the MGMT methylation status in gliomas and other tumors. J Mol Diagn. 2016;18(3):350–361. [DOI] [PubMed] [Google Scholar]
- 60. Nassiri F, Chakravarthy A, Feng S, et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat Med. 2020;26(7):1044–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Grabovska Y, Mackay A, O’Hare P, et al. Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nat Commun. 2020;11(1):4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pardue ML, Gall JG.. Molecular hybridization of radioactive DNA to the DNA of cytological preparations. Proc Natl Acad Sci USA. 1969;64(2):600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jones DT, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68(21):8673–8677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Andreiuolo F, Varlet P, Tauziède-Espariat A, et al. Childhood supratentorial ependymomas with YAP1-MAMLD1 fusion: an entity with characteristic clinical, radiological, cytogenetic and histopathological features. Brain Pathol. 2019;29(2):205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tauziede-Espariat A, Siegfried A, Nicaise Y, et al. ; RENOCLIP-LOC, the BIOMECA (Biomarkers for Ependymomas in Children, Adolescents) consortium. Supratentorial non-RELA, ZFTA-fused ependymomas: a comprehensive phenotype genotype correlation highlighting the number of zinc fingers in ZFTA-NCOA1/2 fusions. Acta Neuropathol Commun. 2021;9(1):135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sanger F, Nicklen S, Coulson AR.. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74(12):5463–5467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Nyren P, Pettersson B, Uhlen M.. Solid phase DNA minisequencing by an enzymatic luminometric inorganic pyrophosphate detection assay. Anal Biochem. 1993;208(1):171–175. [DOI] [PubMed] [Google Scholar]
- 68. Appay R, Fina F, Macagno N, et al. Duplications of KIAA1549 and BRAF screening by droplet digital PCR from formalin-fixed paraffin-embedded DNA is an accurate alternative for KIAA1549-BRAF fusion detection in pilocytic astrocytomas. Mod Pathol. 2018;31(10):1490–1501. [DOI] [PubMed] [Google Scholar]
- 69. Appay R, Fina F, Barets D, et al. Multiplexed droplet digital PCR assays for the simultaneous screening of major genetic alterations in tumors of the central nervous system. Front Oncol. 2020;10:579762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wolter M, Felsberg J, Malzkorn B, Kaulich K, Reifenberger G.. Droplet digital PCR-based analyses for robust, rapid, and sensitive molecular diagnostics of gliomas. Acta Neuropathol Commun. 2022;10(1):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nadauld L, et al. Quantitative and sensitive detection of cancer genome amplifications from formalin fixed paraffin embedded tumors with droplet digital PCR. Transl Med. 2012;2(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Miotke L, Lau BT, Rumma RT, Ji HP.. High sensitivity detection and quantitation of DNA copy number and single nucleotide variants with single color droplet digital PCR. Anal Chem. 2014;86(5):2618–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Hegi ME, Diserens A-C, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997–1003. [DOI] [PubMed] [Google Scholar]
- 74. Malley DS, Hamoudi RA, Kocialkowski S, et al. A distinct region of the MGMT CpG island critical for transcriptional regulation is preferentially methylated in glioblastoma cells and xenografts. Acta Neuropathol. 2011;121(5):651–661. [DOI] [PubMed] [Google Scholar]
- 75. Tesileanu CMS, Gorlia T, Golfinopoulos V, French PJ, van den Bent MJ.. MGMT promoter methylation determined by the MGMT-STP27 algorithm is not predictive for outcome to temozolomide in IDH-mutant anaplastic astrocytomas. Neuro Oncol. 2022;24(4):665–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Hegi ME, Genbrugge E, Gorlia T, et al. MGMT Promoter methylation cutoff with safety margin for selecting glioblastoma patients into trials omitting temozolomide. A pooled analysis of four clinical trials. Clin Cancer Res. 2019;25(6):1809–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Quillien V, Lavenu A, Ducray F, et al. Validation of the high-performance of pyrosequencing for clinical MGMT testing on a cohort of glioblastoma patients from a prospective dedicated multicentric trial. Oncotarget. 2016;7(38):61916–61929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Preusser M, Charles Janzer R, Felsberg J, et al. Anti-O6-methylguanine-methyltransferase (MGMT) immunohistochemistry in glioblastoma multiforme: observer variability and lack of association with patient survival impede its use as clinical biomarker. Brain Pathol. 2008;18(4):520–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Esteller M, Garcia-Foncillas J, Andion E, et al. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343(19):1350–1354. [DOI] [PubMed] [Google Scholar]
- 80. Quillien V, Lavenu A, Ducray F, et al. Clinical validation of the CE-IVD marked Therascreen MGMT kit in a cohort of glioblastoma patients. Cancer Biomark. 2017;20(4):435–441. [DOI] [PubMed] [Google Scholar]
- 81. Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG.. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res. 1999;59(4):793–797. [PubMed] [Google Scholar]
- 82. Platten M, Bunse L, Wick A, et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature. 2021;592(7854):463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Mellinghoff IK, Ellingson BM, Touat M, et al. Ivosidenib in isocitrate dehydrogenase 1-mutated advanced glioma. J Clin Oncol. 2020;38(29):3398–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Wiestler B, Capper D, Hovestadt V, et al. Assessing CpG island methylator phenotype, 1p/19q codeletion, and MGMT promoter methylation from epigenome-wide data in the biomarker cohort of the NOA-04 trial. Neuro Oncol. 2014;16(12):1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. McAleenan A, Jones HE, Kernohan A, et al. Diagnostic test accuracy and cost-effectiveness of tests for codeletion of chromosomal arms 1p and 19q in people with glioma. Cochrane Database Syst Rev. 2022;3(3):CD013387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Le Rhun E, Preusser M, Roth P, et al. Molecular targeted therapy of glioblastoma. Cancer Treat Rev. 2019;80:101896. [DOI] [PubMed] [Google Scholar]
- 87. Drilon A. TRK inhibitors in TRK fusion-positive cancers. Ann Oncol. 2019;30(Suppl 8):viii23–viii30. [DOI] [PubMed] [Google Scholar]
- 88. Izquierdo E, Carvalho DM, Mackay A, et al. DIPG harbors alterations targetable by MEK inhibitors, with acquired resistance mechanisms overcome by combinatorial inhibition. Cancer Discov. 2022;12(3):712–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kim PL. Targeting gene fusions in glioma. Curr Opin Neurol. 2021;34(6):840–847. [DOI] [PubMed] [Google Scholar]
- 90. Synhaeve NE, van den Bent MJ, French PJ, et al. Clinical evaluation of a dedicated next generation sequencing panel for routine glioma diagnostics. Acta Neuropathol Commun. 2018;6(1):126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Nabors LB, Portnow J, Ahluwalia M, et al. Central nervous system cancers, version 3.2020, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Netw. 2020;18(11):1537–1570. [DOI] [PubMed] [Google Scholar]
- 92. Weller M, van den Bent M, Preusser M, et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat Rev Clin Oncol. 2021;18(3):170–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wen PY, Weller M, Lee EQ, et al. Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020;22(8):1073–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. https://eur-lex.europa.eu/eli/reg/2017/746/oj.
- 95. Regulation (EU) 2017/746 of the European Parliament and of the council of 5 April 2017 on in vitro diagnostic medical devices and repealing directive 98/79/EC and commission decision 2010/227/EU. Off J Eur Union. 2017;117:176–332. [Google Scholar]
- 96. Bank PCD, et al. The end of the laboratory developed test as we know it? Recommendations from a national multidisciplinary taskforce of laboratory specialists on the interpretation of the IVDR and its complications. Clin Chem Lab Med. 2020. [DOI] [PubMed] [Google Scholar]
- 97. Vermeersch P, Van Aelst T, Dequeker EMC.. The new IVD Regulation 2017/746: a case study at a large university hospital laboratory in Belgium demonstrates the need for clarification on the degrees of freedom laboratories have to use lab-developed tests to improve patient care. Clin Chem Lab Med. 2020;59(1):101–106. [DOI] [PubMed] [Google Scholar]
- 98. Schoot VV, Viellevoije SJ, Tammer F, et al. The impact of unsolicited findings in clinical exome sequencing, a qualitative interview study. Eur J Hum Genet. 2021;29(6):930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Winkler EC, Knoppers BM.. Ethical challenges of precision cancer medicine. Semin Cancer Biol. 2022;84:263–270. [DOI] [PubMed] [Google Scholar]
- 100. Matrana MR, Campbell B.. Precision Medicine and the Institutional Review Board: ethics and the Genome. Ochsner J. 2020;20(1):98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Method of the Year 2020: spatially resolved transcriptomics. Nat Methods. 2021;18(1):1. [DOI] [PubMed] [Google Scholar]
- 102. Lundberg E, Borner GHH.. Spatial proteomics: a powerful discovery tool for cell biology. Nat Rev Mol Cell Biol. 2019;20(5):285–302. [DOI] [PubMed] [Google Scholar]
- 103. Kumar M, Nanga RPR, Verma G, et al. Emerging MR imaging and spectroscopic methods to study brain tumor metabolism. Front Neurol. 2022;13:789355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sabedot TS, Malta TM, Snyder J, et al. A serum-based DNA methylation assay provides accurate detection of glioma. Neuro Oncol. 2021;23(9):1494–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Miller AM, Shah RH, Pentsova EI, et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature. 2019;565(7741):654–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.