

Abstract
The lysosome is responsible for protein and organelle degradation and homeostasis and the cathepsins play a key role in maintaining protein quality control. Cathepsin D (CTSD), is one such lysosomal protease, which when deficient in humans lead to neurolipofuscinosis (NCL) and is important in removing toxic protein aggregates. Prior studies demonstrated that CTSD germ-line knockout-CtsdKO (CDKO) resulted in accumulation of protein aggregates, decreased proteasomal activities, and postnatal lethality on Day 26 ± 1. Overexpression of wildtype CTSD, but not cathepsin B, L or mutant CTSD, decreased α-synuclein toxicity in worms and mammalian cells. In this study we generated a mouse line expressing human CTSD with a floxed STOP cassette between the ubiquitous CAG promoter and the cDNA. After crossing with Nestin-cre, the STOP cassette is deleted in NESTIN + cells to allow CTSD overexpression-CTSDtg (CDtg). The CDtg mice exhibited normal behavior and similar sensitivity to sub-chronic 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced neurodegeneration. By breeding CDtg mice with CDKO mice, we found that over-expression of CTSD extended the lifespan of the CDKO mice, partially rescued proteasomal deficits and the accumulation of Aβ42 in the CDKO. This new transgenic mouse provides supports for the key role of CTSD in protecting against proteotoxicity and offers a new model to study the role of CTSD enhancement in vivo.
Key words: Cathepsin D transgenic mouse, Mitochondrial bioenergetics, Apoptosis, Cathepsin D knockout mice, Dopamine, Autophagy, Lysosome, Life expectancy, Behavior, Neuronal ceroid lipofuscinosis
Graphical abstract
Over-expression of human Cathepsin D in central nervous system partially rescued postnatal lethality of Ctsd germline knockout (CDKO) mice, and the accumulation of Aβ42 and α-synuclein in the CDKO.
1. Introduction
Lysosomes play a major role in degradation of proteins and organelles through autophagy. At least 11 genes out of 24 loci identified to be associated with Parkinson's disease (PD) are involved in or disrupt the autophagy–lysosome pathway1,2. Insufficient activity of the autophagy–lysosome system may be involved in the formation of the amyloid plaques and tau aggregates which occur in Alzheimer's disease (AD)3,4. Cathepsin D (CTSD) is the principal lysosomal aspartate protease, and is activated in the acidic lysosomal environment5. In support of a unique role for CTSD, its deficiency causes human congenital neuronal ceroid lipofuscinosis (NCL). CTSD homozygous inactivation caused human NCL6 and a patient with CTSD deficiency developed motor disturbances7. PD substantia nigra neurons exhibit decreased CTSD protein levels compared to age-matched controls8. PD and α-synuclein accumulation are associated with lysosomal deficits9, 10, 11, arguing for an important role of the lysosomes in α-synuclein degradation and their dysfunction in Parkinsonisms. Ctsd germline knockout mice (CDKO) exhibit neuronal, inflammatory, and systemic pathologies and die at around postnatal Day 26 (P26) due to intestinal necrosis12, 13, 14. In support of an important role of CTSD in autophagy, we and others found that CDKO mice exhibit accumulation of autophagosomes and α-synuclein14, 15, 16. Furthermore, CDKO mice also accumulate insoluble Aβ4217,18.
The ultimate goal of neurodegenerative disease research is to provide better treatment strategies that attenuate pathogenic progression of neurodegenerative diseases. We previously overexpressed human CTSD in worms and mammalian cells and found that CTSD decreased α-synuclein aggregation in mammalian cells, and decreased α-synuclein toxicity both in worms and in mammalian cells15. This observation suggested the possibility of enhancing CTSD activity as a means for therapy against α-synucleinopathy in PD and perhaps also Aβ and tau accumulation in AD. The advantage of enhancing CTSD activity over enhancement of non-selective autophagy in surveillance of cellular damage, is that it only increases the efficiency of degrading the damaged cellular materials that have already been engulfed by autophagosomes, while sparing the healthy cellular content. However, whether CTSD enhancement in vivo protects against endogenously generated toxic protein species or environmental toxins is unknown. Furthermore, there have been reports of adverse effects of CTSD overexpression in vitro. For example, oxidative stress generated by redox cycling of naphthazarin increased CTSD activity and apoptosis in human fibroblast AG-1518 cells, both the CTSD activation and apoptosis can be prevented by the CTSD inhibitor pepstatin A19. In primary human T lymphocytes, pepstatin A inhibits staurosporine-induced cell death20. In 3Y1-Ad12 cancer cells, overexpression of either wildtype or catalytically inactive CTSD enhanced tumor growth in xenographs21, as well as the apoptotic response to etoposide22. Whether these studies in cell culture translate to the more complex interactions in vivo is unknown. This is particularly important in the context of AD and PD.
The testing of these concepts in neurodegenerative diseases is hampered by the lack of appropriate models. Accordingly, we developed a transgenic model overexpressing CTSD in the brain under the control of the nestin promoter. We found that CTSD is highly expressed in neurons and colocalizes with other lysosomal markers. Furthermore, enhancing lysosomal CTSD did not have any adverse effects on neuronal bioenergetics and survival, did not affect dopamine levels or behavior, and did not change the levels of other autophagy proteins under normal conditions. Using this model, we found that nervous system CTSD elevation did not change dopaminergic neurodegeneration in response to the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) albeit in a short term sub-chronic treatment. However, it partially rescued CTSD deficient phenotypes brought about by deletion of the endogenous Ctsd gene. With direct relevance to AD it attenuated the accumulation of Aβ42 in the CDKO. This new transgenic mouse will enable further studies of the role of CTSD enhancement in vivo in tissue and temporal specific manner, as well as further investigations of the potential of elevating CTSD in PD and AD therapeutic development.
2. Materials and methods
2.1. Mice
Mice are all on the C57BL/6 background. Cathepsin D (Ctsd) germline knockout mice (CDKO) were genotyped as described before15,23. Nestin-cre:CTSDfloxedstop mice were generated by GenOway using homologous recombination by inserting a floxed stop cassette with CTSD cDNA into the Hprt locus. Nestin-cre:CTSDfloxedstop mice genotyping primers are: PCR#1 (2240 bp): F: CATGGTAAGTAAGCTTGGGCTGCAGG; R: ACGTCAGTAGTCATAGGAACTGCGGTCG. PCR#2 (cre-excised product: 325 bp): F: AGCCTCTGCTAACCATGTTCATGCC; R: GCGGATGGACGTGAACTTGTGC.cre: (480 bp) F: TCGCGATTATCTTCTATATCTTCAG; R: GCTCGACCAGTTTAGTTACCC. At least 3 mice per group were used for biochemistry and immunohistochemistry studies. Mice of both sexes were used and we did not observe sex disaggregation in any of our assays. Behavioral studies were first performed with n = 5–6 mice both sexes at 1 year of age, and then repeated at 1.5 and 2 year of age with male only (n = 6–9), and at 4 months of age after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) 5 daily sub-chronic treatment as we previously published23. As described before, MPTP-HCl (Sigma–Aldrich) was diluted in saline and injected i.p. 30 mg/kg for 5 consecutive days. We injected saline i.p. for 5 consecutive days as a control. Twenty-one days after the last injection we sacrificed the mice for biochemical analyses. All experiments were in compliance with the University of Alabama at Birmingham Institutional Animal Care and Use Committee guidelines.
2.2. Chemicals
Chloroquine (CQ) (C-6628-25G), MPTP (M0896-10 MG), 1-methyl-4-phenylpyridinium (MPP+) (D048-100 MG), and staurosporine (S4400-0.5 MG) were purchased from Sigma–Aldrich. Oligomycin, FCCP, antimycin were from Agilent/Seahorse Bioscience.
2.3. Primary neuron cultures
Primary cortical neurons were obtained from Day 0 pups24, 25, 26. We dissected the mouse brain in ice cold Hanks’ balanced sodium salts (without Ca2+ and Mg2+). Cerebral cortices were incubated for 15 min at 37 °C with papain (Worthington). The tissues were triturated and cells were concentrated by centrifugation at 25 °C for 5 min at 1000×g. We then resuspended the cells in Neurobasal medium containing 2% B27 supplement (Invitrogen 17504-044), 1% Pen-Strep (10,000 U/mL, 10,000 μg/mL) and 0.5 mmol/L l-glutamine, and plated them in 24-well or 6-well plates coated with 0.1 mg/mL poly-l-lysine (Sigma–Aldrich, P1274). The cultures were kept in a humid incubator (5% CO2, 37 °C). We have used NeuN and GFAP antibodies to perform immunocytochemistry with these cultures, and found that DIV7-14 cultures consistently consist of >80% neurons.
2.4. Measurement of mitochondrial function
Parameters of mitochondrial function in mouse primary cortical neurons were measured using a Seahorse Bioscience XF24 Extracellular Flux Analyzer24,27, 28, 29, 30, 31, 32, 33. Cells were seeded at 80,000 cells per well, and concentrations of oligomycin, FCCP, and antimycin A were used at 1 μg/mL, 1 μmol/L and 10 μmol/L respectively. After measurements, total protein in each well was determined by the DC protein assay (Bio-Rad) and the oxygen consumption rate (OCR pmol/min) was normalized to μg protein in each well.
2.5. Assessment of cell viability
2.6. Immunohistochemistry
Brains were placed in 10% buffered formalin (Fisher Scientific) overnight at 4 °C followed by paraffin embedding. 5 μm thick sections were used for hematoxylin and eosin (H&E) and immunofluorescent staining. The following antibodies were used: anti-CTSD (Santa Cruz SC-6486), anti-CTSB (Santa Cruz SC-13985), anti-NeuN (Millipore MAB377), anti-GFAP (Dako Z033429), anti-LAMP1 [1D4B] (Abcam ab25245), anti-LAMP2 antibody (Abcam AB37024), α-synuclein (Invitrogen 328200), p-α-synuclein (Covance MMS-5091-100), and anti-Amyloid β4243 (Fuji Wako: Distributor010-26903 Barcode No4548995059260).
2.7. Electron microscopy
P25 mouse frontal cortex was dissected and freshly fixed in 6:2 (paraformaldehyde:gluteraldehyde). After osmium tetroxide staining and embedding in propylene oxide, they were sectioned onto a copper grid. Sections were then counter stained with lead citrate and imaged on a Tecnai Spirit Twin 20-120kv, FEI, Hillsboro, OR, USA.
2.8. Western blot analysis
Mouse brain cortex was collected for Western blot analysis. Briefly, mouse brain cortex was dissected and homogenized in 1.5 mL centrifuge tubes in cell lysis buffer (50 mmol/L Tris, 150 mmol/L NaCl, 2 mmol/L EDTA, 1% Triton X-100, pH to 7.8) containing protease cocktail inhibitors (Roche 4693132001). After 15 min on ice, samples were centrifuged at 16,873×g for 15 min. Supernatants were used for BCA assay to determine protein quantification and 20 μg protein per lane was loaded onto 7.5%–15% SDS-PAGE gels, then were transferred to nitrocellulose membranes (Fisher Scientific, EP2HY450F5) to probe for protein levels using one of the following antibodies: CTSD (Santa Cruz, SC-6486), LC3 (Sigma–Aldrich, L8918), P62 (Abnova, H00008878-M01), Beclin1 (Santa Cruz, SC-11427), HSC70 (Abcam, 19136-100), LAMP1 (Abcam, 1D4B), LAMP2a (Abcam, 37024), GRP78 (Santa Cruz, SC-1050, SOD2 (Abcam, ab86087), G6PD (Novus Biologicals, NB-100-236), α-synuclein (Santa Cruz, SC-7011-R), ubiquitin (Dako, Z0458), S6K (Cell signaling, 9202), pS6K (Cell signaling, 9205L), complex I-NDUFA9 (Invitrogen, 459100), complex V-subunit α (Invitrogen, 459240), complex III-core I (Invitrogen, 459140), complex IV-subunit I (Invitrogen, 459600), MFN2 (Santa Cruz, SC-100560), DRP1 (Abcam, 56788), GAPDH (Cell Signaling, 2118L; Millipore, MAB374), and β-actin (Sigma–Aldrich, A5441). To analyze Western blot membranes, ECL reagent (Fisher Scientific, PI-32106) and Image J software were used.
2.9. CTSD and CTSB activity assays
We performed CTSD activity measurements using the CTSD activity assay kit (Sigma–Aldrich, CS0800-1 KT)34. Briefly, mouse brain cortex was dissected and homogenized in 1.5 mL centrifuge tubes with a small pestle in MES lysis buffer (20 mmol/L MES pH 6.8, 20 mmol/L NaCl, 1 mmol/L MgCl2, 2 mmol/L EDTA, 10 mmol/L NaH2PO4; protease inhibitor cocktail (Roche) and phosphatase inhibitor (Sigma–Aldrich, P5726) were added before use. Samples were incubated for 30 min on ice, followed by 10 min of centrifugation at 15,000 × g. We performed BCA protein assay (BIORAD, 500-0116) on the supernatant, and used 50 μg of lysate for activity assays with and without pepstatin A (2 mg/mL) at 37 °C in a black 96-well plate with clear top and bottom. The values in the presence of pepstatin A were subtracted from the values without pepstatin A to determine CTSD activity in fluorescence units (FLU). Similarly, we performed CTSB activity measurements using the CTSB Activity Assay Kit (Abnova, KA0766)34. Briefly, mouse brain cortex was dissected and homogenized in the CTSB cell lysis buffer (kit) with protease and phosphatase inhibitors. Samples were then placed on ice 30 min and centrifuged at 15,000 × g for 5 min. We combined 50 μg of cell lysate with CTSB reaction buffer (kit) with and without E64 (Sigma–Aldrich, E3132) at 37 °C in a 96-well black plate with clear top and bottom. The values with E64 were subtracted from the values without E64 to determine CTSB activity in fluorescence units (FLU).
2.10. Quantitative real-time PCR analyses
We prepared RNA using Trizol (Invitrogen, 15596-026), synthesized cDNA using iScript™ cDNA Synthesis Kit (Bio-Rad, 170-8891). We performed quantitative real-time PCR using SYBR Green Mastermix (Invitrogen, 4364346) with the following conditions: 95 °C, 5 min; (95 °C, 10 s; 60 °C, 10 s; 72 °C, 15 s) × 40 cycles. Results were normalized against β-actin as a control.
Forward (F) and reverse (R) primer sequences are as the follows:
Ctsd (F) CCGGTCTTTGACAACCTGAT, (R) TCAGTGCCACCAAGCATTAG;
Ctsb (F) TGAAGGAGATCATGGCAGAA, (R) ATATCACCGGCTTCATGCTT;
Lamp1 (F) CTGTCGAGTGGCAACTTCAG, (R) GGATACAGTGGGGTTTGTGG;
Map1-lc3 (F) GTGGAAGATGTCCGGCTCAT, (R) TGGTCAGGCACCAGGAACTT;
Sqstm1/p62 (F) CGAGTGGCTGTGCTGTTC, (R) TGTCAGCTCCTCATCACTGG;
Ppagc1a (F) ACAGCTTTCTGGGTGGATTG; (R) TCTGTGAGAACCGCTAGCAA;
Tfeb1 (F) GCGGACAGATTGACCTTCAG; (R) CTCTCGCTGCTCCTCCTG;
Actb (F) GACGGCCAGGTCATCACTAT, (R) AAGGAAGGCTGGAAAAGAGC.
2.11. Proteasome activity assays
We used 40 μg of cortical extracts (in triplicate, and with n = 3 mice each) and 50 μmol/L substrate in the assay buffer consists of 50 mmol/L Tris (pH 7.5), 2.5 mmol/L EGTA, 20% glycerol, 1 mmol/L DTT, 0.05% NP-40 in the presence and absence of MG132 (Selleck Chemicals, S2619) at a final concentration of 200 μmol/L to block proteasome activities34,35. We measured fluorescence at an excitation wavelength of 380 nm and an emission wavelength of 460 nmol/L every 5 min for 2 h.
2.12. Behavioral tests
For the open field test23,24, we used the Ethovision software to perform this test for a period of 5 min for each mouse individually. The mouse's movements were tracked, total distance traveled (cm), position inside the field (time in center versus against the wall in seconds), and mean velocity (cm/s) were recorded.
For Zero Maze (70 cm diameter) which has two parts which have 15 cm high sides, and two parts which have only a 0.5 cm high wall. Each mouse is put in the arena, and Ethovision camera tracked for 4 min. The position of the animal in the arena at 5 frames per second, and time spent in open versus closed arm was recorded.
The SDI Grip Strength System (San Diego Instruments) was used to measures hind limb and force limb grip strength in mice according to the Meyer Method36.
For Tail Flick we used an overhead halogen light source which supplies an area of 4 mm × 6 mm heat stimulation to the tail. A sensor detects the tail flick of the animal and automatically record the time when the mouse's tail flicks out of the beam of light and displays the reaction time in 0.01 s increments.
For the rotarod test23,24, the paradigm involved 3 days of testing the mice on the rotating rod (San Diego Instruments) which gradually accelerated from 4 to 40 rotation per minute over a period of 5 min. The latency to fall was recorded for each mouse on each day.
For the Morris water maze test23,24,37,38, we use a pool of 120 cm in diameter and a 10 cm diameter platform which is located 0.5 cm below the water surface. For Days 1–5, four trials a day are run, so that all starting positions are equally used (in a random order). The mice are given 60 s to find the platform and 10 s to stay on the platform.
The wheel-running assay was used using running wheel cages (Lafayette Instrument). Mice were single-housed in specialized wheel-running cages with food and water provided ad libitum. Distances during night and day were measured by the Activity Wheel Monitoring (AWM) system software.
2.13. HPLC analysis of striatal monoamines
Striatal biogenic amine analysis was performed at the Vanderbilt Neurochemistry Core facility23. The brain sections are homogenized and biogenic amines extracted and determined by a specific HPLC assay utilizing an Antec Decade II (oxidation: 0.4) electrochemical detector.
2.14. mtDNA quantification
Mitochondrial copy number was determined real-time PCR using forward primer 5′-ccccagccataacacagtatcaaac-3′ and reverse primer 5′-gcccaaagaatcagaacagatgc-3′ in an ABI 7500 (Applied Biosystems)25,39, 40, 41, 42. Real time PCR conditions were as follows: 94 °C for 2 min, followed by 40 cycles of denaturation at 94 °C for 15 s, annealing and extension at 60 °C for 1 min mtDNA copy number was normalized to real-time PCR of 18S nuclear sequence, using forward primer 5′-aaacggctaccacatccaag-3′ and reverse primer 5′-caattacagggcctcgaaag-3′.
2.15. TUNEL staining
Cortex of control, CDKO, CDtg, and CDKO::CDtg mice at p23–25 were used for TUNEL staining (Invitrogen™ Click-iT™ Plus TUNEL Assay Kits for In Situ Apoptosis Detection, Catalog: C10617), DNase treated sections was used for positive control, no TdT treatment was used as a negative control, n = 3 each group.
2.16. Statistical analysis
We used one and two-way ANOVA, and Student t-test. P values < 0.05 was considered statistically significant.
3. Results
3.1. Generation of the nervous system CTSD transgenic (CDtg) mice
We have generated CTSD transgenic (CTSDfloxedstop) mice that are capable of overexpressing CTSD in Cre-expressing tissues. When they are bred into Nestin-cre transgenic animals, the double transgenic animals have elevated CTSD mRNA expression. Fig. 1A shows the wildtype genomic structure, the knock-in of pCAG-loxp-STOP-loxp-hCTSD in the Hprt site, and the genomic structure in Nestin-cre expressing cells after breeding with Nestin-cre mice. Fig. 1B shows the genotyping results from 4 offspring of the CTSDfloxedstop and Nestin-cre mice, using PCR#1 in the diagram, indicating that hCTSD is inserted into the Hprt allele. PCR#1 products (lanes 1 and 4) indicate that the mice carry CTSDfloxedstop knock-in. PCR products using primers that matches Cre recombinase cDNA to indicate that the mouse carries the Nestin-cre transgene. Hence lane 1 is a double transgenic animal with both CTSDfloxedstop and Nestin-cre. After PCR with PCR#1 and cre primers, DNA extracts from 6 offsprings with cre + results and either + or – PCR#1 results were subjected to PCR#2 (Fig. 1C). Positive PCR#2 products indicate the cre mediated recombination between the loxp sites and the deletion of the STOP cassette and therefore the expression of Ctsd mRNA. Fig. 1D indicates that the cortical tissues from CDtg (CTSDfloxedstop knock-in positive and Nestin-cre positive) mice express exogenous human CTSD mRNA using RT-PCR analyses compared to the control without CTSDfloxedstop knock-in.
Figure 1.

Generation of Nestin-cre::CDtg mice. (A) Genome structures of control and Nestin-cre::CDtg mice. a. The wildtype Hprt locus. b. Human CTSD cDNA was inserted into the Hprt allele. A floxed STOP cassette is included between the ubiquitous CAG promoter and the cDNA to allow the Cre-dependent expression of the CTSD gene. c. The CTSD expressing allele. When CTSD transgenic mice are bred with Nestin-cre mice, the STOP cassette between the loxP sequences is then deleted to allow CTSD expression. Wildtype mouse either has the wildtype Hprt allele (a), or the non-expressing Hprt allele with CTSD transgene but negative cre expression (b). PCR#1 was designed to distinguish a (no product) and b (2240 bp) gene configurations. PCR#2 was designed to distinguish b (no product) and c (480 bp) gene configurations. (B) PCR#1 genotyping is used to distinguish a and b (2240 bp) in tail biopsies. cre transgenic primers used are: 5′-AGA TGT TCG CGA TTA TC and 5′-AGC TAC ACC AGA GAC GG. Appearance of a PCR product indicates cre+. Mouse 1 was CDtg. Mouse 1, 2, and 3 were WT. (C) For mice with indicated PCR#1 and cre results, PCR#2 genotyping is used to distinguish b and c (480 bp) in P25 brains. (D) RT-PCR of human CTSD mRNA from P25 brains of indicated genotypes. Primers used for human CTSD mRNA: F: TTCCCGAGGTGCTCAAGAACTACA, R: TGTCGAAGACGACTGTGAAGCACT.
3.2. CTSD overexpression in primary neurons increases autophagic flux while neurons exhibit normal bioenergetics and sensitivities to apoptotic and neurotoxic stress
To determine whether CTSD overexpression has any adverse effects on neuronal survival and function, we cultured primary cortical neurons from postnatal Day 0 (P0) wildtype and CDtg mice. We found that at 7 days in vitro (DIV7), there were increased CTSD protein levels in CDtg neurons compared to controls, as determined by Western blot analyses (Fig. 2A). To determine whether CTSD increase affects overall autophagic flux, we measured LC3II levels in the presence or absence of chloroquine (CQ, 4 h at 40 μmol/L). There was an increase of LC3II in CDtg neurons both in the absence and in the presence of CQ, as well as an increase of autophagic flux as assessed by LC3II with CQ–LC3II without CQ (Fig. 2B).
Figure 2.

Primary cortical neurons from CDtg mice exhibit similar mitochondrial bioenergetics and susceptibility to apoptotic cell death stimuli as wildtype mice despite of increased autophagic flux. (A) Western blot of CTSD in DIV7 primary cortical neurons from WT and CDtg mice. β-Actin Western blot was used as loading control. Data = mean ± SEM, n = 3. (B) Autophagic flux was assessed in wildtype and CDtg neurons by measuring LC3-II levels in the presence and absence of 40 μmol/L chloroquine (CQ) for 4 h. (C) Seahorse XF24 bioanalyzer was used to analyze parameters of mitochondrial functions (2–4). Oxygen consumption rate (OCR) was measured and normalized to the protein amount per well. DIV7 primary cortical neurons WT and Nestin-cre::CDtg mice exhibited similar basal OCR, ATP-linked OCR, proton leak OCR, maximal OCR and non-mitochondrial OCR, at basal conditions and after oligomycin (O) which inhibits ATP synthase, FCCP (F) which induces maximum OCR, and antimycin (A) which eliminates mitochondrial electron transfer chain associated OCR. Cultures from 3 different WT and 3 different CDtg mouse brains were used in the analyses. Shown are representative mitochondrial activities assays from 2 WT to 2 CDtg primary neuron cultures. (D) DIV7 primary neurons were exposed to different concentrations of MPP+ for 24 h. (E) DIV7 primary neurons were exposed to different concentrations of staurosporine for 24 h. Cell viability was assessed by trypan blue exclusion method. Data = mean ± SEM, n = 3. ∗P < 0.05 compared to wildtype. Student t-test.
As mitochondrial quality control is regulated by lysosomal mediated degradation of mitochondria, we examined whether CTSD overexpression alters mitochondrial function. We compared cellular bioenergetics of primary neurons from wildtype and CDtg mice. We used the Seahorse XF24 analyzer to measured basal oxygen consumption rate (OCR) for 30 min before sequential injections of mitochondrial inhibitors oligomycin, FCCP, and antimycin at 1, 1 and 10 μmol/L24,27, 28, 29, 30, 31, 32, 33. These inhibitors allowed us to differentiate ATP-linked, proton-leak-linked and maximal OCR. We found that primary neurons from CDtg mice exhibited similar mitochondrial bioenergetics to that of wildtype mice (Fig. 2C).
Even though CTSD overexpression did not change mitochondrial function, we examined whether it changes mitochondrial damage mediated cell death. To do that, we treated CDtg neurons to mitochondrial toxin 1-methyl-4-phenylpyridinium (MPP+), the active metabolite of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), as well as the apoptotic inducer staurosporine. We found that CDtg neurons did not exhibit an altered sensitivity to MPP+ or staurosporine toxicity over a range of concentrations (Fig. 2D and E). These results indicate that CTSD overexpression does not change cellular bioenergetics nor exacerbate cell death in response to the well-known neurotoxin or a well-known apoptosis stimulus.
3.3. CTSD overexpressing mice are normal compared to wildtype mice
To further characterize the CDtg mice we allowed them to age for 1–2 years over which time they remained tumor-free, with normal appearance, reproductive behaviors, and body weight (Supporting Information Fig. S1A). Comparing CDtg and wildtype mice, there were no significant changes in longevity (Fig. S1A). Moreover, we performed extensive behavior studies at 1, 1.5 and 2 year of age, with open field to test general motor activities (Fig. S1B), zero maze to test anxiety (Fig. S1C), grip strength test to assess muscular strength (Fig. S1D); tail flick to assess pain perception (Fig. S1E), rotarod tests to assess motor coordination (Fig. S1F), and Morris water maze to assess learning and memory (Fig. S1G), and found that CDtg mice exhibited similar behaviors in these tests as the wildtype mice. We found essentially normal behavior in CDtg mice compared to wildtype mice, except a slight increase of rotarod latency to fall (Fig. S1F), and a slight decrease of escape latency on Day 4 of the water maze at 1 year of age (Fig. S1G).
Dopaminergic neurons project from substantia nigra to the striatum and degeneration of these neurons leads to depletion of striatal dopamine. As part of a thorough basal characterization of the CDtg mice, we assessed the neurochemical function of dopaminergic neurons by performing striatal monoamine content analyses of wildtype and CDtg mice from p25 to 1 year of age. We found similar striatal noradrenaline, dopamine, dopamine metabolites DOPAC and HVA, as well as other neurotransmitters such as 5-HIAA, 5-HT and 3-MT levels (Supporting Information Fig. S2).
As CTSD is a lysosomal enzyme, we examined whether CTSD elevation changes the levels of those genes related to autophagy and lysosomal pathways, and mitochondrial content as it is regulated by both mitophagy and mitochondrial biogenesis. CTSD elevation in CDtg mice is consistent from P0 to 1 year of age (∼4–6-fold) compared to control mice (Supporting Information Fig. S3A). There were no significant differences between wildtype and CDtg mice with regard to selective mRNAs related to autophagy and lysosome pathways, nor Pgc1α, a transcription cofactor involved in mitochondrial biogenesis or mtDNA copy numbers compared to wildtype mice from P0 to 1 year of age (Fig. S3B and S3C). There were no significant differences between wildtype and CDtg mice with regard to average mitochondrial DNA copy number from P1 to 1 year of age (Fig. S3C). There was also no significant difference between wildtype and CDtg mice with regard to endogenous α-synuclein from P0 to 1 year of age (Supporting Information Fig. S4).
CTSD elevation in CDtg mice remains higher than wildtype mice (∼4–6-fold) at 2 years of age (Supporting Information Fig. S5A). There are no significant differences between CDtg and wildtype mice in autophagy–lysosomal pathway protein levels as shown by Western blot analyses of P62, LC3-I, and LC3-II (Fig. S5B), Beclin (Fig. S5C), HSC70, LAMP1 or LAMP2a (Fig. S5D and S5E). In addition, levels of α-synuclein, ubiquitinated proteins and pS6K are similar in wildtype and CDtg mouse cortex (Fig. S5F–S5H). There are no significant differences between CDtg and wildtype mice in mitochondrial electron transport chain proteins, or fission/fusion proteins at 2 years of age (Supporting Information Fig. S6).
3.4. CTSD overexpression did not significantly change sensitivity to MPTP in vivo
MPTP selectively targets the nigrostriatal dopaminergic pathway, causing dopaminergic neurodegeneration in vivo. CTSD overexpression does not change basal behavior and striatal monoamine levels. We then investigated whether CTSD overexpression impacts pathology in response to sub-chronic MPTP (5 daily i.p. injection at 30 mg/kg). We assessed behaviors at 1 month after last injection, and sacrificed mice 2 months after last injection to assess striatal monoamine and proteins. Both wildtype and CDtg mice exhibited higher locomotor activities (Fig. 3A). There were no changes in zero maze, grip strength, or tail flick time (Fig. 3B–D). There were also no changes in rotarod or water maze activities comparing CDtg and wildtype mice (Fig. 3E and F). In the wheel running activity tests, there was higher night activity in the CDtg mice which was decreased by MPTP (Fig. 3G).
Figure 3.

Behavioral assessment after sub-chronic MPTP in CDtg mice compared to wildtype (WT) mice. Mice were tested 1 month after the last of the 5 daily 30 mg/kg i.p. injection (All male at the age of 3–4 months, n = 10, 16, 8, and 11 for WT + vehicle, WT + MPTP, CDtg + Vehicle and CDtg + MPTP, respectively). (A) There was an increased motor activity between vehicle versus MPTP by 2-way ANOVA in Open field, but there were no difference in each group in (B) Zero maze, (C) Grip strength, (D) Tail flick, (E) Rotarod, or (F) Morris water maze behavioral assessment. (G) There was a difference between CDtg vehicle versus CDtg MPTP for total distance and night distance& by 2-way ANOVA for genotype × treatment interaction and post hoc multiple comparisons in wheel running activities. Data = mean ± SEM.
As shown in Fig. 4A, striatal dopamine (DA) and metabolite DOPAC were significantly decreased in wildtype and CDtg mice to similar extent. There was a decrease of noradrenaline in the striatum in the WT mice after MPTP, while the CDtg after vehicle was lower than WT + vehicle but did not decrease with MPTP (Fig. 4B). Striatal p62 and LC3 levels were assessed by Western blot analyses. While LC3II is similar between wildtype and CDtg mice in both saline and MPTP administered animals, P62 appeared to be moderately decreased in CDtg + MPTP compared to WT + MPTP (Fig. 4C).
Figure 4.

Levels of striatal monoamine, LC3, and P62 after sub-chronic MPTP in CDtg mice compared to wildtype (WT) mice. Mice were sacrificed 2 months after the last of the 5 daily 30 mg/kg i.p. injection. (A) Both DOPAC and DA were significantly decreased by MPTP by 2-way ANOVA. There was a difference between WT vehicle versus WT MPTP∗, and between CDtg vehicle versus CDtg MPTP&. (B) Only noradrenaline was decreased in CDtg compared to wildtype mice prior to MPTP (numbers of animals as in panel A). There was a genotype significance by 2-way ANOVA, and post hoc multiple comparison showed significant difference compared to WT vehicle∗. (C) similar P62 and LC3II before and after MPTP in the striatum, with P62 decreased in CDtg + MPTP group compared to WT + MPTP group (n = 3) by 2-way ANOVA showing difference for genotype × treatment interaction and post hoc multiple comparisons of difference between WT MPTP versus CDtg MPTP#. Quantification of LC3I was from image ∗I, and LC3II from image ∗II. Data = mean ± SEM. ∗P < 0.05 compared to WT of similar treatment.
3.5. CTSD overexpression in the nervous system partially rescues CDKO phenotype in vivo
CDtg mice appear normal and have normal brain structure as shown by H&E staining in paraffin embedded sections, and electron microscopy (Supporting Information Fig. S7A and S7B) at P25. CDKO mice exhibit significant TUNEL positive cells in the cortex, while in CDtg mice there was no detectable TUNEL staining (Fig. S7C), indicating CTSD overexpression does not lead to an overt increase in cell death.
Immunohistochemistry analyses demonstrated that increased CTSD is mainly in the neuronal populations, as they are expressed in NeuN positive cells (Supporting Information Fig. S8A). Quantification of CTSD level in NeuN positive cells showed a 3–4-fold increase in neurons. Co-immunohistochemistry studies demonstrated that CTSD colocalizes with another lysosomal protein CTSB (Fig. S8B), as well as with LAMP1 and LAMP2 (Fig. S8A and S8B). This study indicates that CTSD overexpression in CDtg is predominantly in neuronal lysosomes. Furthermore, the increase of LAMP1 in CDKO mice was restored to baseline in CDtg:CDKO mice (Fig. S8A).
Interestingly, CTSD overexpression in the central nervous system mediated by nestin promoter attenuated the postnatal lethality of Ctsd whole body knockout (CDKO). CDKO mice lose body weight beginning at P21 and die ∼ P26 while CDtg mice exhibiting similar body weight as wildtype mice, and CDtg:CDKO mice have a lower body weight as wildtype or CDtg mice but most survive more than 6 months of age (Fig. 5A and B). Intestinal necrosis occurred in all CDKO at P2520 but not CDtg:CDKO at this age (Fig. 5C). Likewise, brain gliosis (more GFAP + cells) was present in all CDKO mice but absent in CDtg or CDtg:CDKO mice (Fig. 5D). The increase in LAMP1 that occurs in CDKO mice is also absent in CDtg or CDtg:CDKO mice (Supporting Information Fig. S9A), while LAMP2 immunoreactivity does not appear to be changed by CDKO or CDtg (Fig. S9B). CDtg:CDKO mice eventually succumb to intestinal starting from around P150 days (Supporting Information Fig. S10).
Figure 5.

CTSD overexpression in Nestin-cre::CDtg (CDtg) mice rescues the early lethality of CD−/− mice. (A) CDKO mice lose body weight, develop blindness and seizure, and die of intestinal necrosis, thromboembolia, lymphopenia, and massive neurodegeneration (9–14). CDKO mice die p25 ± 1. CDKO::Nestin-cre::CDtg mice survive beyond P60, some beyond P200. This chart represent n = 9 and 12 each genotype. (B) Nestin driven CTSD transgenic expression attenuates the weight loss of CDKO starting from P21. Nine or more mice each genotype were weighed at the indicated days after birth. Significant differences were observed between WT or CDtg versus CDKO mice at P21. CDKO::CDtg mice exhibited gradual weight loss compared to WT mice beginning from P24. n > 9. (C) CDtg expression rescues tissue necrosis and brain inflammation of CDKO mice at P25. Tissue appearance is shown from representative dissections of mice of the 4 indicated genotypes. CDKO mice exhibited decreased brain, thymus, heart, liver, spleen (sp), stomach, intestine and kidney sizes. WT, CDtg and CDKO::CDtg exhibit similar organ integrity at P25. n > 3. (D) Gliosis in P25 CDKO mouse cortex is rescued by CTSD overexpression in the nervous system. Co-immunostaining of GFAP and Ctsd demonstrated significant increase of GFAP staining in CDKO brains, whereas WT, CDtg and CDKO::CDtg brains exhibited similar GFAP staining. Scale bar: 50 μm. (D) Quantification of immunostaining of CTSD and GFAP. Data = mean ± SEM, ∗P < 0.05 compared to WT, #P < 0.05 compare to CDKO (n = 3).
Using cortical extracts we found higher CTSD enzymatic activities at postnatal day 25 (P25). Extracts from CDKO cortex were used to assess the specificity of the assay and as expected exhibited diminished activity (Fig. 6A). Consistent with prior studies12,14, 15, 16,43, there was an increase of Lamp1 and Lc3b mRNA as well as LAMP1 and LC3II protein, and an increase of CB activities in CDKO mice (Fig. 6B–D). In contrast, CDtg mice exhibit no such increase while CDtg:CDKO phenotypes are similar to wildtype and CDtg in these parameters. As we previously found, cortical extract from CDKO mice exhibit decreased proteasomal activities15 (Fig. 6E), in this study we found that proteasomal activities from cortical extract of the CDtg:CDKO mice are increased compared to CDKO mice, but remain lower than wildtype or CDtg mice, indicating that CDtg partially reverse CDKO proteasomal deficits in the cortex.
Figure 6.

CTSD overexpression in Nestin-cre::CDtg (CDtg) mice rescues LAMP1, LC3II, and CB accumulation and partially rescues proteasomal deficits in CD−/− mice. (A) Using cortical extracts we found higher CTSD enzymatic activities at postnatal day 25 (P25) in Cdtg mice cortex compare to WT (normalized to WT). CDKO extracts were used as a negative control. CDKO::CDtg brains exhibit similar CTSD activities as CDtg mice. (B) Similar mRNA expression of endogenous mouse Ctsd, Lamp1, Map1-lc3, and Sqstm1/P62 in CDtg mice compared to WT mice. CDKO mice do not exhibit detectable levels of endogenous mouse Ctsd mRNA, while Lamp1 and Map1-lc3 levels are increased. In CDKO::CDtg mice, Lamp1 and Map1-lc3 mRNA are comparable to wildtype and CDtg mice. Endogenouse Ctsd mRNA is absent. (C) Similar LC3, LAMP1, and P62 protein levels in CDtg mice compared to WT mice at P25, as assessed by Western blot analyses. CDKO mice do not exhibit detectable levels of CTSD protein, while LAMP1 and LC3-II levels are increased. In CDKO::CDtg mice, LAMP1 and LC3-II protein levels are comparable to those in wildtype and CDtg mice. (D) CB activity is increased in the CDKO cortical extract at P25, but not in CDtg or CDKO::CDtg. E) Tissue lysates from CDKO cortex exhibit decreased proteasome activities compared to WT, CDtg and CDKO::CDtg as assayed with chymotrypsin-like fluorigenic substrate (Suc-LLVY-AMC). The activities that are inhibitable by the proteasome inhibitor MG132 were quantified. CDtg:CDKO exhibit intermediate proteasomal activities compared to wildtype and CDtg versus CDKO mice. Data = mean ± SEM, n = 3 mice each genotype at P25, P < 0.05 (∗ compared to WT; # compared to CDKO) by Student t-test.
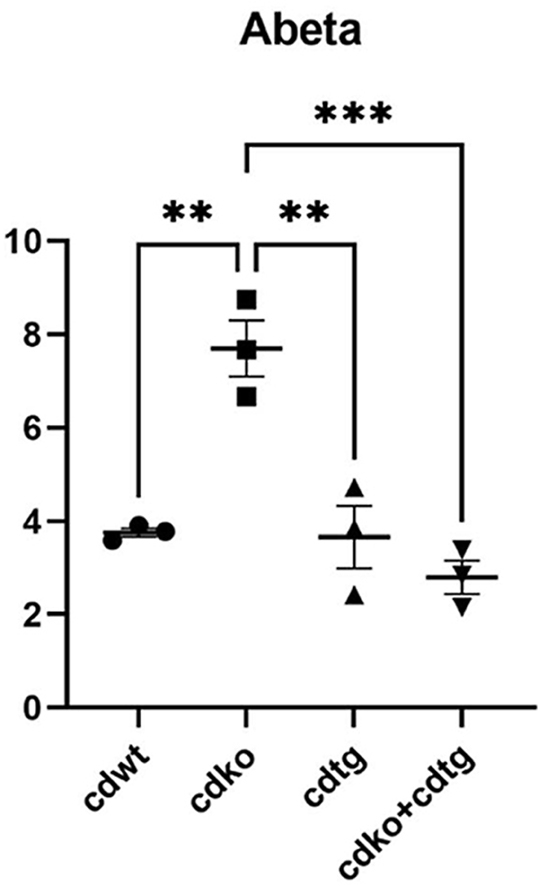
We previously demonstrated that CDKO mice accumulate α-synuclein15. Here we found that such α-synuclein accumulate was suppressed by CDtg in CDKO:CDtg mice at P25 days (Fig. 7A). We did not detect p-α-synuclein in CDKO mice (data not shown). Previous studies demonstrated that CDKO mice also accumulate insoluble Aβ42 and Aβ4017,18. We had previously found that Ctsd+/− haplodeficiency did not alleviate the APP/PS1 pathological phenotypes44. Here we found that Aβ42 level was increased in CDKO mice, and suppressed by CDtg in CDKO:CDtg mice at P25 days (Fig. 7B).
Figure 7.

CTSD overexpression in Nestin-cre::CDtg mice rescues α-synuclein and Aβ42 accumulation due to CDKO. (A) α-synuclein, n = 3 each, (B) Aβ42 immunohistochemistry was performed n = 3 each group. Negative control was performed for wildtype and CDKO mice without primary antibodies (-ctl). Quantification was performed in Image J. One-way ANOVA followed by Tukey post-hoc test. ∗∗P < 0.01 compared to WT, ∗∗∗P < 0.001.
4. Discussion
In summary, we have generated a CDtg mouse that overexpresses human CTSD in the Nestin-expressing cells. We found, as expected, that the CTSD protein is located mainly in the lysosomes. The mice are free of detectable tumors, as well as any overt changes in appearance, reproductive activities, body weight, neurochemical contents, behavior and sensitivity to neurotoxin MPTP. Primary neurons exhibit increased autophagic flux, but normal bioenergetics and sensitivity to apoptotic stimulus. Furthermore, overexpresses human CTSD in the Nestin-expressing cells can rescue systemic deficit of DKO mice by extending lifespan from P26 to > P150, and decrease α-synuclein and Aβ42 that accumulated in CDKO.
We chose targeted insertion into the Hprt locus because it helps to avoid unpredictable position effects with copy number and relative expression levels affected by chromosomal locations, which accompany other transgenic methods that rely on random integration. The Hprt locus insertion strategy has been frequently used45, because of its consistency. Hprt is a housekeeping gene and thus the expression of inserted genes are unlikely affected by developmental and environmental conditions. Hprt deficient mice appear normal even though that Hprt mutation in humans are linked to the Lesch-Nyhan syndrome46. Furthermore, these mice are phenotypically normal with all the tests we performed.
Elevated CTSD levels have been found in different types of cancers35, 36, 37. We did not observe any tumors in any of our 2 year old CDtg mice with CTSD overexpression under the control of Nestin promoter. However, tissue specific transgenic Cre expressing mice may be used in future studies to investigate the impact of CTSD overexpression in tumor formation and progression. In addition, our observation that CTSD elevation also impacts autophagic flux in primary neurons is consistent with the previous observation that lysosomal degradation is rate limiting in autophagic flux4. It is possible that the elevation of CTSD impacts the lysosomal docking of MTOR complexes as well as autophagosome-lysosome fusion38.
Remarkably, overexpressing human CTSD in the Nestin-expressing cells partially rescues the systemic phenotypes of CDKO mice. The mechanism of this is currently unclear. It has been shown that AAV-CTSD delivery to the brain extended survival of CDKO mice to P70, and AAV-CTSD delivery to the brain + liver + stomach extended survival of CDKO mice to P200. The observation that we have even longer survival (some of the mice > P240) may be due to nestin expression in the peripheral nervous system, or potential drainage of central nervous system protein to the periphery39.
Studies have shown that MPTP crosses blood brain barrier, and metabolized to MPP+ by endogenous monoamine oxidase. Then MPP+ can be taken-up by the dopaminergic neurons, inhibits mitochondrial complex I and induces parkinsonism. Previous studies have shown that with 5 consecutive day injection of 30 mg/kg MPTP, nigrostriatal lesion is stable 21 days after the last injection47. In our study CTSD elevation does not protect against MPTP induced neurodegeneration as assessed by striatal dopamine content at 21 days post last MPTP injection. However, our experimental paradigm focused on long term consequence of MPTP and not the peak of MPTP caused lesion, thus we are not able to assess whether CTSD elevation attenuated the transient maximum lesion. Future studies with both the duration of MPTP administration and time course after MPTP are needed to determine impact of CTSD elevation on chronic MPTP toxicity.
5. Conclusions
In this study, we have demonstrated that CTSD elevation can rescue the phenotype of Aβ42 accumulation exhibited in the CDKO. CTSD overexpression is 4–6-fold based on Western blot analyses in primary neurons (Fig. 2), 3–4-fold based on immunohistochemistry in the p25 cortex (Fig. 5), ∼4-fold based on enzymatic activity assays (p25 cortex), ∼7 fold increase based on Western blot analyses in p25 cortex (Fig. 6), and 3–4-fold in NeuN positive neurons at p25 (Fig. S8). It is currently unclear whether higher levels of CTSD might be protective against neurotoxin or neurotoxic protein induced neurodegeneration. Future studies will breed CTSDf/f mice with cre driven by different promoters, including Actin-cre, to determine effect of therapeutic potential of CTSD overexpression in different disease models. For example, CTSDfloxedstop mice can be bred with transgenic cre mice where cre is under the control of inducible and cell type-specific promoters to determine impact of CTSD overexpression in PD models which overexpress α-synuclein and AD models that are based on APP or tau mutations.
Acknowledgments
We thank Dr. Paul Saftig for CDKO mice, Ashish Kumar for assistance of behavioral assessment, Dr. Qiuli Liang for performing some of the Western blot analyses, Dr. Terry Lewis and the histology core (P30 NS47466), members of the UAB EM Core, Ed Phillips and Melissa Chimento, as well as members of Zhang laboratory for technical assistance and scientific discussions. This work was partially supported by NIHR01-NS064090, R56AG060959, R01AG072895-01, I01 BX-003792, and I01 BX-004251 (to Jianhua Zhang), UAB Neuroscience Core Facilities (NS47466 and NS57098), UAB Blue Sky program, UAB Nathan Shock Center P30 AG050886.
Footnotes
Peer review under the responsibility of Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences.
Supporting data to this article can be found online at https://doi.org/10.1016/j.apsb.2023.07.015.
Author contributions
Xiaosen Ouyang, Willayat Y Wani, Gloria A Benavides, Matthew J Redman, and Hai Vo performed experiments. Thomas van Groen helped with behavior studies. Victor Darley-Usmar and Jianhua Zhang directed the research. Xiaosen Ouyang and Jianhua Zhang wrote the manuscript. All authors read/edited/approved the manuscript.
Conflicts of interest
The authors declare that they have no competing interests.
Appendix A. Supplementary data
The following is the Supplementary data to this article.
References
- 1.Gan-Or Z., Dion P.A., Rouleau G.A. Genetic perspective on the role of the autophagy–lysosome pathway in Parkinson disease. Autophagy. 2015;11:1443–1457. doi: 10.1080/15548627.2015.1067364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang J., Culp M.L., Craver J.G., Darley-Usmar V. Mitochondrial function and autophagy: integrating proteotoxic, redox, and metabolic stress in Parkinson's disease. J Neurochem. 2018;144:691–709. doi: 10.1111/jnc.14308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colacurcio D.J., Pensalfini A., Jiang Y., Nixon R.A. Dysfunction of autophagy and endosomal–lysosomal pathways: roles in pathogenesis of Down syndrome and Alzheimer's disease. Free Radic Biol Med. 2018;114:40–51. doi: 10.1016/j.freeradbiomed.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nixon R.A. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. doi: 10.1038/nm.3232. [DOI] [PubMed] [Google Scholar]
- 5.Schneider L., Zhang J. Lysosomal function in macromolecular homeostasis and bioenergetics in Parkinson's disease. Mol Neurodegener. 2010;5:14. doi: 10.1186/1750-1326-5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siintola E., Partanen S., Stromme P., Haapanen A., Haltia M., Maehlen J., et al. Cathepsin D deficiency underlies congenital human neuronal ceroid-lipofuscinosis. Brain. 2006;129:1438–1445. doi: 10.1093/brain/awl107. [DOI] [PubMed] [Google Scholar]
- 7.Steinfeld R., Reinhardt K., Schreiber K., Hillebrand M., Kraetzner R., Bruck W., et al. Cathepsin D deficiency is associated with a human neurodegenerative disorder. Am J Hum Genet. 2006;78:988–998. doi: 10.1086/504159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu Y.D.H., Aebischer P., Olanow C.W., Kordower J.H. Alterations in lysosomal and proteasomal markers in Parkinson's disease: relationship to alpha-synuclein inclusions. Neurobiol Dis. 2009;35:385–398. doi: 10.1016/j.nbd.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 9.Dehay B., Bove J., Rodriguez-Muela N., Perier C., Recasens A., Boya P., et al. Pathogenic lysosomal depletion in Parkinson's disease. J Neurosci. 2010;30:12535–12544. doi: 10.1523/JNEUROSCI.1920-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Peng W., Minakaki G., Nguyen M., Krainc D. Preserving lysosomal function in the aging brain: insights from neurodegeneration. Neurotherapeutics. 2019;16:611–634. doi: 10.1007/s13311-019-00742-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Usenovic M., Tresse E., Mazzulli J.R., Taylor J.P., Krainc D. Deficiency of ATP13A2 leads to lysosomal dysfunction, alpha-synuclein accumulation, and neurotoxicity. J Neurosci. 2012;32:4240–4246. doi: 10.1523/JNEUROSCI.5575-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saftig P., Hetman M., Schmahl W., Weber K., Heine L., Mossmann H., et al. Mice deficient for the lysosomal proteinase cathepsin D exhibit progressive atrophy of the intestinal mucosa and profound destruction of lymphoid cells. EMBO J. 1995;14:3599–3608. doi: 10.1002/j.1460-2075.1995.tb00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakanishi H., Zhang J., Koike M., Nishioku T., Okamoto Y., Kominami E., et al. Involvement of nitric oxide released from microglia–macrophages in pathological changes of cathepsin D-deficient mice. J Neurosci. 2001;21:7526–7533. doi: 10.1523/JNEUROSCI.21-19-07526.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koike M., Shibata M., Waguri S., Yoshimura K., Tanida I., Kominami E., et al. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease) Am J Pathol. 2005;167:1713–1728. doi: 10.1016/S0002-9440(10)61253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qiao L., Hamamichi S., Caldwell K.A., Caldwell G.A., Yacoubian T.A., Wilson S., et al. Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain. 2008;1:17. doi: 10.1186/1756-6606-1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cullen V., Lindfors M., Ng J., Paetau A., Swinton E., Kolodziej P., et al. Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain. 2009;2:5. doi: 10.1186/1756-6606-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suire C.N., Abdul-Hay S.O., Sahara T., Kang D., Brizuela M.K., Saftig P., et al. Cathepsin D regulates cerebral Abeta42/40 ratios via differential degradation of Abeta42 and Abeta40. Alzheimers Res Ther. 2020;12:80. doi: 10.1186/s13195-020-00649-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suire C.N., Leissring M.A. Cathepsin D: a candidate link between Amyloid beta-protein and Tauopathy in Alzheimer disease. J Exp Neurol. 2021;2:10–15. [PMC free article] [PubMed] [Google Scholar]
- 19.Roberg K., Johansson U., Ollinger K. Lysosomal release of cathepsin D precedes relocation of cytochrome c and loss of mitochondrial transmembrane potential during apoptosis induced by oxidative stress. Free Radic Biol Med. 1999;27:1228–1237. doi: 10.1016/s0891-5849(99)00146-x. [DOI] [PubMed] [Google Scholar]
- 20.Bidere N., Lorenzo H.K., Carmona S., Laforge M., Harper F., Dumont C., et al. Cathepsin D triggers Bax activation, resulting in selective apoptosis-inducing factor (AIF) relocation in T lymphocytes entering the early commitment phase to apoptosis. J Biol Chem. 2003;278:31401–31411. doi: 10.1074/jbc.M301911200. [DOI] [PubMed] [Google Scholar]
- 21.Beaujouin M., Liaudet-Coopman E. Cathepsin D overexpressed by cancer cells can enhance apoptosis-dependent chemo-sensitivity independently of its catalytic activity. Adv Exp Med Biol. 2008;617:453–461. doi: 10.1007/978-0-387-69080-3_44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Beaujouin M., Baghdiguian S., Glondu-Lassis M., Berchem G., Liaudet-Coopman E. Overexpression of both catalytically active and -inactive cathepsin D by cancer cells enhances apoptosis-dependent chemo-sensitivity. Oncogene. 2006;25:1967–1973. doi: 10.1038/sj.onc.1209221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Crabtree D., Boyer-Guittaut M., Ouyang X., Fineberg N., Zhang J. Dopamine and its metabolites in cathepsin D heterozygous mice before and after MPTP administration. Neurosci Lett. 2013;538:3–8. doi: 10.1016/j.neulet.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ouyang X., Ahmad I., Johnson M.S., Redmann M., Craver J., Wani W.Y., et al. Nuclear receptor binding factor 2 (NRBF2) is required for learning and memory. Lab Invest. 2020;100:1238–1251. doi: 10.1038/s41374-020-0433-4. [DOI] [PubMed] [Google Scholar]
- 25.Redmann M., Benavides G.A., Wani W.Y., Berryhill T.F., Ouyang X., Johnson M.S., et al. Methods for assessing mitochondrial quality control mechanisms and cellular consequences in cell culture. Redox Biol. 2018;17:59–69. doi: 10.1016/j.redox.2018.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Redmann M., Darley-Usmar V., Zhang J. The role of autophagy, mitophagy and lysosomal functions in modulating bioenergetics and survival in the context of redox and proteotoxic damage: implications for neurodegenerative diseases. Aging Dis. 2016;7:150–162. doi: 10.14336/AD.2015.0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dranka B.P., Benavides G.A., Diers A.R., Giordano S., Zelickson B.R., Reily C., et al. Assessing bioenergetic function in response to oxidative stress by metabolic profiling. Free Radic Biol Med. 2011;51:1621–1635. doi: 10.1016/j.freeradbiomed.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giordano S., Dodson M., Ravi S., Redmann M., Ouyang X., Darley-Usmar V.M., et al. Bioenergetic adaptation in response to autophagy regulators during rotenone exposure. J Neurochem. 2014;131:625–633. doi: 10.1111/jnc.12844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benavides G.A., Liang Q., Dodson M., Darley-Usmar V., Zhang J. Inhibition of autophagy and glycolysis by nitric oxide during hypoxia—reoxygenation impairs cellular bioenergetics and promotes cell death in primary neurons. Free Radic Biol Med. 2013;65:1215–1228. doi: 10.1016/j.freeradbiomed.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giordano S., Lee J., Darley-Usmar V.M., Zhang J. Distinct effects of rotenone, 1-methyl-4-phenylpyridinium and 6-hydroxydopamine on cellular bioenergetics and cell death. PLoS One. 2012;7 doi: 10.1371/journal.pone.0044610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schneider L., Giordano S., Zelickson B.R., Johnson S., Benavides A., Ouyang X., et al. Differentiation of SH-SY5Y cells to a neuronal phenotype changes cellular bioenergetics and the response to oxidative stress. Free Radic Biol Med. 2011;51:2007–2017. doi: 10.1016/j.freeradbiomed.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dodson M., Wani W.Y., Redmann M., Benavides G.A., Johnson M.S., Ouyang X., et al. Regulation of autophagy, mitochondrial dynamics, and cellular bioenergetics by 4-hydroxynonenal in primary neurons. Autophagy. 2017;13:1828–1840. doi: 10.1080/15548627.2017.1356948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wani W.Y., Ouyang X., Benavides G.A., Redmann M., Cofield S.S., Shacka J.J., et al. O-GlcNAc regulation of autophagy and alpha-synuclein homeostasis; implications for Parkinson's disease. Mol Brain. 2017;10:32. doi: 10.1186/s13041-017-0311-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crabtree D., Dodson M., Ouyang X., Boyer-Guittaut M., Liang Q., Ballestas M.E., et al. Over-expression of an inactive mutant cathepsin D increases endogenous alpha-synuclein and cathepsin B activity in SH-SY5Y cells. J Neurochem. 2014;128:950–961. doi: 10.1111/jnc.12497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qiao L., Zhang J. Inhibition of lysosomal functions reduces proteasomal activity. Neurosci Lett. 2009;456:15–19. doi: 10.1016/j.neulet.2009.03.085. [DOI] [PubMed] [Google Scholar]
- 36.Meyer O.A., Tilson H.A., Byrd W.C., Riley M.T. A method for the routine assessment of fore- and hindlimb grip strength of rats and mice. Neurobehav Toxicol. 1979;1:233–236. [PubMed] [Google Scholar]
- 37.Slane J.M., Lee H.S., Vorhees C.V., Zhang J., Xu M. DNA fragmentation factor 45 deficient mice exhibit enhanced spatial learning and memory compared to wild-type control mice. Brain Res. 2000;867:70–79. doi: 10.1016/s0006-8993(00)02258-7. [DOI] [PubMed] [Google Scholar]
- 38.Zhang J., McQuade J.M., Vorhees C.V., Xu M. Hippocampal expression of c-fos is not essential for spatial learning. Synapse. 2002;46:91–99. doi: 10.1002/syn.10115. [DOI] [PubMed] [Google Scholar]
- 39.Wright J.N., Benavides G.A., Johnson M.S., Wani W., Ouyang X., Zou L., et al. Acute increases in O-GlcNAc indirectly impair mitochondrial bioenergetics through dysregulation of LonP1-mediated mitochondrial protein complex turnover. Am J Physiol Cell Physiol. 2019;316:C862–C875. doi: 10.1152/ajpcell.00491.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boyer-Guittaut M., Poillet L., Liang Q., Bole-Richard E., Ouyang X., Benavides G.A., et al. The role of GABARAPL1/GEC1 in autophagic flux and mitochondrial quality control in MDA-MB-436 breast cancer cells. Autophagy. 2014;10:986–1003. doi: 10.4161/auto.28390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mitchell T., Johnson M.S., Ouyang X., Chacko B.K., Mitra K., Lei X., et al. Dysfunctional mitochondrial bioenergetics and oxidative stress in Akita(+/Ins2)-derived beta-cells. Am J Physiol Endocrinol Metab. 2013;305:E585–E599. doi: 10.1152/ajpendo.00093.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Higdon A.N., Benavides G.A., Chacko B.K., Ouyang X., Johnson M.S., Landar A., et al. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: the protective role of autophagy. Am J Physiol Heart Circ Physiol. 2012;302:H1394–H1409. doi: 10.1152/ajpheart.00584.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koike M., Nakanishi H., Saftig P., Ezaki J., Isahara K., Ohsawa Y., et al. Cathepsin D deficiency induces lysosomal storage with ceroid lipofuscin in mouse CNS neurons. J Neurosci. 2000;20:6898–6906. doi: 10.1523/JNEUROSCI.20-18-06898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng S., Wani W.Y., Hottman D.A., Jeong A., Cao D., LeBlanc K.J., et al. Haplodeficiency of Cathepsin D does not affect cerebral amyloidosis and autophagy in APP/PS1 transgenic mice. J Neurochem. 2017;142:297–304. doi: 10.1111/jnc.14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bronson S.K., Plaehn E.G., Kluckman K.D., Hagaman J.R., Maeda N., Smithies O. Single-copy transgenic mice with chosen-site integration. Proc Natl Acad Sci U S A. 1996;93:9067–9072. doi: 10.1073/pnas.93.17.9067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Engle S.J., Womer D.E., Davies P.M., Boivin G., Sahota A., Simmonds H.A., et al. HPRT-APRT-deficient mice are not a model for lesch-nyhan syndrome. Hum Mol Genet. 1996;5:1607–1610. doi: 10.1093/hmg/5.10.1607. [DOI] [PubMed] [Google Scholar]
- 47.Tatton N.A., Kish S.J. In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice using terminal deoxynucleotidyl transferase labelling and acridine orange staining. Neuroscience. 1997;77:1037–1048. doi: 10.1016/s0306-4522(96)00545-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.