

Abstract
Eukaryotic organisms adapt to environmental fluctuations by altering their epigenomic landscapes and transcriptional programs. Nucleosomal histones carry vital epigenetic information and regulate gene expression, yet the mechanisms underlying chromatin‐bound histone exchange remain elusive. Here, we found that histone H2Bs are globally degraded in Caenorhabditis elegans during starvation. Our genetic screens identified mutations in ubiquitin and ubiquitin‐related enzymes that block H2B degradation in starved animals, identifying lysine 31 as the crucial residue for chromatin‐bound H2B ubiquitination and elimination. Retention of aberrant nucleosomal H2B increased the association of the FOXO transcription factor DAF‐16 with chromatin, generating an ectopic gene expression profile detrimental to animal viability when insulin/IGF signaling was reduced in well‐fed animals. Furthermore, we show that the ubiquitin‐proteasome system regulates chromosomal histone turnover in human cells. During larval development, C. elegans epidermal cells undergo H2B turnover after fusing with the epithelial syncytium. Thus, histone degradation may be a widespread mechanism governing dynamic changes of the epigenome.
Keywords: Histone degradation, Insulin/IGF signaling, Ubiquitination
Subject Categories: Chromatin, Transcription & Genomics; Development; Post-translational Modifications & Proteolysis
Ubiquitin‐proteasome system regulates histone H2B exchange in mammalian cells and in C. elegans during development and starvation.

Introduction
Histone post‐translational modifications play a crucial role in establishing and maintaining active and repressive chromatin domains. They are essential for specifying transcriptional programs that govern cell fates and control organismal adaptation (Cavalli & Heard, 2019; Elsherbiny & Dobreva, 2021). Although nucleosomal histones have been long regarded as highly stable with slow turnover kinetics in the post‐mitotic cells, emerging evidence has revealed rapid nucleosomal histone exchanges in response to developmental cues or environmental stimuli (Deal et al, 2010; Maze et al, 2015; Goldman et al, 2017).
Incorporating nascent histone variants into chromatin promotes dynamic changes in the epigenome and gene expression. Aberrant histone turnover has been associated with developmental defects, neurological disorders, and tumorigenesis (Wenderski & Maze, 2016; Martire & Banaszynski, 2020). The most dramatic histone replacement occurs during the histone‐to‐protamine exchange, where spermatids systematically remove their nucleosomal histone octamers. Subsequently, they re‐package, and further condense the parental genome using small arginine‐rich protein known as protamines (Oliva, 2006; Qian et al, 2013). Somatic cells undergo the exchange of subpopulations of histones at the transcription start sites of pluripotency genes upon exiting mitosis (Oh et al, 2020). DNA damage causes histone turnover at damaged chromosome regions, resulting in a genome‐wide loss of nucleosome levels by approximately 30–40%. This process induces enhanced flexibility and mobility of the DNA fiber, facilitating strand invasion (Hauer et al, 2017; Cheblal et al, 2020; Challa et al, 2021).
In response to variations in nutrient conditions, living organisms typically adjust their gene expression profile to enhance survive. When the nematode C. elegans grows under optimal nutrient conditions, high insulin/IGF signaling inactivates and sequesters the transcription factor DAF‐16 in the cytoplasm. However, under stress conditions such as starvation, reduced insulin/IGF signaling triggers the translocation of DAF‐16 to the nucleus, where it engages in an alternative transcriptional program that confers a wide variety of beneficial effects on the organism, including stress resistance and longevity (Henderson & Johnson, 2001; Lin et al, 2001). Currently, it remains unclear whether and how histone turnover is involved in reduced insulin/IGF signaling. In our study, we present evidence that somatic cells in C. elegans undergo systematic degradation of their chromatin‐bound histone H2B upon starvation or reduced insulin/IGF signaling, resulting in changes to transcription profiles that promote animal survival under stress conditions. Furthermore, we have observed similar large‐scale histone turnover also occurs in human cells and developing C. elegans epidermal cells, suggesting that histone turnover may serve as a common strategy to modify the epigenome.
Results
Starvation caused global histone H2B degradation
We found that under the conditions of starvation (Fig 1A; Materials and Methods), the fluorescence of green fluorescent protein (GFP)‐tagged histone H2B (HIS‐58) was completely removed from the nuclei of C. elegans epidermal seam cells (Fig 1A and Appendix Fig S1A and B). In contrast, the GFP‐tagged pleckstrin homology (PH) domain that marks the plasma membrane, remained unchanged in the same starved animals (Fig 1A and Appendix Fig S1A). By examining over 3,000 epidermal seam cells in more than 300 animals living without bacteria as a food source, we detected a 100% loss of the GFP::H2B signal, indicating that the starvation induced a global elimination of HIS‐58/H2B.
Figure 1. Global degradation of histone H2B proteins during starvation.
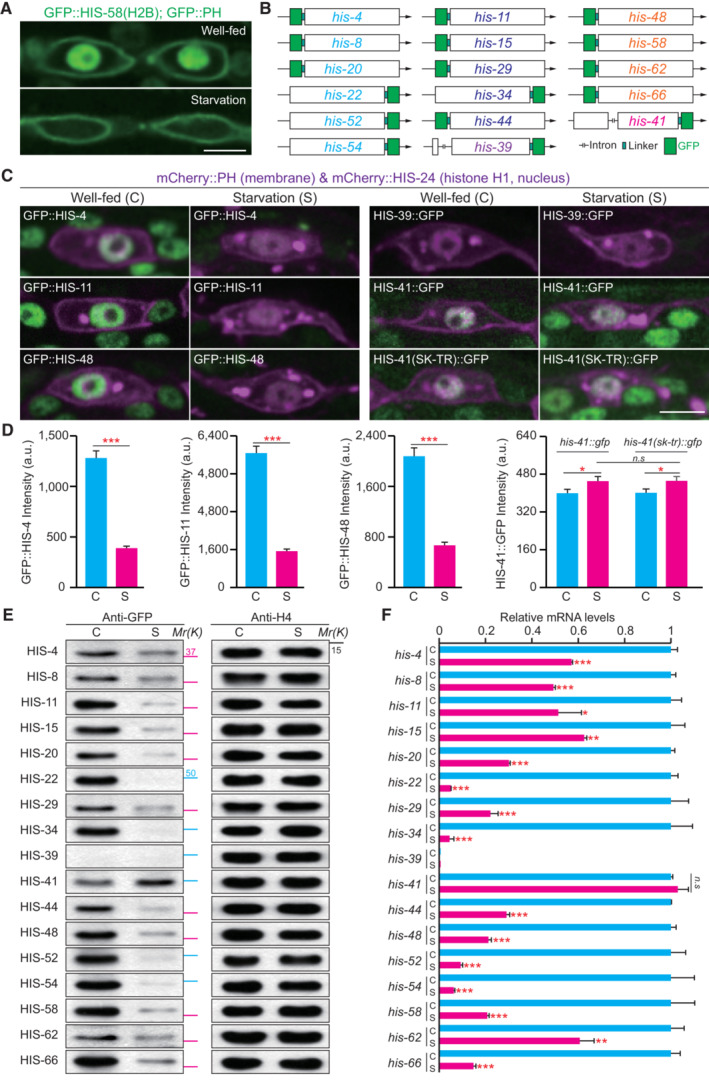
- Fluorescence images of the epidermal seam cells in well‐fed and starved Caenorhabditis elegans larvae expressing GFP‐tagged pleckstrin homology domain (PH) and H2B (HIS‐58) under the control of a seam cell‐specific wrt‐2 promoter. Scale bar, 5 μm. The starved worms were prepared as follows: Isolated worm eggs were transferred to NGM plates with an OP50 bacterial lawn. After 12 h, the hatched L1 larvae were collected and washed with the M9 buffer to remove the bacteria. These worms were then transferred to empty NGM plates without bacterial food and cultured for 6 days.
- Schematic illustration of GFP‐tagged endogenous H2B KI by CRISPR‐Cas9‐based genome editing. Genes with the same color encode an identical amino acid sequence.
- Fluorescence images of the endogenous GFP::H2B in seam cells under well‐fed or starved conditions with different genotypes. HIS‐41(SK‐TR)::GFP indicates mutation of the serine 18 and lysine 27 residues of HIS‐41 to threonine and arginine, respectively. The mCherry::PH (membrane) and mCherry::HIS‐24 (histone H1, nucleus) were expressed under the control of a seam cell‐specific ceh‐16 promoter. Scale bar, 5 μm.
- Quantification of the endogenous GFP::H2B fluorescence in seam cells under well‐fed (C) and starved (S) conditions with different genotypes (each genotype n = 100). Data shown are means ± SEM; P values were determined by a two‐tailed unpaired t‐test, *P < 0.05, ***P < 0.001, n.s., no significance.
- Immunoblot of the endogenous H2B protein in well‐fed (C) and starved (S) animals. Histone H4 was used as the loading control. GFP‐tag was used to identify individual H2B proteins. The blue line indicates AGSGSG as the linker between GFP and H2B, and the pink line indicates TEV‐S (TEV indicating TEV protease cleavage site, and S indicating S‐tag) as the linker between GFP and H2B. Mr, relative molecular weight; K, thousands.
- qRT–PCR showing relative expression levels of the endogenous H2B mRNA in well‐fed (C) and starved (S) animals. GFP‐tag was used to identify individual H2B proteins. Relative RNA levels (normalized to rps‐4) were normalized to the well‐fed condition. In each experiment, we analyzed three biological replicas and three technical replicas. Data shown are means ± SEM; P values were determined by a two‐tailed unpaired t‐test, *P < 0.05, **P < 0.01, ***P < 0.001, n.s., no significance.
Source data are available online for this figure.
Which H2B isoform was incorporated into chromatin to substitute the lost one? The C. elegans genome encodes 17 histone H2B genes, categorized into five subfamilies with identical amino acid sequences (Fig 1B and Appendix Fig S1C). To distinguish individual H2B isoforms, we inserted a GFP tag into each endogenous H2B gene locus (Fig 1B). Despite their high DNA sequence similarity, we generated knock‐in (KI) strains for all 17 GFP::H2B isoforms (Fig 1B; Materials and Methods). We did not observe any apparent developmental defects in these KI animals (N > 100 for each genotype). Except for the his‐39 gene, a putative pseudogene, the remaining strains exhibited the expected H2B fluorescence in the nuclei (representative images from each H2B subfamily in Fig 1C). We found that HIS‐41::GFP was the only H2B isoform that maintained its fluorescence during starvation (Fig 1C and D). GFP signals from other H2B isoforms became undetectable or substantially reduced (Fig 1C and D), and the loss of fluorescence was not limited to the epidermis but extended to other tissues (Fig 1C), indicating a systemic H2B loss in the organism. We validated the microscopic results by performing a Western blot using an anti‐GFP antibody. Histone H4 was used as the loading control since all the 16 C. elegans H4 isoforms share an identical protein sequence. Indeed, the protein level of HIS‐41 increased, while other H2B isoforms were significantly reduced or undetectable (Fig 1E), further confirming the degradation of H2B proteins in starved animals.
What distinguishes HIS‐41 from the other H2B isoforms that were replaced? The serine 18 and lysine 27 residues of HIS‐41 differ from those found in other H2B proteins (Appendix Fig S1C). Using genome editing to change both residues to those found in other H2B proteins, we created the HIS‐41S18T&K27R::GFP knock‐in animal. However, we did not observe any significant loss of fluorescence in the starved animals (Fig 1C and D), which suggests that the difference in degradation between HIS‐41 and other H2B isoforms may not be due to variations in the protein sequence. The distinctive behavior of HIS‐41 could potentially be elucidated if the his‐41 gene encodes the sole DNA replication‐independent H2B variant in C. elegans (Marzluff et al, 2002; Pettitt et al, 2002; Keall et al, 2007). The results of quantitative reverse transcription PCR (qRT–PCR) using random primers showed that the relative expression level of gfp::his‐4 mRNA was eight times higher compared to that detected by Oligo dT primers (Appendix Fig S1D). Conversely, for his‐41::gfp mRNA, the relative expression level detected by random primers was only 0.7 times that of Oligo dT primers (Appendix Fig S1D). The discrepancy could be attributed to differences in their 3′ UTR sequences, providing additional evidence for categorizing his‐4 as a replication‐dependent histone and his‐41 as a replication‐independent histone.
Because starvation halts the progression of the cell cycle, including DNA replication, as well as the transcription and synthesis of replication‐dependent histone genes (Baugh & Sternberg, 2006; Marzluff & Koreski, 2017), we hypothesized that the mRNA of replication‐dependent H2B might be degraded under these conditions. This would leave the his‐41 mRNA as the only available H2B RNA molecules to translate H2B protein, thereby replacing the lost replication‐dependent H2Bs. Our qRT–PCR analyses showed that the level of his‐41 mRNA remained unchanged during starvation, compared to the well‐fed controls (Fig 1F). In contrast, the mRNA levels of replication‐dependent H2B genes in the starved animals were significantly lower compared to those in animals provided with ample food (Fig 1F). Collectively, these results revealed that the replication‐dependent H2B proteins were replaced under the conditions of starvation due to the cooperative action of transcription regulation and protein degradation. We propose that the replication‐independent histone H2B, HIS‐41, may also undergo degradation similar to the other H2Bs. However, the abundance of his‐41 transcripts allows continuous translation, thereby maintaining protein levels and allowing HIS‐41 to predominantly replace the other H2Bs.
H2B degradation requires components in the ubiquitin‐proteasome system (UPS)
We conducted a visual forward genetic screen to identify essential factors for H2B degradation in response to starvation. The C. elegans strain expressing the epidermal GFP::H2B reporter did not produce detectable GFP signals 6 days after starvation (Fig 2A and Appendix Fig S1B). After starving the mutagenized F2 progeny for 6 days, we isolated 10 mutant alleles that retained GFP::HIS‐58(H2B) fluorescence in all the examined animals (Fig 2A–C, genetic mapping and cloning in Materials and Methods). We identified cas921 and cas924 as point mutations in the ubiquitin gene ubq‐1 (Fig 2B), presumably disrupting ubiquitin production (Jonnalagadda et al, 1989). The cas905 allele introduced a codon change in the C. elegans E1 ubiquitin‐activating enzyme UBA‐1 (Fig 2B). Although null alleles defective in the ubiquitin or E1 gene are embryonic lethal (Jones et al, 2002), our isolated allele is viable under the conditions of starvation and does not show apparent developmental abnormalities, which suggests that it might be a weak loss‐of‐function allele blocking H2B degradation rather than causing severe proteotoxicity. The cas901, cas913, cas916, and cas920 mutants caused missense mutations or early stop codons in the E2 ubiquitin‐conjugating enzyme UBC‐20, an ortholog of human UBE2K (Ubiquitin‐conjugating enzyme E2 K; Fig 2B). The cas903, cas908, and cas909 alleles generated missense mutations or early stop codons in the E3 ubiquitin ligase HECD‐1, an ortholog of human HECTD1 (HECT domain E3 ubiquitin‐protein ligase 1; Fig 2B). By constructing GFP KI animals, we showed that UBC‐20 and HECD‐1 resided in the nuclei throughout the body (Appendix Fig S2A–D). Considering that these mutations did not affect the transcriptional pattern of gfp::his‐58 mRNA upon starvation stress (Fig 2D), we suggest that the ubiquitin‐proteasome system (UPS) regulates H2B degradation in the nuclei during starvation.
Figure 2. UPS components are essential for H2B degradation.
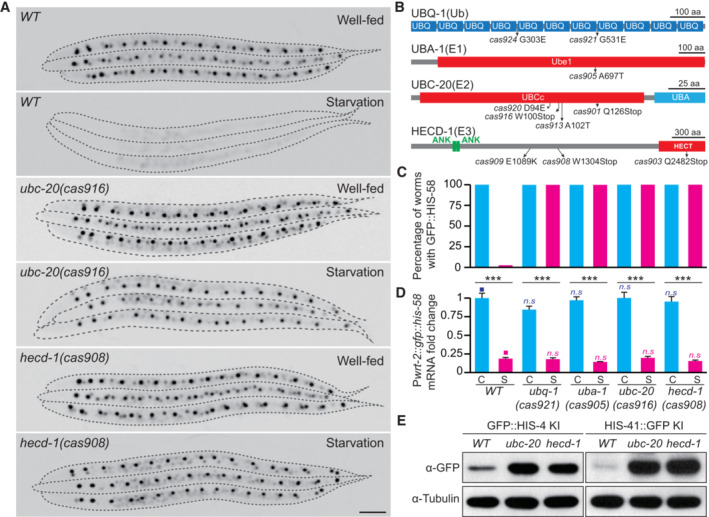
- Fluorescence inverted images of seam cells in WT, ubc‐20(cas916), and hecd‐1(cas908) animals under well‐fed or starved conditions. Seam cells were visualized using Pwrt‐2::gfp::his‐58. Images were inverted so that the high GFP fluorescence intensity was black. Dashed lines show the animal periphery. Scale bar, 50 μm.
- Schematics of the Caenorhabditis elegans UBQ‐1, UBA‐1, UBC‐20, and HECD‐1 proteins. UBQ, ubiquitin domain; Ube1, ubiquitin‐activating enzyme e1 domain; UBCc, ubiquitin‐conjugating enzyme catalytic domain; UBA, ubiquitin associated domain; ANK, ankyrin repeat domain; HECT, homologous to the E6‐AP carboxyl‐terminus domain; Ub, ubiquitin; E1, ubiquitin‐activating enzyme; E2, ubiquitin‐conjugating enzyme; E3, ubiquitin ligase; The amino acid changes in different alleles are indicated.
- Quantification of transgenic animals with visible GFP::HIS‐58 fluorescence under well‐fed (C) or starved (S) conditions for 6 days. Different genotypes (n = 300) are indicated at the bottom of (D).
- qRT–PCR showing relative expression levels of the Pwrt‐2::gfp::his‐58 mRNA under well‐fed (C) or starved (S) conditions with different genotypes. Relative RNA levels (normalized to rps‐4) are normalized to the well‐fed condition. In each experiment, we analyzed three biological replicates and three technical replicates. Data shown are means ± SEM; statistical significance compared with the control (■) with a matching color; P values were determined by a two‐tailed unpaired t‐test, **P < 0.01, ***P < 0.001, n.s., no significance.
- Immunoblot of the endogenous GFP::HIS‐4 and HIS‐41::GFP protein levels in WT, ubc‐20(cas916), and hecd‐1(cas908) mutants in well‐fed animals.
Source data are available online for this figure.
To assess whether the putative ubiquitination components regulate endogenous H2B degradation, we examined the protein levels of endogenous replication‐dependent HIS‐4 or replication‐independent HIS‐41 in wild‐type (WT), ubc‐20(cas916), and hecd‐1(cas908) animals and found that the levels of GFP‐tagged HIS‐4 and HIS‐41 were significantly increased in ubc‐20(cas916) and hecd‐1(cas908) animals compared with the WT animals under well‐fed conditions (Fig 2E). In addition, we conducted an H2B dosage analysis during starvation by genetically crossing GFP::HIS‐4 KI into ubc‐20(cas916) and hecd‐1(cas908) mutants, and synchronized the worms, subjecting them to 6 days of starvation before imaging them using an identical setup. We show that depletion of UBC‐20 and HECD‐1 significantly inhibited GFP::HIS‐4 degradation during starvation (Appendix Fig S2E and F). Furthermore, we performed RNA‐seq and data analysis to examine a possible contribution of mRNA level regulation to the observed decrease in H2B proteins upon starvation. In the wild‐type animal, the his‐4 gene was expressed with ample food and sharply decreased upon starvation stress, but the expression level of his‐41 mRNA remained unchanged during starvation (Appendix Fig S2G). Importantly, the depletion of UBC‐20 and HECD‐1 did not alter the transcriptional pattern of his‐4 and his‐41 mRNA upon starvation stress (Appendix Fig S2G). Thus, the ubiquitin‐proteasome system (UPS) regulates H2B degradation in both well‐fed and starved animals.
To identify the lysine (K) residue(s) crucial for H2B ubiquitination and degradation, we initially isolated the nuclei from the 1‐day starved GFP‐H2B KI C. elegans larvae. Subsequently, we purified their nucleosomes and prepared the H2B isoform‐specific nucleosomes through GFP‐affinity purification (Fig 3A). Employing GFP KI strains of HIS‐4 and HIS‐41, mass spectrometry (MS) successfully identified K31 and K117 as the possible ubiquitination sites, which correspond to K34 and K120 in the human H2B protein (Fig 3B–D and Appendix Fig S3A). Monoubiquitination of human H2BK120 serves as an epigenetic modulator for gene expression (Pavri et al, 2006; Kim et al, 2013), whereas H2BK34 ubiquitination remains less extensively studied. To assess their significance in H2B degradation, we introduced K31A or K117A mutations into HIS‐4::GFP and K31A mutation into HIS‐41::GFP KI strains. Western blots showed that K31, but not K117, is the crucial residue for H2B degradation in well‐fed animals (Fig 3E and Appendix Fig S3B). In addition, mutations in his‐41 K31A inhibited its own degradation during starvation (Appendix Fig S3C–E). Furthermore, we performed transfection of human HEK293T cells with the EGFP‐tagged HIS‐41 or HIS‐41K31A and employed the proteasome inhibitor MG‐132 to block USP‐mediated protein degradation. After GFP‐affinity purification from the isolated HEK293T nuclei, we detected the K48‐linked polyubiquitination in HIS‐41, but substantially less in HIS‐41K31A (Fig 3F). By generating UBE2K/UBC‐20 or HECTD1/HECT‐1 knockout human cell lines, we showed that depletion of E2 or E3 reduced the K48‐linked polyubiquitination of H2B in HEK293T cells (Fig 3F). These findings strongly suggest that K31 mediates H2B polyubiquitination and degradation.
Figure 3. Ubiquitination at K31 promotes H2B degradation.
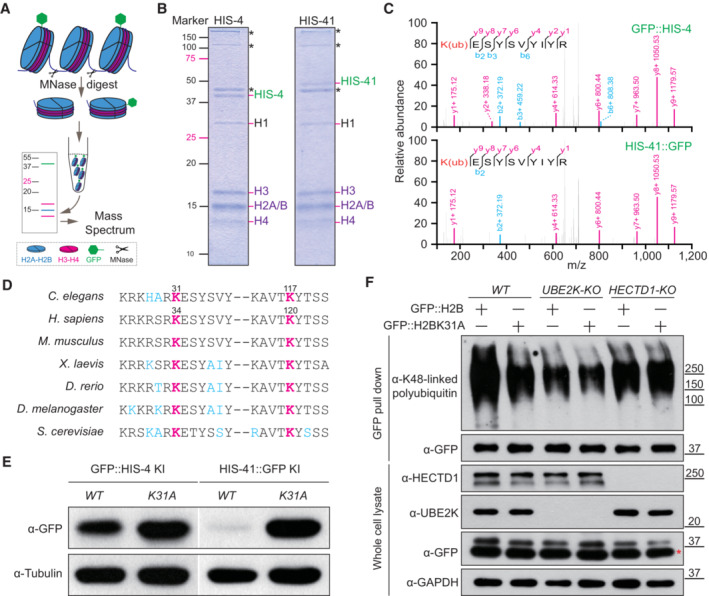
- Schematic of the purification of GFP::H2B associated nucleosomes from Caenorhabditis elegans chromatin. MNase, micrococcal nuclease.
- SDS–PAGE analysis of GFP::HIS‐4 and HIS‐41::GFP associated nucleosomes. The asterisks indicate the non‐nucleosomal proteins (i.e., UNC‐54, UNC‐15, and actin from top to bottom) identified by mass spectrometry.
- Tandem mass spectrum of the K31ub‐modified peptide from GFP::HIS‐4 and HIS‐41::GFP. The y and b series indicate fragments at amide bonds of the peptide.
- Alignment of the region encompassing K31 and K117 in C. elegans H2B from multiple species. K31 and K117 in C. elegans H2Bs correspond to K34 and K120 in human H2Bs, respectively.
- Immunoblot of endogenous GFP::HIS‐4 and HIS‐41::GFP protein levels in WT and K31A mutants under the well‐fed condition.
- Immunoblot of the K48‐linked polyubiquitin of GFP::H2B and GFP::H2BK31A in WT, UBE2K‐KO, and HECTD1‐KO human cells treated with 1 μM MG‐132 for 16 h. The red asterisk indicates the nonspecific protein.
Source data are available online for this figure.
H2B degradation regulates insulin/IGF signaling‐mediated nutrient stress
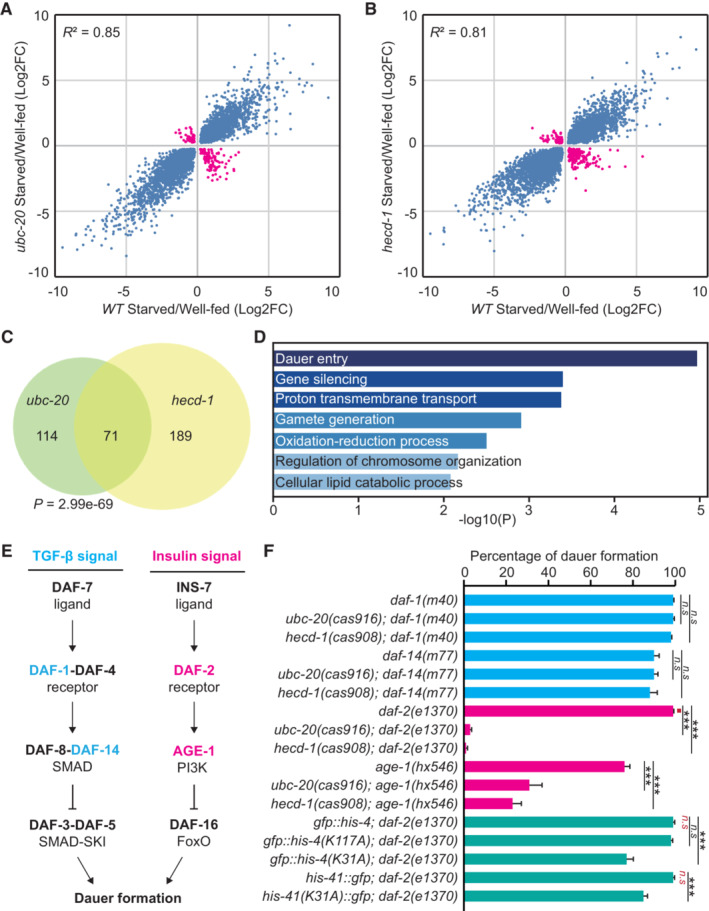
To identify molecular pathways that are potentially affected by inhibiting H2B degradation, we performed RNA sequencing (RNA‐seq) analyses to compare the gene expression profiles of starved WT and mutant C. elegans L1 larvae. Consistent with earlier results (Baugh et al, 2009), starvation significantly altered the transcription program in WT animals. However, we found that a subset of starvation‐responsive genes displayed the opposite expression patterns in ubc‐20 or hecd‐1 mutant animals (Fig 4A–C and Dataset EV1). Gene ontology (GO) term analysis revealed enrichment for genes regulating dauer entry (Fig 4D), a process through which animals arrest development and alter gene expression and metabolism to survive starvation (Riddle & Albert, 1997).
Figure 4. H2B degradation is essential for insulin/IGF‐mediated dauer formation.
-
A, BStarvation‐responsive genes in WT with ubc‐20 (A) or hecd‐1 (B) mutants. Shown are significantly induced and repressed genes in WT, ubc‐20, and hecd‐1 mutants during starvation. The blue or pink points indicate whether upregulated or downregulated genes in mutants are similar or opposite to WT during starvation. FC, fold change.
-
COverlap of abnormally expressed starvation‐responsive genes in ubc‐20 and hecd‐1 mutant animals. The P‐value was calculated using a hypergeometric probability test.
-
DGene enrichment analysis of abnormally expressed starvation‐responsive genes in ubc‐20 and hecd‐1 mutants.
-
ETwo signaling pathways regulate C. elegans dauer formation.
-
FGenetic analysis of dauer formation with different genotypes. Six independent assays were performed, n > 100. Data shown are means ± SEM; P values were determined by two‐tailed unpaired t‐test, ***P < 0.001, n.s., no significance.
Source data are available online for this figure.
While the single mutations of ubc‐20 or hecd‐1 result in viable animals with normal development, our RNA‐seq results inspired us further to investigate the impact of blocking H2B degradation on animal adaptation associated with dauer formation (DAF). To explore this, we introduced H2B degradation mutations into the daf‐1 or daf‐2 mutant animals. DAF‐1 is an ortholog of the human transforming growth factor‐beta receptor (TGF‐beta) (Georgi et al, 1990), and inhibition of H2B degradation in daf‐1 did not cause any DAF abnormalities (Fig 4E and F), suggesting that aberrant H2B retention may not significantly affect TGF‐beta signaling‐mediated dauer formation. daf‐2 encodes the human ortholog of insulin/IGF receptor, and the daf‐2(e1370) allele is a temperature‐sensitive mutant with reduced insulin/IGF receptor activity (Kimura et al, 1997). At the restrictive temperature of 25°C, 100% of daf‐2(e1370) animals entered the dauer stage (Fig 4E and F). Interestingly, the daf‐2; ubc‐20 and daf‐2; hecd‐1 double mutant larvae exhibited a strikingly different outcome, as they failed to enter dauer and all died on the third day following the shift to 25°C (Fig 4F). Notably, no growth abnormalities were observed in the ubc‐20 or hecd‐1 single mutants at 25°C (N > 100 for each genotype). To elucidate the direct contribution of H2B regulation in dauer formation or larval arrest, we conducted RNAi experiments in the daf‐2; daf‐2, ubc‐20; and daf‐2, hecd‐1 mutant animals. RNAi against his‐41/H2B did not reveal novel phenotypes in the daf‐2 background. However, the knockdown of his‐41/H2B partially suppressed larval lethality and rescued dauer formation in daf‐2, ubc‐20 and daf‐2, hecd‐1 (Appendix Fig S4A). These results suggest that maintaining appropriate levels of histone H2B is critical for animal development and survival under conditions of reduced insulin/IGF receptor activity.
We observed that the H2BK31A mutation in daf‐2 at 25°C also inhibited dauer entry and caused animal lethality, albeit to a lesser extent (Fig 4F). To explore the potential redundancy of other WT H2B isoforms in regulating daf‐2‐mediated dauer formation, we generated his‐4 K31A ; his‐41 K31A double mutations in daf‐2(e1370) animals. However, this did not enhance the DAF phenotype (Appendix Fig S4B). Considering that ubiquitination‐mediated degradation of a protein complex may occur at multiple sites of its different subunits, we wondered whether other subunits in the histone octamer were also ubiquitinated. Indeed, our MS experiments revealed that HIS‐4 or HIS‐41 bound core histones (H2A, H2B, H3, H4) carry additional ubiquitinated sites (Appendix Fig S4C and D). Echoing the single H2BK31A mutation, only H4K31A slightly inhibited its own degradation and daf‐2‐mediated dauer formation (Appendix Fig S4B and E). To examine whether H2BK31A and H4K31A have redundant functions, we generated the H4 mutation HIS‐37K31A into the H2B HIS‐41K31A mutant animal, resulting in a double mutant animal that further hindered GFP::HIS‐41K31A degradation (Appendix Fig S4F and G). Essentially, the H2B HIS‐41K31A and H4 HIS‐37K31A double mutant animals exhibited enhanced defects in daf‐2‐mediated dauer formation, reaching a penetrance of approximately 70%, which was significantly higher than the 15% observed in the single mutant animals (Appendix Fig S4B). These findings indicate that UPS‐dependent H2B and H4 degradation collectively contributes to animal survival under reduced insulin/IGF signaling.
H2B retention‐induced lethality requires the FOXO transcription factor DAF‐16
To understand how H2B degradation regulates insulin/IGF signaling, we performed a genetic suppressor screen to identify mutations that rescued animal lethality in the daf‐2; hecd‐1 double mutant. We isolated genetic suppressors that restored larval viability and allowed progeny production in daf‐2; hecd‐1 at 25°C (Fig 5A). Notably, we identified 71 mutations in the human pioneer transcription factor FOXO ortholog daf‐16 (Fig 5B, the complete list of daf‐16 mutations in Dataset EV2). DAF‐16 functions downstream of the DAF‐2 receptor in the insulin/IGF signaling pathway (Lin et al, 1997; Ogg et al, 1997). We confirmed this finding by introducing the reference loss‐of‐function allele of daf‐16(mu86) into daf‐2; hecd‐1 double mutants, and the resulting triple mutant animals survived at the restrictive temperature (Fig 5C). These data indicate that DAF‐16 is involved in aberrant H2B retention‐induced animal lethality under reduced insulin/IGF signaling, suggesting that DAF‐16 might cause aberrant transcription detrimental to animal viability.
Figure 5. Aberrant H2B retention causes DAF‐16‐dependent ectopic transcription.
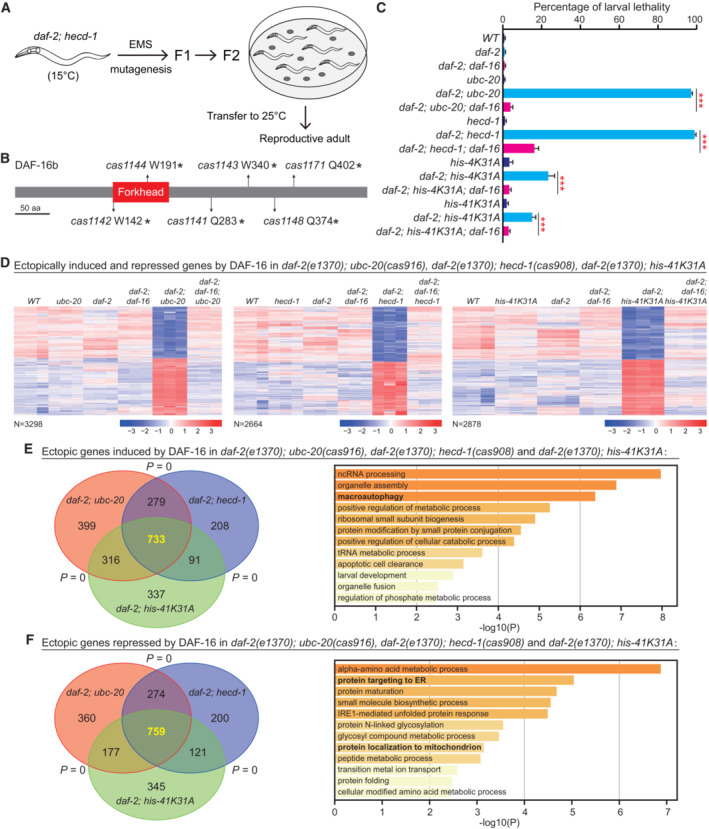
-
AFlowchart of the genetic suppressor screen. daf‐2; hecd‐1 animals are viable and reproductive at 15°C but all die at 25°C. Ethyl methanesulfonate (EMS)‐mutagenized F2 progeny were transferred to 25°C, and reproductive animals were selected as suppressors.
-
BSchematic of the Caenorhabditis elegans DAF‐16 isoform b protein. Amino acid changes in different alleles are indicated.
-
CGenetic analysis of larval lethality in animals with different genotypes. Six independent assays were performed, n > 100. Data shown are means ± SEM; P values were determined by two‐tailed unpaired t‐test, ***P < 0.001.
-
DTranscriptomic analysis of aberrant gene expression by DAF‐16 in animals with different genotypes. Shown are genes induced or repressed (Fold Change > 2) in daf‐2; ubc‐20, daf‐2; hecd‐1, and daf‐2; his‐41 K31A mutants.
-
E, FOverlap and gene enrichment analysis of ectopically upregulated (E) and downregulated (F) genes by DAF‐16 in daf‐2; ubc‐20, daf‐2; hecd‐1, and daf‐2; his‐41 K31A mutants. Gene enrichment analysis was performed using Metascape bioinformatics resources, and statistical analysis was conducted using the Fisher exact test. The P value was calculated using a hypergeometric probability test.
Source data are available online for this figure.
We performed RNA‐seq for the double mutants defective in DAF‐2 and histone degradation at the restrictive temperature. Inhibiting histone degradation did not affect DAF‐2‐dependent gene expression (Appendix Fig S5A–C). However, compared to the daf‐2 single mutant, we observed abnormal expression in 3,298, 2,664, or 2,878 genes in the daf‐2; ubc‐20, daf‐2; hecd‐1, daf‐2; his‐41 K31A double mutants, respectively (Figs 5D and 6A, and Datasets [Link], [Link]). Among these, 733 genes showed ectopic upregulation in all the double mutants, including some involved in macroautophagy (Fig 5E and Dataset EV6). Another 759 genes were commonly downregulated (Fig 5F and Dataset EV6). Notably, 172 of the downregulated genes were previously shown to cause embryonic or larval lethality when compromised by RNAi. Using GO term analysis, we found that these essential genes are associated with the endoplasmic reticulum, mitochondrion, and protein synthesis, and this overlap reflects a statistically significant enrichment of genes with the phenotype (Appendix Fig S5D and Dataset EV7). Remarkably, the loss of DAF‐16 in the double mutants rescued animal viability and restored the abnormally induced or repressed genes to the wild‐type expression level (Fig 5D and Datasets [Link], [Link]). These findings suggest that aberrant H2B retention induces ectopic DAF‐16‐dependent transcription, leading to animal lethality.
Figure 6. H2B degradation prevents ectopic binding of DAF‐16 to chromatin.

- Genome browser tracks of DAF‐16::GFP ChIP‐seq and RNA‐seq peaks at the C18B12.4 and W08E12.7 loci in different genotypes.
- The average and heatmap of HIS‐41::GFP ChIP signals at all the HIS‐41::GFP‐binding sites in daf‐2, daf‐2; hecd‐1, and daf‐2; his‐41 K31A mutants.
- The average and heatmap of DAF‐16::GFP ChIP signals at the DAF‐16‐dependent normal or ectopic gene expression in daf‐2 and daf‐2; hecd‐1 mutants.
H2B degradation prevents an ectopic increase of DAF‐16 binding to chromatin
We aimed to unravel the mechanism by which H2B degradation impacts DAF‐16‐mediated transcription. To achieve this, we determined the genome‐wide distribution of HIS‐41, HIS‐41K31A in daf‐2 animals, and HIS‐41 in daf‐2; hecd‐1 animals using chromatin immunoprecipitation followed by high‐throughput sequencing (ChIP‐seq). As expected, the hecd‐1 null or HIS‐41K31A mutation resulted in increased HIS‐41 signal at genes with the HIS‐41 domain (Fig 6B and Appendix Fig S6A and B), providing further evidence for the involvement of the E3 ligase HECD‐1 and H2BK31 ubiquitination in chromosomal H2B elimination.
Next, we conducted ChIP‐seq analysis using GFP::DAF‐16 KI animals to examine the endogenous DAF‐16 in daf‐2 and daf‐2; hecd‐1 mutant animals, and identified the previously documented DAF‐16‐binding loci (Appendix Fig S5A and B; Murphy et al, 2003; Riedel et al, 2013). In the daf‐2; hecd‐1 double mutants, DAF‐16 exhibited increased association with the transcription start site (TSS) of the genes that displayed aberrant induction or repression of their expression (Fig 6A and C). This observation aligns with the concept that pioneer factors such as FOXO/DAF‐16 preferentially target chromatin domains enriched with nucleosomal histones and interact with histone H3 and H4 (Appendix Fig S6C and D) (Hatta & Cirillo, 2007; Soufi et al, 2015). We also detected an increase in DAF‐16 binding to the TSS of genes that exhibited similar expression levels in daf‐2 and daf‐2; hecd‐1 mutant animals, indicating that increased DAF‐16 association with these loci is insufficient to alter their transcription (Fig 6C and Appendix Fig S5C). By further analyzing DAF‐16::GFP binding sites and DAF‐16‐mediated gene expression in daf‐2, hecd‐1 double mutants, we identified 255 genes bound by DAF‐16::GFP but exhibiting normal gene expression. Intriguingly, we found 652 genes bound by DAF‐16::GFP and displaying ectopic gene expression, suggesting that the inhibition of histone degradation may impact the transcriptional activity of DAF‐16 (Appendix Fig S6E). These findings provide evidence that aberrant nucleosomal H2B retention leads to enhanced association of DAF‐16 with chromatin, some of which may induce a deleterious transcriptional program under reduced insulin/IGF signaling.
UPS‐dependent histone turnover in human cells
To examine whether human cells also undergo UPS‐dependent H2B turnover, we utilized stable isotope labeling with amino acids in cell culture (SILAC)‐based quantitative proteomics (Xu et al, 2010; Maze et al, 2015). We first cultured HEK293T cells in a medium containing light‐lysine until they reached 100% confluency, thereby minimizing the cell‐cycle effects compared to sub‐confluent cells. Subsequently, we transferred the cells to a medium containing heavy‐lysine. After isolating the nuclei, we purified histones and subjected them to analysis using reverse‐phase liquid chromatography coupled with tandem mass spectrometry (LC‐MS/MS) (Fig 7A and Appendix Fig S7A and B). H2B peptides containing heavy‐lysine residues were identified as “new” due to an eight‐dalton mass increase (Fig 7A and Appendix Fig S7A). Our findings revealed that H2B exchange occurred in a time‐dependent manner, which was effectively blocked by MG‐132 (Fig 7B). Similar turnover patterns were observed in H1, H2A, H3, and H4 histones (Fig 7C and Appendix Fig S7A and D), indicating that UPS activity is crucial for the turnover of other histone subunits as well.
Figure 7. UPS‐dependent histone turnover in human cells.
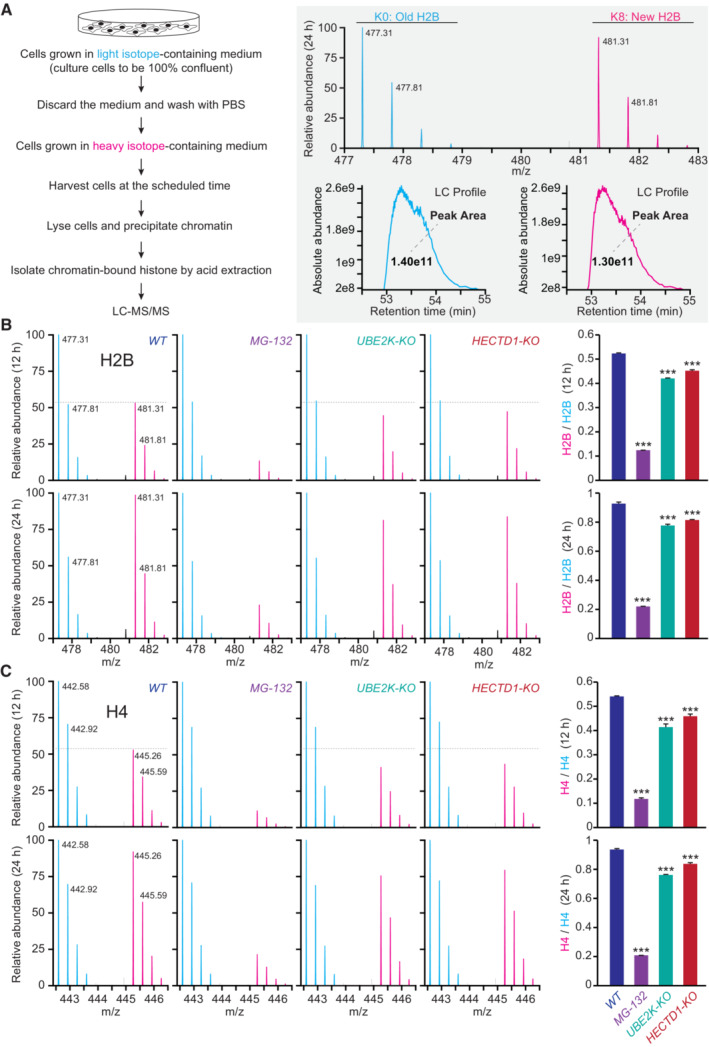
-
ASILAC‐based LC–MS/MS strategy for analyzing H2B turnover in HEK293T cells. The old H2B (K0) was labeled with light amino acids, and the newly synthesized H2B (K8) was labeled with heavy amino acids. K0 peptide and K8 peptide with identical amino acid sequences were co‐eluted from the reverse‐phased HPLC column, ionized into the mass spectrometer, and displayed in the MS spectrum as a peptide pair with a defined mass difference of 8 Da. The first peak areas (absolute peptide abundance) of the K8/K0 peptide pair in each MS spectrum were plotted and calculated against the elution time to generate the extracted ion chromatograms.
-
B, CRepresentative mass spectra and quantification of H2B (B) and H4 (C) turnover in WT cells treated with DMSO or MG‐132, UBE2K‐KO, and HECTD1‐KO cells. Three independent assays were performed; data shown are means ± SEM; P values determined by two‐tailed unpaired t‐test, ***P < 0.001.
Source data are available online for this figure.
Through examination of endogenous H2B levels in wild‐type, UBE2K‐KO, and HECDT1‐KO cells, we observed that the depletion of UBE2K or HECDT1 resulted in increased levels of endogenous H2B compared to the WT controls (Appendix Fig S7C). Using the established SILAC and LC‐MS/MS assays, we found that depletion of E2/UBE2K reduced the replacement of old histones with newly synthesized ones for all the examined histones, and E3/HECTD1 knockout also reduced histone replacement, except for H1 or H2A.Z (Fig 7B and C, and Appendix Fig S7D). These findings highlight the critical roles of UPS activity, UBE2K, and HECTD1 in regulating histone turnover in human cells under nutrient‐rich conditions.
Histone H2B turnover during the C. elegans epithelial development
In addition to starvation or reduced insulin/IGF signaling, we showed that histone H2B turnover also occurs in C. elegans epidermal cells during larval development under nutrient‐rich conditions. During larval development, the epidermal seam cells divide to generate an anterior daughter V.a and a posterior daughter V.p cell, with V.a subsequently fusing with the epithelial syncytium (Fig 8A and B, L1 larvae). We observed that the V.a cell undergoes a loss of green fluorescence from the GFP‐tagged H2B, while its sister V.p cell does not (Fig 8C). The loss of GFP::HIS‐58/H2B is completed about 8 h after the divisions of the seam cells, which corresponds to the birth of V.a and V.p cells (Fig 8B–D). Strikingly, this process is even faster than histone degradation observed during starvation (Appendix Fig S1A and B). To explore whether endogenous HIS‐58 is replaced by HIS‐41 in the anterior daughter, we introduced GFP::HIS‐58 KI or HIS‐41::GFP KI into transgenic strains expressing mCherry::PH and mCherry::H1, which label the membrane and nucleus of epidermal seam cells, respectively. Consistent with our transgenic observations, endogenous GFP::HIS‐58 was highly expressed in epidermal seam cells and underwent rapid degradation in the anterior daughter upon fusion with hyp 7 (Appendix Fig S8A, indicated with yellow arrowhead). Conversely, HIS‐41::GFP showed relatively low expression in epidermal seam cells but became enriched in the nucleus of hyp 7 (Appendix Fig S8B, indicated with yellow and blue arrowhead). These results collectively suggest that HIS‐58 may be replaced by HIS‐41 in the anterior daughter upon fusion with hyp 7, indicating an active and global H2B exchange process involved in epidermal cell differentiation.
Figure 8. H2B loss during Caenorhabditis elegans epidermal development.
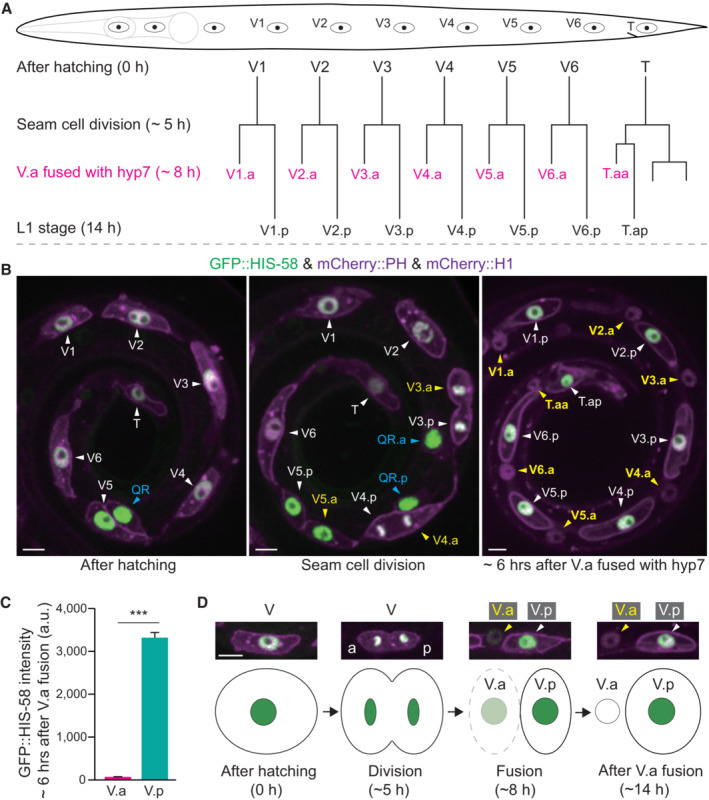
- Seam cell (V) lineages in well‐fed C. elegans L1 larvae. A seam cell (V) divides to generate the anterior daughter cell (V.a) and the posterior daughter (V.p). V.a cells fuse with the epithelial hyp 7 syncytium, whereas V.p cells do not fuse and keep dividing in the following larval stages.
- Fluorescence images of seam cells. The GFP::HIS‐58 (H2B) was expressed under the control of the seam cell‐specific wrt‐2 promoter, and the mCherry::PH (membrane) and mCherry::HIS‐24 (H1, nucleus) were expressed under the control of another seam cell‐specific ceh‐16 promoter. The white arrowheads indicate the seam cell V or V.p nuclei, whereas the yellow arrowheads indicate V.a nuclei. The blue arrowheads indicate the Q cells. Because the Q cell and V5 seam cell are produced by the same mother epidermal cell, wrt‐2 and ceh‐16 promoters are expressed in Q cells. The right image shows that V.p (white arrowheads and cell names) maintained GFP::H2B fluorescence, but V.a lost the GFP signal (yellow arrowheads and cell names) 6 h after V.a fused with hyp 7. Scale bar, 5 μm.
- Quantification of GFP::HIS‐58 in the seam cell nuclei (n = 48) and hyp 7 cell nuclei (n = 48) after the seam cell V.a fused with hyp 7 in L1 larvae. Data shown are means ± SEM; P values were determined by a two‐tailed unpaired t‐test, ***P < 0.001.
- Representative images and schematics of H2B loss after seam cells fused with hyp 7. a, anterior; p, posterior. Scale bar, 5 μm.
Source data are available online for this figure.
To investigate the occurrence of histone degradation in other lineages during development, we performed Western blotting to determine the levels of endogenous GFP::HIS‐4 (a replication‐dependent H2B) and HIS‐41::GFP (the unique replication‐independent H2B) in larvae and adult worms. Our results reveal that GFP::HIS‐4 is almost entirely degraded and replaced by HIS‐41::GFP in adult animals (Appendix Fig S8C and D), which indicates that H2B replacement and degradation are widespread processes that occur in various tissues during development.
Discussion
Historically, the first identified ubiquitinated protein was histone H2A (Goldknopf & Busch, 1977), and chromosomal histones have long been recognized as among the most abundant ubiquitination substrates (West & Bonner, 1980). However, the extensively studied histone ubiquitination primarily involves monoubiquitination, which does not mediate protein degradation but rather regulates gene expression. For instance, monoubiquitination of histone H2A at K119 at the gene promoter regions suppresses transcription (Wang et al, 2004; Tamburri et al, 2020), while H2B monoubiquitination at K120 in the gene body promotes transcriptional elongation (Pavri et al, 2006; Kim et al, 2013). Our study reveals that global histone H2B degradation plays essential roles in gene expression and animal survival under reduced insulin/IGF signaling. We provide compelling evidence that human cells growing in nutrient‐rich environments also utilize the UPS system to exchange their histones (Fig 7 and Appendix Fig S7), though the underlying physiological importance is unclear. It is worth considering that the state of confluence in human cells might be more relevant than merely being well‐fed since confluence induces quiescence, resembling starvation. While the regulation of H2B levels in C. elegans may differ from mammalian cells, our results emphasize the widespread nature of UPS‐based global histone degradation.
The roles of global histone degradation in somatic tissues and spermatids must be different. In spermatids, the replacement of nucleosomal histones with protamines leads to a more condensed chromatin structure, which is essential for improving sperm motility and providing DNA protection during sperm development (Oliva, 2006; Qian et al, 2013). In starved somatic cells, the process of replacing replication‐dependent H2B isoforms with the replication‐independent variant might not significantly alter chromatic architecture or nuclear morphology to the same extent as protamines do in spermatids. Instead, the aberrant retention of chromosomal H2B in somatic cells recruits pioneer factors, such as DAF‐16, which induces an abnormal transcription program detrimental to animal survival under stress conditions. Therefore, nuclear UPS‐dependent global histone degradation maintains the appropriate level of chromosomal histones and regulates gene expression in somatic cells.
Global nucleosomal histone degradation can facilitate the systematic exchange of the epigenome, allowing for efficient switches in transcriptional programs that initiate organismal adaptation to environmental changes. Both C. elegans and human nucleosomes possess additional ubiquitination sites, such as K37 in H2A, K56, K79, K122 in H3, and K31 in H4 (Appendix Fig S4C and D; Mattiroli & Penengo, 2021), some of which may function similarly to K31 in H2B degradation. These multiple ubiquitination sites around the histone core can promote histone eviction, accelerating the dynamic changes in the epigenome. If these post‐translationally modified histones are not degraded after ejection, they may be reincorporated into nucleosomes, potentially not into the original position, thereby scrambling the epigenome. Moreover, chromatin‐unbound histones are potentially harmful due to their abnormally high content of basic amino acids (Singh et al, 2009), and their degradation prevents cellular toxicity. Thus, proper histone degradation is essential for maintaining the appropriate chromatin landscape, allowing for precise regulation of gene expression and ultimately ensuring the survival and adaptation of the organism to changing environmental conditions.
Isolating cells of the same type from C. elegans poses a significant challenge, making it a daunting task to accurately depict the precise nucleosome composition and chromatin landscape in a specific region. However, our imaging and biochemistry data demonstrate the replacement and degradation of all H2B variants during development and under starvation stress (Figs 1 and 8, and Appendix Fig S8). Notably, the replication‐dependent his‐4 was largely replaced by the replication‐independent his‐41 in adult and starved animals, indicating significant changes in chromatic composition. Future studies will develop tools to gain a more in‐depth understanding of the specific changes of nucleosome composition and the epigenome in C. elegans.
We have observed that under conditions of starvation, the replication‐independent histone H2B HIS‐41 increases its abundancy by about two‐fold, as revealed by Western blot analysis. In contrast, the other H2Bs show more substantial decreases in their protein levels. One possibility is that starvation causes an overall decrease in the total H2B protein level. Alternatively, HIS‐41 might be very abundant, possibly even the most abundant of the H2Bs in fed larvae. In such a scenario, a relatively modest increase in its abundance could compensate for the significant decreases in other isoforms. In support of the latter idea, replication‐dependent histones, which are produced in a cell cycle‐regulated manner, provide the histones to support DNA replication. Once this process is complete, these histones are no longer produced. Although all the examined replication‐dependent H2B proteins undergo degradation, the regulation of HIS‐41 protein level appears to be unique in response to starvation, suggesting that a generalized global degradation of H2B may not be applicable. Particularly concerning HIS‐41 protein levels, it remains elusive whether nutrient deprivation, and consequently insulin/IGF signaling, are critical physiological parameters in this regulation.
The nuclear localization of E2 and E3 and the ubiquitination of nucleosomal histones suggest a direct action on chromatin‐bound histones. Recent cryo‐electron tomography studies have detected 26S proteasomes in the nuclei (Yasuda et al, 2020), and the proteasome interacts with transcriptionally active chromatin (Geng & Tansey, 2012). While it remains possible that nuclear proteasomes might degrade histones while still bound to chromatin, in vitro reconstitution experiments have demonstrated that monoubiquitination of H2BK31 is sufficient to evict the H2A‐H2B dimer from the nucleosome (Krajewski et al, 2018). Therefore, we propose a two‐step model for proteasome‐based nucleosomal histone elimination: (i) the nuclear ubiquitin ligation system installs mono‐ubiquitin onto the chromatin‐bound H2B at the K31 site, leading to disassembly of the nucleosome; (ii) subsequently, the evicted histones are further polyubiquitinated and degraded by proteasomes. This model provides insights into the mechanism through which nucleosomal histones are turned over and regulated by the proteasome machinery within the nucleus.
Histone degradation is facilitated by additional E2 and E3 enzymes, as evidenced by the fact that the putative null alleles of ubc‐20 and hecd‐1 in C. elegans did not completely block H2B degradation, and the UBE2K or HECTD1 knockout only partially reduced histone turnover in HEK293T cells (Figs 3F and 7, and Appendix Fig S7). Notably, the human E3 ligase MSL2 adds polyubiquitin chains to H2BK31, although its precise physiological impact remains unclear (Wu et al, 2011). Given that the C. elegans genome encodes 22 E2 and 171 E3 genes (Kipreos, 2005), it is likely that different E2 or E3 enzymes are involved in attaching ubiquitin to the nucleosomes at different genomic regions within the same cell or to all nucleosomes but in different cell types. This diversity of E2 and E3 enzymes highlights the complexity of the ubiquitin‐proteasome system and its role in regulating histone turnover and gene expression in various cellular contexts.
Additional signals can induce histone degradation. For instance, hyperosmotic stress on the human HCT116 colon cancer cell line causes the ubiquitination of the linker histone H1 (Yasuda et al, 2020). During mitotic exit, the APC/C complex mediates histone degradation to upregulate pluripotency gene expression, thereby coordinating transcription changes with cell division regulation (Oh et al, 2020). In post‐mitotic neurons, proteasome degradation‐associated histone turnover plays a crucial role in exchanging the histone H3.3 variant, which is essential for neuronal plasticity and cognition in the mammalian brain (Maze et al, 2015).
Our observation of global H2B degradation relied on the “pulse‐chase” histone labeling strategy, where the epidermal‐specific promoter Pwrt‐2 expressing H2B turns off in starved animals (Fig 2D). The full inventory of GFP::H2B knock‐in C. elegans provides unprecedented resolution at the single‐isoform level, allowing us to track all individual H2B proteins in a metazoan genome. This level of specificity offers great potential to elucidate the precise roles of each H2B isoform in cellular processes and how they respond to different environmental stimuli.
Mutations in UBE2K or HECTD1 have been associated with Alzheimer's disease or congenital heart disease (Song & Jung, 2004; Li et al, 2015), suggesting that disease progression may involve UPS‐dependent nucleosomal histone degradation. The dysregulation of histone turnover could potentially play a role in the pathogenesis of these diseases. In combination with pulse‐chase labeling and genome‐wide histone isoform tagging, future studies will undoubtedly yield valuable insights into systematic nucleosomal histone exchange in somatic tissues across species under specific developmental, physiological, or pathological conditions.
Materials and Methods
Caenorhabditis elegans maintenance, DNA manipulation, and CRISPR‐Cas9 genome editing
Strains were maintained on Nematode Growth Medium (NGM) plates seeded with E. coli OP50 at 20°C (unless otherwise stated), following the standard protocols (Brenner, 1974). The wild‐type strain was Bristol N2. Some strains were provided by the Caenorhabditis Genetics Center (CGC), which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440).
We generated genome‐edited animals using the established CRISPR‐Cas9 strategy (Dickinson et al, 2013). The target sequence conforms to G(N)N19NGG (N = A, C, T, or G). Dataset EV8 lists the targeting sequences used in this study. Target sites in the templates were modified with synonymous mutations. For knock‐in point mutations, we constructed the mutation sites with synonymous mutations to introduce specific restriction enzyme recognition sites on the homologous recombination (HR) templates using the In‐Fusion Advantage PCR cloning kit. The genome‐editing donor plasmids were constructed by amplifying the 1.5 kb upstream and downstream homology arms from N2 genomic DNA and inserting them into the pPD95.77 vector using the In‐Fusion HD Cloning Kit (Clontech, Cat# 639619). Dataset EV8 summarizes primers, plasmids, and strains used in this study.
CRISPR‐Cas9 constructs and HR templates were purified using the AxyPrep Plasmid Purification Miniprep Kit (Axygen, Cat# AP‐MN‐P‐250) and PureLink Quick PCR purification Kit (Invitrogen, Cat# K310001). The sgRNA plasmids, donor plasmids, and the rol‐6(su1006) and Podr‐1::dsRed selection markers were co‐injected into the gonads of young adult worms. F1 transgenic progeny were singled and screened by PCR using the EasyTaq® 2× Super Mix (TransGen Biotech, Cat# AS111‐14). We performed an additional restriction enzyme digestion step before gel electrophoresis in our screen for knock‐in animals carrying the point mutation. We verified all the variations by Sanger sequencing of the entire genes and confirmed that no other mutations were inserted in the target genes. To generate GFP‐tagged H2B knock‐in animals, we sequenced all the H2B genes in each edited worm after backcrossing them with the wild‐type N2 worm. After screening several knock‐in lines, we obtained precisely edited strains that do not mutate other H2B genes.
Genetic mapping and gene cloning
Using single nucleotide polymorphisms (SNPs), we mapped cas924 (ubq‐1 allele) at −3.86 to 1.66 cM on chromosome III, cas905 (uba‐1 allele) at 3.49 to 7.46 cM on chromosome IV, cas901 (ubc‐20 allele) to the left of −20.16 cM on chromosome III, and cas908 (hecd‐1 allele) at 1.78 to 7.46 cM on chromosome IV. We identified mutations using whole‐genome sequencing and confirmed gene cloning using multiple alleles.
Cell culture, DNA manipulation, and transfection
HEK293T cells were obtained from the American Type Culture Collection (ATCC) and were authenticated by morphology and SV40 large T antigen testing. HEK293T cells were grown at 37°C under 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) (Corning, Cat# 10‐013‐CV) supplemented with 10% FBS (HyClone, Cat# WS500T), 50 units/ml penicillin, and 50 μg/ml streptomycin (HyClone, Cat# SV30010). UBE2K or HECTD1 knockout in HEK293T cells was generated according to the established CRISPR‐Cas9 method (Ran et al, 2013) and confirmed by PCR, Sanger sequencing, and Western blot. The targeting sequences of UBE2K and HECTD1 are listed in Dataset EV8. All the cells were regularly tested to be mycoplasma‐free.
The C. elegans H2B (HIS‐41) coding sequence was amplified and cloned into the pcDNA3.1 vector, which encodes an N‐terminal EGFP tag, using the In‐Fusion HD Cloning Kit (Clontech). The H2BK31A mutation was introduced into pcDNA3.1‐EGFP‐H2B by site‐directed mutagenesis. Transfection was performed in a 100 mm cell culture dish when the cell confluence reached 50% using the Lipofectamine 3000 reagent (Invitrogen, Cat# L3000‐015). Primers and plasmids are listed in Dataset EV8.
Starvation stress assay
We transferred the isolated eggs from adult worms onto NGM plates with an OP50 bacterial lawn. After 12 h, we collected the hatched larvae (the L1 larval stage, referred to as “well‐fed worms”) and washed them with M9 buffer to remove the bacteria. Half of these worms were transferred to NGM plates without OP50 bacterial lawn and kept starved for 6 days, referred to as “starved worms”.
RNA isolation and real‐time PCR
The “well‐fed” and “starved” worms were washed off the plates using M9 buffer and resolved in 0.4 ml TRIzol (Invitrogen, Cat# 15596018). Total RNAs were extracted according to the manufacturer's protocol. SUPERase In™ RNase Inhibitor (Invitrogen, Cat# AM2696) was used in each step to prevent RNA degradation. RNA concentration was quantified using the Qubit RNA High Sensitivity Assay Kit (Invitrogen, Cat# Q32855). The cDNAs were synthesized using the RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Cat# K1622). Gene expression levels were determined by real‐time PCR using PowerUp™ SYBR™ Green Master Mix (Applied Biosystems™, Cat# A25742) and the QuantStudio™ 1 Real‐Time PCR System (Thermo Fisher Scientific). Data were analyzed using the standard curve method, and relative gene expressions were normalized to the rps‐4 gene. In each experiment, we analyzed three biological replicates and three technical replicates. The primers used in the Real‐Time PCRs are shown in Dataset EV8.
Dauer formation and larval arrest assays
Caenorhabditis elegans young adult worms were allowed to lay eggs at 20°C for 2 h on NGM plates with an OP50 bacterial lawn. We counted the eggs and transferred the plates to 25°C for 3 days. Dauer, adult, and larval arrest animals were counted based on their morphology. The age‐1(hx546) allele‐associated mutants were tested at 27°C.
Immunoprecipitation and Western blotting
We performed affinity purification of C. elegans nucleosomes using an established protocol (Ooi et al, 2010). Briefly, GFP::HIS‐4 or HIS‐41::GFP knock‐in worms were cultured at 20°C on two hundred 60‐mm NGM plates seeded with OP50 bacteria until starved for 1 day. Starved animals were harvested, 4 ml of packed worms were mixed with 4 ml of lysis buffer [15 mM Tris–HCl pH 7.5, 2 mM MgCl2, 0.34 mM sucrose, 0.15 mM spermine and 0.5 mM spermidine, 0.25% NP‐40, 0.1% Triton X‐100, 1 mM DTT, 1 mM PMSF, 1× Protease inhibitor cocktail (Roche, 04693159001), 1× Phosphatase inhibitor cocktail (Roche, 04906845001)] and 4 ml of 0.5‐mm diameter glass beads. The mixture was lysed using FastPrep‐24 (MP Biomedicals) at 6.5 m/s, 15 s/pulse, 5 pulses with 5‐min intervals on ice. We pelleted the crude nuclei at 4,000 rpm for 10 min, resuspended them in 5 ml digestion buffer (20 mM Tris–HCl pH 7.5, 2 mM MgCl2, 0.2% Triton X‐100, 2 mM CaCl2, 1× Protease inhibitor cocktail), and incubated them at 37°C for 30 min with 20 μl of 50 U/μl MNase (Sigma, Cat# N3755). We pelleted the digested nuclei by spinning them at 1,503 g and resuspended them in 10 ml of nuclear co‐immunoprecipitation lysis buffer (20 mM Tris–HCl pH 7.5, 5% glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 0.5% NP‐40, 1× EDTA‐free protease inhibitor cocktail, 1× Protease inhibitor cocktail). The mixture was incubated on a rocker at 4°C for 4 h. We collected the supernatant after spinning them at 12,000 g for 30 min at 4°C. The supernatant was mixed with the GFP‐antibody conjugated beads (GFP‐Trap beads, ChromoTek, Cat# GTA‐20) at 4°C for 1 h. The GFP‐antibody conjugated beads were washed three times with the nuclear co‐immunoprecipitation wash buffer (20 mM Tris–HCl pH 7.5, 12.5% glycerol, 0.15 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 0.1% NP‐40, 1× EDTA‐free protease inhibitor cocktail, 1× Protease inhibitor cocktail), and boiled in 1× SDS loading buffer for SDS–PAGE gel analysis.
Worm immunoblotting was performed using an established protocol (Segref & Hoppe, 2012). Briefly, well‐fed and starved worms were collected and resuspended in 1× SDS loading buffer. We heated them at 95°C for 5 min, sonicated the lysate twice for 30 s each time, and heated them at 95°C for 5 min again. We performed SDS–PAGE and immunoblotting using an anti‐GFP antibody (Abcam, Cat# ab290) for GFP‐labeled H2B, anti‐H2BK120Ub (Cell Signaling, Cat# 5546S) for ubiquitinated Histone H2B (Lys120), anti‐H4 antibody (Abcam, Cat# ab10158) for endogenous H4, and anti‐tubulin antibody (Abcam, Cat# ab6161) for tubulin.
To detect H2B and H2BK31A ubiquitination, we transiently co‐transfected HEK293T cells with constructs encoding EGFP::HIS‐41(H2B) or EGFP::HIS‐41K31A into wild‐type, UBE2K‐KO, or HECTD1‐KO cells. Transfected cells were treated with 1 μM MG‐132 (Sigma, Cat# M7449) for 16 h and harvested. Nuclei were collected and washed three times in the hypotonic buffer (20 mM Tris–HCl pH 7.5, 10 mM NaCl, 5 mM MgCl2, supplemented with 1× EDTA‐free protease inhibitor cocktail), sonicated, and lysed in the nuclear co‐immunoprecipitation lysis buffer (20 mM Tris–HCl pH 7.5, 5% glycerol, 0.42 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 0.5% NP‐40, 1× EDTA‐free protease inhibitor cocktail). Cleared lysates were incubated with GFP‐antibody conjugated beads (GFP‐Trap Agarose beads, ChromoTek, Cat# GTA‐20), followed by extensive washing in the nuclear co‐immunoprecipitation wash buffer (20 mM Tris–HCl pH 7.5, 12.5% glycerol, 0.15 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 0.1% NP‐40, 1× EDTA‐free protease inhibitor cocktail). Beads were boiled with 1× SDS loading buffer before analysis by SDS–PAGE and immunoblotting using an anti‐K48 linked polyubiquitination antibody (Cell Signaling, Cat# 8081S) for H2B or H2BK31A ubiquitination, anti‐GFP antibody (Abcam, Cat# ab290) for EGFP‐labeled H2B and H2BK31A, anti‐HIP2 antibody (Cell Signaling, Cat# 8226S) for endogenous UBE2K, anti‐HECTD1 antibody (Abcam, Cat# ab101992) for endogenous HECTD1, and anti‐GAPDH antibody (Cell Signaling, Cat# 2118S) for GAPDH.
Chromatin immunoprecipitation
Chromatin immunoprecipitation was performed as previously described (Zhong et al, 2010) with modifications. The daf‐2(e1370) allele‐associated mutants were semi‐synchronized to the L1 larval stage at 20°C and then transferred to 25°C for 24 h. We harvested and washed them three times in M9 buffer and washed them twice in phosphate‐buffered saline (PBS) (Gibco, Cat# C10010500BT). Collected worms were fixed in the crosslinking buffer (1% formaldehyde (Sigma, Cat# 252549) in PBS with 1× EDTA‐free protease inhibitor cocktail) for 15 min at room temperature, quenched with 0.125 M glycine, and washed in the cold PBS buffer with EDTA‐free protease inhibitors three times. Fixed worms were resuspended in ice‐cold FA buffer (50 mM HEPES‐KOH pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% sodium deoxycholate, 1% Triton X‐100, 1 mM PMSF, and 1× EDTA‐free protease inhibitor cocktail) with 0.1% SDS, and sonicated using a Covaris S220 at the following settings: 200 W Peak Incident Power, 20% Duty Factor, 200 Cycles per Burst for 8 min. Samples were transferred to centrifuge tubes and spun at 18,407 g for 15 min at 4°C. The supernatant was transferred to a new tube, and 5% of the material was saved as input and stored at −20°C. The remainder was incubated with GFP‐antibody conjugated beads (GFP‐Trap Dynabeads, ChromoTek, Cat# GTA‐20) at 4°C overnight. The beads were then washed at 4°C twice with 150 mM NaCl FA buffer (5 min each) and once with 1 M NaCl FA buffer (5 min). The beads were transferred to a new centrifuge tube and washed twice with 500 mM NaCl FA buffer (10 min each), once with TEL buffer (0.25 M LiCl, 1% NP‐40, 1% sodium deoxycholate, 1 mM EDTA, 10 mM Tris–HCl pH 8.0) for 10 min, and twice with TE buffer (5 min each). The immunocomplex was then eluted in 200 μl elution buffer (1% SDS in TE with 250 mM NaCl) by incubating the tube at 65°C for 30 min, with a brief vortex every 15 min. We repeated this step and combined the elution as “ChIP Sample”. The saved input samples were thawed. We added 2 μl 10 mg/ml RNase A to the input and ChIP sample, and digested RNA at 37°C for 1 h. We added 2 μl 20 mg/ml Proteinase K and incubated them at 65°C overnight to digest protein and reverse crosslink. According to the manufacturer's instructions, we purified the immunoprecipitated DNA with AMPure XP beads (Beckman, Cat# A63881).
ChIP‐seq library preparation and data analysis
To prepare the ChIP‐seq library, we used 50 ng of purified DNA samples. According to the manufacturer's recommendations, library construction was performed using the KAPA Hyper Prep Kit (Kapa Biosystems, Cat# KR0961). DNA libraries were sequenced on an Illumina HiSeq instrument with 150‐bp paired‐end sequencing. Two batches of worms were collected for each ChIP‐seq experiment. Raw reads were trimmed by cutadapt (version 1.18) with parameter ‐m 20 ‐e 0.1 ‐O 3 ‐q 20 ‐‐quality‐base = 33, removing the low‐quality bases and adaptor sequences. Clean reads were aligned to the C. elegans reference genome (ce10) using bowtie2 aligner (version 2.3.3.1) with parameters ‐N 1 ‐L 25 ‐‐no‐mixed ‐‐no‐discordant. Anomalous genomic regions were blacklisted by the ENCODE blacklist. Duplicates were labeled by Picard MarkDuplicates (version 2.20.4) and removed by the samtools view –F 1024. Biological replicates for each ChIP‐seq were merged in the following analysis. The binding sites of transcriptional factors (TFs) and histones were identified by the MACS2 call peak (version 2.1.2). Histone peaks were identified with parameter ‐‐broad ‐g ce ‐‐extsize 10000 ‐nomodel ‐p 0.1. TF peaks were identified with auto parameters. To normalize the ChIP control, we generated bigwig files using the deeptools (version 3.1.3) bamCompare. ChIP signal out of peak region was assumed to be equal in histone ChIP‐seq. The background noise factor in each histone ChIP‐seq data was calculated by averaging ChIP signal per bin (10 bp) out of peak, and histone ChIP signal in all the bins was divided by the background noise factor. Heatmap and linear map plot were generated using deeptools (version 3.1.3).
RNA‐seq library preparation and data analysis
We prepared the RNA‐seq library using the following protocol. The samples were prepared as described in the “Starvation Stress Assay” section. We collected other samples for RNA‐seq as follows: young adult worms laid eggs at 20°C for 4 h on NGM plates with OP50 bacterial lawn. We then removed the adult worms and transferred the plates to 25°C for 24 h. The hatched or arrested worms were washed off the plates using M9 buffer and resolved in 0.4 ml TRIzol (Invitrogen). Total RNAs were extracted as described in the “RNA Isolation and Real‐Time PCR” section. RNA quality was assessed with the Agilent 2100 bioanalyzer system, and samples with an RNA integrity number (RIN) above 6.0 were used to construct the library. 100 to 500 ng of the total RNA was used for library preparation using the KAPA RNA HyperPrep Kit (KAPA Biosystems, Cat# KR1352). Library samples were analyzed by Agilent 2100 bioanalyzer system and Qubit for quality control and quantification. The samples were sequenced on an Illumina HiSeq platform with 150‐bp paired‐end sequencing. Three batches of worms were collected for mRNA sequencing.
The resulting raw reads of RNA‐seq were assessed for quality, adaptor content, and duplication rates with FastQC. The raw sequencing reads were trimmed using Trim_galore (version 0.4.4) to remove the low‐quality bases and adaptor sequences. Paired‐end reads with at least 20 nucleotides in length were aligned to C. elegans reference genome (ce10) using STAR (2.5.4b) with the parameter ‘‐sjdbOverhang 139’. The numbers of reads aligned to genes were quantified by HTSeq (version 0.9.1). Only uniquely mapped reads were used to calculate the relative expression level of the gene. Reads overlapping with multiple genes or aligning to multiple regions were excluded. Gene names were annotated on the DAVID website (https://david.ncifcrf.gov/). Differentially expressed genes were identified using the DESeq2 package in the R programming language. Differentially expressed genes were defined with the following criteria: upregulated genes (false discovery rate (FDR) less than 0.05, log2‐transformed fold change greater than 1, unnormalized counts from HTSeq greater than 3); downregulated genes (FDR less than 0.05, log2‐transformed fold change less than −1, and unnormalized counts from HTSeq greater than 3). Metascape (http://metascape.org/gp/index.html) was used to analyze gene enrichment terms in upregulated or down‐regulated genes (Zhou et al, 2019). The R packages of Corrplot, heatmap, and VennDiagram were used to analyze data and plot figures.
Stable isotope labeling by amino acids in cell culture (SILAC) assay
HEK293T (wild‐type, UBE2K‐KO, and HECTD1‐KO) cells were cultured at 37°C under 5% CO2 in RPMI 1640 (ThermoFisher, Cat# 88365) supplemented with 10% FBS (HyClone), 50 units/ml penicillin, and 50 μg/ml streptomycin (HyClone), 0.1 mg/ml Lysine (ThermoFisher, Cat# 89987), 0.1 mg/ml Arginine (ThermoFisher, Cat# 89989). To minimize the influence of the cell cycle, we exchanged the medium until the cell confluence reached 100%. We added the fresh RPMI 1640 (ThermoFisher) supplemented with 10% FBS (HyClone), 50 units/ml penicillin, and 50 μg/ml streptomycin (HyClone), 0.1 mg/ml 13C6 15N2‐lysine (ThermoFisher, Cat# 88209), 0.1 mg/ml Arginine (ThermoFisher). Cells were collected and washed with PBS (Gibco) at the following time points: 0, 12, and 24 h.
Acid extraction of the total histone
Chromatin extraction was performed as previously described (Maze et al, 2015). Briefly, pelleted HEK293T cells (~ 107) were resuspended in 200 μl buffer A (10 mM Tris–HCl pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 0.1% Triton X‐100, 1× Protease inhibitor cocktail, 1× Phosphatase inhibitor cocktail) for 10 min at 4°C before being centrifuged at 1,500 g for 5 min to separate the supernatant (cytosol) from the pellet (nuclei). We performed an additional wash of the nuclear pellet with buffer A. To minimize sample complexity, we suspended the pelleted chromatin in 0.4 M H2SO4 and incubated it on a rotator for 1 h at 4°C. After incubation, samples were centrifuged at 16,000 g for 10 min to remove debris. Supernatant histones were precipitated with 1/3 volume of trichloroacetic acid (TCA) (Sigma, Cat# T0699) overnight at 4°C. Histones were then pelleted at 16,000 g for 10 min at 4°C. After removing the supernatant, the pellets were washed twice with ice‐cold acetone to remove acids from the solution without dissolving the histone. After washing, histones were air‐dried and resuspended in nuclease‐free water for mass spectrometry analyses.
Mass spectrometry analysis
To identify putative histone ubiquitination sites in C. elegans, we separated the purified C. elegans nucleosomes by SDS–PAGE and visualized them by Coomassie blue staining. Individual histone bands were cut, reduced, alkylated, propionylated, and digested with trypsin overnight. The resulting tryptic peptides were analyzed using an UltiMate 3000 RSLC nano System (Thermo Scientific) which was directly interfaced with a Thermo Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific). The putative histone ubiquitination sites were searched against the selected database using an in‐house PEAKS searching algorithm and manually validated.
We analyzed histone turnover in HEK293T cells. The total histone extracted from the SILAC assay was reduced, alkylated with Tris(2‐carboxyethyl) phosphine (TCEP) and chloroacetamide, digested with LysC (Wako) and trypsin (Promega) overnight at 37°C, and desalted by Sep‐Pak tC18 columns. The eluted peptides were dried by a speedvac and reconstituted in water with 0.1% formic acid. An UltiMate™ 3000 RSLCnano system, directly interfaced with a Q Exactive HF‐X mass spectrometer, was used for LC–MS/MS analysis. Peptides were loaded to a trap column (75 μm × 20 mm, 3 μm C18100 Å, 164535, Thermo Fisher Scientific) with a max pressure of 620 bar using mobile phase A (0.1% formic acid in H2O) and separated on an analytical column (75 μm × 500 mm, 3 μm C18,100 Å, 164570, Thermo Fisher Scientific) with a gradient of 8–60% mobile phase B (80% acetonitrile and 0.08% formic acid) at a flow rate of 250 nl/min for 120 min. The MS analysis was operated in data‐dependent acquisition (DDA) mode, with one full scan (300–1,800 m/z, R = 60,000 at 200 m/z) at automatic gain control (AGC) of 3e6, followed by top 40 MS/MS scans with high energy collision dissociation (maximum injection time (IT) 100 ms, isolation window 2 m/z, normalized collision energy of 27%). The MS/MS data were searched against the homemade histone database (54 sequences from Uniprot) using the SEQUEST searching engine in Proteome Discoverer 2.3 software (PD2.3). The search criteria included: complete tryptic specificity was required; two missed cleavages were allowed; carbamidomethylation (C) was set as the fixed modifications; oxidation (M) and label 13C6 15N2‐Lys were set as the variable modification; precursor ion mass tolerances were set at 20 ppm for all MS acquired in an Orbitrap mass analyzer, and the fragment ion mass tolerance was set at 20 mmu for all the acquired MS2 spectra. The peptide false discovery rate was calculated using Fixed Validator provided by PD2.3. Relative protein quantification was performed using PD2.2 based on the intensities of heavy and light peptides and confirmed manually.
Microscopy and image analysis
Worms were anesthetized with 1 mg/ml levamisole and mounted on 3% agarose pads. The immobilized larvae were imaged with an Axio Observer Z1 microscope (Carl Zeiss MicroImaging, Inc.) equipped with a 100×, 1.45N.A. objective, an EM CCD camera (Andor iXon+ DU‐897D‐C00‐#BV‐500), and the 488 and 568 nm lines of a Sapphire CW CDRH USB Laser System attached to a spinning disk confocal scan head (Yokogawa CSU‐X1 Spinning Disk Unit). Images were acquired by μManager (https://www.micro‐manager.org) with identical settings. After background fluorescence subtraction, we used ImageJ software (http://rsbweb.nih.gov/ij/) to measure and analyze the GFP‐labeled H2B fluorescent intensity.
Data reporting and statistics
No statistical methods were used to predetermine the sample size. The sample sizes in our experiments were determined from early publications. The experiments were not randomized. We used GraphPad Prism 7 (GraphPad Software, Inc.) for statistical analyses. Independent Student's t‐tests were performed to compare the mean values between two groups. The variance of graphs is means ± SEM. N refers to the number of worms or independent experiments. The significance of statistical differences is indicated as: *P < 0.05; **P < 0.01; ***P < 0.001.
Author contributions
Zhiwen Zhu: Conceptualization; data curation; formal analysis; funding acquisition; validation; investigation; methodology; writing – original draft; writing – review and editing. Dongdong Li: Data curation; funding acquisition; validation; investigation; methodology; writing – original draft; writing – review and editing. Zeran Jia: Data curation; validation; writing – original draft. Wenhao Zhang: Data curation; validation; methodology; writing – original draft. Yuling Chen: Data curation; validation; methodology; writing – original draft. Ruixue Zhao: Resources. Yan‐Ping Zhang: Resources. Wen‐Hong Zhang: Resources. Haiteng Deng: Supervision; validation. Yinqing Li: Funding acquisition; validation. Wei Li: Funding acquisition; validation. Shouhong Guang: Supervision; validation. Guangshuo Ou: Conceptualization; data curation; formal analysis; supervision; funding acquisition; validation; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interest statement
The authors declare no competing interests.
Supporting information
Appendix
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Dataset EV5
Dataset EV6
Dataset EV7
Dataset EV8
Source Data for Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 7
Source Data for Figure 8
Acknowledgements
We thank Drs. G. Li, X. Shen, X. Yang, B. Zhu, W. Xie, F. Lai, Y. Dang, Q. Li, M. Wang, D. Chen, and S. Cai for discussion and comments, and ChatGPT for improving the language of the manuscript. This study was supported by the National Key R&D Program of China (2022YFA1302700, 2019YFA0508401, 2019YFA09004400, 2019YFA09004402), the National Natural Science Foundation of China (grants 31991190, 31730052, 31525015, 31861143042, 31561130153, 31671444, 31871352, 32100538, 32171448, and 31900535), and the China Postdoctoral Science Foundation (2022M711844), and the Beijing Natural Science Foundation (Z210010).
The EMBO Journal (2023) 42: e113328
Data availability
All data is available in the main text or supplementary materials. All sequencing data from this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) with BioProject accession number PRJNA786721 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA786721).
References
- Baugh LR, Sternberg PW (2006) DAF‐16/FOXO regulates transcription of cki‐1/Cip/Kip and repression of lin‐4 during C. elegans L1 arrest. Curr Biol 16: 780–785 [DOI] [PubMed] [Google Scholar]
- Baugh LR, Demodena J, Sternberg PW (2009) RNA Pol II accumulates at promoters of growth genes during developmental arrest. Science 324: 92–94 [DOI] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans . Genetics 77: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli G, Heard E (2019) Advances in epigenetics link genetics to the environment and disease. Nature 571: 489–499 [DOI] [PubMed] [Google Scholar]
- Challa K, Schmid CD, Kitagawa S, Cheblal A, Iesmantavicius V, Seeber A, Amitai A, Seebacher J, Hauer MH, Shimada K et al (2021) Damage‐induced chromatome dynamics link Ubiquitin ligase and proteasome recruitment to histone loss and efficient DNA repair. Mol Cell 81: 811–829.e6 [DOI] [PubMed] [Google Scholar]
- Cheblal A, Challa K, Seeber A, Shimada K, Yoshida H, Ferreira HC, Amitai A, Gasser SM (2020) DNA damage‐induced nucleosome depletion enhances homology search independently of local break movement. Mol Cell 80: 311–326.e4 [DOI] [PubMed] [Google Scholar]
- Deal RB, Henikoff JG, Henikoff S (2010) Genome‐wide kinetics of nucleosome turnover determined by metabolic labeling of histones. Science 328: 1161–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson DJ, Ward JD, Reiner DJ, Goldstein B (2013) Engineering the Caenorhabditis elegans genome using Cas9‐triggered homologous recombination. Nat Methods 10: 1028–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsherbiny A, Dobreva G (2021) Epigenetic memory of cell fate commitment. Curr Opin Cell Biol 69: 80–87 [DOI] [PubMed] [Google Scholar]
- Geng F, Tansey WP (2012) Similar temporal and spatial recruitment of native 19S and 20S proteasome subunits to transcriptionally active chromatin. Proc Natl Acad Sci USA 109: 6060–6065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgi LL, Albert PS, Riddle DL (1990) daf‐1, a C. elegans gene controlling dauer larva development, encodes a novel receptor protein kinase. Cell 61: 635–645 [DOI] [PubMed] [Google Scholar]
- Goldknopf IL, Busch H (1977) Isopeptide linkage between nonhistone and histone 2A polypeptides of chromosomal conjugate‐protein A24. Proc Natl Acad Sci USA 74: 864–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman JA, Kuzu G, Lee N, Karasik J, Gemberling M, Foglia MJ, Karra R, Dickson AL, Sun F, Tolstorukov MY et al (2017) Resolving heart regeneration by replacement histone profiling. Dev Cell 40: 392–404.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatta M, Cirillo LA (2007) Chromatin opening and stable perturbation of core histone: DNA contacts by FoxO1. J Biol Chem 282: 35583–35593 [DOI] [PubMed] [Google Scholar]
- Hauer MH, Seeber A, Singh V, Thierry R, Sack R, Amitai A, Kryzhanovska M, Eglinger J, Holcman D, Owen‐Hughes T et al (2017) Histone degradation in response to DNA damage enhances chromatin dynamics and recombination rates. Nat Struct Mol Biol 24: 99–107 [DOI] [PubMed] [Google Scholar]
- Henderson ST, Johnson TE (2001) daf‐16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans . Curr Biol 11: 1975–1980 [DOI] [PubMed] [Google Scholar]
- Jones D, Crowe E, Stevens TA, Candido EP (2002) Functional and phylogenetic analysis of the ubiquitylation system in Caenorhabditis elegans: ubiquitin‐conjugating enzymes, ubiquitin‐activating enzymes, and ubiquitin‐like proteins. Genome Biol 3: RESEARCH0002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonnalagadda S, Butt TR, Monia BP, Mirabelli CK, Gotlib L, Ecker DJ, Crooke ST (1989) Multiple (alpha‐NH‐ubiquitin)protein endoproteases in cells. J Biol Chem 264: 10637–10642 [PubMed] [Google Scholar]
- Keall R, Whitelaw S, Pettitt J, Muller B (2007) Histone gene expression and histone mRNA 3′ end structure in Caenorhabditis elegans . BMC Mol Biol 8: 51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim JA, McGinty RK, Nguyen UT, Muir TW, Allis CD, Roeder RG (2013) The n‐SET domain of Set1 regulates H2B ubiquitylation‐dependent H3K4 methylation. Mol Cell 49: 1121–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G (1997) daf‐2, an insulin receptor‐like gene that regulates longevity and diapause in Caenorhabditis elegans . Science 277: 942–946 [DOI] [PubMed] [Google Scholar]
- Kipreos ET (2005) Ubiquitin‐mediated pathways in C. elegans . WormBook 1–24 10.1895/wormbook.1.36.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski WA, Li J, Dou Y (2018) Effects of histone H2B ubiquitylation on the nucleosome structure and dynamics. Nucleic Acids Res 46: 7631–7642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Klena NT, Gabriel GC, Liu X, Kim AJ, Lemke K, Chen Y, Chatterjee B, Devine W, Damerla RR et al (2015) Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature 521: 520–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, Dorman JB, Rodan A, Kenyon C (1997) daf‐16: an HNF‐3/forkhead family member that can function to double the life‐span of Caenorhabditis elegans . Science 278: 1319–1322 [DOI] [PubMed] [Google Scholar]
- Lin K, Hsin H, Libina N, Kenyon C (2001) Regulation of the Caenorhabditis elegans longevity protein DAF‐16 by insulin/IGF‐1 and germline signaling. Nat Genet 28: 139–145 [DOI] [PubMed] [Google Scholar]
- Martire S, Banaszynski LA (2020) The roles of histone variants in fine‐tuning chromatin organization and function. Nat Rev Mol Cell Biol 21: 522–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzluff WF, Koreski KP (2017) Birth and death of histone mRNAs. Trends Genet 33: 745–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzluff WF, Gongidi P, Woods KR, Jin J, Maltais LJ (2002) The human and mouse replication‐dependent histone genes. Genomics 80: 487–498 [PubMed] [Google Scholar]
- Mattiroli F, Penengo L (2021) Histone ubiquitination: an integrative signaling platform in genome stability. Trends Genet 37: 566–581 [DOI] [PubMed] [Google Scholar]
- Maze I, Wenderski W, Noh KM, Bagot RC, Tzavaras N, Purushothaman I, Elsasser SJ, Guo Y, Ionete C, Hurd YL et al (2015) Critical role of histone turnover in neuronal transcription and plasticity. Neuron 87: 77–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C (2003) Genes that act downstream of DAF‐16 to influence the lifespan of Caenorhabditis elegans . Nature 424: 277–283 [DOI] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, Ruvkun G (1997) The Fork head transcription factor DAF‐16 transduces insulin‐like metabolic and longevity signals in C. elegans . Nature 389: 994–999 [DOI] [PubMed] [Google Scholar]
- Oh E, Mark KG, Mocciaro A, Watson ER, Prabu JR, Cha DD, Kampmann M, Gamarra N, Zhou CY, Rape M (2020) Gene expression and cell identity controlled by anaphase‐promoting complex. Nature 579: 136–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva R (2006) Protamines and male infertility. Hum Reprod Update 12: 417–435 [DOI] [PubMed] [Google Scholar]
- Ooi SL, Henikoff JG, Henikoff S (2010) A native chromatin purification system for epigenomic profiling in Caenorhabditis elegans . Nucleic Acids Res 38: e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavri R, Zhu B, Li G, Trojer P, Mandal S, Shilatifard A, Reinberg D (2006) Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell 125: 703–717 [DOI] [PubMed] [Google Scholar]
- Pettitt J, Crombie C, Schumperli D, Muller B (2002) The Caenorhabditis elegans histone hairpin‐binding protein is required for core histone gene expression and is essential for embryonic and postembryonic cell division. J Cell Sci 115: 857–866 [DOI] [PubMed] [Google Scholar]
- Qian MX, Pang Y, Liu CH, Haratake K, Du BY, Ji DY, Wang GF, Zhu QQ, Song W, Yu Y et al (2013) Acetylation‐mediated proteasomal degradation of core histones during DNA repair and spermatogenesis. Cell 153: 1012–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F (2013) Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 8: 2281–2308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle DL, Albert PS (1997) Genetic and environmental regulation of dauer larva development. In C. elegans II, Riddle DL, Blumenthal T, Meyer BJ, Priess JR (eds), 2nd edn. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; [PubMed] [Google Scholar]
- Riedel CG, Dowen RH, Lourenco GF, Kirienko NV, Heimbucher T, West JA, Bowman SK, Kingston RE, Dillin A, Asara JM et al (2013) DAF‐16 employs the chromatin remodeller SWI/SNF to promote stress resistance and longevity. Nat Cell Biol 15: 491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segref A, Hoppe T (2012) Analysis of ubiquitin‐dependent proteolysis in Caenorhabditis elegans . Methods Mol Biol 832: 531–544 [DOI] [PubMed] [Google Scholar]
- Singh RK, Kabbaj MH, Paik J, Gunjan A (2009) Histone levels are regulated by phosphorylation and ubiquitylation‐dependent proteolysis. Nat Cell Biol 11: 925–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song S, Jung YK (2004) Alzheimer's disease meets the ubiquitin‐proteasome system. Trends Mol Med 10: 565–570 [DOI] [PubMed] [Google Scholar]
- Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, Zaret KS (2015) Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161: 555–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamburri S, Lavarone E, Fernandez‐Perez D, Conway E, Zanotti M, Manganaro D, Pasini D (2020) Histone H2AK119 mono‐ubiquitination is essential for polycomb‐mediated transcriptional repression. Mol Cell 77: 840–856.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Wang L, Erdjument‐Bromage H, Vidal M, Tempst P, Jones RS, Zhang Y (2004) Role of histone H2A ubiquitination in polycomb silencing. Nature 431: 873–878 [DOI] [PubMed] [Google Scholar]
- Wenderski W, Maze I (2016) Histone turnover and chromatin accessibility: critical mediators of neurological development, plasticity, and disease. Bioessays 38: 410–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MH, Bonner WM (1980) Histone 2B can be modified by the attachment of ubiquitin. Nucleic Acids Res 8: 4671–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Zee BM, Wang Y, Garcia BA, Dou Y (2011) The RING finger protein MSL2 in the MOF complex is an E3 ubiquitin ligase for H2B K34 and is involved in crosstalk with H3 K4 and K79 methylation. Mol Cell 43: 132–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Long C, Chen X, Huang C, Chen S, Zhu B (2010) Partitioning of histone H3‐H4 tetramers during DNA replication‐dependent chromatin assembly. Science 328: 94–98 [DOI] [PubMed] [Google Scholar]
- Yasuda S, Tsuchiya H, Kaiho A, Guo Q, Ikeuchi K, Endo A, Arai N, Ohtake F, Murata S, Inada T et al (2020) Stress‐ and ubiquitylation‐dependent phase separation of the proteasome. Nature 578: 296–300 [DOI] [PubMed] [Google Scholar]
- Zhong M, Niu W, Lu ZJ, Sarov M, Murray JI, Janette J, Raha D, Sheaffer KL, Lam HY, Preston E et al (2010) Genome‐wide identification of binding sites defines distinct functions for Caenorhabditis elegans PHA‐4/FOXA in development and environmental response. PLoS Genet 6: e1000848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, Chanda SK (2019) Metascape provides a biologist‐oriented resource for the analysis of systems‐level datasets. Nat Commun 10: 1523 [DOI] [PMC free article] [PubMed] [Google Scholar]