

Abstract
The NLRP3 inflammasome plays a key role in responding to pathogens, and endogenous damage and mitochondria are intensively involved in inflammasome activation. The NLRP3 inflammasome forms multiprotein complexes and its sequential assembly is important for its activation. Here, we show that NLRP3 is ubiquitinated by the mitochondria‐associated E3 ligase, MARCH5. Myeloid cell‐specific March5 conditional knockout (March5 cKO) mice failed to secrete IL‐1β and IL‐18 and exhibited an attenuated mortality rate upon LPS or Pseudomonas aeruginosa challenge. Macrophages derived from March5 cKO mice also did not produce IL‐1β and IL‐18 after microbial infection. Mechanistically, MARCH5 interacts with the NACHT domain of NLRP3 and promotes K27‐linked polyubiquitination on K324 and K430 residues of NLRP3. Ubiquitination‐defective NLRP3 mutants on K324 and K430 residues are not able to bind to NEK7, nor form NLRP3 oligomers leading to abortive ASC speck formation and diminished IL‐1β production. Thus, MARCH5‐dependent NLRP3 ubiquitination on the mitochondria is required for NLRP3‐NEK7 complex formation and NLRP3 oligomerization. We propose that the E3 ligase MARCH5 is a regulator of NLRP3 inflammasome activation on the mitochondria.
Keywords: MARCH5, mitochondria, NEK7, NLRP3 inflammasome, ubiquitination
Subject Categories: Immunology, Organelles
The mitochondrial‐associated E3 ligase MARCH5 regulates NLRP3‐NEK7 complex formation on mitochondria and the activation of NLRP3 inflammasome.
Introduction
Inflammation is a vital physiological response against endogenous and exogenous noxious stimuli derived from pathogens and tissue damage (Takeuchi & Akira, 2010). Among sensor proteins, NLRP3 (NOD‐like receptor family pyrin domain containing 3) is characterized as a global immune‐sensor protein that recognizes a wide range of PAMP (pathogen‐associated molecular patterns) and DAMPs (damage‐associated molecular patterns) (Chen & Nunez, 2010), rather than a specific ligand (Ratsimandresy et al, 2013). Upon activation signal, NLRP3 interacts with an adapter protein, ASC (apoptosis‐associated speck‐like protein) and subsequently, pro‐caspase‐1 to form the large multiprotein complex of the NLRP3 inflammasome (Guo et al, 2015). Apparently, two sequential steps are needed for the formation of active NLRP3 inflammasome: priming and activation. The priming step involves transcriptional upregulation of TNF‐α, IL‐6, and NLRP3 genes as well as the cytokine precursor genes of pro‐IL‐1β and pro‐IL‐18. The priming step also involves post‐translational modifications (PTMs) of NLRP3, which converts it to signal‐competent state. Multiple PTMs on NLRP3 such as ubiquitylation and phosphorylation have been described (McKee & Coll, 2020). The activation step occurs following the recognition of diverse NLRP3 stimuli, including potassium efflux, ATP, nigericin and monosodium urate (MSU) and assembly of the NLRP3 inflammasome leads to caspase‐1‐dependnet release of mature IL‐1β and IL‐18 (Guo et al, 2015). Notably, none of the stimuli bind to NLRP3 directly (Kelley et al, 2019). Dysregulation or persistent NLRP3 signaling underlies various inflammatory diseases and autoimmune disorders (Fusco et al, 2020).
Mitochondria are intensely involved in inflammasome activation. Mitochondrial ROS (reactive oxygen species) induces the NLRP3 inflammasome activation (Zhou et al, 2011). And oxidized mitochondrial DNA released into the cytosol is also recognized by NLRP3 (Cruz et al, 2007; Shimada et al, 2012; Heid et al, 2013). In addition, new mitochondrial DNA synthesis is necessary for the production of oxidized mtDNA fragments that bind to NLRP3 (Zhong et al, 2018). Increasing pieces of evidence have shown how damaged mitochondrial DNA and RNAs are released into the cytosol and stimulate NLRP3 as well as other inflammatory signaling (Shimada et al, 2012; Tigano et al, 2021). NLRP3 is found in the cytoplasm, mitochondria, ER, and Golgi complex. The mitochondria‐associated adaptor molecule MAVS mediates the recruitment of NLRP3 to the mitochondria (Subramanian et al, 2013), whereas phosphatidylinositol‐4‐phosphate (PtdIns4P) is important for NLRP3 recruitment to the dispersed trans‐Golgi network (dTGN) (Chen & Chen, 2018). In addition, mouse NLRP3 and caspase‐1 independently interact with the mitochondrial lipid cardiolipin (Iyer et al, 2013). So far, it is unclear why mitochondrial translocation of NLRP3 is important for the NLRP3 inflammasome activation. Another important step for the NLRP3 activation is NLRP3 oligomerization. In the resting state, NLRP3 retains its folded structure hiding the pyrin domain, but it converts to the oligomers during activation. Accumulating evidences suggest that NLRP3 oligomerization requires its binding to NEK7 (NIMA‐related kinase 7), which forms a bridge between adjacent NLRP3 subunits (Sharif et al, 2019). It is still unknown how NLRP3‐NEK7 interaction is operated in cells.
MARCH5 (also known as MITOL) is an E3 ubiquitin ligase that locates on the mitochondrial outer membrane (Bauer et al, 2017). MARCH5 plays a central role in maintaining mitochondrial homeostasis by eliminating protein aggregates accumulated on the mitochondria (Yonashiro et al, 2006; Nagashima et al, 2014). MARCH5‐mediated ubiquitination on target proteins facilitates the ER‐mitochondria contact and protein import into the mitochondria (Sugiura et al, 2013; Takeda et al, 2019; Phu et al, 2020). Loss of MARCH5 induces cellular senescence or affects cell survival (Park et al, 2010; Shiiba et al, 2021). Its absence also aggravates neuronal pathogenesis (Takeda et al, 2021). In addition, MARCH5 serves as a positive regulator of TLR7 signaling (Shi et al, 2011), whereas it binds a huge complex of activated RIG‐I‐MAVS oligomers and degrades them to prevent persistent immune activation (Yoo et al, 2015; Park et al, 2020).
In this study, we show MARCH5 as an onset regulator of NEK7 binding to NLRP3. MARCH5 interacts with the NLRP3‐MAVS complex and transfers K27‐linked polyubiquitin to the NACHT domain of NLRP3. MARCH5‐mediated ubiquitination in the NACHT domain is required for NEK7 binding and NLRP3 oligomerization. Accordingly, macrophages derived from March5 cKO mice did not produce IL‐1β and IL‐18 after microbial infection. Our findings highlight the mitochondria as an onset platform for NEK7‐NLRP3 oligomeric complex formation.
Results
Myeloid cell‐specific March5 conditional knockout (March5 cKO) mice exhibit an attenuated mortality rate and inflammatory response to septic shock
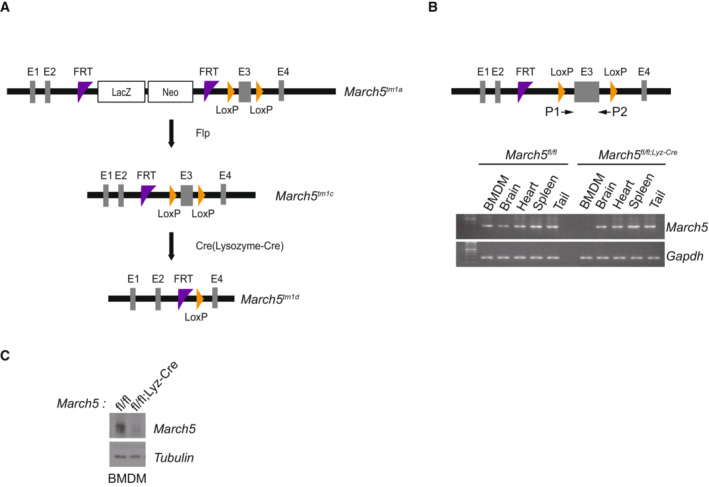
Mitochondria are involved in the cellular innate immune response against viruses. The mitochondrial outer membrane provides a surface platform for RLR signaling against the cytosolic viral RNA genome (Banoth & Cassel, 2018). Our previous studies revealed that MARCH5 degrades the active form of prion‐like MAVS aggregates and oligomerizes RIG‐I, preventing persistent immune reactions (Park et al, 2020). Here, we addressed whether MARCH5 has any immunoregulatory effects upon bacterial infection. We generated myeloid cell‐specific March5 knockout C57BL/6 mice to address this issue using the Cre‐loxP recombination system. To selectively delete the March5 gene in myeloid cells, we crossbred March5 fl/fl mice with Lys2‐Cre mice, deleting exon 3 of the March5 gene with Cre recombinase (Fig EV1A). We examined the mRNA levels using primers targeting exon 3 of the March5 gene in several tissues, namely the brain, heart, spleen, and tail as well as in bone marrow‐derived macrophages (BMDMs) derived from March5 fl/fl and March5 fl/fl;Lyz‐Cre (March5 cKO) mice. All tissues except BMDMs from March5 cKO expressed March5 mRNA (Fig EV1B). Similarly, BMDMs derived from March5 cKO mice did not express the MARCH5 protein (Fig EV1C).
Figure EV1. Generation of March5 fl/fl and March5 fl/fl; Lyz‐Cre mice.
-
AA schematic drawing of the March5 locus and targeting vector. C57BL/6 March5 tm1a mice harboring the FRT and Lox P sites. generates floxed March5 mice, Flp recombinase targets FRT sites (March5 tm1c ). For MARCH5 gene deletion, Cre recombinase targets Lox P sites (March5 tm1d ).
-
BPCR analysis to confirm the deletion of MARCH5 exon 3 by targeted Lysozyme‐Cre recombinase. Arrows indicate the sites recognized by the primers. Total mRNA was extracted from the indicated organs from March5 fl/fl and March5 fl/fl;Lyz‐Cre mice.
-
CBone marrow cells were collected from March5 fl/fl and March5 fl/fl;Lyz‐Cre mice and differentiated for 6 days. Western blot analysis of MARCH5 protein expression. Tubulin was used as a loading control.
Source data are available online for this figure.
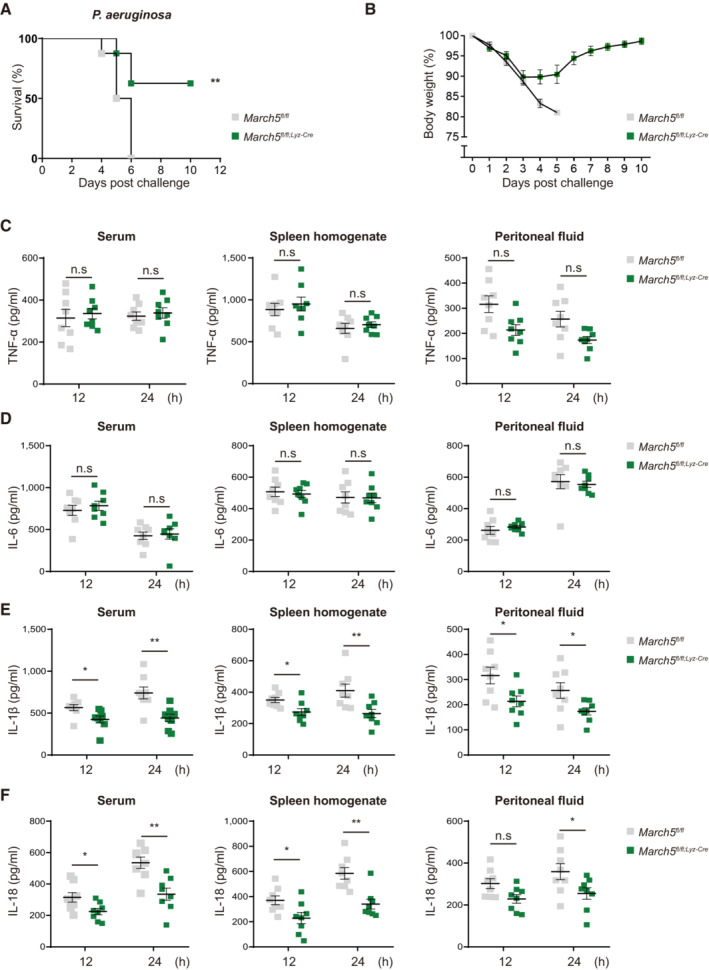
To investigate whether MARCH5 plays an immunomodulatory function in response to bacterial infection, we challenged March5 fl/fl and March5 cKO mice with a lethal dose of Pseudomonas aeruginosa intraperitoneally. Pseudomonas aeruginosa is an opportunistic Gram‐negative pathogen that can cause infections in humans, mainly in hospital patients, and trigger sepsis (Deng et al, 2016). The mice were monitored daily after P. aeruginosa infection, and we observed that all March5 fl/fl control mice died in 6 days. In contrast, 62.5% of March5 fl/fl;Lyz‐Cre mice survived up to 10 days, showing a significant difference in survival (Fig 1A). Pseudomonas aeruginosa infection also led to significant weight loss in mice. In just 5 days, March5 fl/fl mice lost 20% of their body weight and died, whereas March5 fl/fl;Lyz‐Cre mice gained weight rapidly 4 days after P. aeruginosa infection (Fig 1B). Thus, it appeared that March5 cKO mice were resistant to P. aeruginosa infection. Next, individual mice were sacrificed at 12 and 24 h after P. aeruginosa injection, and the inflammatory cytokine levels in the serum, spleen homogenates, and peritoneal fluid were determined using ELISA. The basal levels of inflammatory cytokines were low (< 50 pg/ml) (Chathuranga et al, 2020). Upon P. aeruginosa infection, significant production of TNF‐α and IL‐6 was observed in both March5 fl/fl and March5 cKO mice (Fig 1C and D), but the levels between these groups of mice were not different. Interestingly, however, the production of IL‐1β and IL‐18 after the P. aeruginosa challenge was different between March5 fl/fl and March5 cKO mice; the March5 cKO mice showed significantly lower levels of IL‐1β (Fig 1E) and IL‐18 (Fig 1F) production in three different types of samples after bacterial injection.
Figure 1. MARCH5 is required for the in vivo expression of IL‐1β and IL‐18 and lethality in response to bacterial infection.
-
A, BMarch5 cKO mice are resistant to P. aeruginosa infection (A) survival rates (n = 8) and (B) changes in the body weight of March5 fl/fl and March5 fl/fl;Lyz‐Cre (n = 8) mice after intraperitoneal injection with 1 × 107 CFU Pseudomonas aeruginosa.
-
C–FTNF‐α (C), IL‐6 (D), IL‐1β (E) and IL‐18 (F) from serum, spleen homogenate and peritoneal fluid collected from mice (n = 8) that were sacrificed 12 and 24 h following the bacterial infection. Each cytokine was analyzed by ELISA.
Data information: Values, *P < 0.05, **P < 0.01 (two‐tailed Student's t‐test or Mantel–Cox test). Data were expressed as the mean ± SEM. See also Fig EV2.
Source data are available online for this figure.
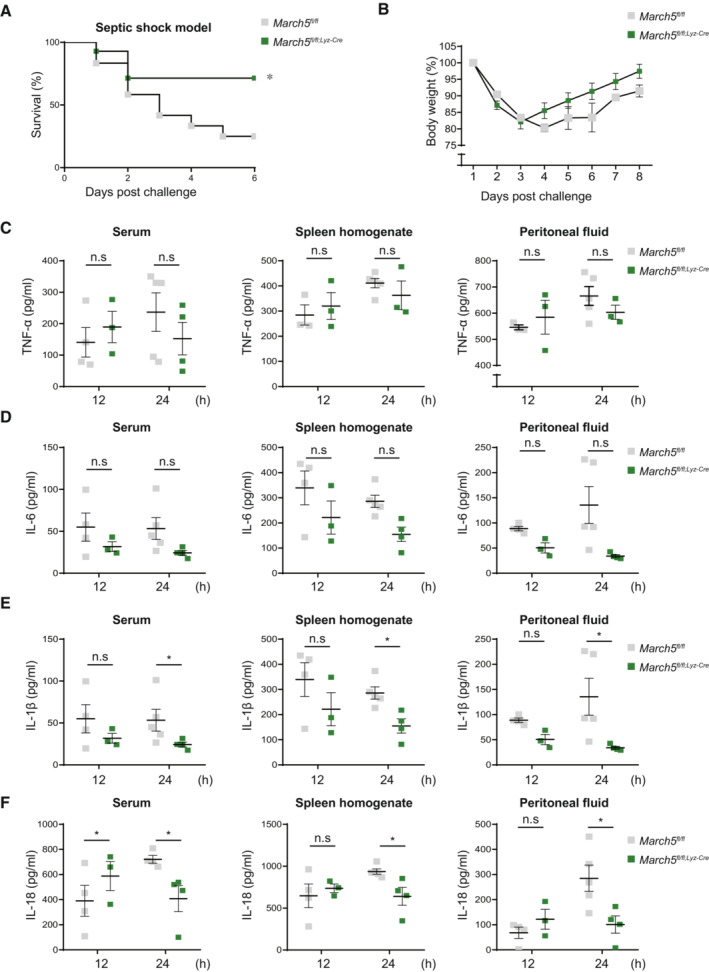
To verify the role of MARCH5 in the inflammatory response in mice, an endotoxemia‐induced septic shock model was used by intraperitoneally injecting lipopolysaccharide (LPS) into mice (Raduolovic et al, 2018). When the mice were monitored after LPS injection (28 mg/kg body weight), 80 percent of March5 fl/fl control mice died 6 days after LPS injection, whereas 70% of March5 cKO mice survived up to 6 days (Fig EV2A). Lipopolysaccharide injection caused a significant weight loss in both March5 fl/fl and March5 cKO mice, showing a 20% reduction in their body weight within 3 days. However, March5 cKO mice tended to regain their body weight earlier than March5 fl/fl mice (Fig EV2B). Consistent with the observation in P. aeruginosa infection (Fig 1C and D), the ability to produce TNF‐α and IL‐6 in March5 fl/fl and March5 cKO mice were not different after LPS injection (Fig EV2C and D). However, levels of IL‐1β and IL‐18 after the LPS challenge were different; the March5 cKO mice showed significantly lower levels of IL‐1β (Fig EV2E) and IL‐18 (Fig EV2F) production. The data showed that March5 cKO mice exhibited a significantly attenuated inflammatory response to septic shock. The data suggested that MARCH5 has a substantial impact on inflammasome activation responsible for the proteolytic processing and secretion of IL‐1β and IL‐18, but not the TLR4 signaling per se.
Figure EV2. MARCH5 is required for survival and in vivo cytokine secretion in response to LPS.
-
A, B(A) Survival rates (n = 12–14) and (B) variation of body weight of March5 fl/fl and March5 fl/fl;Lyz‐Cre (n = 7–9) mice after intraperitoneal injection with 28 mg/kg body weight of LPS.
-
C–FELISA of a TNF‐α (C), IL‐6 (D), IL‐1β (E) and IL‐18 (F) from serum, spleen homogenate and peritoneal fluid from mice (n = 5), sacrificed 12 and 24 h after LPS injection. Values, *P < 0.05 (two‐tailed Student's t‐test or Mantel–Cox test.). Data were expressed as the mean ± SEM.
Source data are available online for this figure.
MARCH5 potentiates NLRP3‐mediated antimicrobial immunity
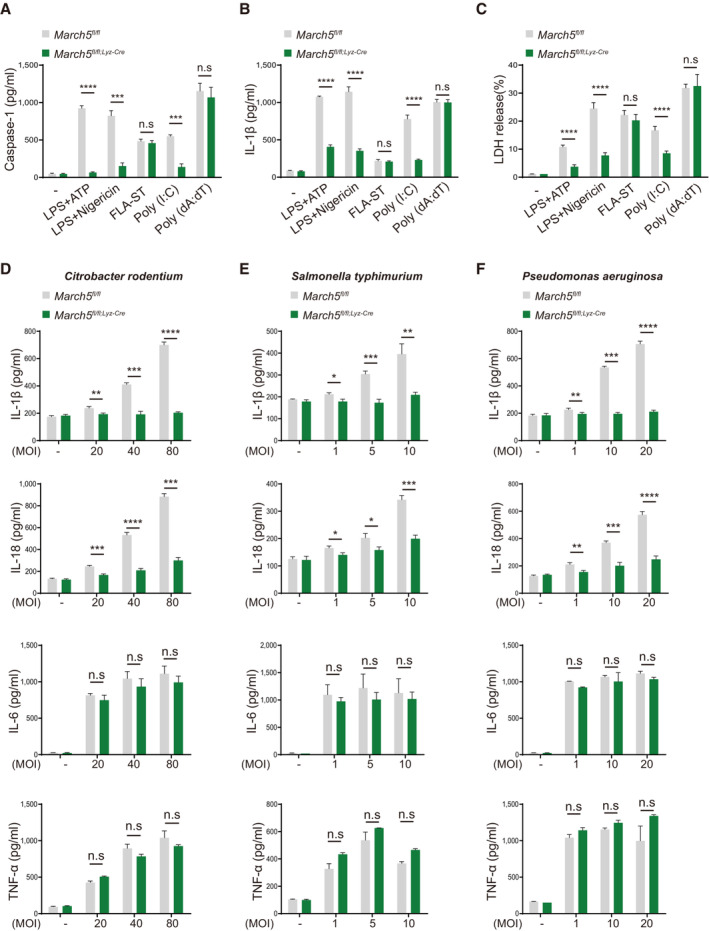
A diverse array of stimuli derived from microbial products and damaged host cells can induce inflammasome activation. Several cytoplasmic pattern recognition receptors recognize different types of DAMPs and PAMPs (Zheng et al, 2020). To identify which receptor is involved in the regulation of the inflammasome by the MARCH5 protein, BMDMs obtained from March5 fl/fl and March5 cKO mice were activated with several different stimulators. To activate the NLRP3, NLRC4, and AIM2 inflammasomes in macrophages, we treated macrophages with LPS plus ATP or nigericin, flagellin (FLA‐ST), and poly(dA:dT), respectively, and then measured caspase‐1 and IL‐1β levels using ELISA and cell death by a lactic acid dehydrogenase (LDH) release assay. Cytosolic poly(I:C) has also been used to activate the NLRP3 inflammasome independently of TLR signaling. We found that treatment with LPS (200 ng/ml) alone did not change caspase‐1 and IL‐1β secretion or cell death in BMDMs derived from March5 fl/fl mice, whereas LPS plus inflammasome activators elicited robust inflammasome activation in these cells (Fig 2A–C). Notably, we found that IL‐1β secretion in BMDMs derived from March5 cKO mice was significantly diminished in the context of NLRP3‐mediated inflammasome signaling activation (LPS plus ATP or LPS plus Nigericin), but not NLRC4 and AIM2 inflammasome signaling activation (Fig 2B). Similarly, LDH release in BMDMs derived from March5 cKO mice was significantly attenuated after stimulation with LPS plus ATP or LPS plus nigericin (Fig 2C), suggesting that NLRP3 inflammasome activation is specifically deficient in macrophages from March5 cKO mice. We also obtained similar results in THP1 cells (Fig EV3A–C). To verify the regulatory role of MARCH5 in NLRP3 inflammasome activation, we infected BMDMs with three different bacteria (Citrobacter rodentium, Salmonella typhimurium, and Pseudomonas aeruginosa) known to stimulate the NLRP3 inflammasome (Deng et al, 2016; Pu et al, 2017; Humphries et al, 2018). Infection with these bacteria induced solid inflammatory responses and a substantial increase in the inflammatory cytokine release of TNF‐α, IL‐6, IL‐1β, and IL‐18 in macrophages derived from March5 fl/fl mice (Fig 2D–F). Consistent with the finding in Fig 1, both TNF‐α and IL‐6 levels were comparable between March5 fl/fl and March5 cKO mice (Fig 2D–F), whereas the production of IL‐1β, IL‐18 as well as caspase‐1 was significantly diminished in BMDMs from March5 cKO mice (Figs 2D–F and EV3D–F). In these experiments, we noticed that IL‐18 levels appeared to be increased at the condition of high MOI of bacterial infection. Because mature IL‐18 can be produced through a Fas‐dependent pathway (Tsutsui et al, 1999), we interpreted that it is possible that the gram‐negative bacteria used in our study also activate the Fas‐dependent pathway or other noncanonical NLRP3 inflammasome (Humphries et al, 2018). The inability to activate NLRP3 inflammasome by March5 cKO mice was further confirmed by western blotting. Upon bacterial infection, March5 fl/fl BMDM promoted pro‐caspase‐1 cleavage and pro‐IL‐1β cleavage, whereas March5 cKO BMDM did not (Fig EV3G). These results collectively indicated that the MARCH5 protein specifically regulates NLRP3‐mediated antimicrobial immunity.
Figure 2. MARCH5 potentiates NLRP3‐mediated antimicrobial immunity.
-
A–C(A) Caspase‐1, (B) IL‐1β secretion and (C) LDH release were measured in the supernatants of March5 fl/fl and March5 fl/fl;Lyz‐Cre BMDMs subjected to the indicated stimuli followed by material method. Values are the mean ± SD. Experiments (A–C) were performed in triplicate and repeated at least three times. ***P < 0.001 (two‐tailed Student's t‐test)
-
D–FMARCH5 mediates activation of the NLRP3 pathway in response to bacterial infection. March5 fl/fl and March5 fl/fl;Lyz‐Cre BMDMs were infected with (D) Citrobacter rodentium (20 MOI, 40 MOI and 80 MOI), (E) Salmonella typhimurium (1 MOI, 5 MOI and 10 MOI), and (F) Pseudomonas aeruginosa (1 MOI, 10 MOI and 20 MOI). Secretion of IL‐1β, IL‐18, IL‐6 and TNF‐α in BMDMs infected for 12 h was measured by ELISA. Values, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 (two‐tailed Student's t‐test). Data are expressed as the mean ± SEM. Experiments (D–F) were performed in triplicate and repeated three times. See also Fig EV3.
Source data are available online for this figure.
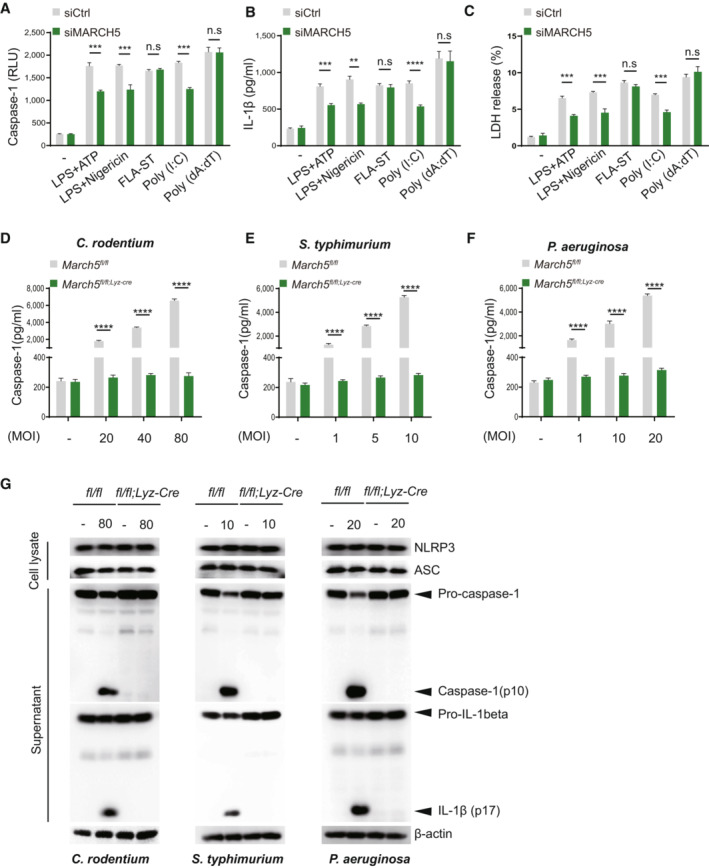
Figure EV3. MARCH5 potentiates NLRP3‐mediated antimicrobial immunity.
-
A–C(A) Activated caspase‐1, (B) IL‐1β, and (C) LDH release were measured in the supernatants of siControl (siCtrl) or siMARCH5 THP‐1 cells and were subjected to the indicated stimuli. Independent experiments were repeated at least three times. Values are the mean ± SD. **P < 0.01, ***P < 0.001 (two‐tailed student's t‐test).
-
D–GMarch5 fl/fl and March5 fl/fl;Lyz‐Cre BMDMs were infected with (D) Citrobacter rodentium (20 MOI, 40 MOI and 80 MOI), (E) Salmonella typhimurium (1 MOI, 5 MOI and 10 MOI), and (F) Psedumonas aeruginosa (1 MOI, 10 MOI and 20 MOI). Secretions of caspase‐1 in BMDMs infected for 12 h were measured (D–F), and the cell pellet was used for western blotting to detect the activation of NLRP3 inflammasome (G) in response to bacterial infection. Values, ***P < 0.001, ****P < 0.0001 (two‐tailed Student's t‐test). Data were expressed as the mean ± SEM.
Source data are available online for this figure.
MARCH5 is indispensable for NLRP3 inflammasome activation
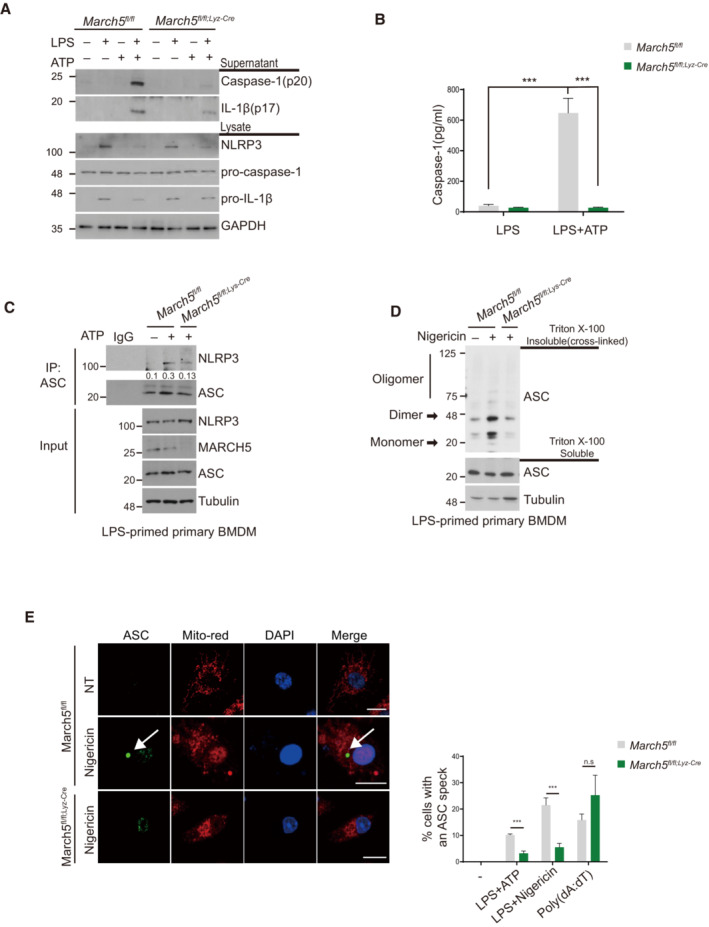
The NLRP3 inflammasome is composed of NLRP3, ASC and procaspase‐1, and inflammasome activation promotes autoproteolytic cleavage of procaspase‐1, the mature form of which executes the proteolytic cleavage of pro‐IL‐1β and pro‐IL‐18 (McKee & Coll, 2020). To verify whether MARCH5 is necessary for the NLRP3 inflammasome activation, we determined the cleaved IL‐1β as well as caspase‐1 (p20) levels in the supernatants of March5 cKO BMDMs and MARCH5‐depleted THP‐1 cells after LPS primed ATP stimulation (Figs 3A and EV4A). Upon treatment with LPS, the NLRP3 and pro‐IL‐1β levels were elevated in the cellular lysates of BMDMs and THP‐1 cells. However, ATP stimulation of LPS‐primed BMDMs and THP‐1 cells showed a substantial difference in the levels of cleaved IL‐1β and caspase‐1, which were decreased in the supernatant from cultured BMDMs of March5 cKO mice and THP‐1 cells depleted of MARCH5 by siRNA. Likewise, caspase‐1 activity was significantly lower in the BMDMs of March5 cKO mice than in those of March5 fl/fl mice (Fig 3B). Since caspase‐1 activation was deficient in macrophages from March5 cKO mice and MARCH5‐deficient THP‐1 cells, we next investigated whether inflammasome assembly was affected by the absence of MARCH5 in these cells. In a co‐immunoprecipitation assay using cell extracts of BMDMs stimulated with LPS and ATP, we found that the interaction between endogenous NLRP3 and ASC was weakened in macrophages of March5 cKO mice (Figs 3C and EV4B), suggesting that MARCH5 is indeed involved in NLRP3 inflammasome assembly. ASC forms oligomers in the activated inflammasome complex. To determine whether the oligomerization of ASC was affected by the presence of MARCH5, cell lysates of LPS‐primed BMDMs costimulated with nigericin were separated into Triton X‐100 soluble and insoluble fractions. Triton X‐100 insoluble pellets were cross‐linked with DSS and subjected to western blotting. As shown in Fig 3D, ASC dimerization and oligomerization in LPS‐primed March5 fl/fl macrophages were markedly boosted upon nigericin treatment, whereas they remained unchanged in BMDMs of March5 cKO (Fig 3D). Similar results were observed in MARCH5‐depleted THP‐1 cells (Fig EV4C). The ASC speck formation assay also supported this finding. ASC assembles into a large protein complex, or “speck,” that is considered to be an upstream readout for inflammasome activation (Stutz et al, 2013; Hoss et al, 2017). Immunofluorescence staining revealed that 10–20% of March5 fl/fl macrophage cells showed ASC specks after stimulation with LPS followed by ATP or nigericin and poly(dA:dT) alone. On the contrary, ASC specks were almost abolished in LPS plus ATP/nigericin‐stimulated BMDMs of March5 cKO mice (Figs 3E and EV4D). The number of ASC specks remained elevated in March5 cKO BMDMs stimulated with poly(dA:dT). These data indicate that MARCH5 specifically promotes the NLRP3 inflammasome assembly and activation.
Figure 3. MARCH5 is essential for activating the NLRP3 inflammasome.
-
AMarch5 fl/fl and March5 fl/fl;Lyz‐Cre BMDMs were untreated or treated with 200 ng/ml LPS for 4 h followed by 5 mM ATP for 45 min. The indicated proteins in the cell lysate and supernatants were immunoblotted.
-
BThe activity of caspase‐1 was detected using ELISA. March5 fl/fl and March5 fl/fl;Lyz‐Cre BMDMs were treated with 200 ng/ml LPS alone for 4 h or with an additional 5 mM ATP for 45 min. Supernatants of stimulated cells were collected and analyzed by ELISA. Each experiment was carried out in triplicate three times.
-
C, DBMDMs from March5 fl/fl and March5 fl/fl;Lyz‐cre mice were primed with LPS (200 ng/ml) for 4 h. Cells were subsequently treated with 5 mM ATP (C) or with 15 μM nigericin (D). (C) Cell lysates were then immunoprecipitated with ASC antibody. Proteins were analyzed by immunoblotting with the indicated antibodies. The immunoprecipitated NLRP3 levels were quantified by using image J. (D) Triton X‐100 insoluble pellets were cross‐linked with 2 mM DSS and immunoblotted for ASC oligomerization. Analysis of Triton X‐100 soluble and insoluble proteins was performed by immunoblotting.
-
EMarch5 fl/fl and March5 fl/fl;Lyz‐cre BMDMs were either transfected with 2 μg/ml poly(dA:dT) for 6 h or treated stimulated with 200 ng/ml LPS for 4 h along with 5 mM ATP or 15 μM nigericin for 30 min subsequently. Representative confocal images show ASC speck formation in BMDMs. The right graph shows the percentage of cells containing ASC specks. At least 100 BMDMs were analyzed in three independent experiments, showing representative data. White arrows indicate ASC speck.
Data information: Values are the mean ± SD. ***P < 0.001 (two‐tailed Student's t‐test). Bars, 10 μm. See also Fig EV4.
Source data are available online for this figure.
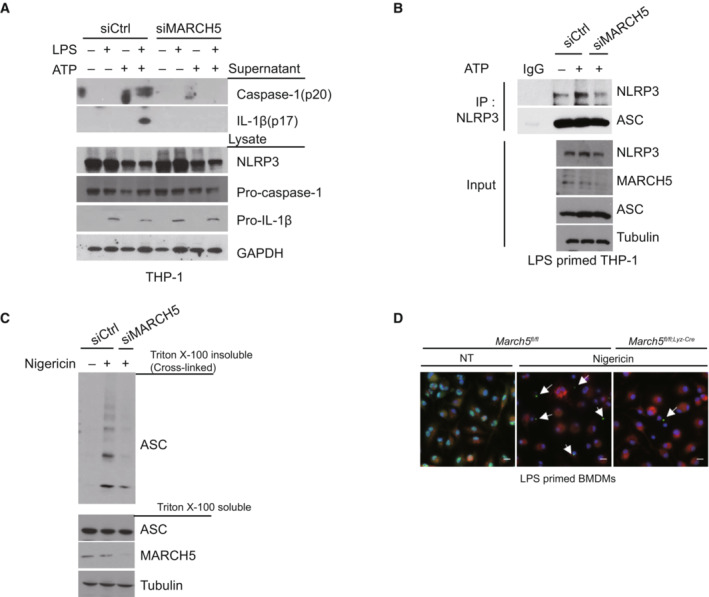
Figure EV4. MARCH5 is essential for activating the NLRP3 inflammasome.
-
ATHP‐1 cells transfected with siControl or siMARCH5 were untreated or treated with LPS alone for 4 h, ATP alone for 45 min, or LPS and ATP together.
-
BTHP‐1 cells transfected with siControl or siMARCH5 were treated with ATP for 30 min and then subjected to immunoprecipitation with ASC antibody. The levels of the indicated proteins were assessed by western blotting.
-
CTHP‐1 cells transfected with siControl or siMARCH5 were primed with LPS for 4 h and treated with 15 μM nigericin for 30 min. Triton X‐100 insoluble pellets were cross‐linked with DSS and immunoblotted with the indicated antibodies to assess ASC oligomerization.
-
DFluorescence microscopy images of ASC specks in LPS‐primed March5fl/fl and March5 fl/fl;Lyz‐Cre BMDMs treated with nigericin (15 μM) for 15–20 min. ASC, green; Mitochondria, red; Nuclei, blue. White arrows indicate ASC specks. Bars, 10 μm. Independent experiments were repeated at least three times.
Source data are available online for this figure.
MARCH5 interacts with the NACHT domain of NLRP3
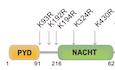
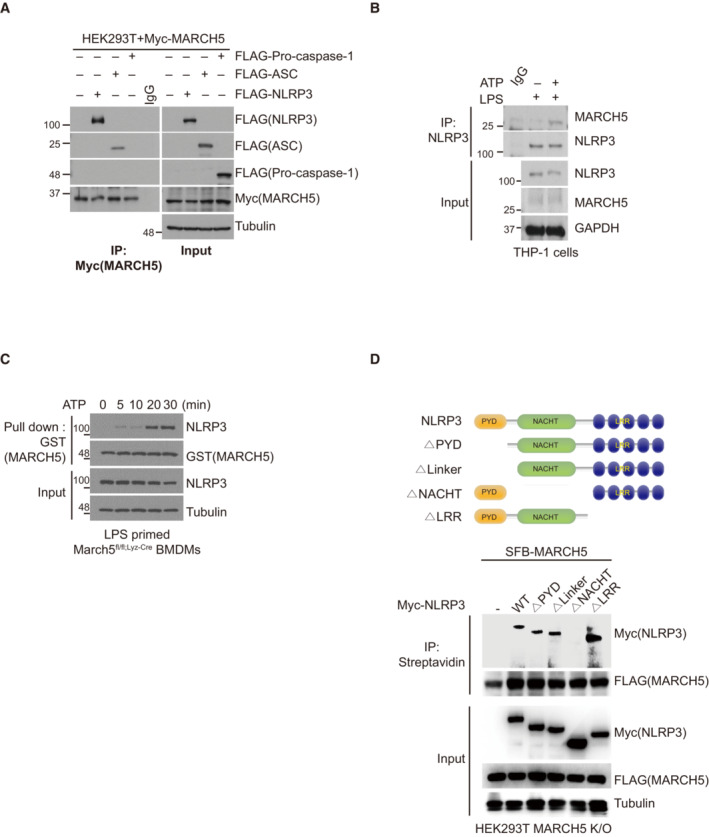
To address the underlying mechanisms by which MARCH5 activates the NLRP3 inflammasome, we examined whether MARCH5 binds any NLRP3 inflammasome components. We transfected HEK293T cells with each Flag‐tagged NLRP3 inflammasome components; Flag‐ NLRP3, ASC, or pro‐caspase‐1 along with Myc‐MARCH5 and performed coimmunoprecipitation experiments with an anti‐Myc antibody. Western blotting revealed that MARCH5 strongly interacted with NLRP3, coprecipitating essentially equal amounts of each inflammasome component (Fig 4A). A portion of ASC was also immunoprecipitated with MARCH5, but no pro‐caspase‐1 was found in this immunoprecipitant. Because overexpressed MARCH5 and NLRP3 strongly interact in HEK293T cells, we next examined whether these interactions occurred in THP‐1 cells after stimulation. In LPS‐primed THP‐1 cells, NLRP3 specifically interacted with endogenous MARCH5 after ATP stimulation (Fig 4B). These findings were also examined using a semi‐in vitro immunoprecipitation assay. The LPS‐primed BMDMs derived from March5 cKO mice were costimulated with ATP, and cell lysates from each time point were incubated with GST‐MARCH5 for the indicated durations. In the pull‐down assay, MARCH5 did not bind to LPS‐stimulated NLRP3 without ATP stimulation; rather, it firmly bound NLRP3 at 20 and 30 min after ATP stimulation (Fig 4C). These data suggested that MARCH5 actively interacts with NLRP3, probably in the context of the NLRP3 inflammasome. Next, we examined which domains of NLRP3 are responsible for the interaction with MARCH5. A series of deletion mutants of Myc‐NLRP3 were transfected into HEK293T MARCH5 KO cells along with SFB (S‐tag, Flag, and a streptavidin‐binding tag)‐MARCH5, and cell lysates were co‐immunoprecipitated with streptavidin beads, followed by western blotting. We found that MARCH5 binding was maintained by NLRP3 mutants lacking the PYD, linker, or LRR domain, but was lost in the mutant lacking the NACHT domain (Fig 4D). Taken together, these data indicate that MARCH5 interacts with the NACHT domain of NLRP3.
Figure 4. MARCH5 interacts with the NACHT domain of NLRP3.
-
AHEK293T cells were cotransfected with Myc‐MARCH5 and FLAG‐tagged NLRP3, ASC, or pro‐caspase‐1. Cell lysates were immunoprecipitated with anti‐FLAG‐M2 beads and detected with the indicated antibodies.
-
BTHP‐1 cells primed with 200 ng/ml LPS for 4 h were subsequently treated with or without 5 mM ATP for 30 min. Cell lysates were immunoprecipitated with IgG or anti‐NLRP3 antibody, and the indicated proteins were detected by immunoblotting.
-
CMarch5 fl/fl BMDMs were treated with 200 ng/ml LPS for 4 h followed by 5 mM ATP for different durations. Cell lysates mixed with GST‐MARCH5 protein were pulled down with GST‐tagged beads, and the indicated proteins were detected by immunoblotting.
-
DSchematic representation of NLRP3 wild‐type and truncated mutants (Upper). HEK293T cells were cotransfected with NLRP3 WT or NLRP3 mutants and SFB‐MARCH5 WT. Treatment with LPS (200 ng/ml for 4 h) and nigericin (15 μM for 60 min). Cell lysates were immunoprecipitated with streptavidin beads and immunoblotted with the indicated antibodies (Lower).
Source data are available online for this figure.
MARCH5 transfers K27‐linked polyubiquitin chains to the K324 and K430 residues of NLRP3
MARCH5 maintains cellular and mitochondrial homeostasis by linking ubiquitin to target proteins (Park et al, 2014). To determine whether MARCH5 ubiquitinates NLRP3, we transfected FLAG‐tagged NLRP3 and HA‐tagged ubiquitin construct into MARCH5 WT and MARCH5 KO HEK293T cells. Immunoprecipitation with an anti‐FLAG antibody revealed that the ubiquitination of NLRP3 was reduced in MARCH5 KO cells (Fig 5A). We previously showed that MARCH5 often promotes degradation of its target proteins through Lys (K) 48‐linked ubiquitination (Park et al, 2010, 2020). Thus, we determined whether MARCH5 alters the protein level of NLRP3 in the NLRP3 inflammasome complex. BMDMs obtained from March5 fl/fl and March5 cKO mice were stimulated with LPS plus ATP and the protein levels of the NLRP3 inflammasome component were compared (Fig EV5A). We found that NLRP3 level was not elevated in macrophages of March5 cKO mice, suggesting that MARCH5‐dependent ubiquitination on NLRP3 does not promote the proteasome‐dependent degradation pathway. Next, we carried out a ubiquitin assay after overexpression of HA‐ub‐K48R to exclude K48‐linked polyubiquitination in MARCH5 KO HEK293T cells. We utilized the MARCH5 K40/54R (MARCH5 2KR) and MARCH5 H43W mutant constructs in this assay (Karbowski et al, 2007; Kim et al, 2016). In MARCH5 2KR, two Lys residues in MARCH5 were switched to Arg to prevent autoubiquitination and degradation of MARCH5 (Kim et al, 2016). The MARCH5 H43W mutant lacks the catalytic activity of MARCH5 and was used as a negative control (Karbowski et al, 2007). In this experiment, we found that MARCH5 2KR but not MARCH5 H43W enhanced NLRP3 polyubiquitination independent of K48‐linked ubiquitin (Fig EV5B). The kinetics of MARCH5‐mediated NLRP3 ubiquitination during inflammasome activation was determined in cells transfected with HA‐ub‐K48R. NLRP3 ubiquitination by MARCH5 in LPS‐primed MARCH5 KO HEK293T cells could be observed at 30 min after nigericin stimulation and became stronger at 60 min (Fig 5B).
Figure 5. MARCH5 ubiquitinates NLRP3 on K324 and K430 via K27‐linked polyubiquitination.
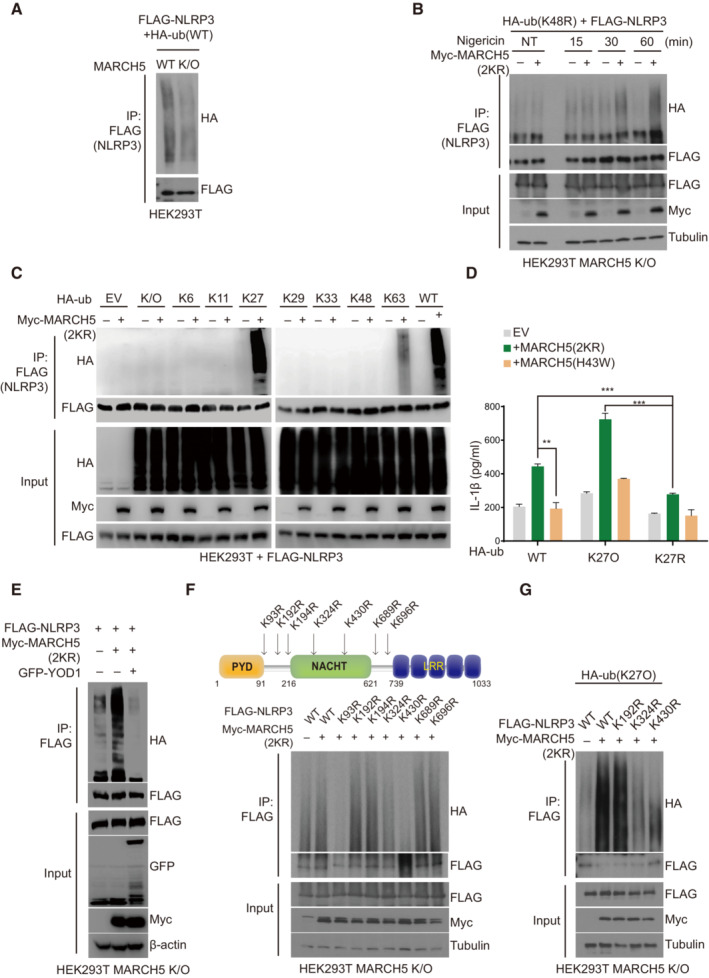
-
AMARCH5 WT and KO HEK293T cells were transfected with FLAG‐NLRP3 and HA‐ub. After the cells were stimulated with LPS (200 ng/ml for 4 h) and nigericin (15 μM for 60 min), the cell lysates were immunoprecipitated with FLAG‐M2 beads. Ubiquitinated NLRP3 was detected by anti‐HA antibody.
-
BFLAG‐NLRP3 and HA‐ub K48R mutant plasmids were transfected into MARCH5 KO HEK293T cells with or without Myc‐MARCH5 2KR. Then, the cells were stimulated with 200 ng/ml LPS followed by 15 μM nigericin for the indicated durations. The cell lysates were immunoprecipitated with FLAG‐M2 beads and immunoblotted by using indicated antibodies.
-
CMARCH5 KO HEK293T cells were transfected with FLAG‐NLRP3, Myc‐MARCH5 2KR, and each HA ‐ ubiquitin, shown by the number of the remaining single Lys residue with the other Lys changed to Arg. For stimulation, the cells were treated with 200 ng/ml LPS for 4 h, followed by 15 μM nigericin for 30–60 min. Cell lysates were immunoprecipitated with FLAG‐M2 beads and analyzed by immunoblotting with the indicated antibodies.
-
DThe NLRP3 inflammasome was reconstituted in HEK293T MARCH5 KO cells expressing ASC, pro‐caspase‐1, and IL‐1β with HA‐ub WT, K27 only (K27O) mutant, or K27R mutant. Additionally, cells were cotransfected with or without Myc‐MARCH5 2KR or the Myc‐MARCH5 H43W mutant. Following stimulation with LPS for 4 h and nigericin for 30 min, IL‐1β secretion was quantitated by ELISA. Values are the mean ± SD. **P < 0.01, ***P < 0.001 (two‐tailed Student's t‐test). All the experiments were carried out in triplicate three times.
-
EHEK293T MARCH5 K/O cells were transfected with FLAG‐NLRP3, Myc‐MARCH5 (2KR), HA‐ubiquitin and GFP‐YOD1. Transfected cells were stimulated with 200 ng/ml LPS for 4 h, followed by 15 μM nigericin for 60 min. Cell lysates were immunoprecipitated with FLAG‐M2 beads and analyzed by ubiquitination with HA‐antibody. The other proteins were detected by indicated antibodies.
-
FSchematic representation of NLRP3 Lys mutants (Upper). HEK293T MARCH5 KO cells were cotransfected with FLAG‐NLRP3 WT or indicated Lys point mutants, HA‐ubiquitin, and Myc‐MARCH5 (2KR). Cells were stimulated with 200 ng/ml LPS for 4 h followed by 15 μM nigericin treatment for 30 min. The cell lysates were immunoprecipitated with FLAG‐M2 beads. NLRP3 ubiquitination was assessed via western blotting using anti‐HA. And each protein was detected by indicated antibodies (Lower).
-
GFLAG‐NLRP3 WT or indicated Lys point mutants, HA‐ub K27 only mutant and Myc‐MARCH5 2KR were transfected into MARCH5 KO HEK293T cells. Cell lysates were immunoprecipitated with FLAG‐M2 beads. Ubiquitinated NLRP3 was detected by immunoblotting using an HA antibody. Representative data are shown from independent experiments that were repeated at least three times. See also Fig EV5.
Source data are available online for this figure.
Figure EV5. K27 ubiquitination and activation of NLRP3 by MARCH5.
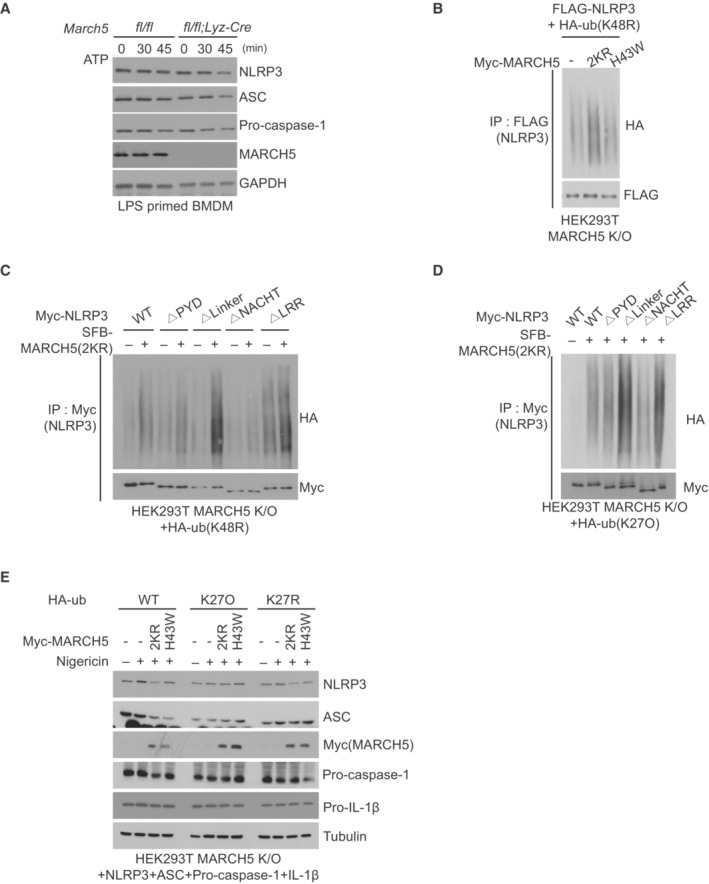
-
AMarch5 fl/fl and March5fl/fl;Lyz‐Cre BMDMs were primed with LPS for 4 h and were stimulated without or with 5 mM ATP at the indicated time points. Whole‐cell lysates were analyzed via western blotting using the indicated antibodies.
-
BMARCH5 KO HEK293T cells were cotransfected with HA‐ub (K48R) mutant and NLRP3 without or with Myc‐MARCH5 2KR or Myc‐MARCH5 H43W. After treatment with nigericin for 30 min of the LPS treatment for 4 h, the cell lysates were subjected to immunoprecipitation with anti‐FLAG M2 beads. An HA antibody was used to assess NLRP3 ubiquitination.
-
CHEK293T MARCH5 KO cells were cotransfected with HA‐ub (K48R) and NLRP3 WT or NLRP3 truncated mutants with or without SFB‐MARCH5 (2KR). After treatment with LPS and nigericin, the cell lysates were subjected to immunoprecipitation with an anti‐Myc antibody overnight, and NLRP3 ubiquitination was assessed via western blotting by using an anti‐HA antibody.
-
DHEK293T MARCH5 KO cells were transfected HA‐ub (K27O) with NLRP3 WT and truncated mutants. After stimulation with LPS and nigericin, cell lysates were immunoprecipitated with anti‐Myc antibody. The HA antibody detected ubiquitination in the subjects.
-
EThe NLRP3 inflammasome was reconstituted in HEK293T MARCH5 KO cells expressing ASC, pro‐caspase 1, and IL‐1β with HA‐ub WT, K27O mutant, or K27R mutant NLRP3. Additionally, cells were cotransfected with or without Myc‐MARCH5 2KR or the Myc‐MARCH5 H43W mutant. After stimulation with LPS for 4 h and nigericin for 30 min, IL‐1β secretion was quantitated using ELISA, and the lysates were detected by western blotting with the indicated antibodies.
Source data are available online for this figure.
Next, we determined which domain of NLRP3 was ubiquitinated by MARCH5 by cotransfecting each truncated NLRP3 construct with HA‐ub‐K48R and MARCH5 in MARCH5 KO HEK293T cells. The data showed that the ubiquitination level of NLRP3 ΔNACHT remained unchanged upon overexpression of MARCH5. In contrast, the polyubiquitination levels of WT NLRP3, NLRP3 ΔLinker, and other truncation mutants were increased by MARCH5 (Fig EV5C). Thus, we concluded that the NACHT domain of NLRP3 is the major ubiquitination target domain of MARCH5.
Next, we addressed which ubiquitination chain is mainly attached to NLRP3 among seven lysine residues (K6, K11, K27, K29, K33, K48, and K63) on ubiquitin. To do this, we utilized different ubiquitin constructs that contained only one intact Lys residue, and the other six Lys residues were mutated to Arg. We cotransfected each ubiquitin mutant with Myc‐MARCH5 and FLAG‐NLRP3, and immunoprecipitation with an anti‐FLAG antibody showed that K27‐only ubiquitin (K27O ub) was the most effective NLRP3 ubiquitination (Fig 5C). We then examined whether K27‐linked polyubiquitination occurs on the NACHT domain of NLRP3 by transfecting NLRP3 truncation mutants with K27O ub. We also found that K27‐linked polyubiquitination of NLRP3 ΔNACHT did not occur under the condition of MARCH5 overexpression (Fig EV5D). Thus, we concluded that MARCH5 promotes NLRP3 activation via K27‐linked polyubiquitination. To investigate the physiological importance of the K27‐linked polyubiquitination of NLRP3 in inflammasome activation, we compared the secretion of IL‐1β in the NLRP3 inflammasome reconstitution system after cotransfection with K27O ub or K27R ub; K27O ub contains only one intact Lys residue at K27, and the other six Lys residues were mutated to Arg whereas K27R ub contains Arg residue at K27 and the other six Lys residues are intact. Indeed, a significant increase in IL‐1β secretion was found in cells transfected with K27O ub and MARCH5, whereas this increase was not observed in cells transfected with K27R ub or MARCH5 H43W, although the expression of the transfected proteins was comparable in this system (Figs 5D and EV5E). To verify the K27‐linked ubiquitination on NLRP3, we utilized an OTU deubiquitinase, YOD1, which targets the K6, K11, K27 and K33‐linked ubiquitin chains of substrates (Mevissen et al, 2013). We observed that YOD1 efficiently removed the ubiquitin chain of NLRP3 induced by MARCH5 (Fig 5E). Taken together, these results indicate that MARCH5 activates the NLRP3 inflammasome via K27‐linked polyubiquitination of the NACHT domain of NLRP3.
We next addressed which Lys residues of NLRP3 are ubiquitinated by searching for putative ubiquitination sites on NLRP3 using the UbPred program (http://ubpred.org). This analysis predicted seven ubiquitination sites on Lys (K93, K192, K194, K324, K430, K689, and K696) of NLRP3. Based on this analysis, we generated 7 Lys point mutants in which each Lys was replaced with Arg (Fig 5F, upper). We cotransfected each FLAG‐tagged NLRP3 Lys mutant with the MARCH5 2KR and HA‐ub constructs in MARCH5 KO HEK293T cells, and after cotreatment with LPS plus nigericin, polyubiquitination was examined in the immunoprecipitant obtained using anti‐FLAG beads. We found that polyubiquitination of NLRP3 was significantly reduced in the cells transfected with K93R, K324R, and K430R mutants, whereas other Lys mutants of K192R, K194R, K689R, and K696R of NLRP3 showed polyubiquitination levels equivalent to those of the WT (Fig 5F, lower). Since MARCH5 targeted the NLRP3 NACHT domain, we focused on the two Lys sites that were in the NACHT domain, K324 and K430. To further confirm the lysine sites on NLRP3 targeted by MARCH5 for K27‐linked polyubiquitination, we carried out a ubiquitination assay after transfection with K27O ub, where the K192R mutant of NLRP3 was used as a positive control. Consistent with Fig 5E, K27‐linked polyubiquitination of NLRP3 was reduced in the K324 and K430 mutants of NLRP3 (Fig 5G). Our data suggested that K27‐linked polyubiquitination by MARCH5 occurred at the K324 and K430 sites of NLRP3.
MARCH5‐dependent NLRP3 ubiquitination allows NEK7 binding to NLRP3
To address the functional role of NLRP3 ubiquitination by MARCH5, we examined NLRP3 oligomerization through fluorescence microscopy. Unlike the ASC specks, the NLRP3 puncta were barely visible, probably due to prompt inflammasome assembly with ASC and pro‐caspase‐1. We utilized immortalized BMDM (iBMDM) cells expressing GFP‐tagged NLRP3 and knocked down ASC by siRNA. Upon treatment with LPS plus nigericin, oligomerized NLRP3 puncta were found in ~20% of cells, but these puncta were significantly diminished in March5‐depleted iBMDMs (Fig 6A and Appendix Fig S1A). In SDD–AGE (semi‐denaturing detergent agarose gel electrophoresis), the high‐molecular‐weight oligomers of NLRP3 were diminished in March5 cKO BMDMs stimulated with LPS and nigericin or ATP at two different time points (Fig 6B), suggesting that MARCH5‐mediated ubiquitination facilitates NLRP3 self‐oligomerization. MAVS recruits NLRP3 to the mitochondria for inflammasome activation (Park et al, 2013; Subramanian et al, 2013). It also shows that NEK7 directly binds NLRP3, and these interactions are essential for NLRP3 oligomerization (He et al, 2016; Sharif et al, 2019). As shown in Appendix Fig S1B, co‐immunoprecipitation with anti‐NLRP3 antibody revealed that NLRP3 interacts with MAVS in BMDMs treated with LPS plus nigericin, and the absence of MARCH5 did not disrupt these interactions. Consistently, ubiquitination‐defective NLRP3 K324R and K430R mutants remained bound to MAVS (Appendix Fig S1C). On the contrary, the depletion of MAVS by siRNA disrupted the interaction of MARCH5 with NLRP3, which furthermore disrupted the interaction of ASC with NLRP3 (Appendix Fig S1D). Thus, the data suggest that MAVS mediates the mitochondrial recruitment of NLRP3 that is subsequently ubiquitinated by MARCH5, which is essential for the inflammasome assembly. Next, we addressed whether MARCH5 affects NEK7 binding to NLRP3. We knocked down MARCH5 (MARCH5 KD) in THP‐1 cells using siRNA, followed by stimulation with LPS and ATP. Co‐immunoprecipitation with an anti‐NLRP3 antibody revealed that the interaction between NLRP3 and NEK7 was impaired in these cells. Accordingly, the association of NLRP3 with ASC was also diminished (Fig 6C). To examine the multimeric complex formation of NLRP3 oligomers with NEK7, stimulated iBMDM cell lysates were separated on a blue native gel, followed by SDS–PAGE in a second dimension. As expected, NLRP3 and NEK7 were found in a high‐molecular‐weight region (Fig 6D, yellow circle). In contrast, these signals were not present in lysates from MARCH5 KD iBMDM cells, suggesting that MARCH5 is indispensable for forming NEK7‐NLRP3 multimeric oligomers. Indeed, wild‐type NLRP3 bound endogenous NEK7 in HEK293T cells, whereas the binding of NLRP3‐K324R and NLRP3‐K430R mutants to NEK7 was strongly reduced (Fig 6E). Consistent with previous reports, the NLRP3‐S198A mutant, which is defective in JNK1‐dependent phosphorylation (Song et al, 2017), also barely binds NEK7. In addition, the ability to form multimeric oligomers of NEK7‐NLRP3 was impaired in cells with NLRP3‐K324R and NLRP3‐K430R mutants, as shown in a two‐dimensional polyacrylamide gel electrophoresis (2D‐PAGE) (Fig 6F). Finally, the colocalization of NEK7 with NLRP3 puncta was significantly reduced, as shown by immunofluorescence staining (Fig 6G). Together, these data indicate that MARCH5‐dependent NLRP3 ubiquitination is necessary for the binding of NEK7 and the formation of NEK7‐NLRP3 oligomers.
Figure 6. MARCH5‐dependent NLRP3 ubiquitination allows the binding of NEK7 to NLRP3.
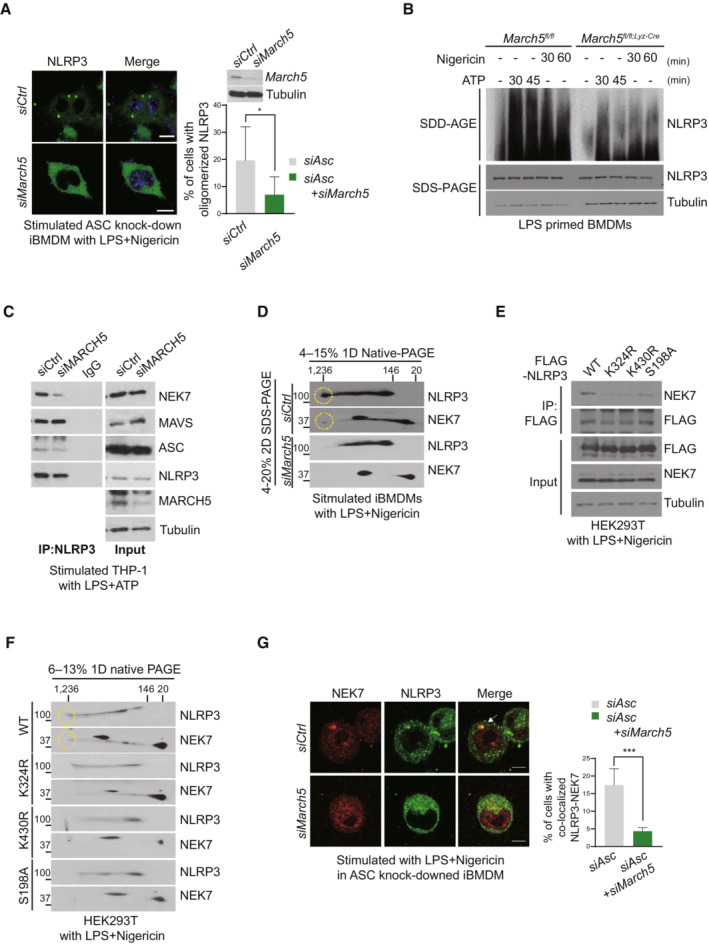
-
ARepresentative confocal images of oligomerized NLRP3. ASC knock‐downed immortalized BMDMs expressing GFP‐tagged NLRP3 WT were transfected using indicated siRNAs, and treated with 200 ng/ml LPS for 4 h followed by 15 μM nigericin for 30 min. White arrows indicate oligomerized NLRP3. The percentage of cells with oligomerized NLRP3 was quantified. At least 100 cells were analyzed. Bar, 10 μm. Values are the mean ± SD. *P < 0.05 (two‐tailed Student's t‐test).
-
BMarch5 fl/fl and March5 fl/fl;Lyz‐Cre BMDMs were stimulated for 4 h with LPS followed by 5 mM ATP or 15 μM nigericin for the indicated durations. SDD–AGE and SDS–PAGE were performed to separate cell lysates, followed by immunoblotting using indicated antibodies.
-
CTHP‐1 cells transfected with siControl or siMARCH5 were stimulated with 200 ng/ml LPS for 4 h followed by 5 mM ATP for 30 min. Cell lysates were subjected to immunoprecipitation by using NLRP3 antibody. Western blotting was performed using the indicated antibodies.
-
DImmortalized BMDMs were transfected with siControl or siMARCH5 and stimulated with LPS and nigericin. Blue native PAGE was used first to separate the cell lysates, and then SDS–PAGE was used to separate them further. Western blotting was performed using the indicated antibodies. Each yellow circle represents the oligomerized form of indicated protein.
-
EHEK293T cells were transfected with FLAG‐tagged NLRP3 WT or indicated mutants and stimulated with LPS and nigericin. Cell lysates were immunoprecipitated with FLAG‐M2 beads and detected with the indicated antibodies.
-
FHEK293T cells were transfected with FLAG‐tagged NLRP3 WT or indicated mutants. Cells were stimulated with LPS and nigericin. Cell lysates were separated by blue‐native PAGE (first dimension) and SDS–PAGE (second dimension). Representative data from independent experiments that were repeated at least three times are shown. Each yellow circle represents the oligomerized form of indicated protein.
-
GRepresentative confocal image showing NLRP3‐NEK7 colocalization. siControl‐ and siMarch5‐ transfected ASC knock‐downed iBMDMs expressing GFP‐tagged NLRP3 WT cells were stimulated with 200 ng/ml LPS for 4 h followed by 15 μM nigericin for 30 min. The percentage of cells with NLRP3‐NEK7 colocalization was quantified. At least 100 cells were analyzed. Values are shown as the mean ± SD. ***P < 0.001 (two‐tailed student's t‐test). See also Appendix Figure S1. Bar, 10 μm.
Source data are available online for this figure.
MARCH5‐mediated ubiquitination of NLRP3 is an essential step for inflammasome activation
Next, we examined the physiological relevance of these two Lys residues of NLRP3 by assessing their roles in NLRP3 inflammasome activation. We transfected ASC with NLRP3 WT or NLRP3 Lys mutant in HEK293T cells and treated them with nigericin. As shown in Fig 7A, approximately 30% of the cells expressing WT NLRP3 formed ASC specks, while cells expressing NLRP3‐K324R and NLRP3‐K430R showed less ASC specks. Consistent with this result, we confirmed ASC oligomerization by SDD‐AGE. We overexpressed the NLRP3‐K324R and NLRP3‐K430R constructs with ASC in HEK293T cells. After treatment with LPS plus nigericin, cell lysates were treated with DSS and divided into a Triton X‐100 soluble fraction and Triton X‐100 insoluble pellet. Immunoblotting showed that NLRP3‐WT and NLRP3‐K93R were able to form ASC oligomers (Fig 7B). In contrast, NLRP3‐K324R and NLRP3‐K430R failed to stimulate ASC oligomerization, similar to NLRP3‐S198A. In the reconstituted NLRP3 inflammasome complex, the mature form of IL‐1β was reduced in the cells transfected with NLRP3‐K324R or NLRP3‐K430R, unlike those transfected with NLRP3‐WT and NLRP3‐K93R, which exhibited a normal level of mature IL‐1β (Fig 7C). The NLRP3‐S198A mutant also formed a nonfunctional NLRP3 inflammasome, as expected. Similarly, IL‐1β secretion was substantially diminished in cells expressing NLRP3‐K324R, NLRP3‐K430R, or NLRP3‐S198A (Fig 7D). These results suggest that MARCH5‐mediated ubiquitination on the K324 and K430 residues of NLRP3 is an important event in NLRP3 inflammasome assembly and activation.
Figure 7. MARCH5‐mediated ubiquitination of NLRP3 is essential for inflammasome activation.
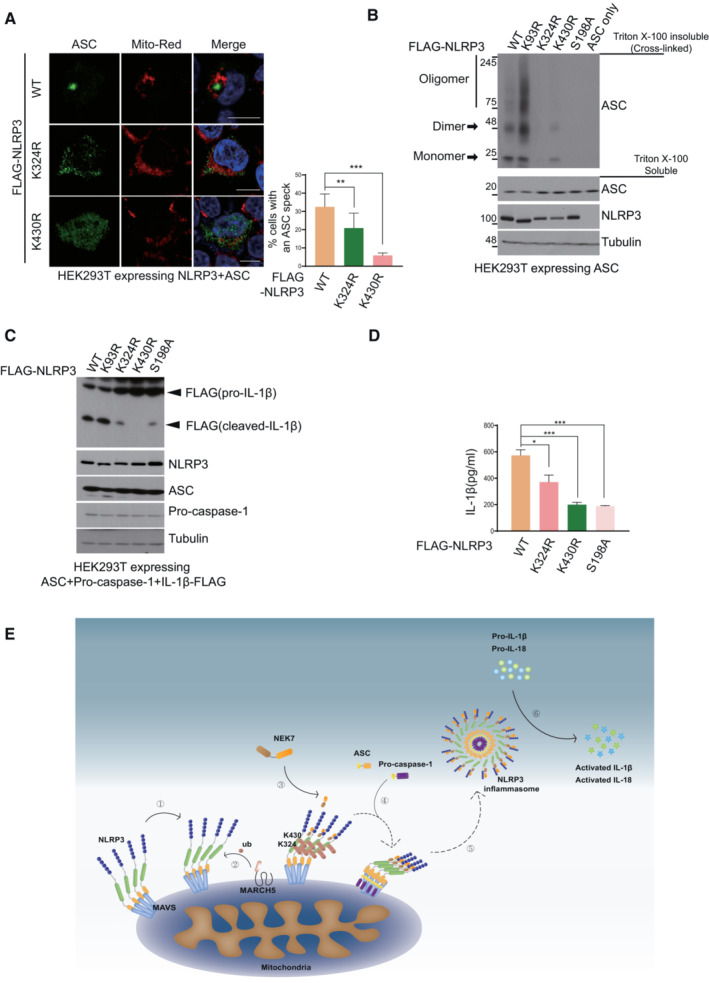
-
AHEK293T cells were transfected with NLRP3 WT or indicated Lys mutants and ASC. Cells were stimulated with 200 ng/ml LPS for 4 h followed by 15 μM nigericin for 30 min. The percentage of cells with an ASC speck was quantified after Confocal microscopy. At least 100 cells were analyzed. Values are the mean ± SD. Values, **P < 0.01, ***P < 0.001 (two‐tailed Student's t‐test). Bar, 10 μm.
-
BHEK293T cells were transfected with ASC and NLRP3 (WT or individual NLRP3 mutants). Cells were treated for 4 h with 200 ng/ml LPS and 30 min with 15 μM nigericin. Harvested cell lysates were incubated with 2 mM DSS for cross‐linking. Triton X‐100 insoluble pellets and soluble fraction lysates were detected by immunoblotting with the indicated antibodies.
-
CNLRP3 inflammasome‐reconstituted HEK293T cells with NLRP3 WT or indicated mutants were stimulated for 4 h with 200 ng/ml LPS and 30 min with 15 μM nigericin. Harvested cells were detected by immunoblotting with the indicated antibodies.
-
DCulture supernatants were obtained from (C) and subjected to ELISA to quantify secreted IL‐1β. Representative data are shown from independent experiments that were repeated at least three times. Values are the mean ± SD. *P < 0.05, ***P < 0.001 (two‐tailed Student's t‐test)
-
ESchematic diagram of NLRP3 inflammasome activation by MARCH5. Upon activation by a priming signal, NLRP3 associates with MAVS on mitochondria ①. MARCH5 recognizes and ubiquitinates two lysine sites, K324 and K430, on the NLRP3 NACHT domain via K27‐linked polyubiquitination ②. NLRP3 ubiquitination by MARCH5 promotes the recruitment of NEK7 and enables self‐oligomerization ③. As a result, ASC and pro‐caspase‐1 are recruited to oligomerized NLRP3 ④ for NLRP3 inflammasome assembly ⑤ and trigger the activation of caspase‐1 and pro‐cytokine cleavage ⑥.
Source data are available online for this figure.
Discussion
NLRP3 responds to various stimuli, and aberrant NLRP3 activation underlies autoimmune and degenerative diseases such as atherosclerosis, Alzheimer's disease, type 2 diabetes, and lupus (Fusco et al, 2020). A multilayered regulatory mechanism ensures accurate NLRP3 inflammasome activation, which has the benefit of presenting multiple specific molecular targets for disease therapy (McKee & Coll, 2020). In the present study, we uncovered the sequence of steps of NLRP3 assembly on the mitochondrial outer membrane (Fig 7E). When the activation signal is received, primed NLRP3 moves to mitochondria and binds MAVS (①). The mitochondrial‐resident E3 ligase MARCH5 delivers ubiquitin to the NACHT domain of NLRP3 through K27‐linked polyubiquitination (②). This modification triggers NEK7 binding to ubiquitinated NLRP3, forming the NEK7‐NLRP3 multimeric complex (③). Consequently, ASC and pro‐caspase‐1 associate with NLRP3 oligomers to complete inflammasome assembly (④.⑤). Finally, the pro‐inflammatory cytokines IL‐1β and IL‐18 are cleaved by activated caspase‐1 (⑥). Thus, our data show that mitochondrial‐resident MARCH5 E3 ligase is an essential regulator that initiates NEK7 binding to NLRP3. It also highlights the mitochondria as an onset platform for NLRP3 immune complex formation.
NLRP3 has been observed in the cytosol as well as in membranous organelles (the ER, mitochondria, and Golgi) during inflammasome activation (Hamilton & Anand, 2019). In resting state cells, the inactive state of NLRP3 could be monomeric or oligomeric double‐ring cages embedded in the organelle membrane such that its pyrin domains are hidden inside (Andreeva et al, 2021). NLRP3 undergoes dynamic relocation in cells for inflammasome activation (Wang et al, 2013), and several activation steps are necessary for inflammasome assembly and activation (Paik et al, 2021). MAVS mediates NLRP3 recruitment to mitochondria through direct interaction, and this step is indispensable for NLRP3 inflammasome activation (Park et al, 2013; Subramanian et al, 2013). Indeed, the absence of MAVS in stimulated THP‐1 cells disturbed the interaction of NLRP3 with ASC and abolished NLRP3 binding to MARCH5 (Appendix Fig S1D). MARCH5‐dependent ubiquitination of NLRP3 did not affect the MAVS‐NLRP3 interaction, but it severely damaged its interaction with NEK7 (Fig 6E). Thus, the data suggest that NLRP3 recruitment to mitochondria by MAVS precedes its interaction with MARCH5. NLRP3 phosphorylation at Ser198 is a key priming event during LPS stimulation (Song et al, 2017). We observed that cells expressing the NLRP3‐S198A mutant also failed to form the multimeric NEK7‐NLRP3 complex (Fig 6E and F). Thus, at least two preceding events may occur before MARCH5 ubiquitinates NLRP3: phosphorylation of NLRP3 by JNK1 and mitochondrial localization of NLRP3 by MAVS. In ASC knock down cells, fluorescence microscopy using GFP‐NLRP3 revealed oligomerized NLRP3 puncta upon stimulation with LPS plus nigericin (Fig 6A and Appendix Fig S1A). ASC specks are limited to one per cell near the MTOC, but more NLRP3 puncta per cell can form in ASC KD cells.
Interestingly, microtubule affinity regulating kinase 4 (MARK4) interacts with NLRP3 and facilitates NLRP3 localization to the mitochondria. MARK4 promotes the relocation of NLRP3 to the MTOC in a microtubule‐dependent manner (Li et al, 2017). Thus, our data suggest that mitochondria serve as an onset platform for NLRP3‐NEK7 complex formation and that subsequent association with ASC and pro‐caspase‐1 occurs near the MTOC. Recently, it has been reported that NLRP3 recruited to the Golgi complex did not require NEK7 for its activation (Schmacke et al, 2022). Thus, there are, at least, two main pathways for NLRP3 activation in cells; NEK7‐dependent and ‐independent NLRP3 activation in mitochondria and Golgi apparatus, respectively.
NLRP3 forms an autoinhibitory conformation in a resting state and undergoes a conformational change upon inflammasome stimulation. Self‐oligomeric assembly of NLRP3 requires its NACHT domain, which directly interacts with the neighboring NLRP3. The NACHT domain comprises four subdomains, NBD, HD1, WHD (winged helix domain), and HD2, and a drastic conformational rearrangement among these subdomains occurs, positioning the NLRP3 NBD‐HD1 module to a direct interacting interface between neighboring NLRP3 molecules (Sharif et al, 2019). Notably, NEK7 is a crucial upstream mediator of NLRP3 oligomerization. Among the NLR receptors, NEK7 specifically binds NLRP3 (He et al, 2016). A cryo‐EM structure study revealed that NEK7 binds to the LRR domain and NACHT domain (HD2) of inactive NLRP3 (Sharif et al, 2019). An as‐yet unidentified priming or activation step is required to induce rotational activation of the NACHT domain. This rotational activation step is necessary for uncovering part of the NACHT surface, enabling NLRP3 oligomerization. We demonstrated that MARCH5 ubiquitinates the NACHT domain of NLRP3 and targets two lysine residues (K324 and K430) of NLRP3 in the NBD and HD1 subdomains, respectively. Multimeric NEK7‐NLRP3 complex formation was abrogated in cells depleted of MARCH5 (Fig 6D) or cells expressing NLRP3 mutants that are defective for ubiquitination at these lysine residues (Fig 6F). Under these conditions, the NLRP3‐NEK7 interaction was impaired (Fig 6C and E). Thus, it can be proposed that upon inflammasome stimulation, NLRP3 attaches to MAVS on the mitochondrial outer membrane, and MARCH5‐mediated ubiquitination of the NLRP3 NACHT domain might trigger its rotational switch to allow NEK7 to bind NLRP3, forming a large oligomeric complex of NEK7 and NLRP3. Meanwhile, we noticed that K93 residue of NLRP3 is likely to be a target ubiquitination site by MARCH5 (Fig 5F) and however, a ubiquitination‐defective mutant of NLRP3 K93R neither interfered with ASC oligomerization (Fig 7B) nor promoted the production of active IL‐1β (Fig 7C). It is shown that mitochondria‐associated NLRP3 oligomers recruits ASC via homotypic PYD‐PYD domain interaction during the process of NLRP3 inflammasome assembly (Vajjhala et al, 2012; Subramanian et al, 2013). Thus, we speculate that ubiquitination at NLRP3 K93 next to PYD (aa 1–91) may interfere with these interactions, avoiding premature association between NLRP3 and ASC as well as ASC oligomerization before NEK7‐NLRP3 binding
Taken together, our data show that MARCH5 plays a key role in regulating NLRP3 inflammasome activation. The mechanism of MARCH5‐mediated NLRP3 inflammasome activation provides new insight into the NEK7‐NLRP3 binding signal and NLRP3 assembly for inflammasome activation on the mitochondria. Uncontrolled NLRP3 inflammasome activation is involved in several human diseases. Although blocking the cytokines or molecules downstream of the inflammasome is a common strategy for treating several diseases, cytokine secretion and pyroptosis by inflammasome activation are both pathogenic. Therefore, targeting inflammasome assembly would be a more effective therapy for these diseases. Targeting the interaction of NLRP3 and MARCH5 or regulating MARCH5 activity may represent new therapeutic strategies for NLRP3‐associated inflammatory disease.
Materials and Methods
Reagents and Tools table
| Reagent/resource | Reference or source | Identifier or catalog number |
|---|---|---|
| Experimental models | ||
| HEK293T | ATCC | Cat# ACS‐4500 |
| THP‐1 | ATCC | Cat# TIB‐202 |
| L929 | Je‐Wook Yoo | N/A |
| iBMDM | Je‐Wook Yoo | N/A |
| iBMDM NLRP3‐GFP | Je‐Wook Yoo | N/A |
| Citrobacter rodentium | ATCC | Cat# 51459 |
| Salmonella typhimurium | ATCC | Cat# 14028 |
| Pseudomonas aeruginosa | ATCC | Cat# BAA‐1744 |
| March5 fl/fl mice | This paper | N/A |
| March5 fl/fl;Lyz‐cre mice | This paper | N/A |
| Recombinant DNA | ||
| pcDNA3.1 | Invitrogen Thermo Fisher | Cat# V79020 |
| pcDNA3.1‐myc‐MARCH5 | This paper | N/A |
| pcDNA3.1‐myc‐MARCH5 (2KR) | This paper | N/A |
| pcDNA3.1‐myc‐MARCH5 (H43W) | This paper | N/A |
| pcDNA6‐SFB‐MARCH5 WT | This paper | N/A |
| pcDNA6‐SFB‐MARCH5 (2KR) | This paper | N/A |
| pCMV6‐AC‐cMyc‐DDK | This paper | N/A |
| pCMV6‐NLRP3 WT‐MycDDK | This paper | N/A |
| pCMV6‐NLRP3 K93R‐MycDDK | This paper | N/A |
| pCMV6‐NLRP3 K192R‐MycDDK | This paper | N/A |
| pCMV6‐NLRP3 K194R‐MycDDK | This paper | N/A |
| pCMV6‐NLRP3 K324R‐MycDDK | This paper | N/A |
| pCMV6‐NLRP3 K430R‐MycDDK | This paper | N/A |
| pCMV6‐NLRP3 K689R‐MycDDK | This paper | N/A |
| pCMV6‐NLRP3 K696R‐MycDDK | This paper | N/A |
| pCMV6‐NLRP3 S194A‐MycDDK | This paper | N/A |
| pcDNA3.1/Myc‐NLRP3 WT | Ren et al 2019 | N/A |
| pcDNA3.1/Myc‐NLRP3 ▵PYD | Ren et al 2019 | N/A |
| pcDNA3.1/Myc‐NLRP3 ▵Linker | Ren et al 2019 | N/A |
| pcDNA3.1/Myc‐NLRP3 ▵NACHT | Ren et al 2019 | N/A |
| pcDNA3.1/Myc‐NLRP3 ▵LRR | Ren et al 2019 | N/A |
| pCMV3‐C‐FLAG‐IL‐1β | Sino Biological | Cat# HG10139‐CF |
| pCMV3‐C‐HA‐NEK7 | Sino Biological | Cat# HG11534‐CY |
| Antibodies | ||
| Mouse monoclonal anti‐NLRP3 (Cryo2‐) |
Adipogen 1:5,000 |
Cat# AG‐20B‐0014 |
| Rabbit polyclonal anti‐NLRP3 |
Cell Signaling Technology 1:1,000 |
Cat# 15101 |
| Goat polyclonal anti‐NLRP3 |
Abcam 1:1,000 |
Cat# ab4207 |
| Rabbit polyclonal anti‐ASC |
Adipogen 1:1,000 |
Cat# AG‐25B‐0006 |
| Mouse monoclonal anti‐ASC |
Santa Cruz Biotechnology 1:1,000 |
Cat# sc‐514414 |
| Rabbit polyclonal anti‐ASC |
Santa Cruz Biotechnology 1:1,000 |
Cat# sc‐22514‐R |
| Mouse monoclonal anti‐IL‐1β |
Cell Signaling Technology 1:1,000 |
Cat# 12242 |
| Goat Polyclonal anti‐IL‐1β (p20) |
Adipogen 1:1,000 |
Cat# AF‐401‐NA |
| Mouse monoclonal anti‐IL‐1β |
Santa Cruz Biotechnology 1:1,000 |
Cat# 12742 |
| Mouse monoclonal anti‐mouse caspase‐1 |
Adipogen 1:1,000 |
Cat# AG‐20B‐0042 |
| Rabbit polyclonal anti‐human caspase‐1 |
Cell Signaling Technology 1:1,000 |
Cat# 3866 |
| Mouse monoclonal anti‐human caspase‐1 |
Santa Cruz Biotechnology 1:1,000 |
Cat# sc‐56036 |
| Mouse monoclonal anti‐mouse MAVS |
Cell Signaling Technology 1:1,000 |
Cat# 83000 |
| Rabbit polyclonal anti‐human MAVS |
Cell Signaling Technology 1:1,000 |
Cat# 3993 |
| Rabbit polyclonal anti‐MARCH5 |
Shigeru YANAGI 1:1,000 |
N/A |
| Mouse monoclonal anti‐NEK7 |
Santa Cruz Biotechnology 1:1,000 |
Cat# sc‐393539 |
| Rabbit monoclonal anti‐NEK7 |
Abcam 1:1,000 |
Cat# ab133514 |
| Mouse monoclonal anti‐FLAG |
Sigma‐Aldrich 1:1,000 |
Cat# F1804 |
| Mouse monoclonal anti‐c‐Myc |
Santa Cruz Biotechnology 1:1,000 |
Cat# sc‐40 |
| Mouse monoclonal anti‐HA |
Santa Cruz Biotechnology 1:1,000 |
Cat# sc‐7392 |
| Mouse monocclonal anti‐Tubulin |
Santa Cruz Biotechnology 1:1,000 |
Cat# sc‐73242 |
| Rabbit polyclonal anti‐GAPDH |
Cell Signaling Technology 1:1,000 |
Cat# 5174 |
| Goat anti Mouse IgG (H+L)‐HRP conjugate |
Bio‐rad 1:5,000 |
Cat# 170516 |
| Goat anti Rabbit IgG (H+L)‐HRP conjugate |
Bio‐rad 1:5,000 |
Cat# 1706515 |
| Donkey anti Goat IgG (H+L)‐HRP conjugate |
Bio‐rad 1:5,000 |
Cat# 642005 |
| Oligonucleotides and other sequence‐based reagents | ||
|
Human MARCH5 targeted siRNA 5′‐GG GUG GAA UUG CGU UUG UU‐3′ |
Bioneer | This paper |
|
Mouse March5 targeted siRNA 5′‐GGU UGU AGG CCA UAA AGA A‐3′ |
Bioneer | This paper |
|
Human MAVS targeted siRNA 5′‐CAG GUU GGC CUC AUG AGA U‐3′ |
Bioneer | Cat# 57506 |
|
Mouse Asc targeted siRNA 5′‐GCU CUU CAG UUU CAC ACC A‐3′ |
Bioneer | This paper |
| Chemicals, Enzymes and other reagents | ||
| Lipopolysaccharides from Escherichia coli O26:B6 | Sigma‐Aldrich | Cat# L8274 |
| ATP | Sigma‐Aldrich | Cat# A6419 |
| Nigericin | Invivogen | Cat# tlrl‐nig |
| FLA‐ST | Invivogen | Cat# tlrl‐stfla |
| Poly(I:C) | Invivogen | Cat# tlrl‐pic |
| Poly(dA:dT) | Invivogen | Cat# tlrl‐patn‐1 |
| Recombinant Human MARCH5 GST Protein | Abnova | Cat# H00054708‐P0 |
| Anti‐FLAG M2 affinity Gel | Sigma‐Aldrich | Cat# A2220 |
| DSS (disuccinimidyl suberate) | Invitrogen Thermo Fisher | Cat# A39267 |
| Software | ||
| GraphPad Prism 8.0 | GraphPad, San Diego, CA | https://www.graphpad.com/scientific‐software/prism/ |
| ImageJ | National Institues of Health | https://imagej.nih.gov/nih‐image/ |
| Biorender | N/A | https://biorender.com/ |
| Ubpred | N/A | http://ubpred.org/ |
| Other | ||
| LDH‐Glo™ Cytotoxicity Assay | Promega | Cat# J2380 |
| Caspase‐Glo® 1 Inflammasome Assay | Promega | Cat# G9951l |
| Lipofectamine 2000 | Invitrogen | Cat# 11668‐019 |
| ELISA MAX™ Deluxe Set Mouse IL‐1β | Biolegend | Cat# 432616 |
| Mouse Caspase‐1 ELISA Kit | Novus biologicals | Cat# NBP2‐75014 |
| BD OptEIA™ Mouse TNF (Mono/Mono) ELISA Set | BD OptEIA | Cat# 555268 |
| BD OptEIA™ Mouse IL‐6 ELISA Set | BD OptEIA | Cat# 555240 |
| IL‐1 beta Human Uncoated ELISA Kit | Invitrogen | Cat# 88‐7261‐22 |
| Recombinant Mouse IL‐18 ELISA Set | MBL | Cat# B002‐5 |
Methods and Protocols
Mice
March5 tm1a mice on a C57BL/6 background harboring LoxP sites flanking March5 exon3 were purchased from the European Mouse Mutant Archive. We cross‐bred Lysozyme M‐Cre mice to generate March5 cKO mice (March5 tm1c ). All mice were maintained in a specific pathogen‐free animal facility. The genotypes of March5 animals were determined by PCR using a specific primer: forward 5′‐GTGACACTACTTTTGATGTGAAG‐3′, reverse 5′‐ATGCTACAGCTCATGTGTAAG‐3′. For the mice experiment, 12–20 weeks‐old male mice were used. Chungnam National University's Institutional Animal Use and Care Committee approved all animal experiments (CNU‐00777, 202109A‐CNU‐168). They were performed in biosafety level BSL‐2 laboratory facilities with the Guide for the Care and Use of Laboratory Animals (published by the US National Institutes of Health).
LPS septic shock model
Lipopolysaccharide (28 mg/kg body weight) was given to mice aged 6–8 weeks via intraperitoneal injection. Serum, spleen, and peritoneal lavage were obtained after 12 and 24 h, and cytokine levels were measured using ELISA. For survival analysis, mice were administered LPS (28 mg/kg body weight) intraperitoneally and monitored for 8 days.
P. aeruginosa infection
Six‐ to eight‐week‐old March5 fl/fl and March5 cKO mice were inoculated intraperitoneally with P. aeruginosa (ATCC, BAA‐1744) (100 μl; 1 × 107 CFU) suspended in sterile endotoxin‐free PBS. The animals were sacrificed at 12 and 24 h of postinoculation, and blood, spleen, and peritoneal lavage was collected. Coagulated blood was centrifuged at 16,000 g for 15 min, and the supernatants were collected as serum. Spleen homogenate, serum, and peritoneal fluid samples were assayed for TNF‐α, IL‐6, IL‐1β, and IL‐18. For survival analysis, mice were administered with P. aeruginosa (100 μl; 1 × 107 CFU) intraperitoneally and monitored for 10 days.
Cell culture
THP‐1 cells were cultured in RPMI1640 with 10% heat‐inactivated FBS and 1% antibiotic‐antimycotic. For differentiation, THP‐1 cells were treated with 100 nM Phorbol‐12‐myristate‐13‐acetate (PMA) for 72 h. Bone marrow‐derived macrophages (BMDMs) were isolated from femurs and tibias of 12–20‐week‐old mice. After collection of bone marrow, red blood cells were removed by ACK lysis buffer (GIBCO BRL), and cells were cultured in RPMI1640 supplemented with 10% heat‐inactivated FBS and 1% antibiotic–antimycotic containing 20% L929 cell‐conditioned medium. HEK293T (ATCC, ACS‐4500) cells were cultured in DMEM supplemented with 10% heat‐inactivated FBS and 1% antibiotic–antimycotic. Immortalized BMDMs (iBMDMs) were kindly provided by Prof. Je‐Wook Yu and cultured in DMEM with 10% heat‐inactivated FBS and 1% antibiotic–antimycotic with 2% L929 cell‐conditioned medium. All cells were cultured at 37°C in a 5% CO2 incubator.
Western blotting
Cells were lysed in an SDS sample buffer and boiled for 10 min. The cell lysate was separated by 4–20% gradient SDS–PAGE and transferred to the nitrocellulose membrane. Immunoblots were detected by ECL system (GE Healthcare) and analyzed by using the following antibodies: anti‐NLRP3 (1:10,000, AG‐20B‐0014, Adipogen), anti‐NLRP3 (1:1,000, #15101S, Cell Signalling), anti‐mouse ASC (1:1,000 AG‐25B‐0006, Adipogen), anti‐human ASC (1:1,000, sc‐22514‐R, Santa Cruz Biotechnology), anti‐ASC (1:1,000, sc‐514414, Santa Cruz Biotechnology), anti‐human IL‐1β (1:1,000, #12242, Cell Signaling Technology), anti‐IL‐1β (1:1,000, sc‐12742, Santa Cruz Biotechnology), anti‐mouse caspase‐1 (1:1,000, AG‐20B‐0042, Adipogen), anti‐human caspase‐1 (1:1,000, #3866, Cell Signaling Technology), anti‐caspase‐1 (1:1,000, sc‐56036, Cell Signalling Technology), anti‐mouse IL‐1β (1:1,000, AF‐401‐NA, R&D System), anti‐human MAVS (1:1,000, #3993, Cell Signaling Technology), anti‐FLAG (1:10,000, F1804, Sigma Aldrich), anti‐c‐Myc (9E10) (1:1,000, sc‐40, Santa Cruz Biotechnology), anti‐Tubulin (1:10,000, sc‐73232, Santa Cruz Technology), anti‐GAPDH (1:5,000, #2188, Cell Signaling Technology), anti‐HA (1:1,000, sc‐7392, Santa Cruz Technology), anti‐Ub (1:1,000, sc‐8017, Santa Cruz Technology). To detect the release of caspase‐1 (p20) and IL‐1β (p18), a conditioned medium was collected and incubated with the same volume of methanol and 1/4 volume of chloroform. The mixture was centrifuged at 16,000 × g for 10 min at RT. The top layer (500 μl) was removed and mixed with 1 ml of methanol by vortexing. Then, the mixture was centrifuged at 16,000 × g for 10 min at RT. The supernatant was removed and dried at RT for 15 min. The protein pellet was dissolved in 2× SDS sample buffer and boiled at 100°C for 15 min, and subjected to SDS–PAGE.
Reconstitution of NLRP3 inflammasome in HEK293T cells
HEK293T cells were cultured in 12‐well plates and transfected by polyethyleneimine (PEI, Polysciences) with following constructs: FLAG‐NLRP3 or Flag‐NLRP3 K mutants (100 ng/well) or Myc‐NLRP3 (10 ng/well), Flag‐ASC (10 ng/well), Flag‐pro‐caspase‐1 (50 ng/well), pro‐IL‐1β‐Flag (400 ng/well). After 24–36 h, cells were lysed with 1× sample buffer and sonicated. The expression of the protein was assessed by immunoblotting.
Immunoprecipitation
Cells were lysed in lysis buffer (50 mM Tris, pH 7.8, 50 mM NaCl, 1% NP‐40, 5 mM EDTA, 10% Glycerol, 1 mg/ml Aprotinin, 1 mg/ml Leupeptin, 5 mM NaF, 0.5 mM Na3VO4). Lysates were sonicated and centrifuged at 16,000 × g at 4°C for 20 min. The supernatant was incubated with either anti‐NLRP3 or anti‐Flag‐M2 beads at 4°C overnight with agitation. The next day, samples were incubated more with protein A/G Sepharose beads (GE healthcare) for 2 h washed with lysis buffer and then eluted with 2× SDS sample buffer. Samples were separated by SDS–PAGE and subjected to immunoblotting.
RNA interference
To knock down the protein expression in THP‐1 and iBMDMs, cells were transfected with small interfering RNA (siRNA) oligonucleotides. Following siRNA were custom synthesized by Bioneer; Human‐specific MARCH5 siRNA (5′‐GGGUGGAAUUGCGUUUGUUTT‐3′), mouse‐specific March5 siRNA (5′‐GGU UGU AGG CCA UAA AGA A‐3′), mouse‐specific ASC siRNA (5′‐GCUCUUCAGUUUCACACCA‐3′), and Human‐specific MAVS siRNA was obtained from Bioneer (57506). siRNA was transfected in THP‐1 cells by Lipofectamine 2000 (Invitrogen).
In vivo ubiquitination assay
HEK293T cells were transfected with HA‐ubiquitin, FLAG, or Myc‐tagged constructs by using polyethylenimine (PEI, Polysciences, 23966). After 24 h later, cells were activated by 200 ng/ml LPS for 4 h followed by 15 μM nigericin for 30–60 min. Cell lysates were immunoprecipitated with Flag‐M2 bead (Sigma‐Aldrich, A2220) or anti‐Myc antibody at 4°C overnight with agitation. Beads were washed with lysis buffer four times. They were eluted with 2× SDS sample buffer, and subjected to western blotting.
Inflammasome activation
Primary BMDMs or THP‐1 cells were seeded in the culture dishes overnight. Cells were primed for 4 h with 200 ng/ml LPS (Sigma‐Aldrich, L9274) and then stimulated as follows: 5 mM ATP (Sigma‐Aldrich, A6419) for 30–45 min, 15 μM nigericin (Invivogen, tlrl‐nig) for 30 min ~1 h respectively. Cells were stimulated with 250 ng/ml FLA‐ST (Invivogen, tlrl‐stfla) for 4 h. Cells were transfected with 2 μg/ml poly(I:C) (Invivogen, tlrl‐pic) or 2 μg/ml poly(dA:dT) (Invivogen, tlrl‐patn‐1) by using Lipofectamine 2000 for 6 h. Citrobacter rodentium (ATCC, 51459), Pseudomonas aeruginosa (ATCC, BAA‐1744), and Salmonella typhimurium strain (ATCC, 14028) grown in lysogeny broth in a log phase were resuspended in phosphate‐buffered saline. For the bacterial infection, each bacteria titer was examined in serial dilution inoculating LB Agar plates. Primary BMDMs were seeded (1 × 106 cells/well) in 12‐well culture plates and allowed to rest for 12 h. Cells were then stimulated with the indicated bacteria.
ELISA
Cells were seeded into 12‐well plates and treated with indicated activators and indicated bacteria. The supernatant was collected at the indicated time points and then quantified by using following commercially available ELISA kits: mouse IL‐1β (Biolegend, 432616), Caspase‐1 (Novus biologicals, NBP2‐75014), mouse TNF‐α (BD OptEIA, 555268), mouse IL‐6 (BD OptEIA, 555240), and mouse IL‐18 (MBL, 065FA).
Cytotoxicity assay
Bone marrow‐derived macrophages were seeded into 12‐well plates and treated with activators. Lactate dehydrogenase (LDH) release to the culture supernatant was quantified by using the LDH cytotoxicity assay kit (Promega).
ASC speck staining
Cells were primed with 200 ng/ml LPS for 4 h, and subsequently stimulated with 5 mM ATP or 15 μM nigericin for 30 min. poly(dA:dT) were transfected using Lipofectamine 2000. After stimulation, cells were fixed with 4% paraformaldehyde for 30 min at 37°C and permeabilized with 0.1% Triton X‐100 for 10 min. The slides were blocked with 2% BSA in PBS. Cells were stained with anti‐ASC (1:200) and anti‐NLRP3 (1:300). To stain nuclei, cells were stained with DAPI. Cells were visualized by confocal microscopy (Zeiss LSM 710) at the three‐dimensional immune system imaging core facility of Ajou University.
ASC oligomer cross‐linking
Cells were primed with 200 ng/ml LPS for 4 h, and subsequently stimulated with 5 mM ATP or 15 μM nigericin. Cells were lysed with Triton buffer (50 mM Tris‐HCl, pH 7.5, 150 mM NaCl, 0.5% Triton X‐100, 1 mg/ml Aprotinin, 1 mg/ml Leupeptin, 5 mM NaF, 0.5 mM Na3VO4). The cell lysates were centrifuged at 6,000 × g at 4°C for 15 min. The supernatant was collected as the Triton X‐100 soluble fraction, and the pellet was collected as Triton X‐100 insoluble fraction. The Triton X‐100 soluble fraction was mixed with 6× SDS sample buffer and boiled at 100°C for 15 min. The Triton X‐100 insoluble fraction was cross‐linked for 30 min at 37°C with 2 mM disuccinimidyl suberate (DSS) to cross‐link the ASC oligomer. After centrifuging at 6,000 × g for 15 min at 4°C, the pellets were collected and dissolved in 2× SDS sample buffer and subjected to SDS‐PAGE.
Semidenaturing detergent agarose gel electrophoresis (SDD‐AGE)
Cells were primed with 200 ng/ml LPS for 4 h, and stimulated by 5 mM ATP or 15 μM nigericin for the indicated duration. Cells were lysed with 1× SDD sample buffer (1× TBE buffer, 10% Glycerol, 2% SDS, 25% Bromophenol blue). NLRP3 oligomers were separated by 1% agarose gel in running buffer (1× TBE and 0.1% SDS) for 1 h 30 min with 80 V at 4°C. Samples were transferred to the PVDF membrane (Millipore) and detected by using the anti‐NLRP3 antibody.
Two‐dimensional polyacrylamide gel electrophoresis (2D‐PAGE)
HEK293T cells and MARCH5 knock‐downed iBMDMs were plated in 6‐well plates. Cells were primed with 200 ng/ml LPS for 4 h, and followed by 15 μM nigericin for 60 min. Cells were lysed using native lysis buffer (20 mM Tris‐HCl pH 7.4, 137 mM NaCl, 2 mM EDTA pH 8.0, 10% Glycerol, 0.1% Triton X‐100, 10 μg/ml Aprotinin, 10 μg/ml Leupeptin, 1 mM PMSF, 0.5 mM NaF, 0.5 mM Na3VO4) for 15 min on ice. Cell lysates were centrifuged at 20,000 × g for 30 min at 4°C, and subjected to 4–12% Blue‐Native PAGE. And then, native gels were soaked in 10% SDS for 5 min. For 2D‐PAGE, the natively resolved gel was cut well by well and loaded into 4–12% SDS‐PAGE gel, followed by conventional western blotting.
Statistical analysis
All statistical analyses were performed using the Prism 6 software (GraphPad). For each result, error bars represent the mean ± SD or mean ± SEM from at least three independent experiments. Statistical significance was measured by a two‐tailed unpaired Student's t‐test or Mantel–Cox test. P‐values are indicated in the figures.
Author contributions
Yeon‐Ji Park: Conceptualization; data curation; formal analysis; funding acquisition; investigation; methodology; writing – original draft; project administration; writing – review and editing. Niranjan Dodantenna: Conceptualization; data curation; formal analysis; investigation; methodology. Yonghyeon Kim: Data curation; formal analysis; methodology. Tae‐Hwan Kim: Investigation. Ho‐Soo Lee: Investigation. Young‐Suk Yoo: Investigation. June Heo: Investigation. Jae‐Ho Lee: Writing – review and editing. Myung‐Hee Kwon: Data curation; formal analysis. Ho Chul Kang: Data curation; formal analysis. Jong‐Soo Lee: Supervision; funding acquisition; project administration; writing – review and editing. Hyeseong Cho: Funding acquisition; investigation; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View and Appendix
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank Prof. Je‐Wook Yu (Yonsei University College of Medicine, Korea) for providing iBMDMs and NLRP3‐GFP iBMDMs and Prof. Ho Chul Kang (Ajou University, Korea) for providing HA‐ubiquitin WT and Lys mutants. This work was supported by grants from the National Research Foundation of Korea grants funded by the Korean Government (MSIP) (NRF‐2020R1A2C3011423, NRF‐2022R1I1A1A01071281, NRF‐RS‐2023‐00209214, NRF‐2019R1A2C2008283, NRF‐2021R1A6A1A03045495).
The EMBO Journal (2023) 42: e113481
Contributor Information
Jong‐Soo Lee, Email: jongsool@cnu.ac.kr.
Hyeseong Cho, Email: hscho@ajou.ac.kr.
Data availability
This study includes no data deposited in external repositories.
References
- Andreeva L, David L, Rawson S, Shen C, Pasricha T, Pelegrin P, Wu H (2021) NLRP3 cages revealed by full‐length mouse NLRP3 structure control pathway activation. Cell 184: 6299–6312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banoth B, Cassel SL (2018) Mitochondria in innate immune signaling. Transl Res 202: 52–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer J, Bakke O, Morth JP (2017) Overview of the membrane‐associated RING‐CH (MARCH) E3 ligase family. N Biotechnol 38: 7–15 [DOI] [PubMed] [Google Scholar]
- Chathuranga K, Kim TH, Lee H, Park JS, Kim JH, Chathuranga WAG, Ekanayaka P, Choi YJ, Lee CH, Kim CJ et al (2020) Negative regulation of NEMO signaling by the ubiquitin E3 ligase MARCH2. EMBO J 39: e105139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Chen ZJ (2018) PtdIns4P on dispersed trans‐Golgi network mediates NLRP3 inflammasome activation. Nature 564: 71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Nuñez G (2010) Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol 10: 826–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM (2007) ATP activates a reactive oxygen species‐dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem 282: 2871–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Q, Wang Y, Zhang Y, Li M, Li D, Huang X, Wu Y, Pu J, Wu M (2016) Pseudomonas aeruginosa triggers macrophage autophagy to escape intracellular killing by activation of the NLRP3 inflammasome. Infect Immun 84: 56–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco R, Siracusa R, Genovese T, Cuzzocrea S, Di Paola R (2020) Focus on the role of NLRP3 inflammasome in diseases. Int J Mol Sci 21: 4223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Callaway JB, Ting JP (2015) Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 21: 677–687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton C, Anand PK (2019) Right place, right time: localisation and assembly of the NLRP3 inflammasome. F1000Res 8: 676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Zeng MY, Yang D, Motro B, Nunez G (2016) NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530: 354–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD (2013) Mitochondrial reactive oxygen species induces NLRP3‐dependent lysosomal damage and inflammasome activation. J Immunol 191: 5230–5238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoss F, Rodriguez‐Alcazar JF, Latz E (2017) Assembly and regulation of ASC specks. Cell Mol Life Sci 74: 1211–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphries F, Bergin R, Jackson R, Delagic N, Wang B, Yang S, Dubois AV, Ingram RJ, Moynagh PN (2018) The E3 ubiquitin ligase Pellino2 mediates priming of the NLRP3 inflammasome. Nat Commun 9: 1560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, Sadler JJ, Knepper‐Adrian V, Han R, Qiao L et al (2013) Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39: 311–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karbowski M, Neutzner A, Youle RJ (2007) The mitochondrial E3 ubiquitin ligase MARCH5 is required for Drp1 dependent mitochondrial division. J Cell Biol 178: 71–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley N, Jeltema D, Duan Y, He Y (2019) The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci 20: 3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Park YY, Yoo YS, Cho H (2016) Self‐clearance mechanism of mitochondrial E3 ligase MARCH5 contributes to mitochondria quality control. FEBS J 283: 294–304 [DOI] [PubMed] [Google Scholar]
- Li X, Thome S, Ma X, Amrute‐Nayak M, Finigan A, Kitt L, Masters L, James JR, Shi Y, Meng G et al (2017) MARK4 regulates NLRP3 positioning and inflammasome activation through a microtubule‐dependent mechanism. Nat Commun 8: 15986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee CM, Coll RC (2020) NLRP3 inflammasome priming: a riddle wrapped in a mystery inside an enigma. J Leukoc Biol 108: 937–952 [DOI] [PubMed] [Google Scholar]
- Mevissen TET, Hospenthal MK, Geurink PP, Elliott PR, Akutsu M, Arnaudo N, Ekkebus R, Kulathu Y, Wauer T, El Oualid F et al (2013) OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell 154: 169–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima S, Tokuyama T, Yonashiro R, Inatome R, Yanagi S (2014) Roles of mitochondrial ubiquitin ligase MITOL/MARCH5 in mitochondrial dynamics and diseases. J Biochem 155: 273–279 [DOI] [PubMed] [Google Scholar]
- Paik S, Kim JK, Silwal P, Sasakawa C, Jo E‐K (2021) An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol 18: 1141–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YY, Lee S, Karbowski M, Neutzner A, Youle RJ, Cho H (2010) Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin‐related protein 1 and mitofusin 1. J Cell Sci 123: 619–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes‐Alnemri T, Yu JW, Alnemri ES (2013) The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol 191: 4358–4366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park YY, Nguyen OT, Kang H, Cho H (2014) MARCH5‐mediated quality control on acetylated Mfn1 facilitates mitochondrial homeostasis and cell survival. Cell Death Dis 5: e1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y‐J, Oanh NTK, Heo J, Kim S‐G, Lee H‐S, Lee H, Lee J‐H, Kang HC, Lim W, Yoo Y‐S (2020) Dual targeting of RIG‐I and MAVS by MARCH5 mitochondria ubiquitin ligase in innate immunity. Cell Signal 67: 109520 [DOI] [PubMed] [Google Scholar]
- Phu L, Rose CM, Tea JS, Wall CE, Verschueren E, Cheung TK, Kirkpatrick DS, Bingol B (2020) Dynamic regulation of mitochondrial import by the ubiquitin system. Mol Cell 77: 1107–1123 [DOI] [PubMed] [Google Scholar]
- Pu Q, Gan C, Li R, Li Y, Tan S, Li X, Wei Y, Lan L, Deng X, Liang H et al (2017) Atg7 deficiency intensifies inflammasome activation and pyroptosis in pseudomonas sepsis. J Immunol 198: 3205–3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raduolovic K, Mak'Anyengo R, Kaya B, Steinert A, Niess JH (2018) Injections of lipopolysaccharide into mice to mimic entrance of microbial‐derived products after intestinal barrier breach. J Vis Exp 57610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratsimandresy RA, Dorfleutner A, Stehlik C (2013) An update on PYRIN domain‐containing pattern recognition receptors: from immunity to pathology. Front Immunol 4: 440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren G, Zhang X, Xiao Y, Zhang W, Wang Y, Ma W, Wang X, Song P, Lai L, Chen H et al (2019) ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J 38: e100376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmacke NA, O'Duill F, Gaidt MM, Szymanska I, Kamper JM, Schmid‐Burgk JL, Madler SC, Mackens‐Kiani T, Kozaki T, Chauhan D et al (2022) IKKbeta primes inflammasome formation by recruiting NLRP3 to the trans‐Golgi network. Immunity 55: 2271–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, Hauenstein AV, Wu Z, Nunez G, Mao Y et al (2019) Structural mechanism for NEK7‐licensed activation of NLRP3 inflammasome. Nature 570: 338–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi HX, Liu X, Wang Q, Tang PP, Liu XY, Shan YF, Wang C (2011) Mitochondrial ubiquitin ligase MARCH5 promotes TLR7 signaling by attenuating TANK action. PLoS Pathog 7: e1002057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiiba I, Takeda K, Nagashima S, Ito N, Tokuyama T, Yamashita SI, Kanki T, Komatsu T, Urano Y, Fujikawa Y et al (2021) MITOL promotes cell survival by degrading Parkin during mitophagy. EMBO Rep 22: e49097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36: 401–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song N, Liu ZS, Xue W, Bai ZF, Wang QY, Dai J, Liu X, Huang YJ, Cai H, Zhan XY et al (2017) NLRP3 phosphorylation is an essential priming event for inflammasome activation. Mol Cell 68: 185–197 [DOI] [PubMed] [Google Scholar]
- Stutz A, Horvath GL, Monks BG, Latz E (2013) ASC speck formation as a readout for inflammasome activation. In The Inflammasome, de Nardo CM, Latz E (eds), pp 91–101. Totowa: Springer; [DOI] [PubMed] [Google Scholar]
- Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN (2013) The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153: 348–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, Kudo Y, McBride HM, Fukuda T, Matsushita N et al (2013) MITOL regulates endoplasmic reticulum‐mitochondria contacts via Mitofusin2. Mol Cell 51: 20–34 [DOI] [PubMed] [Google Scholar]
- Takeda K, Nagashima S, Shiiba I, Uda A, Tokuyama T, Ito N, Fukuda T, Matsushita N, Ishido S, Iwawaki T et al (2019) MITOL prevents ER stress‐induced apoptosis by IRE1alpha ubiquitylation at ER‐mitochondria contact sites. EMBO J 38: e100999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Uda A, Mitsubori M, Nagashima S, Iwasaki H, Ito N, Shiiba I, Ishido S, Matsuoka M, Inatome R et al (2021) Mitochondrial ubiquitin ligase alleviates Alzheimer's disease pathology via blocking the toxic amyloid‐beta oligomer generation. Commun Biol 4: 192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140: 805–820 [DOI] [PubMed] [Google Scholar]
- Tigano M, Vargas DC, Tremblay‐Belzile S, Fu Y, Sfeir A (2021) Nuclear sensing of breaks in mitochondrial DNA enhances immune surveillance. Nature 591: 477–481 [DOI] [PubMed] [Google Scholar]
- Tsutsui H, Kayagaki N, Kuida K, Nakano H, Hayashi N, Takeda K, Matsui K, Kashiwamura S‐I, Hada T, Akira S et al (1999) Caspase‐1‐independent, Fas/Fas ligand–mediated IL‐18 secretion from macrophages causes acute liver injury in mice. Immunity 11: 359–367 [DOI] [PubMed] [Google Scholar]
- Vajjhala PR, Mirams RE, Hill JM (2012) Multiple binding sites on the pyrin domain of ASC protein allow self‐association and interaction with NLRP3 protein. J Biol Chem 287: 41732–41743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Yang C, Mao K, Chen S, Meng G, Sun B (2013) Cellular localization of NLRP3 inflammasome. Protein Cell 4: 425–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonashiro R, Ishido S, Kyo S, Fukuda T, Goto E, Matsuki Y, Ohmura‐Hoshino M, Sada K, Hotta H, Yamamura H et al (2006) A novel mitochondrial ubiquitin ligase plays a critical role in mitochondrial dynamics. EMBO J 25: 3618–3626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo YS, Park YY, Kim JH, Cho H, Kim SH, Lee HS, Kim TH, Sun Kim Y, Lee Y, Kim CJ et al (2015) The mitochondrial ubiquitin ligase MARCH5 resolves MAVS aggregates during antiviral signalling. Nat Commun 6: 7910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, Liwinski T, Elinav E (2020) Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell Discov 6: 36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, Liang S, Sanchez‐Lopez E, He F, Shalapour S, Lin XJ, Wong J, Ding S, Seki E, Schnabl B et al (2018) New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 560: 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221–225 [DOI] [PubMed] [Google Scholar]