

Abstract
The regulation of autophagy initiation is a key step in autophagosome biogenesis. However, our understanding of the molecular mechanisms underlying the stepwise assembly of ATG proteins during this process remains incomplete. The Rab GTPase Ypt1/Rab1 is recognized as an essential autophagy regulator. Here, we identify Atg23 and Atg17 as binding partners of Ypt1, with their direct interaction proving crucial for the stepwise assembly of autophagy initiation complexes. Disruption of Ypt1‐Atg23 binding results in significantly reduced Atg9 interactions with Atg11, Atg13, and Atg17, thus preventing the recruitment of Atg9 vesicles to the phagophore assembly site (PAS). Likewise, Ypt1‐Atg17 binding contributes to the PAS recruitment of Ypt1 and Atg1. Importantly, we found that Ypt1 is phosphorylated by TOR at the Ser174 residue. Converting this residue to alanine blocks Ypt1 phosphorylation by TOR and enhances autophagy. Conversely, the Ypt1 S174D phosphorylation mimic impairs both PAS recruitment and activation of Atg1, thus inhibiting subsequent autophagy. Thus, we propose TOR‐mediated Ypt1 as a multifunctional assembly factor that controls autophagy initiation via its regulation of the stepwise assembly of ATG proteins.
Keywords: ATG proteins, autophagy, stepwise assembly, TOR, Ypt1
Subject Categories: Autophagy & Cell Death
The Rab GTPase Ypt1 serves as a bridge that connects environmental sensing to the formation of the autophagy initiation complex.
Introduction
Autophagy is a highly conserved metabolic process among eukaryotes in which superfluous cellular components or damaged organelles are engulfed by the double‐membraned phagophore to form autophagosomes. These are then delivered to vacuoles/lysosomes for degradation (Ohsumi, 2014). Autophagy was initially characterized by its function to protect a cell from stress conditions such as starvation. In this, it promotes adaption to cellular environmental changes through the reprogramming of the cellular metabolism together with the recycling of cellular contents enabling the maintenance of essential protein synthesis or energy production. However, it is now becoming increasingly understood that autophagy plays a significant role in the more regular maintenance of cellular homeostasis, even under normal, nonstarvation, conditions (Feng et al, 2014). Autophagosome biogenesis involves a stepwise sequence involving signal perception, membrane nucleation, growth and extension, and autophagosome closure, each different stage of which being regulated by a distinct sets of proteins (Shibutani & Yoshimori, 2014).
Target of rapamycin (TOR)/Mechanistic target of rapamycin kinase complex 1 (mTORC1), a master modulator of cell growth and metabolism in yeast/mammals, has emerged as a key mediator of multiple steps and stages of autophagy (Noda & Ohsumi, 1998; Kim & Guan, 2015). For example, Atg13 is a direct phosphorylation substrate of TOR, and under rapamycin treatment or starvation conditions, decreased TOR activity leads to Atg13 dephosphorylation, allowing it to bind and form a scaffold with Atg1 and Atg17 (Kamada et al, 2000; Kabeya et al, 2005). In mammals, mTORC1 suppresses ULK1 activation via phosphorylation of its Ser757 residue (Kim et al, 2011); and inhibits PtdIns 3‐kinase activity by phosphorylating ATG14L (Yuan et al, 2013); and phosphorylates Pacer residue Ser157 to block autophagosome maturation by impairing the interaction between Pacer and Stx17 (Cheng et al, 2019). In addition, mTORC1 phosphorylates WIPI2 at Ser395, leading to WIPI2 association with the E3 ubiquitin ligase HUWEI and subsequent proteasomal degradation (Wan et al, 2018). The histone acetyltransferase p300 is a negative regulator of autophagy. Its activity can be induced by mTORC1 phosphorylation where activated p300 then inhibits autophagy by acetylating ATG5, ATG7, and LC3 (Wan et al, 2017). Moreover, the nuclear localization and activity of transcription factor EB (TFEB), a master regulator of autophagy and lysosome biogenesis, is also regulated by mTORC1 phosphorylation (Napolitano et al, 2018).
In Saccharomyces cerevisiae, autophagy‐related (Atg) proteins are hierarchically recruited to the PAS (Ohsumi, 2014). The PAS is a specific perivacuolar location that serves as an organizing center for the recruitment of various Atg proteins (Ohsumi, 2014). The Atg17‐Atg31‐Atg29 complex forms a core component of the PAS and Atg17 is widely used as a PAS marker (Ohsumi, 2014). Upon rapamycin treatment or nitrogen starvation conditions, Atg13 is rapidly dephosphorylated, enhancing its the association with Atg1 to form an Atg1 complex (Atg1‐Atg13‐Atg17‐Atg31‐Atg29; Kamada et al, 2010). Subsequently, Atg9 vesicles are recruited to the PAS via the direct binding of Atg9 with Atg13 and Atg17 (Sekito et al, 2009; Suzuki et al, 2015). However, Atg11 is responsible for the PAS recruitment of Atg9 vesicles through its direct association with Atg9 under nutrient‐rich conditions (He et al, 2006). Next, PI3K complex I is recruited to the PAS in an Atg9‐dependent manner (Shibutani & Yoshimori, 2014), followed by mobilization of the Atg2‐Atg18 complex and Atg5‐Atg12‐Atg16 ternary complex to the PAS in a PI3P‐dependent manner (Sekito et al, 2009; Ohsumi, 2014). Ultimately, the Atg5‐Atg12‐Atg16 ternary complex facilitates Atg8 lipidation at the PAS, resulting in the elongation of the phagophore (Romanov et al, 2012). Thus, the recruitment of Atg9 vesicles to the PAS is a key step in autophagosomal biogenesis. Atg23 is a peripheral membrane protein that forms a complex with Atg9 and Atg27 (Tucker et al, 2003). In nutrient‐rich conditions, Atg23 is crucial for Atg9 vesicle recruitment to the PAS (Backues et al, 2015). Recently, dimerization‐dependent membrane tethering by Atg23 has been found to be crucial for the binding of Atg9 to Atg23 and for autophagy under nitrogen starvation conditions (Hawkins et al, 2022). However, far less is known about the mechanisms through which Atg23 regulates the PAS recruitment of Atg9 vesicles.
Rab GTPases are molecular switches that participate in membrane trafficking and circulate between active GTP‐bound and an inactive GDP‐bound forms (Davis & Ferro‐Novick, 2015). The GTPase Ypt1 (Rab1 in mammals) regulates membrane tethering events through three different pathways, namely ER‐Golgi, intra‐Golgi traffic, and autophagy (Wang et al, 2015a). In yeast, Ypt1 is required for Cvt pathway activity and autophagy. Atg17 has been shown to recruit the autophagy‐specific GEF TRAPPIII complex to the PAS via its Trs85 subunit, after which TRAPPIII recruits Ypt1 to the PAS and activates it (Lynch‐Day et al, 2010). Under nutrient‐rich medium, activated Ypt1 participates in PAS assembly by interacting with Atg11 (Lipatova et al, 2012). Atg1 is then recruited to the PAS via binding with Ypt1 (Wang et al, 2013). Similarly, Hrr25 has been found to be recruited to the COPII vesicles that had migrated to the PAS via their direct binding to Ypt1 (Wang et al, 2015b) with Ypt1 also having been reported to be involved in the PAS recruitment of Atg9 vesicles (Lipatova et al, 2012). Nevertheless, the understanding of how Ypt1 regulates the PAS recruitment of Atg9 vesicles has remained elusive.
In this study, we show that Atg23 and Atg17 are previously unrecognized binding partners of Ypt1 and that Ypt1 is a direct phosphorylation substrate of TOR. The results described here reveal a strong link between Ypt1 and the stepwise assembly of autophagy initiation complexes, demonstrating that Ypt1 is a multifunctional assembly factor required for the initiation of autophagy.
Results
Atg23 is a binding partner of Ypt1
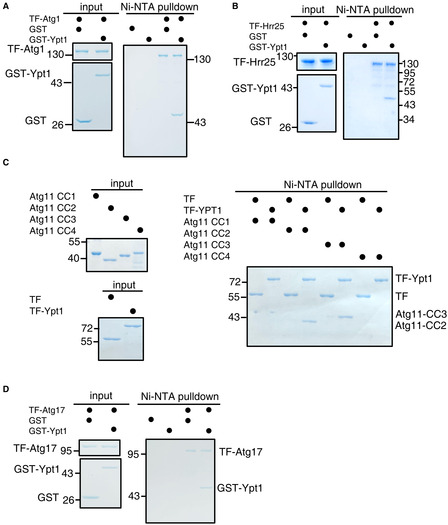
As it is known that Ypt1 regulates the PAS recruitment of Atg9 vesicles (Lynch‐Day et al, 2010; Lipatova et al, 2012), we examined whether Ypt1 could directly bind to Atg9 vesicle components. To this end, we purified nitrogen starvation‐treated FLAG‐Ypt1 protein using anti‐FLAG nanobody agarose beads. Analysis of the copurified proteins by mass spectrometry identified candidate interaction partners of Ypt1, including Atg1, Atg9, Atg11, Atg17, Atg23, Hrr25, Ykt6, and some other selective autophagy receptors (Fig 1A and Dataset EV1). Ypt1 has been previously found to participate in the Cvt pathway and autophagy via its binding with Atg11, Atg1, or Hrr25 (Lipatova et al, 2012; Wang et al, 2013, 2015b). Consistent with previous results, Ni‐NTA pulldown assays indicated that Atg1, Atg11, and Hrr25 could all directly bind to Ypt1 (Fig EV1A–C). We found that Atg17 could also directly associate with Ypt1 (Fig EV1D).
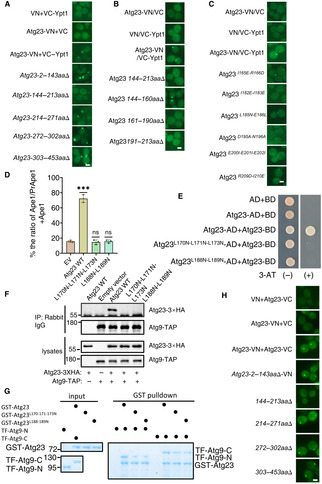
Figure 1. Atg23 is a new binding partner of Ypt1.
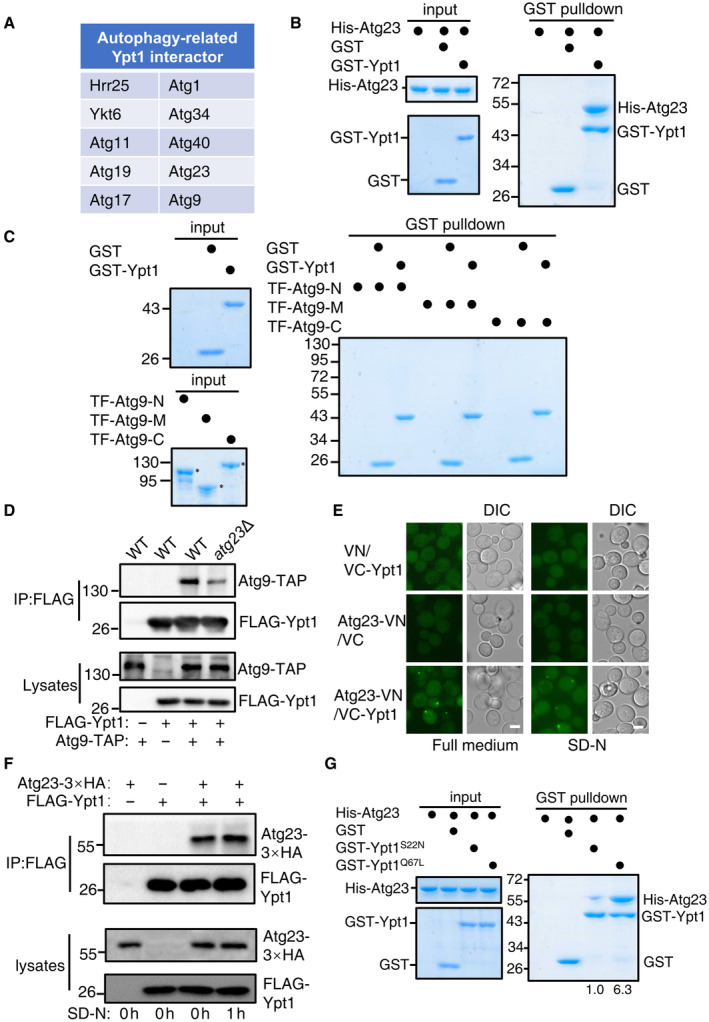
- FLAG‐Ypt1 protein was purified with anti‐FLAG nanobody agarose beads under nitrogen starvation medium and separated by SDS‐PAGE. Ypt1‐associated proteins were then analyzed by LC–MS/MS.
- GST pulldowns were performed using purified His6‐tagged Atg23 with GST or GST‐Ypt1 from E. coli. Protein samples were separated by SDS‐PAGE, and then detected using Coomassie blue staining.
- GST pulldowns were performed using purified His6‐tagged TF‐Atg9 N (2–318aa), TF‐Atg9 M (395–534aa), or TF‐Atg9 C (747–997aa) with GST or GST‐Ypt1 from E. coli. Protein samples were separated by SDS‐PAGE, and then detected using Coomassie blue staining. The asterisk represents the target protein.
- Atg9‐TAP (Tandem Affinity Purification tag, CBP‐TEV‐PA) and FLAG‐Ypt1 were co‐expressed in wild‐type or atg23∆ yeast strains. Cells were grown to the log‐growth phase. Cell lysates were immunoprecipitated with anti‐FLAG agarose beads and then analyzed with anti‐Protein A antibody.
- Fluorescence microscopy images of wild‐type cells expressing the BiFC constructs Atg23‐VN and VC‐Ypt1 cultured in nutrient rich or nitrogen starvation medium for 1 h. Scale bar: 2 μm.
- Atg23‐3×HA and FLAG‐Ypt1 were co‐expressed in wild‐type yeast strains. Cells were grown to the log‐growth phase and then subjected to nitrogen starvation for 0 or 1 h. Cell lysates were immunoprecipitated with anti‐FLAG agarose beads and then analyzed with anti‐HA antibody.
- GST pulldowns were performed using purified His6‐tagged Atg23 with GST, GST‐Ypt1 S22N , or GST‐Ypt1 Q67L proteins from E. coli. Protein samples were separated by SDS‐PAGE, and then detected using Coomassie blue staining. The binding strengths of His‐Atg23 with GST‐Ypt1 S22N or Ypt1 Q67L were quantified by calculating the ratio of His‐Atg23/GST‐Ypt1 S22N or His‐Atg23/Ypt1 Q67L from pulldown assays.
Source data are available online for this figure.
Figure EV1. (corresponding to Fig 1). Ypt1 binds directly to Atg1, Hrr25, Atg11, or Atg17 in vitro .
-
A–DNi‐NTA pulldowns were performed by using purified His6‐tagged TF‐Atg1, Atg17, Hrr25, Atg11 CC1(2–321aa), CC2(322–576aa), CC3(577–858aa), or CC4(859–1178aa) with GST or GST‐Ypt1 from E. coli. Protein samples were separated by SDS–PAGE, and then detected using Coomassie blue staining.
Source data are available online for this figure.
One previous study has highlighted potential binding between Atg23 and Ypt1 (Graef et al, 2013). Given that Ypt1 is known to be recruited to the PAS and can directly interact with Atg11 (Lipatova et al, 2012), plus with our finding of its interaction with Atg23 and Atg9 led us to hypothesis that Ypt1 may participates in the PAS recruitment of Atg9 vesicles via the direct binding of Ypt1 with Atg23 or Atg9. To assess this possibility, GST pulldown assays were performed and were able to verify that Ypt1 could indeed directly bind to Atg23 (Fig 1B). Using the same approach, we then tested whether Atg9 could bind to Ypt1 and found that, the N‐terminal (2–318aa), M (395–534aa), and C‐terminal (747–997aa) regions of Atg9 did not bind to Ypt1 (Fig 1C). To rule out whether this result is due to the loss of biological activity of the TF (trigger factor)‐fused Atg9‐N, M, or C proteins, we conducted GST pulldown assays using these proteins with GST‐Atg23 and found that TF‐Atg9‐N and TF‐Atg9‐C have a direct interaction with Atg23 (Appendix Fig S1A). This result is also consistent with the yeast two‐hybrid experiment results between them (Appendix Fig S1B), indicating that these fused proteins with TF tag have the normal biological activity of Atg9. Concurrently, the deletion of ATG23 significantly decrease the binding of Ypt1 to Atg9 (Fig 1D). Collectively, these data suggested that Atg9 is not likely to directly interact with Ypt1 and the pulldown observed between Ypt1 and Atg9 in the absence of Atg23 could be the result of an indirect interaction.
To further validate whether Atg23 was a bona fide Ypt1 binding protein, bi‐molecular fluorescence complementation (BiFC) assays were conducted to monitor their direct interaction (Feng et al, 2021). We constructed fusion reporters in which the VN component of the Venus yellow fluorescent protein(vYFP) was fused to the C terminus of Atg23 while the VC component was fused to the N terminus of Ypt1. Co‐expression of these fusion reporters in yeast cells grown under nutrient‐rich or nitrogen starvation conditions resulted in a detectable vYFP signals (Fig 1E), suggesting that Ypt1 could indeed interact with Atg23 in vivo. Since Atg23 has been reported to form many puncta at the PAS and peripheral sites, we further tested where Atg23 and Ypt1 interact, by observing the location of the Atg23‐vYFP‐Ypt1 binding signal relative to the PAS, as marked by RFP‐Ape1. As expected, we observed highly colocalized signaling between Atg23‐vYFP‐Ypt1 and RFP‐Ape1 (Appendix Fig S1C), indicating that this colocalization event had occurred at the PAS. Furthermore, Ypt1 and Atg23 could be co‐immunoprecipitated (Fig 1F). This further supported the suggestion of their direct interaction in vivo. In addition, we generated Ypt1 variants with disrupted ability to bind GTP or GDP to test whether their binding was depends on Ypt1 GDP‐ or GTP‐bound forms. Results showed Atg23 preferential pulldown with GTP‐bound form of Ypt1 (Ypt1 Q67L ), and not its GDP‐bound form (Ypt1 S22N ; Fig 1G), suggesting that the binding of Ypt1 to Atg23 depends on its GTP‐bound form. Taken together, these results led us to conclude that Atg23 is a new effector of Ypt1 (Grosshans et al, 2006).
The dimerization of Atg23 is a prerequisite for the Atg23‐Ypt1 binding
To explore the role and molecular mechanism of Atg23‐Ypt1 binding during autophagy, we sought to determine the specific regions required for the Atg23 association with Ypt1. Atg23 contains two coil–coil domains, which locate at 144–213aa and 272–302aa, and are designated as the CC1 and CC2 domains, respectively (Fig 2A). To delineate which of the regions of Atg23 were responsible for its binding with Ypt1, we generated a series of Atg23 deletion variants lacking regions 2–143aa, 144–213aa, 214–271aa, 272–302aa, or 303–453aa, respectively. Subsequent BiFC assays, using yeast strains co‐expressing VC‐Ypt1 with each one of the respective Atg23 deletion variants fused to the VN reporter, showed that no vYFP signal was detectable in cells co‐expressing VC‐Ypt1 with Atg23 144–213aa∆‐VN, the region which harbored the CC1 domain (Fig EV2A).
Figure 2. Dimerization of Atg23 is a prerequisite for the binding of Atg23 to Ypt1.
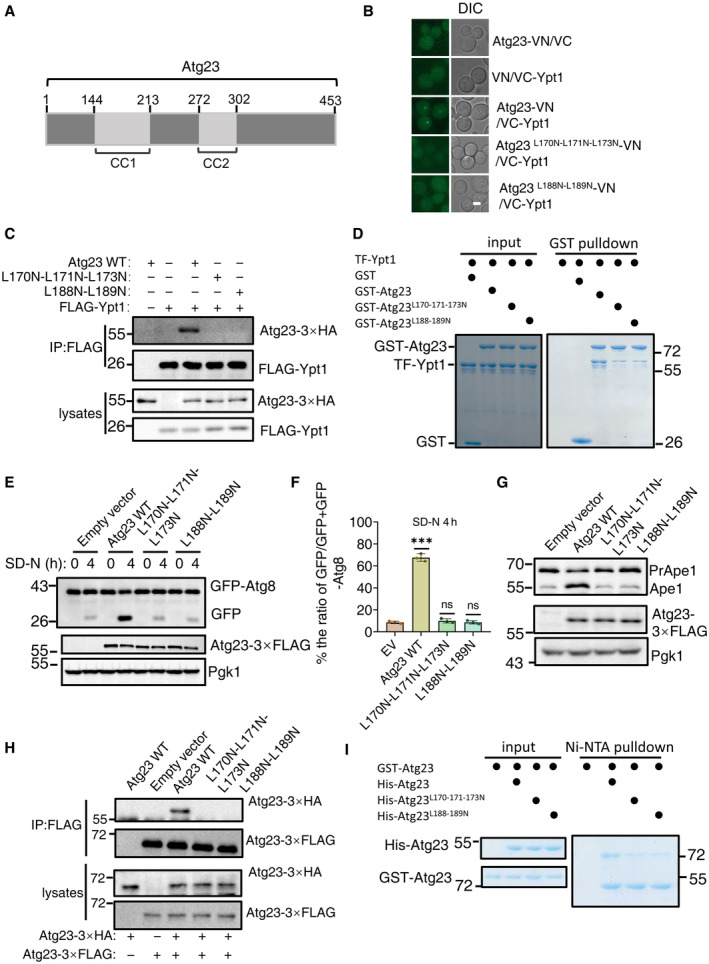
- The scheme of the Atg23 domains.
- Fluorescence microscopy images of wild‐type cells expressing the BiFC constructs VC‐Ypt1 and Atg23‐VN or the indicated Atg23‐VN variants cultured in SD‐N medium for 1 h. Scale bar: 2 μm.
- atg23∆ cells co‐expressing Flag‐Ypt1 and the indicated mutants with Atg23‐3×HA were subjected to nitrogen starvation for 1 h. Cell lysates were immunoprecipitated with anti‐FLAG agarose beads and then analyzed by western blot using an anti‐HA antibody.
- GST pulldowns were performed by using purified His6‐tagged TF‐Ypt1 with GST, GST‐Atg23, GST‐Atg23 L170N‐L171N‐L173N , or GST‐ Atg23 L188N‐L189N from E. coli. Protein samples were separated by SDS‐PAGE, and then detected using Coomassie blue staining.
- atg23∆ yeast strains co‐expressing GFP‐Atg8, Vph1‐mCherry and empty vector, wild‐type Atg23(Atg23 WT), Atg23 L170N‐L171N‐L173N , or Atg23 L188N‐L189N plasmids were cultured in SD‐N for 0 and 4 h. The autophagic activity of cells were analyzed by western blot for GFP‐Atg8 cleavage. Pgk1 served as a loading control.
- The cleavage of GFP‐Atg8 from (E) were quantified and presented as mean ± SD (n = 3). ***P < 0.001; NS, no significance; two‐tailed Student's t‐tests were used.
- atg23∆ yeast strains expressing empty vector, wild‐type Atg23 (Atg23 WT), Atg23 L170N‐L171N‐L173N , or Atg23 L188N‐L189N plasmids were grown to the early log phase. The activity of Cvt pathway were analyzed by western blot for the maturation of precursor Ape1. Pgk1 served as a loading control.
- atg23∆ yeast strains co‐expressing Atg23‐3×HA and empty vector, wild‐type Atg23(Atg23 WT)‐3×FLAG, Atg23 L170N‐L171N‐L173N ‐3×FLAG, or Atg23 L188N‐L189N ‐3×FLAG plasmids were grown to the early log phase. Cell lysates were immunoprecipitated with anti‐FLAG agarose beads and then analyzed by western blot using an anti‐HA antibody.
- Ni‐NTA pulldowns were performed by using purified GST‐tagged Atg23 with His‐Atg23, His‐Atg23 L170N‐L171N‐L173N , or His‐Atg23 L188N‐L189N from E. coli. Protein samples were separated by SDS–PAGE, and then detected using Coomassie blue staining.
Source data are available online for this figure.
Figure EV2. (corresponding to Fig 2). The Atg23 CC1 domain is required for Ypt1‐Atg23 binding and Atg23 L170N‐L171N‐L173N and Atg23 L188N‐L189N mutants impaired the dimerization of Atg23 and its binding with Atg9.
-
A–CFluorescence microscopy images of wild‐type cells expressing the BiFC constructs VC‐Ypt1 and Atg23‐VN or the indicated Atg23‐VN variants cultured in nitrogen starvation for 1 h. Scale bar: 2 μm.
-
DThe maturation of PrApe1 from Fig 2G were quantified by the ratio of Ape1/Ape1 + PrApe1 and presented as mean ± SD (n = 3). ***P < 0.001; NS, no significance; two‐tailed Student's t‐tests were used.
-
EThe AH109 strain was transformed with plasmids expressing BD‐fused with Atg23 and plasmids expressing AD‐fused with Atg23, Atg23 L170N‐L171N‐L173N , or Atg23 L188N‐L189N . These strains were grown on SD‐Leu‐Trp or SD‐His‐Leu‐Trp (3‐AT) agar plates at 30°C for 3 days.
-
FAtg9‐TAP (Tandem Affinity Purification tag, CBP‐TEV‐PA) and Atg23‐3×HA, Atg23 L170N‐L171N‐L173N ‐3×HA, or Atg23 L188N‐L189N ‐3×HA were co‐expressed in atg23∆ yeast strains. Cells were grown to the log‐growth phase and then subjected to nitrogen starvation for 1 h. Cell lysates were immunoprecipitated with anti‐Rabbit IgG agarose beads and then analyzed with anti‐HA antibody.
-
GGST pulldowns were performed using purified His6‐tagged TF‐Atg9 N (2–318aa), or TF‐Atg9 C (747–997aa) with GST, GST‐Atg23, GST‐Atg23 L170N‐L171N‐L173N , or GST‐Atg23 L188N‐L189N from E. coli. Protein samples were separated by SDS‐PAGE, and then detected using Coomassie blue staining.
-
HFluorescence microscopy images of wild‐type cells expressing the BiFC constructs Atg23‐VC and Atg23‐VN or the indicated Atg23‐VN variants cultured in nitrogen starvation for 1 h. Scale bar: 2 μm.
Source data are available online for this figure.
We then generated three deletion variants individually lacking portions of the Atg23 CC1 domain (144–160aa, 161–190aa, and 191–213aa), and fused each of these mutants to VN for further BIFC assays. Results showed that the vYFP signal was only detectable in cells co‐expressing Atg23 144–160aa∆‐VN and VC‐Ypt1, suggesting that the sites on Atg23 necessary for binding with Ypt1 were contained in the 161–213aa region of the CC1 domain (Fig EV2B). To identify the specific residues required for the association of Atg23 with Ypt1, we created a series of random amino acid conversions in this region for BiFC screening, the results revealed that the vYFP signal was absent in the Atg23 L170N‐L171N‐L173N , Atg23 L188N‐L189N , Atg23 L200N‐L201N‐L202N , or Atg23 L182N‐L183N mutants fused with VN, suggesting that the interaction with Ypt1 was dependent on these residues in Atg23 (Figs 2B and EV2C). Two of these variants, Atg23 L170N‐L171N‐L173N and Atg23 L188N‐L189N were subsequently selected to test their Ypt1 binding using Co‐IP assays. To this end, atg23∆ yeast strain expressing FLAG‐Ypt1 was transformed with either Atg23‐3×HA WT, Atg23‐3×HA L170N‐L171N‐L173N , Atg23‐3×HA L188N‐L189N , or an empty vector control. Consistent with results of BiFC assays, neither of the Atg23 mutants were observed to be coprecipitated with FLAG‐Ypt1, indicating some or all the L170‐L171‐L173 and L188‐L189 residues to be essential for interactions with Ypt1 (Fig 2C). These results were further supported by GST pulldown, which showed that Atg23‐Ypt1 binding was almost completely abolished in the Atg23 L170N‐L171N‐L173N and Atg23 L188N‐L189N backgrounds (Fig 2D). Taken together, these data indicated that the Atg23 CC1 domain is required for the direct Atg23‐Ypt1 interaction.
To investigate the role of Atg23‐Ypt1 binding in autophagy, Atg23 L170N‐L171N‐L173N and Atg23 L188N‐L189N mutants were used for testing their autophagic activity by transforming atg23∆ yeast cells co‐expressing GFP‐Atg8 and Vph1‐mCherry with either Atg23 mutants, Atg23 WT, or empty vector controls, respectively. Results of the GFP‐Atg8 cleavage indicated that the Atg23 L170N‐L171N‐L173N and Atg23 L188N‐L189N mutants had significantly lower levels of free GFP under autophagic induction by nitrogen starvation conditions, thus showing comparable autophagic inhibition to that observed in ATG23 knockout (Fig 2E and F). We then examined the activity of the Cvt pathway in two mutant cells under nutrient‐rich medium using Ape1 precursor cleavage assays. Results showed that mature Ape1 was almost absent in cells expressing either Atg23 L170N‐L171N‐L173N or Atg23 L188N‐L189N mutant (Figs 2G and EV2D), suggesting that Atg23 L170‐L171‐L173 residues or L188‐L189 residues are required for the activity of the Cvt pathway.
Since Atg23 has been reported to be able to form dimers with Atg23, and that its dimerization is important for the binding of Atg23 to Atg9 (Hawkins et al, 2022), we then examined whether Atg23 L170N‐L171N‐L173N or Atg23 L188N‐L189N mutants affect the dimerization of Atg23 and its association with Atg9. Y2H analysis showed that AH109 strains co‐expressing Atg23‐BD with Atg23 L170N‐L171N‐L173N ‐AD or Atg23 L188N‐L189N ‐AD could not grow on SD‐His‐Leu‐Trp + 3‐AT plate (Fig EV2E). Both Co‐IP and in vitro pulldown assays showed that Atg23 L170N‐L171N‐L173N or Atg23 L188N‐L189N mutants blocked the dimerization of Atg23 and the subsequent association of Atg23 with Atg9 (Figs 2H and I, and EV2F and G). We next wanted to identify which of the amino acid residues on Atg23 impair the binding of Atg23‐Ypt1 but do not affect the dimerization of Atg23. BiFC assays indicated that the Atg23 CC1 domain is required for the dimerization of Atg23 (Fig EV2H). We then conducted the Ni‐NTA pulldown experiments on a series of the indicated five amino acid residue deletion mutants in the Atg23 CC1 domain and found mutants on the CC1 domain that not only impaired the binding of Atg23 to Ypt1, but also blocked the formation of Atg23 dimerization (Appendix Fig S2). As disruption of Atg23 dimerization has been shown to cause mislocalization of the protein (Hawkins et al, 2022), these data indicated that the failure of monomeric Atg23 to bind Ypt1 could be an indirect effect of Atg23 failing to colocalize with Ypt1. Taken together, these data led to the conclusion that the dimerization of Atg23 is a prerequisite for the binding of Atg23 to Ypt1.
Ypt1‐Atg23 binding is required for autophagy
We next sought to identify the specific regions required for on Ypt1 to associate with Atg23. Using same approach, we identified the Ypt1 W62 residue as required for the binding of Ypt1 to Atg23, both in vivo and in vitro (Fig 3A–D and Appendix Fig S3A). To test whether Ypt1 W62A affects the binding of Ypt1 to Atg17, Atg1, Hrr25, or Atg11, as well as its dimerization, GST or Ni‐NTA pulldown assays were performed. Ypt1 W62A displayed no impaired their associations or any negative effects upon its dimerization (Appendix Fig S3B–G). These data indicated that Ypt1 W62 residue specifically regulates its binding to Atg23.
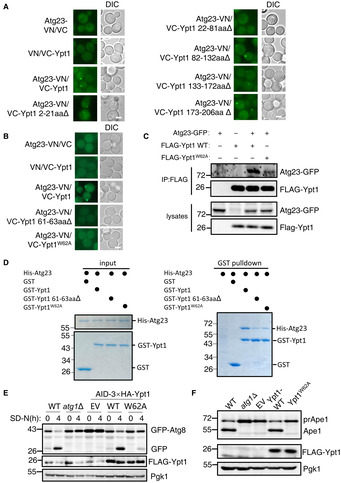
Figure 3. Ypt1‐Atg23 binding is required for autophagy.
-
A, BFluorescence microscopy images of wild‐type cells expressing the BiFC constructs in Atg23‐VN and VC‐Ypt1 or the indicated VC‐Ypt1 variants cultured in nitrogen starvation medium for 1 h. Scale bar: 2 μm.
-
CYeast cells co‐expressing Atg23‐GFP and FLAG‐Ypt1 were subjected to SD‐N for 1 h. Cell lysates were immunoprecipitated with anti‐FLAG agarose beads and then analyzed with anti‐GFP antibody.
-
DGST pulldowns were performed by using purified His6‐tagged Atg23 with GST, GST‐Ypt1, GST‐Ypt1 61‐63aa∆, or GST‐Ypt1 W62A from E. coli.
-
EWild‐type or atg1∆ yeast cells co‐expressing GFP‐Atg8 and Vph1‐mCherry, or AID‐3×HA‐Ypt1 yeast strains co‐expressing GFP‐Atg8 and Vph1‐mCherry transformed with empty vector, FLAG‐Ypt1, or FLAG‐Ypt1 W62A plasmids were treated with IAA for 2 h to degrade endogenous AID‐3×HA‐Ypt1, and then subjected to nitrogen starvation for 0 or 4 h. Autophagic activity was analyzed by western blot for GFP‐Atg8 cleavage. Pgk1 served as a loading control.
-
FBY4741(WT) or atg1∆ cells, or AID‐3×HA‐Ypt1 yeast cells transformed with empty vector, FLAG‐Ypt1, or FLAG‐Ypt1 W62A plasmids were grown to early log phase, and then subjected to IAA for 2 h to degrade endogenous AID‐3×HA‐Ypt1. The activity of the Cvt pathway were analyzed by western blot for the maturation of PrApe1. Pgk1 served as a loading control.
Source data are available online for this figure.
We then set out to investigate whether Ypt1 W62 is required for autophagy and the Cvt pathway. Since Ypt1 is an essential gene for cell viability, the biological function of Ypt1 has formerly been studied primarily by using a temperature‐sensitive strain (Lynch‐Day et al, 2010; Lipatova et al, 2012; Wang et al, 2013, 2015b). However, as growth at 37°C will induce a heat‐shock stress responses in temperature sensitive strains, phenotypic analysis may therefore not accurately or entirely reflect its essential biological functions. To circumvent such a heat‐shock response, we used an auxin‐inducible degron system to degrade Ypt1 (Nishimura et al, 2009). As shown in Appendix Fig S4A, Ypt1 protein degraded rapidly after the addition of indoleacetic acid (IAA). However, a spotting assay indicated that cells expressing AID‐3×HA‐Ypt1 exhibited severely impaired growth irrespective of IAA treatment, suggesting that the addition of the AID‐3×HA tag to the N terminus of Ypt1 had affected its normal function (Appendix Fig S4B). We next examined whether Ypt1 protein degradation blocked autophagy. We found that in response to nitrogen starvation, autophagy was completely abolished in yeast cells co‐expressing AID‐3×HA‐Ypt1 with GFP‐Atg8 and Vph1‐mCherry, with or without IAA treatment. These results were consistent with the previous finding that Ypt1 is necessary for autophagy (Appendix Fig S4C). Furthermore, Cvt pathway activity also appeared to be completely blocked in AID‐3×HA‐Ypt1 yeast cells, regardless of IAA treatment (Appendix Fig S4D). We then used this yeast strain to study whether Ypt1‐Atg23 binding regulates autophagy. As shown in Fig 3E and F, and Appendix Fig S4E and F, Ypt1 W62 is required for autophagy and the Cvt pathway. To rule out the possibility that Ypt1 W62A affects autophagy via other means beyond its interaction with Atg23, GFP‐Ypt1 or GFP‐Ypt1 W62A plasmid were transformed into WT or atg23∆ yeast strains co‐expressing AID‐3×HA‐Ypt1 and Atg17‐2×mCherry. Fluorescence microscopy and image analysis showed that there was no significant difference in the number of cells colocated with Atg17 and Ypt1 between WT cells co‐expressing GFP‐Ypt1 W62A and Atg17‐2×mCherry and atg23∆ cells co‐expressing GFP‐Ypt1 and Atg17‐2×mCherry, although their colocalization was moderately decreased compared with WT cells co‐expressing GFP‐Ypt1 and Atg17‐2×mCherry (Appendix Fig S4G–I). Collectively, these results indicated that the PAS recruitment of Ypt1 does not rely on the binding of Ypt1‐Atg23 to any great extent and strong evidence is provided that Ypt1‐Atg23 binding is required for autophagy.
Ypt1‐Atg23 binding is required for the PAS recruitment of Atg9 vesicles
In light of the above findings, we next investigated how Ypt1‐Atg23 interactions may regulate autophagy. Given that both Ypt1 and Atg23 are required for the PAS recruitment of Atg9 vesicles, we next examined whether Ypt1‐Atg23 interaction was required for Atg9 vesicle recruitment to the PAS. To test this hypothesis, we transformed the AID‐3×HA‐Ypt1 yeast strain co‐expressing Atg9‐2×GFP and Atg17‐2×mCherry or RFP‐Ape1 with Ypt1 WT, Ypt1 W62A , or empty vector control plasmids. Imaging and statistical analysis consistently indicated that the Ypt1 W62A variant blocked the PAS recruitment of Atg9 vesicles under either nutrient‐rich or nitrogen starvation conditions (Fig 4A and B, and Appendix Fig S5A–D). Furthermore, to examine whether Ypt1 W62A variant affects the biogenesis of peripheral Atg9 vesicles, we quantified the number of Atg9 puncta and the formation of mobile Atg9 vesicles in the presence of Ypt1 W62A variant. Results indicated that the number of Atg9 puncta and the formation of mobile Atg9 vesicles was not altered in the Ypt1 W62A mutant (Fig 4C and Appendix Fig S5E, and Movies [Link], [Link]). Collectively, these data suggested that the Ypt1‐Atg23 interaction is involved in the PAS recruitment of Atg9 vesicles, but not in its biosynthesis.
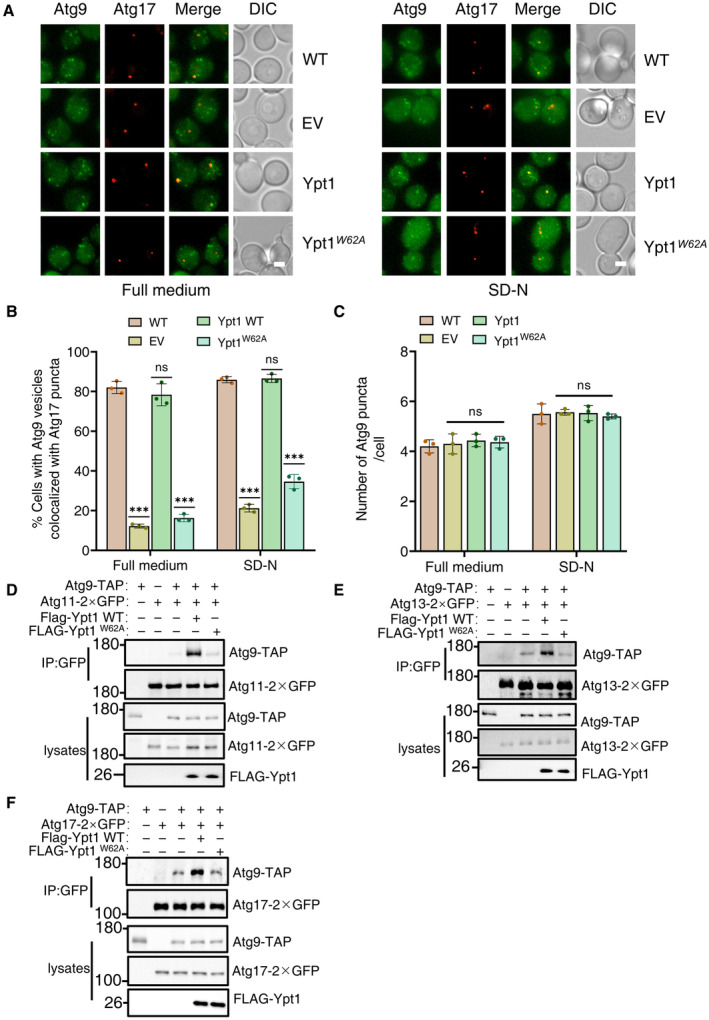
Figure 4. Ypt1‐Atg23 binding is required for the PAS localization of Atg9 vesicles.
-
AWild‐type yeast strains co‐expressing Atg17‐2×mCherry and Atg9‐2×GFP or AID‐3×HA‐Ypt1 yeast strains co‐expressing Atg17‐2×mCherry and Atg9‐2×GFP transformed with empty vector, FLAG‐Ypt1, or FLAG‐Ypt1W62A plasmids were treated with IAA for 2 h to degrade endogenous AID‐3×HA‐Ypt1, and then subjected to SD‐N for 0 or 1 h. Images of cells were obtained using fluorescence microscopy. Scale bar, 2 μm.
-
BCells from (A) were quantified for the number of cells in which Atg9‐2 × GFP colocalized with Atg17‐2 × mCherry puncta. n = 300 cells were pooled from three independent experiments. Data are shown as mean ± SD. ***P < 0.001; ns, no significance; two‐tailed Student's t‐tests were used.
-
CCells from (A) were quantified for the number of Atg9‐2 × GFP. n = 300 cells were pooled from three independent experiments. Data are shown as mean ± SD. ns, no significance; two‐tailed Student's t‐tests were used.
-
D–FAID‐3×HA‐Ypt1 cells co‐expressing Atg9‐TAP and Atg11‐2×GFP, Atg13‐2×GFP, or Atg17‐2×GFP transformed with empty vector, FLAG‐Ypt1, or FLAG‐Ypt1 W62A plasmids were grown to log phase, and then subjected to SD‐N for 1 h. Cell lysates were immunoprecipitated with anti‐GFP agarose beads and then analyzed by western blot using the indicated antibody.
Source data are available online for this figure.
Previous reports have shown that the recruitment of Atg9 vesicles to the PAS requires the direct binding of Atg9‐Atg13, Atg9‐Atg17, and Atg9‐Atg11 (He et al, 2006; Sekito et al, 2009; Suzuki et al, 2015). We therefore tested whether deficiency in Ypt1‐Atg23 binding was required for Atg9 association with Atg11, Atg13, or Atg17 by transforming Ypt1 WT‐3×FLAG, Ypt1 W62A ‐3×FLAG, or the empty vector plasmids into an AID‐3×HA‐Ypt1 yeast strain co‐expressing Atg9‐TAP and Atg11‐2×GFP, Atg13‐2×GFP, or Atg17‐2×GFP. Subsequent Co‐IP assays revealed a dramatic decrease in Atg9 binding with Atg11, Atg13, and Atg17 in the absence of Ypt1‐Atg23 interaction under nitrogen starvation conditions (Fig 4D–F). Taken together, these results led us to conclude that Ypt1‐Atg23 required for the recruitment of Atg9 vesicles to the PAS through the modulation of the Atg9 association with Atg11, Atg13, and Atg17.
Ypt1‐Atg17 binding is required for the PAS recruitment of Ypt1 and Atg1
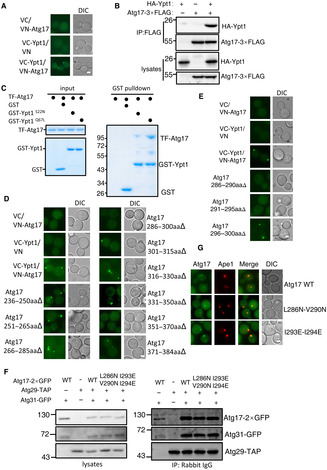
Atg17 is widely used as a PAS marker because it forms a stable ternary complex with Atg29 and Atg31 that serves as a scaffold for the initiation of autophagy (Ohsumi, 2014). This property of Atg17 during the initiation of autophagy led us to focus on the potential function of Ypt1‐Atg17 binding. To further validate the binding of Ypt1‐Atg17 in vivo, we performed Co‐IP and BIFC assays, which respectively showed that Atg17‐3×FLAG could associate with HA‐Ypt1 and that the vYFP signal was only observable in cells co‐expressing VC‐Ypt1 and Atg17‐VN (Fig EV3A and B). Additionally, we found that the binding of Ypt1 to Atg17 depends on its GTP‐bound form (Fig EV3C). Collectively, these data showed that Ypt1 can directly bind to Atg17 and that such binding is GTP‐bound form dependent.
Figure EV3. (corresponding to Fig 5). Ypt1 binds directly with Atg17.
-
AFluorescence microscopy images of wild‐type cells expressing the BiFC VC‐Ypt1 and Atg17‐VN, cultured in nutrient‐rich medium. Scale bar: 2 μm.
-
BAtg17‐3×FLAG and HA‐Ypt1 were co‐expressed in wild‐type yeast strains. Cells were grown to the log‐growth phase. Cell lysates were immunoprecipitated with anti‐FLAG agarose beads and then analyzed with anti‐HA antibody.
-
CGST pulldowns were performed using purified His6‐tagged TF‐Atg17 with GST, GST‐Ypt1 S22N , or Ypt1 Q67L from E. coli. Protein samples were separated by SDS–PAGE, and then detected using Coomassie blue staining.
-
D, EFluorescence microscopy images of wild‐type cells expressing the BiFC VC‐Ypt1 and the indicated Atg17‐VN variants, cultured in nutrient‐rich medium. Scale bar: 2 μm.
-
FAtg29‐TAP, Atg31‐GFP and Atg17‐2×GFP, Atg17 L286N‐V290N ‐2×GFP, or Atg17 I293E‐I294E ‐2×GFP were co‐expressed in atg17∆ yeast strains. Cells were grown to the log‐growth phase. Cell lysates were immunoprecipitated with anti‐Rabbit IgG agarose beads and then analyzed with anti‐GFP antibody.
-
GFluorescence microscopy images of atg17∆ cells co‐expressing the RFP‐Ape1 and Atg17‐2×GFP, Atg17 L286N‐V290N ‐2×GFP, or Atg17 I293E‐I294E ‐2×GFP, cultured in nutrient‐rich medium. Scale bar: 2 μm.
Source data are available online for this figure.
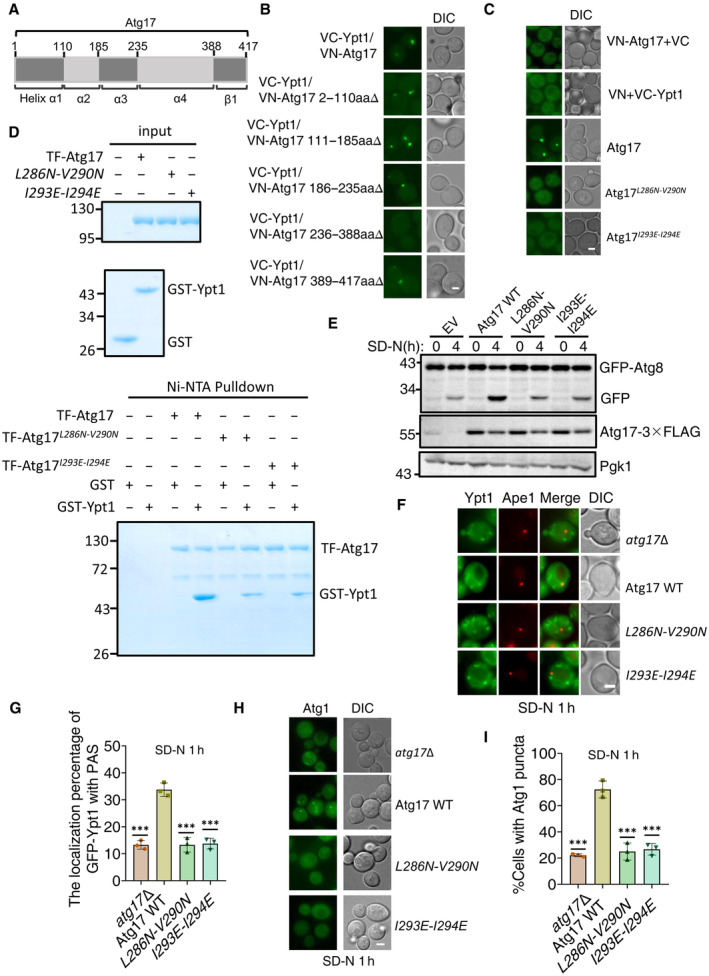
To better understand the effects of Ypt1‐Atg17 binding on autophagy, we first sought to identify which Atg17 regions that were responsible for binding with Ypt1. Since Atg17 contains four α helixes and a β strand located at various positions 2–110aa (α1), 111–185aa (α2), 186–235aa (α3), 236–388aa (α4), and 389–417aa (β1) (Fig 5A; Ragusa et al, 2012), we introduced a series of individual deletions at 2–110 aa, 111–185aa, 186–235aa, 236–388aa, and 389–417aa and used these deletion variants for BiFC assays. Transformation of these Atg17 variants fused to VN and VC‐Ypt1 in yeast cells, resulted in vYFP signal in all cultures except for those with cells harboring 236–388aa∆, which indicated that the Atg17 helix α4 was required for Atg17‐Ypt1 binding (Fig 5B). A series of nine deletions were then individually introduced into the helix α4 domain. Expression of these deletion mutants in BiFC assays indicated that the vYFP signal was abolished in the cells expressing Atg17 286–300aa∆ or 351–370aa∆ (Fig EV3D). Because the Atg17 351–370aa region had been previously reported to be required for the dimerization of Atg17 (Ragusa et al, 2012), we selected the Atg17 286–300aa region to further identify the binding region of Ypt1‐Atg17. To this end, we further introduced a series of three deletions in this region, which revealed that vYFP signal was lost in cells expressing either Atg17‐VN 286–290aa∆ or 291–295aa∆ with VC‐Ypt1 in BiFC assays (Fig EV3E), thus indicating some or all these residues to be required for the Ypt1‐Atg17 interaction. Subsequently, we generated a series of random amino acid conversions in this region for BiFC screening. The results showed that the vYFP signal was absent in the Atg17 L286N‐V290N or Atg17 I293E‐I294E mutants fused with VN, indicating that their binding was dependent on these residues in Atg17 (Fig 5C). Atg17 L286N‐V290N and Atg17 I293E‐I294E mutants were subsequently tested for their Ypt1 binding using Ni‐NTA pulldown assays. These results further confirmed that Atg17‐Ypt1 binding was almost entirely lost in the Atg17 L286N‐V290N or Atg17 I293E‐I294E mutants (Fig 5D). These data indicated that Atg17 L286‐V290 or I293‐I294 residues are required for the binding of Ypt1‐Atg17.
Figure 5. Ypt1‐Atg17 binding is required for autophagy.
-
AThe scheme for the Atg17 domains.
-
B, CFluorescence microscopy images of wild‐type cells expressing the BiFC constructs VC‐Ypt1 and Atg17‐VN or the indicated Atg17‐VN variants cultured in nutrient‐rich medium. Scale bar: 2 μm.
-
DNi‐NTA pulldowns were performed by using purified His6‐tagged TF‐Atg17 variants with GST, or GST‐Ypt1 from E. coli. Protein samples were separated by SDS‐PAGE, and then detected using Coomassie blue staining.
-
Eatg17∆ yeast strains co‐expressing GFP‐Atg8, Vph1‐mCherry and empty vector, wild‐type Atg17(Atg17 WT), or the indicated Atg17 mutant plasmids were cultured in SD‐N medium for 0 or 4 h. The autophagic activity of cells were analyzed by western blot for GFP‐Atg8 cleavage. Pgk1 served as a loading control.
-
F, Gatg17∆ yeast strains co‐expressing GFP‐Ypt1, RFP‐Ape1 and empty vector, wild‐type Atg17(Atg17 WT), or the indicated Atg17 mutant plasmids were cultured in SD‐N medium for 1 h. Images of cells were obtained using inverted fluorescence microscopy. Scale bar, 2 μm. Cells were quantified for the number of cells in which GFP‐Ypt1 colocalized with RFP‐Ape1 puncta. n = 300 cells were pooled from three independent experiments. Data are shown as mean ± SD. ***P < 0.001; two‐tailed Student's t‐tests were used.
-
H, Iatg17∆ yeast strains co‐expressing Atg1‐GFP and empty vector, wild‐type Atg17(Atg17 WT), or Atg17 mutant plasmids were cultured in SD‐N medium for 1 h. Images of cells were obtained using the inverted fluorescence microscopy. Scale bar, 2 μm. Cells were quantified for the number of cells in which Atg1‐GFP puncta appeared. n = 300 cells were pooled from three independent experiments. Data are shown as mean ± SD. ***P < 0.001; two‐tailed Student's t‐tests were used.
Source data are available online for this figure.
We then used these mutants in a GFP‐Atg8 cleavage assay to investigate whether Atg17‐Ypt1 binding was required for autophagic activity. Empty vector, Atg17 WT, Atg17 L286N‐V290N , or Atg17 I293E‐I294E plasmids were separately transformed into an atg17∆ yeast strain co‐expressing GFP‐Atg8 and Vph1‐mCherry. As shown in Fig 5E, the cleavage of the GFP‐Atg8 processing assay showed that Atg17 L286N‐V290N or Atg17 I293E‐I294E mutants all led to free GFP levels under nitrogen starvation conditions becoming decreased to similar levels to those observed in atg17∆ yeast cells, suggesting that Ypt1‐Atg17 binding is required for autophagy. Since Atg17 forms a complex with Atg29 and Atg31, we next wanted to examine whether deficiency in the binding of Ypt1‐Atg17 impairs the formation of the Atg17‐Atg29‐Atg31 complex by transforming Atg17 WT‐2×GFP, Atg17 L286N‐V290N ‐2×GFP, Atg17 I293E‐I294E ‐2×GFP, or the empty vector plasmids into an atg17∆ yeast strain co‐expressing Atg29‐TAP and Atg31‐GFP. Subsequent Co‐IP assays revealed no significant changes in the association of Atg17 with Atg29 and Atg31 in the absence of Ypt1‐Atg17 binding (Fig EV3F). Next, we tested whether deficiency in the binding of Ypt1‐Atg17 affects PAS formation by transforming Atg17 WT‐2×GFP, Atg17 L286N‐V290N ‐2×GFP, Atg17 I293E‐I294E ‐2×GFP, or the empty vector plasmids into an atg17∆ yeast strain expressing RFP‐Ape1. As shown in Fig EV3G, Atg17‐2×GFP, as the puncta form, was well co‐localized with RFP‐Ape1 in cells expressing the Atg17 L286N‐V290N or Atg17 I293E‐I294E mutants under nutrient‐rich medium. Taken together, these data suggested that the Atg17 L286N‐V290N or Atg17 I293E‐I294E mutants do not display any affects upon the formation of the Atg17‐Atg29‐Atg31 complex or the PAS recruitment of Atg17.
Based on this evidence, we then examined whether disruption of Ypt1‐Atg17 interaction impaired the PAS recruitment of Ypt1. Transformation of 3×FLAG‐tagged WT Atg17, Atg17 L286N‐V290N or Atg17 I293E‐I294E into an atg17∆ yeast strain co‐expressing RFP‐Ape1(PAS marker) and GFP‐Ypt1, under nitrogen starvation conditions showed that recruitment of Ypt1 to the PAS was significantly decreased in cells expressing Atg17 L286N‐V290N or Atg17 I293E‐I294E mutants (Fig 5F and G, and Appendix Fig S6A). These findings suggested that the binding of Ypt1‐Atg17 provided a substantial contribution to the PAS recruitment of Ypt1 in response to nitrogen starvation. Given that Ypt1 is required for Atg1 puncta formation and that the absence of Ypt1‐Atg17 binding adversely affects PAS recruitment of Ypt1, we next examined whether the Ypt1‐Atg17 interaction was required for Atg1 puncta formation. To test this hypothesis, 3×FLAG‐tagged plasmids carrying WT Atg17, Atg17 L286N‐V290N , or Atg17 I293E‐I294E plasmids were transformed into an atg17∆ yeast cells expressing Atg1‐GFP. As shown in Fig 5H and I, and Appendix Fig S6B, the formation of Atg1 puncta was significantly impaired in cells expressing Atg17 L286N‐V290N or Atg17 I293E‐I294E mutants under nitrogen starvation conditions. Collectively, these data demonstrated Ypt1‐Atg17 interaction is required for autophagy by regulating the PAS recruitment of Ypt1 and Atg1.
Since Ypt1 is a newly identified interactor of Atg17, we examined whether the PAS recruitment of ATG proteins is regulated by Ypt1. We constructed yeast strains that co‐expressing Atg17‐2×mCherry and the indicated ATG proteins with GFP tag in wild‐type or AID‐3×HA‐Ypt1 cells. IAA was added to induce the degradation of endogenous AID‐3×HA‐Ypt1 protein. Image data analysis found that IAA‐mediated Ypt1 degradation severely affects both the PAS recruitment of Atg9 vesicles, and the puncta formation of Atg1, Atg2, Atg5, Atg8, or Atg14. However, Ypt1 degradation does not impair PAS recruitment of Atg11 and Atg13 (Appendix Fig S6C). These results indicated that Ypt1 regulates the PAS recruitment of Atg9 vesicles, PI3K complex I, the Atg2‐Atg18 complex, and the two ubiquitin‐like systems under nitrogen starvation conditions.
Ypt1 is directly phosphorylated by TOR
Given that phosphorylation is already known as the most common post‐translational modification involved in the regulation of autophagy (Kim & Guan, 2015), coupled with our evidence that Ypt1 is a key regulator of autophagy, we next tested whether Ypt1 function in autophagy is affected by phosphorylation. To this end, we purified FLAG‐Ypt1 from cells grown in nutrient‐rich conditions using anti‐FLAG nanobody agarose beads (Appendix Fig S7A). Protein samples separated by SDS–PAGE were then analyzed by LC–MS, which identified phosphorylation at the S174 residue of Ypt1 (Fig 6A and Appendix Fig S7B).
Figure 6. Ypt1 is directly phosphorylated by TOR.
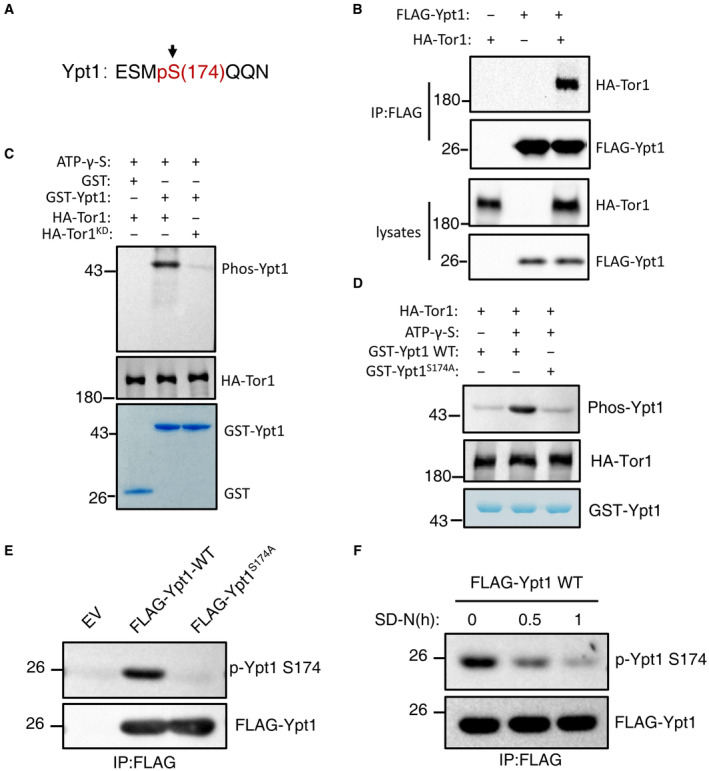
- MS identified that Ypt1 S174 residue was phosphorylated.
- Cells co‐expressing HA‐Tor1 and Flag‐Ypt1 were grown to log phase. Cell lysates were immunoprecipitated with anti‐FLAG agarose beads and then analyzed by western blot using an anti‐HA antibody.
- In vitro kinase assays were performed with purified GST or GST‐Ypt1 from E. coli as substrates and purified nutrient‐rich medium‐treated HA‐Tor1 or HA‐Tor1 KD (kinase dead) from yeast cells as protein kinase. The phosphorylation level of GST‐Ypt1 was detected using an anti‐thioP antibody.
- In vitro kinase assays were performed with purified GST‐Ypt1 WT or S174A from E. coli as substrates and purified nutrient‐rich medium‐treated HA‐Tor1 from yeast cells as protein kinase. The phosphorylation level of GST‐Ypt1 was detected using anti‐thioP antibody.
- Yeast cells expressing empty vector, FLAG‐Ypt1, or FLAG‐Ypt1 S174A were grown to early log phase. Cell lysates were immunoprecipitated with anti‐FLAG agarose beads and then analyzed with anti‐p‐Ypt1 S174 antibody.
- Yeast cells expressing FLAG‐Ypt1 were subjected to nitrogen starvation for 0, 0.5, or 1 h. Cell lysates were immunoprecipitated with anti‐FLAG agarose beads and then analyzed with anti‐p‐Ypt1 S174 antibody.
Source data are available online for this figure.
We next sought to identify the specific protein kinase responsible for Ypt1 phosphorylation. In yeast and mammalian cells, TOR/mTOR, Snf1/AMPK and Atg1/ULK1 are three major protein kinases regulating autophagy (Kim et al, 2011; Ohsumi, 2014). However, activation conditions differ for each. TOR is active in nutrient‐rich medium, Snf1/AMPK is activated under glucose starvation, while Atg1/ULK1 is activated under autophagy induction conditions (Kim et al, 2011). Therefore, given that the S174 residue of Ypt1 is phosphorylated in nutrient‐rich medium, we speculated that Ypt1 would be likely to be phosphorylated by TOR. Co‐IP assays to detect whether Ypt1 could bind to TOR showed that Ypt1 indeed interacted with Tor1 (Fig 6B). Subsequent in vitro kinase assays using WT or KD (kinase dead) HA‐Tor1 purified by immunoprecipitation as kinase, and with Ypt1 as a substrate, showed that Ypt1 was phosphorylated by Tor1 in the WT but not in the KD samples (Fig 6C), thus revealing Ypt1 as a direct phosphorylation substrate of TOR. To verify that the Ypt1 S174 residue was the phosphorylation site of TOR, we generated an Ypt1 S174A conversion mutant and found that phosphorylation thereby decreased to similar levels as those of the negative control in in vitro kinase assays (Fig 6D). A specific phosphor‐antibody recognizing phosphorylated S174 of yeast Ypt1 (p‐Ypt1 S174) was then generated to detect the phosphorylation level of Ypt1 S174 residue under nutrient‐rich conditions. The result showed that mutating S174 to alanine (S174A) completely abolished the phosphorylation signal (Fig 6E). Due to the decreased kinase activity of TOR during nitrogen starvation, we subsequently used this antibody to test whether the phosphorylation level of Ypt1 S174 residue dramatically reduced under nitrogen starvation conditions. Compared with nutrient‐rich conditions, the phosphorylation level of Ypt1 S174 residue in cells was indeed significantly decreased in response to nitrogen starvation (Fig 6F). These results provided direct evidence that Ypt1 is directly phosphorylated by TOR.
Ypt1 is an autophagy determinant controlled by TOR
Given that Ypt1 is known to be essential for autophagy, we next tested whether its phosphorylation by TOR regulates its function in autophagy. To examine this, we constructed an HA‐Ypt1S174A plasmid, which could not be phosphorylated by TOR, as well as an HA‐Ypt1 S174D plasmid, which mimics Ypt1 phosphorylation by TOR. As TOR negatively regulates autophagic activity, we speculated that the Ypt1 S174A mutant would show enhanced, and the Ypt1 S174D show inhibited, autophagy. To test this hypothesis, HA‐Ypt1, HA‐Ypt1 S174D , HA‐Ypt1 S174A , or empty vector plasmids were separately transformed into AID‐3×HA‐Ypt1 yeast cells co‐expressing GFP‐Atg8 and Vph1‐mCherry. As shown in Fig 7A and B, around 30% cells showed vacuole‐localized GFP‐Atg8 in HA‐Ypt1 S174A yeast cells, while approximately 13% HA‐Ypt1 WT cells displayed vacuole‐localized GFP‐Atg8 at 1 h after nitrogen starvation. We then tested the cleavage efficiency of GFP‐Atg8 in these yeast strains. Consistent with imaging data, HA‐Ypt1 S174A markedly enhanced free GFP levels compared with cells carrying WT HA‐Ypt1 (Fig 7C and D), indicating that the dephosphorylation of Ypt1 at the S174 residue promotes autophagy. Concurrently, we examined whether a mimic of Ypt1 phosphorylation by TOR could inhibit autophagy. Imaging data and the cleavage of GFP‐Atg8 assays showed that HA‐Ypt1 S174D prevented the localization of GFP‐Atg8 to the vacuole and accordingly reduced the generation of free GFP at 1 and 4 h after nitrogen starvation (Fig 7E–H), indicating the phosphorylation of Ypt1 at S174 by TOR inhibits autophagy. Similarly, under rapamycin treatment conditions, HA‐Ypt1 S174A and HA‐Ypt1 S174D variants showed the same phenotypes under nitrogen starvation (Fig EV4A–D). To better assess the overall effect of Ypt1 S174A or Ypt1 S174D on autophagy, we performed an ALP assay to examine the autophagic activity of Ypt1 S174A and Ypt1 S174D mutants at multiple starvation time points. As shown in Fig EV4E, compared with the wild‐type Ypt1, the Ypt1 S174D mutant showed significantly decreased autophagic activity, while the Ypt1 S174A mutant showed accelerated autophagy in response to nitrogen starvation. Taken together, we therefore concluded that TOR‐mediated Ypt1 phosphorylation is an autophagy determinant.
Figure 7. Ypt1 is an autophagy determinant controlled by TOR.
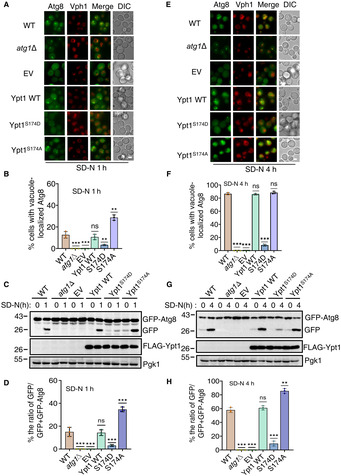
-
A–H(A, E) AID‐3×HA‐Ypt1 yeast strains co‐expressing GFP‐Atg8 and Vph1‐mCherry were transformed into empty vector, FLAG‐Ypt1, FLAG‐Ypt1 S174A , or FLAG‐Ypt1 S174D plasmids. These yeast strains were treated with 0.5 mM IAA for 2 h to degrade endogenous AID‐3×HA‐Ypt1 for 2 h, and then were subject to nitrogen starvation for 1 or 4 h. Images of cells were obtained using an inverted fluorescence microscope. Scale bar, 2 μm. (B, F) Cells from (A, E) were assessed for the translocation of GFP‐Atg8 into vacuoles. n = 300 cells were pooled from three independent experiments. Data are presented as mean ± SD. ***P < 0.001; **P < 0.01; ns, no significance; two‐tailed Student's t‐tests were used. (C, G) The autophagic activity of cells from (A, E) were analyzed by western blot for the cleavage of GFP‐Atg8. Pgk1 served as a loading control. (D, H) The cleavage of GFP‐Atg8 from (C, G) were quantified and presented as mean ± SD (n = 3). ***P < 0.001; **P < 0.01; NS, no significance; two‐tailed Student's t‐tests were used.
Source data are available online for this figure.
Figure EV4. (corresponding to Fig 7). The phosphorylation of Ypt1 S174 by TOR negatively regulates autophagy.
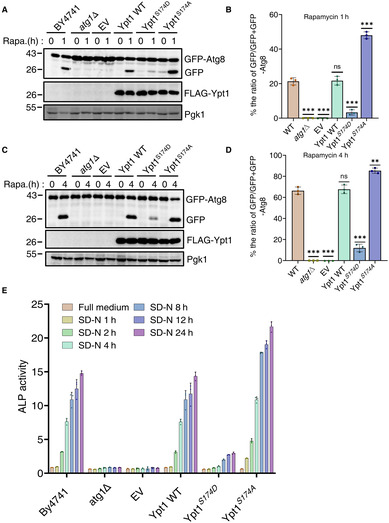
-
A–D(A, C) AID‐3×HA‐Ypt1 yeast strains co‐expressing GFP‐Atg8 and Vph1‐mCherry were transformed into empty vector, FLAG‐Ypt1, FLAG‐Ypt1 S174A , or FLAG‐Ypt1 S174D plasmids. These yeast strains were grown to early log phase, IAA was then added to induce the degradation of AID‐3×HA‐Ypt1 for 2 h. These yeast cells were then subjected to rapamycin treatment for 1 or 4 h. The autophagic activity of cells were analyzed by western blot for the cleavage of GFP‐Atg8. Pgk1 served as a loading control. (B, D) The cleavage of GFP‐Atg8 from (A, C) were quantified and presented as mean ± SD (n = 3). ***P < 0.001; **P < 0.01; NS, no significance; two‐tailed Student's t‐tests were used.
-
EAID‐3×HA‐Ypt1 ALP yeast strain transformed with FLAG‐Ypt1, FLAG‐Ypt1 S174D , Ypt1 S174A , or empty vector were grown to OD600 = 0.6 ~ 0.8. After treatment with 0.5 mM IAA for 2 h, the cells were subjected to nitrogen starvation for the indicated timepoints. Autophagy activity was tested by ALP assay.
Source data are available online for this figure.
TOR‐mediated Ypt1 phosphorylation regulates PAS recruitment and the activation of Atg1
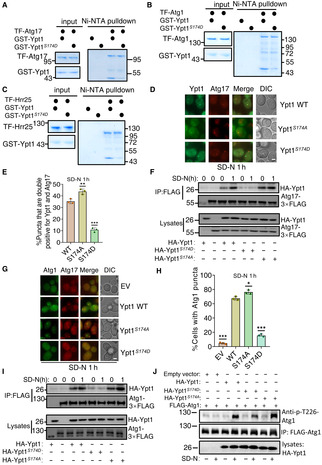
To extend our findings of the regulatory role for TOR‐mediated Ypt1 phosphorylation in autophagy, we next examined whether and how phosphorylation of Ypt1 by TOR affects Ypt1 association with its known autophagy‐related binding partners, Atg1, Atg11, Atg17, Atg23, or Hrr25, as well as its dimerization. In vitro Ni‐NTA pulldown assays illustrated that Ypt1 binding with Atg1, Atg17, and Hrr25 were significantly decreased in the Ypt1 S174D phosphorylation mimic (Fig 8A–C), while association with Atg23, Atg11, and its dimerization remained unaffected (Appendix Fig S8). Subsequently, we measured the binding affinities of Ypt1 WT or Ypt1 S174D with purified TF‐Atg1, Atg17, or Hrr25 using SPR assay (Biacore). K d values showed Ypt1 S174D to have significantly decreased binding to Atg1, Atg17, or Hrr25 (Appendix Fig S9). These results suggested that Ypt1 phosphorylation by TOR could regulate autophagy by modulating the Ypt1 interaction with Atg1, Atg17, and Hrr25.
Figure 8. The phosphorylation of Ypt1 by TOR regulates the PAS recruitment and activation of Atg1.
-
A–CNi‐NTA pulldowns were performed by using purified His6‐tagged TF‐Atg17, TF‐Atg1, or TF‐Hrr25 with GST‐Ypt1 or GST‐Ypt1 S174D from E. coli. Protein samples were separated by SDS‐PAGE, and then detected using Coomassie blue staining.F.
-
DFluorescence microscopy images of AID‐3×HA‐Ypt1 yeast cells co‐expressing Atg17‐2×mCherry and GFP‐Ypt1 WT, S174A, or S174D were subjected to SD‐N for 1 h. Scale bar: 2 μm.
-
ECells from (D) were quantified for the number of cells with puncta that were double positive for Ypt1 and Atg17. n = 300 cells were pooled from three independent experiments. Data are presented as mean ± SD. ***P < 0.001; *P < 0.05; ns, no significance; two‐tailed Student's t‐tests were used.
-
FAID‐3×HA‐Ypt1 yeast cells co‐expressing HA‐Ypt1, HA‐Ypt1 S174A , or HA‐Ypt1 S174D and Atg17‐3×FLAG were grown to log phase and then subjected to SD‐N for 1 h. Cell lysates were immunoprecipitated with anti‐FLAG agarose beads and then analyzed by western blot using an anti‐HA antibody.
-
GAID‐3×HA‐Ypt1 yeast cells co‐expressing Atg17‐2×mCherry and Atg1‐2×GFP were transformed into empty vector, HA‐Ypt1 WT, S174A, or S174D plasmids. These yeast strains were grown to log phase and then subjected to nitrogen starvation for 1 h. Images were obtained using fluorescence microscopy. Scale bar: 2 μm.
-
HCells from (G) were quantified for the number of cells with Atg1 puncta. n = 300 cells were pooled from three independent experiments. Data are presented as mean ± SD. ***P < 0.001; *P < 0.05; ns, no significance; two‐tailed Student's t‐tests were used.
-
IAID‐3×HA‐Ypt1 yeast cells co‐expressing HA‐Ypt1, HA‐Ypt1 S174A , or HA‐Ypt1 S174D and Atg1‐3×FLAG were grown to log phase and then subjected to nitrogen starvation for 0 or 1 h. Cell lysates were immunoprecipitated with anti‐FLAG agarose beads and then analyzed by western blot using an anti‐HA antibody.
-
JAID‐3×HA‐Ypt1 cells co‐expressing HA‐Ypt1, HA‐Ypt1 S174A , or HA‐Ypt1 S174D and Atg1‐3×FLAG were grown to log phase and then subjected to nitrogen starvation for 0 or 1 h, the activity of Atg1 were detected by anti‐phos‐T226‐Atg1 antibody.
Source data are available online for this figure.
Since autophagy initiation requires hierarchical recruitment of autophagy‐related proteins to the PAS (Ohsumi, 2014), and that in vitro Ypt1 interactions with Atg1, Atg17, and Hrr25 are disrupted in the Ypt1 S174D phosphorylation mimic, we then tested the PAS recruitment of Ypt1 in the Ypt1 S174D or Ypt1 S174A mutants under nitrogen starvation. Imaging data and statistical analysis revealed that compared with wild‐type GFP‐Ypt1, the number of cells colocalizing with Atg17‐2×mCherry and GFP‐Ypt1 S174D was significantly decreased, but that this was increased in the GFP‐Ypt1 S174A mutant (Figs 8D and E, and EV5A), suggesting that TOR‐mediated phosphorylation of Ypt1 negatively regulates its recruitment to the PAS. Consistent with that scenario, immunoprecipitation experiments also showed that association with Atg17 was significantly impaired in the Ypt1 S174D phosphorylation mimic, while the binding to Atg17 in the Ypt1 S174A mutant was increased under nitrogen starvation conditions (Fig 8F). We then examined Atg1 recruitment to the PAS in the phosphorylation mimic or mutant by transforming AID‐3×HA‐Ypt1 yeast cells co‐expressing Atg1‐GFP and Atg17‐2×mCherry with HA‐Ypt1, HA‐Ypt1 S174D , HA‐Ypt1 S174A or the empty vector. Fluorescence microscopy and statistical analysis indicated that Atg1 was well‐dispersed in the Ypt1 S174D mutant under nitrogen starvation, while the expression of HA‐Ypt1 S174A enhanced the PAS recruitment of Atg1(Figs 8G and H, and EV5B). Similarly, compared with WT HA‐Ypt1, the association between Ypt1 and Atg1 under nitrogen starvation was increased in the Ypt1 S174A , while their binding was almost blocked in the Ypt1 S174D phosphorylation mimic (Fig 8I). These results further supported the understanding that Ypt1 phosphorylation impairs the PAS recruitment of Atg1.
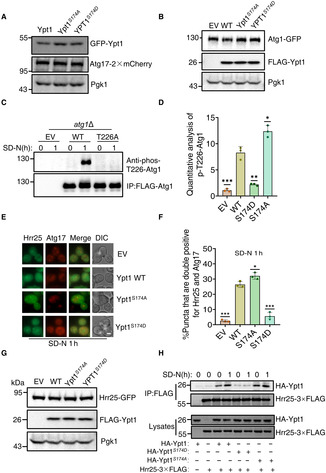
Figure EV5. (corresponding to Fig 8). The phosphorylation of Ypt1 by TOR negatively regulates the PAS recruitment of Hrr25.
-
A, BThe protein samples from Fig 8D (A) and G (B) were analysis by western‐blot using the corresponding antibody. Pgk1 served as a loading control.
-
Catg1∆ yeast cells expressing empty vector, wild‐type FLAG‐Atg1 (WT), or FLAG‐Atg1T226 plasmids were grown to the early log phase, and then subjected to SD‐N for 0 or 1 h. Cell lysates were immunoprecipitated with anti‐FLAG agarose beads and then the phosphorylation level of Atg1 T226 was detected using an anti‐phos‐T226 Atg1 antibody.
-
DThe phosphorylation level of Atg1 T226 from Fig 8J were quantified by calculating the ratio of p‐Atg1‐T226/FLAG‐Atg1 (n = 3). Data are presented as mean ± SD. ***P < 0.001; **P < 0.01; *P < 0.05; two‐tailed Student's t‐tests were used.
-
EAID‐3×HA‐Ypt1 yeast cells co‐expressing Atg17‐2×mCherry and Hrr25‐GFP were transformed into empty vector, HA‐Ypt1 WT, S174A, or S174D plasmids, these yeast strains were grown to log phase and then subjected to nitrogen starvation for 1 h. Images were obtained by fluorescence microscopy. Scale bar: 2 μm.
-
FCells from (E) were quantified for the number of cells with puncta that were double‐positive for Hrr25 and Atg17. n = 300 cells were pooled from three independent experiments. Data are presented as mean ± SD. ***P < 0.001; *P < 0.05; two‐tailed Student's t‐tests were used.
-
GThe protein samples from (E) were analysis by western‐blot using the corresponding antibody. Pgk1 served as a loading control.
-
HAID‐3×HA‐Ypt1 yeast cells co‐expressing HA‐Ypt1, HA‐Ypt1 S174A or HA‐Ypt1 S174D and Hrr25‐3×FLAG were grown to log phase and then subjected to nitrogen starvation for 0 or 1 h. Cell lysates were immunoprecipitated with anti‐FLAG agarose beads and then analyzed by western blot using an anti‐HA antibody.
Source data are available online for this figure.
Previous studies have shown that Atg1 recruitment to the PAS is closely related with Atg1 activation (Kijanska et al, 2010). We then examined Atg1 activation in the Ypt1 S174D or Ypt1 S174A mutants. As Atg1 T226 is phosphorylated in the activated state (Yeh et al, 2010), we generated an anti‐phos‐T226 antibody to detect Atg1 activation (Fig EV5C). Western blot results and quantitative analysis indicated that, compared with wild‐type Ypt1, the phosphorylation of Atg1 T226 in the Ypt1 S174D mutant was significantly reduced, but increased in the Ypt1 S174A mutant, under nitrogen starvation. This implies that the dephosphorylation of Ypt1 is required for the activation of Atg1 under nitrogen starvation conditions (Figs 8J and EV5D). Since Hrr25 has been reportedly to colocalize with the PAS and regulate autophagy via its binding with Ypt1, we also tested whether Hrr25 was recruited to the PAS in the Ypt1 S174D or Ypt1 S174A mutant. Imaging data and statistical analysis showed that Atg17 colocalization with Hrr25 was impaired in the Ypt1 S174D mutant while increased in the Ypt1 S174A mutant under nitrogen starvation (Fig EV5E–G). Co‐IP assays further showed that the association of Ypt1 with Hrr25 was significantly decreased in the Ypt1 S174D phosphorylation mimic while enhanced in the Ypt1 S174A mutant (Fig EV5H). Based on these results, we concluded that Ypt1 phosphorylation is a determinant of autophagy, as controlled by TOR via regulation of Ypt1, Atg1 and Hrr25 recruitment to the PAS, with consequent Atg1 activation then facilitated for the initiation of autophagy.
TOR‐mediated Ypt1 phosphorylation regulating autophagy is conserved in mammals
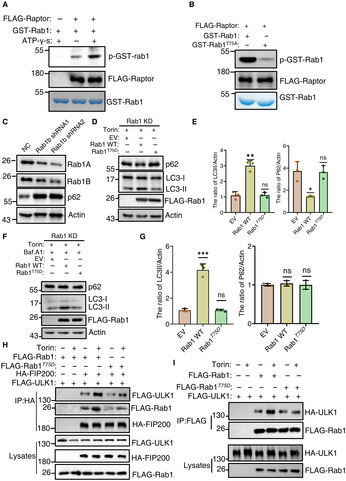
It has been reported that the mammalian Ypt1 homolog, Rab1 is also required for autophagy and ULK1(mammalian Atg1 homolog) can also bind to Rab1 in mammals (Yeh et al, 2010; Wang et al, 2013). We therefore sought to investigate whether Rab1 also shares a conservative mechanism in TOR‐mediated Ypt1 phosphorylation in regulating autophagy in mammals. To this end, in vitro kinase assays were conducted to test whether mTOR could phosphorylate Rab1, and the results showed that mTOR can indeed phosphorylate Rab1 (Fig 9A). To explore which specific sites on Rab1 were phosphorylated by mTOR, anti‐GFP agarose beads were then used to purify GFP‐Rab1 from MEFs under nutrient‐rich medium conditions. Subsequent analysis by LC–MS/MS identified the Rab1 Thr75 residue as the phosphorylation site (Appendix Fig S10A). We then generated T→A conversion mutants for this site to block Rab1 phosphorylation. In vitro mTORC1 kinase assays showed that Rab1 phosphorylation was blocked by the T75A mutation (Fig 9B), suggesting that T75 residue is Rab1 phosphorylation site by mTORC1.
Figure 9. TOR‐mediated Ypt1 phosphorylation regulating autophagy is conserved in mammals.
-
AIn vitro kinase assays were performed using purified GST‐Rab1 from E. coli as substrates, with purified FLAG‐Raptor from nutrient rich‐cultured HEK293T cells as protein kinase. The phosphorylation level of GST‐Rab1A was detected using an anti‐thioP antibody.
-
BIn vitro kinase assays were performed using purified GST‐Rab1 or Rab1A T75A from E. coli as substrates, with purified FLAG‐Raptor from nutrient rich‐cultured HEK293T cells as protein kinase. The phosphorylation level of GST‐Rab1A WT and T75A variants were detected using an anti‐thioP antibody.
-
CThe expression levels of Rab1 in NC (non‐specific control) and Rab1 stable knockdown HEK293T cells were analyzed using western‐blot with an anti‐Rab1A or anti‐Rab1B antibodies, respectively. The expression levels of p62 were detected using anti‐p62 antibody. β‐actin served as a loading control.
-
D–G(D, F) Rab1 KD stable HEK293T cell lines were transfected with the retroviral vectors encoding FLAG‐Rab1A WT, FLAG‐Rab1A T75D , or with the corresponding empty retrovirus (mock). These cells were cultured in complete medium with 500 nM Torin treatment (D), or 250 nM Torin and 100 nM bafilomycin A1 treatment (F) for 120 min. Cell lysates were then analyzed by western‐blot with anti‐p62 and LC3 antibodies. β‐actin served as a loading control. (E, G) Autophagy activity from (D, F) were quantified by the ratio of LC3II/Actin and p62/Actin, and presented as mean ± SD (n = 3). ***P < 0.001; **P < 0.01; *P < 0.05; NS, no significance; two‐tailed Student's t‐tests were used.
-
HRab1 KD stable HEK293T cell lines were co‐transfected with HA‐FIP200, FLAG‐ULK1, and the retroviral vectors encoding FLAG‐Rab1A WT, FLAG‐Rab1A T75D , or with the corresponding empty retrovirus (mock). These cells were cultured in complete medium with 0.25 mM Torin treatment for 0 or 2 h. Cell lyissates were immunoprecipitated with anti‐HA agarose beads and then analyzed by western blot using an anti‐FLAG antibody.
-
IRab1 KD stable HEK293T cell lines were co‐transfected with HA‐ULK1, and the retroviral vectors encoding FLAG‐Rab1A WT, FLAG‐Rab1A T75D , or with the corresponding empty retrovirus (mock). These cells were cultured in nutrient‐rich medium with mTOR inhibitor Torin treatment for 0 or 2 h. Cell lysates were immunoprecipitated with anti‐FLAG agarose beads and then analyzed by western blot using an anti‐HA antibody.
Source data are available online for this figure.
We then tested whether Rab1 phosphorylation by mTORC1 regulates autophagy. To this end, we stably knocked down Rab1A and Rab1B in HEK293T cells. We found significant accumulation of p62 in Rab1 KD cells, indicating that Rab1 positively regulates autophagy in mammals (Fig 9C). We then transfected Rab1 KD cells with FLAG‐Rab1A, FLAG‐Rab1A T75D (mimicking Rab1 phosphorylation by mTORC1), or with an empty vector. As a result, western blot analysis indicated that the generation of LC3II was significantly decreased and p62 was again markedly accumulated in the FLAG‐Rab1A T75D mutant in contrast to that in FLAG‐Rab1A after 2‐h treatment with the mTOR inhibitor Torin (Fig 9D and E). Furthermore, FLAG‐Rab1A T75D mutant showed still decreased LC3 lipidation in Rab1 KD cells cotreated with bafilomycin A1 (Fig 9F and G), indicating that an inhibition role for the phosphorylation of the Rab1 T75 residue on autophagosome formation. To tested whether the reduction in autophagic activity was caused by impaired association of Fip200 with ULK1, we then transfected Rab1 KD cells with HA‐Fip200, FLAG‐ULK1 and FLAG‐Rab1A, FLAG‐Rab1A T75D , or the empty vector. Co‐IP assays demonstrated that the association of FIP200 with FLAG‐Rab1A T75D and FLAG‐ULK1 was significantly decreased compared with Rab1 KD cells carrying WT FLAG‐Rab1A under mTOR inhibitor Torin treatment conditions (Fig 9H), suggesting that dephosphorylation of Rab1 is required for the increase of FIP200‐ULK1 binding. Furthermore, we tested whether Rab1A T75D affects the association of Rab1 with ULK1. As shown in Fig 9I, compare with WT FLAG‐Rab1A, FLAG‐Rab1A T75D significantly impaired the increase of the interaction between Rab1 and ULK1 under autophagy induction condition. This suggests that the association of Rab1 with ULK1 is regulated by its phosphorylation. Taken together, we concluded that TOR‐mediated Ypt1 phosphorylation is conserved in its autophagy regulation.
Discussion
In this study, we identify Atg23 and Atg17 as two previously unrecognized effectors of Ypt1 and confirmed that their interaction with Ypt1 is required for autophagy. Our data showed that disruption of Ypt1‐Atg23 binding blocks the recruitment of Atg9 vesicles to the PAS, while Ypt1‐Atg17 binding is required for the PAS recruitment of Ypt1 and Atg1. Our results also demonstrate that Ypt1 serves as a direct phosphorylation substrate of TOR. Under nutrient‐rich growth conditions, TOR inhibits Atg1 recruitment to the PAS by phosphorylating Ypt1 and Atg13, thereby preventing Ypt1 and Atg13 binding with Atg1, and by doing so ultimately blocking the initiation of autophagy. Conversely, in response to nitrogen starvation, TOR inactivation leads to the dephosphorylation of Ypt1 and Atg13, leading to the subsequent recruitment of Atg1 to the PAS, where it is then activated via the promotion of the association of Ypt1 and Atg13 with Atg1. Activated Atg1 then phosphorylates Atg9 and other autophagy proteins, thus initiating autophagy. We also performed competitive GST pulldown assays using purified TF‐Atg1, TF‐Atg17, and His‐Atg23 with GST‐Ypt1 under different treatment conditions. These confirmed the binding of Ypt1 with Atg1, Atg17, and Atg23 to be competitive, with the priority order of their binding with Ypt1 in vitro being: Atg23 > Atg17 > Atg1 (Appendix Fig S10B–D). Based on these data, we propose that the TOR‐mediated Ypt1 serves as a multifunctional assembly factor controlling the initiation of autophagy its regulation of the stepwise assembly of ATG proteins. (Fig 10).
Figure 10. Model for TOR‐mediated Ypt1 serves as an autophagy initiation determinant regulating the stepwise assembly of ATG proteins.
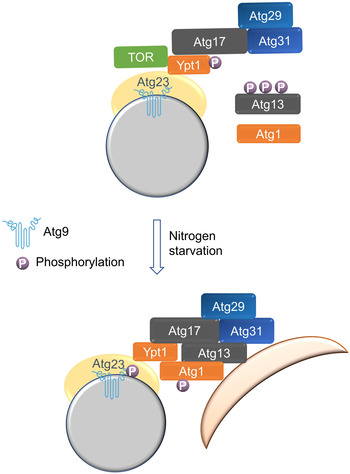
Atg17 recruits Ypt1 and Atg1 to the PAS via the binding of Atg17‐Ypt1, and Atg9 vesicles are recruited to the PAS via the binding of Ypt1‐Atg23. In nutrient‐rich conditions, TOR inhibits the binding of Ypt1 to Atg1 by phosphorylating Ypt1, thereby blocking autophagy. In response to nitrogen starvation, TOR inactivation leads to the dephosphorylation of Atg13 and Ypt1, and then recruits Atg1 to the PAS to activate Atg1. Activated Atg1 phosphorylates Atg9 and other autophagy proteins to initiate the stepwise assembly of ATG proteins and subsequent autophagy. P, phosphorylated.
Atg9 is a multiple transmembrane domain protein that is delivered to the PAS via its association with vesicles that also serve as a resource for autophagosomal membrane formation (Yamamoto et al, 2012). Atg9 vesicle recruitment to the PAS is therefore revealed as a key stage in the formation of autophagosomes. Previous work has shown that Atg9 vesicles are recruited to the PAS through their direct association with Atg11, Atg13, or Atg17 under different cultured conditions. However, so far, the understanding of the molecular mechanism by which Atg9 vesicles are recruited to the PAS has remained incomplete. Particularly unclear is why the Ypt1 temperature sensitive strain affects the PAS recruitment of Atg9 vesicles (Lipatova et al, 2012), and how Atg23 participates in the PAS recruitment of Atg9 vesicles. The data in this study helps answer such questions and indicates that Atg9 vesicle recruitment to the PAS requires not only the interactions of Atg9 with Atg11, Atg17, and Atg13. Our data shows that Atg23 is a new effector of Ypt1, and that their binding is required for autophagy. In the absence of Atg23‐Ypt1 interaction, Atg9 binding to Atg11, Atg13 and Atg17 is impaired, which results in blockading of Atg9 vesicle mobilization to the PAS. Therefore, during the initiation process of autophagy, Ypt1 first recruits Atg9 vesicles to the PAS via interactions with Atg23, after which Atg9 can bind to Atg11, Atg13 and Atg17. In this way, our results provide fresh insight into the mode by which Atg9 vesicles are recruited to the PAS. However, it is worth noting that the formation of Atg9 vesicles is severely impaired in atg23∆ cells (Zoppino et al, 2010), while its formation was not affected in the absence of the binding of Atg23‐Ypt1, suggesting that Atg23 plays another crucial, Ypt1‐independent role in the formation of Atg9 vesicles at the peripheral site.
In this study, Ypt1 not only binds to Atg1, Atg11, Atg17, Atg23, and Hrr25, but also binds to selective autophagic receptors Atg19, Atg34, and Atg40, indicating that Ypt1 may recruit autophagic cargo into autophagosome by enhancing the association of these selective autophagy receptors with Atg11. In addition, we also found that Ypt1 binds to Ykt6. As an autophagy specific t‐SNARE protein, Ykt6 participates in autophagosome and lysosome/vacuole fusion in yeast, Drosophila, and mammalian cells (Kriegenburg et al, 2019). Therefore, the binding of Ykt6‐Ypt1 implies that Ypt1 may also play an important role during the process of autophagosome and lysosome/vacuole fusion through promoting the recruitment of Ykt6 to autophagosomes. The study of these molecular mechanisms will be highlighted in our next work. It has been previously reported that the PAS localization of Atg17‐GFP is not disrupted in the ypt1‐2 mutant (Wang et al, 2013). This is consistent with our finding that Atg17 could direct bind with Ypt1 and that Ypt1 degradation does not affect the formation of Atg17 puncta. One study has reported that Ypt1 is localized to the preautophagosomal structure in an Atg9‐dependent manner (Kakuta et al, 2012). However, another study found that the deletion of ATG9 simply decreases the PAS recruitment of Ypt1(Lynch‐Day et al, 2010). Our research findings are more biased toward the latter study, since the direct binding of Ypt1 with Atg17 and Atg11 provides direct evidence for the PAS recruitment of Ypt1. Linked to this, how the deletion of ATG9 or ATG23 decreases the PAS recruitment of Ypt1 may be a suitable focus for future work.
mTOR is widely understood to serve as a master regulator of autophagy at multiple levels (Kim & Guan, 2015). Our data reveal a new regulatory element, of equal importance to Atg13 and Atg17, that regulates Atg1 kinase activity in response to cellular stress. We also observed that the phosphorylation sites of Ypt1 and Rab1 by mTOR are not conservative in yeast and mammals. We think that the reason for this discrepancy may be caused by the different three‐dimensional structure of mTORC1 complex in mammalian and yeast cells, or the different microenvironment where Ypt1 and Rab1 binds with the mTORC1 complex. In fact, some studies have found that TOR/mTOR phosphorylate the same substrates, despite the sites for such substrates being different in yeast and mammals (Kamada et al, 2010; Kijanska et al, 2010; Yeh et al, 2010; Kim et al, 2011; Puente et al, 2016). For example, Atg13 is reported to be phosphorylated by TOR in both yeast and mammals. However, Atg13 is phosphorylated by mTOR at Ser 258 in mammals (Puente et al, 2016), while in yeast cells, Atg13 S348, S437, S438, S496, S535, S541, S646, and S649 residues were found to be phosphorylated by TOR (Kamada et al, 2010). Regardless of the phosphorylation sites of Atg13 by TOR from yeast and mammals being different, the phosphorylation of Atg13 caused by TOR has a similar function, that of the blocking of autophagy. Dephosphorylation of these sites on Atg13 enhances the formation of Atg1 puncta and the activation of Atg1. Similarly, in this study, although the phosphorylation sites of Ypt1 and Rab1 by TOR are different, the phosphorylation of Ypt1 and Rab1 by mTOR inhibits the binding of Ypt1/Rab1 to Ulk1 thus also inhibiting subsequent autophagy.
In this study, the identification of Atg23 and Atg17 as two new Ypt1 effectors, and of Ypt1 as a substrate of TOR, together helps to elucidate the molecular mechanism of Atg9 and Atg1 recruitment to the PAS, and further expand the functions of TOR in regulating autophagy. Despite these advances, it remains unknown whether there are further undiscovered important effectors of Ypt1 that could contribute to autophagy regulation, or whether Ypt1 is a substrate of other protein kinases. Our ongoing studies will continue to investigate Ypt1 functions in the initiation of autophagy.
Materials and Methods
Yeast strains, constructs, and growth conditions
All yeast strains used in this study are listed in Dataset EV2. The wild‐type (BY4741) and gene deletion library were purchased from Invitrogen (95401.H2). The related strains were constructed as described previously (Janke et al, 2004), and verified by western blot analysis using the corresponding antibody or by polymerase chain reaction (PCR; Vazyme, P505‐d1, Nanjing). All mutant plasmids and related strains in this study were sequenced by Tsingke Biotechnology, Beijing. Yeast cells were cultured at 30°C in synthetic medium (SD; 0.17% yeast nitrogen base without amino acids and ammonium sulfate, 0.5% ammonium sulfate, 2% glucose, and corresponding auxotrophic amino acids and vitamins). For autophagy induction, cells were grown to early‐log phase, and then were treated with nitrogen starvation (SD‐N; 0.17% yeast nitrogen base without amino acids and ammonium sulfate, 2% glucose), or glucose starvation (SD‐G; 0.17% yeast nitrogen base without amino acids and ammonium sulfate, 0.5% ammonium sulfate, 0.5% casamino acid), or 0.2 μg/ml rapamycin (Selleck, AY‐22989) treatment for the corresponding timepoint.
Antibodies
Antibodies used in this study were as follows: anti‐Pgk1 (Nordic Immunology, NE130/7S; 1:10,000), anti‐FLAG (Sigma, F1804; 1:2,500), anti‐HA (Abmart, M20003L; 1:3,000), anti‐thiophosphate ester (Abcam, ab133473; 1:10,000), anti‐GFP (Roche, 11814460001; 1:2,500), anti‐Ape1 antibody was obtained from Dr. Zhiping Xie Lab (Shanghai Jiaotong University, China), anti‐phos‐T226‐Atg1 antibody (Youke, Shanghai), anti‐p62/SQSTM1(Proteintech, 18420‐1‐AP, 1:5,000), anti‐LC3 (MBL, PM036, 1:1,000), anti‐Rab1A (Proteintech, 11671‐1‐AP, 1:1,000), anti‐Rab1B (Proteintech, 17824‐1‐AP, 1:1,000), β‐Actin (Abmart, T40104, 1:5,000); anti‐p‐Ypt1‐S174 (homemade, 1:4,000); goat anti‐mouse IgG1, human ads‐HRP (SouthernBiotech, 1070‐05; 1:10,000); and goat anti‐rabbit, human ads‐HRP (SouthernBiotech, 4010‐05; 1:10,000).
Mammalian cell culture and construction of stable Rab1b knockdown cell lines
HEK293T cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L‐glutamine, and 100 U/ml penicillin–streptomycin in a humidified incubator at 37°C with 5% CO2. Torin1 (S2827) was purchased from Selleck Chemicals. A pGPU6/hygromycin vector was used for the expression of the Rab1b shRNA1 and shRNA2 sequences are “UGCCAGCGAGAACGUCAAUAA” and “CACGUACACAGAGAGCUACAU” in HEK293T cells, respectively. The construction of stable Rab1b knockdown cell lines was performed according to the manufacturer's protocol (Shanghai GenePharma). The efficiency of Rab1A and Rab1B knockdown was tested using western blotting with anti‐Rab1A or anti‐Rab1B antibody.
Imaging, western blots, and immunoprecipitation
Imaging, yeast protein extraction, western blot, and immunoprecipitation experiments were performed as described previously (Yi et al, 2017). Ypt1‐related assays and Rab1‐related immunoprecipitation assays were carried out as detailed previously (Zoppino et al, 2010; Wang et al, 2013).
Auxin treatment
Yeast strains expressing 3×HA‐AID‐Ypt1 were grown to the early logarithmic phase, and then subjected to 0.5 mM IAA (indole‐3‐acetic acid; Sigma, I2886) treatment for 2 h. The degradation of Ypt1 was detected by anti‐HA antibody.
Protein purification from Escherichia coli
Escherichia coli BL21 cells (Tsingke, TSV‐A09) with His/GST‐tag prokaryotic expression plasmid were grown in 500 ml of LB medium containing 50 μg/μl ampicillin (Sangon Biotech, A600064‐0025) to OD600 = 0.5 ~ 0.6 at 37°C, then shaken at 18°C for 24 h in the presence of 0.1 mM IPTG (Sangon Biotech, A600168‐0025), After harvesting by centrifugation and the pellets were suspended in 25 ml of lysis buffer (50 mM Tris.HCl, pH 7.5, 500 mM NaCl, 0.1% Triton X‐100 [Sangon Biotech, A600198‐0500], 0.1 mM PMSF [Sangon Biotech, A610425‐0025], 1 mM DTT, and 20 mM iminazole if the plasmid has His tag) and disrupted by sonication. Subsequently, the lysates were centrifuged at 13,000 g for 30 min. The supernatants were mixed with 1 ml (bed volume) of a GST (GE Healthcare, 17‐0756‐01)/Ni‐NTA agarose resin (GE Healthcare, 17‐5318‐06) and rotated at 4°C for 2 h. The resins were washed with 20 ml of washing buffer (20 mM Tris.HCl, pH 7.5, 500 mM NaCl, 0.1 mM PMSF, 30 mM Imidazole [Sangon Biotech, A600277‐0100] if the protein has His tag) for three successive times. The samples were eluted with 1.5 ml of elution buffer (20 mM Tris.HCl, pH 7.5, 500 mM NaCl, 250 mM Imidazole, 0.1 mM PMSF, 50 mM GSH, pH 7.4 if the protein has GST tag or 250 mM iminazole if the protein has His tag). According to protein purity results, proteins with low purity were then further purified through molecular sieve or anion exchange chromatography. 50% glycerol was added to a final concentration of 10% at −80°C for storage.
In vitro pulldown assay
For in vitro protein pulldown assays, a certain amount of purified protein (3–5 nM, considering the protein purity, a range of protein concentration values is given) was incubated with 10 μl GST beads/Ni‐NTA in 500 μl binding buffer A (50 mM Tris.Hcl pH7.5, 0.5 M NaCl, 1 mM DTT, 1% Triton X‐100, 1% PMSF, and 20 mM iminazole if using Ni‐NTA) on a rotor at 4°C for 1 h. Beads were then washed once with binding buffer A following by washing twice with binding buffer B (100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Tween‐20 in 1×PBS). The beads bound to the purified protein were incubated on a rotor with an equal amount of purified target protein from E. coli with binding buffer B (100/200/300/400 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Tween‐20 in 1×PBS) at 4°C for 3 h. After washing with binding buffer B three times, 50 μl 2×SDS loading buffer was added and boiled at 95°C for 5 min. The protein samples were then separated by SDS‐PAGE and stained with Coomassie brilliant blue dye G250.
ALP assay
AID‐3 × HA‐Ypt1 ALP yeast strain transformed with FLAG‐Ypt1, FLAG‐Ypt1S174D, FLAG‐Ypt1S174A, or empty vector was grown to OD600 = 0.6 ~ 0.8. After treatment with 0.5 mM IAA for 2 h, the cells were subjected to nitrogen starvation. Yeast cells of 4 OD600 were harvested at 0, 1, 2, 4, 8, 12, or 24 h after starvation, and washed with 1 ml of cold 0.85% NaCl. Add 200 μl lysis buffer (20 mM PIPES pH 7.0, 0.5% Triton X‐100, 50 mM KCl, 100 mM potassium acetate, 10 mM MgSO4, 10 μM ZnSO4, 1 mM PMSF) and 100 μl glass beads to disrupt cells at 4°C. After centrifugation at 13,523 g for 10 min at 4°C, the supernatant was determined for protein concentration using the BCA method. After that, the reaction buffer (250 mM Tris–HCl, pH 8.5, 0.4% Triton X‐100, 10 mM MgSO4, 10 μM ZnSO4) was added to the supernatant sample and cultured in a 30°C incubator. After 30 min, a stop buffer (1 M Glycine.KOH, pH 11) was added to stop the reaction. Finally, the absorbance of the sample was measured at OD405 and the ALP activity was calculated.
TIRF microscopy
Yeast strains expressed Atg9‐2 × GFP were grown to logarithmic phase (OD600 = 0.8 ~ 1.2) in nutrient‐rich medium and then subjected to nitrogen starvation for 1 h. Cells were observed at a room temperature using a total internal reflection fluorescent microscope (N‐STORM/A1R, Nikon) equipped with an electron high‐sensitivity camera (DU897E EMCCD, Andor) and 100 × total internal reflection fluorescence objective (100 × TIRF lens, NA 1.49, Nikon). The Imaris software was used to calculate the mean square displacement (MSD) of Atg9 puncta to observe puncta formation and movement. All the images were processed using the Fiji ImageJ software and its plugins.
In vitro phosphorylation assay and alkylation protocol
tor1∆ yeast strains expressing HA‐Tor1 or HA‐Tor1 KD plasmids were grown to the log phase. 50OD600 yeast cells were lysed and the supernatant was immunoprecipitated with anti‐HA agarose beads (Sigma, A2220). Purified HA‐Tor1 or HA‐Tor1 KD were incubated with 5 μg purified GST‐Ypt1 from E. coli and 1.5 μl 10 mM ATP‐γ‐S (Sigma, A1388) in Tor1 kinase buffer (50 mM HEPES.KOH, pH 7.4, 5 mM NaF, 50 mM NaCl, 5 mM MgCl2, 1 mM DTT) at 30°C for 30 min, and then 2 μl 50 mM p‐nitrobenzyl mesylate/PNBM (Abcam, ab138910) was added and left to react for another hour at 30°C. The samples were halted by boiling in protein loading buffer for 5 min. The phosphorylation level of GST‐Ypt1 was then tested using the anti‐thiophosphate ester antibody (Abcam, ab92570).
SPR assay
The purified GST‐Ypt1WT/S174D protein from E. coli was immobilized on the CM5 SPR sensor chip by an amine‐coupling procedure. SPR (Surface plasmon resonance) experiments were conducted on the Biacore X100 instrument (GE Healthcare). The resulting data were fit to a 1:1 binding model using the Biacore Evaluation Software (GE Healthcare). The target proteins‐TF/TF‐Atg1/TF‐Atg17/TF‐Hrr25 were perfused as analytes. Serial dilutions of purified proteins‐TF/TF‐Atg1/TF‐Atg17/TF‐Hrr25 were injected ranging in concentration from 400 to 12.5 nM. The kinetics/affinity in SPR responses were measured and used to determine the KD values. In all experiments, the analysis was performed at 25°C, with an association time of 180 s at a flow rate of 20 μl/min. The resulting data were fit to a 1:1 binding model using the Biacore Evaluation Software. According to the experimental results, there was no specific affinity between protein‐TF and GST‐Ypt1WT or S174D.
Mass spectrometric analysis
The SDS–PAGE was used to separate the protein, and stained with Coomassie Blue G‐250. The gel bands of interest were cut into pieces. Samples were digested by trypsin with prior reduction and alkylation in 50 mM ammonium bicarbonate at 37°C overnight. The digested products were extracted twice with 1% formic acid in 50% acetonitrile aqueous solution and dried to reduce volume by speedvac. For LC–MS/MS analysis, the peptides were separated using 65 min gradient elution at a flow rate 0.300 μl/min with in a Thermo EASY‐nLC1200 integrated nano‐HPLC system directly interfaced to a Thermo Q Exactive HF‐X mass spectrometer. The analytical column was a home‐made fused silica capillary column (75 μm ID, 150 mm length; Upchurch, Oak Harbor, WA) packed with C‐18 resin (300 A, 3 μm, Varian, Lexington, MA). The mobile phase A consisted of 0.1% formic acid, and the mobile phase B consisted of 100% acetonitrile and 0.1% formic acid. The mass spectrometer was operated in the data‐dependent acquisition mode using the Xcalibur 4.1 software with a single full‐scan mass spectrum in the Orbitrap (400–1,800 m/z, 60,000 resolution) followed by 20 data‐dependent MS/MS scans at 30% normalized collision energy. Each mass spectrometry result was analyzed using a Thermo Xcalibur Qual Browser and Proteome Discovery for the database searching.
Quantitative and statistical analysis
Western blot images from three independent experiments were quantified using ImageJ software. Maturation of Ape1 was quantified by the ratio of band intensities of Ape1 to that of total Ape1 (Ape1 + prApe1). GFP intensities were divided by the sum of GFP and the GFP‐Atg8 to calculate the degradation rate of the fused protein. Statistical analyses were presented as mean ± SD (n = 3). ***P < 0.001; **P < 0.01; *P < 0.05; ns, no significance; two‐tailed Student's t‐tests were used.
Author contributions
Zhiping Xie: Resources. Pengwei Zhao: Data curation; formal analysis; investigation. Liqin Zhang: Resources. Yixian Cui: Methodology. Cong Yi: Conceptualization; supervision; funding acquisition; writing – original draft; project administration; writing – review and editing. Choufei Wu: Resources. Yigang Wang: Resources; methodology. Qiming Sun: Resources. Cheng Ma: Resources. Wei Liu: Resources; methodology. Juan Wang: Methodology. Hongguang Xia: Resources. Shan Feng: Resources; formal analysis. Mingzhu Fan: Resources; formal analysis. Xue Bai: Resources; formal analysis. Yuyao Feng: Resources; formal analysis. Yingcong Chen: Data curation; formal analysis; investigation; methodology. Miao Ye: Resources; formal analysis. Yu Xie: Resources; formal analysis. Jing Liu: Resources; formal analysis; investigation. Siyu Fan: Resources; formal analysis. Qiang Jiang: Resources; formal analysis. Weijing Yao: Conceptualization; resources; data curation; software; formal analysis; investigation; methodology; project administration. Yuting Chen: Conceptualization; data curation; formal analysis; investigation; methodology. Yi Zhang: Resources; formal analysis.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Movie EV4
Movie EV5
Movie EV6
Movie EV7
Movie EV8
Dataset EV1
Dataset EV2
Source Data for Expanded View and Appendix
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Source Data for Figure 9
Acknowledgements
We are grateful to Prof. Y. Ohsumi and Prof. H. Nakatogawa for the plasmids, antibodies, and yeast strains. We are also thankful for advice from Prof. Fan Yang in the SPR assays, the support of the Protein facility and Imaging center, Core facilities, Zhejiang University School of Medicine; and from the Mass Spectrometry & Metabolomics Core Facility at the Center for Biomedical Research Core Facilities of Westlake University for sample analysis. The research was supported by National Natural Science Foundation of China (32122028, 92254307, and 32070739), and Zhejiang Provincial Natural Science Foundation of China under Grant No.LR21C070001 to CY; National Natural Science Foundation of China (Grant No: 32100600) to WY; and the National Key Research and Development Program of China (2021YFC2600104) to LZ.
The EMBO Journal (2023) 42: e112814
Data availability
This study includes no data deposited in external repositories. The related strains and plasmids will be made available upon reasonable request.
References
- Backues SK, Orban DP, Bernard A, Singh K, Cao Y, Klionsky DJ (2015) Atg23 and Atg27 act at the early stages of Atg9 trafficking in S. cerevisiae . Traffic 16: 172–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Ma X, Zhu Q, Song D, Ding X, Li L, Jiang X, Wang X, Tian R, Su H et al (2019) Pacer is a mediator of mTORC1 and GSK3‐TIP60 signaling in regulation of autophagosome maturation and lipid metabolism. Mol Cell 73: 788–802 [DOI] [PubMed] [Google Scholar]
- Davis S, Ferro‐Novick S (2015) Ypt1 and COPII vesicles act in autophagosome biogenesis and the early secretory pathway. Biochem Soc Trans 43: 92–96 [DOI] [PubMed] [Google Scholar]
- Feng Y, He D, Yao Z, Klionsky DJ (2014) The machinery of macroautophagy. Cell Res 24: 24–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Ariosa AR, Yang Y, Hu Z, Dengjel J, Klionsky DJ (2021) Downregulation of autophagy by Met30‐mediated Atg9 ubiquitination. Proc Natl Acad Sci USA 118: e2005539118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graef M, Friedman JR, Graham C, Babu M, Nunnari J (2013) ER exit sites are physical and functional core autophagosome biogenesis components. Mol Biol Cell 24: 2918–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosshans BL, Ortiz D, Novick P (2006) Rabs and their effectors: achieving specificity in membrane traffic. Proc Natl Acad Sci USA 103: 11821–11827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins WD, Leary KA, Andhare D, Popelka H, Klionsky DJ, Ragusa MJ (2022) Dimerization‐dependent membrane tethering by Atg23 is essential for yeast autophagy. Cell Rep 39: 110702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Song H, Yorimitsu T, Monastyrska I, Yen W‐L, Legakis JE, Klionsky DJ (2006) Recruitment of Atg9 to the preautophagosomal structure by Atg11 is essential for selective autophagy in budding yeast. J Cell Biol 175: 925–935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno‐Borchart A, Doenges G, Schwob E, Schiebel E et al (2004) A versatile toolbox for PCR‐based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21: 947–962 [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Kamada Y, Baba M, Takikawa H, Sasaki M, Ohsumi Y (2005) Atg17 functions in cooperation with Atg1 and Atg13 in yeast autophagy. Mol Biol Cell 16: 2544–2553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakuta S, Yamamoto H, Negishi L, Kondo‐Kakuta C, Hayashi N, Ohsumi Y (2012) Atg9 vesicles recruit vesicle‐tethering proteins Trs85 and Ypt1 to the autophagosome formation site. J Biol Chem 287: 44261–44269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y (2000) Tor‐mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 150: 1507–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada Y, Yoshino K‐i, Kondo C, Kawamata T, Oshiro N, Yonezawa K, Ohsumi Y (2010) Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol Cell Biol 30: 1049–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kijanska M, Dohnal I, Reiter W, Kaspar S, Stoffel I, Ammerer G, Kraft C, Peter M (2010) Activation of Atg1 kinase in autophagy by regulated phosphorylation. Autophagy 6: 1168–1178 [DOI] [PubMed] [Google Scholar]
- Kim YC, Guan K‐L (2015) mTOR: a pharmacologic target for autophagy regulation. J Clin Invest 125: 25–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, Guan K‐L (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegenburg F, Bas L, Gao J, Ungermann C, Kraft C (2019) The multi‐functional SNARE protein Ykt6 in autophagosomal fusion processes. Cell Cycle 18: 639–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipatova Z, Belogortseva N, Zhang XQ, Kim J, Taussig D, Segev N (2012) Regulation of selective autophagy onset by a Ypt/Rab GTPase module. Proc Natl Acad Sci USA 109: 6981–6986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch‐Day MA, Bhandari D, Menon S, Huang J, Cai H, Bartholomew CR, Brumell JH, Ferro‐Novick S, Klionsky DJ (2010) Trs85 directs a Ypt1 GEF, TRAPPIII, to the phagophore to promote autophagy. Proc Natl Acad Sci USA 107: 7811–7816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano G, Esposito A, Choi H, Matarese M, Benedetti V, Di Malta C, Monfregola J, Medina DL, Lippincott‐Schwartz J, Ballabio A (2018) mTOR‐dependent phosphorylation controls TFEB nuclear export. Nat Commun 9: 3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M (2009) An auxin‐based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 6: 917–922 [DOI] [PubMed] [Google Scholar]
- Noda T, Ohsumi Y (1998) Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 273: 3963–3966 [DOI] [PubMed] [Google Scholar]
- Ohsumi Y (2014) Historical landmarks of autophagy research. Cell Res 24: 9–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puente C, Hendrickson RC, Jiang X (2016) Nutrient‐regulated phosphorylation of ATG13 inhibits starvation‐induced autophagy. J Biol Chem 291: 6026–6035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragusa MJ, Stanley RE, Hurley JH (2012) Architecture of the Atg17 complex as a scaffold for autophagosome biogenesis. Cell 151: 1501–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanov J, Walczak M, Ibiricu I, Schüchner S, Ogris E, Kraft C, Martens S (2012) Mechanism and functions of membrane binding by the Atg5‐Atg12/Atg16 complex during autophagosome formation. EMBO J 31: 4304–4317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekito T, Kawamata T, Ichikawa R, Suzuki K, Ohsumi Y (2009) Atg17 recruits Atg9 to organize the pre‐autophagosomal structure. Genes Cells 14: 525–538 [DOI] [PubMed] [Google Scholar]
- Shibutani ST, Yoshimori T (2014) A current perspective of autophagosome biogenesis. Cell Res 24: 58–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki SW, Yamamoto H, Oikawa Y, Kondo‐Kakuta C, Kimura Y, Hirano H, Ohsumi Y (2015) Atg13 HORMA domain recruits Atg9 vesicles during autophagosome formation. Proc Natl Acad Sci USA 112: 3350–3355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker KA, Reggiori F, Dunn WA, Klionsky DJ (2003) Atg23 is essential for the cytoplasm to vacuole targeting pathway and efficient autophagy but not pexophagy. J Biol Chem 278: 48445–48452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan W, You Z, Xu Y, Zhou L, Guan Z, Peng C, Wong CCL, Su H, Zhou T, Xia H et al (2017) mTORC1 phosphorylates acetyltransferase p300 to regulate autophagy and lipogenesis. Mol Cell 68: 323–335 [DOI] [PubMed] [Google Scholar]
- Wan W, You Z, Zhou L, Xu Y, Peng C, Zhou T, Yi C, Shi Y, Liu W (2018) mTORC1‐regulated and HUWE1‐mediated WIPI2 degradation controls autophagy flux. Mol Cell 72: 303–315 [DOI] [PubMed] [Google Scholar]
- Wang J, Menon S, Yamasaki A, Chou H‐T, Walz T, Jiang Y, Ferro‐Novick S (2013) Ypt1 recruits the Atg1 kinase to the preautophagosomal structure. Proc Natl Acad Sci USA 110: 9800–9805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Cervantes S, Davis S, Ferro‐Novick S (2015a) Identifying a Rab effector on the macroautophagy pathway. Methods Mol Biol 1298: 117–125 [DOI] [PubMed] [Google Scholar]
- Wang J, Davis S, Menon S, Zhang J, Ding J, Cervantes S, Miller E, Jiang Y, Ferro‐Novick S (2015b) Ypt1/Rab1 regulates Hrr25/CK1δ kinase activity in ER‐Golgi traffic and macroautophagy. J Cell Biol 210: 273–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Kakuta S, Watanabe TM, Kitamura A, Sekito T, Kondo‐Kakuta C, Ichikawa R, Kinjo M, Ohsumi Y (2012) Atg9 vesicles are an important membrane source during early steps of autophagosome formation. J Cell Biol 198: 219–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh Y‐Y, Wrasman K, Herman PK (2010) Autophosphorylation within the Atg1 activation loop is required for both kinase activity and the induction of autophagy in Saccharomyces cerevisiae . Genetics 185: 871–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C, Tong J, Lu P, Wang Y, Zhang J, Sun C, Yuan K, Xue R, Zou B, Li N et al (2017) Formation of a Snf1‐Mec1‐Atg1 module on mitochondria governs energy deprivation‐induced autophagy by regulating mitochondrial respiration. Dev Cell 41: 59–71 [DOI] [PubMed] [Google Scholar]
- Yuan H‐X, Russell RC, Guan K‐L (2013) Regulation of PIK3C3/VPS34 complexes by MTOR in nutrient stress‐induced autophagy. Autophagy 9: 1983–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoppino FCM, Militello RD, Slavin I, Alvarez C, Colombo MI (2010) Autophagosome formation depends on the small GTPase Rab1 and functional ER exit sites. Traffic 11: 1246–1261 [DOI] [PubMed] [Google Scholar]