

Abstract
WRKY transcription factors in plants are known to be able to mediate either transcriptional activation or repression, but the mechanism regulating their transcriptional activity is largely unclear. We found that group IId WRKY transcription factors interact with OBERON (OBE) proteins, forming redundant WRKY‐OBE complexes in Arabidopsis thaliana. The coiled‐coil domain of WRKY transcription factors binds to OBE proteins and is responsible for target gene selection and transcriptional repression. The PHD finger of OBE proteins binds to both histones and WRKY transcription factors. WRKY‐OBE complexes repress the transcription of numerous stress‐responsive genes and are required for maintaining normal plant growth. Several WRKY and OBE mutants show reduced plant size and increased drought tolerance, accompanied by increased expression of stress‐responsive genes. Moreover, expression levels of most of these WRKY and OBE genes are reduced in response to drought stress, revealing a previously uncharacterized regulatory mechanism of the drought stress response. These results suggest that WRKY‐OBE complexes repress transcription of stress‐responsive genes, and thereby balance plant growth and stress tolerance.
Keywords: development, histone, stress tolerance, transcription factor, transcriptional repression
Subject Categories: Plant Biology
WRKY and OBERON proteins interact under low‐stress conditions to repress drought stress responses.

Introduction
Because plants cannot move to select suitable growth conditions, they are easily affected by adverse environmental conditions during growth and development. In the face of both biotic and abiotic stresses, plants usually survive by restricting their growth, which is accomplished through stress‐triggered signal transduction (Guo et al, 2018; Gong et al, 2020; Wang et al, 2020; Zhang et al, 2020). In response to stress‐triggered signals, the expression of many stress‐responsive genes is induced to enhance plant stress tolerance (Zhu, 2002; Yamaguchi‐Shinozaki & Shinozaki, 2006; Shi et al, 2018; Kidokoro et al, 2022). A series of transcription factors are known to be critical for the induction of stress‐responsive genes, and most of these transcription factor‐encoding genes are also stress‐responsive genes (Yamaguchi‐Shinozaki & Shinozaki, 2006; Song et al, 2016). The induced expression of stress‐responsive genes not only enhances stress tolerance but also restricts plant growth and development (Jaglo‐Ottosen et al, 1998; Liu et al, 1998; Kim et al, 2012). Therefore, maintaining low expression levels of stress‐responsive genes under non‐stress conditions is critical for normal plant growth and development.
In Arabidopsis thaliana, there are more than 1,500 transcription factors, which form several large transcription factor families including MYB, AP2/ERF, NAC, bHLH, bZIP, MADS, WRKY, and ARF (Riechmann & Ratcliffe, 2000; Ng et al, 2018; Feng et al, 2020). WRKY transcription factors, which are primarily present in plants, are involved in the regulation of multiple biological processes, including development, senescence, hormone signaling, and biotic and abiotic stress responses (Eulgem et al, 2000; Rushton et al, 2010; Phukan et al, 2016; Jiang et al, 2017). WRKY transcription factors contain one or two WRKY domains, which consist of nearly 60 amino acids and are specifically responsible for binding to the W‐box (C/T)TGAC(C/T) at the promoter regions (Eulgem et al, 2000; Ciolkowski et al, 2008). There are 74 Arabidopsis WRKY transcription factors that can be classified into three subfamilies (I, II, and III) based on the number and structure of WRKY domains (Eulgem et al, 2000). Based on additional domains, subfamily II WRKY transcription factors can be further divided into five groups: IIa, IIb, IIc, IId, and IIe (Eulgem et al, 2000). Although WRKY transcription factors are involved in either transcriptional activation or repression (Rushton et al, 2010), little is known about how these WRKY transcription factors obtain the opposite activities in the regulation of transcription.
The Arabidopsis group IId WRKY transcription factors contain six members: WRKY7, WRKY11, WRKY15, WRKY17, WRKY21, and WRKY39 (Eulgem et al, 2000). Previous studies have shown that the group IId WRKY transcription factors are involved in the repression of basal resistance to Pseudomonas syringae (Journot‐Catalino et al, 2006; Arrano‐Salinas et al, 2018) and xylem vessel formation (Ge et al, 2020). Similar to other WRKY transcription factors, the group IId WRKY transcription factors recognize the W‐box motif (Ciolkowski et al, 2008; Brand et al, 2013). Although WRKY transcription factors are involved in both transcriptional activation and repression activities (Rushton et al, 2010), group IId WRKY proteins repress rather than activate the transcription of specific target genes (Arrano‐Salinas et al, 2018; Ge et al, 2020). Further studies are required to investigate whether group IId WRKY proteins mediate transcriptional repression at the whole‐genome level, and how they are involved in transcriptional repression. Interestingly, Arabidopsis WRKY7 was identified as a calmodulin‐binding protein using in vitro assays; a conserved N‐terminal domain of group IId WRKY proteins is responsible for binding to calmodulin (Park et al, 2005). However, it was unknown whether group IId WRKY transcription factors interact with calmodulin to induce calmodulin‐mediated Ca2+ signaling in Arabidopsis plants.
In this study, we found that all group IId WRKY transcription factors interact with PHD finger‐containing proteins: OBE1, OBE2, OBE3, and OBE4, thus forming multiple redundant WRKY‐OBE complexes. Previous studies have shown that OBE1, OBE2, OBE3, and OBE4 redundantly function in the auxin response and embryonic meristem initiation and are essential for early seedling development (Saiga et al, 2008, 2012; Thomas et al, 2009). By generating high‐order wrky mutants using CRISPR‐Cas9‐induced mutagenesis and genetic crossing, we demonstrated that, similar to OBE proteins, group IId WRKY proteins redundantly regulate early seedling development. Furthermore, our observations suggest that the conserved N‐terminal domain of group IId WRKY proteins, previously thought to interact with calmodulin in vitro (Park et al, 2005), is instead responsible for interacting with OBE proteins in Arabidopsis plants. We found that WRKY and OBE proteins co‐occupy target promoter regions at the whole‐genome level and function together to mediate transcriptional repression of numerous stress‐responsive genes under non‐stress conditions. This study indicates that the WRKY‐OBE complexes repress the expression of stress‐responsive genes, especially at non‐stress conditions, and reveals a previously uncharacterized mechanism of the coordination between plant growth and stress tolerance.
Result
Identification of the conserved WRKY‐OBE interaction in higher plants
Although the conserved DNA‐binding WRKY domain of WRKY transcription factors is thought to be responsible for the association of WRKY transcription factors with their target promoter regions (Miao et al, 2004; Ciolkowski et al, 2008; Brand et al, 2013), it is unclear how different subfamilies of WRKY transcription factors affect specific subsets of W‐box‐containing promoters. Given that the unique domain of a given WRKY subfamily is probably responsible for targeting specificity, we performed the sequence alignment and structure prediction for WRKY transcription factors in Arabidopsis thaliana. We found that all group IId Arabidopsis WRKY transcription factors (WRKY7, WRKY11, WRKY15, WRKY17, WRKY21, and WRKY39) contained a conserved WRKY domain and an N‐terminal coiled‐coil (CC) domain composed of two alpha‐helices that are exclusively conserved in group IId WRKY transcription factors (Appendix Fig S1A and B). Because the CC domains usually have the capacity to interact with each other or with other CC domains to form oligomers (Nooren et al, 1999), we predicted that group IId WRKY transcription factors may interact with each other or with other cofactors to regulate transcription in vivo.
Because WRKY7 and WRKY11 belong to two different subgroups of group IId WRKY transcription factors (Appendix Fig S2), we independently introduced native promoter‐driven WRKY7‐Flag and WRKY11‐Flag transgenes into Arabidopsis plants to identify the proteins that interact with the group IId WRKY transcription factors by affinity purification followed by mass spectrometry (AP‐MS). We found that most of the group IId WRKY transcription factors but not any other WRKY proteins were co‐purified with both WRKY7 and WRKY11 (Fig 1A and Dataset EV1), suggesting that group IId WRKY transcription factors can interact with each other. We also found that four PHD finger‐containing proteins, OBE1‐4 (OBE1, OBE2, OBE3, and OBE4), were co‐purified with WRKY7 and WRKY11 (Fig 1A and Dataset EV1). Moreover, we performed AP‐MS using transgenic plants harboring native promotor‐driven OBE1‐Flag and OBE3‐Flag transgenes, and found that the group IId WRKY transcription factors were also co‐purified with OBE1 and OBE3 (Fig 1A and Dataset EV1). These results suggest that group IId WRKY transcription factors interact with each other as well as with OBE1‐4 in Arabidopsis. As indicated by previous studies (Saiga et al, 2008; Thomas et al, 2009), the obe1/2 (obe1 obe2) double mutant showed severe defects in the formation of root and shoot meristems. We transformed the WRKY11‐Flag transgene into obe1 +/− ;obe2 −/− mutant plants and subsequently identified two independent WRKY11‐Flag transgenic alleles with both the obe1/2 double‐mutant and obe2 single‐mutant backgrounds in the progeny of self‐bred WRKY11‐Flag transgenic plants. Although the WRKY11‐Flag transcript level was similar between the obe1/2 double mutant and the obe2 single mutant, the WRKY11‐Flag protein level was markedly reduced in the obe1/2 double mutant relative to the obe2 single mutant (Fig 1B). We also found that the WRKY‐Flag protein level but not the WRKY11‐Flag transcript level was markedly reduced in the obe1/2 mutant relative to the wild type (Fig 1B). These results support the inference that the WRKY‐OBE interaction is required for maintaining the protein level of group IId WRKY transcription factors.
Figure 1. Determination of the interaction between group IId WRKY proteins and OBE proteins in plants.
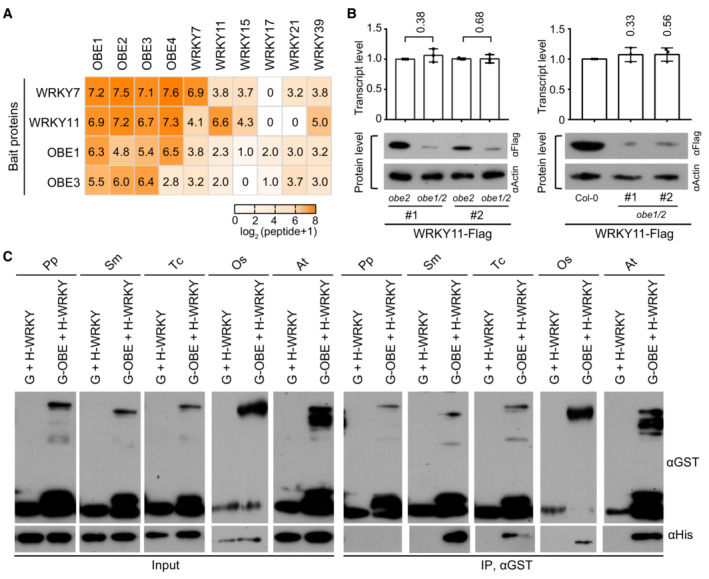
- Heatmap showing the abundance of proteins co‐purified with Flag‐tagged WRKY7, WRKY11, OBE1, and OBE3 as determined by AP‐MS in Arabidopsis plants. The number of matched peptides was used to calculate the abundance of purified proteins. Transgenic plants expressing the indicated bait proteins were used for AP‐MS.
- Effect of obe1/2 on the protein abundance of WRKY11‐Flag in transgenic Arabidopsis plants. The transcript (top) and protein (bottom) levels of WRKY11‐Flag transgene in obe2 and obe1/2 mutants and wild‐type backgrounds are shown. The actin protein level is shown as a loading control. Values are means ± SD of three biological replicates. P‐values were determined by two‐tailed Student's t‐test.
- Determination of the interactions between WRKY11 and OBE1 orthologs from Physcomitrium patens (Pp), Selaginella moellendorffii (Sm), Taxus chinensis (Tc), Oryza sativa (Os), and Arabidopsis thaliana (At) by in vitro pull‐down assays. His‐tagged WRKY11 orthologs and GST‐tagged OBE1 orthologs were expressed and purified from bacteria and then subjected to GST pull‐down assays. G, GST; H, His.
Source data are available online for this figure.
To determine whether the WRKY‐OBE interaction is direct, we performed an in vitro pull‐down assay using bacterially expressed WRKY11 and OBE1, which indicated that WRKY11 directly interacts with OBE1 (Fig 1C). Furthermore, we expressed orthologs of WRKY11 and OBE1 orthologs from other plants, including bryophytes (Physcomitrium patens), ferns (Selaginella moellendorffii), gymnosperms (Taxus chinensis), and monocots (Oryza sativa) (Appendix Figs S2–S4), and investigated whether the WRKY‐OBE interaction is conserved in these plants. As determined by in vitro pull‐down assays, we found that the WRKY‐OBE interaction is conserved in higher plants, including ferns, gymnosperms, and monocots, but not in lower plant bryophytes (Fig 1C). Furthermore, our AP‐MS analysis indicated that the bacterially expressed OsWRKY51, a rice group IId WRKY transcription factor, specifically interacts with OBE proteins in the total protein extract of rice seedlings (Dataset EV1). These results confirm that the interaction between group IId WRKY transcription factors and OBE proteins is conserved in higher plants, suggesting that the interaction is probably important for the survival of these plants.
Characterization of the Arabidopsis WRKY‐OBE complex
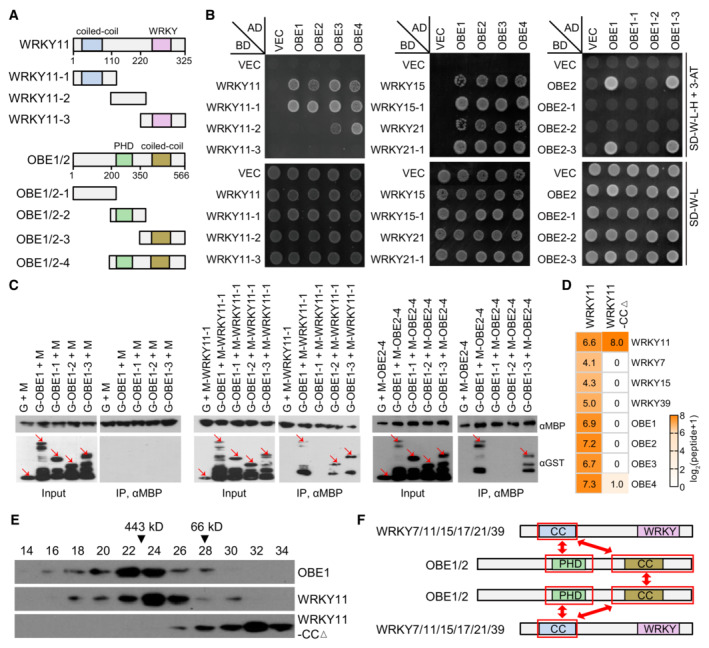
To investigate how the group IId WRKY transcription factors interact with each other and with OBE1‐4, we expressed all of the group IId WRKY transcription factors and the OBE proteins for yeast two‐hybrid (Y2H) assays, and the results indicated that the tested WRKY transcription factors interact with OBE1‐4 but not with each other (Fig 2A and B, and Appendix Fig S5). The Y2H assay also indicated that the OBE proteins interact with each other (Appendix Fig S5), which is consistent with a previous report (Saiga et al, 2012). Furthermore, we selected WRKY40, WRKY36, WRKY12, and WRKY14 as the representatives of groups IIa, IIb, IIc, and IIe of WRKY transcription factors, respectively (Eulgem et al, 2000), and determined whether these WRKY transcription factors also interact with the OBE proteins. As shown in our Y2H results (Appendix Fig S6), unlike group IId WRKY transcription factors, the other group II WRKY transcription factors did not interact with the OBE proteins. These results suggest that the OBE proteins specifically form WRKY‐OBE complexes only with group IId WRKY transcription factors.
Figure 2. Characterization of the protein–protein interactions in the Arabidopsis WRKY‐OBE complex.
-
ADiagram showing full‐length and truncated versions of WRKY11 and OBE1/2 proteins used in Y2H and/or pull‐down assays. Conserved domains are shown.
-
BDetermination of the protein–protein interactions in the WRKY‐OBE complex by Y2H assays. Yeast strains expressing indicated GAL4‐AD‐ and GAL4‐BD‐fused proteins were grown on SD medium lacking Trp and Leu (SD‐W‐L) and SD medium lacking Trp, Leu, and His (SD‐W‐L‐H) supplemented with 3 mM 3‐AT. The yeast strain harboring the empty GAL4‐AD and GAL4‐BD vectors (VEC) was used as a negative control.
-
CDetermination of the protein–protein interactions in the WRKY‐OBE complex by pull‐down assays. The bacterially expressed full‐length and/or truncated versions of WRKY11, OBE1, and OBE2 fused with GST or MBP tags were subjected to MBP pull‐down assays. G, GST; M, MBP. Arrows point to the indicated proteins.
-
D, EDeletion of the CC domain from WRKY11 disrupts the WRKY‐OBE complex formation as determined by AP‐MS and gel filtration. In AP‐MS assays (D), transgenic plants expressing Flag‐tagged wild‐type WRKY11 and WRKY11‐CCΔ proteins were independently used for identifying the WRKY‐OBE complex. The abundance of purified proteins is represented by identified peptides as detected by AP‐MS. For gel filtration (E), proteins extracted from indicated Flag‐tagged transgenic plants were separated in a Superose 6 column (10/300 GL; GE Healthcare Life Sciences) and were detected by immunoblotting.
-
FDiagrams showing the interaction regions of WRKY and OBE proteins in the WRKY‐OBE complex as determined by Y2H and pull‐down assays. The interaction regions are labeled by red frames.
Source data are available online for this figure.
To investigate the details of the interaction, we generated a series of truncated versions of WRKY11 for Y2H and pull‐down assays. Using Y2H assays, we found that the conserved N‐terminal CC domain of WRKY11 (WRKY11‐1) was responsible for the interaction between WRKY11 and OBE proteins. As expected, the conserved N‐terminal CC domain of WRKY15 and WRKY21 (WRKY15‐1 and WRKY21‐1) was also responsible for the interaction with OBE proteins (Fig 2A and B). Consistent with the Y2H results, the CC domain of WRKY11 interacted with OBE1, as determined by pull‐down assays (Fig 2C and Appendix Fig S7). Given that the CC domain is conserved in group IId WRKY transcription factors of land plants (Appendix Fig S4A), we inferred that the CC domain is responsible for the WRKY‐OBE interaction in Arabidopsis as well as in other plants. By performing Y2H and pull‐down assays using a series of truncated versions of OBE1 and OBE2, we found that both the PHD and CC domains of OBE1 were responsible for the interaction between OBE1 and WRKY11 and that the CC domains of OBE1 and OBE2 were also responsible for the interaction of OBE proteins with each other (Fig 2A–C).
Although the N‐terminal CC domain of group IId WRKY transcription factors was identified as an OBE interaction domain by Y2H and pull‐down assays, whether the CC domain is required for the WRKY‐OBE interaction in Arabidopsis plants remained to be determined. Therefore, we obtained transgenic plants that independently expressed wild‐type WRKY11 and CC‐deleted WRKY11 (WRKY11‐CCΔ), and then performed AP‐MS to identify proteins co‐purified with WRKY11 and WRKY11‐CCΔ, respectively. We found that high levels of group IId WRKY and OBE1‐4 proteins were co‐purified with full‐length WRKY11 but not with WRKY11‐CCΔ (Fig 2D and Dataset EV1), suggesting that the conserved CC domain is not only required for the WRKY‐OBE interaction but also for the WRKY‐WRKY interaction in Arabidopsis.
Furthermore, we performed gel filtration to determine whether WRKY and OBE proteins form a high‐molecular‐weight complex in Arabidopsis. As determined by immunoblotting, we found that WRKY11 and OBE1 were mainly eluted in high‐molecular‐weight fractions (~ 443 kDa) but not in the fractions with monomeric WRKY11 or OBE1 sizes (~ 36 kDa for WRKY11 and ~ 66 kDa for OBE1) (Fig 2E), supporting the notion that group IId WRKY transcription factors form a large complex with OBE proteins in Arabidopsis. Subsequently, we determined whether the CC domain of group IId WRKY transcription factors is required for the WRKY‐OBE complex formation by gel filtration, and found that the WRKY11‐CCΔ protein was eluted primarily at the low‐molecular‐weight fractions (< 66 kDa) corresponding to the monomeric WRKY11‐CCΔ size (Fig 2E), suggesting that the CC deletion disrupts the interaction of WRKY11 with other proteins in the WRKY‐OBE complex. Based on these results, we predicted that the OBE proteins form a dimer through their CC domain and that the dimer functions as a bridge to connect two group IId WRKY transcription factors, thus resulting in the formation of WRKY‐OBE‐OBE‐WRKY tetramers in Arabidopsis (Fig 2F).
WRKY‐OBE complex regulates plant development and gene expression
Severe growth retardation was previously observed in root and shoot development in obe1 obe2 (obe1/2) and obe3 obe4 (obe3/4) double mutants (Saiga et al, 2008, 2012; Thomas et al, 2009). Given that group IId WRKY transcription factors interact with OBE1‐4 and form a large complex, we investigated whether these WRKY transcription factors and the OBE proteins have a similar effect on development. We obtained wrky and obe single mutants either from the Arabidopsis Biological Resource Center or from CRIPSR‐Cas9‐induced mutagenesis (Appendix Figs S8 and S9); and generated different orders of wrky or obe multimutants by genetic crossing. Morphological analyses revealed that the wrky7/11/21/39, wrky7/17/21/39, and wrky11/17/21/39 quadruple mutants did not show visible developmental defects, whereas the quintuple mutants wrky11/15/17/21/39 and wrky7/11/17/21/39 exhibited smaller plant size than the wild type (Fig 3A and B and Appendix Fig S10A and B). By genetic crossing between different wrky mutants, we failed to obtain the sextuple wrky7/11/15/17/21/39 mutant but obtained the “wrky7/11/17/21/39 homozygous while wrky15 heterozygous” (wrky7 −/−; wrky11 −/−; wrky17 −/−; wrky21 −/−; wrky39 −/−; wrky15 +/−, referred as wrky‐qm/15 +/−) mutant, and found that the wrky‐qm/15 +/− mutant exhibited similar defects in root and shoot development to the obe1/2 and obe3/4 double mutants (Fig 3A and B). Moreover, the seedling growth was retarded at an early stage in the wrky‐qm/15 +/− mutant and in the obe1/2 or obe3/4 double mutants (Fig 3A and B). The growth retardation was severer in the wrky11/15/17/21/39;wrky7 +/− mutant than in any of the wrky quadruple or quintuple mutants but was weaker than in the wrky‐qm/15 +/− mutant (Appendix Fig S10A–D). These results indicate that, similar to OBE proteins, group IId WRKY transcription factors redundantly regulate root and shoot development.
Figure 3. WRKY and OBE proteins contribute to plant growth and development by regulating gene expression.
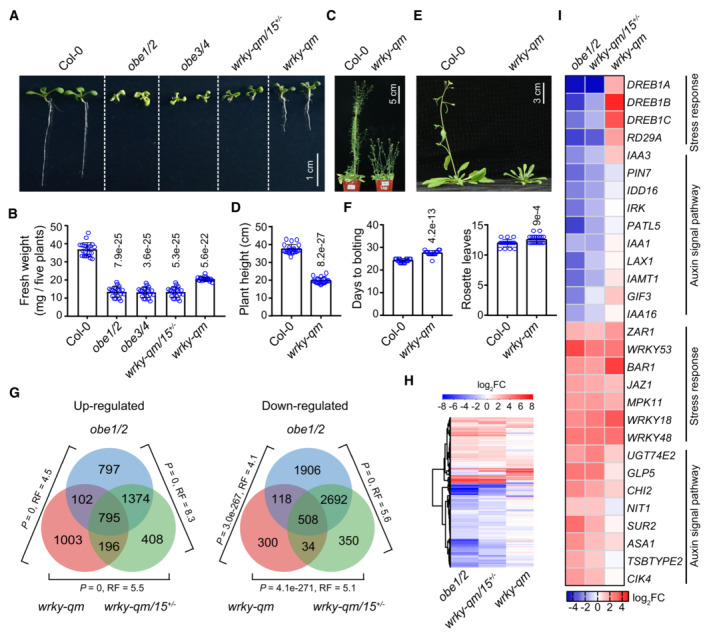
- Morphological phenotypes of wild‐type, obe1/2, obe3/4, wrky‐qm/15 +/−, and wrky‐qm mutant plants. Two‐week‐old plants of the indicated genotypes are shown. The wrky7/11/17/21/39 quintuple mutant is referred to as wrky‐qm.
- Effect of obe1/2, obe3/4, wrky‐qm/15 +/−, and wrky‐qm on plant growth. Fresh weight per five plants as a group was determined. The fresh weight was calculated from a minimum of 20 groups.
- Morphological phenotypes of adult plants of wild type and wrky‐qm mutant.
- Effect of wrky‐qm on plant height. The height was calculated from a minimum of 20 plants.
- Flowering time phenotypes of wild‐type and wrky‐qm mutant plants.
- Flowering time was measured by counting the days to bolting and the number of rosette leaves at bolting. The data were calculated from a minimum of 20 plants.
- The overlap of differentially expressed genes (DEGs) in the wrky‐qm/15 +/−, obe1/2, and wrky‐qm mutants relative to the wild type, as identified by RNA‐seq. RNA was extracted from 11‐day‐old seedlings. P‐values were determined by the hypergeometric test (one‐tailed). RF (representation factor) represents the number of observed overlapping genes divided by the number of expected overlapping genes drawn from two independent groups.
- The expression change (log2FC) of DEGs identified in the wrky‐qm/15 +/−, obe1/2, and wrky‐qm mutants relative to the wild type. Red and blue represent up‐ and downregulated genes, respectively.
- The expression change (log2FC) of representative DEGs in indicated mutants relative to the wild type. Red and blue represent up‐ and downregulated genes, respectively.
Data information: (B), (D), and (F) represent means ± SD. P‐values were determined by two‐tailed Student's t‐test as compared to the wild‐type Col‐0 control and are indicated above columns.
Source data are available online for this figure.
While the growth of the wrky‐qm/15 +/−, obe1/2, and obe3/4 mutant plants was retarded at the early seedling stage, the wrky‐qm and obe1/3 mutant plants could complete the whole life cycle but showed similar developmental defects including shortened root length, reduced plant size, and late flowering (Fig 3C–F, and Appendix Figs S10A–H and S11A–H). Although obe1, obe2, and obe4 single‐mutant plants did not show visible developmental defects, the obe3 single mutant and the obe1/3 double mutants had similar developmental defects and the defects were weaker in the obe3 single mutant than in the obe1/3 double mutant (Appendix Fig S11A and B), indicating that OBE3 has a more important role than OBE1, OBE2, and OBE4 in regulating development. We generated obe1/3/wrky15 and obe1/3/wrky21 triple mutants by CRISPR‐mediated mutagenesis in the obe1/3 double‐mutant background, and found that the triple mutants show similar phenotypes with the obe1/3 double mutant (Appendix Fig S12A and B). These results support the idea that group IId WRKY transcription factors collaborate with OBE proteins to regulate multiple developmental processes.
We performed RNA deep sequencing (RNA‐seq) to determine how group IId WRKY transcription factors and OBEs proteins regulate gene expression at the whole‐genome level. Three biological replicates of RNA‐seq showed that numerous genes were significantly (FDR < 0.05; log2 (fold change) > 1 or < −1) up‐ or downregulated in obe1/2 (3,068 up and 5,224 down), wrky‐qm/15 +/− (2,773 up and 3,584 down), and wrky‐qm (2096 up and 960 down) relative to the wild type (Fig 3G and Dataset EV2). In the obe1/2 mutant, our RNA‐seq analysis identified substantially more differentially expressed genes (DEGs) than those identified by previous microarray analysis (Thomas et al, 2009), and most of the DEGs identified by microarray analysis overlapped with the DEGs identified by our RNA‐seq analysis (Appendix Fig S13), suggesting that our RNA‐seq data were reliable.
The RNA‐seq data showed that both up‐ and downregulated DEGs identified in the obe1/2, wrky‐qm/15 +/−, and wrky‐qm mutants highly overlapped with each other (Fig 3G). The expression changes in DEGs were similar in obe1/2, wrky‐qm, and wrky‐qm/15 +/− (Fig 3H, and Appendix Fig S14A and B, and Dataset EV2). Gene ontology (GO) analyses indicated that biotic and abiotic stress‐responsive genes were enriched in upregulated DEGs in all three mutants, whereas genes related to organ development, auxin signaling, and cell division were enriched in downregulated DEGs in obe1/2 and wrky‐qm/15 +/− but not in wrky‐qm (Fig 3I and Appendix Fig S15). Notably, while the number of upregulated DEGs in wrky‐qm was only slightly lower than that in obe1/2 and wrky‐qm/15 +/−, the number of downregulated DEGs in wrky‐qm was substantially lower than that in obe1/2 and wrky‐qm/15 +/−. Therefore, we predicted that the downregulation of genes related to organ development, auxin signaling, and cell division in obe1/2 and wrky‐qm/15 +/− mutants was probably caused by the absence of well‐developed roots and leaves in these plants. Moreover, the RNA‐seq data identified numerous upregulated DEGs (n = 1,003) in wrky‐qm that were not upregulated in obe1/2 or wrky‐qm/15 +/− mutants (Fig 3G). It is possible that the upregulated DEGs that were specifically identified in wrky‐qm were exclusively expressed in well‐developed roots and leaves and that the absence of well‐developed roots and leaves in obe1/2 or wrky‐qm/15 +/− may affect the identification of these genes. In support of this notion, we found that three genes encoding critical stress‐responsive transcription factors, DREB1A/CBF3, DREB1B/CBF1, and DREB1C/CBF2 (Yamaguchi‐Shinozaki & Shinozaki, 2006; Kidokoro et al, 2022), were upregulated in wrky‐qm but downregulated in obe1/2 and wrky‐qm/15 +/− (Fig 3I).
WRKY‐OBE complex functions as transcriptional repressor
We performed chromatin immunoprecipitation followed by deep sequencing (ChIP‐seq) using WRKY11‐GFP and OBE1‐GFP transgenic plants in the wrky11 and obe1 mutant backgrounds, respectively, to determine the enrichment of WRKY11 and OBE1 at the whole‐genome level. Based on two independent replicates of ChIP‐seq, we identified 11,407 WRKY11 peaks and 9,914 OBE1 peaks at the whole‐genome level (Dataset EV3). The distribution of WRKY11 and OBE1 peaks showed a preference for the promoter and 5’ UTR (untranslated region) compared to random genomic regions (Fig 4A). Metaplots and heatmaps showed that both WRKY11 and OBE1 ChIP‐seq signals formed larger peaks at the transcription start site (TSS)‐proximal promoter region and smaller peaks at the transcription end site (TES)‐proximal region (Fig 4B and C), indicating that WRKY11 and OBE1 are predominantly located at the TSS‐proximal promoter region at the whole‐genome level.
Figure 4. WRKY11 and OBE1 co‐occupy chromatin and repress gene expression.
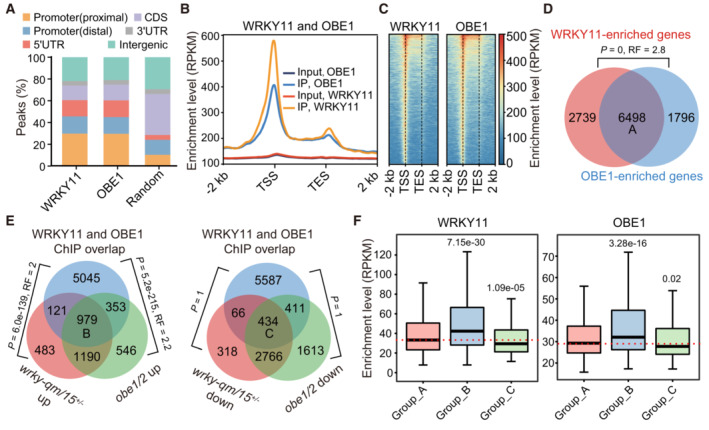
- Distribution of WRKY11 and OBE1 ChIP‐seq peaks in distal promoter, proximal promoter, 5′ UTR, CDS (coding sequence), 3′ UTR, and intergenic regions. Distal and proximal promoters represent 401–1,000 and 0–400 bp upstream of transcription start sites, respectively. The distribution of random genomic regions is shown as a control.
- Metaplots showing WRKY11 and OBE1 ChIP‐seq occupancy over protein‐coding genes. TSS, transcription start site; TES, transcription end site. The ChIP‐seq data of WRKY11 and OBE1 are from two independent biological replicates. The input genomic DNA is shown as a negative control.
- Heatmaps showing the enrichment of WRKY11 and OBE1 ChIP‐seq reads over protein‐coding genes. TSS, transcription start site; TES, transcription end site.
- Venn diagram showing the overlap between WRKY11‐ and OBE1‐enriched genes. The overlapping genes are defined as group A genes. P‐values were determined by the hypergeometric test (one‐tailed). RF (representation factor) represents the number of observed overlapping genes divided by the number of expected overlapping genes drawn from two independent groups.
- Venn diagrams showing the overlap between WRKY11 and OBE1 co‐enriched genes and up‐ or downregulated genes in the wrky‐qm/15 +/− and obe1/2 mutants. Group B and group C genes represent the overlap between WRKY11 and OBE1 co‐enriched genes and co‐upregulated genes (left) or co‐downregulated genes (right) in the wrky‐qm/15 +/− and obe1/2 mutants. P‐values were determined by the hypergeometric test (one‐tailed). RF (representation factor) represents the number of observed overlapping genes divided by the number of expected overlapping genes drawn from two independent groups.
- Box plots of WRKY11‐ and OBE1‐enriched levels at group A, group B, and group C genes. Sample size of each box plot: group A (n = 6,498), group B (n = 979), and group C (n = 434). In box plots, center lines and box edges are medians and the interquartile range (IQR), respectively. Whiskers extend within 1.5 times the IQR. P‐values were determined by two‐tailed Mann–Whitney U‐test (unpaired) for non‐normally distributed data.
Source data are available online for this figure.
By assigning ChIP‐seq peaks to proximal genes, we identified 9,237 WRKY11‐enriched genes and 8,293 OBE1‐enriched genes. We found that the vast majority (6,498/9,237 = 70.3%) of WRKY11‐enriched genes were also OBE1‐enriched genes, and referred to these overlapping genes as group A (Fig 4D). By combinatorically analyzing ChIP‐seq and RNA‐seq data, we noticed that group A genes significantly overlapped with upregulated DEGs in both wrky‐qm/15 +/− and obe1/2 but not with downregulated DEGs in either of these mutants (Fig 4E), implying that WRKY11 and OBE1 are primarily responsible for transcriptional repression. We identified 979 WRKY11 and OBE1 co‐enriched genes that overlapped with upregulated DEGs in both wrky‐qm/15 +/− and obe1/2 (designated group B); and 434 WRKY11 and OBE1 co‐occupied genes that overlapped with downregulated DEGs in both wrky‐qm/15 +/− and obe1/2 (designated group C) (Fig 4E). As indicated by the box plots, the overall enrichment levels of both WRKY11 and OBE1 were significantly higher in group B genes than in groups A and C (Fig 4F), further supporting the notion that WRKY11 and OBE1 mediate transcriptional repression of their common target genes.
The DNA‐binding ability of group IId WRKY is required for the recruitment of WRKY‐OBE complex to chromatin
Considering that the conserved WRKY domain is responsible for the binding of WRKY transcription factors to the W‐box‐containing DNA (Ciolkowski et al, 2008), we performed ChIP‐seq to determine whether the WRKY domain binding to the W‐box motif is responsible for the association of WRKY11 and OBE1 with chromatin. From the ChIP‐seq data, the W‐box motif was the most significantly enriched WRKY11/OBE1‐bound motif, as determined by the Homer de novo motif analysis (Appendix Fig S16). Next, we mapped the enrichment level of WRKY11 and OBE1 in the W‐box motif‐flanking region, and found that both WRKY11 and OBE1 formed a peak centered on the W‐box motif, suggesting that the binding of WRKY11 to the W‐box is responsible for the association of WRKY11 and OBE1 with chromatin (Fig 5A). If the W‐box motif was responsible for the binding of WRKY11 and OBE1 to chromatin, we predicted that the enrichment of WRKY11 and OBE1 was positively associated with the number of W‐box motifs in their target regions. Therefore, we counted the number of W‐box motifs in each of the WRKY11 and OBE1 ChIP‐seq peaks and then compared the enrichment levels of WRKY11 and OBE1 at the ChIP‐seq peaks with different numbers of W‐box motifs. As expected, box plots showed that, as the number of W‐box motifs increased, the enrichment of WRKY11 and OBE1 became stronger (Fig 5B), suggesting that the WRKY domain binding to the W‐box motif is critical for the association of WRKY11 and OBE1 with chromatin in vivo.
Figure 5. Determination of the binding of WRKY11 and OBE1 to W‐box in vivo and in vitro .
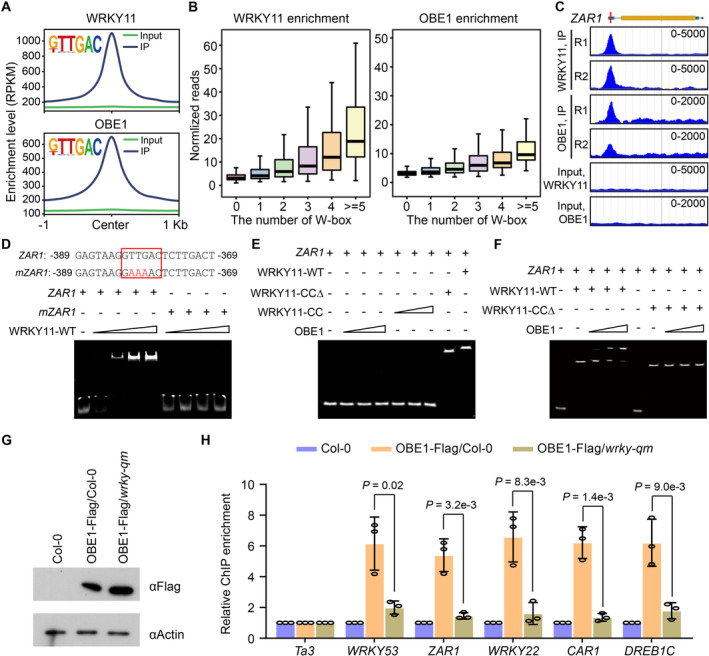
- Metaplots showing WRKY11 and OBE1 ChIP‐seq signals at WRKY11‐ and OBE‐bound regions centered by the W‐box motif. The signals of input genomic DNA are shown as controls.
- Enriched levels of WRKY11 and OBE1 at the WRKY11‐ and OBE1‐bound regions harboring different numbers of W‐box motifs. Relative enrichment level is referred to as the fold change of ChIP‐seq reads and input reads. In box plots, the sample size of WRKY11‐bound regions is n (0) = 5,491, n (1) = 3,972, n (2) = 1,384, n (3) = 369, n (4) = 137, and n (≥5) = 54; the sample size of OBE1‐bound regions is n (0) = 5,554, n (1) = 2,920, n (2) = 1,029, n (3) = 268, n (4) = 103, and n (≥5) = 40). In box plots, center lines and box edges are medians and the interquartile range (IQR), respectively. Whiskers extend within 1.5 times the IQR.
- Genome browser view of WRKY11 and OBE1 ChIP‐seq signals at the representative target gene ZAR1. The scale of RPKM is indicated for each panel.
- Effect of W‐box mutation on binding of the WRKY protein to DNA as determined by EMSA. A W‐box‐containing DNA from ZAR1 was labeled by Cy5 and used as the DNA probe. The W‐box was mutated as indicated. The wild‐type and mutated W‐boxes are labeled by red boxes.
- The binding of full‐length WRKY11, WRKY11‐CCΔ, the CC domain of WRKY11 (WRKY11‐CC), and full‐length OBE1 to DNA as determined by EMSA.
- Determination of the binding of full‐length and CC‐truncated WRKY11 proteins to DNA by EMSA, in the presence or absence of OBE1.
- The protein level of OBE1‐Flag in the wild‐type (Col‐0) and wrky‐qm mutant backgrounds determined by immunoblotting. The actin protein level is shown as a loading control.
- Enrichment of OBE1‐Flag at WRKY53, ZAR1, WRKY22, CAR1, and DREB1C loci as determined by quantitative ChIP–PCR in the wild‐type (Col‐0) and wrky‐qm mutant backgrounds. Values are means ± SD of three independent biological replicates. P‐values were determined by two‐tailed Student's t‐test.
Source data are available online for this figure.
Although it is reasonable to assume that the binding of WRKY11 to the W‐box motif is responsible for the association of WRKY11 with chromatin, it is unknown whether this binding is required for the recruitment of OBE1 to DNA. Therefore, we performed electrophoretic mobility shift assay (EMSA) to determine the DNA‐binding properties of WRKY11 and OBE1 in vitro. Considering that WRKY11 and OBE1 form a sharp ChIP‐seq peak at the promoter region of ZAR1, a well‐characterized disease resistance gene (Lewis et al, 2010; Bi et al, 2021), and that the ZAR1 expression was markedly increased in wrky‐qm/15 +/− and obe1/2 mutants (Figs 3I and 5C), we chose a W‐box motif‐containing promoter sequence of ZAR1 as a DNA probe in EMSA (Fig 5C). The EMSA results showed that the WRKY11 protein strongly binds to the W‐box motif‐containing DNA probe, and this binding was completely disrupted by mutation of the W‐box motif (Fig 5D), supporting the binding specificity of WRKY11 for the W‐box motif. Both the full‐length WRKY11 and the WRKY11‐CCΔ could efficiently bind to DNA, whereas OBE1 and the CC domain of WRKY11 were incapable of binding to DNA (Fig 5E). When we mixed OBE1 and the DNA probe in the presence of full‐length WRKY11, OBE1 was recruited to DNA by WRKY11 to form a high‐molecular‐weight complex (Fig 5F). Because the WRKY11‐CCΔ lacks the OBE1‐interacting ability as previously indicated (Fig 2A‐F), OBE1 could not be recruited to DNA and did not form a high‐molecular‐weight complex (Fig 5F). To determine whether the group IId WRKY transcription factors are required for the recruitment of OBE proteins to chromatin in vivo, we performed ChIP‐PCR for OBE1‐Flag in the wild‐type and wrky‐qm mutant backgrounds. The result indicated that the association of OBE1 with target genes was significantly reduced in the wrky‐qm mutant relative to the wild type (Fig 5G and H). These results strongly suggest that the DNA‐binding ability of group IId WRKY transcription factors is responsible for the recruitment of the WRKY‐OBE complex to its target genes.
The coiled‐coil domain of group IId WRKY proteins regulates target selection
Although the binding of WRKY11 to the W‐box motif provides a plausible explanation for the association of the WRKY‐OBE complex with specific target genes, it is unknown how different subfamilies of WRKY transcription factors affect different subsets of W‐box‐containing promoters. We predicted that the conserved OBE‐interacting domain of group IId WRKY transcription factors may be involved in target selection. Therefore, we performed ChIP‐seq for WRKY11‐CCΔ and determined whether the CC deletion affects the association of WRKY11 with chromatin. By comparing the ChIP‐seq peaks for the wild‐type WRKY11 and WRKY11‐CCΔ, we identified 9,147 specific peaks for WRKY11‐CCΔ (group N), which was markedly higher than the number (1,127) of the wild‐type WRKY11‐specific peaks (group A) (Fig 6A). Interestingly, we found 10,280 overlapping peaks (group A&N) between wild‐type WRKY11 and WRKY11‐CCΔ, which accounted for 90.1 and 53.0% of wild‐type WRKY11 and WRKY11‐CCΔ peaks, respectively (Fig 6A), indicating that WRKY11‐CCΔ binds to numerous target genomic loci that are not bound by wild‐type WRKY11. The presence of specific WRKY11‐CCΔ peaks was verified by comparing the WRKY11 and WRKY11‐CCΔ ChIP‐seq signals at representative genomic loci in the genome browser (Fig 6B). By annotating the W‐box‐containing peaks to different genomic regions, we found that the W‐box motif was enriched not only in the wild‐type WRKY11 and WRKY11‐CCΔ‐overlapping peaks but also in WRKY11‐CCΔ‐specific peaks (Fig 6C). Moreover, the distribution of WRKY11‐CCΔ peaks showed a preference for the promotor and 5’ UTR, which is similar to the distribution of wild‐type WRKY11 peaks (Fig 6C). These analyses suggest that the CC domain is responsible for limiting the binding of the WRKY‐OBE complex to a subset of W‐box‐containing promoters.
Figure 6. The coiled‐coil domain is required for target selection of WRKY11.
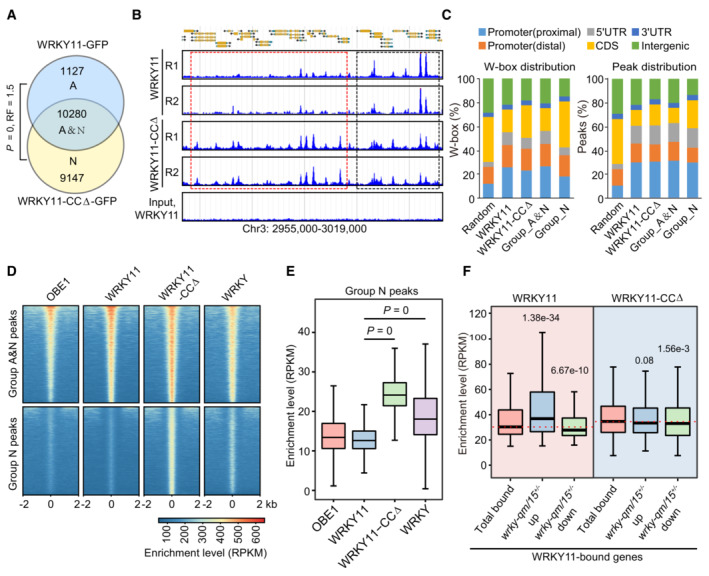
- Venn diagram showing the overlap between WRKY11 and WRKY11‐CCΔ peaks as determined by ChIP‐seq. The overlap between WRKY11 and WRKY11‐CCΔ peaks is defined as group_A&N. WRKY11‐ and WRKY11‐CCΔ‐specific peaks are defined as group_A and group_N peaks, respectively. P‐values were determined by the hypergeometric test (one‐tailed). RF (representation factor) represents the number of observed overlapping genes divided by the number of expected overlapping genes drawn from two independent groups.
- Genome browser view of the ChIP‐seq signals of WRKY11 and WRKY11‐CCΔ at randomly selected genomic regions. WRKY11 and WRKY11‐CCΔ shared peaks are labeled by black boxes, and WRKY11‐CCΔ‐specific peaks are labeled by red boxes. Two replicates of ChIP‐seq are shown.
- Distribution of the WRKY11 and WRKY11‐CCΔ W‐box distribution and peaks distribution in different genomic regions. Distal and proximal promoters represent 401–1,000 and 0–400 bp upstream of transcription start sites, respectively. The distribution of random genomic regions is shown as a control.
- Heatmaps showing the ChIP‐seq signals of WRKY11, WRKY11‐CCΔ, OBE1, and general WRKY TFs over group A&N and group N peaks. The enrichment level is indicated by RPKM as determined by ChIP‐seq.
- Box plots showing the enrichment levels of WRKY11, WRKY11‐CCΔ, OBE1, and general WRKY TFs at group N peaks (n = 9,147).
- Box plots of WRKY11‐ and WRKY11‐CCΔ‐enriched levels at total WRKY11‐bound genes and at the overlap between WRKY11‐bound genes and up‐ or downregulated genes in wrky‐qm/15 +/−. Sample size of each box plot: total WRKY11‐bound genes (n = 9,237), WRKY11‐bound genes and upregulated (n = 1,424), WRKY11‐bound genes and downregulated (n = 773).
Data information: In box plots in (E) and (F), center lines and box edges are medians and the interquartile range (IQR), respectively. Whiskers extend within 1.5 times the IQR. P‐values were determined by two‐tailed, paired Mann–Whitney U‐test in (E) and by two‐tailed, unpaired Mann–Whitney U‐test in (F) for non‐normally distributed data.
Source data are available online for this figure.
To determine how the CC domain affects the target selection, we independently investigated the occupancy of OBE1, WRKY11, and WRKY11‐CCΔ at WRKY11 and WRKY11‐CCΔ overlapping peaks (group A&N peaks) and at WRKY11‐CCΔ‐specific peaks (group N peaks). Although OBE1, WRKY11, and WRKY11‐CCΔ were enriched in group A&N peaks at a comparable level, WRKY11‐CCΔ, but not wild‐type WRKY11 or OBE1, was enriched in group N peaks (Fig 6D and E). We also analyzed the enrichment of general WRKY transcription factors as determined by ChIP‐seq using an antibody of the conserved WRKY domain (Birkenbihl et al, 2018). We found that the enrichment of general WRKY transcription factors was comparable to the enrichment of OBE1, WRKY11, and WRKY11‐CCΔ in group A&N peaks, and was markedly higher than the enrichment of OBE1 and WRKY11 in group N peaks even though it was lower than the enrichment of WRKY11‐CCΔ (Fig 6D and E), suggesting that the CC domain is required for the selection of a subset of WRKY domain‐binding loci as targets of the WRKY‐OBE complex. By integrating the ChIP‐seq and RNA‐seq data, we found that wild‐type WRKY11, but not WRKY11‐CCΔ, was more enriched in upregulated DEGs in the wrky‐qm/15 +/− mutant (Fig 6F). Together, these results support the notion that the CC domain is responsible for preventing the binding of the WRKY‐OBE complex to undesired W‐box‐containing promoters.
OBE1 and OBE2 cooperatively bind to histone H3 with H3K4me2 and H3K4me3 modifications
To determine how the OBE proteins affect the target selection of the WRKY‐OBE complex, we analyzed the domain structures of OBE1‐4 and found that OBE1 and OBE2 contain a PHD finger (Fig 7A), which is similar to the previously reported H3K4me2‐binding PHD domain in VIN3 (Kim & Sung, 2013; Franco‐Echevarria et al, 2022). Consistently, OBE1 was also shown to bind to H3K4me2 (Franco‐Echevarria et al, 2022). Therefore, we purified the PHD domains of OBE1 and OBE2 and determined whether they bind to methylated histone peptides. Histone peptide pull‐down assays indicated that the PHD domain of OBE1 specifically bound to H3K4me3 and to a lesser extent H3K4me2 but not H3 peptides methylated at K9, K27, or K36, while the PHD domain of OBE2 weakly bound to a series of histone H3 peptides without any preferences for methylated histone peptides (Fig 7B). As determined by previous studies (Yang et al, 2018; Tan et al, 2020), aromatic amino acids in the conserved PHD domains of ARID5 and EBS were necessary for their binding ability with methylated histones. By sequence alignment, we found that two conserved aromatic amino acids F234 and W252 may be responsible for binding to methylated histone (Fig 7A). Therefore, we mutated the two aromatic amino acids into alanine and then performed the histone peptide pull‐down assay. We found that the mutations disrupted the binding of the PHD domain to H3K4me2 and H3K4me3 (H3K4me2/3) peptides (Fig 7B), suggesting that the aromatic amino acids F234 and W252 are necessary for the H3K4me2/3‐binding ability of OBE1. The dimerization of OBE proteins in the WRKY‐OBE complex prompted us to investigate whether OBE1 and OBE2 cooperate to bind to histone peptides. By performing histone pull‐down assays using the OBE1 and OBE2 mixture, we found that OBE2 enhanced the binding affinity of OBE1 with H3K4me1/2/3 peptides and that OBE1 enhanced the binding affinity of OBE2 with histone peptides non‐specifically (Fig 7C), suggesting that the two OBE proteins in the WRKY‐OBE complex cooperate to bind to histone.
Figure 7. OBE proteins bind to histone peptides and are responsible for transcriptional repression of WRKY‐OBE complexes.
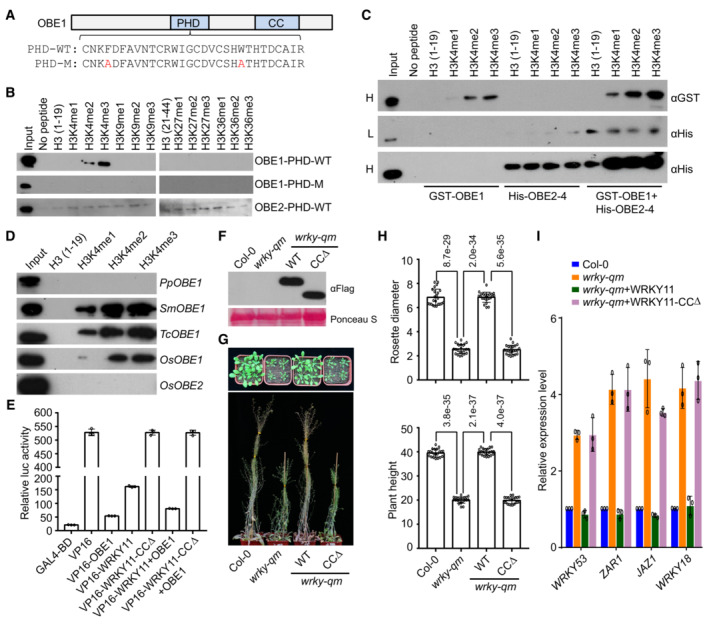
-
ADiagram of the wild‐type and mutated PHD domains in OBE1. Mutated residues in the PHD domain are shown in red.
-
BInteraction of the PHD domains of OBE1 and OBE2 with histone peptides as determined by pull‐down assays. The wild‐type and mutated PHD domains in OBE1 and the wild‐type PHD domain in OBE2 were purified and then mixed with indicated histone peptides for pull‐down assays.
-
CInteraction of GST‐OBE1, His‐OBE2, and the GST‐OBE1 and His‐OBE2 mixture with histone peptides as determined by pull‐down assays. H, high exposure; L, low exposure.
-
DInteraction of OBE1 and/or OBE2 orthologs with histone peptides as determined by pull‐down assays. PpOBE1, SmOBE1, TcOBE1, and OsOBE1 are OBE1 orthologs in Physcomitrium patens (Pp), Selaginella moellendorffii (Sm), Taxus chinensis (Tc), and Oryza sativa (Os), respectively. OsOBE2 is an OBE2 ortholog in Oryza sativa.
-
EDetermination of the transcriptional repression activity of OBE1, WRKY11, WRKY11‐CCΔ, and the mixture of WRKY11 or WRKY11‐CCΔ with OBE1 by a luciferase reporter assay. The transcriptional repression activity of OBE1, WRKY11‐WT, and WRKY11‐CCΔ was determined by fusing with the transcription factor VP16. Values are means ± SD of three independent biological replicates.
-
FThe expression levels of Flag‐tagged wild‐type WRKY11 and WRKY‐CCΔ proteins determined by immunoblotting. The ribosome protein stained by Ponceau S is indicated as a loading control.
-
G, HRestoration of the developmental defects of the wrky‐qm mutant by WRKY11 and WRKY11‐CCΔ transgenes. Morphological phenotype (G) and the statistical data of rosette diameter and plant height (H) are shown. The data were calculated from a minimum of 20 plants. Values are means ± SD. P‐values were determined by two‐tailed Student's t‐test and are indicated above columns.
-
IExpression levels of representative stress‐responsive genes in wild‐type, wrky‐qm, and WRKY11 and WRKY11‐CCΔ transgenic lines. Values are means ± SD of three biological replicates.
Source data are available online for this figure.
Because the WRKY‐OBE interaction is conserved in higher land plants (Fig 1C), we investigated whether the binding of OBE proteins to histone is also conserved. By using purified OBE1 orthologs in Physcomitrium patens (Pp), Selaginella moellendorffii (Sm), Taxus chinensis (Tc), and Oryza sativa (Os), and an OBE2 ortholog in Oryza sativa (Appendix Fig S4B), we performed histone peptide assays to determine whether the OBE orthologs bind to histone H3 with and without H3K4 methylation. We found that the OBE1 orthologs from higher land plants bound to H3K4me2/3 and to a lesser to H3K4me1, and that the OBE1 ortholog from the lower land plant Physcomitrium patens did not bind to any H3 peptides (Fig 7D). It was worth noting that the rice OBE1 ortholog OsOBE1 but not the rice OBE2 ortholog OsOBE2 specifically binds to H3K4me2/3 (Fig 7D). These results suggest that the binding of OBE1 orthologs to H3K4me2/3 is conserved in higher land plants.
Given that the PHD domain of OBE1 is capable of binding to H3K4me3, we determined whether the WRKY‐OBE complex tends to occupy H3K4me3‐enriched genes. By analyzing previous H3K4me3 and H3K27me3 ChIP‐seq data (Shang et al, 2021; Zhao et al, 2022), we found that the H3K4me3 levels are significantly higher in WRKY11‐ and OBE1‐occupied genes than in random genes, while the H3K27me3 levels are significantly lower in WRKY11‐ and OBE1‐occupied genes than in random genes (Appendix Fig S17A and B), supporting the inference that the binding of OBE1 to H3K4me3 is involved in the association of the WRKY‐OBE complex with chromatin. Previous studies showed that some WRKY transcription factors can regulate gene expression by affecting histone modifications (Kim et al, 2008; Hung et al, 2022). Our immunoblotting results indicated that the H3K4me2, H3K4me3, and H3K27me3 levels were not significantly affected by obe1/2, wrky‐qm/15 +/−, or wrky‐qm at the whole‐genome level (Appendix Fig S17C). Moreover, our AP‐MS analysis did not identify that the WRKY‐OBE complex interacts with any histone modifiers. These analyses suggest that the WRKY‐OBE complex is unlikely to repress transcription by affecting histone modifications.
The WRKY‐OBE interaction is required for WRKY‐mediated transcriptional repression
To determine how the WRKY‐OBE complex mediates transcriptional repression, we performed a luciferase reporter assay to investigate the transcriptional repression activity of the WRKY‐OBE complex. While the transcription factor VP16 fused with the GAL4‐binding domain showed a strong transcriptional activation activity as determined by the reporter assay, the fusion of VP16 to OBE1 and, to a lesser extent, to WRKY11 markedly repressed the transcriptional activation activity of VP16, whereas the fusion of VP16 to WRKY11‐CCΔ did not repress the transcriptional activation activity of VP16 (Fig 7E). By combining OBE1 with VP16‐fused WRKY11 and WRKY11‐CCΔ, the reporter assay indicated that OBE1 markedly enhances WRKY11‐mediated transcriptional repression, and showed that the effect of OBE1 on transcriptional repression is disrupted by the deletion of the CC domain in WRKY11 (Fig 7E), suggesting that the WRKY‐OBE interaction is involved in WRKY‐mediated transcriptional repression.
To further investigate whether the WRKY‐OBE interaction contributes to transcriptional repression in Arabidopsis, we generated a WRKY11‐CCΔ construct driven by its native promoter and introduced the construct into the wrky‐qm mutant for complementation testing. We found that, while the wild‐type WRKY11 construct completely complemented the developmental defects in the wrky‐qm mutant, the WRKY11‐CCΔ constructs failed to complement the defects (Fig 7F and H), suggesting that the CC domain is required for the function of group IId WRKY transcription factors in Arabidopsis. Furthermore, we performed quantitative RT‐PCR to determine whether the CC domain is required for transcriptional repression. We randomly selected four representative WRKY11 and OBE1 co‐occupied genes and found that their expression levels were significantly increased in the wrky‐qm mutant, as determined by RNA‐seq, which included WRKY53, ZAR1, JAZ1, and WRKY18 (Fig 3I). We found that the increased levels of these genes in the wrky‐qm mutant were restored by the wild‐type WRKY11 transgene, but not by the WRKY11‐CCΔ transgene (Fig 7I). This result indicated that the expression of these genes can be repressed by wild‐type WRKY11 but not by WRKY11‐CCΔ, suggesting that the WRKY‐OBE interaction is required for transcriptional repression in Arabidopsis. Given that the WRKY‐OBE complex binds to the TSS‐flanking promoter region proximal to the +1 nucleosome, we speculated that the OBE proteins bind to the +1 nucleosome and thereby enable the complex to fulfill its transcriptional repression function by anchoring the promoter to the +1 nucleosome.
WRKY‐OBE complex negatively regulates drought resistance
The increased expression of stress‐responsive genes in the wrky and obe mutants prompted us to determine whether these mutants show an increase in stress tolerance. We selected the wrky‐qm and obe1/3 mutants with a weak growth retardation for stress tolerance analyses. Nine‐day‐old plants grown in soil were subjected to drought treatment until the mortality of Col‐0 occurred, followed by rehydration, and the survival rate of the plants was subsequently determined. We found that 100% of the wrky‐qm mutant plants and 93.1% of the obe1/3 mutant plants survived and continued to grow after rehydration, whereas the lower‐order wrky and obe mutant plants without an obvious growth retardation and the wild‐type plants showed low survival rates (10%) after rehydration (Fig 8A and B), suggesting that the drought stress tolerance of the wrky‐qm and obe1/3 mutants is associated with growth retardation. Given that the prevention of water loss is crucial for the plant drought tolerance (Farooq et al, 2009), we determined the rate of leaf water loss in wild type, wrky‐qm and obe1/3 mutants. The result indicated that the water loss rate was substantially reduced in wrky‐qm and to a lesser extent in obe1/3 compared with the wild type (Fig 8C), which is consistent with the increased drought tolerance of these mutants.
Figure 8. The WRKY‐OBE complex regulates the expression of stress‐responsive genes and drought tolerance in response to drought stress.
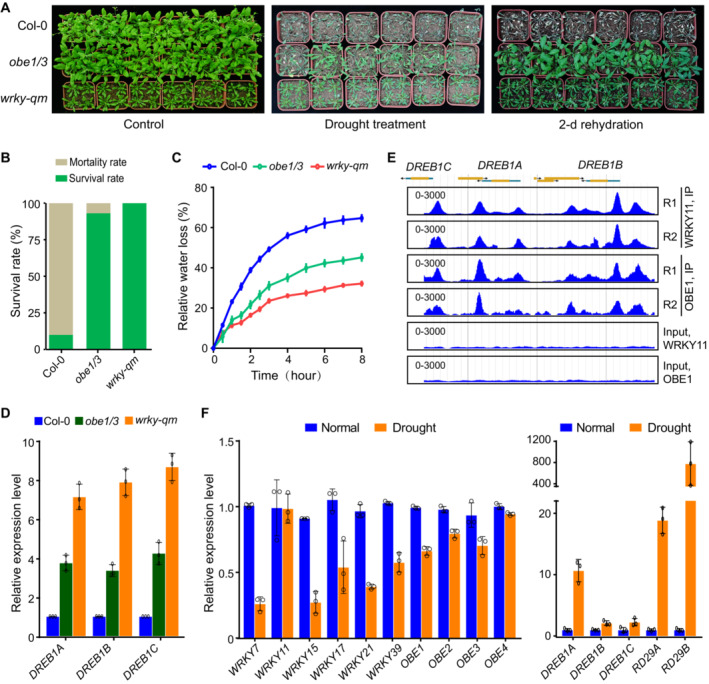
- The morphological phenotype of wild type and obe1/3 and wrky‐qm mutants under well‐watered and drought stress conditions. Nine‐day‐old seedlings were transferred from MS medium to soil and then subjected to drought treatment until the mortality of Col‐0 occurred. The morphological phenotype is shown before and 2 days after rehydration. The well‐watered plants are shown as a control.
- The survival rate of wild type and obe1/3 and wrky‐qm mutants under drought stress conditions. The survival rate was calculated from a minimum of 25 plants after rehydration.
- Relative water loss of detached leaves from wild type and obe1/3 and wrky‐qm mutants. Values are means ± SD of three biological replicates.
- Effect of wrky‐qm and obe1/3 on the expression of DREB1A, DREB1B, and DREB1C as determined by quantitative RT‐PCR. Values are means ± SD of three biological replicates.
- Genome browser view of WRKY11 and OBE1 ChIP‐seq peaks at DREB1A, DREB1B, and DREB1C. The scale of RPKM is indicated in each panel. Two replicates are shown.
- The expression levels of indicated genes under non‐stress and drought stress conditions as determined by RNA‐seq. Well‐watered and drought‐treated Arabidopsis Col‐0 plants were grown at 22°C under long‐day conditions (16 h light/8 h dark) for 28 days. Values are means ± SD of three biological replicates.
Source data are available online for this figure.
In the increased stress‐responsive genes identified by RNA‐seq in the wrky‐qm mutant (Fig 3I), DREB1A, DREB1B, and DREB1C encode critical transcription factors that are responsible for drought and cold stress tolerance (Yamaguchi‐Shinozaki & Shinozaki, 2006; Shi et al, 2018). Quantitative RT‐PCR analysis indicated the expression levels of DREB1A, DREB1B, and DREB1C were increased in the wrky‐qm mutant and to a lesser extent in the obe1/3 mutant (Fig 8D), which is consistent with the finding that the growth retardation and stress tolerance were weaker in the obe1/3 mutant than in the wrky‐qm mutant (Fig 8A–C). As determined by ChIP‐seq, both WRKY11 and OBE1 were enriched at the TSS‐proximal promoter regions of the three DREB1 genes (Fig 8E), suggesting that the WRKY‐OBE complex directly mediates transcriptional repression of these genes. Considering that overexpression of DREB1A, DREB1B, and DREB1C in Arabidopsis plants not only enhances tolerance to drought and/or cold conditions but also causes growth retardation (Liu et al, 1998), we suspected that the growth retardation in the wrky‐qm mutant is associated with increased expression of DREB1A, DREB1B, and DREB1C.
Considering that the WRKY‐OBE complexes are responsible for repressing the expression of stress‐responsive genes, we asked whether the repressive effect is reduced when stress‐responsive genes need to be highly expressed under stress conditions. Based on previous transcriptome analyses (Kilian et al, 2007), the expression levels of WRKY and OBE genes were not significantly reduced by any short‐term stress treatments. Our RNA‐seq data showed that the expression levels of most group IId WRKY and OBE genes were markedly reduced in plants grown under long‐term drought stress conditions (Fig 8F). As a control, the expression levels of DREB1A, DREB1B, DREB1C, RD29A, and RD29B were markedly increased in plants grown under the long‐term drought stress conditions in our RNA‐seq data (Fig 8F). Furthermore, we observed a time‐dependent reduction in the expression level of WRKY17 upon exposure to drought, whereas the expression level of DREB1A was induced over time (Appendix Fig S18). We predicted that the expression levels of these WRKY and OBE genes are reduced in plants that are subjected to a long‐term drought treatment and that the reduced expression of these WRKY and OBE genes contributes to releasing the transcription of stress‐responsive genes (Fig 9). Therefore, this study revealed a previously uncharacterized regulatory mechanism responsible for balancing plant growth and stress tolerance.
Figure 9. The WRKY‐OBE complex represses the expression of stress‐responsive genes to coordinate plant growth and drought tolerance.
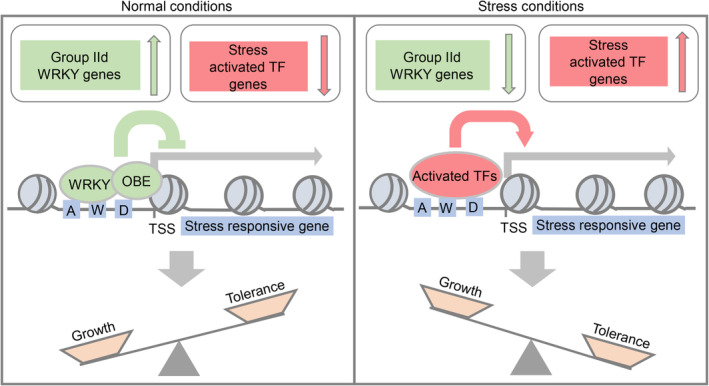
Under normal growth conditions, the group IId WRKY genes are well expressed, and the WRKY‐OBE complexes repress the transcription of stress‐responsive genes in order to prevent the inhibitory effect of the stress‐responsive genes on growth. Under drought stress conditions, the transcription of stress‐responsive genes is initially induced by stress‐activated transcription factors, and subsequently, the expression of group IId WRKY genes is repressed to further release the transcription of stress‐responsive genes, thereby enhancing the drought stress tolerance. The boxes A, W, and D shown in the promoter regions represent ABA‐responsive elements (ABREs), WRKY‐binding elements, and dehydration‐responsive elements (DREs), respectively.
Discussion
Land plants evolved from ancestral charophycean alga 450 mya, which is an important event for plant expansion on Earth (Bowman et al, 2017). To survive on land, the higher land plants have evolved a multicellular diploid sporophyte generation and many specialized tissues and organs, such as vascular tissues, roots, leaves, seeds, and flowers (Ishizaki, 2017). Although WRKY transcription factors exist in both lower and higher plants, they are greatly expanded in higher land plants (Zhang & Wang, 2005), suggesting that WRKY transcription factors are involved in the adaptation of plants to land. Our study indicated that group IId WRKY transcription factors interact with PHD‐containing proteins and form redundant protein complexes that are required for root and shoot development. Previous studies have consistently shown that OBE proteins are required for root and shoot development and vascular formation (Saiga et al, 2008, 2012; Thomas et al, 2009). These results suggest that the expansion of WRKY transcription factors contributes to root and shoot development and vascular tissue formation, thereby facilitating the adaptation of higher plants to terrestrial environments. Because the WRKY‐OBE complexes function as transcriptional repressors and prevent the overexpression of numerous stress‐responsive genes under non‐stress conditions, we predicted that transcriptional repression of stress‐responsive genes mediated by the WRKY‐OBE complexes is required for the maintenance of normal plant growth under non‐stress conditions. The WRKY‐OBE interaction is conserved in higher plants, including ferns, gymnosperms, and monocots, but not in lower land bryophytes, suggesting that the WRKY‐OBE interaction is especially important in higher land plants. Therefore, WRKY‐OBE complexes may have evolved in higher land plants to balance plant growth and tolerance to increased environmental challenges.
Although several WRKY transcription factors have been shown to form dimers (Xu et al, 2006; Liu et al, 2012), dimerization of these WRKY proteins depends on the direct interaction between WRKY proteins. The results of this study indicated that the interaction between group IId WRKY proteins is not direct. In the WRKY‐OBE complex, two copies of OBE proteins directly interact to form a dimer, and the OBE dimer functions as a bridge connecting two copies of WRKY proteins, thus revealing a previously uncharacterized mechanism underlying the dimerization of WRKY proteins. Moreover, we found that the number of W‐box motifs is positively correlated with the enrichment levels of WRKY and OBE proteins at their target promoter regions, implying that two copies of WRKY proteins in the WRKY‐OBE complex probably bind to two adjacent W‐box motifs at its target promoter region and that the dual binding sites cooperate to enhance the binding of WRKY‐OBE complexes to their target promoter regions. Alternatively, the two copies of WRKY proteins in the WRKY‐OBE complex may bind to different target promoters. Previous studies have shown that functionally related genes usually form chromatin clusters and are co‐regulated in eukaryotes (Reimegard et al, 2017; Nutzmann et al, 2020). As the WRKY‐OBE complex co‐regulates numerous stress‐responsive genes, we predicted that two copies of WRKY proteins in the WRKY‐OBE complex probably bind to the promoters of two different stress‐responsive genes, thereby mediating the connection of the two promoters in the chromatin cluster to co‐regulate the expression of stress‐responsive genes.
WRKY transcription factors bind to specific target genes through the recognition of the W‐box by their conserved WRKY domains, which play a major role in the target selection of WRKY proteins (Ciolkowski et al, 2008). Although WRKY domains are highly conserved, the structure of WRKY domains also shows diversity, which is involved in the specific recognition of the W‐box flanking sequence, contributing to the target specificity of WRKY proteins (Miao et al, 2004; Ciolkowski et al, 2008; Brand et al, 2013). Moreover, the dimerization of WRKY proteins and the interaction of WRKY proteins with other cofactors have also been shown to affect the binding affinity of WRKY proteins with the W‐box (Xu et al, 2006; Zou et al, 2008; Liu et al, 2012; Chi et al, 2013). However, these studies cannot completely explain how a given group of WRKY transcription factors targets a subset of W‐box‐containing promoters. This study indicated that the conserved N‐terminal CC domain in group IId WRKY proteins interacts with OBE proteins and is responsible for transcriptional repression. Given that the PHD finger of OBE proteins binds to histones in vitro and that the WRKY‐OBE complexes associate with the promoter proximal to the +1 nucleosome, which forms an obstacle for transcription initiation in vivo, we predicted that the OBE proteins binding to histones in the +1 nucleosome are responsible for the transcriptional repression activity of group IId WRKY proteins. Moreover, considering that deletion of the OBE‐interacting domain in WRKY11 reduces its target specificity, the binding of WRKY‐OBE complexes to the +1 nucleosome also mediates the selection of a subset of W‐box‐containing promoters. Further studies are required to determine how the binding of OBE proteins to the +1 nucleosome contributes to the target selection and transcriptional repression activities of WRKY‐OBE complexes.
We found that WRKY‐OBE complexes are directly responsible for repressing the transcription of stress‐responsive genes rather than for activating the transcription of development‐related genes. As the inhibitory effects of induced expression levels of biotic and abiotic stress‐responsive genes on plant growth have been extensively studied (Guo et al, 2018; Zhang et al, 2020), it is reasonable to predict that the growth defects in the wrky and obe mutants are indirectly caused by the overexpression of stress‐responsive genes. Although multiple mechanisms are responsible for the induction of stress‐responsive genes, a given transcription factor is only responsible for activating a subset of stress‐responsive genes. This study showed that WRKY‐OBE complexes repress the expression of a wide range of biotic and abiotic stress‐responsive genes. Considering that expression levels of group IId WRKY genes are reduced under drought stress conditions, we predicted that the reduced expression of group IId WRKY genes in response to stress conditions represents a previously uncharacterized regulatory mechanism mediating the induction of stress‐responsive genes (Fig 9). Although expression levels of stress‐responsive genes are induced by stress treatments in a short time, the expression levels of group IId WRKY genes are reduced only in long‐term drought stress‐treated plants, suggesting that reduced expression of group IId WRKY genes is responsible for a long‐term stress response. In response to stress conditions, stress‐responsive genes are initially induced by stress‐activated transcription factors, and this induction is subsequently enhanced by reducing the expression of group IId WRKY genes (Fig 9). By combining short‐ and long‐term induction of stress‐responsive genes, plants can obtain sufficient stress tolerance with the least impact on plant growth.
Materials and Methods
Plant materials growth and transformation
Arabidopsis T‐DNA insertion mutants including obe1 (SALK_075710), obe3 (SALK_042597), obe4 (SALK_016218), wrky7 (SALK_093993C), wrky11 (SAIL_349_G09), and wrky17 (SALK_076337C) were obtained from the Arabidopsis Biological Resource Center. The obe2, wrky15, wrky21, and wrky39 mutants were obtained using CRISPR‐Cas9 (Xing et al, 2014). Guide RNAs were designed on the CRISPR‐P 2.0 website. All the mutants used in this study were in the Columbia (Col‐0) background. The obe1/2, obe3/4, obe1/3, and obe1/4 double mutants and wrky multiple mutants were generated by crossing. Given the rootless phenotype of obe1/2, obe3/4, and wrky‐qm/15 +/−, seedlings were generated from the separation of heterozygotes. The seedlings were grown on MS (Murashige and Skoog) medium plates at 22°C under long day (16 h light/8 h dark) conditions. After 11 days of growth, the seedlings were prepared for RNA‐seq and ChIP‐seq, transplanted into soil, and grown under the same conditions for genotyping and phenotype observation. The genomic sequences of OBE1, OBE3, WRKY7, and WRKY11 with their native promoters were inserted into the pCAMBIA1305 vector with a 3 × Flag or GFP tag at their 3′ terminus (Vazyme, C112‐01). The WRKY11‐CCΔ sequence was also inserted into the vector using the multi‐one‐step in‐fusion method (Vazyme, C113‐01). The constructs were transformed into Agrobacterium strain GV3101 and prepared for transgenesis via the floral‐dipping method. Primers used for the construction are listed in Dataset EV4. Transgenic plants were selected using hygromycin (30 mg/l) and ampicillin (50 mg/l).
Drought treatment and water loss rate measurements
Dehydration treatment and water loss rate measurement were performed as described previously with minor modifications (Verslues et al, 2006). To induce dehydration, 9‐day‐old seedlings grown on MS medium plates were transplanted into moist soil and kept under long‐day conditions (16 h light/8 h dark) at 22°C, without any additional water, until the Col‐0 plants exhibited wilting symptoms. The rate of plant survival was assessed 2 days after rehydration. To measure the water loss rate of detached leaves, the second to fourth leaves were cut from 24‐day‐old wild‐type and mutant plants grown under long‐day conditions (16 h light/8 h dark) at 22°C. Equal weights of leaves were cut from the wild‐type and mutant plants and subjected to water loss measurement at different times. The rate of water loss was shown as a percentage of the initial weight of fresh leaves.
Immunoprecipitation, mass spectrometry analysis, and gel filtration
For AP‐MS, a mixture of 2.5 g seedling and 2.5 g flower was collected to extract protein. The materials were then ground in liquid nitrogen and homogenized in 15 ml cold plant lysis buffer (50 mM Tris–HCl [pH 7.5], 150 mM NaCl, 5 mM MgCl2, 10% glycerol, 0.1% NP‐40, 5 mM DTT, 0.1 mM PMSF, and Roche proteinase inhibitor cocktail) and incubated for 20 min with rotation at 4°C. After centrifugation, the supernatant was passed through a micro‐cloth (Millipore, 475855), and anti‐Flag M2 agarose beads (Sigma, A2220) were incubated with supernatant at 4°C for at least 2.5 h, and then the agarose beads were washed six times with cold plant lysis buffer to remove the non‐specific binding proteins. The protein complexes were eluted with 3× FLAG peptide (Sigma, F4799), subjected to SDS–PAGE, and visualized by silver staining (Sigma, PROT‐SIL2). The silver‐stained protein complex was then subjected to MS analysis as previously described (Zhang et al, 2013; Luo et al, 2020).
Gel filtration was performed as previously described (Ning et al, 2015). In brief, the transgenic seedlings of OBE1/WRKY11/WRKY11‐CCΔ‐3 × Flag were ground and suspended in 2.5 ml cold plant lysis buffer. After centrifugation at 14,000 g, the supernatant was filtered through a 0.22 μm filter, and the proteins were loaded into a Superose 6 column (10/300GL) (GE Healthcare, 17‐5172‐01). Next, 500 μl elute was collected and the proteins were immediately run on SDS–PAGE gel and detected by immunoblotting with Flag antibody (Sigma, F7425).
Yeast two‐hybrid assays
For Y2H assays, the full‐length OBE1‐4 and WRKY7/11/15/17/21/39/12/14/36/40 coding sequences and a series of truncated coding sequences were cloned into pGADT7 and pGBKT7 vectors using a cloning kit (Vazyme, C112‐01). Primers used for cloning are listed in the Dataset EV4. The pGADT7 vectors were transformed into the AH109 strain, and the pGBKT7 vectors were transformed into the Y187 strain. The AH109 strain was grown on solid medium lacking Leu (SD‐L), and the Y187 strain was grown on solid medium lacking Trp (SD‐W); both were grown at 28°C. Positive strains on SD‐L and SD‐W were then mated for 16–20 h in YPDA lipid medium. After mating, the mixtures were transferred to solid medium lacking Trp and Leu (SD‐W‐L). Finally, the positive strains grown on SD‐W‐L medium were resuspended in sterile ddH2O and spotted on solid medium lacking Trp, Leu, and His (SD‐W‐L‐H) for Y2H assays. To reduce background growth, different concentrations of 3‐amino‐1,2,4‐triazole (3‐AT) were added to the SD‐W‐L‐H.
Protein purification and pull‐down assay
For protein purification, the full‐length and truncated coding sequences of Arabidopsis WRKY11, OBE1, and OBE2, as well as their orthologs from Physcomitrium patens, Selaginella moellendorffii, Taxus chinensis, and Oryza sativa, were cloned into pGEX‐6p‐1 in fusion with GST tag, pSMT3 in fusion with His tag, and modified pET30a in fusion with the MBP and His tag. Primers used for cloning are listed in Dataset EV4. The constructs were confirmed by Sanger sequencing and transformed into the BL21 strain for protein purification. Protein purification was performed as previously described (Tan et al, 2020). In brief, the protein expression was induced with 0.2 mM IPTG and the E. coli bacteria were precipitated and resuspended in GST‐tag lysis buffer (20 mM Tris–HCl [pH 8.0], 150 mM NaCl, 1 mM DTT, and 1 mM PMSF), His‐tag lysis buffer (20 mM Tris–HCl [pH 8.0], 500 mM NaCl, 20 mM imidazole, 1 mM DTT, and 1 mM PMSF) and MBP‐tag lysis buffer (20 mM Tris–HCl [pH 8.0], 200 mM NaCl, 1 mM EDTA, 1 mM DTT, and 1 mM PMSF), followed by sonication and centrifugation at 30,000 g for 1 h. The supernatant was filtered and incubated with GST beads (GE Healthcare, 17075601), Ni‐NTA Resin (Millipore, 70666‐4), or MBP Resin (Smart‐life sciences, SA077005) for 1 h at 4°C with rotation. The beads were then washed three times with GST‐tag lysis buffer, His‐tag lysis buffer, or MBP‐tag lysis buffer to remove non‐specific proteins. Proteins were eluted with GST elution buffer containing 10 mM glutathione, His‐tag lysis buffer containing 250 mM imidazole, or MBP‐tag lysis buffer containing 10 mM maltose. Purified proteins were confirmed using Coomassie blue staining.
For in vitro pull‐down assays, the MBP‐tagged Arabidopsis OBE2‐4 and WRKY11‐1 proteins were mixed with GST‐tagged full‐length or truncated versions of Arabidopsis OBE1; the GST‐tagged full‐length OBE1 ortholog and the MBP‐tagged full‐length WRKY11 ortholog from each of other plants were also mixed. The mixture was incubated with MBP or GST beads and gently rotated for 1 h at 4°C. After washing at least five times, the proteins were eluted with MBP or GST elution buffer containing 10 mM maltose or glutathione, and input and elution samples were subjected to SDS–PAGE and immunoblotting using GST antibody (Abmart, 12G8) and His antibody (Abmart, 10E2). The rice group IId WRKY protein OsWRKY51 was expressed and purified from bacteria in fusion with the MBP tag. The MBP‐fused OsWRKY51 protein was mixed with the total protein extract of rice seedlings and subjected to pull‐down assays using the MBP beads. The OSWRKY51‐interaction proteins were identified by mass spectrometry.
Electrophoretic mobility shift assay
Purified full‐length and truncated WRKY11 proteins or a mixture of WRKY11 and OBE1 proteins were incubated with 1.25 μM double‐stranded probe DNA labeled with Cy5 fluorescent dyes in binding buffer (25 mM HEPES–KOH [pH 7.6], 50 mM KCl, 0.1 mM EDTA, 12.5 mM MgCl2, 0.5% (w/v) BSA, 5% glycerol, and 1 mM DTT) at room temperature for 30 min. The binding reaction mixture was resolved with 10% native PAGE in 0.5 × TBE at 4°C for 150 min at 80 V. The interaction between the proteins and DNA was detected using a Bio‐Rad scanner.
Domain prediction, sequence alignment, and phylogenetic analysis
Protein sequences were downloaded from the TAIR and NCBI databases. Protein structures were predicted using Alphafold (https://www.alphafold.ebi.ac.uk/). The domains of each protein were predicted using the NCBI database (https://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi). Multiple protein sequence alignments were performed using DNAMAN (version 7) software. A phylogenetic tree was generated by MEGA (version 7) using the neighbor‐joining method. Bootstrapping was performed using 500 replicates.
Quantitative RT‐PCR, RNA‐seq, and data analysis
Total RNA was extracted using TRIzol reagent (Invitrogen, 15596018) from Arabidopsis plants. For quantitative RT‐PCR, cDNA was synthesized using 5×All‐In‐One RT Master Mix (Abm, G492), and quantitative PCR was performed using the Bio‐Rad CFX96 Real‐Time System. For RNA‐seq, 11‐day‐old seedlings and 28‐day‐old well‐watered plants and drought‐treated plants were subjected to RNA extraction. RNA libraries were built by Novogene and sequenced with Illumina NovaSeq 6000 using a paired‐end scheme (PE150). After adaptors and low‐quality reads were filtered, clean reads were mapped to the Arabidopsis genome (TAIR10) using the default parameters of HISAT2 (v2.2.0) (Kim et al, 2019). Unique reads mapped to the transcripts were counted using the featureCounts algorithm (v2.0.2) (Liao et al, 2014). The differentially expressed genes (DEGs) were defined as those with |log2(fold change)| > 1 and FDR < 0.05 with the R package edgeR (v3.32.1) (Robinson et al, 2010). GO analyses were performed using the DAVID 2021 website (https://david.ncifcrf.gov/tools.jsp). The heatmaps of DEGs and GO analysis were drawn using R package gplots (v3.1.1). Box plots and scatter plots were drawn using the R package ggplot2 (v3.3.5). Venn diagrams were drawn using the R package ggvenn (v0.1.9). RNA‐seq data were obtained from three independent biological replicates.
ChIP‐seq and quantitative ChIP‐PCR
ChIP assays were performed as described previously with some modifications (Guo et al, 2022). Briefly, OBE1‐GFP, WRKY11‐GFP, and WRKY11‐CCΔ‐GFP transgenic plants were subjected to ChIP‐seq. In brief, 6 g of 11‐day‐old seedlings were fixed with 1% formaldehyde under vacuum for 12 min (four times at 3 min each time), and the reaction was stopped by the addition of 0.125 M glycine under vacuum for 5 min (twice for 2.5 min each time). The cross‐linked seedlings were ground into a powder in liquid nitrogen. The nuclei were extracted using 30 ml cold lysis buffer (0.4 M sucrose, 10 mM Tris–HCl [pH8.0], 10 mM MgCl2, 0.25% Triton X‐100, 1 mM DTT, 0.1 mM PMSF, and Roche protease inhibitor cocktail) for 30 min at 4°C. The sample was then filtered through two layers of microcloth (Millipore, 475855) to remove debris and pelleted by centrifuging at 1,500 g at 4°C for 20 min. The pellet was washed with buffer II (10 mM Tris–HCl [pH8.0], 10 mM MgCl2, 0.25 M sucrose, and 1% Triton X‐100, 1 mM DTT, 0.1 mM PMSF, and Roche protease inhibitor cocktail) until the pellet became gray. The pellet was resuspended in buffer III (10 mM Tris–HCl [pH8.0], 10 mM MgCl2, 1.7 M sucrose, and 0.25% Triton X‐100, 1 mM DTT, 0.1 mM PMSF, and Roche protease inhibitor cocktail) and centrifuged at 14,000 g at 4°C for 1 h. Chromatin was sonicated to a size from 100 to 500 bp using a Bioruptor (Diagenode). After the sample was centrifuged at 13,000 g at 4°C for 15 min, dilution buffer (20 mM Tris–HCl [pH8.0], 2 mM EDTA, 200 mM NaCl, 1 mM PMSF, and Roche protease inhibitor cocktail) was added to dilute the supernatant. GFP antibody (Abcam, ab290) was used to immunoprecipitate the protein–DNA complex overnight at 4°C. Next, 40 μl protein A beads (Thermo Scientific, 10001D) were added to immunoprecipitate the protein–DNA complex by interacting with GFP antibody at 4°C for 2 h with rotation. The beads were then washed with low salt buffer (20 mM Tris–HCl [pH8.0], 150 mM NaCl, 0.1% SDS, 1% Triton X‐100, and 2 mM EDTA), high salt buffer (20 mM Tris–HCl [pH8.0], 500 mM NaCl, 0.1% SDS, 1% Triton X‐100, and 2 mM EDTA), LiCl buffer (0.25 M LiCl, 1% NP‐40, 1% SDS, 1 mM EDTA, and 10 mM Tris–HCl [pH8.0]), and TE buffer (10 mM Tris–HCl [pH8.0] and 1 mM EDTA), and the protein–DNA complex was reverse crosslinked at 65°C overnight. DNA was extracted from the eluted sample with phenol/chloroform/isoamyl (Biorigin; 25:24:1) reagent, and the purified DNA was sent to Novogene (Beijing, China) for library generation and paired‐end (PE150) sequencing on the Illumina NovaSeq 6000 platform.
After adaptors and low‐quality reads were filtered, the clean data were mapped to the Arabidopsis TAIR10 genome using Bowtie2 (v2.3.4) with parameters ‐k 1 and ‐N 1, allowing one mismatch (Langmead & Salzberg, 2012). ChIP‐seq peaks were identified against input‐controlled peaks using MACS2 (v2.2.7.1) with the parameter ‐f BAMPE (Zhang et al, 2008). PCR duplicates were removed by using Picard Tools (v2.23.0) with MarkDuplicates. The read counts were normalized to RPKM (reads per kilobase per million mapped reads) by the number of clean reads mapped to the genome in each library. The ChIP‐seq results in this study were obtained from two independent biological replicates. The profile plot and heatmap were drawn using the plotProfile and plotHeatmap functions in DeepTools (v3.5.1). Box plots, scatter plots, and stacked histograms were drawn using the R package ggplot2 (v3.3.5). Venn diagrams were drawn using the ggvenn R package (v0.1.9). For quantitative ChIP–PCR, 2 g of 11‐day‐old seedlings and anti‐Flag antibody (Sigma, F1804) were used. Primers used for quantitative ChIP–PCR are listed in Dataset EV4.
Peptide pull‐down assay
For peptide pull‐down assays, 1 μg of purified proteins were incubated with 1 μg of different biotinylated histone peptides in binding buffer (50 mM Tris–HCl [pH 7.5], 150 mM NaCl, and 0.05% NP‐40) at 4°C for 4 h. The protein mixture was then incubated with 40 μl of Streptavidin MagneSphere Para‐Magnetic Particles (Promega, Z5481) for 1 h with rotation at 4°C. The particles were washed and boiled in SDS sample buffer and were subjected to immunoblotting using anti‐GST antibody. Histone peptides used in this study were previously described (Qian et al, 2021). For determining the cooperation between OBE1 and OBE2 in binding to histone peptides, the purified GST‐OBE1 and His‐OBE2‐4 proteins were pre‐incubated overnight in binding buffer. Then, the protein mixture was incubated with 1 μg of biotinylated histone peptides at 4°C for 4 h, followed by 40 μl of Streptavidin MagneSphere Para‐Magnetic Particles for 1 h with rotation at 4°C. After washing with binding buffer, the protein‐bound particles were subjected to SDS–PAGE and immunoblotting.
Dual luciferase reporter assay
The luciferase reporter assay was performed as described with minor modifications (Ning et al, 2015). In brief, the transcriptional activator VP16 fused to the GAL4 DNA‐binding domain activates the transcription of the reporter gene. OBE1, WRKY11, and truncated WRKY11 sequences were ligated in frame with the GAL4‐BD‐VP16 fusion sequence in the effector construct. The reporter construct and each of the effector constructs were co‐transformed into protoplast cells for the luciferase activity assay as previously reported (Yoo et al, 2007). The luciferase activity was measured with Dual‐Luciferase® Reporter Assay System (Promega, E1910).
Author contributions
Ping Du: Conceptualization; data curation; formal analysis; investigation; methodology; writing – original draft. Qi Wang: Data curation; formal analysis; investigation; methodology. Dan‐Yang Yuan: Data curation; formal analysis. Shan‐Shan Chen: Investigation; methodology. Yin‐Na Su: Data curation; formal analysis. Lin Li: Investigation; methodology. She Chen: Investigation; methodology. Xin‐Jian He: Conceptualization; resources; data curation; formal analysis; supervision; funding acquisition; investigation; writing – original draft; project administration; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Appendix S1
Dataset EV1
Dataset EV2
Dataset EV3
Dataset EV4
Source Data for Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Source Data for Figure 8
Acknowledgement
This work was supported by the National Natural Science Foundation of China (grant number: 32025003).
The EMBO Journal (2023) 42: e113639
Data availability
Raw RNA‐seq and ChIP‐seq data were deposited in the Gene Expression Omnibus (GEO) database under the accession code GSE221660 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE221660). Accession numbers: WRKY7 (AT4G24240), WRKY11 (AT4G31550), WRKY15 (AT2G23320), WRKY17 (AT2G24570), WRKY21 (AT2G30590), WRKY39 (AT3G04670), OBE1 (AT3G07780), OBE2 (AT5G48160), OBE3 (AT1G14740), OBE4 (AT3G63500), ZAR1 (AT3G50950), DREB1A (AT4G25480), DREB1B (AT4G25490), and DREB1C (AT4G25470).
References
- Arrano‐Salinas P, Dominguez‐Figueroa J, Herrera‐Vasquez A, Zavala D, Medina J, Vicente‐Carbajosa J, Meneses C, Canessa P, Moreno AA, Blanco‐Herrera F (2018) WRKY7, ‐11 and ‐17 transcription factors are modulators of the bZIP28 branch of the unfolded protein response during PAMP‐triggered immunity in Arabidopsis thaliana . Plant Sci 277: 242–250 [DOI] [PubMed] [Google Scholar]
- Bi G, Su M, Li N, Liang Y, Dang S, Xu J, Hu M, Wang J, Zou M, Deng Y et al (2021) The ZAR1 resistosome is a calcium‐permeable channel triggering plant immune signaling. Cell 184: 3528–3541 [DOI] [PubMed] [Google Scholar]
- Birkenbihl RP, Kracher B, Ross A, Kramer K, Finkemeier I, Somssich IE (2018) Principles and characteristics of the Arabidopsis WRKY regulatory network during early MAMP‐triggered immunity. Plant J 96: 487–502 [DOI] [PubMed] [Google Scholar]
- Bowman JL, Kohchi T, Yamato KT, Jenkins J, Shu S, Ishizaki K, Yamaoka S, Nishihama R, Nakamura Y, Berger F et al (2017) Insights into land plant evolution garnered from the Marchantia polymorpha genome. Cell 171: 287–304 [DOI] [PubMed] [Google Scholar]
- Brand LH, Fischer NM, Harter K, Kohlbacher O, Wanke D (2013) Elucidating the evolutionary conserved DNA‐binding specificities of WRKY transcription factors by molecular dynamics and in vitro binding assays. Nucleic Acids Res 41: 9764–9778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi Y, Yang Y, Zhou Y, Zhou J, Fan B, Yu JQ, Chen Z (2013) Protein‐protein interactions in the regulation of WRKY transcription factors. Mol Plant 6: 287–300 [DOI] [PubMed] [Google Scholar]
- Ciolkowski I, Wanke D, Birkenbihl RP, Somssich IE (2008) Studies on DNA‐binding selectivity of WRKY transcription factors lend structural clues into WRKY‐domain function. Plant Mol Biol 68: 81–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulgem T, Rushton PJ, Robatzek S, Somssich IE (2000) The WRKY superfamily of plant transcription factors. Trends Plant Sci 5: 199–206 [DOI] [PubMed] [Google Scholar]
- Farooq M, Wahid A, Kobayashi NSMA, Fujita DBSMA, Basra SMA (2009) Plant drought stress: effects, mechanisms and management. Sustain Agric 153–188 [Google Scholar]
- Feng K, Hou XL, Xing GM, Liu JX, Duan AQ, Xu ZS, Li MY, Zhuang J, Xiong AS (2020) Advances in AP2/ERF super‐family transcription factors in plant. Crit Rev Biotechnol 40: 750–776 [DOI] [PubMed] [Google Scholar]
- Franco‐Echevarria E, Rutherford TJ, Fiedler M, Dean C, Bienz M (2022) Plant vernalization proteins contain unusual PHD superdomains without histone H3 binding activity. J Biol Chem 298: 102540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S, Han X, Xu X, Shao Y, Zhu Q, Liu Y, Du J, Xu J, Zhang S (2020) WRKY15 suppresses tracheary element differentiation upstream of VND7 during xylem formation. Plant Cell 32: 2307–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Z, Xiong L, Shi H, Yang S, Herrera‐Estrella LR, Xu G, Chao DY, Li J, Wang PY, Qin F et al (2020) Plant abiotic stress response and nutrient use efficiency. Sci China Life Sci 63: 635–674 [DOI] [PubMed] [Google Scholar]
- Guo Q, Major IT, Howe GA (2018) Resolution of growth‐defense conflict: mechanistic insights from jasmonate signaling. Curr Opin Plant Biol 44: 72–81 [DOI] [PubMed] [Google Scholar]
- Guo J, Cai G, Li YQ, Zhang YX, Su YN, Yuan DY, Zhang ZC, Liu ZZ, Cai XW, Guo J et al (2022) Comprehensive characterization of three classes of Arabidopsis SWI/SNF chromatin remodelling complexes. Nat Plants 8: 1423–1439 [DOI] [PubMed] [Google Scholar]
- Hung FY, Shih YH, Lin PY, Feng YR, Li C, Wu KQ (2022) WRKY63 transcriptional activation of COOLAIR and COLDAIR regulates vernalization‐induced flowering. Plant Physiol 190: 532–547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizaki K (2017) Evolution of land plants: insights from molecular studies on basal lineages. Biosci Biotechnol Biochem 81: 73–80 [DOI] [PubMed] [Google Scholar]
- Jaglo‐Ottosen KR, Gilmour SJ, Zarka DG, Schabenberger O, Thomashow MF (1998) Arabidopsis CBF1 overexpression induces COR genes and enhances freezing tolerance. Science 280: 104–106 [DOI] [PubMed] [Google Scholar]
- Jiang J, Ma S, Ye N, Jiang M, Cao J, Zhang J (2017) WRKY transcription factors in plant responses to stresses. J Integr Plant Biol 59: 86–101 [DOI] [PubMed] [Google Scholar]
- Journot‐Catalino N, Somssich IE, Roby D, Kroj T (2006) The transcription factors WRKY11 and WRKY17 act as negative regulators of basal resistance in Arabidopsis thaliana . Plant Cell 18: 3289–3302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidokoro S, Shinozaki K, Yamaguchi‐Shinozaki K (2022) Transcriptional regulatory network of plant cold‐stress responses. Trends Plant Sci 27: 922–935 [DOI] [PubMed] [Google Scholar]
- Kilian J, Whitehead D, Horak J, Wanke D, Weinl S, Batistic O, D'Angelo C, Bornberg‐Bauer E, Kudla J, Harter K (2007) The AtGenExpress global stress expression data set: protocols, evaluation and model data analysis of UV‐B light, drought and cold stress responses. Plant J 50: 347–363 [DOI] [PubMed] [Google Scholar]
- Kim DH, Sung S (2013) Coordination of the vernalization response through a VIN3 and FLC gene family regulatory network in Arabidopsis . Plant Cell 25: 454–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KC, Lai Z, Fan B, Chen Z (2008) Arabidopsis WRKY38 and WRKY62 transcription factors interact with histone deacetylase 19 in basal defense. Plant Cell 20: 2357–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Mizoi J, Kidokoro S, Maruyama K, Nakajima J, Nakashima K, Mitsuda N, Takiguchi Y, Ohme‐Takagi M, Kondou Y et al (2012) Arabidopsis growth‐regulating factor7 functions as a transcriptional repressor of abscisic acid‐ and osmotic stress‐responsive genes, including DREB2A. Plant Cell 24: 3393–3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Paggi JM, Park C, Bennett C, Salzberg SL (2019) Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nat Biotechnol 37: 907–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD, Wu R, Guttman DS, Desveaux D (2010) Allele‐specific virulence attenuation of the Pseudomonas syringae HopZ1a type III effector via the Arabidopsis ZAR1 resistance protein. PLoS Genet 6: e1000894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2014) featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30: 923–930 [DOI] [PubMed] [Google Scholar]
- Liu Q, Kasuga M, Sakuma Y, Abe H, Miura S, Yamaguchi‐Shinozaki K, Shinozaki K (1998) Two transcription factors, DREB1 and DREB2, with an EREBP/AP2 DNA binding domain separate two cellular signal transduction pathways in drought‐ and low‐temperature‐responsive gene expression, respectively, in Arabidopsis . Plant Cell 10: 1391–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZQ, Yan L, Wu Z, Mei C, Lu K, Yu YT, Liang S, Zhang XF, Wang XF, Zhang DP (2012) Cooperation of three WRKY‐domain transcription factors WRKY18, WRKY40, and WRKY60 in repressing two ABA‐responsive genes ABI4 and ABI5 in Arabidopsis . J Exp Bot 63: 6371–6392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo YX, Hou XM, Zhang CJ, Tan LM, Shao CR, Lin RN, Su YN, Cai XW, Li L, Chen S et al (2020) A plant‐specific SWR1 chromatin‐remodeling complex couples histone H2A.Z deposition with nucleosome sliding. EMBO J 39: e102008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao Y, Laun T, Zimmermann P, Zentgraf U (2004) Targets of the WRKY53 transcription factor and its role during leaf senescence in Arabidopsis . Plant Mol Biol 55: 853–867 [DOI] [PubMed] [Google Scholar]
- Ng DW, Abeysinghe JK, Kamali M (2018) Regulating the regulators: the control of transcription factors in plant defense signaling. Int J Mol Sci 19: 3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning YQ, Ma ZY, Huang HW, Mo H, Zhao TT, Li L, Cai T, Chen S, Ma L, He XJ (2015) Two novel NAC transcription factors regulate gene expression and flowering time by associating with the histone demethylase JMJ14. Nucleic Acids Res 43: 1469–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nooren IM, Kaptein R, Sauer RT, Boelens R (1999) The tetramerization domain of the Mnt repressor consists of two right‐handed coiled coils. Nat Struct Biol 6: 755–759 [DOI] [PubMed] [Google Scholar]
- Nutzmann HW, Doerr D, Ramirez‐Colmenero A, Sotelo‐Fonseca JE, Wegel E, Di Stefano M, Wingett SW, Fraser P, Hurst L, Fernandez‐Valverde SL et al (2020) Active and repressed biosynthetic gene clusters have spatially distinct chromosome states. Proc Natl Acad Sci USA 117: 13800–13809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CY, Lee JH, Yoo JH, Moon BC, Choi MS, Kang YH, Lee SM, Kim HS, Kang KY, Chung WS et al (2005) WRKY group IId transcription factors interact with calmodulin. FEBS Lett 579: 1545–1550 [DOI] [PubMed] [Google Scholar]
- Phukan UJ, Jeena GS, Shukla RK (2016) WRKY transcription factors: molecular regulation and stress responses in plants. Front Plant Sci 7: 760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian F, Zhao QY, Zhang TN, Li YL, Su YN, Li L, Sui JH, Chen S, He XJ (2021) A histone H3K27me3 reader cooperates with a family of PHD finger‐containing proteins to regulate flowering time in Arabidopsis . J Integr Plant Biol 63: 787–802 [DOI] [PubMed] [Google Scholar]
- Reimegard J, Kundu S, Pendle A, Irish VF, Shaw P, Nakayama N, Sundstrom JF, Emanuelsson O (2017) Genome‐wide identification of physically clustered genes suggests chromatin‐level co‐regulation in male reproductive development in Arabidopsis thaliana . Nucleic Acids Res 45: 3253–3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riechmann JL, Ratcliffe OJ (2000) A genomic perspective on plant transcription factors. Curr Opin Plant Biol 3: 423–434 [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26: 139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rushton PJ, Somssich IE, Ringler P, Shen QJ (2010) WRKY transcription factors. Trends Plant Sci 15: 247–258 [DOI] [PubMed] [Google Scholar]
- Saiga S, Furumizu C, Yokoyama R, Kurata T, Sato S, Kato T, Tabata S, Suzuki M, Komeda Y (2008) The Arabidopsis OBERON1 and OBERON2 genes encode plant homeodomain finger proteins and are required for apical meristem maintenance. Development 135: 1751–1759 [DOI] [PubMed] [Google Scholar]
- Saiga S, Moller B, Watanabe‐Taneda A, Abe M, Weijers D, Komeda Y (2012) Control of embryonic meristem initiation in Arabidopsis by PHD‐finger protein complexes. Development 139: 1391–1398 [DOI] [PubMed] [Google Scholar]
- Shang JY, Lu YJ, Cai XW, Su YN, Feng C, Li L, Chen S, He XJ (2021) COMPASS functions as a module of the INO80 chromatin remodeling complex to mediate histone H3K4 methylation in Arabidopsis. Plant Cell 33: 3250–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Ding Y, Yang S (2018) Molecular regulation of CBF signaling in cold acclimation. Trends Plant Sci 23: 623–637 [DOI] [PubMed] [Google Scholar]
- Song L, Huang SC, Wise A, Castanon R, Nery JR, Chen H, Watanabe M, Thomas J, Bar‐Joseph Z, Ecker JR (2016) A transcription factor hierarchy defines an environmental stress response network. Science 354: agg1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan LM, Liu R, Gu BW, Zhang CJ, Luo J, Guo J, Wang Y, Chen L, Du X, Li S et al (2020) Dual recognition of H3K4me3 and DNA by the ISWI component ARID5 regulates the floral transition in Arabidopsis . Plant Cell 32: 2178–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CL, Schmidt D, Bayer EM, Dreos R, Maule AJ (2009) Arabidopsis plant homeodomain finger proteins operate downstream of auxin accumulation in specifying the vasculature and primary root meristem. Plant J 59: 426–436 [DOI] [PubMed] [Google Scholar]
- Verslues PE, Agarwal M, Katiyar‐Agarwal S, Zhu J, Zhu JK (2006) Methods and concepts in quantifying resistance to drought, salt and freezing, abiotic stresses that affect plant water status. Plant J 45: 523–539 [DOI] [PubMed] [Google Scholar]
- Wang Q, Yu F, Xie Q (2020) Balancing growth and adaptation to stress: crosstalk between brassinosteroid and abscisic acid signaling. Plant Cell Environ 43: 2325–2335 [DOI] [PubMed] [Google Scholar]
- Xing HL, Dong L, Wang ZP, Zhang HY, Han CY, Liu B, Chen QJ (2014) A CRISPR/Cas9 toolkit for multiplex genome editing in plants. BMC Plant Biol 14: 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Chen C, Fan B, Chen Z (2006) Physical and functional interactions between pathogen‐induced Arabidopsis WRKY18, WRKY40, and WRKY60 transcription factors. Plant Cell 18: 1310–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi‐Shinozaki K, Shinozaki K (2006) Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annu Rev Plant Biol 57: 781–803 [DOI] [PubMed] [Google Scholar]
- Yang Z, Qian S, Scheid RN, Lu L, Chen X, Liu R, Du X, Lv X, Boersma MD, Scalf M et al (2018) EBS is a bivalent histone reader that regulates floral phase transition in Arabidopsis. Nat Genet 50: 1247–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SD, Cho YH, Sheen J (2007) Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc 2: 1565–1572 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wang L (2005) The WRKY transcription factor superfamily: its origin in eukaryotes and expansion in plants. BMC Evol Biol 5: 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W et al (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biol 9: R137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CJ, Zhou JX, Liu J, Ma ZY, Zhang SW, Dou K, Huang HW, Cai T, Liu R, Zhu JK et al (2013) The splicing machinery promotes RNA‐directed DNA methylation and transcriptional silencing in Arabidopsis. EMBO J 32: 1128–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Zhao Y, Zhu JK (2020) Thriving under stress: how plants balance growth and the stress response. Dev Cell 55: 529–543 [DOI] [PubMed] [Google Scholar]
- Zhao N, Su XM, Liu ZW, Zhou JX, Su YN, Cai XW, Chen L, Wu Z, He XJ (2022) The RNA recognition motif‐containing protein UBA2c prevents early flowering by promoting transcription of the flowering repressor FLM in Arabidopsis. New Phytol 233: 751–765 [DOI] [PubMed] [Google Scholar]
- Zhu JK (2002) Salt and drought stress signal transduction in plants. Annu Rev Plant Biol 53: 247–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou X, Neuman D, Shen QJ (2008) Interactions of two transcriptional repressors and two transcriptional activators in modulating gibberellin signaling in aleurone cells. Plant Physiol 148: 176–186 [DOI] [PMC free article] [PubMed] [Google Scholar]