

Abstract
Crystal structures of ligand-bound G-protein-coupled receptors provide tangible templates for rationally designing molecular probes. Herein, we report the structure-based design, chemical synthesis, and biological investigations of bivalent ligands targeting putative mu opioid receptor C─C motif chemokine ligand 5 (MOR-CCR5) heterodimers. The bivalent ligand VZMC013 possessed nanomolar level binding affinities for both the MOR and CCR5, inhibited CCL5-stimulated calcium mobilization, and remarkably improved anti-HIV-1BaL activity over previously reported bivalent ligands. VZMC013 inhibited viral infection in TZM-bl cells coexpressing CCR5 and MOR to a greater degree than cells expressing CCR5 alone. Furthermore, VZMC013 blocked human immunodeficiency virus (HIV)-1 entry in peripheral blood mononuclear cells (PBMC) cells in a concentration-dependent manner and inhibited opioid-accelerated HIV-1 entry more effectively in phytohemagglutinin-stimulated PBMC cells than in the absence of opioids. A three-dimensional molecular model of VZMC013 binding to the MOR-CCR5 heterodimer complex is constructed to elucidate its mechanism of action. VZMC013 is a potent chemical probe targeting MOR-CCR5 heterodimers and may serve as a pharmacological agent to inhibit opioid-exacerbated HIV-1 entry.
Graphical Abstract
INTRODUCTION
The opioid epidemic in the United States is a significant public health crisis. A recent report suggested that opioids were involved in 69.5% of all drug overdose deaths in the United States during 2018.1 Acquired immune deficiency syndrome (AIDS), caused by the human immunodeficiency virus (HIV) infection, remains another major public health issue with approximately 37.9 million people infected globally in 20182 and an estimated 38,000 new cases reported in the United States each year.3 Currently, the opioid abuse epidemic is closely associated with AIDS/HIV infection. Opioid use disorder (OUD) increases the risk of transmission of HIV infection directly through sharing needles, syringes, or other drug injection materials.4 It was reported that injection drug abuse accounted for 7% of all new HIV diagnoses in 2018 in the United States.5 Conversely, HIV infection can elevate the susceptibility to drug addiction as well.6 It was estimated that HIV-infected individuals prescribed opioids were 21–53% more likely to have OUD than uninfected individuals.7 Moreover, long-term opioid use increases the risk of death among the HIV-infected population compared to uninfected individuals.8 Therefore, it is important to uncover the relationship between opioid use and enhanced HIV infectivity with the goal of limiting the spread and pathophysiological consequences of HIV.
Chronic opioid exposure tends to severely suppress both innate and adaptive immune functions, resulting in significantly increased vulnerability to infectious pathogens, such as HIV.9,10 Opioids can increase HIV replication and exacerbate the progression of AIDS.11-13 One possible pathway for opioid-mediated increased HIV replication occurs through the opioid-dependent upregulation of the major chemokine coreceptor CCR5.14-17 As is well-known, opioid addiction liability is predominantly mediated via actions at the mu opioid receptor (MOR). Mounting studies have shown that MOR agonists, such as [d-Ala2-MePhe4-Gly(ol)5]enkephalin (DAMGO),14 morphine,15,18-21 and methadone,22 can induce elevated CCR5 receptor expression in different immune and nonimmune cells. An increase in CCR5 expression furnishes additional viral entry sites and thus promotes HIV-1 infection/replication.
Accumulating in vitro studies suggest there are functional crosstalks between the mu opioid receptor (MOR) and C─C motif chemokine ligand 5 (CCR5), both belonging to the G-protein-coupled receptor (GPCR) superfamily. Previous reports have revealed that MOR-CCR5 crosstalk is, in part, mediated by the formation of putative MOR-CCR5 heterodimers.23-27 For example, using a coimmunoprecipitation approach, Suzuki et al. demonstrated that the MOR and CCR5 could form oligomers at the cell membrane of human or monkey lymphocytes and the oligomerization can modulate the function of both receptors.23 In addition, Chen and co-workers investigated possible mechanisms of cross-desensitization between the MOR and CCR5 coexpressed in Chinese hamster ovary (CHO) cells. They proposed that MOR-CCR5 heterodimerization may contribute to the observed cross-desensitization.26 Thus, putative MOR-CCR5 heterodimerization affects immune cell function and appears to contribute to the synergistic effects of opioids and HIV coexposure in neuroHIV progression.
Bivalent ligands containing two discrete pharmacophores tethered by an appropriate spacer are powerful chemical probes to characterize GPCR dimerization.28-31 Hence, bivalent ligands that are capable of interacting simultaneously with both receptors would help facilitate the study of the putative MOR-CCR5 heterodimer and its role in opioid accelerated HIV entry/replication. We hypothesized that bivalent ligands containing both an MOR and a CCR5 antagonist pharmacophore would efficiently inhibit opioid-dependent increases in HIV entry/replication.
To test our hypothesis and to understand how MOR-CCR5 dimerization uniquely alters the function of each receptor, we previously designed and synthesized a bivalent ligand, VZMC001 (Figure 1), containing the MOR antagonist pharmacophore naltrexone (1) and the CCR5 antagonist pharmacophore maraviroc (2).32-34 Naltrexone, a selective MOR antagonist, has been used in the treatment of OUD,35 and maraviroc is the only marketed anti-HIV drug that blocks the CCR5 coreceptor function.36 VZMC001 exhibited a 7- or 3.3-fold higher inhibition of HIV entry than maraviroc in primary human astrocytes with or without morphine. Nevertheless, VZMC001 possessed less favorable binding affinity profiles for both the MOR and CCR5 compared to the corresponding monomeric ligands. Moreover, results from our molecular dynamics (MD) simulation showed that the maraviroc portion of VZMC001 was partially dislodged from the CCR5 binding pocket. We speculated that this limitation stemmed from our original molecular design that was based on the homology model of the CCR5.34 On the other hand, as a proof-of-concept, VZMC001 served as a promising lead to further design and develop more potent bivalent ligand(s) targeting the putative MOR-CCR5 heterodimer. Herein, we report the structure-based molecular design, synthesis, and comprehensive biological investigations of the second-generation bivalent ligand VZMC013.
Figure 1.

Chemical structures of naltrexone (red) and maraviroc (blue) within the previously reported bivalent ligand VZMC001 (the structure of the intervening spacer is shown in black).
RESULTS AND DISCUSSION
Structure-Based Molecular Design
When we first initiated this project utilizing naltrexone and maraviroc as pharmacophores, neither of the ligand-bound crystal structures of the MOR and CCR5 were available for structure-based molecular design. Given these constraints, it was critical to choose appropriate spacers as well as the attachment points on each pharmacophore to establish a desirable bivalent ligand. Based on several successful cases,37-39 the C6-position of naltrexone was selected as the attachment point that could be readily transformed to the 6β-amino group. Additionally, molecular docking of maraviroc into a previously reported CCR5 homology model by our group40 revealed that the tropane core and the difluorocyclohexyl moiety established interactions with Glu283 and Ile198, respectively, in the binding pocket. The 4′-position of the terminal phenyl ring in maraviroc was therefore selected as the attachment point to avoid disruption of these interactions. Furthermore, several studies indicated that a spacer with 21 atoms may be ideal to yield the optimal pharmacological effects.37,38,41,42 Therefore, the bivalent ligand VZMC001 with a 21-atom spacer linked through the corresponding attachment points on both pharmacophores was designed and prepared.32 Two monovalent ligands VZMC002 (containing the MOR antagonist pharmacophore only, Figure S1) and VZMC003 (containing the CCR5 antagonist pharmacophore only, Figure S1) were also constructed as controls to examine the potential influence of the spacer on the pharmacological effects on each pharmacophore.
Currently, there are mainly two approaches to characterize putative GPCR dimerization: chemistry-based bivalent ligand targeting approach and structural biology-based GPCR crystal structure determination approach.31 For the former approach, bivalent ligands targeting putative GPCR dimers are commonly synthesized empirically, regardless of the choices of the attachment points on each monomeric pharmacophore and the length and chemical composition of spacers.31 For the latter one, GPCR homodimer constructs have been observed in some crystal structures,43,44 which provide further evidence to support GPCR dimerization. To our knowledge, few bivalent ligands reported have been designed in a rational way, that is, with the support of structural biology, since the concept of bivalent ligands was first introduced in 1982.45
High-resolution GPCR-ligand cocrystal structures offer new opportunities for structure-based molecular design of bivalent ligands. In 2012, the crystal structure of the MOR in complex with an epoxymorphinan antagonist β-funaltrexamine (β-FNA, Figure S1) (PDB ID: 4DKL) was determined.46 It was shown that the C6-position of β-FNA in the cocrystal structure pointed toward the extracellular end of transmembrane helix 5 (TM5) and TM6 (Figure 2a) of the MOR. Considering the high structural similarity between naltrexone and β-FNA, we believed that the C6-position of naltrexone may still be the optimal attachment point on the MOR pharmacophore. One year later, the crystal structure of CCR5 complexing with maraviroc (CCR5/maraviroc, PDB ID: 4MBS) was reported.44 Maraviroc was determined to bind within the pocket such that the 3′-methyl group on the 1,2,4-triazol moiety is directed away from the extracellular domains of TM1, 2, and 3 (Figure 2b). This observation revealed that the 3′-methyl group on the 1,2,4-triazol moiety, rather than the 4′-position of the phenyl ring in maraviroc, may be more accessible from the extracellular side and was therefore selected as the attachment point for the CCR5 pharmacophore for this round of design. With both attachment points designated, we utilized the same 21-atom length spacer as in VZMC001 to construct the novel bivalent ligand, VZMC013 (Figure 2c). A new CCR5 monovalent control VZMC014 (containing the CCR5 antagonist pharmacophore only, Figure S1) was proposed, while the MOR monovalent control ligand VZMC002 was adopted from the previous study.34
Figure 2.

(a) The binding mode of β-FNA in the MOR (adapted from PDB ID: 4DKL).46 (b) The binding mode of maraviroc in the CCR5 (adapted from PDB ID: 4MBS).44 (c) Structure-based molecular design of the second generation bivalent ligands VZMC013, VZMC017, and VZMC019.
Meanwhile, to further investigate how the spacer length affects biological activities, two additional bivalent ligands VZMC017 and VZMC019 (with their corresponding CCR5 monovalent controls VZMC018 and VZMC020, Figure S1) bearing longer (23-atom) or shorter (19-atom) spacers relative to VZMC013 were also designed.
Chemical Synthesis
We report the chemical syntheses of the newly designed bivalent ligands (VZMC013, VZMC017, and VZMC019) and corresponding monovalent control ligands (VZMC014, VZMC018, and VZMC020) in the present study. The synthetic routes for these compounds are depicted in Schemes 1-5. Synthesis of the 2′-aminoethyl maraviroc precursor 24 was first carried out step-wise (Schemes 1-3). First, to prepare compound 9, a similar synthetic route as reported was employed (Scheme 1).47 The benzaldehyde 3 was condensed with ammonium acetate and malonic acid to yield the amino acid 4, which was esterified in methanol to give β-amino ester 5. The racemic β-amino ester 5 was resolved by using l-(+)-tartaric acid to afford 6 as the l-(+)-tartaric acid salt.48 Protection of 6 using benzyl chloroformate was carried out under modified Schotten-Baumann conditions,49 followed by hydrolysis to furnish the Cbz-protected acid 7. The β-aminoaldehyde 9 was obtained from 7 through borane-mediated reduction and subsequent oxidation of the resulting alcohol under Parikh–Doering conditions.
Scheme 1. Synthesis of Intermediate 9a.

aReagents and conditions: (a) Malonic acid (1.5 equiv), ammonium formate (2.5 equiv), EtOH, reflux, 59%; (b) (i) conc. H2SO4, MeOH, 0 °C to r.t.; (ii) 2 M NaOH, pH 10, 0 °C. Two steps 99%; (c) l-(+)-tartaric acid (1 equiv), MeOH, 45 °C to r.t., 25%; (d) (i) benzyl chloroformate (1.2 equiv), Na2CO3(aq) (3.3 equiv), dichloromethane, 0 °C to r.t.; (ii) 2 M NaOH, pH 13, MeOH, 0 °C to r.t.; (iii) 2 M HCl, pH 1, 0 °C. Three steps 80%; (e) BH3·THF (4 equiv), anhydrous THF, N2, 0 °C to r.t., 58%; (f) sulfur trioxide pyridine complex (5 equiv), Et3N (10 equiv), DMSO/dichloromethane = 1/1, 0 °C, 87%.
Scheme 5. Synthesis of the Monovalent Ligands VZMC014, VZMC018, and VZMC020a.

a Reagents and conditions: (a) 31 (1.2 equiv), EDCI (1.5 equiv), HOBt (1.2 equiv), Et3N (2 equiv), 4 Å MS, anhydrous DMF, 0 °C to r.t., 27%–47%; (b) 10% Pd/C, MeOH, 60 psi H2, r.t., 69%–100%; (c) diglycolic anhydride (1 equiv), DMF, r.t., 92%–100%; (d) 24 (1.2 equiv), EDCI (4 equiv), HOBt (4 equiv), Et3N (4 equiv), 4 Å MS, anhydrous DMF, 0 °C to r.t., 21%–46%.
Scheme 3. Synthesis of 2′-Aminoethyl Maraviroc Precursor (24)a.

a Reagents and conditions: (a) Tetrahydro-2,5-dimethoxyfuran (1 equiv), benzyl amine (1.2 equiv), NaOAc, HCl(aq), 0 °C to r.t., 48%; (b) hydroxylamine hydrochloride (1 equiv), pyridine (1.1 equiv), EtOH, reflux, 51%; (c) sodium (10 equiv), n-pentanol, 120 °C, 72%; (d) isobutyryl chloride (1.2 equiv), Na2CO3(aq) (2.8 equiv), dichloromethane, 0 °C to r.t., 49%; (e) (i) PCl5 (1.5 equiv), dichloromethane, 0 °C; (ii) 13 (0.77 equiv), tert-amyl alcohol, 0 °C to r.t.; (iii) AcOH, tert-amyl alcohol, 85 °C. Three steps 29%; (f) p-TsOH monohydrate (1 equiv), 30% Pd/C, MeOH, 60 psi H2, r.t.; (g) 9 (1.1 equiv), NaBH(OAc)3 (1.1 equiv), AcOH, dichloromethane, r.t. Two steps 53%; (h) 30% Pd/C, MeOH, 60 psi H2, r.t., 67%; (i) 4,4-difluorocyclohexanecarboxylic acid (2 equiv), EDCI (2 equiv), HOBt (2 equiv), Et3N (4 equiv), 4 Å molecular sieves (MS), dichloromethane, 0 °C to r.t., 54%; (j) 20% piperidine/DMF, r.t., 42%.
Then, commercially available β-alanine 10 was converted to corresponding methyl ester hydrochloride 11,50 which was protected using fluorenylmethyloxycarbonyl chloride (Fmoc-Cl) to provide 12.51 Hydrazinolysis of 12 led to the Fmoc-protected hydrazide 13 (Scheme 2).52
Scheme 2. Synthesis of Intermediate 13a.

aReagents and conditions: (a) Thionyl chloride (2.5 equiv), MeOH, 0 °C to r.t., 62%; (b) Fmoc-Cl (1.05 equiv), Et3N (3.75 equiv), dichloromethane, 0 °C to r.t., 81%; (c) hydrazine monohydrate (5 equiv), MeOH, r.t., 49%.
Next, N-benzylnortropinone 15 was obtained from acetonedicarboxylic acid 14, tetrahydro-2,5-dimethoxyfuran, and benzylamine following the literature procedures53 and was then transformed to oxime 16 using hydroxylamine hydrochloride. The oxime 16 was reduced by sodium in n-pentanol to give amine 17.54 Acylation of 17 with isobutyryl chloride supplied the subunit 18. Activation of 18 to its corresponding imidoyl chloride followed by trapping with hydrazide 13 and acetic acid-catalyzed thermal cyclization in three successive steps provided the triazole 19.47 Subsequent Bn-deprotection of compound 19 by hydrogenolysis with p-toluenesulfonic acid (p-TsOH) and 30% Pd/C afforded the key intermediate 20, which was directly used as a tosylate salt for the next step. Reductive amination of amine 20 with the β-aminoaldehyde 9 was facilitated using sodium triacetoxyborohydride as a reducing reagent to give compound 21.55 Removal of the Cbz group of 21 by hydrogenolysis with 30% Pd/C in methanol offered free amine 22, which was then coupled with the 4,4-difluorocyclohexanecarboxylic acid to yield amide 23. Treatment of 23 in 20% piperidine/DMF finally provided the precursor 2′-aminoethyl maraviroc 24 (Scheme 3).56
With intermediate 24 in hand, we sought to synthesize the naltrexamine-containing acids 30a–c to prepare the bivalent ligands as in our previous reports32,34 with only minor modifications. The bivalent ligands VZMC013, VZMC017, and VZMC019 were obtained by coupling amine 24 with corresponding acids 30a–c via the HOBt/EDCI method (Scheme 4).57 In addition, the monovalent ligands VZMC014, VZMC018, and VZMC020 were furnished following similar synthetic protocols (Scheme 5).
Scheme 4. Synthesis of the Bivalent Ligands VZMC013, VZMC017, and VZMC019a.

a Reagents and conditions: (a) Benzyl chloroformate (1 equiv), dichloromethane/MeOH = 1/1, 0 °C to r.t., 30%–49%; (b) diglycolic anhydride (1.05 equiv), THF, r.t., 70%–86%; (c) 6β-naltrexamine hydrochloride (0.5 equiv), EDCI (1.25 equiv), HOBt (1.05 equiv), Et3N (2 equiv), 4 Å MS, anhydrous DMF, 0 °C to r.t., 36%–61%; (d) 30% Pd/C, MeOH, 60 psi H2, r.t., 90%–94%; (e) diglycolic anhydride (1 equiv), DMF, r.t., 72%–81%; (f) 24 (1.2 equiv), EDCI (4 equiv), HOBt (4 equiv), Et3N (4 equiv), 4 Å MS, anhydrous DMF, 0 °C to r.t., 20%–55%.
In Vitro MOR Radioligand Binding Studies
All bivalent ligands were first tested for their binding affinity to the MOR. The competitive radioligand binding assay was conducted using monoclonal mouse MOR expressed in CHO cell lines (mMOR-CHO). VZMC013 showed high MOR binding affinity (Ki value of 6.05 nM; Table 1), which was 8.6-fold higher than that of VZMC001 (Ki = 51.8 nM), suggesting that the structure-based reselection of the attachment point on the CCR5 pharmacophore exerted partial influence on MOR binding as well. Moreover, VZMC013 possessed relatively higher binding affinity than that of VZMC017 (Ki = 11.2 nM), while similar to that of VZMC019 (Ki = 4.23 nM), indicating that the chosen spacer length range was suitable to preserve ligand binding affinity to the MOR. The monovalent ligand VZMC002 displayed a slightly lower MOR affinity compared to VZMC013 (Table S1). VZMC013 possessed a relatively lower binding affinity for the MOR compared to the parent pharmacophore naltrexone (Ki = 0.7 nM), which was not unusual based on previous reports from other research groups.58-60 Briefly, VZMC013 effectively recognized the MOR.
Table 1.
MOR Radioligand Binding Affinitya
| compounds | Ki (nM) |
|---|---|
| VZMC013 | 6.05 ± 0.22b |
| VZMC017 | 11.2 ± 1.92b |
| VZMC019 | 4.23 ± 0.27b |
| VZMC001 | 51.8 ± 7.9c |
| Naltrexone | 0.7 ± 0.1c |
[3H]Naloxone was used as the radioligand in the binding assay.
The values are the mean ± SEM of at least three independent experiments.
Data have been reported in ref 33 and are presented here for comparison.
MOR [35S]GTPγS Functional Studies
The [35S]GTPγS functional assay was carried out in mMOR-CHO cells to define the relative efficacy of these bivalent ligands to activate the MOR, as previously illustrated.57,61 Emax values of all ligands were measured relative to that of the maximal stimulated response produced by the full MOR agonist DAMGO. VZMC013 produced minimal stimulation of the MOR and displayed insignificant apparent efficacy (%Emax = 9.22%), similar to those of the previously designed bivalent ligand VZMC001 and the parent compound naltrexone (Table 2). VZMC017 also showed minimal efficacy (%Emax = 4.27%), whereas VZMC019 exhibited slightly higher efficacy (%Emax = 19.4%) but was still a low-efficacy partial agonist relative to DAMGO. Overall, the results indicated that VZMC013 may act as an MOR antagonist as designed, comparable to the MOR neutral antagonist naltrexone.
Table 2.
[35S]GTPγS Binding Results
Calcium Mobilization Assay Results in mMOR-CHO Cells
The pharmacological profiles of the three newly prepared MOR bivalent ligands were further characterized for their effects on MOR-dependent intracellular Ca2+ signaling in an mMOR-CHO cell line that was transfected with chimeric Gqi5 protein as previously described.61 No agonism was observed for VZMC013 at varying concentrations (Figure S2a), compared with the known MOR agonist DAMGO (EC50 = 36.3 ± 1.85 nM). Meanwhile, VZMC013 inhibited DAMGO-induced increases in intracellular Ca2+ concentration [Ca2+]i effectively with moderate potency (Figure S2b and Table 3). Additionally, VZMC013 showed comparable potency to the previously developed bivalent ligand VZMC001 (IC50 = 40.0 nM) in inhibiting DAMGO-stimulated Ca2+ mobilization which conformed to our molecular design. Moreover, VZMC013 was more potent than VZMC017 (IC50 = 74.4 nM) and VZMC019 (IC50 = 1103 nM), further suggesting the critical role of spacer length in designing bivalent ligands. In summary, the results of the [35S]GTPγS functional assay and the ability to inhibit DAMGO-stimulated calcium mobilization demonstrated that VZMC013 is a potent MOR antagonist.
Table 3.
Inhibition of DAMGO Induced Ca2+ Mobilization
| compounds | IC50 (nM) |
|---|---|
| VZMC013 | 50.0 ± 2.45a |
| VZMC017 | 74.4 ± 3.20a |
| VZMC019 | 1103 ± 7.11a |
| VZMC001 | 40.0 ± 4.8b |
| naltrexone | 6.62 ± 1.45a |
The values are the mean ± SEM of at least three independent experiments.
Data have been reported in ref 33 and are presented here for comparison purposes.
In Vitro CCR5 Radioligand Binding Studies
To verify the binding affinity of the bivalent ligands to the CCR5, the competitive radioligand binding assay was conducted in recombinant rhesus macaque CCR5-expressing Chem-1 cells, and macrophage inflammatory protein 1 beta (MIP-1β) was used as a control. VZMC013 exhibited a reasonably high binding affinity with a Ki value of 3.29 nM (Table 4 and Figure S3a), which was 12-fold greater than the monovalent compound VZMC014 (Ki = 41 nM) (Table S2 and Figure S3b). In addition, VZMC013 possessed a relatively higher binding affinity for the CCR5 than VZMC017 and VZMC019. Most importantly, VZMC013 showed a dramatically improved binding affinity for the CCR5 than VZMC001,33 demonstrating the success of the attachment point relocation on the CCR5 pharmacophore via the structure-based molecular design approach.
Table 4.
CCR5 Radioligand Binding Affinitya
| compounds | Ki (nM)b |
|---|---|
| VZMC013 | 3.29 ± 0.29 |
| VZMC017 | 5.70 ± 0.23 |
| VZMC019 | NDc |
| VZMC001 | 239 ± 56d |
| MIP-1β | 0.056 ± 0.006 |
[125I]MIP-1α was used as the radioligand in the binding assay.
Ki values were calculated using the Cheng–Prusoff equation. The values are the mean ± SEM of at least three independent experiments.
Its IC50 value is higher than 10,000 nM, the highest concentration tested.
Data have been reported in ref 33 and are presented here for comparisons. It should be noted that the assay methods employed in both studies are similar to minor differences.
Calcium Mobilization Assay Results in HOS-CCR5 Cells
We then conducted the calcium mobilization assay to further test both the agonist and antagonist properties of the newly synthesized compounds in the HOS-CCR5 (stably transfected for the expression of CCR5) cells using our reported protocol,33,34 as calcium mobilization is associated with the activation of CCR5. Prior to the assay, the HOS-CCR5 cells were transiently transfected with a Gqi5 to boost calcium signaling levels. As expected, none of these compounds appeared to activate CCR5 within the range of concentrations tested (represented by VZMC013 and VZMC014, Figure S4a) compared to the control C─C motif chemokine ligand 5 (CCL5/RANTES) under the same conditions (CCL5 exhibited an agonism profile with an EC50 value of 155 ± 6.58 nM, Figure S4a). In the antagonism assay, these compounds were assessed for their ability to inhibit CCL5-stimulated Ca2+ mobilization. As shown in Table 5 and Figure S4b, VZMC013 demonstrated two-digit nanomolar inhibitory potency (IC50 = 57.5 nM) and was 2.2-fold more potent than VZMC001 (IC50 = 126 nM).33 Thus, as predicted, the positional switch in the spacer attachment on maraviroc in VZMC013 improved its inhibition of CCR5 agonist-induced calcium mobilization relative to VZMC001. This prediction was further supported by the enhanced potency of VZMC014 (IC50 = 116 nM) compared to the monovalent ligand VZMC003 (IC50 = 622 nM)33 (Table S3 and Figure S4b). VZMC013 also demonstrated a much higher potency than those of VZMC017 and VZMC019, indicating the importance of spacer length. In brief, VZMC013 acted as a potent CCR5 antagonist.
Table 5.
Inhibition of CCL5-Stimulated Intracellular Ca2+ Mobilizationa
| compounds | IC50 (nM) |
|---|---|
| VZMC013 | 57.5 ± 4.87 |
| VZMC017 | 965 ± 30.5 |
| VZMC019 | 1260 ± 77.6 |
| VZMC001 | 126 ± 28b |
| maraviroc | 0.77 ± 0.20 |
The values are the mean ± SEM of at least three independent experiments.
Data have been reported in ref 33 and are presented here for comparison purposes.
Anti-HIV-1BaL Activity and Cytotoxicity of VZMC013 in GHOST CCR5 Cells
To further characterize the capacity of VZMC013 to block HIV-1 entry by occupying the HIV binding site on the CCR5, we utilized a well-established HIV-1 entry assay, in which the ability for small molecules to inhibit HIV-1 entry is measured as a decrease in HIV-1 reverse transcriptase (RT) activity that is equal to a decrease in radioactivity output after standard radioactive incorporation of tritiated thymidine triphosphate (needed for the synthesis of viral DNA).62 This assay was run in GHOST-CCR5 cells using the CCR5-tropic viral strain HIV-1BaL. Compounds VZMC001, VZMC002, VZMC013, and VZMC014 (maraviroc tested as the control) were subjected to this assay. The treatment concentrations for each compound were 0 and from 0.001 to 100 μM; in which the RT activity at 0 μM of tested compound was defined as 100%, and the results were expressed as EC50 values. As shown in Table 6, the previously developed bivalent ligand VZMC001 did not show any significant inhibition of HIV-1 entry at concentrations up to 100 μM (RT activity: 107.6–226.1%), whereas the newly designed bivalent ligand VZMC013 acted as a potent inhibitor in preventing HIV-1 entry, with an EC50 value of 0.093 μM. The results from this assay clearly indicated that our structure-based design strategy has yielded a bivalent ligand with a markedly improved ability to inhibit HIV-1 entry in cells compared to the previously designed bivalent compound, and the proper selection of an attachment site on the CCR5 pharmacophore was critical for inhibiting HIV-1 entry.
Table 6.
Inhibition of HIV-1BaL and Cytotoxicity in GHOST CCR5 Cellsa
| compounds | EC50 (μM) | TC50 (μM) | TI |
|---|---|---|---|
| VZMC013 | 0.093 ± 0.004 | >100 | >1075 |
| VZMC001 | >100 | >100 | – |
| maraviroc | 0.018 ± 0.002 | >0.5 | >28 |
Measured in triplicate.
These compounds were concurrently assessed for cytotoxicity in GHOST-CCR5-expressing cells using the XTT assay. The treatment concentrations for each compound were the same as in the anti-HIV assay in which the viability of untreated cells was defined as 100%, and the results were interpreted as TC50 (a 50% reduction in cell viability). The therapeutic index (TI, TC50/EC50) values were calculated if applicable. As depicted in Table 6 and Table S4, neither VZMC013 nor other compounds exhibited cytotoxicity at any concentration ≤100 μM (cell viability ~100%). The bivalent ligand VZMC013 possessed a TI value >1075.
Inhibitory Effect of VZMC013 on HIV-1BaL Entry into MOR-CCR5 Coexpressed TZM-bl Cells
CCR5-expressing TZM-bl cells coexpress an HIV-1 long-terminal repeat (LTR)-firefly luciferase reporter that can be activated by trans-activator of transcription (Tat) in cells expressing HIV. Therefore, increases or decreases in pro-viral gene expression coincide with increases or decreases, respectively, in luciferase activity. To initially determine whether the coexpression of MOR affected HIV-1 entry/expression, OPRM1- or control plasmid-transfected TZM-bl cells were exposed to varying concentrations of HIV-1BaL, and HIV-1 entry was calculated based on LTR-driven luciferase activity. As seen in Figure S5, viral entry in TZM-bl cells expressing MOR was enhanced compared to non-MOR-expressing TZM-bl cells upon exposure to identical amounts of virus. Our findings indicate that MOR coexpression by itself can be sufficient to enhance the infectivity of R5-tropic HIV in CCR5-expressing cells, which could possibly result from a putative MOR-CCR5 heterodimer in addition to the CCR5 mediating the virus entry into host cells.
Next, the inhibitory effect of HIV-1BaL infection of the bivalent compound VZMC013 in OPRM1 and control plasmid transfected TZM-bl cells was measured and compared. VZMC013 demonstrated submicromolar potency in inhibiting fold higher inhibitory potency of virus infection in TZM-bl cells containing OPRM1 (EC50 = 368 nM) than that of those cells containing only control plasmid (EC50 = 610 nM). The results further suggested that the CCR5 may heterodimerize with the MOR, and the putative MOR-CCR5 complex may play an essential role in facilitating viral entry. In turn, our bivalent ligand VZMC013 may efficiently bind to the putative MOR-CCR5 heterodimers expressed in TZM-bl cells and more effectively block HIV entry.
Furthermore, when morphine (100 μM) was added (Figure 3c), the ability of VZMC013 to inhibit HIV infectivity was enhanced in TZM-bl cells containing OPRM1 (EC50 = 247 nM) compared to OPRM1-expressing TZM-bl cells without morphine (EC50 = 368 nM). Therefore, this finding indicates that morphine can exacerbate HIV-1 entry in an MOR-dependent manner and that our bivalent ligand VZMC013 is more potent at suppressing viral entry in the presence of morphine. This further supported our hypothesis that functional MOR-CCR5 dimers may be inhibited effectively by a properly designed bivalent ligand.
Figure 3.

Comparison of % inhibition of infection by VZMC013 in (a) control plasmid, (b) OPRM1, and (c) OPRM1 (with 100 μM of morphine added) transfected TZM-bl cells. % Inhibition of infection was related to the decrease of RLU. Data analysis was performed using GraphPad Prism version 8.0.1 for Windows.
Inhibitory Effect of VZMC013 on HIV-1 Entry to PBMC Cells
Peripheral blood mononuclear cells (PBMCs) include large numbers of CCR5-expressing leukocytes and are susceptible to infection by R5-tropic HIV-1 strains. To determine whether VZMC013 would alter HIV infectivity in nonstimulated PBMCs, we used an HIV-1BaL Env-pseudotyped virus encoding a recombinant firefly luciferase gene (HIV-1-Luc). VZMC013 inhibited HIV-1-Luc infectivity in a concentration-dependent manner in PBMCs with a nearly complete blockade at a concentration of 1000 nM (Figure 4a). These results were as expected since VZMC013 has shown potent CCR5 antagonism profiles in the radioligand binding and calcium mobilization assays.
Figure 4.

(a) Varying concentrations of VZMC013 were added before PBMC cells were infected by HIV-1BaL Env-pseudotyped Luc-expressing virus (HIV-1-Luc, 1.5 ng p24/well). Luciferase content was expressed as RLU. Null: PBMC cells were infected by HIV-1-Luc, with no ligand added. (b) PHA-stimulated PBMCs were exposed to opioids 3 days prior to infection with HIV-1BaL Env-pseudotyped Luc-expressing virus. Morphine (Morph) and DAMGO were used at a concentration of 10 nM. Bivalent compound VZMC013 (100 nM) was added 1 h before the cultures were infected with HIV and maintained in vitro. RLU values of each treatment group were normalized based on the null group (RLU values of the null group were defined as “1”). Null: PHA-stimulated PBMC cells were infected by HIV-1-Luc, with no ligand added. Statistical analysis was performed using GraphPad Prism version 8.0.1 for Windows. Data were analyzed according to one-way ANOVA followed by Newman–Keuls posthoc test; *p < 0.05 and **p < 0.01 are considered as statistically significant and ns: not significant.
Inhibitory Effect of VZMC013 on HIV-1 Entry into PHA-Stimulated PBMCs
Phytohemagglutinin (PHA) can stimulate PBMCs into cell cycle, and PHA-stimulated PBMCs have been widely utilized for investigating the replication of HIV primary isolates in vitro.63 The ability of VZMC013 to inhibit HIV-1 entry into PHA-stimulated PBMCs was further tested. As seen in Figure 4b, exposure to VZMC013 (100 nM) inhibited viral entry, with a 22% reduction in luminescence. While exposure to morphine (10 nM) or DAMGO (10 nM) led to remarkable increases in HIV expression in PHA-stimulated PBMCs, VZMC013 (100 nM) was more effective in preventing viral entry, displaying 55% or 69% decreases in HIV expression, respectively, in cells coexposed to morphine or DAMGO and HIV. These results were in agreement with our findings evaluating HIV-1BaL entry into MOR-CCR5 coexpressing TZM-bl cells discussed above. That is, MOR agonists may enhance HIV invasion through activation of the MOR-CCR5 dimer, while a bivalent ligand specifically inhibiting the heterodimer may effectively block the viral invasion.
Molecular Dynamics Simulation Studies on VZMC013 with the MOR-CCR5 Heterodimer
To delineate the possible binding mode between the bivalent ligand VZMC013 and the putative MOR-CCR5 heterodimer, we implemented molecular modeling studies including molecular docking and MD simulation. Previous studies of the crystal packing interactions of GPCRs revealed that the postulated and observed homodimer interfaces of GPCRs involved TM1/TM2/TM7, TM4/TM5, and TM5/TM6.64 The crystal structure study of the MOR indicated that two different interfaces, TM1/TM2/helix 8 and TM5/TM6, were involved in the dimerization of the MOR and TM5/TM6 is a more prominent interface observed in the ligand-bound MOR crystal structure.46 Moreover, TM5/TM6 as the interface of the MOR was also observed in the MD simulation studies performed by Meral et al.65 In our case, the attachment point for the spacer of the bivalent ligands designed, that is, C6-position of naltrexone pointed toward the TM5/TM6 in the MOR binding pocket. Based on such observations, TM5/TM6 was selected as the interface of the MOR to dimerize with the CCR5. For the CCR5, Jin et al. conducted a site-direct mutagenesis study and found that mutations with lysine on TM5 and TM6 did not completely prevent the dimerization of CCR5. Thus, they speculated that other interfaces have a higher possibility to be the interface of the CCR5.66 Moreover, the computational study together with the site-direct mutagenesis study performed by Zhang et al. revealed that the CCR5 homodimerization involved TM1, TM2, TM3, and TM4.67 In the present study, considering the fact that the attachment point for the spacer of the bivalent ligands designed, that is, the 3′-methyl group on the 1,2,4-triazol moiety of maraviroc pointed toward TM1, TM2, and TM3, and the possible steric hindrance of extracellular loop 1 (ECL1) between TM2 and TM3, TM1/TM2 was therefore selected as the plausible dimer interface of the CCR5. An MOR-CCR5 heterodimer model in which TM5/6 of the MOR and TM1/2 of the CCR5 were selected as dimer interfaces in complexing with VZMC013 (Figure S6a) was inserted into a membrane-aqueous sodium chloride solution system (Figure S6b) and subjected to further MD simulations.
After 100 ns MD simulations, the root-mean-square deviation (RMSD) of the backbone atoms of the proteins was applied to evaluate the dynamic equilibrium of the ligand–receptor complex. As shown in Figure 5a, for the MOR-CCR5_VZMC013 complex, the average RMSD value for backbone atoms in the protein during the 50–100 ns MD simulation was 2.84 Å. An average RMSD value of <3.0 Å for the backbone atoms of the protein is reported to indicate stable binding.68 Therefore, the average RMSD value confirmed the thermodynamic equilibrium of the MOR-CCR5_VZMC013 complex after 100 ns MD simulations.
Figure 5.

(a) RMSD and (b) RMSF of the backbone atoms of the proteins in the MOR-CCR5_VZMC013 complex. The CCR5 portion was labeled in green text, and the MOR portion was labeled in blue text.
Furthermore, the root-mean-square fluctuation (RMSF) values of the backbone atoms of the protein in the MOR-CCR5_VZMC013 complex are plotted in Figure 5b. Apparently, residues located in the seven TMs of the MOR-CCR5 dimer displayed a more stable conformation than other domains of the two proteins, for example, intracellular loops (iCLs) and extracellular loops (ECLs). This observation agreed with the fact that the transmembrane spanning domains of GPCRs generally form more rigid and stable conformations than those of ICLs or ECLs. RMSF values obtained from 100 ns MD simulations may further substantiate the stability of the MOR-CCR5_VZMC013 complex. Therefore, the conformations of the MOR-CCR5_VZMC013 complex after 100 ns MD simulations were selected for further analyses.
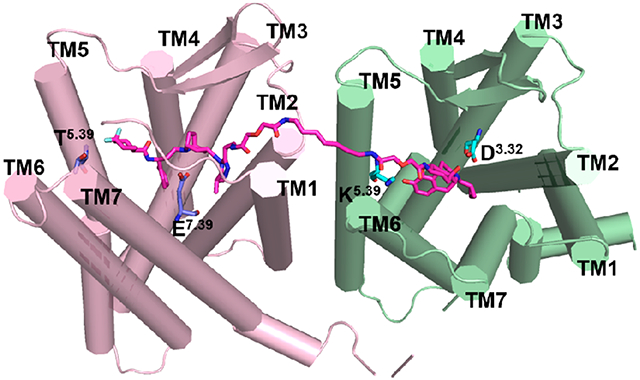
After MD simulations, the stable binding mode of the MOR-CCR5_VZMC013 complex showed that there were mainly 39 amino acid residues from the two proteins within a 5 Å proximity of the bivalent ligand VZMC013 (Figure 6 and Table S5). Among them, 19 residues were associated with the CCR5 pharmacophore, and 10 of them were in juxtaposition to the MOR pharmacophore and the remaining 10 to the spacer. As shown in Figure 6, the CCR5 pharmacophore portion of VZMC013 interacted with amino acid residues from all seven TM helices of CCR5, while the MOR pharmacophore portion of VZMC013 occupied the bottom of a hydrophobic pocket formed mainly by residues from TM3, TM6, and TM7 of the MOR. Residues from TM1 and TM2 of the CCR5 and TM5 and TM6 of the MOR contributed mainly hydrophobic interactions with the spacer.
Figure 6.

Binding mode of VZMC013 in the MOR-CCR5 heterodimer complex after MD simulations. The MOR-CCR5 heterodimer (top figure) is shown as a cylinder model. The MOR-CCR5 heterodimer (bottom figure) is shown as a cartoon model. The key residues within a 5 Å proximity of VZMC013 are shown as surface models in the top figure and stick models in the bottom figure, respectively. Carbon atom: The key residues directly interacted with the MOR were colored in cyan; and the key residues directly interacted with the CCR5 were colored in light-blue. Compound VZMC013 is shown as a stick and ball model (magenta).
For comparison purposes, the docking mode of naltrexone, the MOR pharmacophore of VZMC013, in the inactive MOR was depicted first (Figure S7a and Table S6). It seemed that naltrexone may form hydrophobic interactions with residues M1513.36, W2936.48, I2966.51, H2976.52, I3227.39, and Y3267.43. The protonated nitrogen atom at the 17-position of naltrexone formed an ionic interaction with the oxygen atom at the side chain of D1473.32. A hydrogen-bonding interaction was formed between the dihydrofuran oxygen atom of naltrexone and the phenolic group of Y1483.33. Similar interactions were also observed in the crystal structure of β-FNA bound to the MOR.46 In the binding of VZMC013 with the MOR-CCR5 heterodimer, the MOR pharmacophore moved closer to TM5 and TM6 of the MOR. This conformational change may somehow weaken the ionic interaction with D1473.32, the hydrogen-bonding interaction with Y1483.33, and the hydrophobic interactions with residues W2936.48 and Y3267.43. The distance analyses in Table S7 further supported these observations. On the other hand, residues L2325.38, V2365.42, V3006.55, and W3187.35 seemed to form additional hydrophobic interactions with the MOR pharmacophore, which partially compensated for those weakened interactions. This provided a plausible explanation for the reasonably high binding affinity of VZMC013 to the MOR.
From the crystal structure of maraviroc bound to CCR5, maraviroc binds to the pocket formed by residues from TM1–TM3 and TM5–TM7 of CCR5 (Figure S7b and Table S6).44 E2837.39 formed an ionic interaction with the protonated nitrogen atom of the tropane moiety. Five hydrogen bonds were formed between maraviroc and the CCR5: two hydrogen bonds between 1′-N, 2′-N of the triazole moiety and residues Y371.39, Y892.63 respectively, one hydrogen bond between the nitrogen atom of the carboxamide group and Y2516.51, and dual hydrogen bonds between one fluorine atom on the cyclohexane ring and residues T1955.39, T2596.59. Moreover, the phenyl group of maraviroc formed hydrophobic interactions with Y1083.32, F1093.33, F1123.36, W2486.48, and Y2516.51. In comparison, an examination of the VZMC013 and MOR-CCR5 heterodimeric binding complex revealed that the spacer induced a rotation of the triazole moiety (the dihedral angle changed from 177.7° shown in Figure S7b to 102.0° shown in Figure S7c). Due to this rotation, the two hydrogen-bonding interactions between the triazole moiety of the CCR5 pharmacophore and residues Y371.39 and Y892.63 of CCR5 seemed no longer possible. Moreover, the CCR5 pharmacophore moved closer to the TM1 and TM2 of the CCR5, which may slightly move the CCR5 pharmacophore away from its original binding position thereby decreasing the hydrophobic interactions between the phenyl group of maraviroc and residues Y1083.32, F1093.33, F1123.36, W2486.48, and Y2516.51. Therefore, the relatively lower binding affinity of VZMC013 than maraviroc to the CCR5 seemed reasonable.
On the other hand, the putative movement of the CCR5 pharmacophore toward TM1 and TM2 of the CCR5 may result in amino acid residues C178ECL2, S179ECL2, S180ECL2, and F182ECL2 from ECL2 to form stronger interactions with the CCR5 pharmacophore, whereas those interactions seemed less obvious in the crystal structure of maraviroc binding to CCR5 (Table S6). Among these, residues C178ECL2 and F182ECL2 formed hydrophobic interactions, and residues S179ECL2 and S180ECL2 formed polar interactions with the CCR5 pharmacophore. These interactions were also supported by the distance analyses listed in Table S7. In previous studies, it has been demonstrated that the interaction between the ECL2 of CCR5 and the V3 loop of HIV gp120 was critical to the process of HIV accessing the host cell membrane.44,69 Particularly, residues R11 and S13 of the V3 loop were involved in polar interactions with residues S179ECL2 and S180ECL2 from the ECL2 of CCR5 (Figure S8, which showed the putative binding between V3 loop of HIV gp120 and CCR5).70,71 Therefore, the CCR5 pharmacophore interacted with the ECL2 of CCR5, which may effectively inhibit the V3 loop of gp120 binding to the ECL2 of CCR5 and further block the attachment of HIV to the host cell.
CONCLUSIONS
Utilizing the available ligand-bound crystal structures of the MOR and CCR5, we successfully designed and developed a new bivalent ligand VZMC013 targeting putative MOR-CCR5 heterodimers. VZMC013 demonstrated prominent binding affinities for both MOR and CCR5 at nanomolar levels, which were much higher affinities than those of our previously reported bivalent ligand VZMC001. [35S]GTPγS and calcium mobilization assays of MOR function confirmed VZMC013 to be a potent MOR antagonist as designed. In addition, VZMC013 acted as a CCR5 antagonist and inhibited CCL5-stimulated Ca2+ transients in HOS-CCR5 cells more potently than VZMC001, and this finding was further supported by its significantly improved anti-HIV-1BaL activity at the CCR5 HIV coreceptor. Moreover, VZMC013 showed more potent inhibitory activity of viral infection in TZM-bl cells coexpressing CCR5 and MOR than in TZM-bl cells expressing CCR5 alone, implying that the presence of MOR and putative MOR-CCR5 heterodimeric complexes enhances HIV entry and suggesting that MOR complexation with CCR5 fundamentally alters the functional properties of CCR5 as an HIV coreceptor. Most importantly, VZMC013 was able to block opioid-accelerated HIV-1 invasion more effectively in TZM-bl cells and PHA-stimulated PBMC cells than in controls (lacking opioids), further suggesting its promising role in inhibiting opioid exacerbated HIV-1 infectivity. Utilizing molecular docking and MD simulation approaches, a possible binding mode of VZMC013 in the newly constructed MOR-CCR5 heterodimer model was postulated and helped explain the underlying mechanism of inhibition of viral infection by VZMC013. In summary, VZMC013 is a potent chemical probe that can be used to investigate the specific functional role of putative MOR-CCR5 heterodimers in viral entry and may also serve as a pharmacological agent to alleviate opioid-dependent increases in HIV entry. We believe that the knowledge retrieved from this practice may be applicable to designing novel bivalent chemical probes targeting other GPCR dimerization and characterizing their function and pharmaceutical applications.
EXPERIMENTAL SECTION
Chemistry
All reagents were purchased from commercial suppliers and with no further purification when used. TLC analyses were performed on the Analtech Uniplate F254 plates. Spots were visualized by irradiation with UV light (λ 254 nm) and iodine vapor. Flash column chromatography was carried out on columns packed with silica gel (230–400 mesh, Merck). 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were obtained at ambient temperature with tetramethylsilane (TMS) as the internal standard on a Bruker Ultrashield 400 Plus spectrometer (Bruker, Germany). 19F NMR (376 MHz) spectra were not externally calibrated, and chemical shifts are given as received from the automatic data processing with MestReNova. Chemical shifts were expressed in δ units (ppm), and J values were reported in hertz (Hz). HRMS spectra were acquired from a PerkinElmer Flexar UHPLC with AxION 2 time of flight (TOF) mass spectrometer (PerkinElmer, USA). Analysis of the sample purity was performed on a Varian Prostar 210 high-performance liquid chromatography (HPLC) system using a column Agilent Microsorb-MV 100-5 C18 column (250 × 4.6 mm). HPLC eluent conditions: acetonitrile/water (with 0.1% trifluoroacetic acid), fixed at 40%/60%, or acetonitrile increased from 40% to 100% in gradient within 20 min of test. Flow rate, 0.5 mL/min; UV detection, 210 nm; temperature, ambient; injection volume, 5 μL. The purity of all final compounds was identified as ≥95%.
Preparation of Compound 9
3-Amino-3-phenylpropanoic acid (4)
To a mixture of malonic acid (1.56 g, 15 mmol) and ammonium formate (1.58 g, 25 mmol) was added a solution of benzaldehyde 3 (1.06 g, 10 mmol) in 30 mL of ethanol. The resulting mixture was refluxed for 5 h and then cooled to ambient temperature. The mixture was stirred overnight and filtered. The precipitate was washed with cool ethanol to give 4 as a white solid (0.97 g, 59%). 1H NMR (400 MHz, D2O): δ 7.52–7.44 (m, 5H), 4.66–4.63 (m, 1H), 2.94–2.79 (m, 2H). C9H11NO2 (165.0790).
Methyl 3-Amino-3-phenylpropanoate (5)
To a solution of 3-amino-3-phenylpropanoic acid 4 (7.74 g, 46.8 mmol) in MeOH (45 mL) cooled in an ice–water bath was dropwise added 5 mL of concentrated sulfuric acid. The resulting mixture was warmed up to ambient temperature and stirred for 6.5 h. The excess solvent was removed under reduced pressure, 100 mL of dichloromethane was added to the residue, and then the mixture was cooled to 0 °C. The pH of the solution was adjusted to 10 using 2 M NaOH. After separation and further extraction with dichloromethane, the organic layers were combined, washed with water and brine, dried over Na2SO4, and filtered. The filtrate was concentrated to provide 5 as a yellow oil (8.31 g, 99%). 1H NMR (400 MHz, CDCl3): δ 7.38–7.32 (m, 4H), 7.28–7.24 (m, 1H), 4.42 (t, J = 6.4 Hz, 1H), 3.68 (s, 3H), 2.67 (d, J = 7.6 Hz, 2H), 1.73 (s, 2H). C10H13NO2 (179.0946).
Methyl (S)-3-Amino-3-phenylpropanoate l-(+)-Tartaric Acid Salt (5·l-(+)-Tartaric Acid) (6)
A solution of l-(+)-tartaric acid (5.43 g, 36.2 mmol) in MeOH (42 mL) was heated to 45 °C. Then, a solution of methyl 3-amino-3-phenylpropanoate 5 (6.48 g, 36.2 mmol) in MeOH (13 mL) was added. The resulting mixture was cooled to ambient temperature and was stirred overnight. The mixture was filtered, and the precipitate was washed with cool MeOH and recrystallized in MeOH for two times, affording 6 as a white crystalline solid (2.98 g, 25%). (c 1.12, CHCl3). C14H19NO8 (329.1111).
(S)-3-(((Benzyloxy)carbonyl)amino)-3-phenylpropanoic Acid (7)
To a solution of 6 (0.33 g, 1.0 mmol) in dichloromethane (1.6 mL) was added sodium carbonate aqueous solution (0.35 g of sodium carbonate dissolved in 1.5 mL of H2O) at 0 °C. Benzyl chloroformate (0.21 g, 1.2 mmol) was then dropwise added in 1 min, and upon completion of the addition, the reaction mixture was warmed up to ambient temperature and was stirred at room temperature for 5 h. The water phase was extracted with dichloromethane (5 mL × 3), and the combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated to provide a colorless oil. The oil was dissolved in MeOH (4 mL), and the pH was adjusted to 13 with 2 M NaOH at 0 °C. The resulting mixture was stirred at this temperature for 4 h. Five mL of water was added, and then the solution was acidified to pH 1 with 2 M HCl. The mixture was filtered, and the precipitate was washed with water and then redissolved in dichloromethane. The organic layer was dried over Na2SO4, filtered, and concentrated to offer 7 as a white solid (0.24 g, 80%). 1H NMR (400 MHz, DMSO-d6): δ 7.90 (d, J = 8.60 Hz, 1H, exchangeable), 7.38–7.28 (m, 10H, Ph-H), 5.00–4.96 (m, 3H), 2.74–2.64 (m, 2H). 1H NMR (400 MHz, CDCl3): δ 7.33–7.25 (m, 10H, Ph-H), 5.71–5.70 (m, 1H, exchangeable), 5.17–5.03 (m, 3H), 2.97–2.84 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 175.9, 155.7, 140.4, 136.2, 128.8, 128.5, 128.2, 127.8, 126.2, 67.1, 51.4, 40.2. HRMS (ESI) m/z calcd for C17H16NO4 [M − H]−: 298.1079, found: 298.1081.
Benzyl (S)-(3-Hydroxy-1-phenylpropyl)carbamate (8)
To a solution of 7 (1.87 g, 6.24 mmol) in anhydrous THF (10 mL) was added borane tetrahydrofuran complex solution (1.0 M in THF, 24.9 mmol) under N2 protection at 0 °C. The resulting mixture was stirred for an additional 1 h at room temperature. Two mL of acetone and 20 mL of water were added. The excess solvent was removed under reduced pressure, and 50 mL of saturated sodium bicarbonate solution was then added. The mixture was extracted with ethyl acetate (30 mL × 3). The combined organic extracts were washed with saturated sodium bicarbonate solution (50 mL), followed by 0.1 M HCl (30 mL) and brine (50 mL). After being dried over Na2SO4 and filtered, the filtrate was concentrated to yield a crude product, which was recrystallized with ethyl acetate and hexane to furnish 8 as a white powder (1.03 g, 58%). 1H NMR (400 MHz, CDCl3): δ 7.36–7.27 (m, 10H, Ph-H), 5.39–5.38 (m, 1H), 5.15–5.03 (m, 2H), 4.96–4.95 (m, 1H), 3.70–3.66 (m, 2H), 2.63 (brs, 1H, OH), 2.12–2.04 (m, 1H), 1.92–1.84 (m, 1H). 13C NMR (100 MHz, CDCl3): δ 156.6, 141.7, 136.3, 128.8, 128.7, 128.6, 128.2, 127.6, 126.4, 67.0, 59.2, 52.6, 39.1. HRMS (ESI) m/z calcd for C17H19NNaO3 [M + Na]+: 308.1263, found: 308.1277.
Benzyl (S)-(3-Oxo-1-phenylpropyl)carbamate (9)
Sulfur trioxide pyridine complex (1.73 g, 10.85 mmol) was added to a solution of 8 (0.619 g, 2.17 mmol) and triethylamine (2.196 g, 21.7 mmol) in DMSO/dichloromethane (10 mL/10 mL) at 0 °C. The mixture was stirred for 0.5 h. Water (100 mL) was added, and the mixture was extracted with ethyl acetate (30 mL × 3). The combined organic layers were washed with 0.5 M HCl and brine, dried over Na2SO4, filtered, and concentrated. Further purification of the residue by column chromatography led to 9 as a yellowish solid (0.53 g, 87%). 1H NMR (400 MHz, CDCl3): δ 9.72 (s, 1H, CHO), 7.35–7.27 (m, 10H, Ph-H), 5.45–5.43 (m, 1H, NH), 5.26–5.24 (m, 1H), 5.12–5.00 (m, 2H), 3.03–2.88 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 199.9, 155.6, 140.6, 136.2, 128.9, 128.5, 128.2, 128.1, 127.9, 126.3, 67.0, 50.6, 49.5. IR (diamond, cm−1): 3317.00, 3031.58, 2922.38, 1693.15, 1585.06, 1525.06, 1496.85, 1454.47, 1405.38, 1338.29, 1242.68, 1049.96, 1027.24, 913.83, 738.70. HRMS (ESI) m/z calcd for C17H17NNaO3 [M + Na]+: 306.1106, found: 306.1105; calcd for C18H21NNaO4 [M + MeOH + Na]+: 338.1368, found: 338.1395.
Preparation of Compound 13
Methyl 3-Aminopropanoate Hydrochloride (11)
Thionyl chloride (10.12 g, 75 mmol) was slowly added to methanol (25 mL) at 0 °C. The resulting mixture was stirred for 20 min. After the addition of 3-aminopropanoate 10 (2.67 g, 30 mmol), the mixture was stirred at room temperature for 14 h. The solvent was evaporated to give a crude salt, which was further recrystallized using methanol/ether to afford 11 as a white solid (2.60 g, 62%). C4H10ClNO2 (139.0400).
Methyl 3-((((9H-Fluoren-9-yl)methoxy)carbonyl)amino)-propanoate (12)
Methyl 3-aminopropanoate hydrochloride 11 (0.56 g, 4 mmol) was placed in 10 mL of dichloromethane, and the mixture was cooled in an ice–water bath. Triethylamine (1.50 g, 15 mmol) and a solution of Fmoc-Cl (1.09 g, 4.2 mmol) in dichloromethane (10 mL) were added in sequence. The resulting mixture was stirred at ambient temperature for 1.5 h, and then the mixture was treated with 0.5 N HCl. The organic layer was washed with saturated sodium bicarbonate solution (30 mL), brine (30 mL), dried over Na2SO4, filtered, and concentrated. The crude product was purified by column chromatography to supply 12 as a white solid (1.05 g, 81%). 1H NMR (400 MHz, CDCl3): δ 7.77 (d, J = 7.52 Hz, 2H, fluorene-H), 7.59 (d, J = 7.40 Hz, 2H, fluorene-H), 7.40 (t, J = 7.40 Hz, 2H, fluorene-H), 7.32 (td, J1 = 7.44 Hz, J2 = 0.72 Hz, 2H, fluorene-H), 5.30 (brs, 1H, NH), 4.39 (d, J = 6.96 Hz, 2H, OCH2), 4.21 (t, J = 6.92 Hz, 1H, CH), 3.71 (s, 3H, OCH3), 3.48 (q, J = 6.00, 2H, CH2), 2.57 (t, J = 5.84, 2H, COCH2). 13C NMR (100 MHz, CDCl3): δ 172.9, 156.3, 143.9, 141.3, 127.7, 127.1, 125.1, 120.0, 66.8, 51.8, 47.3, 36.6, 34.2. C19H19NO4 (325.1314).
(9H-Fluoren-9-yl)methyl (3-hydrazinyl-3-oxopropyl)carbamate (13)
The intermediate 12 (0.32 g, 1.0 mmol) was suspended in methanol (2.5 mL). A solution of hydrazine monohydrate (0.25 g, 5 mmol) in methanol (2.5 mL) was added, and the mixture was stirred at room temperature for 30 h. The solvent was removed, and the residue was reslurried in ethyl acetate and filtered to afford 13 as a white solid (0.16 g, 49%). 1H NMR (400 MHz, DMSO-d6): δ 9.01 (s, 1H), 7.89 (d, J = 7.48 Hz, 2H), 7.69 (d, J = 7.36 Hz, 2H), 7.42 (t, J = 7.36 Hz, 2H), 7.37–7.30 (m, 3H), 4.28 (d, J = 6.76 Hz, 2H), 4.23–4.19 (m, 1H), 3.22–3.17 (m, 2H), 2.21 (t, J = 7.36 Hz, 2H). 13C NMR (100 MHz, DMSO-d6): δ 169.8, 157.1, 142.5, 139.4, 137.4, 128.9, 127.3, 121.3, 120.0, 109.7, 37.0, 34.2. C18H19N3O3 (325.1426).
Preparation of the 2′-Aminoethyl Maraviroc Precursor 24
8-Benzyl-8-azabicyclo[3.2.1]octan-3-one (15)
Tetrahydro-2,5-dimethoxyfuran (1.32 g, 10 mmol) was placed in 10 mL of 0.1 M HCl and the mixture was stirred at room temperature for 6 h. Then benzylamine (1.29 g, 12 mmol) was mixed with 10 mL of water and 1 mL of concentrated hydrochloric acid, which was added to the above mixture at 0 °C. The resulting mixture was adjusted to pH 4 using 1 M sodium acetate solution. After the introduction of 1,3-acetonedicarboxylic acid 14 (1.46 g, 10 mmol), the resulting mixture was stirred at room temperature overnight before being filtered. The filtrate was washed with ether, followed by being adjusted to pH 10 with 2 N NaOH. The mixture was extracted with ethyl acetate (50 mL × 3). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was further purified by column chromatography to supply 15 as an oil (1.03 g, 48%). 1H NMR (400 MHz, CDCl3): δ 7.43–7.41 (m, 2H), 7.36–7.32 (m, 2H), 7.29–7.25 (m, 1H), 3.75 (s, 2H), 3.51–3.48 (m, 2H), 2.69 (dd, J1 = 16.08 Hz, J2 = 4.44 Hz, 2H), 2.23–2.18 (m, 2H), 2.14–2.07 (m, 2H), 1.66–1.60 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 210.8, 139.9, 128.9, 127.6, 59.1, 55.7, 48.8, 28.3. C14H17NO (215.1310).
8-Benzyl-8-azabicyclo[3.2.1]octan-3-one oxime (16)
Hydroxylamine hydrochloride (1.03 g, 14.9 mmol) and pyridine (1.29 g, 16.3 mmol) were added to a solution of 15 (3.20 g, 14.9 mmol) in 60 mL of ethanol. The resulting mixture was heated to reflux overnight and allowed to cool to room temperature. The mixture was then diluted with saturated sodium carbonate solution. After filtration, the filtrate was evaporated under reduced pressure to remove the excess solvent. The residue was treated with water (100 mL) and extracted with dichloromethane (60 mL × 3). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated. The residue was recrystallized with ethanol to afford 8-benzyl-8-azabicyclo[3.2.1]octan-3-one oxime 16 as a crystalline solid (1.74 g, 51%). 1H NMR (400 MHz, DMSO-d6): δ 10.31 (s, 1H, OH), 7.40–7.38 (m, 2H), 7.34–7.30 (m, 2H), 7.26–7.22 (m, 1H), 3.61 (s, 2H, NCH2), 3.26–3.22 (m, 2H), 2.85 (d, J = 15.16 Hz, 1H), 2.44 (dd, J1 = 14.40 Hz, J2 = 3.04 Hz, 1H), 2.06–2.00 (m, 2H), 1.96–1.90 (m, 2H),1.49–1.45 (m, 1H), 1.36–1.29 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 153.3, 139.8, 128.3, 128.1, 126.7, 58.0, 57.3, 54.6, 36.9, 31.0, 27.4, 26.5. HRMS (ESI) m/z calcd for C14H19N2O [M + H]+: 231.1497, found: 231.1502.
8-Benzyl-8-azabicyclo[3.2.1]octan-3-amine (17)
To a stirring solution of 16 (0.46 g, 2 mmol) in 8 mL of 1-pentanol was added sodium (0.46 g, 20 mmol) in portions at 120 °C. The resulting mixture was stirred at this temperature for 5 h and then allowed to cool to 0 °C in an ice–water bath. The mixture was acidified to pH = 2 with 6 M HCl and then extracted with 6 M HCl. The combined aqueous layers were basified with 5 M NaOH to pH 10. The resulting mixture was extracted with ethyl acetate (20 mL × 3). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated to provide 17 as an oil (0.31 g, 72%), which was used for the next step without further purification. 1H NMR (400 MHz, DMSO-d6): δ 8.07 (brs, 2H, NH2), 7.36–7.30 (m, 4H), 7.26–7.21 (m, 1H), 3.58 (s, 2H, NCH2), 3.31–3.22 (m, 1H), 3.17 (s, 2H), 1.97–1.94 (m, 2H), 1.73–1.70 (m, 4H), 1.57–1.52 (m, 2H). 13C NMR (100 MHz, DMSO-d6): δ 139.8, 128.3, 128.1, 126.7, 57.0, 54.0, 43.0, 34.1, 26.3. HRMS (ESI) m/z calcd for C14H21N2 [M + H]+: 217.1705, found: 217.1710.
N-(8-Benzyl-8-azabicyclo[3.2.1]octan-3-yl)isobutyramide (18)
To a stirring mixture of 8-benzyl-8-aza-bicyclo[3.2.1]octan-3-amine 17 (0.22 g, 1.0 mmol) in dichloromethane (6 mL) was added sodium carbonate solution (0.30 g, 2.8 mmol, in 6 mL of H2O). A solution of isobutyryl chloride (0.13 g, 1.2 mmol) in dichloromethane (3 mL) was then added dropwise at 0 °C. The resulting mixture was warmed up to room temperature and stirred for 0.5 h. The mixture was adjusted to pH = 9 with saturated sodium bicarbonate solution and extracted with dichloromethane (5 mL × 3). The combined organic layers were washed with 1 M NaOH (5 mL), brine, dried over Na2SO4, filtered, and concentrated. The residue was recrystallized with ethyl acetate/hexane to furnish 18 as a white solid (0.14 g, 49%). 1H NMR (400 MHz, CDCl3): δ 7.37–7.36 (m, 2H, Ph-H), 7.33–7.29 (m, 2H, Ph-H), 7.26–7.22 (m, 1H, Ph-H), 5.25 (d, J = 6.64 Hz, 1H, NH), 4.20–4.09 (m, 1H), 3.53 (s, 2H, CH2), 3.22 (brs, 2H), 2.27 (hept, J = 6.88 Hz, 1H, CH), 2.05–2.02 (m, 2H), 1.83–1.78 (m, 2H), 1.75–1.70 (m, 2H), 1.53–1.47 (m, 2H), 1.12 (d, J = 6.88 Hz, 6H, CH3 × 2). 13C NMR (100 MHz, CDCl3): δ 176.4, 140.0, 128.7, 128.3, 127.0, 59.0, 56.5, 41.2, 38.7, 35.9, 26.5, 19.7. HRMS (ESI) m/z calcd for C18H27N2O [M + H]+: 287.2123, found: 287.2132.
(9H-Fluoren-9-yl)methyl 2-(4-(8-benzyl-8-azabicyclo[3.2.1]-octan-3-yl)-5-isopropyl-4H-1,2,4-triazol-3-yl)ethyl)carbamate (19)
To a solution of 18 (0.69 g, 2.4 mmol) in 15 mL of anhydrous dichloromethane over 1 h. After being stirred at room temperature for 5 h, the mixture was treated with addition of a solution of (9H-fluoren-9-yl)methyl(3-hydrazinyl-3-oxopropyl)carbamate 13 (0.60 g, 1.85 mmol) in tert-amyl alcohol (15 mL) over 1 h at 0 °C. The resulting mixture was stirred at room temperature for 16 h. The mixture was then treated with saturated sodium bicarbonate solution (15 mL) and extracted with dichloromethane (30 mL × 3). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated to ca. 10 mL. After the addition of acetic acid (1.2 mL), the mixture was heated at 85 °C for 2 h. The reaction was quenched by the addition of saturated sodium bicarbonate solution (50 mL) and extracted with ethyl acetate (50 mL × 3). The combined organic phases were washed with brine, dried over Na2SO4, and concentrated. The residue was further purified by column chromatography to give 19 as a white crystalline solid (0.31 g, 29%). 1H NMR (400 MHz, DMSO-d6): δ 7.88 (d, J = 7.52 Hz, 2H), 7.67 (d, J = 7.44 Hz, 2H), 7.55 (t, J = 5.68 Hz, 1H, exchangeable), 7.43–7.38 (m, 4H), 7.33–7.27 (m, 4H), 7.20 (t, J = 7.24 Hz, 1H), 4.33–4.30 (m, 2H), 4.29–4.20 (m, 2H), 3.55 (brs, 2H), 3.40–3.36 (m, 2H), 3.23–3.17 (m, 3H), 2.96 (t, J = 7.48 Hz, 2H), 2.12–2.03 (m, 4H), 1.73–1.68 (m, 4H), 1.28 (d, J = 6.76 Hz, 6H). 13C NMR (100 MHz, DMSO-d6): δ 158.3, 156.1, 151.0, 143.8, 140.6, 139.8, 128.1, 128.0, 127.5, 126.9, 126.5, 125.0, 120.0, 65.4, 58.3, 55.4, 46.6, 36.5, 26.3, 25.8, 25.0, 21.9. IR (Diamond, cm−1): 3320.83, 2933.60, 2875.13, 2160.60, 1979.45, 1712.86, 1507.73, 1449.25, 1349.55, 1312,41, 1247.11, 1138.87, 1099.09, 1071.14, 1029.43, 969.64, 920.53, 876.36, 844.60, 797.40, 758.76, 738.58, 696.58. C36H41N5O2 (575.3260).
(9H-Fluoren-9-yl)methyl 2-(4-(8-azabicyclo[3.2.1]octan-3-yl)-5-isopropyl-4H-1,2,4-triazol-3-yl)ethyl)carbamate (20·Tosylate)
A solution of compound 19 (0.46 g, 0.80 mmol) in anhydrous methanol (10 mL) was hydrogenated in the presence of 30% Pd─C (0.14 g) and p-toluenesulfonic acid monohydrate (0.15 g, 0.80 mmol) under a hydrogen atmosphere (60 psi) at room temperature for 24 h. The mixture was filtered through Celite, and the filtrate was concentrated to afford 20 as a tosylate salt, which was directly used for the next step due to a stability issue of the free amine. HRMS (ESI) m/z calcd for C29H36N5O2 [M + H]+: 486.2869, found: 486.2881; calcd for C29H35N5NaO2 [M + Na]+: 508.2688, found: 508.2701.
Benzyl((S)-3-(3-(3-(2-((((9H-Fluoren-9-yl)methoxy)carbonyl)-amino)ethyl)-5-isopropyl-4H-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]-octan-8-yl)-1-phenylpropyl)carbamate (21)
To a mixture of the 20· tosylate salt from the last step and compound 9 (0.25 g, 0.88 mmol) in dichloromethane (10 mL) were added acetic acid (0.28 mL) and sodium triacetoxyborohydride (0.19 g, 0.88 mmol) in sequence. After being stirred at room temperature overnight, the reaction was quenched by addition of saturated sodium bicarbonate solution, and the mixture was extracted with dichloromethane (30 mL × 3). The combined organic layers were washed with brine, dried over Na2SO4, and concentrated. The residue was purified by column chromatography to supply 21 as a white solid (0.32 g, two-step yield: 53%). 1H NMR (400 MHz, DMSO-d6): δ 7.88 (d, J = 7.48 Hz, 2H), 7.86–7.81 (m, 1H), 7.65 (d, J = 7.36 Hz, 2H), 7.46–7.39 (m, 3H), 7.32–7.19 (m, 12H), 5.06–4.94 (m, 2H), 4.80–4.74 (m, 1H), 4.30 (d, J = 6.96 Hz, 2H), 4.23–4.17 (m, 1H), 3.42–3.35 (m, 2H), 3.30–3.28 (m, 1H), 3.25–3.22 (m, 2H), 3.20–3.17 (m, 1H), 2.88 (t, J = 7.12 Hz, 2H), 2.34–2.89 (m, 2H), 2.05–1.99 (m, 2H), 1.92–1.75 (m, 4H), 1.67–1.65 (m, 4H), 1.27–1.25 (m, 6H). 13C NMR (100 MHz, DMSO-d6): δ 158.5, 156.1, 155.5, 150.9, 143.84, 143.83, 140.7, 128.25, 128.20, 128.15, 127.7, 127.61, 127.55, 127.0, 126.6, 126.3, 125.1, 120.1, 79.3, 78.9, 78.6, 66.4, 65.4, 65.1, 58.7, 58.3, 53.0, 46.8, 46.7, 36.5, 25.2, 22.0. HRMS (ESI) m/z calcd for C46H53N6O4 [M + H]+: 753.4128, found: 753.4135; calcd for C46H52N6NaO4 [M + Na]+: 775.3948, found: 775.3945.
(9H-Fluoren-9-yl)methyl(2-(4-(8-((S)-3-amino-3-phenylpropyl)-8-azabicyclo[3.2.1]octan-3-yl)-5-isopropyl-4H-1,2,4-triazol-3-yl)-ethyl)carbamate (22)
To a solution of compound 21 (0.10 g, 0.13 mmol) in anhydrous methanol (8 mL) was added 30% Pd/C (0.03 g). The mixture was hydrogenated under a hydrogen atmosphere (60 psi) at room temperature for 20 h. The mixture was filtered through Celite and washed with methanol. The combined filtrate was concentrated under reduced pressure to provide 22 as a white solid (55 mg, 67%). Compound 22 could be used for next-step reaction without any further purification. 1H NMR (400 MHz, DMSO-d6): δ 7.90–7.84 (m, 3H), 7.59 (d, J = 7.36 Hz, 1H), 7.44–7.26 (m, 8H), 7.20 (t, J = 7.36 Hz, 1H), 6.29 (s, 1H), 4.31–4.13 (m, 1H), 3.95 (q, J = 7.52 Hz, 1H), 3.14–3.06 (m, 2H), 2.97–2.81 (m, 3H), 2.47–2.46 (m, 1H), 2.38–2.29 (m, 2H), 2.13–2.00 (m, 2H), 1.94–1.89 (m, 2H), 1.83 (brs, 2H), 1.76–1.64 (m, 6H), 1.47 (d, J = 7.40 Hz, 1H), 1.26 (d, J = 6.72 Hz, 6H). HRMS (ESI) m/z calcd for C38H47N6O2 [M + H]+: 619.3760, found: 619.3725.
(9H-Fluoren-9-yl)methyl (2-(4-(8-((S)-3-(4,4-difluorocyclohexane-1-carboxamido)-3-phenylpropyl)-8-azabicyclo[3.2.1]octan-3-yl)-5-isopropyl-4H-1,2,4-triazol-3-yl)ethyl)carbamate (23)
A mixture of 4,4-difluorocyclohexanecarboxylic acid (27 mg, 0.16 mmol), EDCI (31 mg, 0.16 mmol), HOBt (22 mg, 0.16 mmol), trimethylamine (33 mg, 0.32 mmol), and 4 Å molecular sieves in anhydrous dichloromethane (3 mL) was stirred at 0 °C for 0.5 h. Then, compound 22 (50 mg, 0.08 mmol) in dichloromethane (1 mL) was slowly added to the mixture. The reaction temperature was warmed to room temperature and stirred overnight. The mixture was filtered, and the filtrate was concentrated under reduced pressure to get crude residue. The residue was further purified by column chromatography to get compound 23 as a white foam solid (33 mg, 54%). 1H NMR (400 MHz, CD3OD): δ 7.68 (d, J = 7.60 Hz, 2H), 7.49 (d, J = 7.48 Hz, 2H), 7.29–7.11 (m, 9H), 4.97 (t, J = 7.48 Hz, 1H), 4.79 (d, J = 3.08 Hz, 1H), 4.36–4.27 (m, 1H), 4.19 (d, J = 6.96 Hz, 1H), 4.05 (t, J = 6.84 Hz, 1H), 3.54–3.53 (m, 1H), 3.41–3.37 (m, 1H), 3.26–3.23 (m, 2H), 2.95 (t, J = 7.04 Hz, 2H), 2.36–2.31 (m, 1H), 2.29–2.19 (m, 1H), 2.15–2.07 (m, 2H), 2.00–1.83 (m, 6H), 1.75–1.59 (m, 9H), 1.25 (d, J = 6.84 Hz, 6H), 1.19 (s, 2H), 0.82–0.75 (m, 1H). 13C NMR (100 MHz, CD3OD): δ 176.7, 145.3, 144.0, 142.6, 129.7, 128.8, 128.3, 128.2, 127.73, 127.66, 126.2, 121.0, 78.3, 71.6, 67.9, 61.1, 60.8, 60.4, 56.3, 52.7, 52.6, 44.3, 43.7, 43.6, 40.5, 40.4, 38.4, 37.7, 37.6, 37.54, 37.48, 36.4, 36.3, 36.2, 34.14, 34.09, 34.07, 33.90, 33.89, 33.86, 33.85, 33.81, 33.7, 33.61, 33.58, 27.3, 27.2, 27.04, 26.98, 26.9, 26.8, 22.3, 22.23, 22.20. 19F NMR (376 MHz, CD3OD): δ −103.31, −102.67, −93.67, −93.04. HRMS (ESI) m/z calcd for C45H55F2N6O3 [M + H]+: 765.4304, found: 765.4280; calcd for C45H54F2N6NaO3 [M + Na]+: 787.4123, found: 787.4097.
N-((S)-3-(3-(3-(2-Aminoethyl)-5-isopropyl-4H-1,2,4-triazol-4-yl)-8-azabicyclo[3.2.1]octan-8-yl)-1-phenylpropyl)-4,4-difluorocyclohexane-1-carboxamide (24)
A mixture of compound 23 (0.10 g, 0.13 mmol) in 20% piperidine/DMF (4 mL) was stirred at room temperature for 1 h. The mixture was then concentrated under reduced pressure to get a crude residue. The residue was further purified by column chromatography to afford 24 as a white solid (30 mg, 42%). 1H NMR (400 MHz, CDCl3): δ 7.38–7.34 (m, 2H), 7.29–7.27 (m, 3H), 6.38 (d, J = 7.36 Hz, 1H), 5.16 (q, J = 7.32 Hz, 1H), 4.35–4.26 (m, 1H), 3.49 (s, 1H), 3.40–3.35 (m, 2H), 3.23 (t, J = 6.24 Hz, 2H), 3.05–2.93 (m, 3H), 2.43 (t, J = 6.72 Hz, 2H), 2.29–2.12 (m, 5H), 2.07–1.93 (m, 4H), 1.90–1.77 (m, 6H), 1.66–1.61 (m, 4H), 1.39 (dd, J1 = 6.80 Hz, J2 = 1.20 Hz, 6H), 1.26 (s, 1H). 13C NMR (100 MHz, CDCl3): δ 173.2, 159.1, 141.9, 128.9, 127.6, 126.5, 77.2, 59.1, 58.3, 52.0, 50.9, 48.0, 47.2, 42.9, 39.9, 36.0, 35.8, 34.9, 33.08, 33.05, 32.9, 32.83, 32.81, 32.79, 32.60, 32.56, 30.5, 26.8, 26.7, 26.1, 26.0, 25.9, 21.8, 1.0. 19F NMR (376 MHz, CDCl3): δ −92.83, −93.46, −100.30, −100.93. HRMS (ESI) m/z calcd for C30H45F2N6O [M + H]+: 543.3623, found: 543.3630; calcd for C30H44F2N6NaO [M + Na]+: 565.3442, found: 565.3438.
Preparation of the Bivalent Ligands VZMC013, VZMC017, and VZMC019
Benzyl-(7-aminoheptyl)carbamate (26a)
To a stirring solution of 1,7-diaminoheptane 25a (0.90 g, 6.91 mmol) in MeOH (80 mL) at 0 °C was dropwise added the solution of benzyl chloroformate (1.18 g, 6.91 mmol) in dichloromethane (80 mL) within 5 h while keeping the temperature at 0 °C. The reaction mixture was stirred at room temperature overnight, and then the excess solvent was removed under reduced pressure. Dichloromethane (100 mL) and water (100 mL) were added, and the mixture was adjusted to pH 2 with 6 N HCl. The layers were separated. The aqueous layer was washed with dichloromethane (50 mL × 2), then adjusted to pH 12 using 10 N NaOH, and extracted with dichloromethane (50 mL × 3). The combined organic layers were dried over Na2SO4, concentrated, and recrystallized with methanol to give 26a as a white solid (0.55 g, 30%). 1H NMR (400 MHz, DMSO-d6): δ 7.38–7.28 (m, 5H), 7.20 (brs, 1H, exchangeable), 5.00 (s, 2H), 2.98 (q, J = 2.4 Hz, 2H), 2.53–2.51 (m, 2H), 2.04 (brs, 2H, exchangeable), 1.41–1.38 (m, 2H), 1.34–1.31 (m, 2H), 1.24 (s, 6H). 13C NMR (100 MHz, DMSO-d6): δ 156.0, 137.3, 128.2, 127.6, 127.6, 65.0, 41.4, 33.0, 29.3, 28.6, 26.3, 26.2. C15H24N2O2 (264.1838).
Benzyl (9-Aminononyl)carbamate (26b)
This compound was prepared in a similar way as 26a, using 1,9-diaminononane as the starting material. White solid. Yield: 49%. 1H NMR (400 MHz, CDCl3): δ 7.36–7.29 (m, 5H, Ph-H), 5.09 (s, 2H, CH2), 4.74 (brs, 1H, exchangeable), 3.18 (q, J = 6.76 Hz, 2H), 2.68 (t, J = 6.96 Hz, 2H), 1.50–1.40 (m, 5H), 1.28 (brs, 11H). 13C NMR (100 MHz, CDCl3): δ 157.7, 138.9, 130.0, 129.4, 129.3, 66.7, 42.5, 41.7, 33.7, 31.0, 30.6, 30.5, 30.3, 28.0, 27.8. HRMS (ESI) m/z calcd for C17H29N2O2 [M + H]+: 293.2229, found: 293.2228; calcd for C17H28N2NaO2 [M + Na]+: 315.2048, found: 315.2038.
Benzyl (5-Aminopentyl)carbamate (26c)
This compound was prepared in a similar way as 26a, using cadaverine as the starting material. White solid. Yield: 31%. 1H NMR (400 MHz, CDCl3): δ 7.36–7.28 (m, 5H, Ph-H), 5.09 (s, 2H), 4.90 (brs, 1H, exchangeable), 3.19 (q, J = 6.44 Hz, 2H), 2.69 (t, J = 6.80 Hz, 2H), 2.21 (brs, 2H, exchangeable), 1.55–1.43 (m, 4H), 1.38–1.31 (m, 2H). HRMS (ESI) m/z calcd for C13H21N2O2 [M + H]+: 237.1603, found: 237.1609.
3,13-Dioxo-1-phenyl-2,15-dioxa-4,12-diazaheptadecan-17-oic Acid (27a)
To a stirring solution of benzyl-(7-aminoheptyl)-carbamate 26a (2.76 g, 10.46 mmol) in THF (20 mL) was added diglycolic anhydride (1.27 g, 10.98 mmol) in three portions. The resulting mixture was stirred at room temperature for 21 h. The excess solvent was removed under reduced pressure, and the residue was recrystallized by ethyl acetate/hexane to provide 27a as a white solid (3.10 g, 78%). 1H NMR (400 MHz, DMSO-d6): δ 12.80 (s, 1H, exchangeable), 7.80 (s, 1H, exchangeable), 7.38–7.28 (m, 5H), 7.19 (s, 1H, exchangeable), 5.00 (s, 2H), 4.10 (s, 2H), 3.94 (s, 2H), 3.08 (q, J = 6.80 Hz, 2H), 2.98 (q, J = 6.80 Hz, 2H), 1.42–1.37 (m, 4H), 1.24 (s, 6H). HRMS (ESI) m/z calcd for C19H28N2NaO6 [M + Na]+: 403.1845, found: 403.2027.
3,15-Dioxo-1-phenyl-2,17-dioxa-4,14-diazanonadecan-19-oic Acid (27b)
This compound was prepared in a similar way as 27a. White solid. Yield: 86%. 1H NMR (400 MHz, DMSO-d6): δ 7.85 (t, J = 5.60 Hz, 1H, exchangeable), 7.39–7.29 (m, 5H), 7.22 (t, J = 5.52 Hz, 1H, exchangeable), 5.00 (s, 2H), 4.10 (s, 2H), 3.94 (s, 2H), 3.09 (q, J = 6.72 Hz, 2H), 2.98 (q, J = 6.72 Hz, 2H), 1.42–1.39 (m, 4H), 1.24 (brs, 10H). 13C NMR (100 MHz, DMSO-d6): δ 171.5, 168.5, 156.0, 137.3, 128.3 (Ph─C × 2), 127.7, 127.7 (Ph─C × 2), 70.2, 67.9, 65.0, 40.2, 38.1, 29.4, 29.1, 28.9, 28.7 (CH2 × 2), 26.3, 26.2. HRMS (ESI) m/z calcd for C21H31N2O6 [M − H]−: 407.2182, found: 407.2179.
3,11-Dioxo-1-phenyl-2,13-dioxa-4,10-diazapentadecan-15-oic Acid (27c)
This compound was prepared in a similar way as 27a. White solid. Yield: 70%. 1H NMR (400 MHz, DMSO-d6): δ 7.87 (t, J = 5.28 Hz, 1H, exchangeable), 7.39–7.29 (m, 5H), 7.22 (t, J = 5.52 Hz, 1H, exchangeable), 5.01 (s, 2H), 4.10 (s, 2H), 3.94 (s, 2H), 3.08 (q, J = 6.72 Hz, 2H), 2.98 (q, J = 6.72 Hz, 2H), 1.45–1.37 (m, 4H), 1.27–1.20 (m, 2H). HRMS (ESI) m/z calcd for C17H23N2O6 [M − H]−: 351.1556, found: 351.1551.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(3′,13′-dioxo-1′-phenyl-2′,15′-dioxa-4′,12′-diazaheptadecanamido)-morphinan (28a)
The title compound was prepared following the general amide coupling procedure by reacting acid 27a with 6β-naltrexamine hydrochloride (prepared according to the method reported by our group)61 in DMF overnight. The crude product was further purified by column chromatography to furnish 28a as a white solid. Yield: 36%. 1H NMR (400 MHz, CDCl3): δ 7.48 (s, 1H), 7.30–7.35 (m, 5H), 6.82 (s, 1H), 6.73 (d, J = 8.04 Hz, 1H), 6.56 (d, J = 8.08 Hz, 1H), 5.30 (s, 1H), 5.09 (s, 2H), 4.84 (s, 1H), 4.43 (s, 1H), 4.06–4.05 (m, 5H), 3.35–3.27 (m, 2H), 3.21–3.16 (m, 2H), 3.11–3.06 (m, 1H), 3.01 (s, 1H), 2.66 (m, 2H), 2.37 (m, 2H), 2.21 (m, 2H), 1.68–1.63 (m, 3H), 1.53–1.48 (m, 7H), 1.33 (s, 7H), 0.82 (m, 1H), 0.55–0.54 (m, 2H), 0.15–0.14 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 168.4, 168.2, 156.0, 142.1, 140.4, 137.3, 128.3, 127.7, 127.6, 118.4, 117.1, 90.5, 70.4, 70.4, 69.6, 65.0, 61.8, 58.4, 50.6, 47.0, 38.2, 30.3, 30.0, 29.3, 29.2, 28.4, 26.4, 26.2, 24.5, 22.2, 9.2, 3.7, 3.5. HRMS (ESI) m/z calcd for C39H53N4O8 [M + H]+: 705.3863, found: 705.3982.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(3′,15′dioxo-1′-phenyl-2′,17′-dioxa-4′,14′-diazanonadecanamido)-morphinan (28b)
This compound was prepared in a similar way as 28a by coupling acid 27b with 6β-naltrexamine hydrochloride. White solid. Yield: 61%. 1H NMR (400 MHz, CD3OD): δ 7.34–7.27 (m, 5H), 6.63 (d, J = 8.12 Hz, 1H), 6.57 (d, J = 8.16 Hz, 1H), 5.06 (s, 2H), 4.53 (d, J = 7.60 Hz, 1H), 4.06 (s, 2H), 4.05 (s, 2H), 3.80–3.74 (m, 1H), 3.26 (t, J = 7.20 Hz, 2H), 3.14–3.06 (m, 4H), 2.70–2.61 (m, 2H), 2.46–2.36 (m, 2H), 2.29–2.22 (m, 1H), 2.18–2.12 (m, 1H), 1.95–1.86 (m, 1H), 1.61–1.43 (m, 8H), 1.32 (brs, 10H), 0.93–0.84 (m, 1H), 0.58–0.49 (m, 2H), 0.20–0.12 (m, 2H). 13C NMR (100 MHz, CD3OD): δ 171.5, 171.4, 158.9, 143.7, 141.9, 138.6, 132.5, 129.5, 128.9, 128.8, 125.4, 120.1, 118.6, 92.9, 71.7, 71.6, 71.5, 67.3, 63.7, 60.3, 52.5, 48.9, 45.3, 41.8, 40.1, 31.9, 31.2, 30.9, 30.6, 30.5, 30.4, 28.0, 27.8, 25.5, 23.5, 10.3, 4.5, 4.2. HRMS (ESI) m/z calcd for C41H57N4O8 [M + H]+: 733.4176, found: 733.4187; calcd for C41H56N4NaO8 [M + Na]+: 755.3996, found: 755.3992.
17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-(3′,11′-dioxo-1′-phenyl-2′,13′-dioxa-4′,10′-diazapentadecanamido)-morphinan (28c)
This compound was prepared in a similar way as 28a by coupling acid 27c with 6β-naltrexamine hydrochloride. White solid. Yield: 60%. 1H NMR (400 MHz, CD3OD): δ 7.37–7.27 (m, 5H), 6.65 (d, J = 8.12 Hz, 1H), 6.59 (d, J = 8.16 Hz, 1H), 5.09 (s, 2H), 4.57 (d, J = 7.56 Hz, 1H), 4.11–4.05 (m, 4H), 3.82–3.76 (m, 1H), 3.32–3.28 (m, 2H), 3.17–3.12 (m, 3H), 2.99 (q, J = 7.24 Hz, 1H), 2.76–2.70 (m, 2H), 2.53–2.45 (m, 2H), 2.36–2.23 (m, 2H), 2.00–1.90 (m, 1H), 1.65–1.32 (m, 10H), 0.98–0.89 (m, 1H), 0.61–0.58 (m, 2H), 0.23–0.22 (m, 2H). HRMS (ESI) m/z calcd for C37H49N4O8 [M + H]+: 677.3550, found: 677.3562; calcd for C37H48N4NaO8 [M + Na]+: 699.3370, found: 699.3369.
Compounds 29a–c and 30a–c were prepared as previously reported.32,34
Bivalent Ligand VZMC013
The target compound was prepared following the general amide coupling procedure reported by our group72 by reacting the 2′-aminoethyl maraviroc precursor 24 with the acid 30a in DMF overnight. The crude product was further purified by column chromatography to afford VZMC013 as a white solid. Yield: 20%. Compound VZMC013 was converted to its hydrochloride salt for biological assays. 1H NMR (400 MHz, CDCl3): δ 8.18–8.16 (m, 1H), 7.56–7.52 (m, 1H), 7.34–7.27 (m, 4H), 7.04 (brs, 1H), 6.74–6.71 (m, 1H), 6.54 (d, J = 8.16 Hz, 1H), 6.49–6.47 (m, 1H), 5.12–5.07 (m, 1H), 4.43 (d, J = 6.08 Hz, 1H), 4.37–4.28 (m, 1H), 4.05 (s, 1H), 4.02 (s, 2H), 3.98 (s, 3H), 3.74–3.64 (m, 3H), 3.40 (brs, 2H), 3.34–3.25 (m, 5H), 3.11–3.09 (m. 1H), 3.04–3.00 (m, 4H), 2.65–2.60 (m, 2H), 2.43–2.37 (m, 4H), 2.25–2.11 (m, 8H), 2.08–2.05 (m, 2H), 2.02–1.97 (m, 2H), 1.92–1.85 (m, 4H), 1.84–1.74 (m, 5H), 1.67–1.65 (m, 6H), 1.62–1.60 (m, 1H), 1.57–1.52 (m, 5H), 1.48–1.44 (m, 2H), 1.38 (d, J = 8.84 Hz, 6H), 1.33 (brs, 4H), 1.26 (s, 2H), 0.90–0.79 (m, 2H), 0.54 (d, J = 8.12 Hz, 2H), 0.13 (d, J = 4.56 Hz, 2H). 13C NMR (100 MHz, CDCl3): δ 173.6, 169.0, 168.9, 168.8, 168.6, 159.4, 152.8, 143.5, 142.1, 140.0, 131.0, 129.0, 127.7, 126.7, 119.3, 118.2, 92.1, 71.3, 71.2, 71.1, 70.9, 70.2, 62.6, 59.5, 58.9, 51.9, 50.3, 48.7, 47.7, 47.5, 44.21, 44.19, 44.17, 43.0, 39.2, 39.1, 36.5, 33.0, 29.9, 29.4, 29.3, 28.7, 26.7, 26.6, 26.2, 26.15, 26.07, 26.0, 23.7, 22.9, 21.85, 21.83, 9.6, 4.2, 4.1, 4.0. 19F NMR (376 MHz, CDCl3): δ −92.75, −92.38, −100.27, −100.89. IR (diamond, cm−1): 3246.08, 2536.28, 2159.05, 2027.61, 1976.79, 1648.71, 1545.36, 1450.68, 1373.09, 1325.41, 1232.43, 1126.20, 1033.83, 962.12, 936.21, 916.27, 877.82, 856.61, 747.87, 702.53. HRMS (ESI) m/z calcd for C65H93F2N10O10 [M + H]+: 1211.7044, found: 1211.7048; calcd for C65H94F2N10O10 [M + 2H]2+: 606.3561, found: 606.3527. HPLC purity: 99.43%. Rt: 6.768 min.
Bivalent Ligand VZMC017
This target compound was prepared in a similar way as VZMC013 by coupling the 2′-aminoethyl maraviroc precursor 24 with 30b. White solid. Yield: 45%. Compound VZMC017 was converted to its hydrochloride salt for biological assays. 1H NMR (400 MHz, CDCl3): δ 8.21–8.15 (m, 1H), 7.57–7.55 (m, 1H), 7.37–7.27 (m, 4H), 7.17–7.12 (m, 1H), 6.94 (brs, 1H), 6.75–6.71 (m, 1H), 6.56–6.53 (m, 1H), 6.44–6.40 (m, 1H), 5.24, (brs, 1H), 5.14–5.07 (m, 2H), 4.41 (dd, J1 = 5.52 Hz, J2 = 2.00 Hz, 1H), 4.36–4.27 (m, 1H), 4.07–4.00 (m, 4H), 3.98 (s, 3H), 3.80–3.70 (m, 2H), 3.40 (brs, 2H), 3.36–3.29 (m, 3H), 3.28–3.21 (m, 3H), 3.19–3.15 (m, 2H), 3.11–3.10 (m. 1H), 3.06–2.98 (m, 4H), 2.68–2.60 (m, 2H), 2.41–2.38 (m, 3H), 2.21–2.15 (m, 5H), 2.09–1.98 (m, 5H), 1.92–1.76 (m, 8H), 1.67–1.64 (m, 5H), 1.56–1.47 (m, 8H), 1.38–1.36 (m, 6H), 1.29 (brs, 9H), 0.85–0.79 (m, 1H), 0.56–0.52 (m, 2H), 0.15–0.11 (m, 2H). 13C NMR (100 MHz, CDCl3): δ 173.5, 168.9, 168.63, 168.57, 168.4, 159.3, 159.1, 152.6, 143.5, 142.0, 139.94, 139.90, 130.7, 128.84, 128.82, 127.58, 127.55, 126.5, 124.32, 124.30, 119.2, 118.1, 118.0, 91.8, 71.0, 70.9, 70.7, 70.1, 62.4, 59.4, 59.3, 59.1, 58.7, 58.3, 57.3, 51.8, 49.80, 49.76, 48.6, 48.3, 47.5, 47.3, 45.2, 43.9, 42.9, 39.6, 39.2, 39.1, 36.3, 36.2, 35.4, 35.0, 33.13, 33.09, 32.9, 32.8, 32.64, 32.60, 31.9, 29.4, 29.1, 28.9, 27.3, 26.70, 26.65, 26.6, 26.5, 26.1, 26.00, 25.96, 25.9, 23.4, 22.6, 21.7, 15.5, 9.5, 4.0, 3.8. 19F NMR (376 MHz, CDCl3): δ −92.60, −93.23, −100.30, −100.93. HRMS (ESI) m/z calcd for C67H97F2N10O10 [M + H]+: 1239.7357, found: 1239.7338; calcd for C67H96F2N10NaO10 [M + Na]+: 1261.7177, found: 1261.7164; calcd for C67H98F2N10O10 [M + 2H]2+: 620.3718, found: 620.3594; calcd for C67H97F2N10NaO10 [M + H + Na]2+: 631.3627, found: 631.3517. HPLC purity: 99.96%. Rt: 7.300 min.
Bivalent Ligand VZMC019
This target compound was prepared in a similar way as VZMC013 by coupling the 2 ′-aminoethyl maraviroc precursor 24 with 30c.White solid. Yield: 55%. Compound VZMC019 was converted to its hydrochloride salt for biological assays. Hydrochloride salt. 1H NMR (400 MHz, DMSO-d6): δ 11.27 (s, 1 H), 10.10 (s, 1H), 9.34 (s, 1H), 8.84 (s, 1H), 8.54 (d, J = 8.36 Hz, 1H), 8.28 (d, J = 8.40 Hz, 1H), 8.14 (t, J = 5.64 Hz, 1H), 8.07 (t, J = 5.80 Hz, 1H), 7.62 (d, J = 8.56 Hz, 2H), 7.33 (d, J = 8.52 Hz, 2H), 6.72 (d, J = 8.12 Hz, 1H), 6.65 (d, J = 8.24 Hz, 1H), 6.18 (s, 1H), 4.88–4.82 (m, 1H), 4.76 (d, J = 7.72 Hz, 1H), 4.50–4.40 (m, 1H), 4.23–4.21 (m, 1H), 4.16 (s, 2H), 4.05 (s, 2H), 3.97 (d, J = 3.16 Hz, 3H), 3.85 (d, J = 5.60 Hz, 1H), 3.73 (brs, 1H), 3.57–3.48 (m, 4H), 3.18–3.08 (m, 6H), 3.05–3.03 (m, 2H), 2.97–2.92 (m, 3H), 2.89–2.83 (m, 2H), 2.75–2.69 (m, 5H), 2.47–2.42 (m, 3H), 2.40–2.34 (m, 2H), 2.28–2.21 (m, 3H), 2.17–2.09 (m, 4H), 2.05–2.02 (m, 1H), 1.89–1.71 (m, 5H), 1.67–1.55 (m, 2H), 1.53–1.43 (m, 6H), 1.29–1.27 (m, 8H), 1.08–1.04 (m, 1H), 0.70–0.65 (m, 1H), 0.62–0.57 (m, 1H), 0.53–0.48 (m, 1H), 0.44–0.38 (m, 1H). 13C NMR (100 MHz, DMSO-d6): δ 173.5, 168.8, 168.4, 168.3, 167.8, 159.7, 151.5, 142.1, 141.3, 138.0, 137.1, 129.6, 126.6, 120.6, 119.8, 119.2, 117.9, 89.7, 70.7, 70.6, 70.32, 70.28, 69.7, 61.5, 61.2, 60.3, 56.6, 50.3, 49.8, 48.6, 47.0, 46.7, 46.5, 45.6, 40.8, 40.1, 38.14, 38.09, 32.9, 32.2, 32.0, 30.6, 29.4, 28.9, 28.8, 25.59, 25.50, 25.47, 24.33, 24.31, 23.8, 23.7, 23.49, 23.47, 23.0, 21.5, 11.7, 11.5, 5.7, 5.1, 2.6. 19F NMR (376 MHz, DMSO-d6): δ −89.77, −90.40, −98.68, −99.30. 19F NMR (376 MHz, CDCl3): δ −92.66, −93.29, −100.32, −100.95. HRMS (ESI) m/z calcd for C63H89F2N10O10 [M + H]+: 1183.6731, found: 1183.6692; calcd for C63H88F2N10NaO10 [M + Na]+: 1205.6551, found: 1205.6507; calcd for C63H90F2N10O10 [M + 2H]2+: 592.3405, found: 592.3318; calcd for C63H89F2N10NaO10 [M + H + Na]2+: 603.3314, found: 603.3225. HPLC purity: 99.96%. Rt: 6.867 min.
Preparation of the Monovalent Ligands VZMC014, VZMC018, and VZMC020
2-(2-(Methylamino)-2-oxoethoxy)-acetic Acid (31)
To a 2 M solution of methanamine in THF (27.5 mL, 55 mmol) was added diglycolic anhydride (5.80 g, 50 mmol) in portions. After being stirred for 15 h at room temperature, the reaction mixture was concentrated under reduced pressure. The residue was further recrystallized with methanol to afford 31 as a faint yellow solid (6.11 g, 83%). 1H NMR (400 MHz, DMSO-d6): δ 12.80 (brs, 1H, exchangeable), 7.76 (s, 1H, exchangeable), 4.09 (s, 2H), 3.94 (s, 2H), 2.62 (d, J = 4.80 Hz, 3H). 13C NMR (100 MHz, DMSO-d6): δ 171.3, 169.1, 70.1, 67.8, 25.1. C5H9NO4 (147.0532).
Benzyl (7-(2-(2-(Methylamino)-2-oxoethoxy)acetamido)heptyl)-carbamate (32a)
The title compound was prepared following the general amide coupling procedure by reacting acid 31 with the Cbz-protected amine 26a. White solid. Yield: 47%. 1H NMR (400 MHz, DMSO-d6): δ 7.99–7.96 (m, 2H, exchangeable), 7.38–7.28 (m, 5H), 7.19 (brs, 1H, exchangeable), 5.00 (s, 2H), 3.91 (s, 2H), 3.90 (s, 2H), 3.10 (q, J = 6.80 Hz, 2H), 2.98 (q, J = 6.80 Hz, 2H), 2.65 (d, J = 8.80 Hz, 3H), 1.46–1.38 (m, 4H), 1.25 (brs, 6H). 13C NMR (100 MHz, DMSO-d6): δ 168.8, 168.2, 156.0, 137.3, 128.2, 127.6, 70.2, 65.0, 38.1, 29.3, 29.1, 28.4, 26.3, 26.1, 25.0. C20H31N3O5 (393.2264).
Benzyl (9-(2-(2-(Methylamino)-2-oxoethoxy)acetamido)nonyl)-carbamate (32b)
This title compound was prepared in a similar way as 32a, by reacting acid 31 with amine 26b. White solid. Yield: 36%. 1H NMR (400 MHz, DMSO-d6): δ 8.02–7.97 (m, 2H, exchangeable), 7.39–7.29 (m, 5H, Ph-H), 7.21 (t, J = 5.44 Hz, 1H, exchangeable), 5.01 (s, 2H), 3.92 (s, 2H), 3.91 (s, 2H), 3.11 (q, J = 6.76 Hz, 2H), 2.98 (q, J = 6.68 Hz, 2H), 2.66 (d, J = 4.72 Hz, 3H), 1.45–1.38 (m, 4H), 1.25 (brs, 10H). HRMS (ESI) m/z calcd for C22H35N3NaO5 [M + Na]+: 444.2474, found: 444.2461.
Benzyl (5-(2-(2-(Methylamino)-2-oxoethoxy)acetamido)pentyl)-carbamate (32c)
This title compound was prepared in a similar way as 32a, by reacting acid 31 with amine 26c. White solid. Yield: 27%. 1H NMR (400 MHz, DMSO-d6): δ 8.02–7.97 (m, 2H, exchangeable), 7.39–7.29 (m, 5H, Ph-H), 7.23 (t, J = 5.44 Hz, 1H, exchangeable), 5.01 (s, 2H), 3.92 (s, 2H), 3.91 (s, 2H), 3.11 (q, J = 6.64 Hz, 2H), 2.99 (q, J = 6.68 Hz, 2H), 2.66 (d, J = 4.68 Hz, 3H), 1.47–1.38 (m, 4H), 1.29–1.21 (m, 2H). C18H27N3O5 (365.1951).
Compounds 33a–c and 34a–c were prepared as previously reported.32,34
Monovalent Ligand VZMC014
The target compound was prepared following the general amide coupling procedure by reacting the 2′-aminoethyl maraviroc precursor 24 with 34a. White solid. Yield: 46%. Hydrochloride salt. 1H NMR (400 MHz, CD3OD): δ 7.88 (brs, 1H), 7.57 (brs, 1H), 7.25–7.21 (m, 2H), 7.15 (brs, 1H), 4.99–4.95 (m, 1H), 4.43–4.30 (m, 1H), 4.24–4.13 (m, 1H), 3.92 (s, 3H), 3.90–3.88 (m, 2H), 3.57–3.48 (m, 2H), 3.38–3.29 (m, 2H), 3.16–3.08 (m, 7H), 3.03–2.98 (m, 2H), 2.89–2.84 (m, 2H), 2.76 (s, 1H), 2.69 (s, 3H), 2.41–2.32 (m, 3H), 2.26 (s, 4H), 2.19–2.02 (m, 2H), 1.96–1.87 (m, 4H), 1.76–1.67 (m, 4H), 1.64–1.61 (m, 2H), 1.43 (brs, 3H), 1.25 (s, 7H), 1.12–1.19 (m, 1H), 1.02 (t, J = 6.68 Hz, 4H). 13C NMR (100 MHz, CD3OD): δ 176.6, 172.1, 171.9, 171.5, 171.4, 153.4, 144.2, 129.6, 128.3, 127.7, 112.6, 71.55, 71.52, 71.48, 70.1, 66.9, 60.9, 60.4, 59.8, 58.1, 52.7, 50.1, 45.0, 43.7, 42.2, 40.1, 39.7, 38.2, 37.72, 37.69, 36.4, 33.9, 33.8, 30.40, 30.38, 30.0, 28.5, 27.9, 27.3, 27.2, 27.15, 27.06, 27.0, 25.9, 22.3, 15.4, 14.0. 19F NMR (376 MHz, CD3OD): δ −93.06, −93.68, −102.67, −103.26. HRMS (ESI) m/z calcd for C46H72F2N9O7 [M + H]+: 900.5523, found: 900.2709. HPLC purity: 99.72%. Rt: 6.583 min.
Monovalent Ligand VZMC018
This target compound was prepared in a similar way as VZMC014 by coupling the 2′-aminoethyl maraviroc precursor 24 with 34b. White solid. Yield: 35%. Hydrochloride salt. 1H NMR (400 MHz, CDCl3): δ 8.19 (t, J = 5.76 Hz, 1H), 7.38–7.35 (m, 2H), 7.30–7.28 (m, 3H), 7.11 (t, J = 5.72 Hz, 1H), 6.68–6.50 (m, 2H), 6.31–6.29 (m, 1H), 5.17–5.11 (m, 1H), 4.36–4.26 (m, 1H), 4.05 (s, 2H), 4.03 (s, 2H), 3.99 (s, 3H), 3.77 (q, J = 5.88 Hz, 2H), 3.39–3.34 (m, 4H), 3.29 (q, J = 6.80 Hz, 3H), 3.03–2.98 (m, 3H), 2.89–2.86 (m, 3H), 2.42–2.38 (m, 2H), 2.27 (s, 1H), 2.23–2.11 (m, 6H), 2.07–1.93 (m, 6H), 1.93–1.83 (m, 4H), 1.38 (d, J = 6.80 Hz, 6H), 1.29–1.21 (m, 16H). 13C NMR (100 MHz, CDCl3): δ 173.4, 173.1, 169.2, 168.7, 168.6, 168.5, 168.4, 128.9, 127.7, 126.5, 77.2, 71.3, 71.24, 71.22, 70.8, 70.7, 58.4, 51.8, 47.4, 42.9, 39.20, 39.18, 39.1, 36.3, 36.1, 32.8, 29.71, 29.68, 29.50, 29.45, 29.1, 28.94, 28.92, 26.7, 26.64, 26.62, 26.58, 26.02, 25.95, 25.9, 25.7, 21.7. 19F NMR (376 MHz, CDCl3): δ −92.70, −93.33, −100.45, −101.05. HRMS (ESI) m/z calcd for C48H76F2N9O7 [M + H]+: 928.5836, found: 928.5843. HPLC purity: 95.89%. Rt: 8.062 min.
Monovalent Ligand VZMC020
This target compound was prepared in a similar way as VZMC014 by coupling the 2′-aminoethyl maraviroc precursor 24 with 34c. White solid. Yield: 21%. Hydrochloride salt. 1H NMR (400 MHz, DMSO-d6): δ 11.45 (brs, 1H), 10.11 (s, 1H), 8.56–8.54 (m, 1H), 8.14 (s, 1H), 8.06–8.01 (m, 2H), 7.62 (d, J = 8.60 Hz, 2H), 7.35–7.32 (m, 3H), 7.22 (brs, 2H), 7.10 (brs, 2H), 4.88–4.82 (m, 1H), 4.49 (brs, 1H), 4.24–4.23 (m, 1H), 4.15 (s, 2H), 4.05 (s, 2H), 3.92 (s, 3H), 3.16–3.09 (m, 4H), 2.97–2.92 (m, 2H), 2.76 (brs, 3H), 2.66 (d, J = 4.68 Hz, 2H), 2.56–2.55 (m, 1H), 2.46–2.33 (m, 3H), 2.27–2.13 (m, 6H), 2.08–2.00 (m, 2H), 1.90–1.75 (m, 3H), 1.70–1.56 (m, 2H), 1.51–1.42 (m, 3H), 1.30–1.25 (m, 7H), 1.05 (d, J = 6.12 Hz, 1H). 13C NMR (100 MHz, DMSO-d6): δ 168.9, 168.4, 168.3, 142.1, 141.3, 129.6, 120.5, 89.7, 70.4, 70.3, 69.6, 64.9, 61.6, 56.7, 50.3, 46.5, 45.6, 38.1, 38.04, 38.01, 29.4, 28.90, 28.89, 28.8, 25.1, 23.79, 23.75, 23.7, 23.4, 23.0, 21.2, 15.1, 11.5, 5.7, 5.1, 3.2, 2.6. 19F NMR (376 MHz, DMSO-d6): δ −89.81–90.42, −98.82, −99.46. C44H67F2N9O7 (871.5132). HPLC purity: 98.62%. Rt: 6.589 min.
In Vitro MOR Radioligand Binding Assay
The competition binding assay was conducted using monoclonal mouse MOR expressed in Chinese hamster ovary (CHO) cell lines. In this assay, 30 μg of membrane protein was incubated with the radioligand [3H]naloxone in the presence of different concentrations of tested compounds in TME buffer (50 mM Tris, 3 mM MgCl2, and 0.2 mM EGTA, pH 7.4) for 1.5 h at 30 °C. The bound radioligand was separated by filtration using a Brandel harvester. Specific (i.e., opioid receptor-related) binding to the MOR was determined as the difference in binding obtained in the absence and presence of 5 μM of DAMGO. The IC50 values were determined and converted to Ki values using the Cheng–Prusoff equation: Ki = IC50/[1 + ([L*]/KD)], where [L*] is the concentration of the radioligand and KD is the KD of the radioligand was determined.73
[35S]-GTPγS Functional Assay
[35S]-GTPγS functional assays were conducted in the mouse MOR cell membrane used for the receptor binding assays. Membrane protein (10 μg) was incubated with varying concentrations of test compounds, GDP (15 μM), and 80 pM [35S]-GTPγS in assay buffer for 1 h at 30 °C. Nonspecific binding was determined with 10 μM unlabeled GTPγS. DAMGO (5 μM) was included in the assay for a maximally effective concentration of a full agonist for the MOR. After incubation, the bound radioactive ligand was separated from the free radioligand by filtration through a GF/B glass fiber filter paper and rinsed three times with ice-cold wash buffer (50 mM Tris-HCl, pH 7.2) using the Brandel harvester. The results were determined by utilizing a scintillation counter. All assays were determined in triplicate and repeated at least three times. Percent DAMGO stimulated [35S]-GTPγS binding was defined as (net-stimulated binding by ligand/net-stimulated binding by 3 μM DAMGO) × 100%.
Calcium Mobilization Assay in mMOR-CHO Cells
A CHO cell line stably expressing the mouse mu opioid receptor (mMOR-CHO) was used for this assay. The cells were transfected with Gαqi5 for 4 h and then plated (10,000 cells/well) to black 96-well plates with clear bottoms (Greiner Bio-One). After 24 h of incubation, the culture medium was removed, and the cells were washed with assay buffer (50 mL of HBSS, 1 mL of HEPES, 250 μL of probenecid, 50 μL of 1 mM CaCl2, and 50 μL of 1 mM MgCl2). The tested compounds were dissolved in DMSO as a stock solution for the assay (1 M). For agonist assays, cells were then incubated with 50 μL/well loading buffer (6 mL of assay buffer, 24 μL of Fluo4-AM solution (Invitrogen), and 12 μL of probenecid solution) for 60 min. Following incubation, different concentrations of the tested compounds were added by a FlexStation3 microplate reader (Molecular Devices) and read at ex494/em516. Each concentration was run in triplicate. For antagonism studies, the cells were incubated with the same loading buffer as the agonist assay for 60 min. Then, different concentrations of the tested compounds (20 μL/well) were manually added to each well followed by another 15 min of incubation. After that, the solution of DAMGO in assay buffer (500 nM) or just assay buffer (blank) was added by a FlexStation3 microplate reader (Molecular Devices) and read at ex494/em516. Each concentration was run in triplicate. The corresponding IC50 value of each compound was calculated by nonlinear regression using GraphPad Prism 8.0.1 (GraphPad Software, San Diego, CA).
In Vitro CCR5 Radioligand Binding Assay
The CCR5 competitive radioligand binding assay was conducted following previously reported studies.74,75 Rhesus macaque chemokine CCR5 receptors expressed in Chem-1 cells are assayed in modified HEPES buffer (50 mM HEPES, 1 mM CaCl2, 5 mM MgCl2, 0.2% BSA, pH 7.4). Three μg of membrane protein was incubated with 0.10 nM [125I]MIP-1α in the presence of six concentrations of tested compounds (10 μM, 1 μM, 0.1 μM, 10 nM, 1 nM, and 0.1 nM) for 90 min at 25 °C. Nonspecific binding is estimated in the presence of 0.1 μM MIP-1β. Membranes are filtered and washed, and the filters are then counted to determine [125I]MIP-1α specifically bound, which specific binding was defined as 85% as a historical value. The IC50 values were determined and converted to Ki values using the Cheng–Prusoff equation: Ki = IC50/[1 + ([L*]/KD)], where [L*] is 0.10 nM and KD is 0.31 nM (historical value).
Calcium Mobilization Assay in HOS-CCR5 Cells
HOS-CCR5 cells were transfected with Gqi5 pcDNA16 using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s recommended procedure and maintained in RPMI 1640 supplemented with 10% fetal bovine serum, 100 μg/mL penicillin, 100 μg/mL streptomycin, and 1 mg/mL G418 at 37 °C and 5% CO2. 48 h after transfection, a total of 2,500,000 cells were spun down and brought back up in 8 mL of 50:1 HBSS:HEPES assay buffer. Cells were then plated at 25,000 cells per well into a clear bottom, black 96-well plate (Greiner Bio-one) and 50 μL of Fluo4 loading buffer [40 μL of 2 μM Fluo4-AM (Invitrogen), 100 μL of 2.5 mM probenecid, in 5 mL of assay buffer] was added to bring the volume up to 130 μL. After incubating for 45 min, 50 μL of varying concentrations of tested compounds was added, and the plate was incubated for an additional 15 min. Plates were then read on a FlexStation3 microplate reader (Molecular Devices) at 494/516 ex/em for a total of 120 s. After 16 s of reading, 20 μL of 200 nM CCL5 (Biosource) in assay buffer, or assay buffer alone, was added to the wells to bring the total volume up to 200 μL. The changes in calcium mobilization were monitored, and peak height values were obtained using SoftMaxPro software (Molecular Devices). Nonlinear regression curves and IC50 values were generated using GraphPad Prism 8.0.1 (GraphPad Software, San Diego, CA). All experiments were repeated at least three times.
Anti-HIV-1BaL Activity (Reverse Transcriptase Activity) and Cytotoxicity Assays in GHOST CCR5 Cells
100 μL of GHOST CCR5 cells at a density of 1 × 104 cells/well in 10% complete DMEM (10% FBS with 1% l-glutamine, and 1% penicillin/streptomycin) media were plated in a 96-well flat bottom plate and incubated for 24 h at 37 °C/5% CO2. Following the incubation, 100 μL of each compound at 6 concentrations were added in triplicate to both efficacy and toxicity plates followed by 100 μL of HIV-1BaL at a predetermined titer to the plates being evaluated for efficacy and 50 μL of complete DMEM to the plates being evaluated for cytotoxicity. The cultures were incubated for 6 days at 37 °C/5% CO2. Following the incubation, 50 μL of cell culture supernatant was removed from the wells of the efficacy plates for subsequent evaluation of virus content by reverse transcriptase activity assay. The toxicity plates being evaluated for cytotoxicity were stained with 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-2H-tetrazolium hydroxide (XTT).
RT activity (EC50 values) was measured using standard radioactivity incorporation polymerization assay. One μL of titrated thymidine triphosphate (TTP) at 1 Ci/mL was used per enzyme reaction. Poly rA and oligo dT were prepared at concentrations of 0.5 mg/mL and 1.7 units/mL, respectively, from a stock solution which was kept at −20 °C. The RT reaction buffer was prepared fresh and consists of 125 μL of 1 M EGTA, 125 μL of dH2O, 125 μL of 20% Triton X-100, 50 μL of 1 M Tris (pH 7.4), 50 μL of 1 M DTT, and 40 μL of 1 M MgCl2. For each reaction, 1 μL of TTP, 4 μL of dH2O, 2.5 μL of rAdT ,and 2.5 μL of reaction buffer were mixed. Ten μL of this reaction mixture was placed in a round-bottom microtiter plate with 15 μL of virus containing supernatant. The plate was incubated at 37 °C in a humidified incubator for 60–90 min. Following the incubation, 10 μL of the reaction volume was spotted onto a DEAE filter mat (PerkinElmer, catalog no. 1450-522) in the appropriate plate format, washed 5 times for 5 min each in a sodium (150 mM) citrate (15 mM) buffer (Invitrogen, catalog no. 15557-036), 2 times for 1 min each in deionized water (ImQuest), 2 times for 1 min each in 70% reagent alcohol (Fisher, catalog no. L-7168), and then air-dried. The dried filtermat was placed in a plastic sleeve, and 4 mL of Opti-Fluor O (PerkinElmer, catalog no. 1205-440) scintillation fluid was added to each sleeve. Incorporated radioactivity was quantified using a Wallac 1450 Microbeta Trilux liquid scintilation counter.
TC50 values for the tested compounds were derived by measuring the reduction of the tetrazolium dye XTT. XTT in metabolically active cells is metabolized by the mitochondrial enzyme NADPH oxidase to a soluble formazan product. XTT solution was prepared as a stock of 1 mg/mL in RPMI-1640 without additives. Phenazine methosulfate (PMS) solution was prepared at 0.15 mg/mL in DPBS and stored in the dark at −20 °C. XTT/PMS stock was prepared immediately before use by adding 40 μL of PMS per mL of XTT solution. 50 μL of XTT/PMS was added to each well of the plate, and the plate was incubated for 4 h at 37 °C. The plates were sealed and inverted several times to mix the soluble formazan product, and the plate was read at 450 nm (650 nm reference wavelength) with a Molecular Devices SpectraMax Plus 384 96-well format spectrophotometer.
HIV-1BaL Infection Assay in MOR-CCR5 Coexpressed TZM-bl Cells
HIV-1BaL.01 was kindly donated by the NIH AIDS Reagent Program, NIAID, NIH (by Drs. Suzanne Gartner, Mikulas Popovic, and Robert Gallo) and expanded in PHA-activated PBMCs. The amount of viral particles was determined by commercial ELISA for p24 capsid antigen (Zeptometrix), and aliquots were stored at −80 °C. The TZM-bl, a cell line contained integrated copies of luciferase under control of the HIV-1 long-terminal repeat (LTR), were kindly donated by the NIH AIDS Reagent Program, NIAID, NIH (from Drs. John Kappes and Xiaoyun Wu). Cells were transfected with plasmid containing GFP-tagged human opioid receptor mu 1 gene (OPRM1; Origene) or with the original empty vector also containing GFP (control plasmid; Origene) and selected for 2 weeks with 2 mg/mL G418 (Invivogen) in DMEM (Hyclone) supplemented with 1×MEM nonessential amino acids (Gibco), 10% heat-inactivated FBS (Hyclone), and 100 U penicillin-0.1 mg/mL streptomycin (Sigma). Then, GFP+ cells were sorted out twice by flow cytometry (BDFACSAria model, Becton-Dickinson) and maintained in the conditions mentioned above with 800 μ/mL G418. For the infection, cells were detached from 25 cm2 flasks with 0.25% Trypsin (Sigma), washed by centrifugation (400g, 10 min), and viable cells determined by Trypan blue (Sigma) exclusion. Cells were added in 96-well black walled plate at 1.5 × 104 cells/well in 90 μL volume of DMEM containing 10% heat-inactivated FBS and 100 U penicillin-0.1 mg/mL streptomycin. After 6–10 h incubation at 37 °C in 5% CO2:95% air, the bivalent compound VZMC013 was added 1 h before infection with HIV-1BaL at 460 pg p24/well, in a final volume of 100 μL per well. Luciferase activity was evaluated following the addition of 100 μL/well of 2× luciferase substrate containing lysis buffer (Bright-Glo Luciferase Substrate, Promega) after 2–3 days of infection. Luminescence, reported as relative luminescence units (RLU), was evaluated in PHERAstar FS plate reader, and results expressed as percentage of inhibition relative to vehicle-treated, infected cells (100% infection).
HIV-1-Luc Infection Assay in Peripheral Blood Mononuclear Cells
Isolated human PBMCs were obtained commercially from the New York Blood Bank and exempt from Human Subjects criteria (NIH Exemption 4: https://grants.nih.gov/sites/default/files/exemption_infographic_v7_508c-3-21-19.pdf). The adult PBMCs are publicly available, and no information or identifiers are provided so subjects cannot be identified.
HIV-1BaL env-pseudotyped Luciferase viruses were generated by the cotransfecting HEK293TT cell line (kindly donated by Dr. John Schiller, NIAID, NIH) with plasmids containing expression vectors for HIV-1BaL.01 env and for HIV-1NL4–3 delta-env with Luciferase inserted into nef gene. After 3 days in culture, supernatants were harvested, floating cells removed by centrifugation, HIV-1 p24 Ag measured by commercial ELISA (Zeptometrix), and viral aliquots stored at −80 °C. Both plasmids were donated by NIH AIDS Reagent Program, NIAID, NIH (HIV-1BaL.01 env by Dr. John Mascola and HIV-1NL4–3 Luc R−E− by Dr. Nathaniel Landau).
PBMCs were isolated from buffy coats of healthy blood donors by centrifugation in Ficoll gradient (MP Biomedicals). Viable cells, determined by Trypan blue exclusion, were stimulated, or not, for 2 days with 5 μg/mL PHA (Sigma). Non-PHA stimulated cells were distributed in 96-well black walled plates at 2 × 105 per well in RPMI-1640 medium supplemented with 1 mM sodium pyruvate (Gibco), 1× MEM nonessential amino acids, 10% heat-inactivated FBS, 100 U penicillin-0.1 mg/mL streptomycin, and 20 U/mL recombinant human Interleukin-2 (Peprotech). Cells were infected with HIV-1BaL.01 env-pseudotyped Luc virus at 1.5 ng p24/well in a final volume of 100 μL, and uninfected cells served as a negative control. The bivalent compound VZMC013 was added 1 h before infection and kept in the culture. After 2 days of infection, 100 μL of 2× Luciferase substrate containing lysing buffer (Bright-Glo Luciferase Assay System, Promega) was added per well, the plate was agitated for 2 min (700 rpm), and Luminescence was evaluated using a PHERAstar FS plate reader (BMG Labtech).
HIV-1 Infection Assay in PHA-Stimulated PBMC Cells
In a 24-well plate, PHA-stimulated PBMC cells from healthy donors were infected by incubation with the HIV-1BaL.01 env-pseudotyped Luc virus. A concentration of HIV-1 p24 50 pg/106 cells was used, and uninfected cells served as a negative control. Cells were treated with and without morphine or DAMGO (10 nM) along with bivalent compound VZMC013 (100 nM) 3 days before HIV-1 infection. After approximately 18–20 h, the supernatant was removed and stored at −80 °C, and cells were rinsed twice with PBS and lysed. The lysate was subsequently tested for the relative Tat protein expression by using a luciferase assay system (Promega). Luciferase activity was measured using a PHERAstar FS plate reader (BMG Labtech).
Molecular Docking and Molecular Dynamics Simulation
To generate the MOR-CCR5 heterodimer, the monomeric crystal structures of CCR5 (PDB ID: 4MBS)44 and MOR (PDB ID: 4DKL)46 were downloaded from the Protein Data Bank.76 Prior to performing the molecular docking, other parts of the two crystal structures were removed with the exception of the N-terminus, the seven transmembrane helices, and the C-terminus. In addition, the missing residues in intracellular loop 3 (ICL3) of 4MBS and ICL3 of 4DKL were modeled by Sybyl 8.0. Naltrexone was first docked into the MOR to obtain its monomeric complex with MOR, named as MOR_naltrexone complex. The remaining CCR5 and original ligand maraviroc obtained the CCR5 monomer complexing with maraviroc, referred to CCR5_maraviroc complex. Afterward, Hex 8.0.0 was applied to dock the two complexes together to build the heterodimer model.77 The process included four steps: (1) CCR5_maraviroc and MOR_naltrexone complexes were put together and moved close enough to each other; (2) CCR5_maraviroc complex was fixed, and its TM1/2 was selected as the dimer interface; (3) MOR_naltrexone complex was rotated to find its dimer interface at TM5/6 and further to interact with TM1/2 of the CCR5; and (4) the new coordinates of CCR5_maraviroc and MOR_naltrexone complexes were obtained and saved as the starting model of the MOR-CCR5 heterodimer complexing with their respective ligands.
The above heterodimer model was inserted into a 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) lipid membrane and solvated by TIP3 water molecules to conduct an energy minimization determination using Amber14.0. The energy minimization process included three steps: (1) restraining the ligands, the heterodimer complex, and the lipid membrane to minimize the water molecules and ions; (2) restraining the backbone atoms of the heterodimer, the ligands, and the lipid membrane to minimize the protein side chains, the water molecules, and ions; and (3) energy minimizing the whole system without restraint. The energy minimized heterodimer model was applied as the starting structure to build the bivalent ligand VZMC013 into the binding site of the MOR-CCR5 heterodimer.
In the energy minimized heterodimer model, the CCR5 pharmacophore maraviroc and the MOR pharmacophore naltrexone were both in their respective binding pockets. The molecular graphics software Sybyl-X 8.0 (TRIPOS Inc., St. Louis, MO) was applied to connect the two pharmacophores by the spacer displayed in Figure 2c. Finally, the MOR-CCR5 heterodimer complexing with the bivalent ligand VZMC013 was obtained and named as the MOR-CCR5_VZMC013 complex (Figure S6a). With the MOR-CCR5_VZMC013 complex in hand, the bivalent ligand VZMC013 was first extracted from the complex by Sybyl-X 8.0 (TRIPOS Inc., St. Louis, MO), and then VZMC013 was examined with the correct atom type, bond type, and hydrogen atoms. Next, the structure of VZMC013 was optimized to its minimum energy conformation by Gaussian 03. After that, VZMC013 was submitted to the antechamber program in AMBER 14 to form its restrained electrostatic potential charges descriptors and other force field parameters. In the following, a system including the MOR-CCR5_VZMC013 complex, the POPC lipid membrane, the TIP3 water box, and 0.15 M sodium and chloride ions was built by the CHARMM-GUI Web site service.77 Meanwhile, the input files of the MD simulations were also generated. MD simulations were performed by GROMACS (ver. 2018.2) package (www.gromacs.org). The Amber 03 force field was selected for the simulations.78 The 20,000 steps of the steepest-descent energy minimization were initially applied to the system. After that, the system was heated to 310 K via two steps. First, heating from 0 to 100 K under a constant volume ensemble for 0.5 ns. Second, heating from 100 to 310 K under a constant pressure ensemble (NPT) for another 1.0 ns. Subsequently, a 100 ns MD simulation was conducted using the NPT (P = 105 Pa, T = 310 K) ensemble. The long-range electrostatic interactions were computed using the particle mesh Ewald method.79 A smooth cutoff of 10 Å was used to calculate the nonbonded van der Waals interactions. The periodic boundary conditions were applied during the MD simulations. The snapshots were collected at 2 fs intervals.
Supplementary Material
ACKNOWLEDGMENTS
The authors are grateful to NIDA Drug Supply Program for providing the free base of naltrexone. This work was supported by NIH/NIDA grant R01 DA044855 (Y.Z. and K.F.H.) and R01 DA034231 (P.E.K. and K.F.H.).
ABBREVIATIONS USED
- AIDS
acquired immune deficiency syndrome
- Cbz
carboxybenzyl
- CCR5
C─C chemokine receptor type 5
- CCL5
C─C motif chemokine ligand 5
- CHO
Chinese hamster ovary
- 13C NMR
carbon nuclear magnetic resonance
- DAMGO
[d-Ala2-MePhe4-Gly(ol)5]enkephalin
- ECL
extracellular loop
- EDCI
1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide
- Fmoc
fluorenylmethyloxycarbonyl
- β-FNA
β-funaltrexamine
- 19F NMR
fluorine nuclear magnetic resonance
- GPCRs
G-protein-coupled receptors
- HIV
human immunodeficiency virus
- 1H NMR
proton nuclear magnetic resonance
- HOBt
hydroxybenzotriazole
- HPLC
high-performance liquid chromatography
- HRMS
high-resolution mass spectrum
- ICL
intracellular loops
- LTR
long-terminal repeat
- Luc
luciferase gene
- MD
molecular dynamics
- MIP-1β
macrophage inflammatory protein 1 beta
- MOR
mu opioid receptor
- OUD
opioid use disorder
- PBMC
peripheral blood mononuclear cells
- PDB
Protein Data Bank
- PHA
phytohemagglutinin
- RMSD
root-mean-square deviation
- RMSF
root-mean-square fluctuation
- RT
reverse transcriptase
- SEM
standard error of the mean
- Tat
trans-activator of transcription
- TI
therapeutic index
- TM
transmembrane
- TMS
tetramethylsilane
- TOF
time-of-flight
- TsOH
toluenesulfonic acid
Footnotes
Accession Codes
The binding mode of VZMC013 in the MOR-CCR5 heterodimer complex (PDB). The MOR-CCR5_VZMC013 complex in the membrane-aqueous sodium chloride solution system (PDB). The binding mode of VZMC013 in the MOR-CCR5 heterodimer complex after MD simulations (PDB). The docking pose of naltrexone in the inactive MOR (PDB).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute on Drug Abuse or the National Institutes of Health. The authors declare no competing financial interest.
Contributor Information
Boshi Huang, Department of Medicinal Chemistry, Virginia Commonwealth University, Richmond, Virginia 23298, United States.
Huiqun Wang, Department of Medicinal Chemistry, Virginia Commonwealth University, Richmond, Virginia 23298, United States.
Yi Zheng, Department of Medicinal Chemistry, Virginia Commonwealth University, Richmond, Virginia 23298, United States.
Mengchu Li, Department of Medicinal Chemistry, Virginia Commonwealth University, Richmond, Virginia 23298, United States.
Guifeng Kang, Department of Medicinal Chemistry, Virginia Commonwealth University, Richmond, Virginia 23298, United States.
Victor Barreto-de-Souza, Department of Pharmacology and Toxicology, Virginia Commonwealth University, Richmond, Virginia 23298, United States.
Nima Nassehi, Department of Pharmacology and Toxicology, Virginia Commonwealth University, Richmond, Virginia 23298, United States.
Pamela E. Knapp, Department of Pharmacology and Toxicology, Virginia Commonwealth University, Richmond, Virginia 23298, United States; Department of Anatomy and Neurobiology, Virginia Commonwealth University, Richmond, Virginia 23298, United States
Dana E. Selley, Department of Pharmacology and Toxicology, Virginia Commonwealth University, Richmond, Virginia 23298, United States
Kurt F. Hauser, Department of Pharmacology and Toxicology, Virginia Commonwealth University, Richmond, Virginia 23298, United States; Department of Anatomy and Neurobiology, Virginia Commonwealth University, Richmond, Virginia 23298, United States
Yan Zhang, Department of Medicinal Chemistry, Virginia Commonwealth University, Richmond, Virginia 23298, United States.
REFERENCES
- (1).Wilson N; Kariisa M; Seth P; Smith H IV; Davis NL Drug and Opioid-Involved Overdose Deaths - United States, 2017–2018. MMWR Morb. Mortal. Wkly. Rep 2020, 69, 290–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).The Global HIV/AIDS Epidemic; U.S. Department of Health and Human Services: Washington, DC. https://www.hiv.gov/hiv-basics/overview/data-and-trends/global-statistics (accessed 2020-05-20). [Google Scholar]
- (3).Overview: Data and Trends: U.S. Statistics; U.S. Department of Health and Human Services: Washington, DC. https://www.hiv.gov/hiv-basics/overview/data-and-trends/statistics (accessed 2020-05-20). [Google Scholar]
- (4).Injection Drug Use and HIV Risk∣HIV∣CDC; Centers for Disease Control and Prevention: Atlanta, GA. https://www.cdc.gov/hiv/risk/idu.html (accessed 2020-05-20). [Google Scholar]
- (5).HIV in the United States: At A Glance∣Statistics Overview∣Statistics Center∣HIV Public Health Partners∣HIV∣CDC; Centers for Disease Control and Prevention: Atlanta, GA. https://www.cdc.gov/hiv/statistics/overview/ataglance.html (accessed 2020-05-20). [Google Scholar]
- (6).Chang SL; Vigorito M Role of HIV-1 Infection in Addictive Behavior: A Study of a HIV-1 Transgenic Rat Model. Am. J. Infect. Dis 2006, 2, 98–106. [Google Scholar]
- (7).Cunningham CO Opioids and HIV Infection: From Pain Management to Addiction Treatment. Top. Antivir. Med 2018, 25, 143–146. [PMC free article] [PubMed] [Google Scholar]
- (8).Weisberg DF; Gordon KS; Barry DT; Becker WC; Crystal S; Edelman EJ; Gaither J; Gordon AJ; Goulet J; Kerns RD; Moore BA; Tate J; Justice AC; Fiellin DA Long-term Prescription of Opioids and/or Benzodiazepines and Mortality Among HIV-Infected and Uninfected Patients. JAIDS, J. Acquired Immune Defic. Syndr 2015, 69, 223–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Roy S; Ninkovic J; Banerjee S; Charboneau RG; Das S; Dutta R; Kirchner VA; Koodie L; Ma J; Meng J; Barke RA Opioid Drug Abuse and Modulation of Immune Function: Consequences in the Susceptibility to Opportunistic Infections. J. Neuroimmune Pharmacol 2011, 6, 442–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wang X; Ye L; Zhou Y; Liu M-Q; Zhou D-J; Ho W-Z Inhibition of Anti-HIV MicroRNA Expression: A Mechanism for Opioid-Mediated Enhancement of HIV Infection of Monocytes. Am. J. Pathol 2011, 178, 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hauser KF; El-Hage N; Buch S; Berger JR; Tyor WR; Nath A; Bruce-Keller AJ; Knapp PE Molecular Targets of Opiate Drug Abuse in NeuroAIDS. Neurotoxic. Res 2005, 8, 63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Hauser KF; Fitting S; Dever SM; Podhaizer EM; Knapp PE Opiate Drug Use and the Pathophysiology of NeuroAIDS. Curr. HIV Res 2012, 10, 435–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tahamtan A; Tavakoli-Yaraki M; Mokhtari-Azad T; Teymoori-Rad M; Bont L; Shokri F; Salimi V Opioids and Viral Infections: A Double-Edged Sword. Front. Microbiol 2016, 7, 970–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Steele AD; Henderson EE; Rogers TJ Mu-opioid Modulation of HIV-1 Coreceptor Expression and HIV-1 Replication. Virology 2003, 309, 99–107. [DOI] [PubMed] [Google Scholar]
- (15).Mahajan SD; Schwartz SA; Shanahan TC; Chawda RP; Nair MPN Morphine Regulates Gene Expression of α- and β-Chemokines and Their Receptors on Astroglial Cells Via the Opioid μ Receptor. J. Immunol 2002, 169, 3589–3599. [DOI] [PubMed] [Google Scholar]
- (16).Bokhari SM; Yao H; Bethel-Brown C; Fuwang P; Williams R; Dhillon NK; Hegde R; Kumar A; Buch SJ Morphine Enhances Tat-induced Activation in Murine Microglia. J. NeuroVirol 2009, 15, 219–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).El-Hage N; Dever SM; Podhaizer EM; Arnatt CK; Zhang Y; Hauser KF A Novel Bivalent HIV-1 Entry Inhibitor Reveals Fundamental Differences in CCR5-μ-opioid Receptor Interactions between Human Astroglia and Microglia. AIDS 2013, 27, 2181–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Miyagi T; Chuang LF; Doi RH; Carlos MP; Torres JV; Chuang RY Morphine Induces Gene Expression of CCR5 in Human CEM x174 Lymphocytes. J. Biol. Chem 2000, 275, 31305–31310. [DOI] [PubMed] [Google Scholar]
- (19).Suzuki S; Chuang AJ; Chuang LF; Doi RH; Chuang RY Morphine Promotes Simian Acquired Immunodeficiency Syndrome Virus Replication in Monkey Peripheral Mononuclear Cells: Induction of CC Chemokine Receptor 5 Expression for Virus Entry. J. Infect. Dis 2002, 185, 1826–1829. [DOI] [PubMed] [Google Scholar]
- (20).Guo C-J; Li Y; Tian S; Wang X; Douglas SD; Ho W-Z Morphine Enhances HIV Infection of Human Blood Mononuclear Phagocytes through Modulation of β-Chemokines and CCR5 Receptor. J. Invest. Med 2002, 50, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Li Y; Merrill JD; Mooney K; Song L; Wang X; Guo C-J; Savani RC; Metzger DS; Douglas SD; Ho W-Z Morphine Enhances HIV Infection of Neonatal Macrophages. Pediatr. Res 2003, 54, 282–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Li Y; Wang X; Tian S; Guo C-J; Douglas SD; Ho W-Z Methadone Enhances Human Immunodeficiency Virus Infection of Human Immune Cells. J. Infect. Dis 2002, 185, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Suzuki S; Chuang LF; Yau P; Doi RH; Chuang RY Interactions of Opioid and Chemokine Receptors: Oligomerization of mu, kappa, and delta with CCR5 on Immune Cells. Exp. Cell Res 2002, 280, 192–200. [DOI] [PubMed] [Google Scholar]
- (24).Szabo I; Chen X-H; Xin L; Adler MW; Howard OMZ; Oppenheim JJ; Rogers TJ Heterologous Desensitization of Opioid Receptors by Chemokines Inhibits Chemotaxis and Enhances the Perception of Pain. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 10276–10281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Szabo I; Wetzel MA; Zhang N; Steele AD; Kaminsky DE; Chen C; Liu-Chen LY; Bednar F; Henderson EE; Howard OM; Oppenheim JJ; Rogers TJ Selective Inactivation of CCR5 and Decreased Infectivity of R5 HIV-1 Strains Mediated by Opioid-induced Heterologous Desensitization. J. Leukocyte Biol 2003, 74, 1074–1082. [DOI] [PubMed] [Google Scholar]
- (26).Chen C; Li J; Bot G; Szabo I; Rogers TJ; Liu-Chen L-Y Heterodimerization and Cross-desensitization between the μ-Opioid Receptor and the Chemokine CCR5 Receptor. Eur. J. Pharmacol 2004, 483, 175–186. [DOI] [PubMed] [Google Scholar]
- (27).Song C; Rahim RT; Davey PC; Bednar F; Bardi G; Zhang L; Zhang N; Oppenheim JJ; Rogers TJ Protein Kinase Cζ Mediates μ-Opioid Receptor-induced Cross-desensitization of Chemokine Receptor CCR5. J. Biol. Chem 2011, 286, 20354–20365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Pulido D; Casadó-Anguera V; Pérez-Benito L; Moreno E; Cordomí A; López L; Cortés A; Ferré S; Pardo L; Casadó V; Royo M Design of a True Bivalent Ligand with Picomolar Binding Affinity for a G Protein-Coupled Receptor Homodimer. J. Med. Chem 2018, 61, 9335–9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Reinecke BA; Kang G; Zheng Y; Obeng S; Zhang H; Selley DE; An J; Zhang Y Design and Synthesis of a Bivalent Probe Targeting the Putative Mu Opioid Receptor and Chemokine Receptor CXCR4 Heterodimer. RSC Med. Chem 2020, 11, 125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Newman AH; Battiti FO; Bonifazi A 2016 Philip S. Portoghese Medicinal Chemistry Lectureship: Designing Bivalent or Bitopic Molecules for G-Protein Coupled Receptors. The Whole Is Greater Than the Sum of Its Parts. J. Med. Chem 2020, 63, 1779–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Huang B; St. Onge CM; Ma H; Zhang Y Design of bivalent ligands targeting putative GPCR dimers. Drug Discovery Today 2021, 26, 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Yuan Y; Arnatt CK; Li G; Haney KM; Ding D; Jacob JC; Selley DE; Zhang Y Design and Synthesis of a Bivalent Ligand to Explore the Putative Heterodimerization of the Mu Opioid Receptor and the Chemokine Receptor CCR5. Org. Biomol. Chem 2012, 10, 2633–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Yuan Y; Arnatt CK; El-Hage N; Dever SM; Jacob JC; Selley DE; Hauser KF; Zhang Y A Bivalent Ligand Targeting the Putative Mu Opioid Receptor and Chemokine Receptor CCR5 Heterodimer: Binding Affinity Versus Functional Activities. Med-ChemComm 2013, 4, 847–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Arnatt CK; Falls BA; Yuan Y; Raborg TJ; Masvekar RR; El-Hage N; Selley DE; Nicola AV; Knapp PE; Hauser KF; Zhang Y Exploration of Bivalent Ligands Targeting Putative Mu Opioid Receptor and Chemokine Receptor CCR5 Dimerization. Bioorg. Med. Chem 2016, 24, 5969–5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Obeng S; Yuan Y; Jali A; Selley DE; Zhang Y In Vitro and In Vivo Functional Profile Characterization of 17-Cyclo-propylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(isoquinoline-3-carboxamido)morphinan (NAQ) as a Low Efficacy Mu Opioid Receptor Modulator. Eur. J. Pharmacol 2018, 827, 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Peng P; Chen H; Zhu Y; Wang Z; Li J; Luo R-H; Wang J; Chen L; Yang L-M; Jiang H; Xie X; Wu B; Zheng Y-T; Liu H Structure-Based Design of 1-Heteroaryl-1,3-propanediamine Derivatives as a Novel Series of CC-Chemokine Receptor 5 Antagonists. J. Med. Chem 2018, 61, 9621–9636. [DOI] [PubMed] [Google Scholar]
- (37).Bhushan RG; Sharma SK; Xie Z; Daniels DJ; Portoghese PS A Bivalent Ligand (KDN-21) Reveals Spinal δ and κ Opioid Receptors Are Organized as Heterodimers That Give Rise to δ1 and κ2 Phenotypes. Selective Targeting of δ–κ Heterodimers. J. Med. Chem 2004, 47, 2969–2972. [DOI] [PubMed] [Google Scholar]
- (38).Zhang S; Yekkirala A; Tang Y; Portoghese PS A Bivalent Ligand (KMN-21) Antagonist for μ/κ Heterodimeric Opioid Receptors. Bioorg. Med. Chem. Lett 2009, 19, 6978–6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zheng Y; Akgün E; Harikumar KG; Hopson J; Powers MD; Lunzer MM; Miller LJ; Portoghese PS Induced Association of μ Opioid (MOP) and Type 2 Cholecystokinin (CCK2) Receptors by Novel Bivalent Ligands. J. Med. Chem 2009, 52, 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Li G; Haney KM; Kellogg GE; Zhang Y Comparative Docking Study of Anibamine as the First Natural Product CCR5 Antagonist in CCR5 Homology Models. J. Chem. Inf. Model 2009, 49, 120–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Xie Z; Bhushan RG; Daniels DJ; Portoghese PS Interaction of Bivalent Ligand KDN21 with Heterodimeric δ-κ Opioid Receptors in Human Embryonic Kidney 293 Cells. Mol. Pharmacol 2005, 68, 1079–1086. [DOI] [PubMed] [Google Scholar]
- (42).Daniels DJ; Lenard NR; Etienne CL; Law P-Y; Roerig SC; Portoghese PS Opioid-induced Tolerance and Dependence in Mice Is Modulated by the Distance Between Pharmacophores in a Bivalent Ligand Series. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 19208–19213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Wu B; Chien EYT; Mol CD; Fenalti G; Liu W; Katritch V; Abagyan R; Brooun A; Wells P; Bi FC; Hamel DJ; Kuhn P; Handel TM; Cherezov V; Stevens RC Structures of the CXCR4 Chemokine GPCR with Small-Molecule and Cyclic Peptide Antagonists. Science 2010, 330, 1066–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Tan Q; Zhu Y; Li J; Chen Z; Han GW; Kufareva I; Li T; Ma L; Fenalti G; Li J; Zhang W; Xie X; Yang H; Jiang H; Cherezov V; Liu H; Stevens RC; Zhao Q; Wu B Structure of the CCR5 Chemokine Receptor–HIV Entry Inhibitor Maraviroc Complex. Science 2013, 341, 1387–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Erez M; Takemori AE; Portoghese PS Narcotic Antagonistic Potency of Bivalent Ligands Which Contain β-Naltrexamine. Evidence for Bridging between Proximal Recognition Sites. J. Med. Chem 1982, 25, 847–849. [DOI] [PubMed] [Google Scholar]
- (46).Manglik A; Kruse AC; Kobilka TS; Thian FS; Mathiesen JM; Sunahara RK; Pardo L; Weis WI; Kobilka BK; Granier S Crystal Structure of the μ-Opioid Receptor Bound to a Morphinan Antagonist. Nature 2012, 485, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Haycock-Lewandowski SJ; Wilder A; Åhman J Development of a Bulk Enabling Route to Maraviroc (UK-427,857), a CCR-5 Receptor Antagonist. Org. Process Res. Dev 2008, 12, 1094–1103. [Google Scholar]
- (48).Wasserman HH; Berger GD The Use of β-Lactams in the Synthesis of Spermine and Spermidine Alkaloids: Total Synthesis of Homaline. Tetrahedron 1983, 39, 2459–2464. [Google Scholar]
- (49).Nagarajan SR; Devadas B; Malecha JW; Lu H-F; Ruminski PG; Rico JG; Rogers TE; Marrufo LD; Collins JT; Kleine HP; Lantz MK; Zhu J; Green NF; Russell MA; Landis BH; Miller LM; Meyer DM; Duffin TD; Engleman VW; Finn MB; Freeman SK; Griggs DW; Williams ML; Nickols MA; Pegg JA; Shannon KE; Steininger C; Westlin MM; Nickols GA; Keene JL R-Isomers of Arg-Gly-Asp (RGD) Mimics as Potent αvβ3 Inhibitors. Bioorg. Med. Chem 2007, 15, 3783–3800. [DOI] [PubMed] [Google Scholar]
- (50).Gao D-D; Dou H-X; Su H-X; Zhang M-M; Wang T; Liu Q-F; Cai H-Y; Ding H-P; Yang Z; Zhu W-L; Xu Y-C; Wang H-Y; Li Y-X From Hit to Lead: Structure-based Discovery of Naphthalene-1-sulfonamide Derivatives as Potent and Selective Inhibitors of Fatty Acid Binding Protein 4. Eur. J. Med. Chem 2018, 154, 44–59. [DOI] [PubMed] [Google Scholar]
- (51).Danner P; Bauer M; Phukan P; Maier ME Total Synthesis of Cryptophycin 3. Eur. J. Org. Chem 2005, 2005, 317–325. [Google Scholar]
- (52).Poojari S; Parameshwar Naik P; Krishnamurthy G Synthesis of Macrocycles Containing 1,3,4-Oxadiazole and Pyridine Moieties. Tetrahedron Lett. 2014, 55, 305–309. [Google Scholar]
- (53).Zhao G-L; Lin S; Korotvička A; Deiana L; Kullberg M; Córdova A Asymmetric Synthesis of Maraviroc (UK-427,857). Adv. Synth. Catal 2010, 352, 2291–2298. [Google Scholar]
- (54).Armour DR; De Groot MJ; Price DA; Stammen BLC; Wood A; Perros M; Burt C The Discovery of Tropane-derived CCR5 Receptor Antagonists. Chem. Biol. Drug Des 2006, 67, 305–308. [DOI] [PubMed] [Google Scholar]
- (55).Åhman J; Birch M; Haycock-Lewandowski SJ; Long J; Wilder A Process Research and Scale-up of a Commercialisable Route to Maraviroc (UK-427,857), a CCR-5 Receptor Antagonist. Org. Process Res. Dev 2008, 12, 1104–1113. [Google Scholar]
- (56).Atherton E; Fox H; Harkiss D; Sheppard RC Application of Polyamide Resins to Polypeptide Synthesis: an Improved Synthesis of β-Endorphin Using Fluorenylmethoxycarbonylamino-acids. J. Chem. Soc., Chem. Commun 1978, 539–540. [Google Scholar]
- (57).Zheng Y; Obeng S; Wang H; Jali AM; Peddibhotla B; Williams DA; Zou C; Stevens DL; Dewey WL; Akbarali HI; Selley DE; Zhang Y Design, Synthesis, and Biological Evaluation of the Third Generation 17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(4′-pyridyl)carboxamido]morphinan (NAP) Derivatives as μ/κ Opioid Receptor Dual Selective Ligands. J. Med. Chem 2019, 62, 561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Portoghese PS; Larson DL; Sayre LM; Yim CB; Ronsisvalle G; Tam SW; Takemori AE Opioid Agonist and Antagonist Bivalent Ligands. The Relationship between Spacer Length and Selectivity at Multiple Opioid Receptors. J. Med. Chem 1986, 29, 1855–1861. [DOI] [PubMed] [Google Scholar]
- (59).Neumeyer JL; Zhang A; Xiong W; Gu X-H; Hilbert JE; Knapp BI; Negus SS; Mello NK; Bidlack JM Design and Synthesis of Novel Dimeric Morphinan Ligands for κ and μ Opioid Receptors. J. Med. Chem 2003, 46, 5162–5170. [DOI] [PubMed] [Google Scholar]
- (60).Neumeyer JL; Peng X; Knapp BI; Bidlack JM; Lazarus LH; Salvadori S; Trapella C; Balboni G New Opioid Designed Multiple Ligand from Dmt-Tic and Morphinan Pharmacophores. J. Med. Chem 2006, 49, 5640–5643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Ma H; Obeng S; Wang H; Zheng Y; Li M; Jali AM; Stevens DL; Dewey WL; Selley DE; Zhang Y Application of Bivalent Bioisostere Concept on Design and Discovery of Potent Opioid Receptor Modulators. J. Med. Chem 2019, 62, 11399–11415. [DOI] [PubMed] [Google Scholar]
- (62).Ma H; Wang H; Li M; Barreto-de-Souza V; Reinecke BA; Gunta R; Zheng Y; Kang G; Nassehi N; Zhang H; An J; Selley DE; Hauser KF; Zhang Y Bivalent Ligand Aiming Putative Mu Opioid Receptor and Chemokine Receptor CXCR4 Dimers in Opioid Enhanced HIV-1 Entry. ACS Med. Chem. Lett 2020, 11, 2318–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Holl V; Schmidt S; Aubertin AM; Moog C The Major Population of PHA-stimulated PBMC Infected by R5 or X4 HIV Variants after a Single Cycle of Infection Is Predominantly Composed of CD45RO+CD4+ T Lymphocytes. Arch. Virol 2007, 152, 507–518. [DOI] [PubMed] [Google Scholar]
- (64).Meng X-Y; Mezei M; Cui M Computational Approaches for Modeling GPCR Dimerization. Curr. Pharm. Biotechnol 2014, 15, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Meral D; Provasi D; Prada-Gracia D; Möller J; Marino K; Lohse MJ; Filizola M Molecular Details of Dimerization Kinetics Reveal Negligible Populations of Transient μ-Opioid Receptor Homodimers at Physiological Concentrations. Sci. Rep 2018, 8, 7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Jin J; Momboisse F; Boncompain G; Koensgen F; Zhou Z; Cordeiro N; Arenzana-Seisdedos F; Perez F; Lagane B; Kellenberger E; Brelot A CCR5 Adopts Three Homodimeric Conformations That Control Cell Surface Delivery. Sci. Signaling 2018, 11, No. eaal2869. [DOI] [PubMed] [Google Scholar]
- (67).Zhang F; Yuan Y; Xiang M; Guo Y; Li M; Liu Y; Pu X Molecular Mechanism Regarding Allosteric Modulation of Ligand Binding and the Impact of Mutations on Dimerization for CCR5 Homodimer. J. Chem. Inf. Model 2019, 59, 1965–1976. [DOI] [PubMed] [Google Scholar]
- (68).Hou T; McLaughlin W; Lu B; Chen K; Wang W Prediction of Binding Affinities between the Human Amphiphysin-1 SH3 Domain and Its Peptide Ligands Using Homology Modeling, Molecular Dynamics and Molecular Field Analysis. J. Proteome Res 2006, 5, 32–43. [DOI] [PubMed] [Google Scholar]
- (69).Garcia-Perez J; Rueda P; Alcami J; Rognan D; Arenzana-Seisdedos F; Lagane B; Kellenberger E Allosteric Model of Maraviroc Binding to CC Chemokine Receptor 5 (CCR5). J. Biol. Chem 2011, 286, 33409–33421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Shaik MM; Peng H; Lu J; Rits-Volloch S; Xu C; Liao M; Chen B Structural Basis of Coreceptor Recognition by HIV-1 Envelope Spike. Nature 2019, 565, 318–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Tamamis P; Floudas CA Molecular Recognition of CCR5 by an HIV-1 gp120 V3 Loop. PLoS One 2014, 9, No. e95767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Yuan Y; Elbegdorj O; Chen J; Akubathini SK; Zhang F; Stevens DL; Beletskaya IO; Scoggins KL; Zhang Z; Gerk PM; Selley DE; Akbarali HI; Dewey WL; Zhang Y Design, Synthesis, and Biological Evaluation of 17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(4′-pyridyl)carboxamido]morphinan Derivatives as Peripheral Selective μ Opioid Receptor Agents. J. Med. Chem 2012, 55, 10118–10129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Yung-Chi C; Prusoff WH Relationship between the Inhibition Constant (KI) and the Concentration of Inhibitor Which Causes 50% Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- (74).Blanpain C. d.; Migeotte I; Lee B; Vakili J; Doranz BJ; Govaerts C. d.; Vassart G; Doms RW; Parmentier M CCR5 Binds Multiple CC-Chemokines: MCP-3 Acts as a Natural Antagonist. Blood 1999, 94, 1899–1905. [PubMed] [Google Scholar]
- (75).Colin P; Bénureau Y; Staropoli I; Wang Y; Gonzalez N; Alcami J; Hartley O; Brelot A; Arenzana-Seisdedos F; Lagane B HIV-1 Exploits CCR5 Conformational Heterogeneity to Escape Inhibition by Chemokines. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 9475–9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Wu EL; Cheng X; Jo S; Rui H; Song KC; Dávila-Contreras EM; Qi Y; Lee J; Monje-Galvan V; Venable RM; Klauda JB; Im W CHARMM-GUI Membrane Builder toward Realistic Biological Membrane Simulations. J. Comput. Chem 2014, 35, 1997–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Duan Y; Wu C; Chowdhury S; Lee MC; Xiong G; Zhang W; Yang R; Cieplak P; Luo R; Lee T; Caldwell J; Wang J; Kollman P A Point-charge Force Field for Molecular Mechanics Simulations of Proteins Based on Condensed-phase Quantum Mechanical Calculations. J. Comput. Chem 2003, 24, 1999–2012. [DOI] [PubMed] [Google Scholar]
- (79).Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A Smooth Particle Mesh Ewald Method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.