

Abstract
Controlled protein degradation by the ubiquitin-proteasome pathway is critical for almost all cellular processes. E3 ubiquitin ligases are responsible for targeting proteins for ubiquitylation and subsequent proteasomal degradation with spatial and temporal precision. While studies have revealed various E3-substrate pairs involved in distinct biological processes, the complete substrate profiles of individual E3 ligases are largely unknown. Here we report a new approach to identify substrates of an E3 ligase for proteasomal degradation using unnatural amino acid incorporation pulse-chase proteomics (degradomics). Applying this approach, we determine the steady-state substrates of the C-terminal to LisH (CTLH) E3 ligase, a multi-component complex with poorly defined substrates. By comparing the proteome degradation profiles of active and inactive CTLH-expressing cells, we successfully identify previously known and new potential substrates of CTLH ligase. Altogether, degradomics can comprehensively identify degradation substrates of an E3 ligase, which can be adapted for other E3 ligases in various cellular contexts.
Keywords: Ubiquitin, The Ubiquitin Proteasome System, Degradomics, Protein half-life, CTLH
Graphical Abstract
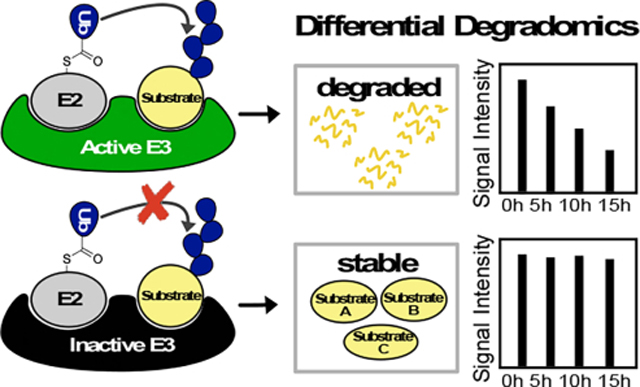
E3 ligases catalyze the ubiquitination of substrates, often resulting in proteasomal degradation. Improving approaches to identify substrates of E3 ligases has been critical to understanding the cellular pathways they regulate. This work proposes a universal approach using differential degradation proteomics (degradomics) to identify the substrates of an E3 ligase in the native cellular context.
Introduction
Ubiquitin (Ub), an 8.6 kDa regulatory protein, plays an important role in many cellular pathways, including protein degradation, trafficking, and DNA repair1. These functions are mediated through the covalent attachment of the Ub to primary amines (in most cases) on substrates, referred to as protein ubiquitylation. The coordination of three consecutive enzymes catalyzes this process: the Ub activating E1 enzyme (2 genes), conjugating E2 enzyme (~40 genes), and the E3 ligase (600~700 genes). Cellular fates of ubiquitylated substrates are mainly determined by their ubiquitin chain linkage types1, 2. Of the eight linkage varieties, the lysine 48-linked polyubiquitin chain is the most abundant, serving as a canonical signal for proteasomal degradation. Through this signal, the ubiquitin proteasomal system (UPS) removes unwanted, defective, or misfolded proteins from cells, degrading ~80% of the human proteome3.
Constituting ~5% of the human genome, the E3 ligase family imparts the most specificity in selecting substrates for ubiquitylation4. As such, identifying the whole substrate landscape of each E3 ligase in different cellular contexts is critical for understanding the various roles and underlying molecular mechanisms of the ubiquitin pathway. Several high-throughput approaches have been developed to discover E3-substrate pairs5–11. However, each has technical limitations, which can result in a low yield of true positives from initial hits, requiring additional low-throughput validation. Luciferase reporting assays such as Yeast Two-Hybrid or in vitro ubiquitylation phage display offers high throughput options to find substrates through gene libraries. Yet, these assays can present irrelevant hits due to the absence of complex regulatory components such as chaperones, binding partners, or post-translational modifications in the assay systems. Alternatively, immunoprecipitation (IP) methods employing either a di-Gly antibody for ubiquitin remnant tryptic peptides (Lys-ϵ-Gly-Gly), tandem ubiquitin-binding entity (TUBE) or proximity labeling is limited by the dynamic kinetics of ubiquitylation, where substrates may quickly be degraded or de-ubiquitylated. In addition, comparing the total proteome between cell lines differentially expressing E3 ligases (i.e., wildtype vs. knock-out) allows for identifying proteins whose abundance is altered by the E3 ligase gene deletion. However, in this case, the data analysis is complicated because the protein abundance changes can be derived from both protein synthesis and degradation. Altogether, developing a generally applicable high-throughput method that can investigate E3-substrate pairs in relevant biological systems while maintaining sensitivity for highly probable hits will expand the current toolbox to identify the UPS-degraded substrates and advance the mechanistic study of an E3 ligase and its biological role.
In this study, we apply unnatural amino acid incorporation and a pulse-chase approach to profile the degradation of substrates without the interfering signal of newly translated proteins (differential degradomics). To our knowledge, this is an unexplored approach to identify bona fide substrates of an E3 ligase for proteasomal degradation. We hypothesize that cells lacking an E3 activity may exhibit significantly reduced degradation kinetics of the substrates, which should be detected by degradation proteomics (Figure 1A). To test this hypothesis, we combine the azidohomoalanine (AHA) pulse-chase method with the multiplexing tandem mass tagging (TMTpro)-mass spectrometry to quantify global protein half-lives in the corresponding cells13. We apply this method to identify degradation substrates of a largely uncharacterized E3 ligase complex, the C-terminal to LisH (CTLH). Comparative degradation profiling of cells expressing active and inactive CTLH E3 ligase identifies previously known substrates of the CTLH complex, MKLN1 and ZMYND19, as well as other novel potential substrates14, 15.
Figure 1.
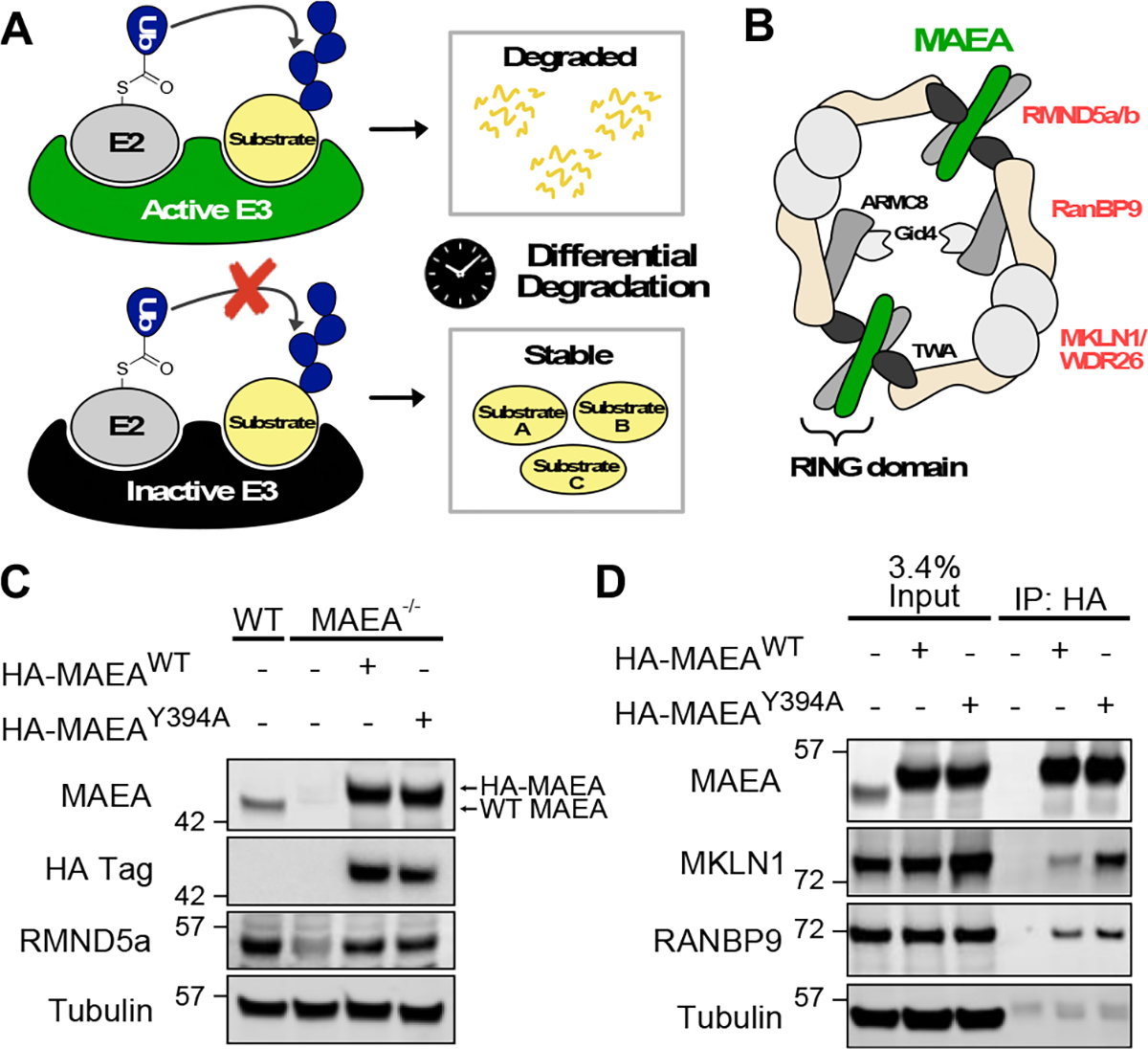
(A) Schematic description of the degradomics approach to identify substrates of an E3 ligase. (B) Cartoon of the CTLH complex based on the structural study12. (C) Validation of the HEK293T MAEA−/− cell lines over-expressing active (WT) or inactive (Y394A) forms of MAEA. (D) Immunoprecipitation of WT or Y394A MAEA to confirm their CTLH complex formation. Wild-type HEK293T cells were used for negative control.
Our results demonstrate several advantages of the differential degradomics approach. First, the unbiased quantitative degradomic analysis provides degradation kinetics of over 80% of the global proteome in a single experiment, thus proving to be truly high throughput. Second, as our readout is the ultimate degradation of proteins, substrate identification is not affected by the transient interaction between the E3 and substrates nor by dynamic ubiquitylation and deubiquitylation processes. Third, the AHA-degradomics workflow can potentially be applied to study E3 substrates under various cellular stimuli. This aspect is crucial as the activity of an E3 ligase and its substrate ubiquitylation are often regulated by special environmental cues. Lastly, in contrast to differential proteomics, differential degradomics specifically measures protein degradation affected by the E3 ligase of interest, thus decoupling the contribution of protein synthesis to the total protein expression. Altogether, differential degradomics can provide a facile, robust, and high-throughput screening of global proteome to define the substrate landscape of an E3 ligase under various cellular contexts.
Results and Discussion
Establishment of cell lines expressing an active or inactive form of CTLH E3 ligase.
The C-terminal to LisH (CTLH) E3 ligase comprises over seven components, forming an over 1 MDa complex (Figure 1B)12, 15, 16. Two of the seven components, MAEA and RMND5 (a or b), contain the RING domain for E3 ligase activity, which form a heterodimeric complex (Figure 1B). While the role of the CTLH ligase has been implicated in various cellular processes such as erythropoiesis, microtubule dynamics, and chromosome segregation, its direct ubiquitylation substrates in the affected pathways are poorly characterized17–20. To determine degradation substrates of the CTLH complex in mammalian cells, we engineered HEK293T cells to delete the MAEA gene (Figure 1C). As the MAEA protein forms a heterodimeric complex with RMND5, the loss of MAEA destabilized RMND5a in HEK293T cells. We then reintroduced the HA-tagged MAEAWT or a catalytically inactive MAEAY394A mutant into the knock-out cells using lentiviral vectors. The Y394A mutation results in a perturbed E2~ubiquitin thioester orientation, thereby inhibiting efficient nucleophilic attack of the epsilon amine on the substrate lysine 12, 21, 22. To confirm the incorporation of the exogenous MAEA into the CTLH complex, we enriched HA-MAEA using an anti-HA antibody. Immunoblotting the elute against antibodies for known CTLH components showed that MAEAWT and MAEAY394A expressing cells reacted with the MKLN1 and RANBP9 antibodies, whereas the control cells deficient in HA-tagged MAEA did not. In contrast, our negative control tubulin band showed equally low intensity across the three samples, representing non-specific bead binding. Of note, we detected more MKLN1 proteins in cells stably expressing MAEAY394A compared to MAEAWT-expressing cells. As MKLN1 is auto-ubiquitylated by the CTLH E3 ligase and subsequently degraded, the accumulation of MKLN1, specifically in MAEAY394A cells, confirms the catalytic inactivity of this mutant14. Altogether, comparing the degradation profiles of the MAEA WT and Y394A cell lines will only identify proteins whose half-lives are altered by the catalytic activity of the CTLH ligase, ruling out potential artifacts caused by the CRISPR-gene editing, clonal selection, or non-catalytic function of MAEA.
Workflow of the AHA-TMT-degradomics using the HEK293T cells expressing MAEAWT or MAEAY394A proteins
After validating the cell lines, we performed an 18-plex TMTpro-degradomics analysis using the MAEAWT and MAEAY394A cells. First, cells were incubated with a culture medium containing either methionine or its homolog azidohomoalanine (AHA) for 12 hours to allow methionine synthetase to incorporate AHA into the global proteome (Figure 2A)23. Following wash-out, the cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) for 5, 10, and 15 hours to pulse-chase the AHA-containing, pre-existing proteome. Each condition was duplicated or triplicated for statistical analysis. Importantly, the lysates from the individual plates were not normalized by the total protein levels to monitor the decay of the AHA-labeled pre-existing proteome over time24, 25. Therefore, testing the sample quality prior to the mass-spectrometry analysis was critical. To this end, proteins extracted from each plate were reacted with biotin-alkyne using copper-catalyzed click chemistry. At this point in the workflow, a western blot experiment using fluorescently tagged streptavidin with the biotinylated samples ensured three quality checkpoints (Figure 2B, C). First, the methionine-treated cell extract (sample1) showed less than 5% of the streptavidin signal compared to the AHA-treated cell extracts, indicating the strong enrichment of the AHA-labeled proteome. Second, there was little variation among the replicates. Third, the gradual decrease of the streptavidin signal over time in both genotypes indicated a successful pulse-chase of the pre-existing global proteome. Following the sample quality western blotting, we enriched the biotinylated proteins with streptavidin beads, digested the elutes with trypsin, and labeled the tryptic peptides with TMT-pro reagents (Figure 2B, see method for detail).
Figure 2.
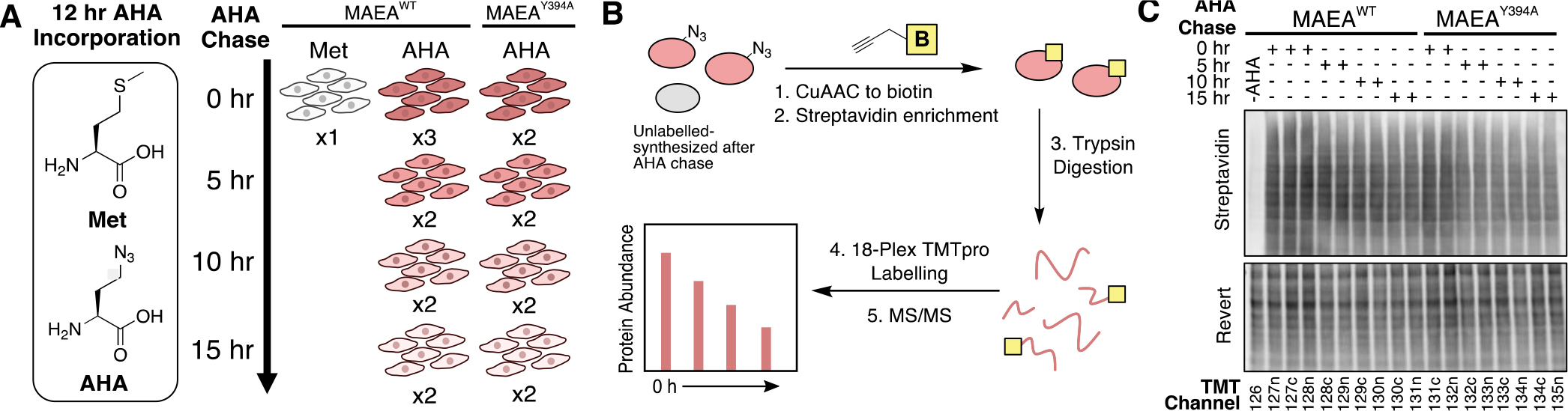
(A) Left: Structure of methionine (Met) and azidohomoalanine (AHA). Right: 18 biological samples were prepared as indicated and assigned to each channel of 18plex TMTpro reagent. (B) Workflow of AHA-TMT degradomics sample preparation. (C) Quality check western blot for AHA-containing proteome using streptavidin before immunoprecipitation.
Analysis of the AHA-TMT-degradomics
The TMTpro-labeled samples were analyzed using a real-time search and Multi-Notch MS3-based method for the highest quantitative accuracy26–28. We quantified 8,600 proteins across all 18 samples at a 1% false discovery rate, demonstrating comprehensive detection (~80% of the proteome coverage considering that the estimated proteome size of a typical cell is ~10,000 proteins)29. When normalized to the top 28 long-lived protein signals observed (see Supp Table 1 for the list), the average TMT signal of each channel showed gradual decay over time in both cell lines, while the first channel showed less than 5% of the TMT signal (Figure 3A), consistent with the streptavidin western blot (Figure 2C). The duplicated or triplicated samples were grouped together in the principal-component analysis (Figure 3B). Interestingly, the MAEAWT and MAEAY394A samples from different time points were grouped by the first principal component while separated by the second principal component. Considering that component 1 contributes 63.5% of the variance, major differences across the samples were derived from the degradation time points rather than the genotypes. Among the 8,600 proteins, 539 showed less than 5-hour half-lives, representing 6.3% of the quantified proteome. The gene ontology (GO) analysis of the 539 proteins for biological processes (BP) using DAVID showed that these proteins were enriched for the ubiquitylation and transcription-related pathways, consistent with a previous report (Figure 3D)30, 31. We concluded that our degradomics data showed excellent quality and coverage.
Figure 3.
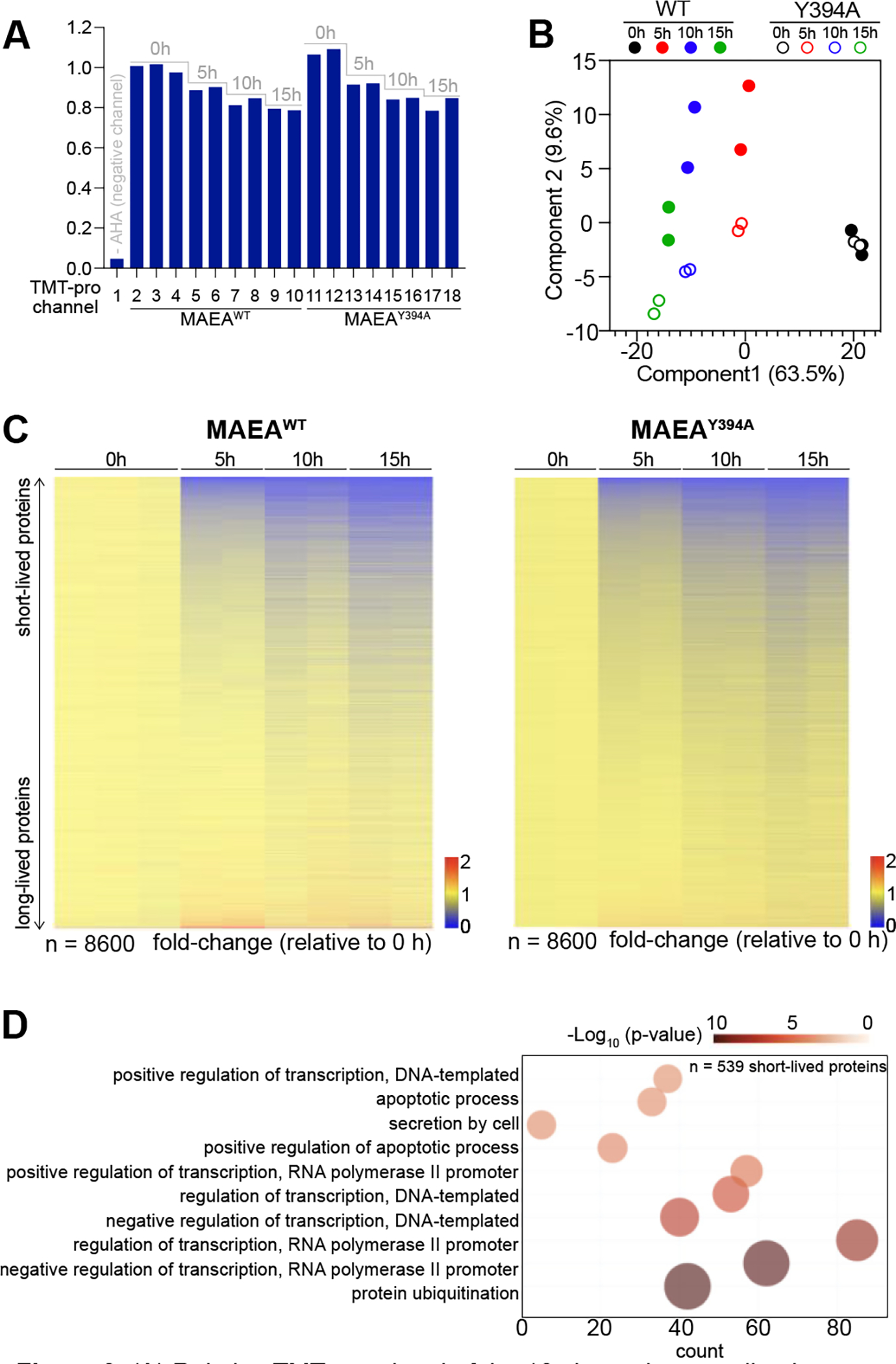
(A) Relative TMT-pro signal of the 18 channels normalized to channel 2. (B) Principal component analysis (PCA) shows the duplicated and triplicated samples grouped together. (C) Heat map of 8600 quantified proteins across samples exhibits gradual decay of AHA-enriched proteins in both cell lines. (D) DAVID GO analysis of the 539 short-lived proteins.
Differential degradomics identifies novel substrates of CTLH E3 ligase.
Next, we focused on identifying potential substrates of the CTLH complex by analyzing the protein half-life profiles. We hypothesized that CTLH degradation substrates would be selectively stabilized in MAEAY394A cells, whereas the AHA-TMT signal would steadily decrease in MAEAWT (Figure 2B). Two previously identified substrates of the CTLH E3 ligase, MKLN1 and ZMYND19, followed this expected pattern (Figure 4A)14, 15. We applied a simple regression analysis to calculate the half-lives of long-lived proteins (T1/2 > 5hrs), such as MKLN1, using the four time points. Using this method, T1/2 of MKLN1 was calculated to be 34.7 h in MAEAWT reconstituted cells; in comparison, MKLN1 became stabilized indefinitely in the absence of the active CTLH E3 ligase in our experimental setup (Figure 4A, top). This data suggests that CTLH mediated ubiquitylation and degradation is likely the only pathway to degrade MKLN1 in the present condition. A second order polynomial was fitted to approximate the half-lives of short-lived proteins (T1/2 < 5hrs), such as ZMYND19. The half-life of ZMYND19 was extended approximately 4-fold (from 4.6 h to 17 h) in the MAEAY394A mutant expressing cells. Notably, the pre-existing ZMYND19 signal was still reduced over time in the absence of the active CTLH, suggesting that an alternative degradation pathway exists to degrade ZMYND19. In summary, the estimated protein half-lives of MKLN1 and ZMYND19 were significantly extended without the active CTLH E3 ligase, supporting our hypothesis that differential degradomics can successfully identify substrates. It is important to note here that these proteins showed elevated expression levels in MAEAY394A cells at the 0h time point, likely due to the imbalance in these proteins’ synthesis and degradation during the 12 hours of AHA incorporation, leading to the steady-state accumulation (Figure 4A, bottom).
Figure 4.
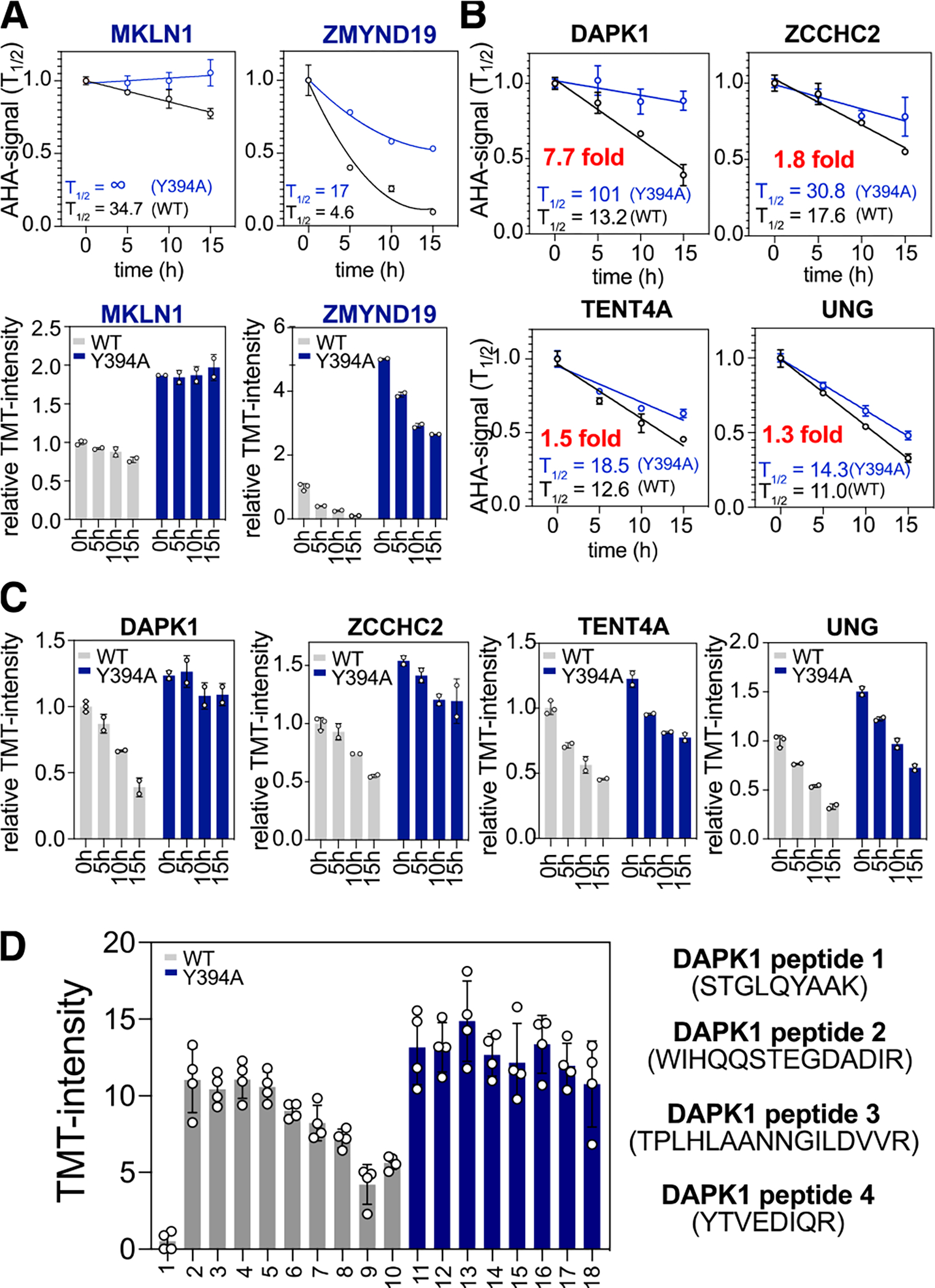
(A) Degradation curves normalized for t=0 h for each cell line (top) and corresponding AHA-TMT signal bar graph normalized to t = 0 h of MAEAWT (bottom) for previously discovered substrates of CTLH E3 ligase. (B) Degradation curves for four newly discovered substrates. (C) AHA-TMT signal bar graphs for the new substrates. (D) TMT signal bar graphs for four quantified DAPK1 peptides.
To identify novel CTLH substrates, we searched for proteins that showed similar degradation patterns to MKLN1 or ZMYND19. Stringent evaluations of the data increase the probability of true positive hits. Thus, we considered proteins with < 0.3 standard deviations across all eight conditions and more than two quantified peptides. The detailed parameters considered in our manual scoring strategy can be found in the method section. Surprisingly, after dataset filtering, only four proteins among the 8,600 quantified, showed consistent stabilization in the MAEAY394A cells at 5, 10, and 15 h time points (Figure 4B, C). These potential hits included DAPK1, ZCCHC2, TENT4A, and UNG, with more than a 1.3-fold extension of their half-lives in the absence of the active CTLH. Notably, ZCCHC2, TENT4A, and UNG signals were still reduced in the absence of functional CTLH, suggesting an involvement of other E3 ligases. For example, previous studies have shown that the UNG protein level is regulated by the cell cycle-dependent phosphorylation on the S23, T60, and S6432. It is plausible that CTLH is an additional E3 ligase that may regulate UNG turnover under a context other than the cell cycle, which requires further validation.
Death-associated protein kinase 1 (DAPK1) showed 13.2 hr of half-life in the MAEAWT reconstituted cells and 101 hours in the MAEAY394A cells (7.7-fold difference). Importantly, four unique peptides were quantified for DAPK1, and the TMTpro ratios of the four peptides show a similar stabilizing pattern in the inactive CTLH cells (Figure 4D).
Validation of the degradomics results with orthogonal approaches.
To validate the degradomics results, we initially performed western blot (WB) analysis by taking samples from 0 and 15 h AHA pulse-chase. We specifically enriched the AHA-labelled proteins using streptavidin beads after the copper-catalyzed alkyne-azide cycloaddition (CuAAC) reaction with biotin alkyne, as described for the proteomic analysis, and the elute analyzed by immunoblotting; samples were not normalized before loading for the reason described earlier, but the relative concentration of total protein in the elute (i.e., the AHA-labeled proteins) is depicted in the Ponceau stain and in the streptavidin WB (Figure 5A). Specifically, as expected, the streptavidin blot showed the reduction of the total AHA-labeled proteins collected after 15 hr pulse-chase. We tested commercially available antibodies for MKLN1, ZMYND19, DAPK1, ZCCHC2, and UNG proteins against HEK293T cell lysates, and we successfully reproduced the respective degradomics results using the antibodies against MKLN1 and UNG (Figure 5A). The other antibodies, however, showed non-specific smears or undetectable signals in the lysates. Promisingly, the two novel hits we were not able to experimentally confirm due to the lack of specific antibodies have been shown to interact with CTLH-complex components: from studies conducted in HEK293T cells, ZCCHC2 interacted with ARMC8, and TENT4A interacted with RMND5a33.
Figure 5.
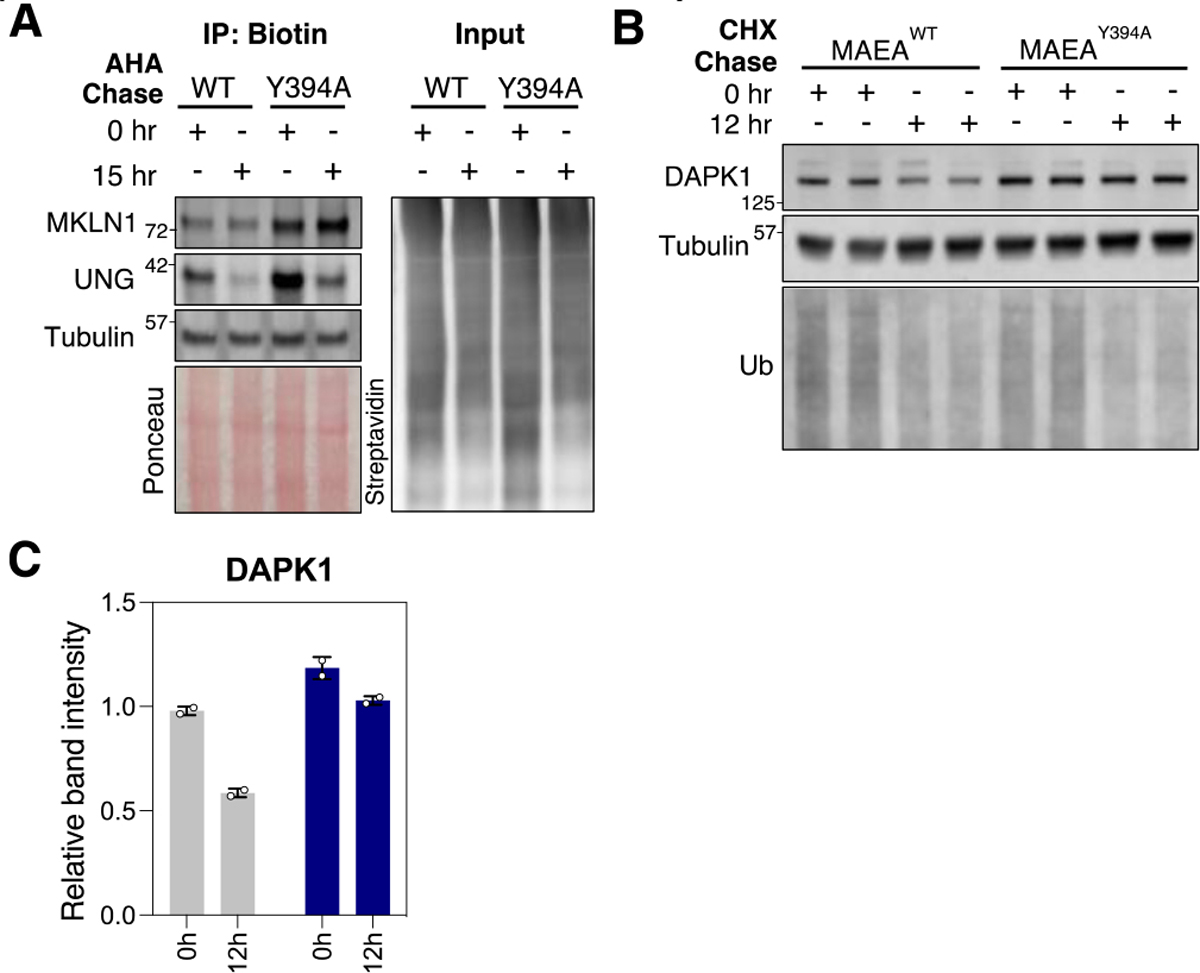
(A) Left: Biotin immunoprecipitation (IP) western blot of MKLN1 and UNG. Right: Streptavidin blot of IP input indicates total biotinylated protein between samples. (B) Western blot of duplicate samples treated with cycloheximide (CHX). (C) Quantification of the WB in panel B.
As an orthogonal approach to observe degradation dependent on CTLH activity, we treated the MAEAWT and MAEAY394A cells with a translation inhibitor, cycloheximide (CHX). Blotting for ubiquitin demonstrated that CHX reduced the global translation of new substrates for ubiquitylation, while long-lived protein tubulin remained stable (Figure 5B). Blocking translation for 12 h confirmed slowed degradation of DAPK1 in cells with inactive CTLH. In the validation experiments, the specific antibodies’ availability and the antibody reaction’s linearity limited the hit validation, highlighting another strength of the proteomic quantification. Given these preliminary findings, further validation of these hits as bona fide substrates of CTLH would involve in vitro ubiquitylation assays.
Degradomics data decouple the contribution of protein synthesis and degradation to the steady-state protein level.
Proteomic analysis of whole cell lysates derived from wild-type and E3 activity deficient samples has been used to identify potential substrates of the E3 ligase34, 35. Similarly, we obtained the steady-state protein expression levels by plotting the log2 ratio of MAEAY394A over MAEAWT at the 0-time point against the −log10 p-value (Figure 6A). In the volcano plot, TCEAL9 and AEN showed a dramatically upregulated expression in the MAEAY394A mutant cells, but their half-lives were not altered based on our degradation profiling (Figure 6B, C). The overall data suggest that the increased synthesis, rather than the decreased degradation, may have contributed to the high expression levels of TCEAL9 and AEN in the MAEAY394A cell line. Indeed, CHX pulse-chase western blotting confirmed that TCEAL9 was more expressed at 0-time point, which became rapidly degraded in both WT and mutant MAEA cell lines (Figure 6D).
Figure 6.
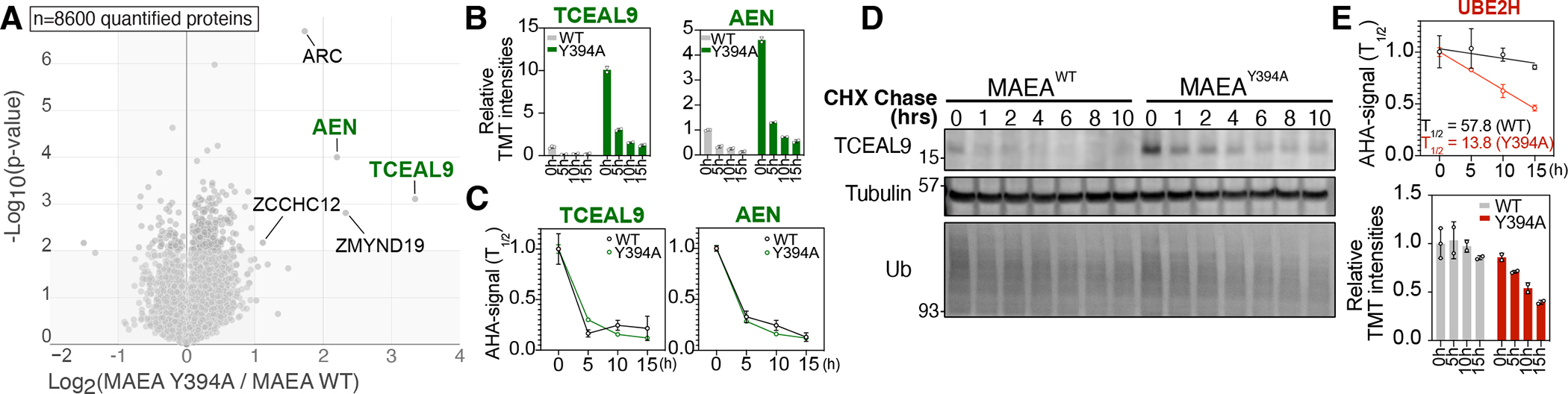
(A) Volcano plot of 0 h samples from each cell line compares steady state protein expression relative to CTLH activity. (B) TMT-AHA signal bar graphs (top) and degradation curves (bottom). (D) Western blot for Tceal9 with cells treated by cycloheximide (CHX) pulse-chase over 10 hours. (E) Degradation curve and AHA-TMT signal for E2 enzyme UBE2H show its destabilization in MAEAY394A cells.
In contrast to the substrates with observed CTLH-dependent degradation, the degradation of the cognate E2 enzyme for CTLH E3 ligase, UBE2H, was 4.2-fold accelerated in the Y394A mutant (from 57.8 h to 13.8 h, Figure 6E). This is due to the inefficient transfer of ubiquitin to the substrates, resulting in the autoubiquitylation of UBE2H and its subsequent degradation, as reported previously36. These examples highlight another advantage of the differential degradomics approach: the differences in both steady-state protein levels and the degradation kinetics can be projected simultaneously, thus providing in-depth information about the homeostasis of the protein of interest.
Conclusion
In this study, we investigated the advantage of AHA-pulse chase proteomics as a novel approach to identifying the degradation substrates of an E3 ligase. We chose CTLH E3 ligase as a case study because its role has been implicated in several cellular pathways, but its direct substrates are poorly characterized. Previous structural studies allowed us to establish a delicate system where the catalytically inactive mutant inhibits degradation of the substrates while maintaining the integrity of the CTLH E3 complex12. This resulted in a subtly perturbed condition to interrogate the catalytic activity of an E3 ligase. Furthermore, using AHA pulse-chase, rather than translation or protease inhibitors, to measure protein degradation limits cytotoxicity. Therefore, a great strength of this approach is the preservation of biological context. Another advantage of this approach is its relatively simple workflow. In our experience, the CRISPR-cell line generation and AHA-proteomic analyses take about eight weeks. This resulted in a high throughput method to discover probable E3-substate pairs, which result in substrate degradation. Furthermore, the degradation kinetics give an insight into whether substrate degradation by the E3 investigated is exclusive or redundant. Overall, the wide range of options to develop differentially expressed E3 ligases, and the ease of applying AHA-degradomics make this method universally applicable to a wide variety of E3 ligases, cell lines, and stimuli conditions.
Moving forward, there are several points to consider when applying this approach to other E3 ligases. First, in this study, we measured degradation substrates without special stimuli. However, substrates of E3 ligases dynamically change depending on cellular environmental changes. For example, the yeast ortholog of the CTLH complex, the GID complex, ubiquitylates specific substrates only during metabolic switches from gluconeogenesis to glycolysis37. Because the stress condition can be easily incorporated during the pulse-chase step of our workflow, the differential degradomics could be used to map E3-substrate pairs important for stress response pathways. Second, the constitutive deletion of the E3 ligase activity can result in global compensatory changes, as observed in the increased translation of AEN and TCEAL9 transcripts in cells with inactive CTLH. A suggestion to minimize this effect would be to induce rapid degradation of the E3 protein via a degradation tag system prior to AHA treatment38. Third, although we have successfully validated two previously identified substrates of CTLH, MKLN1 and ZYMND19, and four new potential substrates, our approach is limited to identifying the degradational substrates. If a substrate is ubiquitylated for one of the multiple proteasomal degradation-independent pathways, we will not identify it as a hit. Therefore, if an E3 ligase is not involved in the protein degradation function, then considering other methods than the differential degradomics would be appropriate. Lastly, if applicable, a direct measure of ubiquitylation, such as in vitro ubiquitylation assay, is required to confirm the degradomics results because the stabilization of proteins can be a secondary effect caused by an E3 ligase activity change.
Experimental Section
Reagents:
The following antibodies and reagents were used: MKLN1 (Santa Cruz, sc-398956), RMND5a (ProteinTech, 17559), MAEA (ProteinTech, 28363), ARMC8 (ProteinTech, 12653), RMND5b (ProteinTech, 25803), HA tag (ThermoFisher Scientific, 26183-HRP), RanBP9 (Cell Signaling, 14638S), TCEAL9 (Sigma Aldrich, HPA011790), Tubulin (Sigma Aldrich, T9026), ZCCHC2 (Abcam, ab88756; Sigma Aldrich, HPA040943), DAPK1 (Cell Signaling, 3008), UNG (Cell Signaling, 12394–1-AP), Ubiquitin (Santa Cruz, sc-8017), IRDye 800CW Streptavidin (LI-COR, 926–32230), IRDye 800CW goat anti-rabbit IgG H+L (LI-COR, 925–32211) and IRDye 680 RD goat anti-mouse IgG H+L (LI-COR, 926–68070). The following chemical reagents were used: azidohomoalanine (Click Chemistry Tools, 1066–1000), Fluor 488-Aklyne (Sigma-Aldrich, 761621), biotin-PEG4-alkyne (Click Chemistry Tools, TA105–25), benzonase nuclease HC (Millipore, 71205–3), sodium dodecyl sulfate (SDS) (Fisher Scientific, PI28364), chloroacetamide (Fisher Scientific, AAA1523830), Revert Total Protein Stain kit (LI-COR, 926–11021), Gibco DMEM, high glucose, no glutamine, no methionine, no cystine (Fisher Scientific, 21–013-024), Sodium pyruvate (Gibco, 11360–070), L-glutamine solution (Sigma-Aldrich, G7513), cycloheximide (Fisher Scientific, AC357420050). TCEP (Gold Biotechnology), Formic acid (Sigma Aldrich, 94318), TMTpro 18plex Label Reagent (Fisher Scientific, ##), InstantBlue (Abcam, ab119211), Protein Assay Dye Reagent Concentrate (Bio-Rad, 5000006), Monoclonal anti-HA agarose (Sigma-Aldrich, A2095), high capacity streptavidin agarose resin (Thermo Fisher Scientific, 20359), hexafluoroisopropanol (HFIP) (Acros Organics, 445820100).
Cell lines:
HEK293T cells, purchased from the American Type Culture Collection (ATCC), were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and kept in an incubator maintaining 5% CO2 at 37°C.
Generation of CRISPR knock-out and stable cell lines:
MAEA gene-knockout cell lines were generated in 293T cells by plasmid-based transient transfection of pX459 plasmid expressing Cas9/gRNA. Briefly, three guide RNAs were selected based on Chop-chop website (https://chopchop.cbu.uib.no) and tested by the pooled-western blot analysis. The following guide was chosen for the clonal selection: 5’-CGCACGCCCTACCTTGTACG-3’. The complete knock-out of MAEA was confirmed by western blotting and mass-spectrometry. The base gene block for MAEA was acquired from the orfeome library v8 (pDORN223 backbone), and further edited to express full-length MAEA WT and Y394A mutant. Gateway cloning method was then used to generate pHAGE-Flag-HA-MAEA plasmids. The MAEA KO HEK293T cell line was transduced with the corresponding lentiviral vector.
Cell lysis and immunoblotting assay:
Cells were harvested from 6-cm or 15-cm dishes not exceeding 70% confluency. RIPA buffer (50 mM HEPES, 150 mM NaCl, 1% sodium deoxycholate, 1% NP-40, 0.1% SDS, 10 mM sodium pyrophosphate, 10 mM β-glycerol phosphate, 2.5 mM MgCl2, 200 μM TCEP, 50 U/mL benzonase, and protease inhibitors) was added to cell pellets or cell plates following scraping cells. Protein concentration of samples were measured with Bradford assay and normalized, followed by addition of LDS supplemented with 50 mM DTT, and incubation for 5 min at 75°C. 30 μg of each lysate was loaded on a 4–12% NuPAGE Bis-Tris gel (Thermo Fisher Scientific) and run with MES SDS running buffer (50 mM MES, 50 mM Tris Base, 1 mM EDTA, and 0.05% SDS). Proteins were transferred to PVDF membranes (0.45 μm, Millipore), and total protein was stained with Ponceau staining (Thermo Fisher Scientific) or Revert total protein stain kit (LI-COR). After blocking with 5% non-fat milk in TBST (30 min, r.t.), the membrane was incubated with primary antibodies overnight at 4°C, washed twice with TBST, and incubated with fluorescent IRDye antibody (1:15000, LI-COR) for 1 h. Biotinylated proteins were directly probed with the Streptavidin-IRDye800 (LI-COR). After 20 min wash with TBST, the membrane was imaged using OdysseyCLx (LI-COR) imager or Chemidoc MP (Bio-Rad).
Immunoprecipitation of HA-MAEA:
Cells were harvested from 15-cm dishes not exceeding 70% confluency by washing 3x with DPBS, adding lysis buffer, and scraping the lysate on ice. Samples were normalized by total protein concentration measured by Bradford assay. For each sample, 2.5 mg of the total lysate was incubated with 15 μL of HA-agarose beads at 4 °C for 2 hr with rotation. After the flowthrough was collected, beads were washed with DPBS 3x, and incubated with 1x LDS for 5 min at 55 °C and proteins eluted through a centrifugation filter cup. Immunoblotting was performed as previously described.
Cyclohexamide treatment experiment:
MAEAWT and MAEAY394A cells were plated at least 12 hours before treatment with cycloheximide (CHX) to a final concentration of 10 μM in media. At corresponding time points post-treatment, the cells were collected (5k rpm, 3 minutes), washed once with DPBS, and pelleted again by centrifugation. Cells were lysed with RIPA buffer, sonicated, and normalized by Bradford assay before loading onto SDS-PAGE gel and performing the immunoblotting assay as described earlier.
Azidohomoalanine (AHA) pulse-chase for degradomics, lysis, and alkylation:
Protocol adapted from previous work25. In general, this proteomic workflow results in the quantification of protein degradation by measuring the quantity of only pre-existing proteins labelled with azidohomoalanine (AHA) over time. Over the pulse-chase wash with methionine (Met)-media, the newly synthesized proteins will not incorporate AHA and, therefore, will be discarded during AHA enrichment. More specifically, methionine (Met) free DMEM base was first prepared by supplementing DMEM without glutamine, pyruvate, methionine, and cystine (ThermoFisher) with 10% dialyzed fetal bovine serum, 1 mM sodium pyruvate, 4 mM L-glutamine, 250 μM cystine. The media was filtered through a 0.2-μm filter and stored at 4°C. Either AHA (250 μM) or Met (250 μM) was added to the Met-free media base within twelve hours of adding to the cells. Cells were plated onto 6 × 15-cm dishes with 60% confluency for 24 h before replacing rich DMEM media with the corresponding AHA-DMEM medium. One additional 10-cm plate for negative control was treated with 250 μM Met media. After 12 h, cells were collected by washing with rich DMEM and divided equally among 8 × 10-cm dishes to ensure equal distribution of AHA incorporation among cells. At this point, 3 (for WT) and 2 (for Y394A) x 10-cm plate portions of cells and the negative control plate were collected for 0 h timepoint. At 5, 10, and 15 h after wash-out, all the cells from two plates were collected into tubes and washed 3 times with DPBS, and the pellet was stored at −80 °C until all samples were collected. Pellets were lysed with equal volumes of RIPA buffer (modified with 2% SDS instead of 0.1%) and sonicated 3x with a tip sonicator. In subsequent quality control experiments or assays, lysates were not normalized by Bradford assay to avoid the dilution effect of AHA-labelled proteome through cell division. Lysates were reduced with 5 mM TCEP 10 min, r.t., and alkylated with 20 mM chloroacetamide, 15 min, r.t. followed by MeOH/CHCl3 protein precipitation resulting in white protein disk. Proteins were fully resuspended in 2% SDS (50 mM HEPES, 150 mM NaCl, pH 7.2, 2.5 mM TCEP) with sonication.
AHA incorporation analysis by in-gel fluorescence:
For quality control check of AHA incorporation into cells, click reagents were added to a small aliquot (10 μM) of 2% SDS resuspended cell lysates with final concentration of 100 μM Tris(benzyltriazolylmethyl)amine (TBTA) ligand, 1 mM CuSO4, 1 mM sodium ascorbate, and 100 μM Alexa Fluor 488-alkyne. The samples were incubated for 1 h at r.t. protected from light. The samples were denatured by adding LDS sample buffer and incubating at 75°C for 5 min and resolved by SDS-PAGE. The Alexa Fluor 488 signal was detected by ChemiDoc MP (Bio-Rad).
Biotin click and streptavidin immunoprecipitation of AHA incorporated proteins:
To remaining proteins resuspended in 2% SDS, 100 μM TBTA ligand, 1 mM CuSO4, 1 mM sodium ascorbate, and 100 μM biotin-alkyne click reagents were added and samples incubated for 2 h r.t. while rotating. Following incubation, proteins were precipitated with MeOH/CHCl3 extraction again to remove excess biotin, and the pellet was resuspended in 2% SDS and sonicated. Equal volumes of lysates were diluted with HEPES buffer (50 mM HEPES, 150 mM NaCl, pH 7.4), so the final SDS concentration is <0.5%. 20 μL of streptavidin agarose beads were added to each sample and incubated for 3 hr at room temperature. Flow through was stored for quality control, and beads were thoroughly washed with RIPA x2, 2% SDS 2x, 3 M Urea 2x, 0.1 M Na2CO3 2x, and RIPA 2x. The beads were washed with water once and transferred onto hydrophilic PTFE membrane filter cups (Millipore Sigma). The beads were washed two more times with water. To elute proteins from agarose beads, 50 μM of hexafluoroisopropanol (HFIP) was added to the dried beads, incubated for 5 min shaking, and eluted by spinning them into a low-binding 1.5 mL tube. Another 50 μM of HFIP was added to beads, combined with first eluate and dried in SpeedVac for 10 min (HFIP bp = 58 °C, volatile). The dried sample was used for either WB or TMTpro-MS analysis.
TMTpro-MS sample preparation and analysis:
The eluted AHA proteome was reconstituted in 100 mM EPPS (pH 8.5) and digested by trypsin (1:100 trypsin/protein ratio) overnight. Samples were TMTpro-labelled for 60 min at room temperature, and the labeling efficiency was checked by mass-spectrometry. The remaining samples were quenched, pooled, C18 desalted, and dried down. The dried TMTpro-labeled sample was resuspended in 100 μl of 10 mM NH4HCO3 pH 8.0 and fractionated using basic pH reverse phase HPLC into 96 fractions pooled in a non-continuous manner into 24 fractions39. Each consolidated fraction was desalted and dried down prior to LC-MS/MS analysis. Mass spectrometry data were collected using an Orbitrap Eclipse Tribrid mass spectrometer coupled to an UltiMate 3000 RSLCnano system liquid chromatography pump. Peptides were separated on a 100 μm inner diameter microcapillary column packed in-house with ~40 cm of HALO Peptide ES-C18 resin (2.7 μm, 160 Å) with a gradient consisting of 5%–24% (0–85 min), 24–36% (85–110min) (ACN, 0.1% FA) over a total 120 min run at ~500 nL/min. Each analysis used the Multi-Notch MS3-based TMT method27, to reduce ion interference compared to MS2 quantification40, to reduce ion interference compared to MS2 quantification40, combined with the FAIMS Pro Interface 41 and combined with newly implemented Real-Time Search analysis software 26, 28. The scan sequence began with an MS1 spectrum (Orbitrap analysis; resolution 120,000 at 200 Th; mass range 400–1500 m/z; automatic gain control (AGC) target 4×105; maximum injection time 50 ms). Precursors for MS2 analysis were selected using a cycle type of 1.25 sec/CV method. MS2 analysis consisted of collision-induced dissociation (quadrupole ion trap analysis; Rapid scan rate; AGC 1.0×104; isolation window 0.5 Th; normalized collision energy (NCE) 35; maximum injection time 35 ms). Monoisotopic peak assignment was used and previously interrogated precursors were excluded using a dynamic window (150 s ±10 ppm). MS3 precursors were fragmented by high energy collision-induced dissociation (HCD) and analyzed using the Orbitrap (NCE 45; AGC 2.5×105; maximum injection time 200 ms, the resolution was 50,000 at 200 Th). The closeout was set at two peptides per protein per fraction so that MS3s were no longer collected for proteins having two peptide-spectrum matches (PSMs) that passed the quality filters 28. Mass spectra were processed using a Comet-based (2019.01 rev. 5) software pipeline 42. Spectra were converted to mzXML, and monoisotopic peaks were re-assigned using Monocle 43. MS/MS spectra were matched with peptide sequences using the Comet algorithm along with a composite sequence database, including the Human Reference Proteome (2020–01 - SwissProt entries only) UniProt database and sequences of common contaminants. Searches were performed using a 50 ppm precursor ion tolerance and the recommended product ion parameters for ion trap. TMTpro tags on lysine residues and peptide N termini (+304.207 Da) and carbamidomethylation of cysteine residues (+57.021 Da) were set as static modifications, while oxidation of methionine residues (+15.995 Da) was set as a variable modification. Peptide-spectrum matches (PSMs) were adjusted to a 1% false discovery rate (FDR) 44 and then collapsed further to a final protein-level FDR of 1%. Moreover, protein assembly was guided by principles of parsimony to produce the smallest set of proteins necessary to account for all observed peptides. For TMTpro-based reporter ion quantitation, we extracted the summed signal-to-noise (S:N) ratio for each TMTpro channel and found the closest matching centroid to the expected mass of the TMT reporter ion (integration tolerance of 0.003 Da). Reporter ion intensities were adjusted to correct for the isotopic impurities of the different TMTpro reagents according to manufacturer specifications. Proteins were quantified by summing reporter ion signal-to-noise measurements across all matching PSMs, yielding a “summed S:N” measurement. PSMs with poor quality, MS3 spectra with four or more TMT reporter ion channels missing, isolation specificity less than 0.5, or TMT reporter summed S:N that was less than 120 or had no MS3 spectra were excluded from quantification. Protein or peptide quantification values were exported for further analysis in Microsoft Excel, GraphPad Prism, and Perseus 45. The supplemental data Table lists all quantified proteins and the associated TMT reporter ratio to control channels used for quantitative analysis.
Data analysis:
The projection of the dataset by PCA was obtained using Perseus software and visualized by the Prism9 software. The DAVID GeneOntology analysis was performed by listing the 539 short-lived proteins (T1/2<5h) and comparing them with the DAVID-default homo sapiens whole genome as background. Individual protein values were normalized to the 0h time point was subjected for PCA analysis with Benjamini-Hochberg cutoff with 0.05 FDR prior to the Log2 transformation. Heatmap was generated using the Morpheus (Broad Institute).
Hit identification strategy:
(1) The initial candidates were first curated by comparing the average TMT ratio between WT and the mutant cells at 15h time point (normalized to the WT 0h). 183 proteins showed more than 30% higher signal in the mutant cells. Our positive controls ZMYND19 ranked 4 and MKLN1 ranked 7 in this filtering. (2) To increase the chance of finding true positives, we used stringent filtering on the standard deviation: we selected proteins with < 0.3 standard deviations across all 8 different conditions, resulting in 91 proteins. (3) Next, we normalized the mutant TMT signals with the mutant 0h signal instead of the WT 0h signal and compared the difference between WT and the mutant values at 15 h values. Only 40 proteins showed > 10% difference. (4) proteins having 1 unique peptide were filtered out, resulting in 22 proteins. The T1/2 of these proteins were finally compared, and proteins showing consistent stabilization at each time point were chosen.
Supplementary Material
Acknowledgements
This work was supported by the Josie Robertson Foundation and Geoffrey Beene Grant (H.A.), as well as NIH T32 GM136640-Tan to V.N.J. This research was also in part supported by the Memorial Sloan Kettering Cancer Center Support Grant P30CA008748 (A.O. and H.A.)
References
- 1.Swatek KN and Komander D, Cell Res, 2016, 26, 399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Komander D and Rape M, Annu Rev Biochem, 2012, 81, 203–229. [DOI] [PubMed] [Google Scholar]
- 3.Collins GA and Goldberg AL, Cell, 2017, 169, 792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng N and Shabek N, Annu Rev Biochem, 2017, 86, 129–157. [DOI] [PubMed] [Google Scholar]
- 5.Bex C, Knauth K, Dambacher S and Buchberger A, Nucleic Acids Res, 2007, 35, e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang H, Jedynak BM and Bader JS, PLoS Comput Biol, 2007, 3, e214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jiang H, Chiang CY, Chen Z, Nathan S, D’Agostino G, Paulo JA, Song G, Zhu H, Gabelli SB and Cole PA, J Biol Chem, 2022, 298, 101854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nardone C, Palanski BA, Scott DC, Timms RT, Barber KW, Gu X, Mao A, Leng Y, Watson EV, Schulman BA, Cole PA and Elledge SJ, Mol Cell, 2023, 83, 57–73 e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Udeshi ND, Mani DR, Eisenhaure T, Mertins P, Jaffe JD, Clauser KR, Hacohen N and Carr SA, Mol Cell Proteomics, 2012, 11, 148–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iconomou M and Saunders DN, Biochem J, 2016, 473, 4083–4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X, Thielert M, Li H and Cravatt BF, Biochemistry, 2021, 60, 637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sherpa D, Chrustowicz J, Qiao S, Langlois CR, Hehl LA, Gottemukkala KV, Hansen FM, Karayel O, von Gronau S, Prabu JR, Mann M, Alpi AF and Schulman BA, Mol Cell, 2021, 81, 2445–2459 e2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dieterich DC, Link AJ, Graumann J, Tirrell DA and Schuman EM, Proc Natl Acad Sci U S A, 2006, 103, 9482–9487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maitland MER, Onea G, Chiasson CA, Wang X, Ma J, Moor SE, Barber KR, Lajoie GA, Shaw GS and Schild-Poulter C, Scientific Reports, 2019, 9, 9864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohamed WI, Park SL, Rabl J, Leitner A, Boehringer D and Peter M, EMBO Rep, 2021, 22, e52981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lampert F, Stafa D, Goga A, Soste MV, Gilberto S, Olieric N, Picotti P, Stoffel M and Peter M, Elife, 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wei Q, Pinho S, Dong S, Pierce H, Li H, Nakahara F, Xu J, Xu C, Boulais PE, Zhang D, Maryanovich M, Cuervo AM and Frenette PS, Nat Commun, 2021, 12, 2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhen R, Moo C, Zhao Z, Chen M, Feng H, Zheng X, Zhang L, Shi J and Chen C, Blood, 2020, 135, 208–219. [DOI] [PubMed] [Google Scholar]
- 19.Heisler FF, Loebrich S, Pechmann Y, Maier N, Zivkovic AR, Tokito M, Hausrat TJ, Schweizer M, Bahring R, Holzbaur EL, Schmitz D and Kneussel M, Neuron, 2011, 70, 66–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sherpa D, Mueller J, Karayel O, Xu P, Yao Y, Chrustowicz J, Gottemukkala KV, Baumann C, Gross A, Czarnecki O, Zhang W, Gu J, Nilvebrant J, Sidhu SS, Murray PJ, Mann M, Weiss MJ, Schulman BA and Alpi AF, Elife, 2022, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiao S, Langlois CR, Chrustowicz J, Sherpa D, Karayel O, Hansen FM, Beier V, von Gronau S, Bollschweiler D, Schafer T, Alpi AF, Mann M, Prabu JR and Schulman BA, Mol Cell, 2020, 77, 150–163 e159. [DOI] [PubMed] [Google Scholar]
- 22.Plechanovova A, Jaffray EG, Tatham MH, Naismith JH and Hay RT, Nature, 2012, 489, 115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kiick KL, Saxon E, Tirrell DA and Bertozzi CR, Proc Natl Acad Sci U S A, 2002, 99, 19–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McShane E, Sin C, Zauber H, Wells JN, Donnelly N, Wang X, Hou J, Chen W, Storchova Z, Marsh JA, Valleriani A and Selbach M, Cell, 2016, 167, 803–815 e821. [DOI] [PubMed] [Google Scholar]
- 25.An H, Ordureau A, Körner M, Paulo JA and Harper JW, Nature, 2020, 583, 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Erickson BK, Mintseris J, Schweppe DK, Navarrete-Perea J, Erickson AR, Nusinow DP, Paulo JA and Gygi SP, J Proteome Res, 2019, 18, 1299–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McAlister GC, Nusinow DP, Jedrychowski MP, Wuhr M, Huttlin EL, Erickson BK, Rad R, Haas W and Gygi SP, Anal Chem, 2014, 86, 7150–7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schweppe DK, Eng JK, Yu Q, Bailey D, Rad R, Navarrete-Perea J, Huttlin EL, Erickson BK, Paulo JA and Gygi SP, J Proteome Res, 2020, 19, 2026–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Harper JW and Bennett EJ, Nature, 2016, 537, 328–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dennis G Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC and Lempicki RA, Genome Biol, 2003, 4, P3. [PubMed] [Google Scholar]
- 31.Li J, Cai Z, Vaites LP, Shen N, Mitchell DC, Huttlin EL, Paulo JA, Harry BL and Gygi SP, Mol Cell, 2021, 81, 4722–4735 e4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hagen L, Kavli B, Sousa MM, Torseth K, Liabakk NB, Sundheim O, Pena-Diaz J, Otterlei M, Hørning O, Jensen ON, Krokan HE and Slupphaug G, Embo j, 2008, 27, 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huttlin EL, Bruckner RJ, Navarrete-Perea J, Cannon JR, Baltier K, Gebreab F, Gygi MP, Thornock A, Zarraga G, Tam S, Szpyt J, Gassaway BM, Panov A, Parzen H, Fu S, Golbazi A, Maenpaa E, Stricker K, Guha Thakurta S, Zhang T, Rad R, Pan J, Nusinow DP, Paulo JA, Schweppe DK, Vaites LP, Harper JW and Gygi SP, Cell, 2021, 184, 3022–3040.e3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nie L, Wang C, Li N, Feng X, Lee N, Su D, Tang M, Yao F and Chen J, Mol Cell Proteomics, 2020, 19, 2015–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burande CF, Heuze ML, Lamsoul I, Monsarrat B, Uttenweiler-Joseph S and Lutz PG, Mol Cell Proteomics, 2009, 8, 1719–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sherpa D, Mueller J, Karayel Ö, Xu P, Yao Y, Chrustowicz J, Gottemukkala KV, Baumann C, Gross A, Czarnecki O, Zhang W, Gu J, Nilvebrant J, Sidhu SS, Murray PJ, Mann M, Weiss MJ, Schulman BA and Alpi AF, Elife, 2022, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Santt O, Pfirrmann T, Braun B, Juretschke J, Kimmig P, Scheel H, Hofmann K, Thumm M and Wolf DH, Mol Biol Cell, 2008, 19, 3323–3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nabet B, Roberts JM, Buckley DL, Paulk J, Dastjerdi S, Yang A, Leggett AL, Erb MA, Lawlor MA, Souza A, Scott TG, Vittori S, Perry JA, Qi J, Winter GE, Wong KK, Gray NS and Bradner JE, Nat Chem Biol, 2018, 14, 431–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Paulo JA, O’Connell JD, Everley RA, O’Brien J, Gygi MA and Gygi SP, J Proteomics, 2016, 148, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paulo JA, O’Connell JD and Gygi SP, J Am Soc Mass Spectrom, 2016, 27, 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schweppe DK, Prasad S, Belford MW, Navarrete-Perea J, Bailey DJ, Huguet R, Jedrychowski MP, Rad R, McAlister G, Abbatiello SE, Woulters ER, Zabrouskov V, Dunyach JJ, Paulo JA and Gygi SP, Anal Chem, 2019, 91, 4010–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eng JK, Jahan TA and Hoopmann MR, Proteomics, 2013, 13, 22–24. [DOI] [PubMed] [Google Scholar]
- 43.Rad R, Li J, Mintseris J, O’Connell J, Gygi SP and Schweppe DK, J Proteome Res, 2021, 20, 591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elias JE and Gygi SP, Nat Methods, 2007, 4, 207–214. [DOI] [PubMed] [Google Scholar]
- 45.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M and Cox J, Nat Methods, 2016, 13, 731–740. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.