

ABSTRACT
Sertoli cells are highly polarized testicular cells that provide a nurturing environment for germ cell development and maturation during spermatogenesis. The class III phosphatidylinositol 3-kinase (PtdIns3K) plays core roles in macroautophagy in various cell types; however, its role in Sertoli cells remains unclear. Here, we generated a mouse line in which the gene encoding the catalytic subunit, Pik3c3, was specifically deleted in Sertoli cells (cKO) and found that after one round of normal spermatogenesis, the cKO mice quickly became infertile and showed disruption of Sertoli cell polarity and impaired spermiogenesis. Subsequent proteomics and phosphoproteomics analyses enriched the F-actin cytoskeleton network involved in the disorganized Sertoli-cell structure in cKO testis which we identified a significant increase of the F-actin negative regulator SCIN (scinderin) and the reduced phosphorylation of HDAC6, an α-tubulin deacetylase. Our results further demonstrated that the accumulation of SCIN in cKO Sertoli cells caused the disorder and disassembly of the F-actin cytoskeleton, which was related to the failure of SCIN degradation through the autophagy-lysosome pathway. Additionally, we found that the phosphorylation of HDAC6 at site S59 by PIK3C3 was essential for its degradation through the ubiquitin-proteasome pathway. As a result, the HDAC6 that accumulated in cKO Sertoli cells deacetylated SCIN at site K189 and led to a disorganized F-actin cytoskeleton. Taken together, our findings elucidate a new mechanism for PIK3C3 in maintaining the polarity of Sertoli cells, in which both its autophagy regulation or protein kinase activities are required for the stabilization of the actin cytoskeleton.
Abbreviations: ACTB: actin, beta; AR: androgen receptor; ATG14: autophagy related 14; BafA1: bafilomycin A1; BECN1: beclin 1, autophagy related; BTB: blood-testis barrier; CASP3: caspase 3; CDC42: cell division cycle 42; CDH2: cadherin 2; CHX: cycloheximide; CTNNA1: catenin (cadherin associated protein), alpha 1; CYP11A1: cytochrome P450, family 11, subfamily A, polypeptide 1; EBSS: Earle’s balanced salt solution; ES: ectoplasmic specialization; FITC: fluorescein isothiocyanate; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GCNA: germ cell nuclear acidic protein; GJA1: gap junction protein, alpha 1; H2AX: H2A.X variant histone; HDAC6: histone deacetylase 6; KIT: KIT proto-oncogene, receptor tyrosine kinase; LAMP1: lysosomal associated membrane protein 1; MAP3K5: mitogen-activated protein kinase kinase kinase 5; MAP1LC3B: microtubule associated protein 1 light chain 3 beta; OCLN: occludin; PIK3C3: phosphatidylinositol 3-kinase catalytic subunit type 3; PIK3R4: phosphoinositide-3-kinase regulatory subunit 4; PNA: arachis hypogaea lectin; RAC1: Rac family small GTPase 1; SCIN: scinderin; SQSTM1/p62: sequestosome 1; SSC: spermatogonia stem cell; STK11: serine/threonine kinase 11; TJP1: tight junction protein 1; TubA: tubastatin A; TUBB3: tubulin beta 3 class III; TUNEL: TdT-mediated dUTP nick-end labeling; UB: ubiquitin; UVRAG: UV radiation resistance associated gene; VIM: vimentin; WT1: WT1 transcription factor; ZBTB16: zinc finger and BTB domain containing 16.
KEYWORDS: Autophagy, cell polarity, F-actin, Sertoli cell, spermatogenesis
Introduction
Cell polarity, characterized by the asymmetric distribution of different morphologies or molecular components across a cell, is necessary for diverse cellular processes, such as adhesion, migration, differentiation and cell fate determination [1,2]. Mature Sertoli cells of the adult mammalian testis are highly polarized supporting cells whose functions are to nurture all stages of germ cells and support their development from prospermatogonia into sperm [3]. They are tall irregular columnar-like cells that are arranged perpendicular to the basement membrane of the seminiferous epithelium and are characterized by many processes that form on their surface and different morphologies from the base of the tubule to its lumen [4]. The complexity and uniqueness of cell shape are consistent with the high degree of apical-basal polarity in Sertoli cells, which allows the cells to compartmentalize various cellular interactions along the seminiferous epithelium and ensures that spermatogenesis occurs in a highly organized manner both spatially and temporally [5].
During spermatogenesis, germ cell transport across the seminiferous epithelium depends on testis-specific anchoring junctions known as ectoplasmic specialization (ES) [6]. There are two types of ES in testes: basal ES is restricted to adjacent Sertoli cells, which together with tight junctions, form the blood-testis barrier (BTB) to provide a unique immune-privileged environment that supports the normal development of spermatocytes and spermatids within the apical compartment; apical ES is limited to the Sertoli cell – elongating spermatid interface, where it facilitates the formation of spermatid heads, promotes spermatid positioning and prevents the release of immature spermatozoa [7,8]. ES consists of testis-specific cell—cell actin-based anchoring junction with bundles of actin microfilaments lining the Sertoli cell plasma membrane in a perpendicular orientation [9]. The activities of these F-actin microfilaments, including bundling and unbundling, are regulated by three main protein polarity complexes: the F2RL2/PAR3-PARD6/PAR6-atypical PRKC complex, the CRB/crumbs-PATJ-PALS1 complex, and the SCRIB-DLG-LGL complex [10]. The PAR3 and Crumbs complexes promote BTB function, while the SCRIB complex functions when the BTB is disassembled to allow the transition of germ cells through the BTB to enter meiosis [11–13]. In addition to the three polarity complexes, the family of Rho GTPases, which have diverse functions in the regulation of the actin cytoskeleton, cell movement and cell adhesion, is also linked to the polarity of Sertoli cells [14,15]. The deletion of Rho GTPase family members CDC42 and RAC1 in Sertoli cells resulted in the loss of cell polarity in adult testes [16,17]. Although the pivotal role of Sertoli cell polarity in spermatogenesis has just been revealed, how polarity is established and maintained in Sertoli cells is still poorly understood.
Macroautophagy (called autophagy hereafter) is an evolutionarily conserved membrane trafficking processes involved in the lysosomal degradation of cytoplasmic protein and damaged organelles, thereby maintaining intracellular metabolic and energetic homeostasis [18]. Stress states such as hypoxia or nutrient deprivation cause the recruitment of autophagy-related (ATG) proteins to the phagophore assembly site (PAS) and nucleation of a phagophore, followed by the gradual expansion and the formation of a double-membrane autophagosome. Autophagosomes sequester cytoplasmic components and deliver them along cytoskeletal structures to lysosomes for degradation [19]. Autophagy is involved in a wide range of cellular physiological processes such as regulation of cell growth and apoptosis, maintenance of epithelial barrier function, and repair of oxidative stress damage, but the relation between autophagy and cell polarity is poorly known [20–22]. PIK3C3/VPS34 is a component of the only class III PtdIns3K; this type of kinase phosphorylates phosphatidylinositol to generate phosphatidylinositol-3-phosphate (PtdIns3P), a phospholipid that is critical for autophagy [23]. In yeast or mammalian cells, PIK3C3 together with PIK3R4 and BECN1 form the core complex, which exerts different functions by binding multiple regulatory proteins [24]. PtdIns3K-C1 contains ATG14, which is considered autophagy specific and is involved in autophagosome formation, while UVRAG-containing complex 2 (PtdIns3K-C2) functions in late stages of autophagy and endocytic trafficking [19,25,26]. Recently, the deletion or knockdown of PIK3C3 or the core components of the PtdIns3K complex, including Drosophila Vps15/PIK3R4 and Atg6/BECN1, each results in severe malformation in wing development, with disrupted epithelial polarity [27–29]. These studies reveal the potential role of PIK3C3 in model organism cell polarity; However, it remains unclear whether PIK3C3 participates in the establishment of highly specialized architecture in mammalian cells, such as Sertoli cells.
In this study, we found that the Sertoli cell-specific knockout of Pik3c3 led to the loss of cell polarity and male sterility in adult mice. Further study showed that the disorganization of the Sertoli cell architecture was related to the defects in clearing SCIN and HDAC6, which negatively regulated the organization of the F-actin cytoskeleton. Our study provides a novel mechanism whereby PIK3C3 contributes to maintaining Sertoli cell polarity and male fertility.
Results
Deletion of the Pik3c3 gene in Sertoli cells results in loss of fertility in adult male mice
To determine the role of PIK3C3 in Sertoli cells, pik3c3 conditional knockout mice (Amhr2Cre/+; Pik3c3flox/flox, referred to as “cKO” hereafter) were generated in a cross involving Amhr2-Cre mice, in which Cre recombinase is expressed in the Sertoli cells of male mice. The efficiency of pik3c3 knockout was first examined via the immunoblotting of primary Sertoli cells isolated from 2-week-old (2 W) mice (Figure 1A and Figure S1A). The fertility test was then performed in control (Pik3c3flox/flox) and cKO mice starting at 6 W of age, and we found that cKO mice were transiently fertile at 6 W and 7W of age, but their fertility declined rapidly at 8 W of age and was completely lost at 9 W of age (Figure 1B). We then collected the testes of the mice at 5 W, 7 W, and 9 W of age, and the testis size and testis:body weight ratio of the cKO mice progressively decreased with advancing age (Figure S1B-C and Figure 1C,D). The abnormal size and weight of the cKO testis indicated a potential disturbance of spermatogenesis; however, we found dramatically reduced sperm counts only in 9 W cKO mice (Figure S1D-E and Figure 1E,F). Subsequent in vitro fertilization (IVF) experiments with sperm collected from 7 W and 9 W cKO mice further demonstrated the inability of sperm from 9 W cKO mice to fertilize oocytes, while sperm from the epididymis of 7 W mice showed normal fertilization, with nearly 80% of fertilized oocytes developing to the 2-cell stage (Figure S1F-G and Figure 1G,H). Similarly, the analysis of sperm morphology in cKO mice at 9 W revealed a significantly higher percentage of spermatozoa with head abnormalities (~30%) (Figure S1H-I and Figure 1I,J). Furthermore, sperm motility analysis demonstrated severe decreases in the percentage of motile sperm, percentage of progressive motile sperm, average path velocity (VAP), curve-linear velocity (VCL), straight line velocity (VSL), straightness (STR) and beat-cross frequency (BCF) (Figure 1K,L and Figure S1J-P). Thus, the decreased sperm count and abnormalities in the sperm head and sperm motility are responsible for the loss of fertility in 9 W cKO mice.
Figure 1.
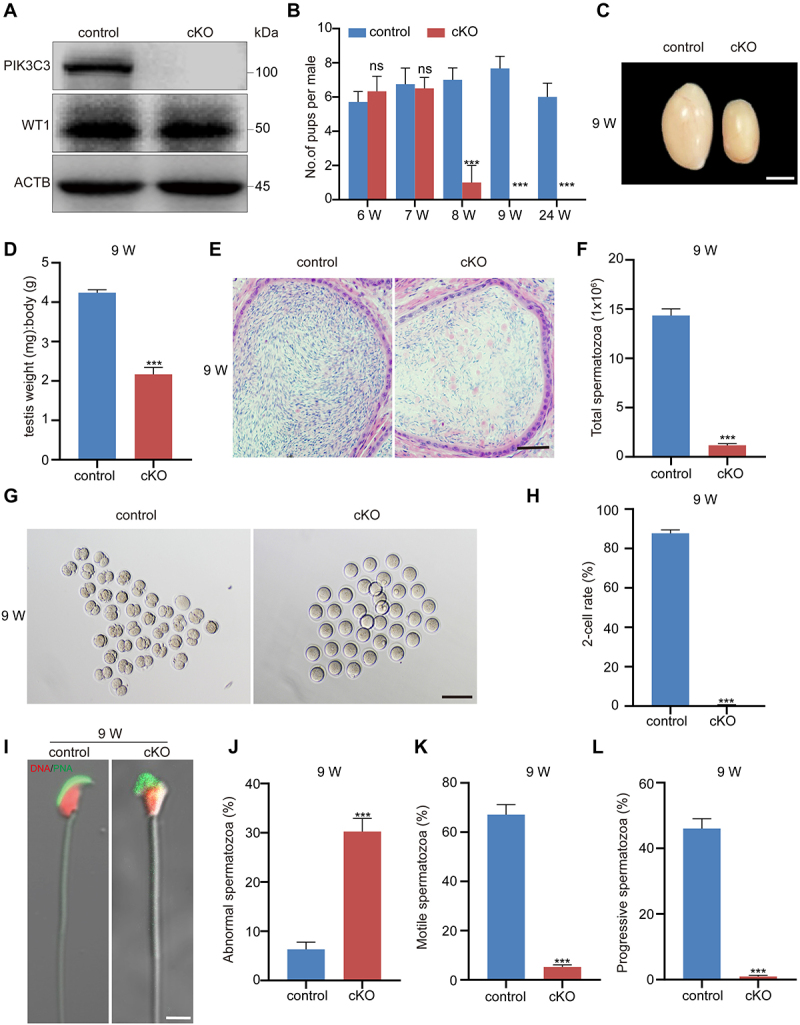
Male sterility in pik3c3 cKO mice. (A) Immunoblotting of PIK3C3 in primary Sertoli cells collected from control and cKO mice at 2 W of age. WT1 was examined as the Sertoli cell marker and the expression of ACTB was used as the internal control. (B) Fertility tests in control and cKO mice. In each group, four male mice were individually mated with one wild-type female mice for one month. (C) Image showing the size of the testes in control and cKO mice. Testes were collected from 9 W of control and cKO mice. Bar: 3 mm. (D) Graphs showing the average ratio of testis weight: body weight in 9 W control and cKO mice (n = 4). (E) Representative cross-sections of the cauda epididymis from 9 W control and cKO mice. Bar: 60 μm. (F) Sperm counts in 9 W control and cKO mice (n = 4). (G) Representative micrograph of 2-cells formed after in vitro fertilization (IVF) with sperm from 9 W control and cKO mice. Bar: 150 μm. (H) Graph showing the 2-cell rate after IVF (n = 4). (I) Sperm morphology after immunofluorescence with the acrosome-specific marker PNA. Bar: 5 μm. (J-L) Graphs showing the percentages of abnormal spermatozoa (J), motile spermatozoa (K) and progressive spermatozoa (L) in 9 W control and cKO mice (n = 4). ***P < 0.01; ns, not significant.
PIK3C3 is required for the progression of spermatogenesis
We next evaluated the structure of the seminiferous tubules during spermatogenesis based on specific markers in control and cKO mice at 5 W, 7 W and 9 W of age. The seminiferous tubule structure was normal in cKO mice at 5 W, while at 7 W, nearly 50% of the seminiferous tubules were disorganized and showed large vacuoles in the seminiferous epithelium. In 9 W cKO testis, vacuolation had extended to nearly all tubules, large numbers of cells were lost, and the tubules decreased in size (Figure S2A-B). Germ cells and the acrosomes of spermatids were then labeled with DDX4 (DEAD box helicase 4) and arachis hypogaea lectin (PNA), respectively, to evaluate the seminiferous epithelium cycle [30,31]. In 7 W cKO testes, spermatogenesis from stage IV-IX could still be well classified in some tubules; however, it was very difficult to distinguish in 9 W cKO testis (Figure S2C-D). To determine the cell type responsible for the large amount of cell loss in cKO mice, we labeled germ cells and Sertoli cells with GCNA (germ cell nuclear acidic protein) and WT1 (WT1 transcription factor), respectively [32,33]. We found that germ cell loss, but not Sertoli cell loss, induced the progressive degeneration of the seminiferous epithelium (Figure S2E-G and Figure 2A–C). Different germ cell types in multiple stages of spermatogenesis were also checked by using ZBTB16 (zinc finger and BTB domain containing 16) and KIT (KIT proto-oncogene receptor tyrosine kinase) to label undifferentiated (spermatogonia stem cell, SSC, ZBTB16+ KIT −) and A1–A4-differentiating spermatogonia (ZBTB16+ KIT+) and using H2AX (H2A.X variant histone) and PNA to label primary spermatocytes (H2AX+) and round spermatids (PNA+ with round nuclei), respectively [34,35]. The results showed that all examined germ cell types displayed a decreasing trend starting at 7 W, especially in the 9 W cKO testes, and we found a significant decrease in primary spermatocytes (H2AX+) and round spermatids (PNA+). Moreover, it was difficult to identify elongated spermatids in the cKO tubules (Figure S2H-M and Figure 2D–I). This was consistent with the dramatic reduction in the sperm count and the abnormalities of sperm morphology and motility observed in the epididymis of 9 W cKO mice. TUNEL and cleaved CASP3 staining further demonstrated a significant increase in cellular apoptosis in cKO mice at 7 W and 9 W of age (Figure S2N-Q and Figure 2J–M). These results suggest that the deletion of Pik3c3 in Sertoli cells causes a progressive loss of germ cells and defective spermiogenesis in mice.
Figure 2.
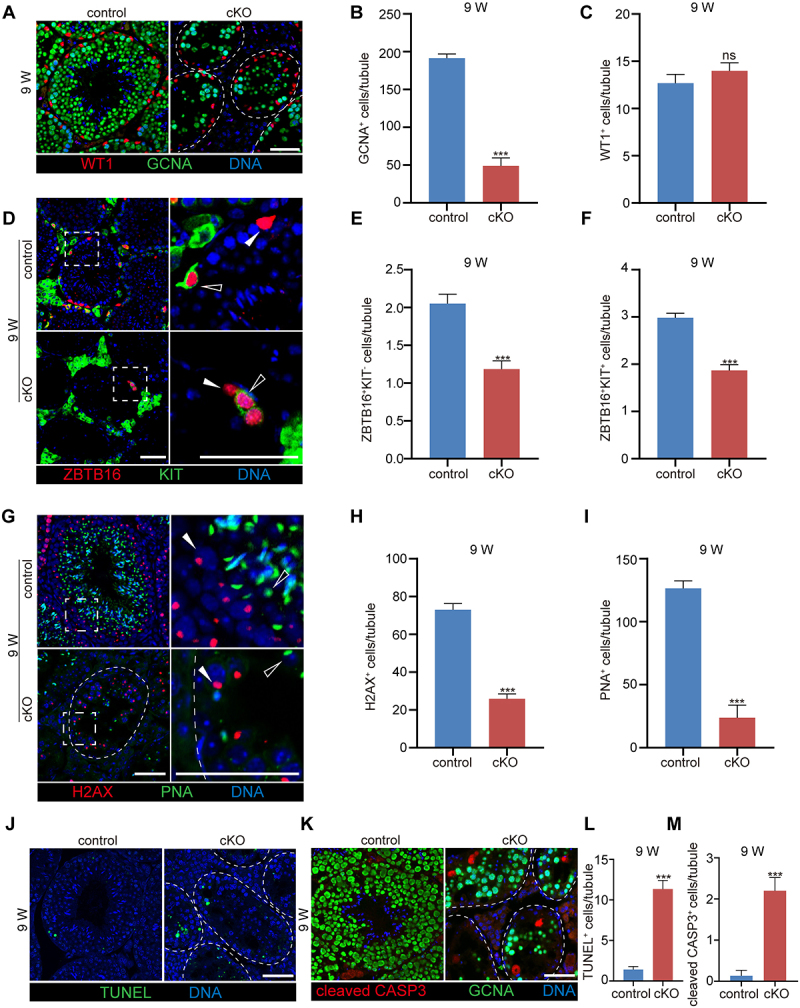
Deletion of Pik3c3 in Sertoli cells resulted in progressive loss of germ cells. (A) Immunofluorescence of WT1 and GCNA in control and cKO mice testis at 9 W of age. (B-C) Graph showing the number of germ cells (B) or Sertoli cells (C) per seminiferous tubule. (D) Immunofluorescence of ZBTB16 and KIT in control and cKO mice testis at 9 W of age. Filled triangles, SSCs; hollow triangles, A1-A4 differentiating spermatogonia. (E-F) Graph showing the number of SSCs (ZBTB16+ KIT −) (E) or differentiating spermatogonia (ZBTB16+ KIT +) (F) per seminiferous tubule. (G) Immunofluorescence of H2AX and PNA in control and cKO mice testis at 9 W of age. Filled triangles, primary spermatocytes; hollow triangles, spermatids. (H-I) Graph showing the number of spermatocytes (H2AX+) (H) or round spermatids (PNA+) (I) per seminiferous tubule. (J-K) Apoptosis evaluated by TUNEL assay and cleaved CASP3 staining in control and cKO mice testis at 9 W of age. (L-M) Graph showing the number of TUNEL+ (L) or cleaved CASP3+ (M) apoptotic cells per seminiferous tubule. The largest cross-section of each testis was used for staining and cell counting (n = 6). All Bar: 50 μm. ***P < 0.01; ns, not significant.
PIK3C3 is essential for the maintenance of Sertoli cell polarity
The loss of Sertoli cell polarity can lead to an impairment of seminiferous tubules and apoptosis of germ cells. Although comparable Sertoli cell numbers were observed in cKO testes, we noted the detachment of Sertoli cells from the basal membrane along with the vacuolation of seminiferous tubules (Figure 2A). This suggests a change in the cellular structure after the deletion of Pik3c3 in Sertoli cells. Based on the differences in spermatogenesis and tubule morphology between 7 W and 9 W cKO testes, we chose the testis at 8 W mice to evaluate the change in Sertoli cell polarity. We examined the specialized structures of Sertoli cells, such as the BTB or aES, and the distribution of the cytoskeleton in cKO testes by using different molecular markers. In mammals, the BTB structure contains tight junctions (TJs), bESs and gap junctions [36]. We first examined the distribution of TJP1 (tight junction protein 1), a marker of TJs, and CTNNA1 (catenin (cadherin associated protein), alpha 1), a marker of bES, in seminiferous tubules. As shown in Figure 3A, both TJP1 and CTNNA1 were limited specifically at the BTB near the basement of the control tubules, but in cKO mice, these proteins were localized throughout the Sertoli cells. We then measured the distance traveled by TJP1 and CTNNA1 beyond the BTB near the basement membrane (D TJP1 and DCTNNA1) and compared the D TJP1 and DCTNNA1 results against the radius of the seminiferous tubules (DSTr) (Figure 3B). The results demonstrated significantly higher DTJP1:DSTr and DCTNNA1:DSTr ratios in cKO testes than in control mice [37]. We also labeled gap junctions with GJA1/connexin 43 (gap junction protein, alpha 1), germ cells with GCNA and Sertoli cells with VIM (vimentin) to observe intercellular communication in seminiferous tubules. In control testes, GJA1 was restricted to the Sertoli cell-Sertoli cell interface, Sertoli cell-germ cell interface and germ cell-germ cell interface, whereas the localization of GJA1 at the Sertoli cell interface completely disappeared in cKO mice (Figure 3C). The results suggest the impairment of intercellular communications both between Sertoli cells themselves and between Sertoli cells and germ cells. Transparent electron microscopy (TEM) showed typical BTB features in control testis, characterized by ordered F-actin between the cisternae of the ER and plasma membrane, with electron-rich areas representing TJs between adjacent Sertoli cell membranes, while in the cKO testis, such typical structures were not observed (Figure 3D). The barrier function of the BTB was also assessed via the tail vein injection of a FITC (fluorescein isothiocyanate) tracer [38]. However, fluorescence was only found to be enriched in the seminiferous tubules of cKO testes (Figure 3E). The results further indicated the disturbance of BTB function after the deletion of Pik3c3 in Sertoli cells. Based on the importance of the cytoskeleton in establishing and maintaining cellular polarity, we examined the distribution of microtubules and microfilaments in Sertoli cells with the markers TUBB3 (tubulin beta 3 class III) and F-actin, respectively. Figure 3F shows TUBB3 oriented in linear arrays parallel to the long axis from the base to the top of the Sertoli cells; however, this polarized distribution of microtubules disappeared in cKO mice, accompanied by detachment of the nucleus from the basement membrane. In control tubules, the microfilament marker F-actin labeled with phalloidin was located in the BTB at the basement membrane and around the spermatid head, where it showed aES (apical ectoplasmic specialization) between elongating/elongated spermatids and Sertoli cells (Figure 3G); however, the characterized features of F-actin in both the BTB and aES disappeared in cKO mice (Figure 3G). The TEM analysis of aES structure in cKO testes also showed diffuse F-actin bundles at the Sertoli cell-spermatid interface (Figure 3H). Immunoblotting further demonstrated altered protein levels of the cellular junction markers TJP1 and OCLN (occludin) (TJ), CDH2 (cadherin 2) and CTNNA1 (bES marker), and GJA1 (gap junction marker) between control and cKO testis (Figure 3I,J). Overall, the deletion of Pik3c3 in Sertoli cells leads to the loss of cell polarity, with extensive impairment of cell junctions and a disordered cytoskeleton. The disorganization of the BTB and aES causes germ cell loss and defective spermatogenesis in cKO mice.
Figure 3.
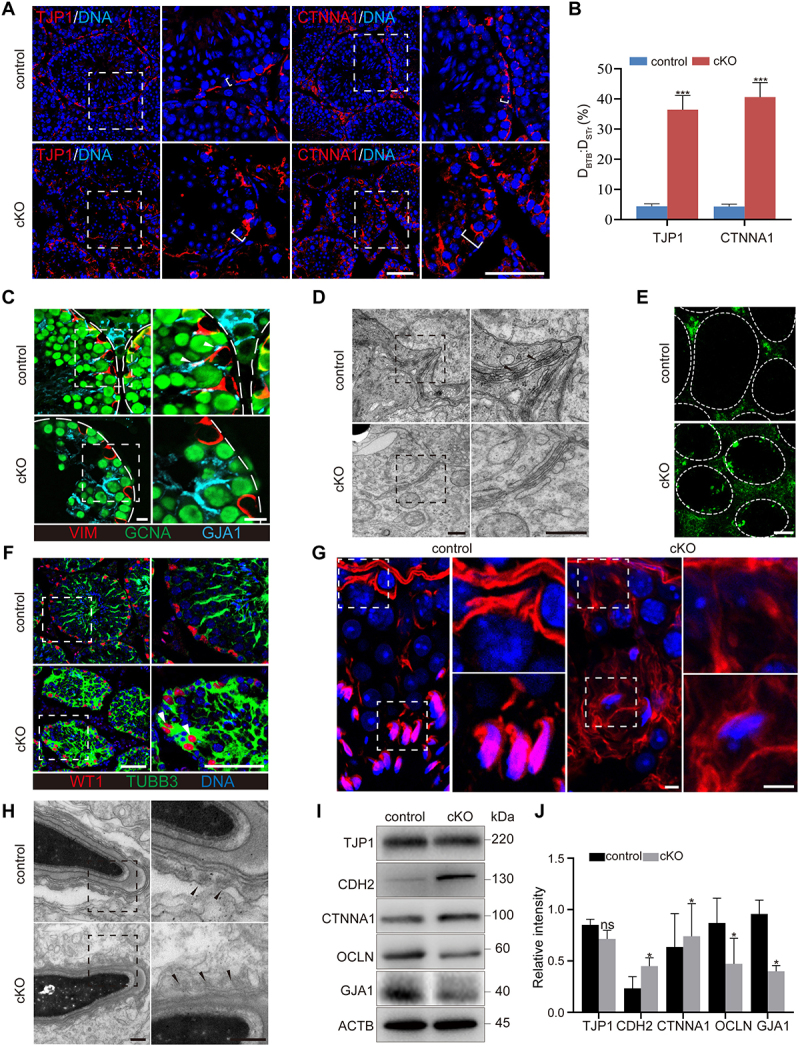
Deletion of Pik3c3 in Sertoli cells resulted in impaired Sertoli cell polarity. (A) Immunofluorescence of TJP1 or CTNNA1 in control and cKO mice testis at 8 W of age. Bar: 50 μm. (B) Graph showing the distance traveled by TJP1 or CTNNA1 beyond the BTB near the basement membrane (DTJP1 or DCTNNA1) versus the radius of the seminiferous tubule (DSTr) per seminiferous tubule. The largest cross-section of each testis was used for staining and cell counting (n = 6). ***P < 0.01. (C) Immunofluorescence of VIM, GCNA and GJA1. Filled triangles, gap junctions localized between Sertoli cells and germ cells. Bars: 10 μm. (D) TEM showed the BTB structure in control and cKO testis. Filled triangles, electron rich areas referred as tight junctions. Bars: 400 nm. (E) Immunofluorescence of FITC showed the disturbed BTB structure in cKO testis. FITC was injected into the control and cKO mice and testes were isolated 1 h later for cryosection. Bar: 50 μm. (F) Immunofluorescence of WT1 and TUBB3. Filled triangles, Sertoli cell nuclei traveled away from the basement membrane of seminiferous tubule. Bar: 50 μm. (G) Immunofluorescence of phalloidin. Bar: 5 μm. (H) TEM of aES structure. Filled triangles, actin bundles. Bar: 300 nm. (I-J) Immunoblotting of TJP1, CDH2, CTNNA1, OCLN and GJA1 proteins. The expression of ACTB was used as internal control (I). The relative intensity of TJP1, CDH2, CTNNA1, OCLN and GJA1 was shown as compared to the expression of ACTB (J).
SCIN accumulation in pik3c3 cKO Sertoli cells leads to the disassembly of the F-actin cytoskeleton
To investigate the potential protein targets through which PIK3C3 regulates the polarity of Sertoli cells, control and cKO testes were collected at 8 W of age to perform TMT-labeled quantitative proteomics. Compared to control testes, a total of 1608 proteins were found to be significantly differentially expressed (≥1.3-fold or ≤ 0.769-fold) in cKO testes, among which 892 proteins were upregulated and 716 were downregulated (Figure 4A and Table S1). KEGG pathway enrichment analysis revealed that the differentially expressed proteins were associated with processes such as the regulation of the actin cytoskeleton, tight junctions, or autophagy, which was consistent with the disorganization of the Sertoli cells observed in cKO testis (Figure 4B). The top 20 differentially expressed proteins enriched in the three pathways are shown in Figure 4C, among which we identified SCIN, a Ca2+-activated actin filament-severing protein that regulates cortical F-actin disassembly [39], showing a 3.871-fold increase in the cKO testis (Figure 4C). Immunostaining further demonstrated increased expression of SCIN in cKO seminiferous tubules (Figure 4D). We next isolated and cultured primary Sertoli cells from control and cKO mice at 8 W of age to determine the effect of SCIN on actin microfilaments. Western blot analysis first showed increased expression of SCIN in primary Sertoli cells isolated from cKO mice (Figure 4E,F). Since AMHR2 is also expressed in Leydig cells of adult mice, we isolated Leydig cells from control and cKO mice at 8 W of age and examined PIK3C3 expression. Similar as previous reports about the cell type that the Amhr2-Cre recombinase functioned in testis [40,41], we did not observe altered PIK3C3 and SCIN expressions between the two groups (Figure S3A). Same result was also observed in germ cells isolated from 8 W control and cKO testis (Figure S3B) which means the specificity of PIK3C3 on the expression of SCIN in Sertoli cells. The disorder and disassembly of the F-actin cytoskeleton was then observed by phalloidin staining (Figure 4G,H) and quantified based on the ratio of polymerized F-actin to monomeric G-actin in 8 W cKO Sertoli cells. After differential sedimentation to separate F- and G-actin, our results revealed a significant reduction in F-actin levels, and the F-:G-actin ratio decreased 31.93 ± 6.5% in cKO Sertoli cells (Figure 4I,J). We further overexpressed SCIN in control primary Sertoli cells and found a significant increase in Sertoli cells with a disordered F-actin cytoskeleton (Figure 4K,L). Moreover, the ratio of F-:G-actin also decreased significantly in SCIN-overexpressing Sertoli cells (Figure 4M,N). Therefore, SCIN accumulation in Sertoli cells leads to the disorder and disassembly of the F-actin cytoskeleton.
Figure 4.
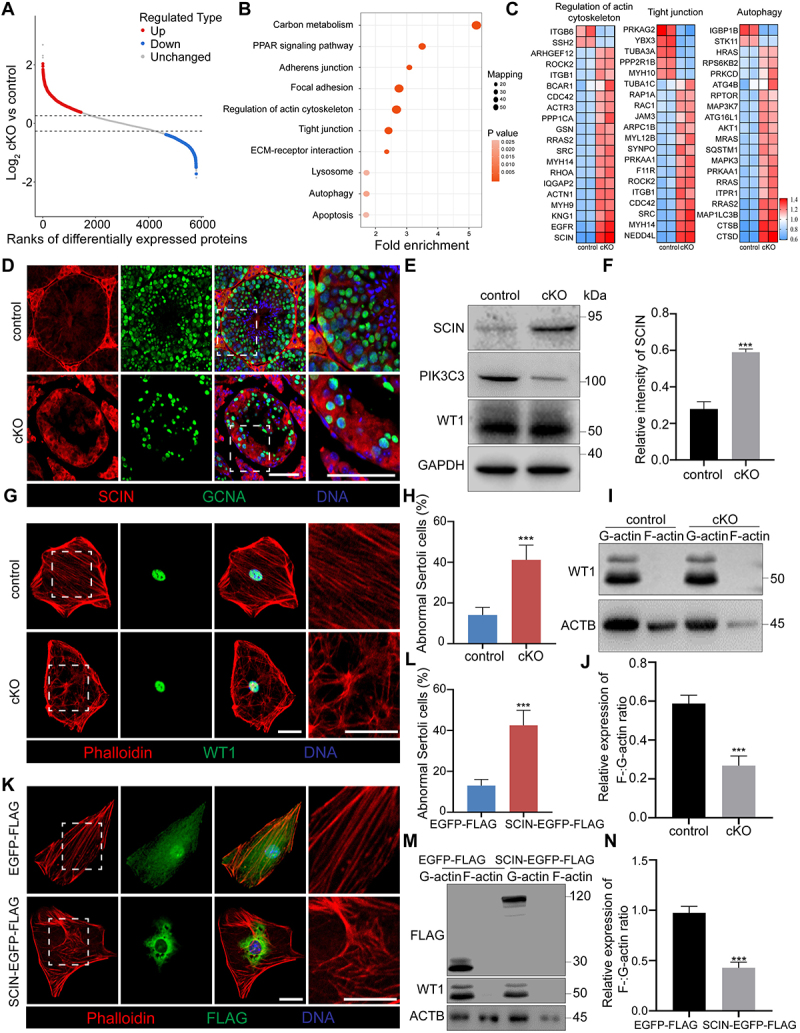
F-actin negative regulator protein SCIN was screened by proteomics. Testes at 8 W of age were collected from control and cKO mice for TMT-labeled quantitative proteomics. (A) Scatter plot displaying differentially expressed proteins (DEPs) comparing control with cKO testes. Horizontal dashed lines indicate cutoff of log21.3FC (0.37851) and -log21.3FC (−0.37851). Red dots: upregulated proteins (FC ≥ 1.3; CV < 0.1). Blue dots: downregulated proteins (FC ≤ 0.769; CV < 0.1). Gray dots: unchanged proteins (0.769 < FC < 1.3; CV ≥ 0.1). FC: Fold Change; CV: Coefficient of Variation. (B) KEGG pathway analysis was performed on DEPs between control and cKO testes. (C) Heatmap showing top 20 DEPs associated with regulation of actin cytoskeleton, tight junctions, and autophagy KEGG pathways. (D) Immunofluorescence of SCIN and GCNA. Bar: 60 μm. (E-F) Increased expression of SCIN in primary Sertoli cells of cKO mice (E). The relative intensity of SCIN was shown as compared to the expression of GAPDH (F). (G-H) Disorganized F-actin cytoskeleton in cKO Sertoli cells. Primary Sertoli cells were isolated from the testes of control and cKO male mice at 8 W of age and cultured on cover slips. Immunofluorescence of phalloidin and WT1 was performed (G). Bar: 30 μm. Graph showing the percentage of Sertoli cells with abnormal F-actin cytoskeleton structure among all Sertoli cells on each cover slip (n = 5) (H). (I-J) F-actin and G-actin from Sertoli cells were segmented and analyzed by immunoblotting using an antibody against ACTB (I). Graph showing the relative expression of F-:G-actin ratio in Sertoli cells (n = 3) (J). (K-L) Disturbance of F-actin cytoskeleton after overexpression of SCIN in primary Sertoli cells. Primary Sertoli cells were plated and cultured on cover glass slips from the testes of male mice at 2 W of age. Cells were transfected with EGFP-FLAG and SCIN-EGFP-FLAG. Immunofluorescence of phalloidin and FLAG was performed (K). Bar: 30 μm. Graph showing the percentage of Sertoli cells with abnormal F-actin cytoskeleton structure among all Sertoli cells on each cover glass slip (n = 5) (L). (M-N) F-actin and G-actin from Sertoli cells were segmented and analyzed by immunoblotting using an antibody against ACTB (M). Graph showing the relative expression of F-:G-actin ratio in Sertoli cells (n = 3) (N). ***P < 0.01.
SCIN is degraded through the autophagy—lysosome pathway in Sertoli cells
To investigate the mechanism of the abnormal accumulation of SCIN induced by pik3c3 deletion, we performed a cycloheximide (CHX) chase assay to measure the half-life of SCIN in control and cKO Sertoli cells, and the results showed delayed degradation of SCIN in cKO Sertoli cells (Figure 5A). In mammalian cells, protein degradation is mainly mediated by two proteolytic systems: the ubiquitin-proteasome system (UPS) and the autophagy-lysosome system [42]. We first treated primary Sertoli cells with MG132, a pivotal inhibitor of the UPS. As shown in Figure 5B, the blockage of the UPS was proven by the gradually increase in UB (ubiquitin) expression with increasing treatment time; however, the protein levels of SCIN were not affected and remained constant during the treatment. This result suggests that the degradation of SCIN in Sertoli cells is independent of the UPS. We then activated the autophagy pathway in Sertoli cells through the starvation of cells with Earle’s balanced salt solution (EBSS) [43]. In contrast to what was observed following the blockage of the UPS pathway, the expression of SCIN and autophagy receptor protein SQSTM1/p62 (sequestosome 1) decreased gradually with the extension of the EBSS incubation time (Figure 5C). To further clarify whether the degradation of SCIN occurs through the autophagy-lysosome pathway, we performed an immunoprecipitation assay to observe the interaction between SCIN and MAP1LC3B (microtubule associated protein 1 light chain 3 beta), an autophagy marker that localizes to the inner sheath of autophagosomes, and the results confirmed their interactions not only in primary Sertoli cells but also in HEK293 cells exogenously expressing SCIN-EGFP-FLAG and MAP1LC3B-HA (Figure S3C and Figure 5D). The deletion of Pik3c3 in MEFs or the liver has been reported to block starvation-induced autophagosome formation [44]. Similarly, we observed the interruption of autophagy flux in pik3c3-deleted Sertoli cells, as shown by the increased expression of SQSTM1 and MAP1LC3B II (Figure 5E). Immunostaining with antibodies against SCIN and LAMP1 (lysosomal-associated membrane protein 1), a lysosome marker, then demonstrated that EBSS starvation induced SCIN aggregation and colocalization with lysosomes in control Sertoli cells, while in cKO Sertoli cells, it was difficult to observe the colocalization of the two proteins (Figure 5F,G). These results suggest that defective autophagy in pik3c3-deleted Sertoli cells leads to the failure of SCIN degradation through the autophagy-lysosome pathway. In addition, when SCIN was knocked down by siScin transfection in cKO Sertoli cells, as shown by F-actin immunofluorescence, the disordered F-actin cytoskeleton could be successfully rescued (Figure 5H,I). The immunoblotting of isolated F-actin and G-actin also demonstrated a dramatic increase in polymerized F-actin in cKO Sertoli cells with reduced expression of SCIN (Figure 5J,K). Taken together, the results indicated that SCIN functions downstream of PIK3C3 to regulate F-actin cytoskeleton organization in Sertoli cells.
Figure 5.
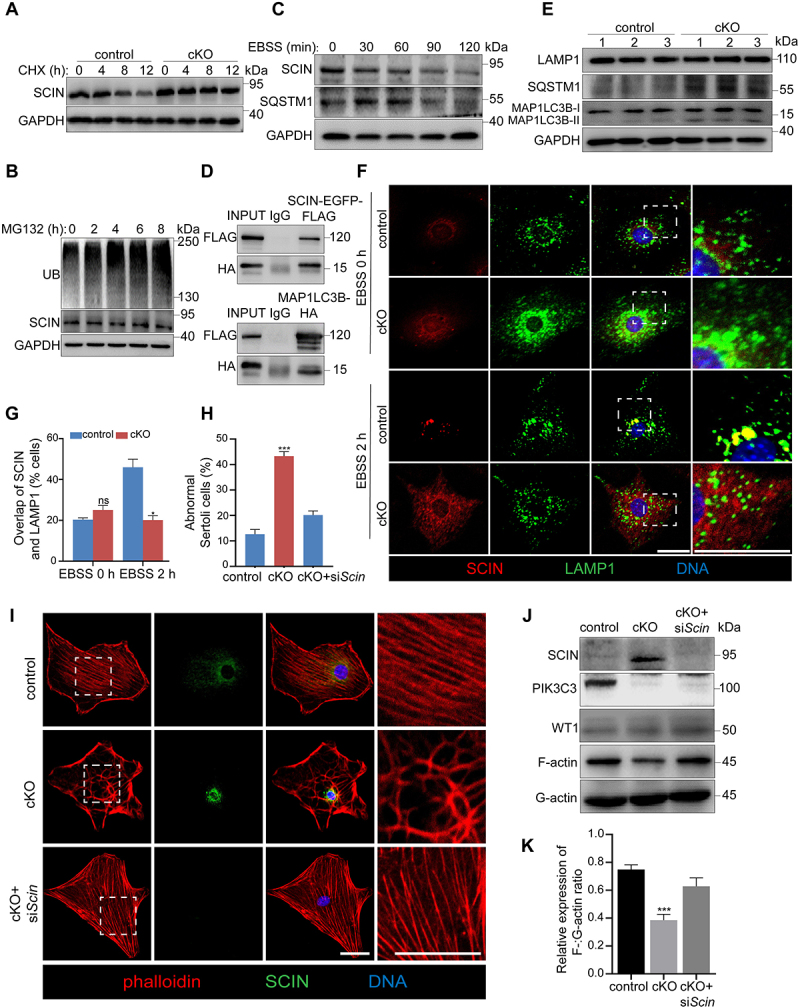
PIK3C3 regulated SCIN degradation through the autophagy pathway. Primary Sertoli cells were isolated from control and cKO mice at 8 W of age. (A-C) Immunoblotting of SCIN in control and cKO primary Sertoli cells after CHX (A), MG132 (B) or EBSS (C) treatment. CHX (100 μg/ml) were treated for 12 h to measure the half-life of SCIN in control and cKO Sertoli cells. MG132 (10 μM), the proteasome inhibitor, was treated on control Sertoli cells for 8 h and cells were collected every 2 h to examine the UB (ubiquitin) and SCIN levels. Autophagy was induced by incubation of Sertoli cells with EBSS for 2 h to examine the protein levels of SCIN and SQSTM1, an autophagy marker. The expression of GAPDH was used as internal control. (D) Immunoprecipitation and immunoblotting showed the interaction of SCIN-EGFP-FLAG with MAP1LC3B-HA in HEK293 cells. (E) Immunoblotting of LAMP1, SQSTM1 and MAP1LC3B proteins in control and cKO Sertoli cells. The expression of GAPDH was used as internal control. (F-G) Immunofluorescence showing the overlap of SCIN and LAMP1 in control and cKO primary Sertoli cells after EBSS treatment (F) Bar: 30 μm. Graph showing the percentage of Sertoli cells with overlapped staining of SCIN and LAMP1 among all Sertoli cells on each cover slip (n = 3) (G). (H-I) the effect of SCIN on F-actin stabilization. Primary Sertoli cells were plated on cover glass slips and transfected with siCtrl and siScin, respectively. Immunofluorescence showed the reorganization of F-actin after knockdown of Scin in cKO Sertoli cells (H). Bar: 30 μm. Graph showing the percentage of Sertoli cells with abnormal F-actin cytoskeleton structure among all Sertoli cells on each cover glass slip. (n = 5) (I). (J) F-actin and G-actin from control, cKO or cKO+siScin primary Sertoli cells were segmented and analyzed by immunoblotting using an antibody against ACTB. (K) Graph showing the relative expression of F-:G-actin ratio in each group (n = 3). ***P < 0.01; ns, not significant.
HDAC6 is a negative regulator of the F-actin cytoskeleton in Sertoli cells
All phosphatidylinositol 3-kinases are dual-specificity enzymes with both lipid and protein kinase activities [45]. When functioning as a lipid kinase, PIK3C3 phosphorylates phosphatidylinositol to produce PtdIns3P, a lipid that has been well documented to be the major effector of PIK3C3 in regulating autophagy [23]. However, its protein kinase activity and downstream effectors have seldom been reported. To investigate the potential role of PIK3C3 in regulating downstream protein phosphorylation, we collected testes from control and cKO mice for TMT-labeled quantitative phosphoproteomics, and a total of 773 differentially regulated phosphosites (≥1.3-fold or ≤ 0.769-fold), which mapped to 638 proteins, were identified in cKO testes, including 209 upregulated and 564 downregulated phosphosites compared to the control testes (Figure 6A and Table S2). Consistent with the proteomics results, KEGG pathway analysis showed the enrichment of processes related to the regulation of the actin cytoskeleton, tight junctions, and autophagy (Figure 6B). A Venn diagram showed a total of 164 overlapping proteins between differentially expressed proteins and phosphorylated proteins, among which twelve proteins were involved in the process of actin binding (Figure 6C). The differentially phosphorylated sites of the twelve identified proteins are shown in Figure 6D among which HDAC6 was the most differentially expressed protein, showing a 1.47-fold increase in cKO testes, while its phosphorylation at the S59 site was 3.37-fold lower than that in control testes. HDAC6 is a class IIb histone deacetylase that regulates multiple biological processes, such as cell motility, cell survival, and cell cycle progression [46]. Increased HDAC6 expression has been reported to impair basal-apical polarity in both mammary epithelium and endothelial cells [47,48]. In control testes, the expression of HDAC6 was limited in elongating or elongated spermatids and was difficult to detect in other cell types. However, stronger HDAC6 staining signals were found in the seminiferous tubules of the cKO mice. Since the HDAC6 signals seldom overlapped with GCNA-positive germ cells, the results suggest the abnormal accumulation of HDAC6 in Sertoli cells of cKO mice (Figure 6E). In primary Sertoli cells isolated from control and cKO mice, immunoblotting further demonstrated a significant increase in HDAC6 protein levels in cKO Sertoli cells (Figure 6F,G). We then examined the effect of HDAC6 on the F-actin cytoskeleton by the overexpression of HDAC6 in primary Sertoli cells. As shown in Figure 6H,I increased disorganization of the F-actin cytoskeleton was observed in HDAC6-overexpressing cells (49.83 ± 6.65% vs. 16.85 ± 2.76% abnormality in cells transfected with or without HDAC6). The immunoblotting of isolated F-actin and G-actin also showed a decreased ratio of F-:G-actin (Figure 6J,K). These results suggest that the abnormal accumulation of HDAC6 in cKO Sertoli cells leads to the disorder and disassembly of the F-actin cytoskeleton.
Figure 6.
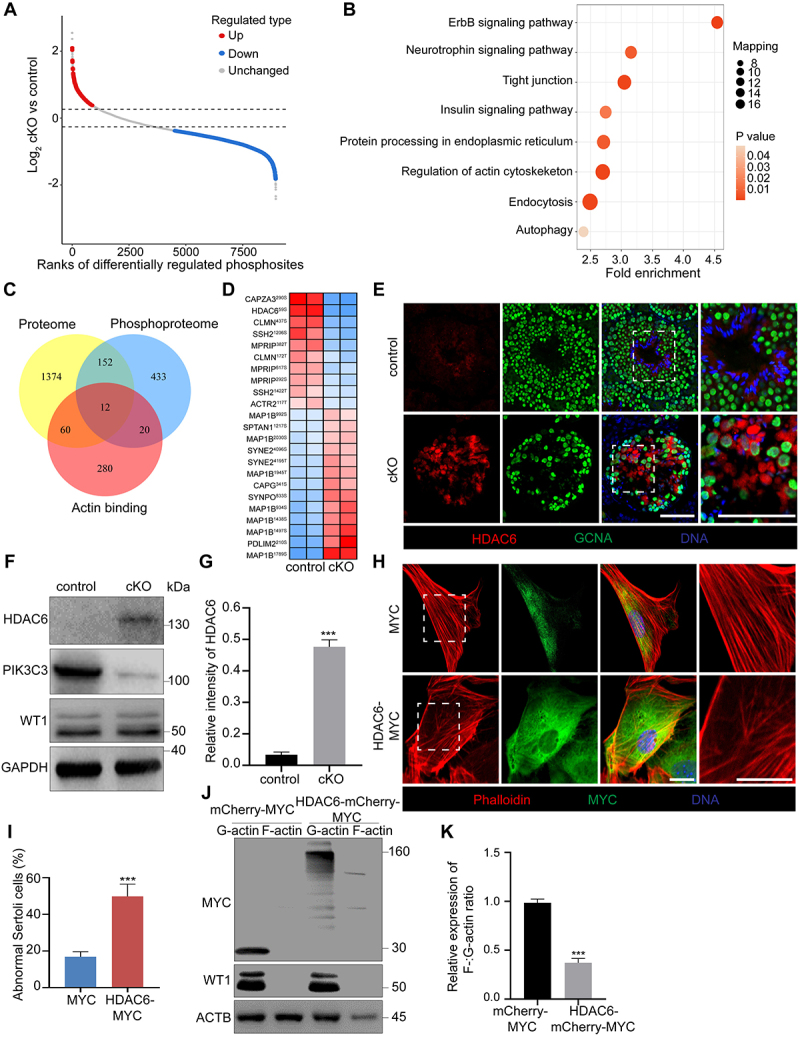
The accumulation of HDAC6 in Sertoli cells led to the disassembly of F-actin. Testes were collected from control and cKO mice at 8 W of age for TMT-labeled quantitative phosphoproteomics. (A) Scatter plot displaying differentially regulated phosphosites comparing control with cKO testes. Horizontal dashed lines indicate cutoff of log21.3FC (0.37851) and -log21.3FC (−0.37851). Red dots: upregulated phosphosites (FC ≥ 1.3; CV < 0.1). Blue dots: downregulated phosphosites (FC ≤ 0.769; CV < 0.1). Gray dots: unchanged phosphosites (0.769 < FC < 1.3; CV ≥ 0.1). FC: Fold Change; CV: Coefficient of Variation. (B) KEGG pathway analysis of differentially phosphorylated proteins. (C) the Venn diagram showing overlapped differential proteins revealed by TMT-labeled quantitative proteomics and phosphoproteomics and actin binding proteins. Twelve actin-binding proteins were identified as differential expressed in both protein levels and phosphorylated status between control and cKO groups. (D) Heatmap of twelve differential actin binding protein with identified phosphorylation sites. (E) Immunofluorescence for HDAC6 and GCNA in seminiferous tubules of control and cKO mice. Bar: 60 μm. (F-G) Immunoblotting of HDAC6, PIK3C3 and WT1 proteins in primary Sertoli cells collected from control and cKO mice at 8 W of age (F). The expression of GAPDH was used as internal control. The relative intensity of HDAC6 was shown as compared to the expression of GAPDH (n = 3) (G). (H-I) Disturbance of F-actin cytoskeleton after overexpression of HDAC6 in primary Sertoli cells. Primary Sertoli cells were plated and cultured on cover slips from the testes of male mice at 2 W of age. Cells were transfected with MYC and HDAC6-MYC. Immunofluorescence of phalloidin and MYC was performed (H). Bar: 30 μm. Graph showing the percentage of Sertoli cells with abnormal F-actin cytoskeleton structure among all Sertoli cells on each cover glass slip (n = 4) (I). (J- K) F-actin and G-actin from Sertoli cells were segmented and analyzed by immunoblotting using an antibody against ACTB (J). Graph showing the relative expression of F-:G-actin ratio in mCherry-MYC or HDAC6- mCherry-MYC overexpressed Sertoli cells (K). ***P < 0.01.
PIK3C3 phosphorylates HDAC6 and promotes its degradation through the UPS pathway
We next investigated the molecular mechanism by which the deletion of Pik3c3 leads to the abnormal accumulation of HDAC6. Since primary Sertoli cells express very low levels of HDAC6, we used HEK293 cells for further studies. We first knocked out Pik3c3 in HEK293 cells and found that HDAC6 expression was significantly increased compared to that in control cells (Figure 7A). In contrast, when exogenous PIK3C3-HA was transfected into HEK293 cells, the expression of endogenous HDAC6 decreased gradually in a dose-dependent manner (Figure 7B). Immunoprecipitation from lysates of HEK293 cells demonstrated interactions between endogenous PIK3C3 and HDAC6, which were further confirmed by the coimmunoprecipitation of exogenously expressed PIK3C3-HA with HDAC6-MYC (Figure S3D and Figure 7C). The activity of PIK3C3 has been reported to be regulated by EP300 (E1A binding protein p300)-mediated acetylation [49]. Due to the characteristics of HDAC6 as a deacetylase, we performed an immunoprecipitation assay to determine whether PIK3C3 can be deacetylated by HDAC6. As shown in Figure S3E, the acetylation level of PIK3C3-HA was not altered, even under increasing doses of HDAC6-MYC, suggesting that PIK3C3 is not a substrate for HDAC6-mediated deacetylation. In contrast to the accumulation of HDAC6 in Sertoli cells after pik3c3 deletion, phosphoproteomics revealed significantly decreased phosphorylation at S59 of HDAC6. To explore whether HDAC6 phosphorylation affects HDAC6 protein levels or functions, we first evaluated the protein kinase activity of PIK3C3 in HDAC6 phosphorylation by exogenously expressing HDAC6-MYC in control and pik3c3 KO HEK293 cells (Figure 7D). After immunoprecipitation with an MYC antibody, immunoblotting revealed a dramatic decrease in HDAC6 Ser/Thr phosphorylation in KO cells. In addition, when HDAC6-MYC was co-expressed with different doses of exogenous PIK3C3-HA in HEK293 cells, a dose-dependent increase in HDAC6 phosphorylation was observed (Figure 7E). By performing in vitro kinase assays using purified GST-HDAC6-MYC, we further found the immunoprecipitate of PIK3C3-HA could phosphorylate HDAC6 in a dose-dependent manner (Figure 7F). However, in HEK293 cells expressing an HDAC6 phosphorylation mutant (HDAC6S59A), exogenous PIK3C3 failed to induce HDAC6 phosphorylation (Figure 7G). Together, these results suggest that PIK3C3 can directly phosphorylate HDAC6 at site S59. Next, we overexpressed HDAC6 in control and pik3c3 KO cells and performed CHX chase assays to determine the relationship between pik3c3 deletion and HDAC6 accumulation. The results showed the accumulation of exogenous HDAC6 in pik3c3 KO cells but not in control cells (Figure 7H). HDAC6 has been reported to function as a receptor for autophagic degradation of protein aggregates [50]. To see which degradation pathway being involved in regulating the degradation of HDAC6, we inhibited or activated the autophagy-lysosome pathway in HEK293 cells with bafilomycin A1 (BafA1) and EBSS treatment, respectively. The blockade of autophagic flux after BafA1 treatment was revealed by the accumulation of SQSTM1 and the increased expression of MAP1LC3B-II, while the activation of autophagy after EBSS starvation was manifested by the rapid degradation of SQSTM1. In both cases, we did not find the altered protein levels of HDAC6 (Figure S3F-G). Since HDAC6 harbors a specific ubiquitin-binding domain at the C-terminal end, to determine whether HDAC6 degradation occurs through the UPS pathway, we treated HEK293 cells with MG132 and found that HDAC6 accumulated gradually with the blockage of the UPS pathway, suggesting that HDAC6 is mainly degraded through the UPS pathway (Figure 7I). Under the control of PIK3C3, HEK293 cells were subjected to exogenous His-FLAG-UB, HDAC6-MYC and PIK3C3-HA expression and treated with the proteasome inhibitor MG132. Immunoprecipitation demonstrated the dose-dependent effect of PIK3C3 on HDAC6 ubiquitination. However, in PIK3C3-deficient HEK293 cells, after MG132 treatment, exogenous HDAC6 showed a dramatic decrease in ubiquitination compared to the expression level in control HEK293 cells (Figure 7J–M). Interestingly, we found that HDAC6 ubiquitination was dependent on its phosphorylation at the S59 site, as different ubiquitination levels were detected after HEK293 cells were transfected with wild-type (WT), phosphorylation-deficient (S59A) or phosphorylation-mimicking (S59D) HDAC6 plasmids. Compared to the ubiquitin levels observed in WT-HDAC6-expressing HEK293 cells, a decrease was observed in the S59A group, while an increase was found in the S59D group (Figure 7N,O). Therefore, the PIK3C3-mediated phosphorylation of HDAC6 is required for its degradation through the UPS pathway.
Figure 7.
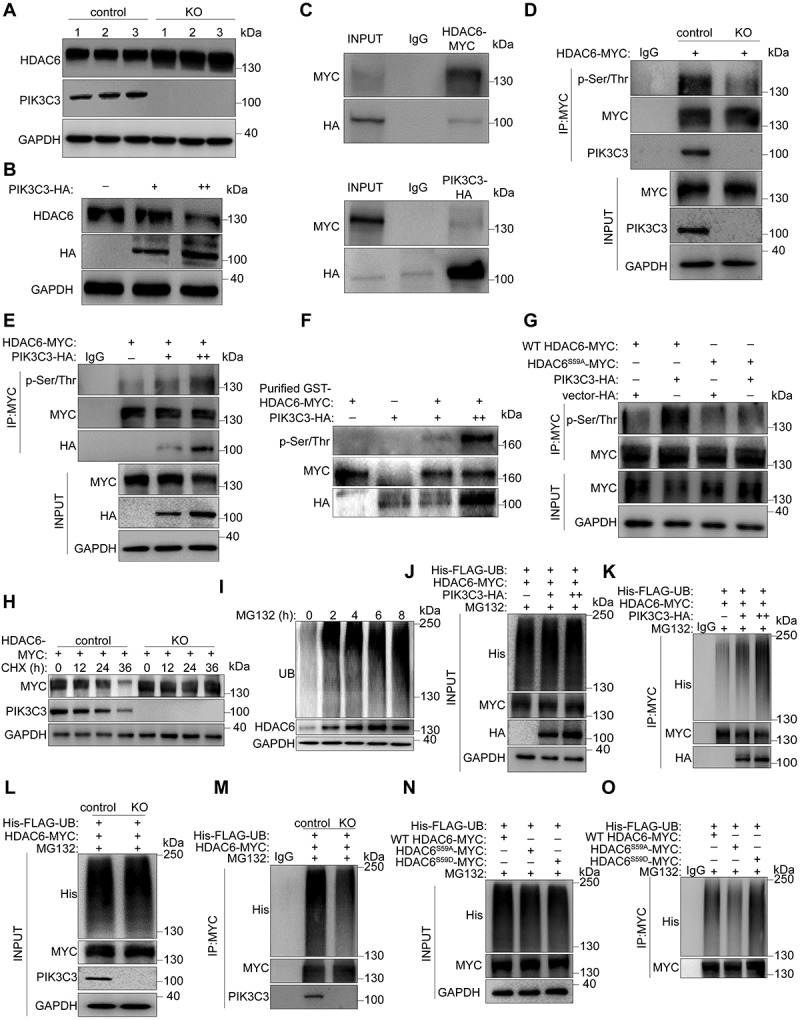
The requirement of PIK3C3 regulated phosphorylation on the degradation of HDAC6. Control or pik3c3 knockout (KO) HEK293 cells were transfected with different plasmids for immunoprecipitation and immunoblotting assays. WT HDAC6-MYC, wild-type HDAC6; HDAC6S59A-MYC, phosphorylation-deficient mutant HDAC6; HDACS59D-MYC, phosphorylation-mimicking mutant HDAC6. Cells were treated with MG132 (10 μM) for 24 h before collection to block proteins degradation through the UPS pathway. (A) Immunoblotting of PIK3C3 and HDAC6 in control and KO HEK293 cells. (B) Dose dependent effect of PIK3C3 on protein levels of HDAC6. Different doses of plasmids loaded with PIK3C3-HA (2 µg or 4 µg/105 cells) were transfected into control HEK293 cells. (C) Interactions between HDAC6 and PIK3C3 in HEK293 cells were shown by immunoprecipitation with HA or MYC antibodies, respectively. (D) Decreased HDAC6 phosphorylation in KO HEK293 cells. Immunoprecipitation was performed with MYC antibody and the phosphorylation levels were checked by antibody against p-Ser/Thr. (E) Dose-dependent effect of HDAC6 phosphorylation after exogenous expression of PIK3C3 in control HEK293 cells. (F) in vitro kinase assays were performed using PIK3C3-HA immunoprecipitated from transfected HEK293 cells and GST-HDAC6-MYC purified from E. coli and the phosphorylation levels were checked by antibody against p-Ser/Thr. (G) PIK3C3 phosphorylated HDAC6 at site S59. PIK3C3-HA and WT HDAC6-MYC or HDACS59A-MYC were co-transfected into HEK293 cells. Immunoprecipitation was performed with MYC antibody and the phosphorylation levels were checked by antibody against p-Ser/Thr. (H) Blockage of HDAC6 degradation after CHX (20 μg/ml) treatment for 36 h. Exogenous HDAC6-MYC was transfected into control and KO HEK293 cells and western blots were performed by using MYC and PIK3C3 antibodies. (I) Immunoblotting of HDAC6 in HEK293 cells after MG132 treatment. MG132 (10 μM) was treated on HEK293 cells for 8 h and cells were collected every 2 h to examine the UB (ubiquitin) and HDAC6 levels. The expression of GAPDH was used as internal control. (J-M) Dose-dependent effect of PIK3C3 on the ubiquitination of HDAC6. His-FLAG-UB, HDAC6-MYC and different dose of PIK3C3-HA were transfected into HEK293 cells. Immunoprecipitation was performed with MYC antibody and the ubiquitination levels of HDAC6 was checked by His antibody. (N-O) Blockage of HDAC6 ubiquitination in HEK293 KO cells. His-FLAG-UB and HDAC6-MYC were transfected into control and KO HEK293 cells. (L-M) the ubiquitin levels of HDAC6 after HEK293 cells being co-transferred with His-FLAG-UB, HDAC6-MYC, HDAC6S59A-MYC, HDAC6S59D-MYC.
HDAC6-regulated deacetylation of SCIN enhances its actin filament-severing ability
HDAC6 has been reported to regulate microtubule- or actin-dependent cell motility by altering the acetylation status of downstream effectors [51,52]. Based on the abnormal accumulation of HDAC6 and SCIN in cKO Sertoli cells and their effects on the disorganization of the F-actin cytoskeleton, we propose possible regulatory effects between HDAC6 and SCIN. Immunoprecipitation first confirmed the interaction between endogenous HDAC6 and SCIN and then that between exogenously expressed HDAC6-MYC and SCIN-EGFP-FLAG in HEK293 cells (Figure S3H and Figure 8A). To determine whether SCIN is the substrate of HDAC6 deacetylase, we expressed SCIN-EGFP-FLAG with different doses of HDAC6-MYC in HEK293 cells and treated them with/without the HDAC6-specific inhibitor tubastatin A (TubA). Immunoprecipitation experiments revealed that HDAC6 inhibition increased the acetylation level of SCIN (Figure 8B) and that the acetylation level of SCIN was downregulated by HDAC6 in a dose-dependent manner (Figure 8C). We then performed a mass spectrometry analysis of SCIN-EGFP-FLAG from TubA-treated HEK293 cells and identified two acetylation sites, at lysines K189 and K495 of SCIN, as potential targets of HDAC6; both of these sites are highly conserved among species (Figure S3I-J, table S3 and Figure 8D). According to the two identified acetylation sites, we constructed three (K189R, K495R and K189R/K495R) deacetylation-mimicking mutants of SCIN, in which lysines were replaced with arginines, and expressed them in HEK293 cells. Immunoprecipitation experiments revealed that each of the two sites was important for SCIN acetylation. When they were both mutated, the acetylation of SCIN was completely blocked (Figure 8E). We next co-expressed HDAC6 with SCIN mutants (K189R and K495R) to determine which acetylation site was regulated by HDAC6. As shown in Figure 8F, exogenous HDAC6 induced SCIN deacetylation in wild-type SCIN- and SCIN-K495R-expressing cells; however, it failed to further deacetylate SCIN when the K189 site was mutated (SCINK189R). This result suggests that the K189 site of SCIN is the target of HDAC6 (Figure 8F). Acetylation can affect protein functions through diverse mechanisms, including the regulation of protein stability and subcellular localization [53]. We first examined whether the deacetylation of SCIN by HDAC6 could affect its stability, and immunoblotting results showed that inhibiting the activity of HDAC6 by either Tub A treatment or exogenous HDAC6 expression did not affect SCIN protein levels (Figure 8G,H). Next, we assessed the effect of the HDAC6-mediated deacetylation of SCIN on its subcellular localization. Wild-type SCIN, deacetylation-mimicking SCINK189R and acetylation-mimicking SCINK189Q (lysine was replaced by glutamine) were expressed in primary Sertoli cells. The immunostaining of F-actin showed a disordered cytoskeleton in Sertoli cells expressing SCIN and SCIN-K189R (~40% or ~ 60% Sertoli cell abnormality, respectively), while in Sertoli cells expressing SCINK189Q, acetylated SCIN failed to sever actin filaments (Figure 8I,J). Immunoblotting results further demonstrated that SCINK189R, but not SCINK189Q, led to a decrease in the F-:G-actin ratio, suggesting that the acetylation status of SCIN at the K189 site is crucial for it to regulate the assembly of the F-actin cytoskeleton (Figure 8K,L).
Figure 8.
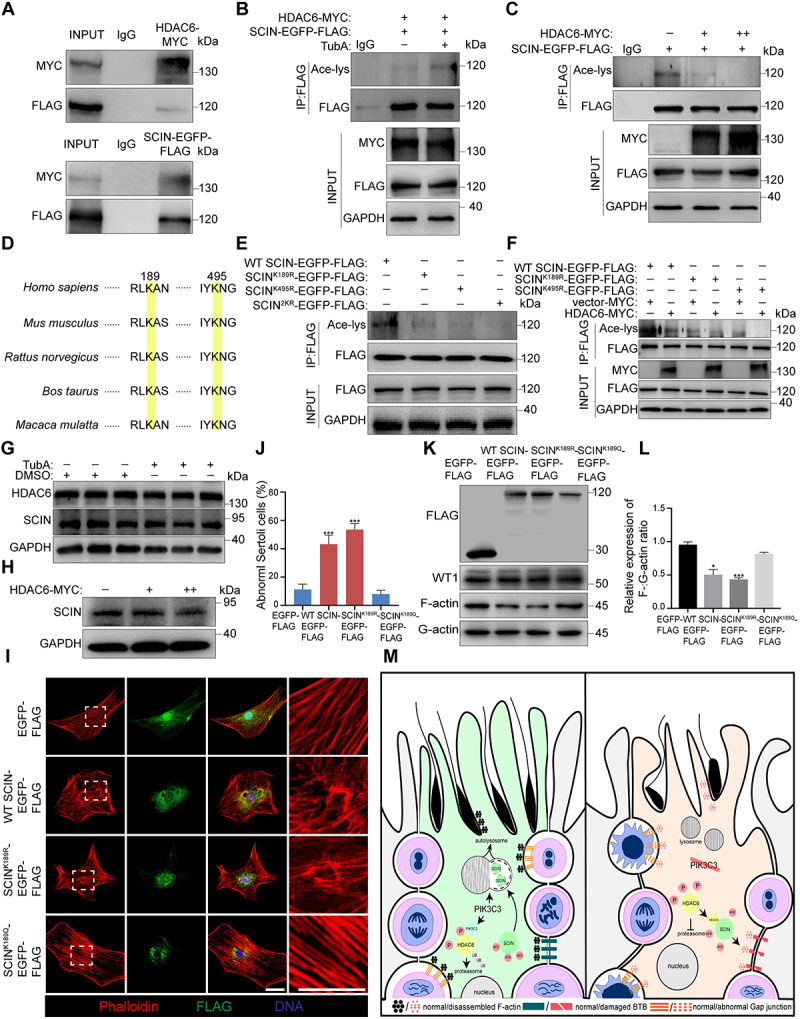
HDAC6 deacetylated SCIN to negatively regulate F-actin cytoskeleton. HEK293 cells were transfected with HDAC6-MYC, SCIN-EGFP-FLAG or SCIN mutant plasmids for immunoprecipitation experiments. WT SCIN-EGFP-FLAG, wild-type SCIN; SCINK189R-EGFP-FLAG, SCIN deacetylation-mimicking mutant; SCINK189Q-EGFP−-FLAG, SCIN acetylation-mimicking mutant; SCINK495R-EGFP-FLAG, SCIN deacetylation-mimicking mutant. SCIN2KR-EGFP-FLAG, SCINK189R-EGFP-FLAG and SCINK495R-EGFP-FLAG plasmids were transfected together. (A) Immunoprecipitation and immunoblotting showed the interaction between HDAC6 and SCIN in HEK293 cells. (B) Increased acetylation of SCIN after blocking the activity of HDAC6. HDAC6-MYC and SCIN-EGFP-FLAG were transfected into HEK293 cells. Cells were treated with TubA for 8 h before collection to inhibit the deacetylase activity of HDAC6. Immunoprecipitation was performed with FLAG antibody and SCIN acetylation levels was checked by Ace-lys antibody. (C) Dose-dependent effect of HDAC6 on the acetylation levels of SCIN. SCIN-EGFP-FLAG and HDAC6-MYC with different doses (2 µg or 4 µg/105 cells) were transfected into HEK293 cells. (D) the K189 and K495 sites of SCIN are highly conserved between species. (E) Acetylation levels of SCIN after HEK293 cells were transfected with different SCIN plasmids. (F) Deacetylated regulation of HDAC6 on SCIN with different site mutants. (G) Immunoblotting of HDAC6 and SCIN in HEK293 cells after treatment with TubA for 8 h. (H) Immunoblotting of SCIN in HEK293 cells after being transfected with different doses of HDAC6-MYC (2 µg or 4 µg/105 cells). (I-J) Primary Sertoli cells were plated on cover slips and transfected with EGFP-FLAG, WT SCIN-EGFP-FLAG, SCINK189R-EGFP-FLAG and SCINK189Q-EGFP-FLAG. Immunofluorescence of phalloidin and FLAG was performed (I). Bar: 30 μm. Graph showing the percentage of Sertoli cells with abnormal F-actin cytoskeleton among all Sertoli cells on each cover slip (n = 5) (J). (K-L) Immunoblotting of F-actin and G-actin in primary Sertoli cells after being transfecting with different SCIN plasmids (K). Graph showing the relative expression of F-:G-actin ratio (n = 3) (L). The expression of WT1 was used as the Sertoli cell marker. *P < 0.05; ***P < 0.01. (M) a schematic diagram depicting the role of PIK3C3 in Sertoli cells. PIK3C3 plays a pivotal role in the establishment of Sertoli cell cytoarchitecture through stabilizing the actin cytoskeleton. Left panel: In normal Sertoli cells, PIK3C3 maintains SCIN in a low level to stabilize the F-actin cytoskeleton and the structure of various anchor junctions between Sertoli cells themselves and between Sertoli cell and germ cell. PIK3C3 regulated autophagy-lysosome pathway is essential for the degradation of SCIN, whereas the phosphorylation of HDAC6 by PIK3C3 is required for its UPS degradation. Right panel: In PIK3C3-deficient Sertoli cells, the degradations of SCIN and HDAC6 were both blocked. The accumulated HDAC6 further deacetylates SCIN and enhances its actin severing activity. The disassembled F-actin cytoskeleton results in the loss of Sertoli cell polarity with disrupted anchoring junctions, such as BTB, aES or gap junctions which ultimately leads to germ cell apoptosis, impaired sperm maturation, and infertility in cKO mice.
Discussion
Sertoli cells are among the cell types with the most complex and dynamic structures known in biology. Their significant spatial differences in cell shape, structure, and function result in highly organized cell polarity [54]. Although previous studies using genetically modified mice have demonstrated that the loss of polarity in Sertoli cells leads to male sterility, the underlying mechanism is not well understood [16,17,55]. In this study, we explored the function of the PI3K family member PIK3C3, also known as VPS34, in Sertoli cells using a conditional knockout mouse model. Our results showed infertility in adult cKO mice and a loss of Sertoli cell polarity, with dysfunctions in membrane specializations such as basal or apical ectoplasmic specification (bES and aES). Further study revealed that the deletion of Pik3c3 in Sertoli cells induced disorder and disassembly of the F-actin cytoskeleton network, which was related to the abnormal accumulation of SCIN, an F-actin-severing protein, and HDAC6, an α-tubulin deacetylase. We found that pik3c3 deletion resulted in the failure of SCIN degradation through the autophagy-lysosome pathway. However, its protein kinase activity was required for the phosphorylation and degradation of HDAC6 through the UPS pathway. Furthermore, SCIN was identified as the target of HDAC6. The increased deacetylation of SCIN induced by the accumulation of HDAC6 in cKO Sertoli cells enhanced its actin filament-severing activity. Herein, we propose a new mechanism of PIK3C3 action in maintaining cell polarity that involves both its function on regulating the autophagy-lysosome pathway and its protein kinase activity (Figure 8M).
In mouse, the first wave of spermatogenesis initiates a few days after birth and along with spermatocytes occurring in the seminiferous epithelium at P14, Sertoli cell gradually differentiates into the highly organized structure in order to support germ-cell differentiation, meiosis and transformation into spermatozoa. The process will be finished around 5 W of age which is marked by the arrangement of spermatozoa on the luminal side of seminiferous epithelium and the completion of aES assembly [56,57]. When Pik3c3 was conditionally knocked out in Sertoli cells by Amhr2-Cre recombinase, our results demonstrated normal first wave of spermatogenesis which was manifested by the morphology of seminiferous tubules and the transient fertility of cKO mice at 6 W and 7 W of age. However, in 7 W testis, vacuole tubules were appeared and after that the mice were shortly infertile with a complete loss of Sertoli cell polarity in nearly all tubules of 9 W testis. It was reported that AMHR2 expression in testis started at P7 and peaked around P12 [58]. This means PIK3C3 in Sertoli cells has no effect on the first wave of spermatogenesis and Sertoli cell differentiation, however, it plays a pivotal role in maintaining the Sertoli cell polarity.
The maintenance of cell-cell adhesion by various junction complexes is considered a prerequisite for the establishment of apical-basal polarity in epithelial cells [59]. Ectoplasmic specialization (ES) is a testis specific phenomenon involving actin-based hybrid anchoring and tight junctions [7]. The disruption of the BTB has been demonstrated to impair apical compartment homeostasis, which results in the apoptosis of large numbers of spermatocytes and round spermatids and the loss of Sertoli cell polarity [60,61]. The disorganization of aES can also lead to impaired Sertoli cell polarity and is a major cause of sperm maturation dysfunction [62]. In pik3c3-deleted Sertoli cells, the disruption of BTB and aES structure was manifested by the markers staining, immunoblotting, TEM and disorganized cytoskeleton network. Furthermore, as the tightest blood-tissue barrier in mammals, the dysfunction of BTB was also shown by the leakage of the FITC tracer in the seminiferous tubules, while the disturbance of aES led to sperm head malformation and abnormal sperm motility. In cKO seminiferous tubules, except a great loss of spermatocytes and spermatids, we also found a decrease in SSC numbers. This is different from what was reported about the maintenance of SSC niche in only BTB-disturbed testes [63]. Among the various junction complexes, gap junctions facilitate cell-cell communication and have been demonstrated to maintain germ cell survival in Sertoli cells [64]. The decrease in the gap junction marker GJA1 and its mislocalization between both Sertoli cells themselves and between Sertoli and germ cells were then observed in cKO seminiferous tubules. Thus, the dysfunctions of different junction complexes in pik3c3-deleted Sertoli cells cause the defect of spermatogenesis and a large loss of germ cells in cKO mice.
In the seminiferous epithelium, tight junctions, ES, and gap junctions are attached to F-actin, and the correct organization of F-actin is required for the establishment of the structure of BTB and aES [65,66]. However, how the actin cytoskeleton is regulated to maintain the cytoarchitechture of Sertoli cells is still unknown. In this study, we found that the disruption of the F-actin cytoskeleton network was related to the abnormal structures of different junction complexes, including the BTB and aES. Moreover, proteomics and phosphoproteomics revealed that the differentially expressed proteins and phosphoproteins were enriched in the regulation of the actin cytoskeleton. We identified SCIN among the most highly accumulated proteins in cKO Sertoli cells, while HDAC6 was among the overlapped actin-binding proteins with differential expression and phosphorylation in both proteomics and phosphoproteomics. The actin-binding protein SCIN belongs to the Gelsolin superfamily and has been implicated in many processes, such as exocytosis, cancer cell invasion and proliferation [67]. Based on the limited reported investigations, SCIN has been implicated in directing goal movement by severing actin filaments; however, how the protein functions in maintaining cell polarity remains unclear. A previous study in mice with Sertoli cell-specific knockout of Ar (androgen receptor) showed the loss of Sertoli cell polarity in cKO mice. Although increased expression of SCIN was found, no connection was established between SCIN and Sertoli cell polarity [68]. In this study, the overexpression of SCIN caused a disordered distribution of F-actin and the decreased G-:F-actin ratio in cultured primary Sertoli cells. This finding was consistent with the control of F-actin stabilization by SCIN in primary chondrocytes [69]. We then found that the failure of SCIN degradation in cKO Sertoli cells was related to the autophagy-lysosome pathway. The blockage of the autophagic flux induced by pik3c3 deletion has been well documented in various cell systems [44,70]. Here, our study indicates that the degradation of SCIN through the autophagy-lysosome pathway is necessary to maintain the stability of the F-actin cytoskeleton. This was further demonstrated by the recovery of F-actin organization after SCIN knockdown in cKO Sertoli cells.
All members of the PtdIns3K family are dual-specificity enzymes, and their catalytic structural domain has the ability to catalyze both protein and lipid phosphorylation [45,71,72]. Previous studies have shown that the protein kinase activity of the class I phosphoinositide 3-kinase (PI3K) is required for agonist-dependent beta-adrenergic receptor (beta AR) internalization [73]. However, for PIK3C3, beyond its well-known lipid kinase activity, its function as a protein kinase and its substrate proteins are seldom reported [74]. In this study, we confirmed that PIK3C3 could interact with HDAC6 and phosphorylate HDAC6 at S59. Our results then demonstrated that phosphorylation at S59 is required for HDAC6 degradation through the UPS pathway. This is different from what was observed in a previous study in which MAP3K5/ASK1-mediated phosphorylation blocked the ubiquitination and degradation of HDAC6 in photoreceptor-connecting cilia [75]. Since different phosphorylation sites were targeted, the studies suggest that posttranslational modification, especially phosphorylation, plays a critical role in the stabilization of HDAC6. HDAC6 is a class II deacetylase that mainly localizes to the cytoplasm and mediates the deacetylation of nonhistone proteins (specifically α-tubulin) to modulate microtubule dynamics [51,76]. Additionally, HDAC6 has been shown to deacetylate cortactin and change its F-actin-binding ability, thereby indirectly influencing actin-dependent cell motility [52]. In this study, the disorganization of both microtubules and actin microfilaments was observed in pik3c3-deleted Sertoli cells. This may be related to the increased deacetylation of target proteins by HDAC6. In HDAC6-overexpressing primary Sertoli cells, we found the disassembly of F-actin and a decreased ratio of F-:G-actin. Moreover, we identified SCIN as a substrate of HDAC6 and the conserved K189 site as the target of HDAC6 deacetylase. The deacetylation of SCIN at K189 has no effect on its stability; however, it is necessary for the regulation of F-actin assembly. The overexpression of an acetylation-deficient mutant of SCIN (K189R) resulted in a disordered F-actin cytoskeleton in primary Sertoli cells, while the acetylation-mimicking mutant (K189Q) had the opposite effect. Until now, there have been no reports addressing the regulation of posttranslational modification on SCIN activity. Here, our study revealed the importance of HDAC6-mediated SCIN deacetylation for F-actin disassembly, and we propose the PIK3C3-HDAC6-SCIN mediated regulation of cytoskeleton stabilization is a new mechanism for maintaining the cell polarity of Sertoli cells.
Cell polarity is one of the most fundamental features of epithelial cells [2,77]. The loss of cell polarity in epithelial cells leads to uncontrolled cell proliferation, enhanced migration and invasion, and ultimately tumorigenesis [78]. In contrast to other types of epithelial cells, Sertoli cells stop dividing in prepubescent mice, and they eventually develop into highly stable and terminally differentiated cells to support spermatogenesis in seminiferous tubules [79]. The inactivation of PtdIns3K activity has been shown to disrupt the epithelial organization of the Drosophila wing, which is caused by the dysregulation of endosome-localized STK11/LKB1 [29]. Moreover, in Caco2 cells, the inhibition of PtdIns3K-C2, but not PtdIns3K-C1, impairs polarity in organoid structures [29,80]. These results suggest the interdependence between PIK3C3-mediated intracellular trafficking and epithelial cell polarity. However, since PtdIns3K-C2 can also regulate the autophagy pathway by promoting the fusion of autophagosomes and lysosomes [81], the involvement of autophagy in the regulation of cell polarity by PIK3C3 cannot be ruled out. In this study, we did not find alterations in STK11 or endosomal-related RAB5 and RAB7 in cKO testes (data not shown). Instead, our study demonstrated the important role of PIK3C3 in stabilizing the F-actin cytoskeleton by modulating the autophagic degradation of SCIN. The blockade of the autophagic flux in Atg5- and Atg7-deleted Sertoli cells has been reported to result in impaired cell polarity with disordered F-actin cytoskeleton [62]. This phenotype is similar to that of pik3c3 deletion and the mechanism is also similar with the failure to degrade another F-actin negative regulator PDLIM1 through the autophagy pathway. However, in addition to the disruption on ES structure, deletion of Pik3c3 also led to abnormal self-renewal of SSC and the damage to seminiferous tubules was more severe. Nearly all tubules were disorganized with large vacuoles in 9 W of cKO testis. This can be explained by the protein kinase activity of PIK3C3. In contrast to previous studies of the requirement of PIK3C3 lipid kinase activity for modulating cell polarity by producing PtdIns3P [24], we propose that PIK3C3 functions as a protein kinase in phosphorylating HDAC6 and stimulating its degradation through the UPS pathway. Thus, together with the regulation of PIK3C3 on SCIN degradation through the autophagy-lysosome pathway, our study implies a new mechanism for maintaining the F-actin cytoskeleton and Sertoli cell polarity.
In conclusion, our study revealed the essential role of PIK3C3 on the establishment and maintenance of Sertoli cell polarity. Compared with the lipid kinase function of PIK3C3, we clarified the protein kinase function of PIK3C3 and deduced that it plays broad biological roles. We propose a detailed molecular mechanism of the regulation of Sertoli cell polarity by PIK3C3, in which both its protein kinase activity and its regulation on autophagy are required to maintain the organization of Sertoli cells with SCIN as the major effector in modulating the stability of the actin cytoskeleton. Since Amhr2-Cre recombinase induces efficient recombination in Sertoli cells of murine testis from P7 days, whereas Amh-Cre functions in fetal gonad as early as E12, future study with Amh-Cre mice will be helpful to delineate the role of PIK3C3 at the early stage of Sertoli cell differentiation [58,82,83].
Materials and methods
Mice
Amhr2-Cre mice and Pik3c3flox/flox mice were obtained from Prof. Youqiang Su (School of life sciences, Shandong University) and the Jackson Laboratory (019081), respectively. Wild-type C57BL/6 mice were purchased from the animal core facility of Nanjing medical university. Mice were maintained under a 12/12 h dark-light cycle at 22°C with free access to food and water. Primary Sertoli cells for plasmid DNA overexpression and RNA interference were collected from 2 W or 8 W C57BL/6 male mice. All experimental protocols and studies were approved by the Committee on the Ethics of Animal Experiments of Nanjing Medical University.
Isolation of mouse primary Leydig cells, Sertoli cells and germ cells
The primary Leydig cells, Sertoli cells, and germ cells were isolated by using the method previously described [84–86]. In brief, testes were removed and the tunica albuginea was stripped under a stereoscope and incubated in 10 ml PBS (Thermo Fisher Scientific, 10010023) containing 2 mg/ml collagenase IV (GLPBIO, GC19591) and 1 mg/ml DNase I (Sigma-Aldrich, DN25) for 25–35 min at 37°C with gentle shaking.
For primary Leydig cells isolation, following the collagenase digestion, PBS was added to tube to bring the final volume up to 50 ml. After 30 min of unit gravity sedimentation, the supernatant with the Leydig cells was collected. The supernatants were centrifuged at 250 × g at 4°C for 10 min. The pelleted cells were re-suspended in 10 ml M199 medium (Thermo Fisher Scientific, 11150067), and added to the top of Percoll (Sigma-Aldrich, P4937) density gradients. Gradient consisted of four 10 ml densities; 1.035, 1.076, 1.085, and 1.095 g/ml Percoll diluted with PBS. Following the addition of cells, gradient tube was centrifuged at 800×g at 4°C for 30 min. The layers between 12 ml and 13 ml marks were retained and pelleted by centrifugation at 250×g at 4°C for 10 min. The purified Leydig cells were then collected and used for immunoblotting.
For primary Sertoli cells and germ cells isolation, the seminiferous tubules without leydig cells were isolated by centrifugation at 100 × g for 30 s at 4°C, and washed three times with PBS. Next, the seminiferous tubules were digested with 1 mg/ml collagenase IV, 1 mg/ml DNase I, and 1 mg/ml hyaluronidase (Sigma-Aldrich, H3506) for 20 min at 37°C with gentle shaking. After allowing the tubules to settle, they were washed three times with PBS and then digested with 2 mg/ml hyaluronidase and 1 mg/ml DNase I at 37°C for 15–20 min to further isolate peritubular myoid cells. After the tubules were allowed to settle, they were washed three times with PBS and finally the seminiferous tubule fragments, which contained only germ cells and Sertoli cells, were digested with 1 mg/ml trypsin (DIFCO,215250) and 1 mg/ml DNase I for 5 min at 37°C and then terminated with fetal bovine serum. Cell suspension was filtered with 70 μm and 40 μm cell strainer. The filtered cell suspension was centrifuged at 600 × g for 5 min at 4°C and the supernatant was discarded. Tissue culture plates for Sertoli cells were prepared by coating each 100-mm plate with 5 mL 5 µg/mL DSA lectin (Sigma-Aldrich, L2766) in PBS, and incubating at 37°C with 5% CO2 for 1 h. The cells were transferred to the lectin-coated plates with fresh DMEM/F12 medium (Cytiva, SH30023.01) containing 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, 10270106) and incubated at 37°C with 5% CO2 for 1 h. After 1 h, germ cells suspended in the culture medium were collected and subsequent immunoblotting was performed. After the germ cells were isolated, fresh culture medium was added to the lectin-coated plates to continue Sertoli cell culture at 37°C with 5% CO2. After 1 day of culture, Sertoli cells were treated with hypotonic solution (20 mM Tris, pH 7.4) for 2 min to remove residual germ cells. Subsequent experiments were performed after 3–4 days of culture.
Plasmid construction and siRNA synthesis
Plasmids (SCIN-EGFP-FLAG, MAP1LC3B-HA, PIK3C3-HA, His-FLAG-UB, HDAC6-MYC, HDAC6-mCherry-MYC, HDAC6S59A-MYC, HDAC6S59D-MYC, SCINK189R-EGFP-FLAG, SCINK189Q-EGFP-FLAG, SCINK495R-EGFP-FLAG and SCIN2KR-EGFP-FLAG cloned in pcDNA3.1 vectors) for transfection of primary Sertoli cells or HEK293 cells were synthesized by Tsingke Biological Technology (Beijing, China). SCIN siRNA were synthesized by RiboBio (Guangzhou, China) and the sequence of siRNA is as follows: siCtrl#F, 5ʹ- CGUACGCGGAAUACUUCGA −3ʹ; siScin-1#F, 5ʹ-AUGAUGACAAUCGGUGUCC-3ʹ; siScin-2#F, 5ʹ -UGGUUCACUUCCUUCUUCC −3ʹ. The siRNA powder was then diluted with DEPC water to a storage concentration of 20 µM and stored at − 80°C for use.
Generation of Pik3c3 knockout cell lines using CRISPR-Cas9 gene editing
Pik3c3 knockout on HEK293 cell lines was carried out by plasmid-based transfection of Cas9 and gRNA using pST1374 and pGL3 plasmid, respectively. Initially, two gRNAs for Pik3c3 were designed using CRISPRdirect website. The sequence of gRNA is as follows: gRNA-1#F, 5ʹ-ACCGGGTAGCATACCTTAACACAA-3ʹ; gRNA-2#F, 5ʹ -ACCGGCTTTGTAGGATGTTCTCAC −3ʹ. 1 μg/ml puromycine (Yeasen Biotechnology, 60209ES60) and 10 μg/ml blasticidin (Yeasen Biotechnology, 60218ES10) were selected for drug screening after transfection. Single cells were sorted into a 96-well plate using a limiting dilution method, and expanded clonal cells were screened by immunoblotting with PIK3C3 antibodies.
Cell culture, transfection and treatment
Primary Sertoli cells and HEK293 cells were cultured in DMEM/F12 medium (Cytiva, SH30023.01) containing 10% FBS and DMEM high glucose medium (Cytiva, SH30243.01) containing 10% FBS, respectively, at 37°C and 5% CO2 in a humidified atmosphere. For plasmid DNA overexpression in primary Sertoli cells and HEK293 cells, plasmid DNA was mixed with Lipofectamine 3000 (Thermo Fisher Scientific, L3000015) in Opti-MEM (Thermo Fisher Scientific, 31985070) and incubated at room temperature for 15 min. The ratio of plasmid DNA to Lipofectamine 3000 was calculated according to the producer’s instruction. The mixture was then added to the cell culture medium and the cells were incubated at 37°C, 5% CO2 for 48 h and samples were collected for immunoblotting, immunoprecipitations, or immunofluorescences. For primary Sertoli cell RNA interference, siRNA was mixed with Lipofectamine RNAiMAX (Thermo Fisher Scientific, 13778075) in Opti-MEM and incubated at room temperature for 5 min. The ratio of siRNA to Lipofectamine RNAiMAX was calculated according to the producer’s instruction. The mixture was then added to the cell culture medium and the cells were incubated at 37°C, 5% CO2 for 48–72 h and samples were collected for immunoblotting or immunofluorescence. To specifically inhibit the function of HDAC6, HEK293 cells were treated with 10 μM tubastatin A (MedChemExpress, HY-13271A) for 8 h before collection. To block the ubiquitin-proteasome pathway or induce autophagy, primary Sertoli cells were treated with 10 μM MG132 (MedChemExpress, HY-13259) or EBSS (Beyotime Biotechnology, C0213) for an appropriate time before collection, respectively. To block the autophagy pathway, HEK293 cells were treated with 100 nM BafA1 (MedChemExpress, HY-100558) for an appropriate time before collection.
Immunoblotting
Testes or cells were lysed with RIPA lysate (Beyotime Biotechnology, P0013D) containing protease inhibitors (MedChemExpress, HY-K0010) and placed on ice for 20 min. Then add protein loading buffer (Beyotime Biotechnology, P0015) and denature in 95°C water for 5 min. Approximately 20 μg proteins in each sample were loaded and separated by electrophoresis (Bio-Rad, USA). After electronic transfer (Bio-Rad, USA), PVDF membranes (Thermo Fisher Scientific, 88518) were blocked in 5% nonfat dry milk for 1 h and then incubated with primary antibodies overnight at 4°C. Membranes were washed 3 times with TBST (Beyotime Biotechnology, ST677) and incubated with secondary antibodies for 1 h at room temperature. Finally, protein bands were visualized using ECL (Beyotime Biotechnology, P0018) and Immunoblotting detection systems (GE Healthcare, USA). Primary and secondary antibodies were obtained from following commercial sources: rabbit monoclonal anti-PIK3C3 (Cell Signaling Technology, 4263), rabbit monoclonal anti-MAP1LC3B (Cell Signaling Technology, 3868), mouse monoclonal anti-ACTB/actin (Proteintech Group, 66009–1-Ig), rabbit polyclonal anti-OCLN (Proteintech Group, 13409–1-AP), rabbit polyclonal anti-CDH2 (Proteintech Group, 22018–1-AP), mouse monoclonal anti-GAPDH (Proteintech Group, 60004–1-Ig), mouse monoclonal anti-His (Proteintech Group, 66005–1-Ig), mouse monoclonal anti-SQSTM1 (Santa Cruz Biotechnology, sc -28,359), mouse monoclonal anti-UB (Santa Cruz Biotechnology, sc-8017), mouse monoclonal anti-HA (Santa Cruz Biotechnology, sc-7392), rabbit polyclonal anti-p-Ser/Thr (ZEN BIOSCIENCE, 530893), rabbit polyclonal anti-Ace-lys (ZEN BIOSCIENCE, 502391), rabbit monoclonal anti-CTNNA1 (Cell Signaling Technology, 36611), rabbit monoclonal anti-GJA1 (Cell Signaling Technology, 83649), rabbit monoclonal anti-HDAC6 (Cell Signaling Technology, 7558), rabbit monoclonal anti-WT1 (Abcam, ab89901), mouse monoclonal anti-TJP1 (Thermo Fisher Scientific, 33–9100), mouse monoclonal anti-SCIN (Santa Cruz Biotechnology, sc -376,136), rat monoclonal anti-LAMP1 (Developmental Studies Hybridoma Bank, 1D4B), mouse monoclonal anti-FLAG (ZEN-BIOSCIENCE, 250111), rabbit polyclonal anti-MYC (Proteintech Group, 16286–1-AP), rabbit polyclonal anti-CYP11A1 (Bioss, bs-3608 R), control polyclonal anti-rabbit IgG (Beyotime Biotechnology, A7016), control polyclonal anti-mouse IgG (Beyotime Biotechnology, A7028), goat anti-mouse IgG H&L (YIFEIXUE Biotechnology, YFSA01), goat anti-rabbit IgG H&L (YIFEIXUE Biotechnology, YFSA02), goat anti-rat IgG H&L (YIFEIXUE Biotechnology, YFSA04).
Immunofluorescence and TUNEL assay
Testes were isolated from control and cKO mice and fixed with 4% paraformaldehyde (PFA) at 4°C for 12 h. Then the testis was cut in half with a razor blade and fixed with 4% PFA at 4°C for another 12 h. Testes were dehydrated in a gradient of 10%, 20%, and 30% sucrose, embedded in OCT (SAKURA,4583), and then frozen sectioned. After cryosectioning at 6 μm, the samples were washed 3 times with pre-cooled PBS. The samples were then penetrated with 0.1% Triton X-100 (Sigma-Aldrich, T8787) for 10 min and blocked in 3% BSA/PBS for 1 h at room temperature. Primary antibodies were diluted in blocking solution and applied to samples overnight at 4°C. After washing 3 times with PBS, as needed for staining, the secondary antibody, phalloidin or PNA were diluted in blocking solution and applied to the samples for 1 h at room temperature. Nuclei were counterstained with Hoechst 33,342 (Life Technologies, H3570) at room temperature for 15 min and immunofluorescence signals were observed using a confocal laser scanning microscope (LSM 710; Zeiss, Germany). For cellular immunofluorescence, Sertoli cells were plated on cover glass slips, and after 24 h, cells were washed 3 times with PBS, fixed with 4% PFA for 40 min at room temperature, and stained as described above. Primary and secondary antibodies were obtained from following commercial sources: mouse monoclonal anti-DDX4 (Abcam, ab27591), rat monoclonal anti-GCNA (Abcam, ab82527), mouse monoclonal anti-ZBTB16 (Abcam, ab104854), rabbit monoclonal anti-KIT (Abcam, ab32363), rabbit monoclonal anti-H2AX (Abcam, ab81299), mouse monoclonal anti-VIM/vimentin (Abcam, ab8978), mouse monoclonal anti-TUBB3 (Cell Signaling Technology, 4466S), rabbit polyclonal anti-cleaved CASP3 (Cell Signaling Technology, 9661), Alexa Fluor 488 donkey anti-mouse IgG (Thermo Fisher Scientific, A21202), Alexa Fluor 594 donkey anti-mouse IgG (Thermo Fisher Scientific, A21203), Alexa Fluor 488 donkey anti-rat IgG (Thermo Fisher Scientific, A21208), Alexa Fluor 594 donkey anti-rabbit IgG (Thermo Fisher Scientific, A21207), Alexa Fluor 488 donkey anti-rabbit IgG (Thermo Fisher Scientific, A32790), Alexa Fluor 647 goat anti- rabbit IgG (Thermo Fisher Scientific, A31573). The other primary antibodies used for immunofluorescence are of the same sources as those used for immunoblotting. The TRITC-phalloidin (Sigma-Aldrich, P1951) and FITC-PNA (Sigma-Aldrich, L7381) were purchased from Sigma-Aldrich. For TUNEL staining, testis samples were labeled with TUNEL Apoptosis Detection Kit (Yeasen Biotechnology, 40307ES60) and TUNEL-positive cells were counted in each seminiferous tubule.
Immunoprecipitation
According to the requirement of sample protein concentration in different experiments, 1 × 106 plasmid-transfected HEK293 cells and 1 × 107 untreated primary Sertoli cells or HEK293 cells were lysed with RIPA lysate (Beyotime Biotechnology, P0013D) containing 1% NP-40, 0.25% deoxycholate and protease inhibitors without triton X-100 and SDS, respectively, and placed on ice for 20 min [87]. The supernatant was retained by centrifugation at 15,000 × g for 20 min at 4°C. A total 3 µg antibody was added to each protein sample and incubated overnight at 4°C in a rotating wheel. Then add 30 µl of protein A/G beads (Abmart, A10001) to each sample and incubate at 4°C for 3 h. After centrifugation at 2000 × g for 20 s, the supernatant was removed and washed twice with PBS. The beads were then added to protein loading buffer and denatured at 95°C for 5 min. After a brief centrifugation, the collected supernatant was used for immunoblotting with specific antibodies.
In vitro protein kinase assays
The HDAC6-MYC was expressed as GST-fusion proteins in E. coli and purified using a GST-fusion protein purification kit (Beyotime Biotechnology, P2262) following the manufacturer’s protocol. The PIK3C3-HA protein was purified from HEK293 cells 48 h after transfection by immunoprecipitation with anti-HA antibody beads. In vitro protein kinase assay was carried out with varying concentration of PIK3C3-HA protein (0, 1 µg or 2 µg) along with or without GST-HDAC6-MYC protein (10 µg) as substrate, ATP (200 μM), and kinase buffer (25 mM Tris-HCl, 5 mM β-Glycerol phosphate disodium salt pentahydrate, 2 mM Dithiothreitol, 0.1 mM Na3VO4, 10 mM MnCl2; pH 7.5) containing protease inhibitors in a total volume of 60 µL. The reaction was carried out at 30°C for 40 min and then stopped by adding 15 µl of 5 X SDS sample buffer. The reaction products were used for immunoblotting with anti-p-Ser/Thr antibody.
In vitro fertilization (IVF)
The cauda epididymis of control and cKO mice were isolated and sperm released in HTF medium (Sigma-Aldrich, MR-070) containing 10% FBS (SORFA, SX1111) for 1 h at 37°C with 5% CO2 to capacitate sperm. Three-week-old female C57BL/6 mice were injected with hCG (Ningbo second hormone factory, 200929) 48 h after PMSG (Ningbo second hormone factory, 11C25) injection, and cumulus-oocyte complexes (COC) were collected from the ampulla of the fallopian tube into M2 medium (Sigma-Aldrich, M7167) 14–16 h after hCG injection. In vitro fertilization was performed by incubating COC and capacitated sperm in HTF medium containing 10% FBS at 37°C with 5% CO2. After 6 h of incubation, fertilized oocytes were transferred to KSOM medium (Sigma-Aldrich, MR-101) containing 10% FBS and incubated at 37°C with 5% CO2. Fertilized oocytes were cultured in KSOM for 24 h followed by observing 2-cells and counting the 2-cell:MII ratio.
Fertility test
Four control and cKO mice at 6 W, 7 W, 8 W, 9 W and 24 W of age were mated one-to-one with 6-week-old wild-type C57BL/6 female mice. Neonatal mice were counted within 2 days of birth. One reproductive cycle was observed for each pair of mice.
Spermatozoa analysis
Spermatozoa were collected from the cauda epididymis, extruded and suspended in HTF medium containing 10% FBS. Spermatozoa samples (10 μl) were subjected to computer-assisted semen analysis (Hamilton-Thorne Research, USA) after 10 min incubation at 37°C. Total spermatozoa counts were counted and spermatozoa motility parameters were analyzed including motile spermatozoa count, forward motile spermatozoa count, average path velocity (VAP), straight line velocity (VSL), curve-linear velocity (VCL), straightness (STR), Beat-cross frequency (BCF). The remaining sperm samples were fixed with 4% PFA for 40 min and plated on glass slides, followed by immunofluorescence according to standard methods to assess spermatozoa morphology and to calculate the percentage of abnormal spermatozoa counts.
H&E staining
Mouse testes and cauda epididymis were isolated and fixed in 10% Hartman’s fixative (Sigma-Aldrich, H0290) for 24 h at room temperature. After paraffin embedding, tissue sections were cut to 6 μm. Sections were deparaffinized with two additional xylene treatments (10 min each) and rehydrated using graded ethanol; sections were treated twice with absolute ethanol (10 min each) and then with 95% ethanol for 10 min, followed by treatment with 70% ethanol for 10 min. After two washes in water, the slides were stained in hematoxylin solution for 5 min. After washing in running water for 10 min, it was treated in 0.1% hydrochloric acid for 5 s. Slides were washed in running water until sections turned blue. The slides were then stained with eosin for 20 s. Treated with 70% and 90% ethanol for 15 s, respectively, followed by two treatments with absolute ethanol (1 min each). Treated twice with xylene (15 min each). Cover with neutral balsam and place in 65°C oven overnight. Finally, the slides were then mounted in mounting media and examined with a Nikon-112 microscope (Nikon, Japan).
Transmission electron microscopy (TEM)
The mouse testes were isolated and fixed in 2.5% glutaraldehyde for 2 h at 4°C, then the testis was cut into small pieces of 1 mm3 and then fixed in 2.5% glutaraldehyde for 24 h at 4°C. The samples were then processed according to standard TEM procedures and embedded in epoxypropane resin. Ultrathin sections were cut on an ultramicrotome and stained with uranyl acetate and lead citrate. Finally, samples were observed by transmission electron microscopy (Tecnai G2 Spirit BioTwin, FEI, USA).
Fluorescein isothiocyanate (FITC) tracer assay
Male mice at 8W were anesthetized by intraperitoneal injection of 0.02 ml/mg chloral hydrate (Sigma-Aldrich, C8383), followed by injection of 200 μl of 5 mg/ml fluorescein isothiocyanate (Sigma-Aldrich, F7250) via the tail vein of the mice. Mice were euthanized 1 h later, and testes isolated and snap frozen in liquid nitrogen. Testes were then OCT-embedded and cryosectioned. Sections were stored at −80°C or observed for immunofluorescence signal using a confocal laser scanning microscope (LSM 710; Zeiss, Germany).
TMT-labeling quantitative proteomics and phosphoproteomics
About 100 mg of testicular tissue from 8-week-old control and cKO mice were collected for proteomics and phosphoproteomics analysis by PTM BIO (Hangzhou, China). Briefly, testes were snap-frozen in liquid nitrogen and ground to a powder, which was subsequently sonicated in lysis buffer (8 M urea, 1% protease inhibitor, 1% phosphatase inhibitor) and trypsinized into peptides. Then the peptides from individual samples were isobaric-mass tagged by TMT6–127, TMT6–128, TMT6–129 and TMT6–130, respectively, according to the manufacturer’s protocol for TMT10plex Isobaric Mass Tag Labeling Kit (Thermo Fisher Scientific, 90113CH). Next, peptides were fractionated by high pH reverse-phase HPLC using a Betasil C18 column (Thermo Fisher Scientific, 70105–052130), then peptide fractions were subjected to a nanospray ionization source followed by tandem mass spectrometry (MS/MS) in Q Exactive Plus mass spectrometer (Thermo Fisher Scientific, IQLAAEGAAPFALGMBDK) coupled online to the UPLC for identification of peptides (for proteomics) or phosphorylation sites (for phosphoproteomics). The resulting MS/MS data were processed using the Maxquant search engine (v.1.6.15.0).
Mass spectrometry analysis
1×107 HEK293 cells were divided into four groups, each group of cells was transfected with a plasmid containing the SCIN-EGFP-FLAG coding sequence and the activity of HDAC6 in each group of cells was inhibited with 10 μM tubastatin A. Cells were continued to be cultured for 48 h and then protein samples were collected for SDS-PAGE to separate the proteins. After developing the protein in the gel with Coomassie Brilliant Blue staining, the gel containing SCIN-EGFP-FLAG was separated with a razor blade. The gel was then analyzed by Bioprofile (Shanghai, China) with the Easy-nLC1200 chromatographic system (Thermo Fisher, LC140) and Q-exactive Plus mass spectrometer. Acetylation modification site identification of SCIN was performed using MaxQuant search engine (v. 1.6.1.0).
Cycloheximide chase (CHX) assay
Primary Sertoli cells and HEK293 cells were plated one day before the experiment, and cycloheximide (Sigma-Aldrich, M8699) was added to the culture at 100 μg/ml or 20 μg/ml to block new protein synthesis in primary cells or HEK293 cells, respectively. Protein levels were determined by immunoblotting.
G-actin:F-actin quantification
G-actin and F-actin were first isolated, with minor modifications as previously described [88]. Sertoli cells were lysed on ice for 20 min with RIPA lysate containing protease inhibitors, followed by centrifugation at 17,000 × g for 30 min at 4°C. Soluble actin (G-actin) was collected in the supernatant. The insoluble actin (F-actin) in the pellet was lysed with equal volumes of mixed RIPA lysate and lysate 2 (1.5 mM guanidine hydrochloride, 1 mM sodium acetate, 1 mM CaCl2, 1 mM adenosine triphosphate [Beyotime Biotechnology, D7378], and 20-mM Tris-HCl, pH 7.5) on ice for 1 h, and pipetted for one min every 20 min to dissolve F-actin into G- actin. The solubilized F-actin was collected in the supernatant by centrifugation at 17,000 × g for 30 min at 4°C. G-actin from the supernatant and F-actin from the pellet were then analyzed by immunoblotting using the anti-ACTB antibody. Finally, the band intensities of actin were determined using ImageJ and the G-actin: F-actin ratios were plotted as column graphs.
In vitro ubiquitination assay
As previously described [89], HDAC6-MYC, HDAC6S59A-MYC, HDAC6S59D-MYC and PIK3C3-HA were transfected into HEK293 cells with His-UB. HEK293 cells were treated with 10 mΜ MG132 for 6 h in advance. Forty-eight hours after transfection, protein extraction was performed with RIPA lysate and the whole-cell lysates were incubated in rotation with anti-MYC antibody or rabbit IgG isotype at 4°C overnight. Precipitation of the antibody-bound proteins was performed by adding protein A/G beads and incubated at 4°C for 3 h. After a brief centrifuge and washing with washing buffer (50 mM Tris pH 8.5), the precipitates were for immunoblotting analysis with specific antibodies.
Statistical analysis
Each experiment was repeated at least three times. Student’s t test or one-way ANOVA were used to compare the statistical differences between experimental groups. ImageJ (NIH, USA) was used to analyze western blot protein bands and confocal images. All data are presented as mean ± SEM. P < 0.05 considered to indicate a significant result.
Supplementary Material
Acknowledgements
We would like to thank Prof. Jiahao Sha and Prof. Mingxi Liu at State key laboratory of reproductive medicine and offspring health, Nanjing medical University for his help with our study design and all the discussions about the project.
Funding Statement
This work was supported by the National Key Research and Development Program of China (2018YFC1004203, 2018YFC1003703) and the National Natural Science Foundation of China (31871513, 8220060142).
Disclosure statement
No potential conflict of interest was reported by the authors.
Data and materials availability
Raw data and extracted text files for quantitative proteomics, phosphoproteomics and SCIN mass spectrometry analysis have been deposited into PRIDE, under accession nos. PXD036240, PXD036241and PXD036242, respectively.
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/15548627.2023.2235195.
References
- [1].Niessen MT, Iden S, Niessen CM.. The in vivo function of mammalian cell and tissue polarity regulators–how to shape and maintain the epidermal barrier. J Cell Sci. 2012 Aug 1;125(Pt 15):3501–3510. doi: 10.1242/jcs.092890 [DOI] [PubMed] [Google Scholar]
- [2].St Johnston D, Ahringer J. Cell polarity in eggs and epithelia: parallels and diversity. Cell. 2010 May 28;141(5):757–774. doi: 10.1016/j.cell.2010.05.011 [DOI] [PubMed] [Google Scholar]
- [3].Franca LR, Hess RA, Dufour JM, et al. The Sertoli cell: one hundred fifty years of beauty and plasticity. Andrology. 2016. Mar;4(2):189–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wong V, Russell LD. Three-dimensional reconstruction of a rat stage V Sertoli cell: I. Methods, basic configuration, and dimensions. Am J Anat. 1983. Jun;167(2):143–161. doi: 10.1002/aja.1001670202 [DOI] [PubMed] [Google Scholar]
- [5].Oatley JM, Brinster RL. The germline stem cell niche unit in mammalian testes. Physiol Rev. 2012. Apr;92(2):577–595. doi: 10.1152/physrev.00025.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yan HH, Mruk DD, Lee WM, et al. Ectoplasmic specialization: a friend or a foe of spermatogenesis? BioEssays. 2007. Jan;29(1):36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wong EW, Mruk DD, Cheng CY. Biology and regulation of ectoplasmic specialization, an atypical adherens junction type, in the testis. Biochim Biophys Acta. 2008. Mar;1778(3):692–708. doi: 10.1016/j.bbamem.2007.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Berruti G, Paiardi C. The dynamic of the apical ectoplasmic specialization between spermatids and Sertoli cells: the case of the small GTPase Rap1. Biomed Res Int. 2014;2014:635979. doi: 10.1155/2014/635979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kopera IA, Bilinska B, Cheng CY, et al. Sertoli-germ cell junctions in the testis: a review of recent data. Philos Trans R Soc Lond B Biol Sci. 1546. [2010 May 27];365:1593–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ellenbroek SI, Iden S, Collard JG. Cell polarity proteins and cancer. Semin Cancer Biol. 2012. Jun;22(3):208–215. doi: 10.1016/j.semcancer.2012.02.012 [DOI] [PubMed] [Google Scholar]
- [11].Wong EW, Mruk DD, Lee WM, et al. Par3/Par6 polarity complex coordinates apical ectoplasmic specialization and blood-testis barrier restructuring during spermatogenesis. Proc Natl Acad Sci U S A. 2008 Jul 15;105(28):9657–9662. doi: 10.1073/pnas.0801527105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gao Y, Lui WY, Lee WM, et al. Polarity protein Crumbs homolog-3 (CRB3) regulates ectoplasmic specialization dynamics through its action on F-actin organization in Sertoli cells. Sci Rep. 2016 Jun 30;6(1):28589. doi: 10.1038/srep28589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Su WH, Mruk DD, Wong EW, et al. Polarity protein complex Scribble/Lgl/Dlg and epithelial cell barriers. Adv Exp Med Biol. 2012;763:149–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Filic V, Mijanovic L, Putar D, et al. Regulation of the actin cytoskeleton via Rho GTPase signalling in dictyostelium and mammalian cells: a parallel slalom. Cells. 2021 Jun 24;10(7):1592. doi: 10.3390/cells10071592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lawson CD, Ridley AJ. Rho GTPase signaling complexes in cell migration and invasion. J Cell Bio. 2018 Feb 5;217(2):447–457. doi: 10.1083/jcb.201612069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Heinrich A, Bhandary B, Potter SJ, et al. Cdc42 activity in Sertoli cells is essential for maintenance of spermatogenesis. Cell Rep. 2021 Oct 26;37(4):109885. doi: 10.1016/j.celrep.2021.109885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Heinrich A, Potter SJ, Guo L, et al. Distinct roles for Rac1 in sertoli cell function during testicular development and spermatogenesis. Cell Rep. 2020 Apr 14;31(2):107513. doi: 10.1016/j.celrep.2020.03.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27(1):107–132. doi: 10.1146/annurev-cellbio-092910-154005 [DOI] [PubMed] [Google Scholar]
- [19].Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018. Jun;19(6):349–364. doi: 10.1038/s41580-018-0003-4 [DOI] [PubMed] [Google Scholar]
- [20].Wirawan E, Vanden Berghe T, Lippens S, et al. Autophagy: for better or for worse. Cell Res. 2012. Jan;22(1):43–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Foerster EG, Mukherjee T, Cabral-Fernandes L, et al. How autophagy controls the intestinal epithelial barrier. Autophagy. 2022. Jan;18(1):86–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Galati S, Boni C, Gerra MC, et al. Autophagy: a player in response to oxidative stress and DNA Damage. Oxid Med Cell Longev. 2019;2019:5692958. doi: 10.1155/2019/5692958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Backer JM. The intricate regulation and complex functions of the class III phosphoinositide 3-kinase Vps34. Biochem J. 2016 Aug 1;473(15):2251–2271. doi: 10.1042/BCJ20160170 [DOI] [PubMed] [Google Scholar]
- [24].Bilanges B, Posor Y, Vanhaesebroeck B. PI3K isoforms in cell signalling and vesicle trafficking. Nat Rev Mol Cell Biol. 2019. Sep;20(9):515–534. doi: 10.1038/s41580-019-0129-z [DOI] [PubMed] [Google Scholar]
- [25].Hurley JH, Young LN. Mechanisms of autophagy initiation. Annu Rev Biochem. 2017 Jun 20;86(1):225–244. doi: 10.1146/annurev-biochem-061516-044820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rostislavleva K, Soler N, Ohashi Y, et al. Structure and flexibility of the endosomal Vps34 complex reveals the basis of its function on membranes. Science. 2015 Oct 9;350(6257):aac7365. doi: 10.1126/science.aac7365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Abe M, Setoguchi Y, Tanaka T, et al. Membrane protein location-dependent regulation by PI3K (III) and rabenosyn-5 in Drosophila wing cells. PLoS One. 2009 Oct 2;4(10):e7306. doi: 10.1371/journal.pone.0007306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lorincz P, Lakatos Z, Maruzs T, et al. Atg6/uvrag/vps34-containing lipid kinase complex is required for receptor downregulation through endolysosomal degradation and epithelial polarity during Drosophila wing development. Biomed Res Int. 2014;2014:851349. doi: 10.1155/2014/851349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].O’Farrell F, Lobert VH, Sneeggen M, et al. Class III phosphatidylinositol-3-OH kinase controls epithelial integrity through endosomal LKB1 regulation. Nat Cell Biol. 2017. Dec;19(12):1412–1423. [DOI] [PubMed] [Google Scholar]
- [30].Fujiwara Y, Komiya T, Kawabata H, et al. Isolation of a DEAD-family protein gene that encodes a murine homolog of Drosophila vasa and its specific expression in germ cell lineage. Proc Natl Acad Sci U S A. 1994 Dec 6;91(25):12258–12262. doi: 10.1073/pnas.91.25.12258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Soderstrom KO, Malmi R, Karjalainen K. Binding of fluorescein isothiocyanate conjugated lectins to rat spermatogenic cells in tissue sections. Enhancement of lectin fluorescence obtained by fixation in Bouin’s fluid. Histochemistry. 1984;80(6):575–579. doi: 10.1007/BF02400975 [DOI] [PubMed] [Google Scholar]
- [32].Tanaka H, Pereira LA, Nozaki M, et al. A germ cell-specific nuclear antigen recognized by a monoclonal antibody raised against mouse testicular germ cells. Int J Androl. 1997. Dec;20(6):361–366. [DOI] [PubMed] [Google Scholar]
- [33].Pelletier J, Schalling M, Buckler AJ, et al. Expression of the Wilms’ tumor gene WT1 in the murine urogenital system. Genes Dev. 1991. Aug;5(8):1345–1356. [DOI] [PubMed] [Google Scholar]
- [34].Niedenberger BA, Busada JT, Geyer CB. Marker expression reveals heterogeneity of spermatogonia in the neonatal mouse testis. Reproduction. 2015. Apr;149(4):329–338. doi: 10.1530/REP-14-0653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ball RL, Fujiwara Y, Sun F, et al. Regulatory complexity revealed by integrated cytological and RNA-seq analyses of meiotic substages in mouse spermatocytes. BMC Genomics. 2016 Aug 12;17(1):628. doi: 10.1186/s12864-016-2865-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wong CH, Cheng CY. The blood-testis barrier: its biology, regulation, and physiological role in spermatogenesis. Curr Top Dev Biol. 2005;71:263–296. [DOI] [PubMed] [Google Scholar]
- [37].Gao Y, Mruk DD, Lui WY, et al. F5-peptide induces aspermatogenesis by disrupting organization of actin- and microtubule-based cytoskeletons in the testis. Oncotarget. 2016 Sep 27;7(39):64203–64220. doi: 10.18632/oncotarget.11887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li MW, Xia W, Mruk DD, et al. Tumor necrosis factor alpha reversibly disrupts the blood-testis barrier and impairs Sertoli-germ cell adhesion in the seminiferous epithelium of adult rat testes. J Endocrinol. 2006. Aug;190(2):313–329. [DOI] [PubMed] [Google Scholar]
- [39].Rodriguez Del Castillo A, Lemaire S, Tchakarov L, et al. Chromaffin cell scinderin, a novel calcium-dependent actin filament-severing protein. The EMBO Journal. 1990. Jan;9(1):43–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kyronlahti A, Euler R, Bielinska M, et al. GATA4 regulates Sertoli cell function and fertility in adult male mice. Mol Cell Endocrinol. 2011 Feb 10;333(1):85–95. doi: 10.1016/j.mce.2010.12.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Boyer A, Girard M, Thimmanahalli DS, et al. mTOR regulates gap junction Alpha-1 protein trafficking in sertoli cells and is required for the maintenance of spermatogenesis in mice. Biol Reprod. 2016. Jul;95(1):13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Wang Y, Le WD. Autophagy and ubiquitin-proteasome system. Adv Exp Med Biol. 2019;1206:527–550. [DOI] [PubMed] [Google Scholar]
- [43].Liu C, DeRoo EP, Stecyk C, et al. Impaired autophagy in mouse embryonic fibroblasts null for Kruppel-like Factor 4 promotes DNA damage and increases apoptosis upon serum starvation. Mol Cancer. 2015 May 6;14:101. doi: 10.1186/s12943-015-0373-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Jaber N, Dou Z, Chen JS, et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci U S A. 2012 Feb 7;109(6):2003–2008. doi: 10.1073/pnas.1112848109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Walker EH, Perisic O, Ried C, et al. Structural insights into phosphoinositide 3-kinase catalysis and signalling. Nature. 1999 Nov 18;402(6759):313–320. doi: 10.1038/46319 [DOI] [PubMed] [Google Scholar]
- [46].Liang T, Fang H. Structure, functions and selective inhibitors of HDAC6. Curr Top Med Chem. 2018;18(28):2429–2447. doi: 10.2174/1568026619666181129141822 [DOI] [PubMed] [Google Scholar]
- [47].Xu W, Gulvady AC, Goreczny GJ, et al. Paxillin-dependent regulation of apical-basal polarity in mammary gland morphogenesis. Development. 2019 May 1;146(9). doi: 10.1242/dev.174367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kim DJ, Martinez-Lemus LA, Davis GE. EB1, p150Glued, and Clasp1 control endothelial tubulogenesis through microtubule assembly, acetylation, and apical polarization. Blood. 2013 Apr 25;121(17):3521–3530. doi: 10.1182/blood-2012-11-470179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Su H, Yang F, Wang Q, et al. VPS34 acetylation controls its lipid kinase activity and the initiation of canonical and non-canonical autophagy. Mol Cell. 2017 Sep 21;67(6):907–921 e7. doi: 10.1016/j.molcel.2017.07.024 [DOI] [PubMed] [Google Scholar]
- [50].Iwata A, Riley BE, Johnston JA, et al. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005 Dec 2;280(48):40282–40292. doi: 10.1074/jbc.M508786200 [DOI] [PubMed] [Google Scholar]
- [51].Hubbert C, Guardiola A, Shao R, et al. HDAC6 is a microtubule-associated deacetylase. Nature. 2002 May 23;417(6887):455–458. doi: 10.1038/417455a [DOI] [PubMed] [Google Scholar]
- [52].Zhang X, Yuan Z, Zhang Y, et al. HDAC6 modulates cell motility by altering the acetylation level of cortactin. Mol Cell. 2007 Jul 20;27(2):197–213. doi: 10.1016/j.molcel.2007.05.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Narita T, Weinert BT, Choudhary C. Functions and mechanisms of non-histone protein acetylation. Nat Rev Mol Cell Biol. 2019. Mar;20(3):156–174. doi: 10.1038/s41580-018-0081-3 [DOI] [PubMed] [Google Scholar]
- [54].Griswold MD. 50 years of spermatogenesis: Sertoli cells and their interactions with germ cells. Biol Reprod. 2018 Jul 1;99(1):87–100. doi: 10.1093/biolre/ioy027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Xiong Z, Wang C, Wang Z, et al. Raptor directs Sertoli cell cytoskeletal organization and polarity in the mouse testis. Biol Reprod. 2018 Dec 1;99(6):1289–1302. doi: 10.1093/biolre/ioy144 [DOI] [PubMed] [Google Scholar]
- [56].Jones SL, Styles J, Geyer CB. A new translation and reader’s guide to the first detailed description of the first wave of spermatogenesis in the mouse. Mol Reprod Dev. 2021. Jul;88(7):473–478. doi: 10.1002/mrd.23519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Busada JT, Velte EK, Serra N, et al. Rhox13 is required for a quantitatively normal first wave of spermatogenesis in mice. Reproduction. 2016. Nov;152(5):379–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Arango NA, Kobayashi A, Wang Y, et al. A mesenchymal perspective of Mullerian duct differentiation and regression in Amhr2-lacZ mice. Mol Reprod Dev. 2008. Jul;75(7):1154–1162. [DOI] [PubMed] [Google Scholar]
- [59].Knust E. Regulation of epithelial cell shape and polarity by cell-cell adhesion (Review). Mol Membr Biol. 2002. Apr-Jun;19(2):113–120. doi: 10.1080/09687680210137219 [DOI] [PubMed] [Google Scholar]
- [60].Zhang H, Chen F, Dong H, et al. Loss of Fbxw7 in Sertoli cells impairs testis development and causes infertility in micedagger. Biol Reprod. 2020 Apr 15;102(4):963–974. doi: 10.1093/biolre/ioz230 [DOI] [PubMed] [Google Scholar]
- [61].Mazaud-Guittot S, Meugnier E, Pesenti S, et al. Claudin 11 deficiency in mice results in loss of the Sertoli cell epithelial phenotype in the testis. Biol Reprod. 2010. Jan;82(1):202–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Liu C, Wang H, Shang Y, et al. Autophagy is required for ectoplasmic specialization assembly in sertoli cells. Autophagy. 2016 May 3;12(5):814–832. doi: 10.1080/15548627.2016.1159377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Hu X, Tang Z, Li Y, et al. Deletion of the tyrosine phosphatase Shp2 in Sertoli cells causes infertility in mice. Sci Rep. 2015 Aug 12;5(1):12982. doi: 10.1038/srep12982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Brehm R, Zeiler M, Ruttinger C, et al. A sertoli cell-specific knockout of connexin43 prevents initiation of spermatogenesis. Am J Pathol. 2007. Jul;171(1):19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Gao Y, Xiao X, Lui WY, et al. Cell polarity proteins and spermatogenesis. Semin Cell Dev Biol. 2016. Nov;59:62–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sakakibara S, Maruo T, Miyata M, et al. Requirement of the F-actin-binding activity of l-afadin for enhancing the formation of adherens and tight junctions. Genes Cells. 2018. Mar;23(3):185–199. [DOI] [PubMed] [Google Scholar]
- [67].Zunino R, Li Q, Rose SD, et al. Expression of scinderin in megakaryoblastic leukemia cells induces differentiation, maturation, and apoptosis with release of plateletlike particles and inhibits proliferation and tumorigenesis. Blood. 2001 Oct 1;98(7):2210–2219. doi: 10.1182/blood.V98.7.2210 [DOI] [PubMed] [Google Scholar]
- [68].Willems A, Batlouni SR, Esnal A, et al. Selective ablation of the androgen receptor in mouse sertoli cells affects sertoli cell maturation, barrier formation and cytoskeletal development. PLoS One. 2010 Nov 30;5(11):e14168. doi: 10.1371/journal.pone.0014168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Chan B, Parreno J, Glogauer M, et al. Adseverin, an actin binding protein, regulates articular chondrocyte phenotype. J Tissue Eng Regen Med. 2019. Aug;13(8):1438–1452. [DOI] [PubMed] [Google Scholar]
- [70].Reifler A, Li X, Archambeau AJ, et al. Conditional knockout of pik3c3 causes a murine muscular dystrophy. Am J Pathol. 2014. Jun;184(6):1819–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Carpenter CL, Auger KR, Duckworth BC, et al. A tightly associated serine/threonine protein kinase regulates phosphoinositide 3-kinase activity. Mol Cell Biol. 1993. Mar;13(3):1657–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Dhand R, Hiles I, Panayotou G, et al. PI 3-kinase is a dual specificity enzyme: autoregulation by an intrinsic protein-serine kinase activity. Embo J. 1994 Feb 1;13(3):522–533. doi: 10.1002/j.1460-2075.1994.tb06290.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Naga Prasad SV, Jayatilleke A, Madamanchi A, et al. Protein kinase activity of phosphoinositide 3-kinase regulates beta-adrenergic receptor endocytosis. Nat Cell Biol. 2005. Aug;7(8):785–796. [DOI] [PubMed] [Google Scholar]
- [74].Stack JH, Emr SD. Vps34p required for yeast vacuolar protein sorting is a multiple specificity kinase that exhibits both protein kinase and phosphatidylinositol-specific PI 3-kinase activities. J Biol Chem. 1994 Dec 16;269(50):31552–31562. doi: 10.1016/S0021-9258(18)31729-0 [DOI] [PubMed] [Google Scholar]
- [75].Ran J, Liu M, Feng J, et al. ASK1-mediated phosphorylation blocks HDAC6 ubiquitination and degradation to drive the disassembly of photoreceptor connecting cilia. Dev Cell. 2020 May 4;53(3):287–299 e5. doi: 10.1016/j.devcel.2020.03.010 [DOI] [PubMed] [Google Scholar]
- [76].Valenzuela-Fernandez A, Cabrero JR, Serrador JM, et al. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008. Jun;18(6):291–297. [DOI] [PubMed] [Google Scholar]
- [77].Royer C, Lu X. Epithelial cell polarity: a major gatekeeper against cancer? Cell Death Differ. 2011. Sep;18(9):1470–1477. doi: 10.1038/cdd.2011.60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Martin-Belmonte F, Perez-Moreno M. Epithelial cell polarity, stem cells and cancer. Nat Rev Cancer. 2011 Dec 15;12(1):23–38. doi: 10.1038/nrc3169 [DOI] [PubMed] [Google Scholar]
- [79].Figueiredo AF, Franca LR, Hess RA, et al. Sertoli cells are capable of proliferation into adulthood in the transition region between the seminiferous tubules and the rete testis in Wistar rats. Cell Cycle. 2016 Sep 16;15(18):2486–2496. doi: 10.1080/15384101.2016.1207835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Zhao S, Xia J, Wu X, et al. Deficiency in class III PI3-kinase confers postnatal lethality with IBD-like features in zebrafish. Nat Commun. 2018 Jul 6;9(1):2639. doi: 10.1038/s41467-018-05105-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Zhang X, Wang L, Ireland SC, et al. GORASP2/GRASP55 collaborates with the PtdIns3K UVRAG complex to facilitate autophagosome-lysosome fusion. Autophagy. 2019. Oct;15(10):1787–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Beau C, Vivian N, Munsterberg A, et al. In vivo analysis of the regulation of the anti-Mullerian hormone, as a marker of Sertoli cell differentiation during testicular development, reveals a multi-step process. Mol Reprod Dev. 2001. Jul;59(3):256–264. [DOI] [PubMed] [Google Scholar]
- [83].Rey R, Josso N. Regulation of testicular anti-M ullerian hormone secretion. Eur J Endocrinol. 1996. Aug;135(2):144–152. doi: 10.1530/eje.0.1350144 [DOI] [PubMed] [Google Scholar]
- [84].Bhushan S, Aslani F, Zhang Z, et al. Isolation of Sertoli Cells and peritubular cells from rat testes. J Vis Exp. 2016. Feb;8(108):e53389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Salva A, Klinefelter GR, Hardy MP. Purification of rat leydig cells: increased yields after unit-gravity sedimentation of collagenase-dispersed interstitial cells. J Androl. 2001. Jul-Aug;22(4):665–671. [PubMed] [Google Scholar]
- [86].Chang YF, Lee-Chang JS, Panneerdoss S, et al. Isolation of Sertoli, Leydig, and spermatogenic cells from the mouse testis. Biotechniques. 2011. Nov;51(5):341–2, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Jiang XJ, Wu YQ, Ma R, et al. PINK1 alleviates cognitive impairments via attenuating pathological tau aggregation in a mouse model of tauopathy. Front Cell Dev Biol. 2021;9:736267. doi: 10.3389/fcell.2021.736267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Yan X, Li M, Luo Z, et al. VIP induces changes in the F-/G-Actin ratio of schlemm’s canal endothelium via LRRK2 transcriptional regulation. Invest Ophthalmol Vis Sci. 2020 Jun 3;61(6):45. doi: 10.1167/iovs.61.6.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Jin X, Wang K, Wang L, et al. RAB7 activity is required for the regulation of mitophagy in oocyte meiosis and oocyte quality control during ovarian aging. Autophagy. 2022. Mar;18(3):643–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.