

Abstract
Protein phosphatase 1 regulatory subunit 3F (PPP1R3F) is a member of the glycogen targeting subunits (GTSs), which belong to the large group of regulatory subunits of protein phosphatase 1 (PP1), a major eukaryotic serine/threonine protein phosphatase that regulates diverse cellular processes. Here, we describe the identification of hemizygous variants in PPP1R3F associated with a novel X-linked recessive neurodevelopmental disorder in 13 unrelated individuals. This disorder is characterized by developmental delay, mild intellectual disability, neurobehavioral issues such as autism spectrum disorder, seizures and other neurological findings including tone, gait and cerebellar abnormalities. PPP1R3F variants segregated with disease in affected hemizygous males that inherited the variants from their heterozygous carrier mothers. We show that PPP1R3F is predominantly expressed in brain astrocytes and localizes to the endoplasmic reticulum in cells. Glycogen content in PPP1R3F knockout astrocytoma cells appears to be more sensitive to fluxes in extracellular glucose levels than in wild-type cells, suggesting that PPP1R3F functions in maintaining steady brain glycogen levels under changing glucose conditions. We performed functional studies on nine of the identified variants and observed defects in PP1 binding, protein stability, subcellular localization and regulation of glycogen metabolism in most of them. Collectively, the genetic and molecular data indicate that deleterious variants in PPP1R3F are associated with a new X-linked disorder of glycogen metabolism, highlighting the critical role of GTSs in neurological development. This research expands our understanding of neurodevelopmental disorders and the role of PP1 in brain development and proper function.
Keywords: X-linked, protein phosphatase 1, PPP1R3F, glycogen metabolism, developmental delay, intellectual disability, seizureautism
Introduction
Protein phosphatase 1 (PP1) is a major eukaryotic serine/threonine protein phosphatase that regulates diverse cellular processes including cell cycle progression, protein synthesis, muscle contraction, carbohydrate metabolism, transcription and neuronal signaling (1). The PP1 holoenzyme is composed of a catalytic subunit and at least one of the over 200 predicted regulatory subunits in vertebrates (2) that determine substrate specificity and target PP1 to specific subcellular compartments (3). Among the regulatory subunits, glycogen-targeting subunits (GTSs) regulate glycogen synthesis by targeting PP1 to dephosphorylate the rate-limiting enzymes glycogen synthase (GS) or glycogen phosphorylase.
There are seven known or putative GTS genes in the human genome: PPP1R3A (R3A, GM) (4–6), PPP1R3B (R3B, GL) (7), PPP1R3C (R3C, PTG/R5) (8), PPP1R3D (R3D, R6) (9), PPP1R3E (R3E) (10), PPP1R3F (R3F) (11) and PPP1R3G (R3G) (10). All GTSs contain a PP1-binding motif, a glycogen-binding domain and a GS-binding domain, and show different expression patterns (Supplementary Material, Fig. S1). GTSs may also contain domains that bind other regulatory proteins and substrates (12,13), and are themselves regulated by phosphorylation at multiple sites (11). R3A and R3F are tail-anchored proteins that have a C-terminal transmembrane domain that targets these proteins to the membrane (6,14). R3F is primarily expressed in the brain and has been suggested to regulate GS in astrocytoma cells in response to glucose and extracellular signals (11).
Here, we report the identification of hemizygous variants in PPP1R3F associated with a novel X-linked recessive neurodevelopmental disorder in 13 unrelated individuals. PPP1R3F variants segregated with disease in affected hemizygous males that inherited the variants from their heterozygous carrier mothers. This disorder is characterized by developmental delay (DD), mild intellectual disability (ID), autism spectrum disorder (ASD), seizures and other neurological findings including tone, gait and cerebellar abnormalities. Functional studies suggest that R3F plays a role in maintaining steady brain glycogen levels under changing glucose conditions. Most patient variants have defects in PP1 binding, protein stability, subcellular localization and regulation of glycogen metabolism. Collectively, the genetic and molecular data indicate that deleterious variants in PPP1R3F are associated with a new X-linked disorder of glycogen metabolism, highlighting the critical role of GTSs in neurological development.
Results
Clinical presentations
Through international collaboration efforts facilitated by the GeneMatcher platform (15), we identified 13 patients from 13 unrelated families of different ancestries (Old Order Amish, Jewish, Asian, Middle Eastern and European) with a similar clinical spectrum. The onset of the clinical manifestations appeared to be around 2–3 years of age for most probands in this series. All the patients were male. Typical disease presentation in this series included DD, ASD, speech delay, behavioral disorders and neurological abnormalities. ID was often mild, usually noted as learning difficulties or concerns for ASD. Expressive language deficiency was particularly prominent in most cases with five individuals reported to be nonverbal. Most patients presented with variable neurological findings, including hypotonia, hyperreflexia, gross motor delay, spasticity in the lower extremities, dystonia and ataxia. Seizures, when present, were heterogeneous in their clinical presentation. Types of seizure include generalized, nocturnal, tonic, atonic, focal myoclonic and atypical absence. Significant behavior issues such as ASD, attention deficit hyperactivity disorder (ADHD) or aggressive behavior were observed in most of the patients. Brain magnetic resonance imaging (MRI) studies showed variable abnormalities in four patients. Microcephaly was observed in three patients. Some facial dysmorphic features were noted in most of the patients although they seemed nonspecific as there is no common gestalt. The phenotypic manifestations are summarized in Table 1 and detailed clinical descriptions of each case are available in the Supplementary Material.
Table 1.
Clinical and genetic information of patient series
| Patient # | 1 | 2 | 3 | 4 |
| Age (years) | 15 years old | 2.5 years old | 14 years old | 5 years old |
| Gender | Male | Male | Male | Male |
| Country | United States | Jordan | USA | USA |
| Ancestry | Old Order Amish | Jordanian | Hispanic | European |
| Variant information | ||||
| PPP1R3F variant | c.140C > A; p.Pro47Gln | c.446C > G; p.Pro149Arg | c.634G > T; p.Gly212Ter | c.910C > T; p.Gln304Ter |
| Variant coordinate (hg38) | chrX:49270009(C > A) | chrX:49270315(C > G) | chrX:49270503(G > T) | chrX:49270779(C > T) |
| Zygosity | Hemizygous | Hemizygous | Hemizygous | Hemizygous |
| Allele Freq | gnomAD 0.00% | gnomAD 0.00% | gnomAD 0.00% | gnomAD 0.00% |
| Polyphen2 | probably_damaging | probably_damaging | na | na |
| Provean | neutral | neutral | na | na |
| SIFT | tolerated | damaging | na | na |
| MutationTaster | polymorphism | disease_causing | disease_causing | disease_causing |
| CADD score | 23.7 | 24 | 37 | 35 |
| Other genetic findings | Normal CMA | Normal karyotyping | SMARCC2 variant (de novo) c.230C > T (p.Pro77Leu), normal CMA and exome array | SMARCC2 variant (de novo) c.230C > T (p.Pro77Leu), normal Fragile X test, normal CMA and exome array |
| Clinical findings | ||||
| Learning disability (HP:0001328) | Yes; ‘slow processing’ | Yes | Yes | Yes |
| ID (HP:0001249) | Yes; mild | Yes; mild | Yes; mild | No |
| Speech delay (HP:0000750) | Yes; especially expressive language | Yes; delayed speech | Yes, expressive language delay | Yes; delayed speech |
| Behavioral disorder | Yes; aggressive behavior (HP:0000718) | NA | Yes; ADHD (HP:0007018) | No |
| Neurological findings | Unsteady gait with increased deep tendon reflexes of lower extremities (HP:0002317, HP:0001347) | Mild hyperkinesia (HP:0002487) | Gross motor DD (HP:0002194), mixed hyper-hypotonia with brisk reflexes and abnormal gait (HP:0001276, HP:0001252, HP:0001348, HP:0001288) | Gross motor DD (HP:0002194); history of hypotonia and reduced core strength (HP:0001252). |
| Seizures | Generalized, mostly nocturnal, onset at 3 years old, controlled with valproic acid (HP:0002197) | No seizures (Normal EEG) | Abnormal EEG with no seizure (HP:0002353) | No seizures (Normal EEG) |
| Brain imaging | Normal MRI at 4 years old | Normal MRI | Normal MRI | Normal MRI at 2 years old. Spine MRI at 4 years old showed hydromyelia (HP:0100565) from T7 to L1, and intrasacral meningocele (HP:0005765) at S3. |
| Dysmorphic Features | No | Uplifted ear lobe (HP:0009909) | Relative microcephaly (HP:0000252), hypotelorism (HP:0000601), broad nasal tip (HP:0000455), hypoplastic philtrum (HP:0005326), narrow philtrum (HP:0011829), posteriorly rotated ears (HP:0000358), everted lower lip vermilion (HP:0000232), thin upper lip vermilion (HP:0000219) | Very sparse hair growth over temples (HP:0008070), hooded eyelids (HP:0030820). |
| Other | Occasional tremors (HP:0001337), nocturnal enuresis (HP:0010677) | Normal hearing assessment | Short stature (HP:0004322), bruising susceptibility (HP:0000978), dental crowding (HP:0000678), dry skin (HP:0000958), hypercalciuria (HP:0002150), joint hypermobility (HP:0001382), ligamentous laxity (HP:0001388), limb pain (HP:0009763), pes planus (HP:0001763), thoracolumbar scoliosis (HP:0002944). | Chronic fatigue (HP:0012432), exercise intolerance (HP:0003546), oculomotor apraxia (HP:0000657). Tethered cord syndrome and syringomyelia (repaired) with residual chronic left sided weakness (leg > arm) (HP:0002144, HP:0003396). Mother with seizures and math learning disabilities. |
| 6 | 7 | 8 | 9 | 10 |
| 3 years old | 15 month | 15 years old | 33 years old | 13 years old |
| Male | Male | Male | Male | Male |
| Germany | China | USA | The Netherlands | Turkey |
| Russian | East Asian | European | Dutch | Turkish |
| c.121G > T;p.Asp41Tyr | c.835C > G; p.Arg279Gly | c.538C > T; p.His180Tyr | c.317C > T, p.Pro106Leu | c.1290dupC; p.Arg431Glnfs*34 |
| chrX:49269990(G > T) | chrX:49270704(C > G) | chrX:49270407(C > T) | chrX:49270186(C > T) | chrX:49285980(insC) |
| Hemizygous | Hemizygous | Hemizygous | Hemizygous | Hemizygous |
| gnomAD 0.00% | gnomAD 0.0006% | gnomAD 0.00% | gnomAD 0.00471%, 1 hemizygote | gnomAD 0.00% |
| probably_damaging | possibly_damaging | probably_damaging | benign | na |
| deleterious | neutral | neutral | neutral | na |
| damaging | tolerated | tolerated | damaging | na |
| disease_causing | polymorphism | polymorphism | disease_causing | na |
| 24.1 | 23.8 | 21.9 | 21.7 | disease_causing |
| Conventional karyotyping, CMA, Fragile X and Angelman syndrome testing are all normal | Normal CMA, a homozygous variant in EFCAB8 and compound heterozygous variants in CBY2 | Exome sequencing detected no known copy number variations. Exome SNP analysis normal | SNP array revealed a paternally inherited 15q11.2 microdeletion. Normal Fragile X test. | Conventional karyotyping and CMA were normal |
| Yes | Yes | Yes; cognitive delay and autism | Yes | Yes |
| (too young to evaluate) | Yes; mild | Yes | Yes; severe | Yes; severe |
| Yes; absent speech, nonverbal | Yes; absent speech | NA | NA | Yes; absent speech, nonverbal (HP:0001344) |
| Yes; ASD (HP:0000729) | NA | Yes; aggressive behavior (HP:0000718) | Yes; aggression fits, bites hands when tense, otherwise social and friendly (HP:0000718) | Yes; hand flapping (HP:0100023) |
| Oral motor weakness (HP:0030190) | Spastic-dystonic quadriplegic cerebral palsy (HP:0002510, HP:0100021) | No | Yes; pyramidal signs (HP:0007256) | Spastic paraplegia (HP:0001258) |
| No seizures (Normal EEG) | No seizures | Abnormal EEG (HP:0002353), variable: generalized tonic, atonic, myoclonic and absence, onset at 2.5 years old (HP:0001250); intractable. | No seizures | Complex focal seizures (HP:0002384), onset at 6 years old, controlled with lamotrigine and perampanel |
| Unremarkable MRI; small subependymal hemorrhage on the right side by brain ultrasound | Normal MRI | Abnormal; interval diffuse volume loss and thinning of the corpus callosum (HP:0033725) | NA | Abnormal; temporal hyperintensity, arachnoid cyst (HP:0410263, HP:0100702) |
| Depressed nasal bridge (HP:0005280), epicanthus (HP:0000286), anteverted nares (HP:0000463), prominent lips (HP:0012471), widely spaced nipples (HP:0006610), mild clinodactyly of the fourth and fifth fingers (HP:0030084) | NA | No | Coarse facial features (HP:0000280), prominent supraorbital ridge (HP:0000336), thick eyebrows (HP:0000574), brachycephaly (HP:0000248), small deep set eyes (HP:0000490), short nose with broad nasal tip (HP:0000455), hypoplastic midface (HP:0011800), short philtrum (HP:0000322), large chin (HP:0011822), small ears with overfolded helix (HP:0008551), coarse hair (HP:0002208), thick neck (HP:0000475), dry skin (HP:0000958) | Microcephaly (HP:0000252), brachycephaly (HP:0000248), coarse facies (HP:0000280) from age 12 years old, bushy eyebrows (HP:0000574), prominent mouth, short philtrum (HP:0000322) |
| Sleep disorder (HP:0002360) | Microcephaly (HP:0000252) | Mother with history of a couple of generalized tonic–clonic and myoclonic seizures as a child, controlled with antiepileptic meds for 3 years, was weaned off meds and no seizures since. | Short stature (HP:0004322), deafness (HP:0000365), myopia (HP:0000545), scoliosis (HP:0002650), progressive flexion contracture of the fingers (HP:0005876), camptodactyly (HP:0012385) which seems progressive, pes planus (HP:0001763) and stiff feet, kyphosis (HP:0002808), scoliosis (HP:0002650), female fat distribution (HP:0007552) | Short stature (HP:0004322), ventricular septal defect (HP:0001629), valvular pulmonary stenosis (HP:0034350) |
| 11 | 12 | 13 | ||
| 5 years old | 5 years old | 6 years old | ||
| Male | Male | Male | ||
| Suriname | Egypt | Germany | ||
| South East Asian | Egyptian | European | ||
| c.244G > T; p.Asp82Tyr | c.1187A > T; p.Asp396Val | c.207_228del, p.Gly70AlafsTer95 | ||
| chrX:49270113(G > T) | chrX:49285877(A > T) | chrX:49270078(del22bp) | ||
| Hemizygous | Hemizygous | Hemizygous | ||
| gnomAD 0.00506% | gnomAD 0.00% | gnomAD 0.004935%, 1 hemizygote | ||
| possibly_damaging | probably_damaging | na | ||
| neutral | deleterious | na | ||
| damaging | damaging | na | ||
| polymorphism | disease_causing | na | ||
| 25.6 | 22.8 | disease_causing | ||
| Normal CMA | No copy number variations in exome analysis | Conventional karyotyping, CMA, Fragile X and a multi-gene panel testing all gave normal results | ||
| Yes; moderate | Yes | Yes | ||
| Not formally tested | Yes; moderate | Yes, mild | ||
| Yes; no speech development (HP:0001344) | Yes; absent speech (HP:0001344) | Yes | ||
| Yes; ASD (HP:0000729) | NA | Yes, ASD (HP:0000729) | ||
| Mild/moderate motor delay (HP:0001270) | Hypertonia mainly in lower limbs and brisk reflexes (HP:0001276, HP:0001348) | Muscular hypotonia (HP:0001252), fatigability (HP:0012378), uses wheelchairs for longer distances | ||
| Atypical febrile seizures with normal EEG, onset at 1.5 years old. No AED required. (HP:0002373) | Focal-myoclonic; Generalized (HP:0011166) | Myoclonic seizures, atypical absences, onset at 1 year 8 months, initially frequent and fever associated, currently controlled with valproic acid and sultiame (HP:0032794) | ||
| Decreased volume of occipital white matter and some signal intensity changes at FLAIR at 2 years old (HP:0410263) | Mild cortical atrophy, deep Sylvian fissures, bilateral high signal at T2 in putamen, mild deep white matter signal, thin body of corpus callosum, cerebellar atrophy mainly vermis (at 2–3 years old) (HP:0410263, HP:0002120, HP:0001272) | Normal MRI at 3 years old | ||
| Microcephaly (HP:0000252), deep set eyes (HP:0000490), prominent cheeks | Long face (HP:0000276), hairy forehead (HP:0011335), straight palpebral fissures (HP:0008050), prominent nasal root (HP:0000426), hypoplastic alae nasi (HP:0000430), long philtrum (HP:0000343), v-shaped upper lip, everted lower lip (HP:0000232), pointed chin (HP:0000307), low set ears (HP:0000369) | Mild dysmorphic features (HP:0001999), macrocephaly (HP:0000256) with prominent forehead (HP:0011220), hypertelorism (HP:0000316), small mouth (HP:0000160), short philtrum (HP:0000322) | ||
| Short stature (HP:0004322), prematurity, growth retardation (HP:0008897), conductive hearing loss (HP:0000405) | Nystagmus at 3 ms old (HP:0000639), occasional tremors (HP:0001337), unsteady, upward gaze, rigidity (HP:0002063), after experienced epilepsy he fairly followed objects or reacted to surrounding, underweight (HP:0004325), right undescended testis (HP:0000028) | Pectus excavatum (HP:0000767), Kyphosis (HP:0002808), Pes planus (HP:0001763), sleep disturbance (HP:0002360) |
CMA, chromosome microarray; N/A, not analyzed.
Identification of hemizygous PPP1R3F variants in patients
Whole-exome sequencing analyses, either clinical or on a research basis, were performed on these probands and their parents. Studies were approved by the Institutional Review Boards (IRBs) of all participating institutions and written informed consent was obtained from each participant or their legal guardian. These analyses revealed hemizygous variants in all the patients in the X-linked gene PPP1R3F (Fig. 1A) as the main candidate from exome sequencing studies. In five patients, Fragile X testing was negative. In other patients, Fragile X was either considered unlikely or the information was not available. Chromosome microarray and karyotyping analyses were performed for most patients with no significant findings (Supplemental Material). All PPP1R3F variants co-segregated with the phenotype in each family as expected based on the pedigrees (Fig. 1B). Affected individuals inherited the PPP1R3F variants from their carrier mothers. Sanger sequencing confirmation and segregation of variants showed that noncarrier male and heterozygous carrier female siblings were unaffected. However, in two instances, carrier mothers were reported to have had a history of childhood seizures (Table 1, Patients 4 and 8).
Figure 1.
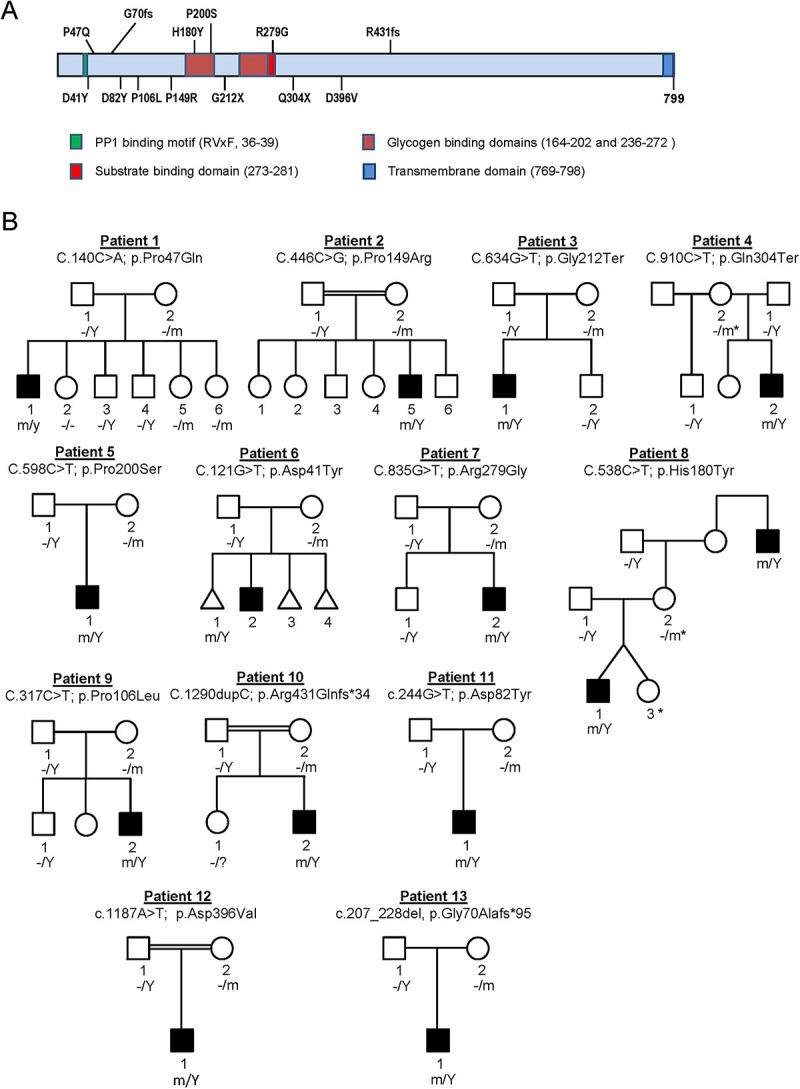
Segregation of PPP1R3F variants in patient families. (A) Schematic diagram showing the domain structure of R3F and locations of identified variants from patient series. (B) Pedigrees and PPP1R3F variant segregation in the 13 families. m, mutant allele, −, normal allele, Y, Y chromosome. Asterisks denote females that have reported childhood seizures or seizure with learning disabilities.
Among the 13 PPP1R3F variants identified, four are predicted to result in loss of function through the introduction of early terminations, including two nonsense variants in Patients 3 (p.Gly212Ter) and 4 (p.Gln304Ter), and two frameshift variants in Patients 10 (p.Arg431Glnfs*34) and 13 (p.Gly70Alafs*95). The remaining variants were missense. PPP1R3F is not a particularly constrained gene (pLI = 0, o/e = 0.73 (0.42–1.31), missense Z score = 0.21). However, being variant permissive does not by itself exclude the gene from being associated with a disease, as other X-linked genes with low pLI scores have been associated with relatively mild diseases (16–20). The tolerance landscape generated by MetaDome showed that, except for p.His180Tyr and p.Asp396Val, most missense variants are at locations ranked from intolerant to neutral (Supplementary Material, Fig. S2). Patient 3 likely has co-occurring disorders (21–23) with both a de novo SMARCC2 variant (p.Pro77Leu) and the PPP1R3F variant. Features like scoliosis and some dysmorphic features may be because of the SMARCC2 variant, whereas ID, motor and speech delays and behavioral problems are shared features of both disorders; gait abnormalities are mainly observed in patients with PPP1R3F variants.
We assessed the effects of missense variants on protein function through bioinformatic prediction programs PolyPhen-2, SIFT, PROVEAN, MutationTaster and CADD. All variants were identified as damaging or disease causing by at least one of these in silico algorithms (Table 1). Most missense variants are located in the highly conserved N-terminal half of the protein, which contains the PP1-binding site, the glycogen-binding site and the substrate-binding site (Fig. 1A). No clear orthologs of PPP1R3F could be identified in invertebrates, suggesting a recent evolutionary origin. Multiple sequence alignment using mammalian R3F protein sequences (Supplementary Material, Fig. S3) revealed that eight of the nine missense variants are at locations fully conserved in all species. The p.Asp396Val variant is at a location that is conserved in all mammals other than the kangaroo rat.
Additional adult patients with PPP1R3F variants
While none of these variants were present at any significant allele frequency in public population databases (<0.005%, Table 1), we did observe the existence of some of the variants in gnomAD (p.Gly70AlafsTer95, p.Asp82Tyr, p.Pro106Leu and p.Arg279Gly, one case of each, including hemizygotes for p.Gly70AlafsTer95 and p.Pro106Leu). The clinical expressivity makes it possible that some adult individuals present in public databases carrying loss-of-function variants in the gene may have eluded a genetic diagnosis to date. Because the phenotypic information of hemizygous individuals in public databases is not available for further interrogation, we explored the Regeneron Genetics Center internal exome database of 173 585 adult participants in the DiscovEHR study (24) and another internal Amish exome database of 10 809 samples with limited electronic health record (EHR) information recorded in the form of ICD-10 codes. We identified five hemizygous males (p.Pro47Gln (x4) and p.Arg279Gly (x1)) and one homozygous female (p.His180Tyr) with PPP1R3F variants of interest, all of them adults older than 49 years of age (Supplementary Material, Table S1). Exploration of the available ICD codes in these individuals’ EHR showed that five of them had ICD codes (Supplementary Material, Table S1) of interest with some overlap with the phenotype we describe here, indicating possible unrecognized cases, and/or variable expressivity of the disease. For the remaining case, incomplete penetrance of the disorder or unrecorded features cannot be discarded.
R3F is predominantly expressed in the brain astrocytes
Little is known about the function of the protein encoded by PPP1R3F, R3F. It has been postulated to regulate glycogen metabolism, based on the presence of glycogen- and GS-binding sites, similar to other glycogen-regulating subunits known to regulate glycogen metabolism (11). Of note, PPP1R3F transcripts have a long isoform (NM_033215.5) and a short isoform of (NM_001184745.2). The latter lacks all N-terminal functional domains and is likely to be nonfunctional. Interestingly, a recent transcriptional network analysis identified PPP1R3F as a candidate master regulator affecting a large body of downstream genes that are associated with ASD (25). This would be consistent with our findings and a previous report of non-synonymous rare variants in PPP1R3F in X-chromosome synaptic genes in ASD and schizophrenia (26). We first investigated the relative abundance of R3F expression in different mouse tissues by immunoblotting. R3F is predominantly expressed in brain tissues, with weak expression in the liver and kidney and no detected expression in the heart and skeletal muscles (Fig. 2A). Among the brain tissues, the cortex has relatively higher levels of R3F than the cerebellum, hippocampus and thalamus (Fig. 2A). Real-time PCR showed that R3F mRNA levels are much higher in mouse astrocytes than those in neurons and oligodendrocytes (Fig. 2B). R3F expression is relatively constant in young mice, with higher expression levels between P30 and P60 (Fig. 2C).
Figure 2.

R3F expression pattern and its role in regulating glycogen levels. (A) Immunoblotting detection of R3F in various mouse tissue lysates with GAPDH as a loading control. (B) Comparison of Ppp1r3f mRNA levels in mouse astrocytes and oligodendrocytes relative to neurons. The expression level in the neuron is set as 1. (C) Immunoblotting detection of R3F in brain lysates prepared from mice of indicated ages. Three mice were analyzed for each time point. (D) Quantification of relative R3F and GAPDH band intensities in D. *P < 0.05. (E) Relative glycogen levels (glycogen/protein) in WT and PPP1R3F KO cells at indicated hours after switching from high glucose medium to low glucose medium. *P < 0.05. (F) Relative glycogen levels (glycogen/protein) in WT and R3F KO cells at indicated minutes after switching from low glucose medium to high glucose medium. *P < 0.05. (G). Immunoblotting analysis of PP1, GS, R3F and β-actin levels in WT and KO H4 cells collected at indicated hours after switching from high glucose medium to low glucose medium.
R3F modulates cellular glycogen levels
Next, we generated a PPP1R3F knockout (KO) strain of the H4 cells, a human astrocytoma cell line. However, we observed no significant differences in steady-state cellular glycogen contents between WT and PPP1R3F KO cells in normal DMEM culture media. Then, we measured changes in cellular glycogen levels in response to changes in extracellular glucose levels. When cells were shifted from high glucose medium to low glucose medium, WT cells showed a significantly slower decrease in glycogen content than KO cells (Fig. 2D). The strongest difference appeared at 8 h after media replacement. When cells were shifted from low glucose medium to high glucose medium, glycogen levels increased faster in WT cells than in KO cells (Fig. 2E). PP1 catalytic subunits bind to regulatory subunits, forming a holoenzyme that phosphorylates substrates. R3F is proposed to recruit PP1 and activate GS by dephosphorylating the enzyme (11). Immunoblotting revealed that when cells were shifted from high glucose to low glucose, total and dephosphorylated GS levels increased in WT cells, but not in PPP1R3F KO cells (Fig. 2F). R3F is also expressed in HEK293T and HepG2 cells, with a higher expression level found in HEK293T cells. Similar changes in total GS were observed in HEK293T cells, but not in HepG2 cells (Supplementary Material, Fig. S5).
R3F is localized to the endoplasmic reticulum via its C-terminal trail anchor
The R3F variants identified in our patients are located primarily in the N-terminal half of the protein that contains the major functional domains (Fig. 1A). We investigated the functional consequences of the variants in the first nine patients identified through the GeneMatcher platform (Patients 1–9, Table 1). To investigate intracellular localization, patient-derived R3F variants were co-expressed with the endoplasmic reticulum (ER) membrane marker mCherry-SEC61-C18 in COS-7 cells. WT R3F co-localized with SEC61-C18, indicating that this protein is localized to the ER (Fig. 3), which is consistent with a previous report (14). However, unlike SEC61, R3F is not uniformly distributed in the reticular structures of the ER, suggesting that there may be factors in the ER constraining R3F localization to specific membrane domains. Deletion of the transmembrane domain (ΔTM variant) disrupted the ER localization pattern and instead led to a particulate distribution pattern in the cytoplasm (Fig. 3), suggesting that soluble R3F is associated with glycogen particles. Missense mutants (p.Pro47Gln is shown) also exhibited an ER localization pattern, similar to the WT protein. The nonsense mutant p.Gln304Ter mostly accumulated in cell nuclei, with only low-level expression detected in the cytoplasm. The nonsense mutant p.Gly212Ter was expressed at a very low level and any detected expression was also found in cell nuclei (Fig. 3).
Figure 3.

COS-7 cells were co-expressed with mCherry-SEC61-C18 and Myc-R3F. Immunofluorescence staining of R3F was performed with an anti-Myc antibody. Cell nuclei were revealed with DAPI staining. Scale bar: 10 μm. The rightmost column shows magnified images from the boxed region of each variant. Experiments were repeated three times.
Effects of R3F variants on interactions with PP1 and GS
To investigate whether these R3F variants affect interactions with PP1 or GS, we carried out co-immunoprecipitation experiments in HEK293T cells. In addition to patient-derived mutations, we also included WT as a positive control and the Phe39Ala point mutation that abrogates the PP1-binding motif as a negative control. To study the role of the transmembrane domain, we included a mutant with the TM deletion (ΔTM) and a mutant with both Phe39Ala and ΔTM mutations. As shown in Fig. 4A, WT R3F could co-IP with both PP1 and GS. The Phe39Ala mutant, the p.Asp41Tyr variant and the Phe39Ala + ΔTM double mutant failed to co-IP with PP1 (Fig. 4B) and pulled down primarily phosphorylated GS (P-GS; Fig. 4C). The p.Pro47Gln and p.Gln304Ter variants pulled down little or no PP1 but retained co-IP with GS (Fig. 4A and D). The relative amount of P-GS that co-IPed with p.Pro47Gln was higher than WT and lower than Phe39Ala and p.Asp41Tyr (Fig. 4C). While WT R3F and other variants were IPed at similar levels (Fig. 4D), the p.Gly212Ter variant expression was barely detectable because of the early truncation of the protein. Treatment of cells with the proteasome inhibitor MG132 increased its level to above the detection limit of immunoblotting (Supplementary Material, Fig. S6). Thus, truncation of R3F caused by the p.Gly212Ter mutation leads to an unstable protein that fails to accumulate in cells. Consequently, the p.Gly212Ter mutant failed to co-IP with PP1 and GS (Fig. 4A–C). Other missense variants (p.Pro106Leu, p.Pro149Arg, p.His180Tyr, p.Pro200Ser and p.Arg279Gly) showed no obvious defects in co-IP with PP1 or GS. No co-IP defects were observed for the ΔTM mutant either (Fig. 4A–C). However, this mutant was expressed at a much higher level than other R3F variants (Fig. 4A), suggesting that TM deletion allows the protein to escape a mechanism that normally limits the steady-state level of WT R3F on the ER membrane.
Figure 4.

Functional characterization of R3F and identified variants. (A) Co-immunoprecipitation of R3F variants with PP1 and GS (upper panel). R3F variants were immunoprecipitated with the anti-Myc antibody from 293 T cells and immunoblotted with antibodies against PP1, GS, GS pSer641 and R3F. Protein levels in cell lysates are detected by immunoblotting and are shown in the lower panel. (B–E) Band intensities of immunoprecipitated PP1 (A), GS pSer641 (B), GS total (C) and R3F variants (E) were quantitated and plotted as ratios to the IgG heavy chain band. *P < 0.05 compared with WT.
To understand the structural basis of the loss on PP1 binding of the R3F variants, we modeled PP1-binding domains of R3F and R3A in complex with PP1. The p.Asp41Tyr and p.Pro47Gln variants are very close to the PP1-binding site. Although Asp41 is not in the core sequence of the PP1-binding domain, the corresponding amino acid residue in Asp70R3A forms a salt-bridge with Arg261PP1 in the crystal structures of PP1 in complex with the PP1-binding domain of R3A (27,28). Changing this residue to Tyr is expected to negatively impact PP1 binding (Supplementary Material, Fig. S8), consistent with our experimental findings (Fig. 4A). The p.Pro47Gln variant may also affect PP1 binding by altering the structure of the PP1-binding domain (Supplementary Material, Fig. S8).
Effects of R3F variants on glycogen metabolism
To test whether R3F variants can affect R3F function in regulating glycogen metabolism, we stably expressed these variants in H4 KO cells. Protein levels were similar among R3F variants except for p.Gly212Ter (Supplementary Material, Fig. S7). These cells were switched to low glucose medium after culturing in high glucose medium and glycogen contents were measured at 8 h after the switch. WT R3F expression increased glycogen content in H4 KO cells compared with vector control, whereas the inactivating mutant Phe39Ala and patient variants p.Asp41Tyr and p.Gly212Ter had no effect (Fig. 5). Variants p.Pro47Gln, p.Pro149Arg and p.Pro200Ser moderately increased glycogen content, but the increases were significantly less than WT R3F. On the other hand, variants p.Pro106Leu, p.His180Tyr, p.Arg279Gly and p.Gln304Ter increased glycogen content to the same extent as WT R3F (Fig. 5). Glycogen synthesis can occur in the nucleus (29), which may explain why the glycogen level is not decreased with the p.Gln304Ter in this experiment.
Figure 5.
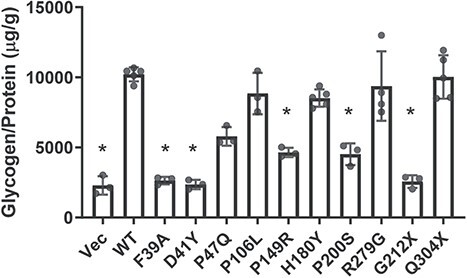
Effects of R3F variants on glycogen metabolism in H4 KO cells. Relative glycogen levels (glycogen/protein) in H4 KO cells stably expressing the indicated R3F variants at 8 h after cells were switched from a high glucose medium to a low glucose medium. Each dot represents the value from an independent replicate. *P < 0.05 compared with WT.
Discussion
We describe a novel neurodevelopmental disorder associated with variants in PPP1R3F, a gene that encodes a PP1 regulatory subunit that likely functions in regulating brain astrocyte glycogen levels in response to changing blood glucose levels. Evidence supporting a causal role of PPP1R3F variants includes segregation of the variants consistent with an X-linked disorder, multiple computational evidence predicting a deleterious effect of variants on the gene and identification of functional defects in most of the variants studied. The individuals in our study demonstrated a wide variability of clinical features, which is not uncommon for neurodevelopmental disorders (30). The most consistent clinical findings in our cohort are developmental, intellectual and speech delays, ranging from mild to severe. Other common symptoms include behavioral disorders and neurological problems in tone and gait. However, because of the limited number of patients deeply phenotyped and clinically characterized documented in this report, a genotype–phenotype correlation cannot be reliably determined at this point.
Although several conserved functional domains of R3F are identified in the N-terminal end of the protein, the sequences between PP1- and glycogen-binding sites and more than half of the protein C-terminal to the GS-binding site contain no known functional domains (Fig. 1A). These parts of the protein contain patches of highly conserved sequences (Supplementary Material, Fig. S3) and could accommodate additional substrate-binding domains and other regulatory motifs. Therefore, we could not rule out the possibility that R3F may function by regulating a different substrate, potentially an ER-localized protein. R3A also contains a long C-terminal sequence with unknown functions. It has recently been reported that R3A can dephosphorylate ryanodine receptor 2 (RyR2) (12). Although GS is a known substrate of R3F, it is possible that other substrates also exist, such as astrocyte-specific RyRs. Indeed, our functional assay did not reveal any defects in glycogen metabolism for p.Pro106Leu, p.His180Tyr and p.Arg279Gly variants (Fig. 4B). The lack of observed functional defects of these variants may be because of limitations in our assays such as the overexpression of R3F variants, the functional defect may be through diminished or aberrant interactions with other regulatory proteins of the holoenzyme, or perhaps these and other pathogenic variants cause defects in regulating the phosphorylation of a yet-to-be-identified substrate. As noted, some of the missense variants described in this report have been observed in population databases like gnomAD, including the p.Pro106Leu and p.Arg279Gly variants. We cannot discard the possibility that the patient phenotype could be caused or confounded by an undetected co-occurring variant. This highlights the challenge in identifying causal variants in a variant permissive gene. Further experiments and characterization of additional patients with variants in PPP1R3F will be necessary to clarify the effect of deleterious variants in this gene.
Although other GTSs are also expressed in the brain to some extent, including R3C, R3D and R3G, R3F is the only subunit that is attached to the ER membrane through a TM tail anchor located in the C-terminus of the protein (Fig. 1A). The ER localization allows R3F to direct the PP1 enzymatic activity to the surface of the ER, and subjects R3F and PP1 to regulation by ER-associated proteins. Deletion of the TM domain leads to the mislocalization of the protein and presumably the loss of interactions with other proteins in its normal subcellular environment. This would be relevant for variants such as p.Arg431Glnfs*34 predicted to escape nonsense mediated decay and potentially result in a truncated protein, yet missing the TM domain. The p.Gln304Ter variant retains all known functional domains in the N-terminal half of the protein, yet it abolishes the co-IP with PP1 and leads to the mislocalization of the protein to the nucleus. Interestingly, most other soluble GTSs are shorter than the truncated protein generated by the p.Gln304Ter mutation (Supplementary Material, Fig. S1), but these proteins are not localized to the nucleus. C-terminal truncation of R3F could expose a nuclear localization signal that directs the protein into the nucleus. R3A is also a membrane-anchored GTS. It is localized to the sarcoplasmic reticulum (SR) in muscle cells and is phosphorylated by SR-localized Ca2+-calmodulin-dependent protein kinase (CaMKII) (31). A C-terminal deletion mutation results in the mislocalization of R3A to the cytosol and is associated with type 2 diabetes (4).
The brain is the highest energy-consuming organ in the human body. Astrocytes are the major site of glycogen synthesis and storage in the brain (32,33). Glucose from capillary vessels is taken up by astrocytes and converted into glycogen storage particles. Although glycogen concentration in the brain is much lower than in muscle or liver, evidence in the recent decade indicates that brain glycogen plays important roles in brain functions such as memory, learning, sleep and prevention of seizures (32–34). Our data show that R3F plays an important role in glycogen synthesis during low glucose conditions (Fig. 2), unlike other glycogen targeting subunits such as R3A, R3B, R3C and R3G, which increase glycogen synthesis with high glucose (35,36). Our results suggest that the main function of R3F is to prevent glycogen depletion in the brain under low glucose conditions. A plausible mechanism for the newly described disorder is the inability of astrocytes to effectively respond to fluctuations in local glucose levels in the absence of functional R3F. Glycogen mobilization is critical for higher brain functions such as learning and memory in different animal models (37–39). Lactate produced in astrocytes provides energy to neurons during periods of hypoglycemia and ischemia and long-term memory formation. It is thought that maintaining a readily releasable glycogen pool is important to sustain astrocyte-dependent neural plasticity (37–39). The majority of identified patients demonstrate learning disability, ID and developmental and speech delays. Several patients are also reported to experience or have experienced seizures. Interestingly, the accumulation of glycogen particles in neurons and astrocytes leads to fatal epilepsy in patients with Lafora disease (40). However, mice with brain-specific deletion of GS showed greater susceptibility to epilepsy (34), highlighting the importance of proper regulation of brain glycogen levels.
In conclusion, our findings provide evidence that deleterious variants in PPP1R3F are associated with a novel neurodevelopmental disorder characterized by DD, mild ID, ASD features, behavioral disorders, seizures and abnormal neurological findings including tone, gait and cerebellar abnormalities. These findings expand our knowledge of neurodevelopmental disorders and highlight the roles of PP1 and glycogen metabolism in brain development and function.
Materials and Methods
Subjects and clinical assessment
The study was approved by the IRBs of all participating institutions, and written informed consent was obtained from each participant or their legal guardian. Thirteen affected individuals of different ancestries were clinically evaluated independently by the referring physicians. Additional clinical information, medical records and neuroimaging studies were reviewed to evaluate the phenotype features and detailed case information is included in the clinical phenotype descriptions in the Supplementary Information.
Genetic analyses
Clinical or research-based exome sequencing was performed on all probands and their parents in most cases using Illumina platforms, as described previously (23). Data analysis pipelines based on each pedigree focused on the presence of (i) rare variants (MAF < 0.01 in population and in-house genomic databases), (ii) deleteriousness based on the effect on the protein product (missense, nonsense, frameshift and splice-site variants) and (iii) segregation with the disease. Candidate variants were validated using Sanger sequencing in affected individuals, parents and siblings whenever it was applicable.
Cell lines and culture conditions
HEK293T, COS-7, human astrocytoma cell line H4 and PPP1R3F CRISPR KO (−2, −4) H4 cells (Synthego, Redwood City, CA) were cultured in Dulbecco Modified Eagle Medium supplemented with 10% FBS and 1% penicillin and streptomycin. All cells were maintained at 37°C and 5% CO2 culture conditions and tested negative upon routine mycoplasma testing with the Universal Mycoplasma Detection Kit (American Type Culture Collection, Manassas, VA).
Mouse primary cells and real-time RT-PCR
C57BL/6 J wild-type male mice (3 months old) were dissected to obtain tissues for protein detection. Primary mouse neurons were isolated from cortical tissues of E14.5 embryos (41). Primary mouse astrocytes and oligodendrocytes were isolated from P0 cortical tissues (42). Real-time RT-PCR primers for Ppp1r3f were ATGGCCCCAGATGACACTTC (sense) and TGGCGGACCTCTGTAAAAGC (antisense). Relative gene expression was calculated by the 2−ΔΔCt method using Gapdh as a reference gene.
Plasmids
N-terminal Myc-tagged R3F expression plasmid (pcDNA-myc-R3F) was a gift from Dr Michael Schrader (14). PPP1R3F cDNA with an N-terminal FLAG tag was cloned into a retroviral vector pMSCV. Mutations were introduced into the wild-type plasmid using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). All constructs were validated by Sanger DNA sequencing to confirm the presence of the mutation and the absence of unintended mutations.
Stable expression cells constructed by retroviral gene transfer
HEK293T cells plated on 60-mm dishes were transfected with a pMSCV-based retroviral expression plasmid (2 μg), together with the pGag-pol (2 μg) and the pVSV-G (0.7 μg) plasmids using FuGENE6 (Promega, Madison, WI). Fifty hours after transfection, virus-containing media were harvested and used to infect cells in the presence of polybrene (8 μg/mL). The infected cells were selected with puromycin (2 μg/mL) for at least 10 days.
Glycogen assay
Cells were harvested on ice after washing two times with ice-cold PBS. The cells were aliquoted into two halves in PBS suspensions and span down as pellets at 4°C. One half of the cell pellet was heated at 100°C for 10 min right after suspension with Milli-Q water to test the glycogen concentration. The other half of the cell pellet was used to detect the protein concentration by adding the same volume of NP40 lysis buffer with protease/phosphatase inhibitor cocktails (Thermo Fisher, Waltham, MA). Glycogen assays were done with the Glycogen Colorimetric/Fluorometric Assay kit (BioVision, Milpitas, CA, Cat. #K646-100). Protein assays were done using the Bio-Rad protein assay dye reagent concentrate (Bio-Rad, Hercules, CA, Cat. #500-0006).
Immunoprecipitation and immunoblotting
HEK293T cells (1X10 (6)) were transfected with the expression plasmids of Myc-tagged PPP1R3F and its variants for 30 h. The transfected cells were lysed on ice for 30 min in 1 mL of lysis buffer (50 mM Tris–HCl, 150 mM NaCl, 1% NP-40, 0.05% SDS, 1 mM EDTA, pH 7.5) with protease/phosphatase inhibitor cocktails (Thermo Fisher). For each immunoprecipitation, a 0.8-mL aliquot of the lysate was incubated with 2.0 μg of the anti-Myc antibody (Santa Cruz Biotechnology, Dallas, TX) for 3 h and incubated another 1 h after adding 40 μL of a 1:1 slurry of Protein A/G Plus-Agarose (Santa Cruz Biotechnology). The agarose beads were washed four times with 1 mL of lysis buffer. The precipitates were added 1 X SDS loading buffer and analyzed by standard immunoblotting procedures. Anti-PPP1R3F rabbit polyclone antibody (206170-T36, Sino Biological), anti-pGS (Ser641) rabbit monoclonal antibody (47043, Cell Signaling, Danvers, MA), anti-GS rabbit monoclonal antibody (3886, Cell Signaling), anti-PP1 mouse monoclonal antibody (SC-7482, Santa Cruz) were applied as 1:1000 dilution for western blot analysis. Anti-β actin mouse monoclonal antibody (MABT523, Sigma-Aldrich, St. Louis, MO) and anti-GAPDH mouse monoclonal antibody (600041, Proteintech, Rosemont, IL) were diluted as 1:2000 for western blot analysis.
Immunofluorescence analysis
COS-7 cells (1X10 (5) cells/mL) were seeded in 12-well plates with 18 mm cover glass and transfected with the indicated plasmids by FuGENE 6 transfection reagent. The transfected cells were fixed with 4% paraformaldehyde for 10 m after 20 h culture and permeabilized with 0.25% Triton X-100 in PBS. The cells were incubated with anti-Myc antibody (1:50, sc-40 Santa Cruz) overnight at 4°C and incubated with the Alexa 488 goat anti-mouse antibody (Thermo Fisher) for 1 h at room temperature. The slides were mounted with the VECTASHIELD medium with DAPI (Vector Laboratories, Newark, CA) and observed with an Olympus confocal microscope as previously described (43).
Protein structure analysis
Sequence alignment between R3A/Gm with R3F was constructed by CLUSTALW (44). The structure of human PP1 and the PP1-binding region from the R3A protein (Protein Data Bank (PDB) ID 6DNO) was downloaded from the PDB (27). The mutant structures were built by the PDB reader of Charmm-Gui (45). Protein structures were visualized by Visual Molecular Dynamics Version 1.9.3 (46).
Statistical analysis
All experimental data are presented as mean ± standard deviation. Statistical significance was calculated using one-way ANOVA test. P-values < 0.05 were considered significant.
Supplementary Material
Acknowledgements
We thank Dr. Michael Schrader (University of Exeter) for providing the R3F expression plasmid.
Conflict of Interest statement. J.R.L. has stock ownership in 23 and Me, is a paid consultant for Regeneron Genetics Center and Genome International, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, genomic disorders, and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics (BG); J.R.L. serves on BG's Scientific Advisory Board (SAB).
Contributor Information
Zhigang Liu, Genomic Medicine Institute, Cleveland Clinic Lerner Research Institute, Cleveland, OH 44195, USA.
Baozhong Xin, DDC Clinic for Special Needs Children, Middlefield, OH 44062, USA.
Iris N Smith, Genomic Medicine Institute, Cleveland Clinic Lerner Research Institute, Cleveland, OH 44195, USA.
Valerie Sency, DDC Clinic for Special Needs Children, Middlefield, OH 44062, USA.
Julia Szekely, DDC Clinic for Special Needs Children, Middlefield, OH 44062, USA.
Anna Alkelai, Regeneron Genetics Center, Regeneron Pharmaceuticals, Tarrytown, NY 10591, USA.
Alan Shuldiner, Regeneron Genetics Center, Regeneron Pharmaceuticals, Tarrytown, NY 10591, USA.
Stephanie Efthymiou, Department of Neuromuscular Disorders, University College London (UCL) Institute of Neurology, London WC1N 3BG, UK.
Farrah Rajabi, Division of Genetics & Genomics, Boston Children’s Hospital, Boston, MA 02115, USA.
Stephanie Coury, Division of Genetics & Genomics, Boston Children’s Hospital, Boston, MA 02115, USA.
Catherine A Brownstein, Division of Genetics & Genomics, Boston Children’s Hospital, Boston, MA 02115, USA; The Manton Center for Orphan Disease Research, Boston Children's Hospital, Boston, MA 02115, USA.
Sabine Rudnik-Schöneborn, Institute for Human Genetics, Medical University Innsbruck, Innsbruck 6020, Austria.
Ange-Line Bruel, Inserm UMR1231 GAD, Génétique des Anomalies du Développement, Fédération Hospitalo-Universitaire Médecine Translationnelle et Anomalies du Développement (FHU TRANSLAD), CHU Dijon Bourgogne, Dijon 21000, France; UF Innovation en diagnostic génomique des maladies rares, CHU Dijon Bourgogne, Dijon 21000, France.
Julien Thevenon, Université Grenoble Alpes, Institute for Advanced Biosciences, Grenoble, France.
Shimriet Zeidler, Department of Clinical Genetics, Erasmus University Medical Center, Rotterdam 3015 GD, The Netherlands.
Parul Jayakar, Division of Genetics and Metabolism, Nicklaus Children's Hospital, Miami, FL 33155, USA.
Axel Schmidt, Institute of Human Genetics, University of Bonn, School of Medicine & University Hospital Bonn, 53105 Bonn, Germany.
Kirsten Cremer, Institute of Human Genetics, University of Bonn, School of Medicine & University Hospital Bonn, 53105 Bonn, Germany.
Hartmut Engels, Institute of Human Genetics, University of Bonn, School of Medicine & University Hospital Bonn, 53105 Bonn, Germany.
Sophia O Peters, Institute of Human Genetics, University of Bonn, School of Medicine & University Hospital Bonn, 53105 Bonn, Germany.
Maha S Zaki, Clinical Genetics Department, Human Genetics and Genome Research Institute National Research Centre, Cairo 12622, Egypt.
Ruizhi Duan, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA.
Changlian Zhu, Center for Brain Repair and Rehabilitation, Institute of Neuroscience and Physiology, University of Gothenburg, Göteborg 417 56, Sweden; Henan Key Laboratory of Child Brain Injury and Henan Pediatric Clinical Research Center, Institute of Neuroscience and Third Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China.
Yiran Xu, Henan Key Laboratory of Child Brain Injury and Henan Pediatric Clinical Research Center, Institute of Neuroscience and Third Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, China.
Chao Gao, Department of Pediatric Rehabilitation Medicine, Children's Hospital Affiliated to Zhengzhou University, Zhengzhou 450012, China.
Tania Sepulveda-Morales, International Laboratory for Human Genome Research, Laboratorio Internacional de Investigación sobre el Genoma Humano, Universidad Nacional Autónoma de México, Juriquilla, Querétaro 76226, México.
Reza Maroofian, Department of Neuromuscular Disorders, University College London (UCL) Institute of Neurology, London WC1N 3BG, UK.
Issam A Alkhawaja, Al-Bashir Hospital, Pediatric Department, Pediatric Neurology Unit, Amman, Jordan.
Mariam Khawaja, Prince Hamzah Hospital, Amman, Jordan; Hospital Clínic and Fundació Hospital Sant Joan de Déu de Martorell/Barcelona, Barcelona, Spain.
Hunaida Alhalasah, Al-Karak Government Teaching Hospital, Al-Karak, Jordan.
Henry Houlden, Department of Neuromuscular Disorders, University College London (UCL) Institute of Neurology, London WC1N 3BG, UK.
Jill A Madden, Division of Genetics & Genomics, Boston Children’s Hospital, Boston, MA 02115, USA; The Manton Center for Orphan Disease Research, Boston Children's Hospital, Boston, MA 02115, USA.
Valentina Turchetti, Department of Neuromuscular Disorders, University College London (UCL) Institute of Neurology, London WC1N 3BG, UK.
Dana Marafi, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Department of Pediatrics, Faculty of Medicine, Kuwait University, Kuwait City 13060, Kuwait.
Pankaj B Agrawal, Division of Genetics & Genomics, Boston Children’s Hospital, Boston, MA 02115, USA; The Manton Center for Orphan Disease Research, Boston Children's Hospital, Boston, MA 02115, USA; Division of Neonatology, Department of Pediatrics, University of Miami School of Medicine and Jackson Health System, Miami, FL 33136, USA.
Ulrich Schatz, Institute for Human Genetics, Medical University Innsbruck, Innsbruck 6020, Austria.
Ari Rotenberg, Houston Specialty Clinic, Houston, TX 77024, USA.
Joshua Rotenberg, Houston Specialty Clinic, Houston, TX 77024, USA.
Grazia M S Mancini, Department of Clinical Genetics, Erasmus University Medical Center, Rotterdam 3015 GD, The Netherlands.
Somayeh Bakhtiari, Pediatric Movement Disorders Program, Division of Pediatric Neurology, Barrow Neurological Institute, Phoenix Children’s Hospital, Phoenix, AZ 85016, USA; Departments of Child Health, Neurology, and Cellular & Molecular Medicine, and Program in Genetics, University of Arizona College of Medicine–Phoenix, Phoenix, AZ 85004, USA.
Michael Kruer, Pediatric Movement Disorders Program, Division of Pediatric Neurology, Barrow Neurological Institute, Phoenix Children’s Hospital, Phoenix, AZ 85016, USA; Departments of Child Health, Neurology, and Cellular & Molecular Medicine, and Program in Genetics, University of Arizona College of Medicine–Phoenix, Phoenix, AZ 85004, USA.
Isabelle Thiffault, Genomic Medicine Center, Children’s Mercy Kansas City, Children's Mercy Research Institute, Kansas City, MO 64108, USA.
Steffen Hirsch, Institute if Human Genetics, Heidelberg University Hospital, 69120 Heidelberg, Germany.
Maja Hempel, Institute if Human Genetics, Heidelberg University Hospital, 69120 Heidelberg, Germany.
Lara G Stühn, Institute of Medical Genetics and Applied Genomics, University of Tübingen, 72076 Tübingen, Germany.
Tobias B Haack, Institute of Medical Genetics and Applied Genomics, University of Tübingen, 72076 Tübingen, Germany.
Jennifer E Posey, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA.
James R Lupski, Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX 77030, USA; Texas Children's Hospital, Houston, TX 77030, USA; Human Genome Sequencing Center, Baylor College of Medicine, Houston, TX 77030, USA; Department of Pediatrics, Baylor College of Medicine, Houston, TX 77030, USA.
Hyunpil Lee, Genomic Medicine Institute, Cleveland Clinic Lerner Research Institute, Cleveland, OH 44195, USA.
Nicholas B Sarn, Genomic Medicine Institute, Cleveland Clinic Lerner Research Institute, Cleveland, OH 44195, USA.
Charis Eng, Genomic Medicine Institute, Cleveland Clinic Lerner Research Institute, Cleveland, OH 44195, USA.
Claudia Gonzaga-Jauregui, International Laboratory for Human Genome Research, Laboratorio Internacional de Investigación sobre el Genoma Humano, Universidad Nacional Autónoma de México, Juriquilla, Querétaro 76226, México.
Bin Zhang, Genomic Medicine Institute, Cleveland Clinic Lerner Research Institute, Cleveland, OH 44195, USA.
Heng Wang, DDC Clinic for Special Needs Children, Middlefield, OH 44062, USA.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary material. Raw data that support these findings are available from the corresponding authors upon reasonable request.
Funding
The research is supported by funding from the National Institutes of Health (NIH) (award R01HL094505 to BZ) and the Lisa Dean Moseley Foundation. INS is funded, in part, by the Ambrose Monell Cancer Genomic Medicine Fellowship and the NIH (K99 GM143552). CE is an American Cancer Society Clinical Research Professor and the Sondra J. and Stephen R. Hardis Endowed Chair of Cancer Genomic Medicine at the Cleveland Clinic. CZ is funded by the National Natural Science Foundation of China (U21A20347) and the Swedish Research Council (2018-02267). JRL is funded by HG006542, R35NS105078, and U01HG011758 from the NIH. DM was supported by a Medical Genetics Research Fellowship Program through the NIH T32 GM007526-42. JEP was supported by NHGRI K08 HG008986.
Author contributions
H.W., B.Z. and C.G. designed the study. Z.L. performed the functional studies and statistical analysis. B.X. and C.G. performed variant analysis and coordinated the patient recruitment. I.N.S., H.L., N.B.S. and C.E. assisted the animal experiments and interpretation of data. A.K and A.S. analyzed internal patient databases. All other authors contributed clinical findings, exome sequencing analysis and genetic testing of patients. Z.L., B.X., C.G., B.Z. and H.W. drafted the manuscript. All authors contributed to the review and critical revisions of the manuscript.
References
- 1. Cohen, P.T. (2002) Protein phosphatase 1--targeted in many directions. J. Cell Sci., 115, 241–256. [DOI] [PubMed] [Google Scholar]
- 2. Hendrickx, A., Beullens, M., Ceulemans, H., Den Abt, T., Van Eynde, A., Nicolaescu, E., Lesage, B. and Bollen, M. (2009) Docking motif-guided mapping of the interactome of protein phosphatase-1. Chem. Biol., 16, 365–371. [DOI] [PubMed] [Google Scholar]
- 3. Verbinnen, I., Ferreira, M. and Bollen, M. (2017) Biogenesis and activity regulation of protein phosphatase 1. Biochem. Soc. Trans., 45, 89–99. [DOI] [PubMed] [Google Scholar]
- 4. Savage, D.B., Agostini, M., Barroso, I., Gurnell, M., Luan, J., Meirhaeghe, A., Harding, A.H., Ihrke, G., Rajanayagam, O., Soos, M.A. et al. (2002) Digenic inheritance of severe insulin resistance in a human pedigree. Nat. Genet., 31, 379–384. [DOI] [PubMed] [Google Scholar]
- 5. Delibegovic, M., Armstrong, C.G., Dobbie, L., Watt, P.W., Smith, A.J. and Cohen, P.T. (2003) Disruption of the striated muscle glycogen targeting subunit PPP1R3A of protein phosphatase 1 leads to increased weight gain, fat deposition, and development of insulin resistance. Diabetes, 52, 596–604. [DOI] [PubMed] [Google Scholar]
- 6. Lerin, C., Montell, E., Nolasco, T., Clark, C., Brady, M.J., Newgard, C.B. and Gomez-Foix, A.M. (2003) Regulation and function of the muscle glycogen-targeting subunit of protein phosphatase 1 (GM) in human muscle cells depends on the COOH-terminal region and glycogen content. Diabetes, 52, 2221–2226. [DOI] [PubMed] [Google Scholar]
- 7. Armstrong, C.G., Doherty, M.J. and Cohen, P.T. (1998) Identification of the separate domains in the hepatic glycogen-targeting subunit of protein phosphatase 1 that interact with phosphorylase a, glycogen and protein phosphatase 1. Biochem. J., 336(Pt 3), 699–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Printen, J.A., Brady, M.J. and Saltiel, A.R. (1997) PTG, a protein phosphatase 1-binding protein with a role in glycogen metabolism. Science, 275, 1475–1478. [DOI] [PubMed] [Google Scholar]
- 9. Armstrong, C.G., Browne, G.J., Cohen, P. and Cohen, P.T. (1997) PPP1R6, a novel member of the family of glycogen-targetting subunits of protein phosphatase 1. FEBS Lett., 418, 210–214. [DOI] [PubMed] [Google Scholar]
- 10. Munro, S., Ceulemans, H., Bollen, M., Diplexcito, J. and Cohen, P.T. (2005) A novel glycogen-targeting subunit of protein phosphatase 1 that is regulated by insulin and shows differential tissue distribution in humans and rodents. FEBS J., 272, 1478–1489. [DOI] [PubMed] [Google Scholar]
- 11. Kelsall, I.R., Voss, M., Munro, S., Cuthbertson, D.J. and Cohen, P.T. (2011) R3F, a novel membrane-associated glycogen targeting subunit of protein phosphatase 1 regulates glycogen synthase in astrocytoma cells in response to glucose and extracellular signals. J. Neurochem., 118, 596–610. [DOI] [PubMed] [Google Scholar]
- 12. Alsina, K.M., Hulsurkar, M., Brandenburg, S., Kownatzki-Danger, D., Lenz, C., Urlaub, H., Abu-Taha, I., Kamler, M., Chiang, D.Y., Lahiri, S.K. et al. (2019) Loss of protein phosphatase 1 regulatory subunit PPP1R3A promotes atrial fibrillation. Circulation, 140, 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kelsall, I.R., Rosenzweig, D. and Cohen, P.T. (2009) Disruption of the allosteric phosphorylase a regulation of the hepatic glycogen-targeted protein phosphatase 1 improves glucose tolerance in vivo. Cell. Signal., 21, 1123–1134. [DOI] [PubMed] [Google Scholar]
- 14. Costello, J.L., Castro, I.G., Camoes, F., Schrader, T.A., McNeall, D., Yang, J., Giannopoulou, E.A., Gomes, S., Pogenberg, V., Bonekamp, N.A. et al. (2017) Predicting the targeting of tail-anchored proteins to subcellular compartments in mammalian cells. J. Cell Sci., 130, 1675–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sobreira, N., Schiettecatte, F., Valle, D. and Hamosh, A. (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat., 36, 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bech-Hansen, N.T., Naylor, M.J., Maybaum, T.A., Pearce, W.G., Koop, B., Fishman, G.A., Mets, M., Musarella, M.A. and Boycott, K.M. (1998) Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat. Genet., 19, 264–267. [DOI] [PubMed] [Google Scholar]
- 17. Bech-Hansen, N.T., Naylor, M.J., Maybaum, T.A., Sparkes, R.L., Koop, B., Birch, D.G., Bergen, A.A., Prinsen, C.F., Polomeno, R.C., Gal, A. et al. (2000) Mutations in NYX, encoding the leucine-rich proteoglycan nyctalopin, cause X-linked complete congenital stationary night blindness. Nat. Genet., 26, 319–323. [DOI] [PubMed] [Google Scholar]
- 18. Doffinger, R., Smahi, A., Bessia, C., Geissmann, F., Feinberg, J., Durandy, A., Bodemer, C., Kenwrick, S., Dupuis-Girod, S., Blanche, S. et al. (2001) X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat. Genet., 27, 277–285. [DOI] [PubMed] [Google Scholar]
- 19. Pusch, C.M., Zeitz, C., Brandau, O., Pesch, K., Achatz, H., Feil, S., Scharfe, C., Maurer, J., Jacobi, F.K., Pinckers, A. et al. (2000) The complete form of X-linked congenital stationary night blindness is caused by mutations in a gene encoding a leucine-rich repeat protein. Nat. Genet., 26, 324–327. [DOI] [PubMed] [Google Scholar]
- 20. Zonana, J., Elder, M.E., Schneider, L.C., Orlow, S.J., Moss, C., Golabi, M., Shapira, S.K., Farndon, P.A., Wara, D.W., Emmal, S.A. et al. (2000) A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO). Am. J. Hum. Genet., 67, 1555–1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. O'Roak, B.J., Vives, L., Girirajan, S., Karakoc, E., Krumm, N., Coe, B.P., Levy, R., Ko, A., Lee, C., Smith, J.D. et al. (2012) Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature, 485, 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Soden, S.E., Saunders, C.J., Willig, L.K., Farrow, E.G., Smith, L.D., Petrikin, J.E., LePichon, J.B., Miller, N.A., Thiffault, I., Dinwiddie, D.L. et al. (2014) Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci. Transl. Med., 6, 265ra168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Strauss, K.A., Gonzaga-Jauregui, C., Brigatti, K.W., Williams, K.B., King, A.K., Van Hout, C., Robinson, D.L., Young, M., Praveen, K., Heaps, A.D. et al. (2018) Genomic diagnostics within a medically underserved population: efficacy and implications. Genet Med, 20, 31–41. [DOI] [PubMed] [Google Scholar]
- 24. Dewey, F.E., Murray, M.F., Overton, J.D., Habegger, L., Leader, J.B., Fetterolf, S.N., O'Dushlaine, C., Van Hout, C.V., Staples, J., Gonzaga-Jauregui, C. et al. (2016) Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science, 354, aaf6814. 10.1126/science.aaf6814. [DOI] [PubMed] [Google Scholar]
- 25. Dorner, A.J., Bole, D.G. and Kaufman, R.J. (1987) The relationship of N-linked glycosylation and heavy chain-binding protein association with the secretion of glycoproteins. J. Cell Biol., 105, 2665–2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Piton, A., Gauthier, J., Hamdan, F.F., Lafreniere, R.G., Yang, Y., Henrion, E., Laurent, S., Noreau, A., Thibodeau, P., Karemera, L. et al. (2011) Systematic resequencing of X-chromosome synaptic genes in autism spectrum disorder and schizophrenia. Mol. Psychiatry, 16, 867–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kumar, G.S., Choy, M.S., Koveal, D.M., Lorinsky, M.K., Lyons, S.P., Kettenbach, A.N., Page, R. and Peti, W. (2018) Identification of the substrate recruitment mechanism of the muscle glycogen protein phosphatase 1 holoenzyme. Sci. Adv., 4, eaau6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yu, J., Deng, T. and Xiang, S. (2018) Structural basis for protein phosphatase 1 recruitment by glycogen-targeting subunits. FEBS J., 285, 4646–4659. [DOI] [PubMed] [Google Scholar]
- 29. Sun, R.C., Dukhande, V.V., Zhou, Z., Young, L.E.A., Emanuelle, S., Brainson, C.F. and Gentry, M.S. (2019) Nuclear glycogenolysis modulates histone acetylation in human non-small cell lung cancers. Cell Metab., 30, 903, e907–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Parenti, I., Rabaneda, L.G., Schoen, H. and Novarino, G. (2020) Neurodevelopmental disorders: from genetics to functional pathways. Trends Neurosci., 43, 608–621. [DOI] [PubMed] [Google Scholar]
- 31. Sacchetto, R., Bovo, E., Donella-Deana, A. and Damiani, E. (2005) Glycogen- and PP1c-targeting subunit GM is phosphorylated at Ser48 by sarcoplasmic reticulum-bound Ca2+−calmodulin protein kinase in rabbit fast twitch skeletal muscle. J. Biol. Chem., 280, 7147–7155. [DOI] [PubMed] [Google Scholar]
- 32. Bak, L.K., Walls, A.B., Schousboe, A. and Waagepetersen, H.S. (2018) Astrocytic glycogen metabolism in the healthy and diseased brain. J. Biol. Chem., 293, 7108–7116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Duran, J., Gruart, A., Lopez-Ramos, J.C., Delgado-Garcia, J.M. and Guinovart, J.J. (2019) Glycogen in astrocytes and neurons: physiological and pathological aspects. Adv. Neurobiol, 23, 311–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lopez-Ramos, J.C., Duran, J., Gruart, A., Guinovart, J.J. and Delgado-Garcia, J.M. (2015) Role of brain glycogen in the response to hypoxia and in susceptibility to epilepsy. Front. Cell. Neurosci., 9, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gasa, R., Jensen, P.B., Berman, H.K., Brady, M.J., DePaoli-Roach, A.A. and Newgard, C.B. (2000) Distinctive regulatory and metabolic properties of glycogen-targeting subunits of protein phosphatase-1 (PTG, GL, GM/RGl) expressed in hepatocytes. J. Biol. Chem., 275, 26396–26403. [DOI] [PubMed] [Google Scholar]
- 36. Zhang, Y., Xu, D., Huang, H., Chen, S., Wang, L., Zhu, L., Jiang, X., Ruan, X., Luo, X., Cao, P. et al. (2014) Regulation of glucose homeostasis and lipid metabolism by PPP1R3G-mediated hepatic glycogenesis. Mol. Endocrinol., 28, 116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Boury-Jamot, B., Carrard, A., Martin, J.L., Halfon, O., Magistretti, P.J. and Boutrel, B. (2016) Disrupting astrocyte-neuron lactate transfer persistently reduces conditioned responses to cocaine. Mol. Psychiatry, 21, 1070–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Newman, L.A., Korol, D.L. and Gold, P.E. (2011) Lactate produced by glycogenolysis in astrocytes regulates memory processing. PLoS One, 6, e28427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Suzuki, A., Stern, S.A., Bozdagi, O., Huntley, G.W., Walker, R.H., Magistretti, P.J. and Alberini, C.M. (2011) Astrocyte-neuron lactate transport is required for long-term memory formation. Cell, 144, 810–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gentry, M.S., Guinovart, J.J., Minassian, B.A., Roach, P.J. and Serratosa, J.M. (2018) Lafora disease offers a unique window into neuronal glycogen metabolism. J. Biol. Chem., 293, 7117–7125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sarn, N., Jaini, R., Thacker, S., Lee, H., Dutta, R. and Eng, C. (2021) Cytoplasmic-predominant Pten increases microglial activation and synaptic pruning in a murine model with autism-like phenotype. Mol. Psychiatry, 26, 1458–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schildge, S., Bohrer, C., Beck, K. and Schachtrup, C. (2013) Isolation and culture of mouse cortical astrocytes. J. Vis. Exp., 50079. 10.3791/50079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wei, W., Liu, Z., Zhang, C., Khoriaty, R., Zhu, M. and Zhang, B. (2022) A common human missense mutation of vesicle coat protein SEC23B leads to growth restriction and chronic pancreatitis in mice. J. Biol. Chem., 298, 101536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thompson, J.D., Gibson, T.J. and Higgins, D.G. (2002) Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformatics. Chapter 2, Unit 2.3. 10.1002/0471250953.bi0203s00. [DOI] [PubMed] [Google Scholar]
- 45. Jo, S., Cheng, X., Islam, S.M., Huang, L., Rui, H., Zhu, A., Lee, H.S., Qi, Y., Han, W., Vanommeslaeghe, K. et al. (2014) CHARMM-GUI PDB manipulator for advanced modeling and simulations of proteins containing nonstandard residues. Adv Protein Chem Struct Biol, 96, 235–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Humphrey, W., Dalke, A. and Schulten, K. (1996) VMD: visual molecular dynamics. J. Mol. Graph., 14, 33–38, 27–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its Supplementary material. Raw data that support these findings are available from the corresponding authors upon reasonable request.