

Abstract
Heparan sulfate (HS) and heparin contain imprinted “sulfation codes”, which dictate their diverse physiological and pathological functions. A group of orchestrated biosynthetic enzymes cooperate in polymerizing and modifying HS chains. The biotechnological development of enzymes that can recreate this sulfation pattern on synthetic heparin is challenging, primarily due to the paucity of quantitative data for sulfotransferase enzymes. Herein, we identified critical structural characteristics that determine substrate specificity and shed light on the catalytic mechanism of sugar sulfation of two HS sulfotransferases, 2-O-sulfotransferase (HS2ST) and 6-O-sulfotransferase (HS6ST). Two sets of molecular clamps in HS2ST recognize appropriate substrates; these clamps flank the acceptor binding site on opposite sides. The hexuronic epimers, and not their puckers, have a critical influence on HS2ST selectivity. In contrast, HS6ST recognizes a broader range of substrates. This promiscuity is granted by a conserved tryptophan residue, W210, that positions the acceptor within the active site for catalysis by means of strong electrostatic interactions. Lysines K131 and K132 act in concert with a second tryptophan, W153, shedding water molecules from within the active site, thus providing HS6ST with a binding preference toward 2-O-sulfated substrates. QM/MM calculations provided valuable mechanistic insights into the catalytic process, identifying that the sulfation of both HS2ST and HS6ST follows a SN2-like mechanism. When they are taken together, our findings reveal the molecular basis of how these enzymes recognize different substrates and catalyze sugar sulfation, enabling the generation of enzymes that could create specific heparin epitopes.
Keywords: heparin, glycosaminoglycan biosynthesis, sulfotransferase, sulfation, HS2ST, HS6ST, sugar ring puckering, active-site solvation
Graphical Abstract
INTRODUCTION
Heparan sulfate (HS) and heparin, complex unbranched sulfated polysaccharides, are both part of a class of molecules named glycosaminoglycans (GAGs), consisting of a repeating disaccharide unit formed of glucosamine (GlcN) and glucuronic acid (GlcA)/iduronic acid (IdoA) residues.1 HS is ubiquitously found in all animal cells and tissues and plays various biological roles in embryonic development, as well as in extracellular signaling, while heparin is expressed primarily in mast cells1–3 and plays an important role in coagulation. Heparin is widely used in the medical field as an anticoagulant, with a global turnover of more than US$ 6 billion in 2018.4 HS and heparin share the same biosynthetic pathway and mainly differ in the degree of chain sulfation; 80% of heparin is sulfated, while ∼50% of the HS chain is unmodified.5 During the polymerization of these GAGs, a series of non-template-driven enzyme-catalyzed modifications take place.6–8 These include cleavage of the acetyl group from GlcNAc and concomitant N-sulfation by the bifunctional enzyme family of N-deacetylases/N-sulfotransferases (NDST isoforms NDST1–4)9 forming N-sulfoglucosamine (GlcNS). Glucuronyl C5-epimerase then converts GlcA (d-glucuronic) to IdoA (l-iduronic).10 A final step is the addition of one or more sulfate groups, carried out by HS 2-, 3-, and 6-O-sulfotransferases (HS2ST, HS3ST and HS6ST, respectively).11
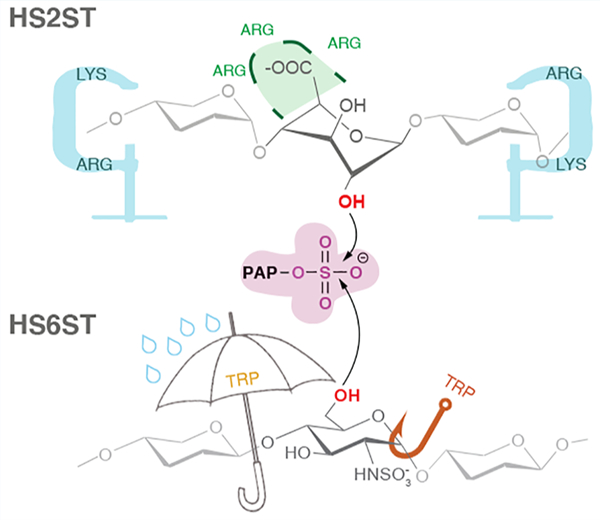
These Golgi sulfotransferases are responsible for transferring the sulfate group from the cofactor molecule PAPS (3′-phosphoadenosine-5′-phosphosulfate) to a specific position of the acceptor unit, specifically: HS2ST sulfates the 2-OH position of GlcA/IdoA, HS6ST the 6-OH position of GlcNS (Scheme 1), and HS3ST the 3-OH position of GlcNS. The density and exact position of sulfate groups constitute specific binding motifs for other biomolecules and, hence, are of paramount importance for the physiological functions of GAGs. Specifically, highly sulfated “patches” are often flanked by less sulfated regions within the HS/heparin chain.12–14 A significant body of work has shown that these sulfation patterns within the polysaccharide chain dictate their physiological functions.13,15–21
Scheme 1. Schematic Representation of IdoA, GlcA, and GlcNS Monosaccharidesa.
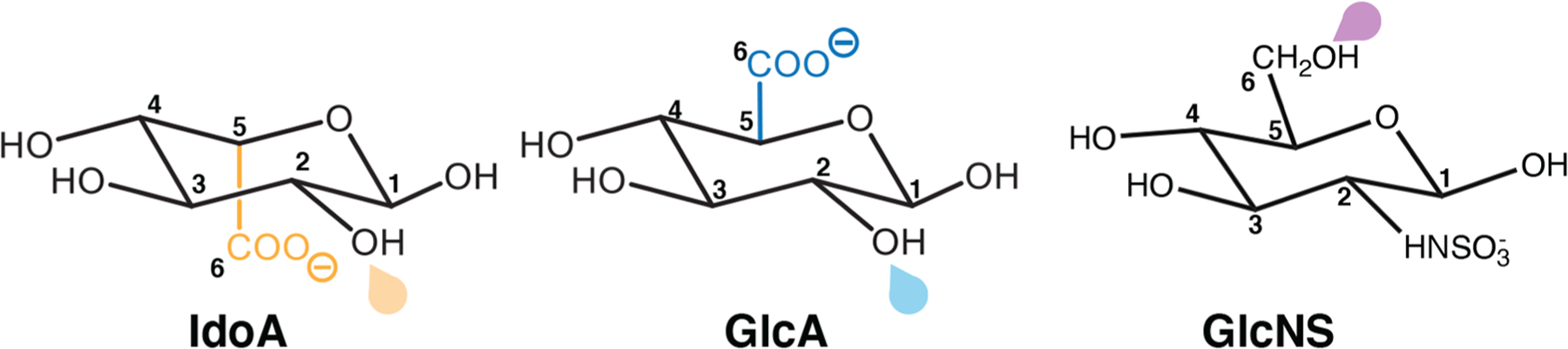
aThe C5 centers of the epimers IdoA and GlcA are shown in orange and blue, respectively. Drops (orange/blue for HS2ST and purple for HS6ST) point toward the sulfation sites.
Sulfation at the 2- and 6-positions are functionally linked, since genetic22 and biochemical23 inhibition of HS2ST12,30,31 sulfotransferase activity causes an increase in 6-O-sulfation, compensating for the loss of 2-O-sulfation. Since a functional link has been shown to exist between HS2ST and HS6ST, we focused on these two enzymes in this study (Figure 1). A single HS2ST isoform has been identified to date in humans which may play an important regulatory role in the biosynthetic process of GAGs.24 Moreover, HS2ST null knockout mice display significant developmental abnormalities that lead to death of the early fetus, presenting bilateral renal agenesis and defects in the eyes and skeleton.25,26
Figure 1.
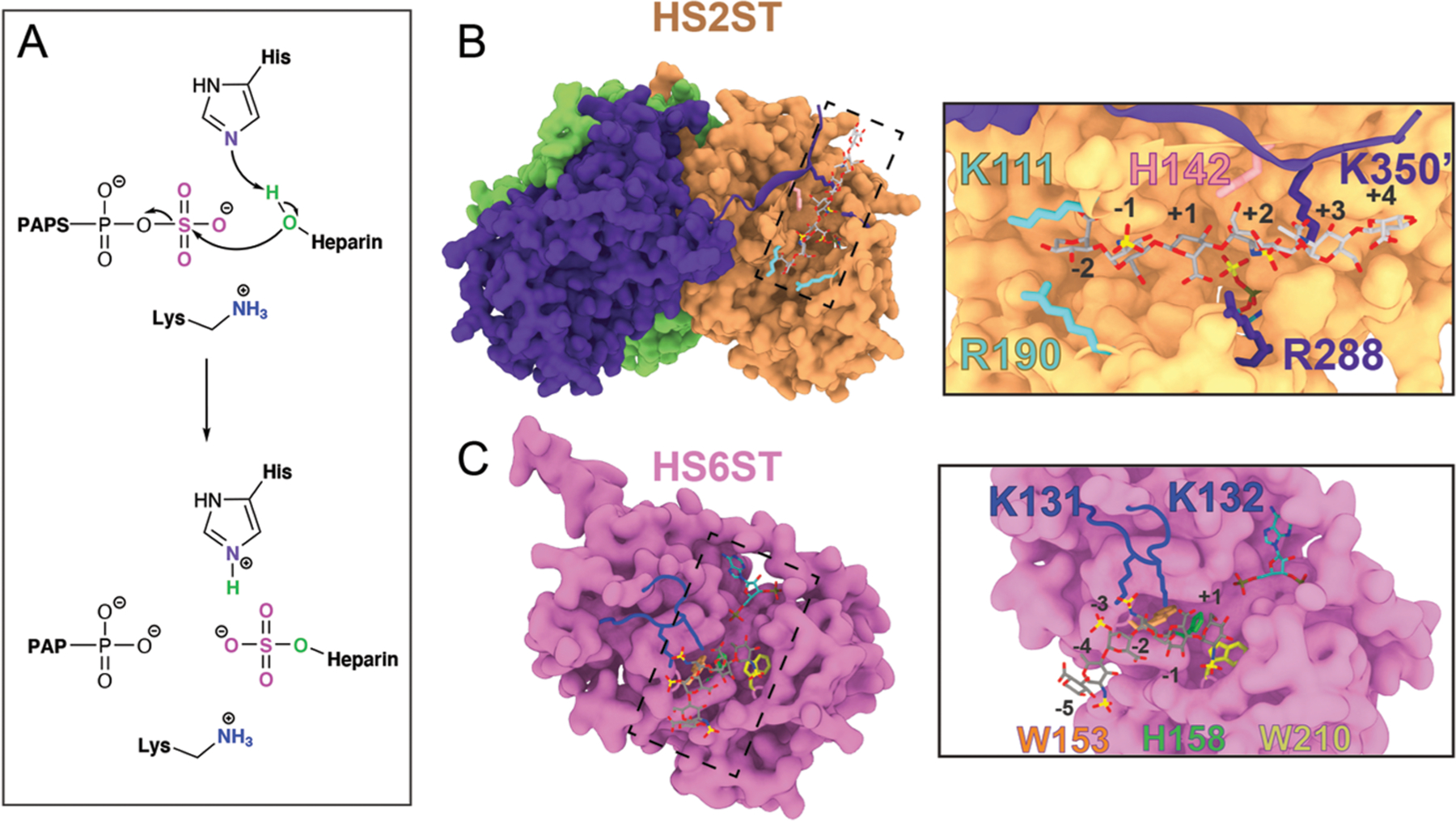
Representation of HS2ST and HS6ST. (A) General proposed catalytic mechanism exerted by HS2ST and HS6ST. (B) Surface representation of the HS2ST trimer. Each monomer is represented with a unique color. The insert highlights the glycan clamps (K111/R190 in cyan and R288/K350′ in purple) and the H142 catalytic base. (C) Surface representation of the HS6ST monomer. The insert highlights W153 and W210 near the active site, catalytic H153, and the loop containing the K131/K132 pair. The numbers indicate the position of the saccharide units in relation to the acceptor unit.
HS2ST preferentially sulfates the C2 position of the hexuronic epimer IdoA, with a much lower sulfation activity toward GlcA: specifically, HS2ST has a 2.5-fold preference toward IdoA substrates.25,27 The next modification occurs at the 6-position of the glucosamine residue by HS6ST, and this sulfation is critical for conferring antithrombin activity to heparin.28 The HS6ST isoforms HS6ST-1, −2, and −3 and an additional alternatively spliced isoform, HS6ST-2S, have been identified in mammals.29 There is evidence that all HS6ST isoforms exhibit a substrate preference for a negatively charged sugar (a uronic acid residue) neighboring an N-sulfoglucosamine. The crystal structure of zebrafish HS6ST isoform 3 has recently been solved, revealing a unique orientation of HS binding.14 In 2-O-, 3-O-, and N-sulfotransferases, the saccharide binds perpendicularly to the sulfate donor PAPS in an open cleft. In contrast, a recent crystallographic study revealed that, for HS6ST, this cleft is blocked by a coil that shallows the binding cleft, suggesting that glycan binding may exert some modulatory function by burying PAPS within the active site, ensuring its proper placement.14 This unusual topology is thought to explain HS6ST promiscuity toward different substrates, and this substrate preference appears to be conserved among HS6ST isoforms.32
HS2ST and HS6ST are central enzymes in HS/heparin biosynthesis, and a precise molecular understanding of the protein:GAG interactions is vital to understanding the mechanisms that control HS and heparin sulfation. By synergistically exploiting in silico and in vitro approaches, we hereby identified structural characteristics that determine substrate specificity for HS2ST and HS6ST and deciphered the mechanism by which these enzymes sulfate their substrates. In particular, we elucidate the contribution and the role of individual residues within the active site to substrate recognition and catalysis for HS2ST and HS6ST. The structural framework presented here opens new avenues for designing substrate-specific sulfotransferases and for building a platform that would enable a cost-effective synthesis of diverse HS oligosaccharides with different sulfation patterns toward heparin-based therapeutics.
METHODS
The Methods section is provided within the Supporting Information and consists of (i) system setup and molecular dynamics (MD) simulations, (ii) the molecular-mechanics generalized-Born surface area (MM-GBSA) method, (iii) multiple sequence alignment, (iv) enzymatic assays, (v) metadynamics simulations for sugar puckering and water coordination, (vi) QM/MM setup and ONIOM calculations, (vii) ring-puckering analysis, (viii) interaction fingerprint analysis, and (ix) grid inhomogeneous solvation theory (GIST) analysis.
RESULTS AND DISCUSSION
Structural Motions of the HS2ST Enzyme Bound to the Crystallographic Substrate.
Our work starts with the analysis of the HS2ST crystal structure (PDB ID 4NDZ), which consists of a trimer that binds the glycan acceptor substrate in the 4C1-IdoA conformation within a groove formed by one subunit complemented by a C-terminal beta-strand that is swapped over from a neighboring subunit (Figure 1). We carried out extensive 2 μs MD simulations of (i) the substrate- and PAPS-free apo HS2ST homotrimer (HS2STapo), (ii) the HS2ST homotrimer complexed to PAPS and GlcA+5-GlcNAc+4-GlcA+3-GlcNS+2-4C1-IdoA+1-GlcNS−1-GlcA−2 (PDB ID: 4NDZ),27 hereafter referred to as HS2ST:4C1-IdoA, and (iii) the HS2ST homotrimer bound to PAPS and GlcA+5-GlcNAc+4-GlcA+3-GlcNS+2-4C1-GlcA+1-GlcNS−1-GlcA−2 (where we changed the sugar to a glucuronic acid at the catalytic position), hereafter referred to as HS2ST:4C1-GlcA. The saccharide unit that undergoes sulfation is in position “+1” and is indicated in boldface. Since the simulated HS2ST is a homotrimer, all of the results reported herein are referred to as the averaged results of a triplicate of two runs. Upon examining HS2ST:glycan simulations, we identified for the first time a group of positively charged residues forming molecular clamps in both the HS2ST:4C1-GlcA and HS2ST:4C1-IdoA complexes that are “open” or “closed” in order to stably position the negatively charged hexasaccharide substrates within the active site. Specifically, these molecular clamps are formed by amino acid residues K111/R190 and R288/K350′ (“prime”, because it is from the C-terminus of the adjacent monomer). The K111/R190 clamp (in cyan in Figure 1B) interacts with the substrate hexasaccharide at the +4 position, while R288/K350′ flanks the acceptor unit (at position +1) and GlcNS2S+2 (in purple in Figure 1B). Intriguingly, we found that these residues are highly conserved among vertebrate HS2ST enzymes (Figure S1), suggesting that they play a crucial role in positioning the ligand precisely within the active site. To follow the movement of the clamps during the MD simulations, the center of mass of both LYS/ARG residue pairs was tracked for each clamp. For HS2STapo, the average distance between the K111/R190 pair was 18 Å (Figure 2A, left panel). In the HS2ST:4C1-GlcA and HS2ST:4C1-IdoA complexes, however, the average clamp distance was reduced to around 13 Å (Figure 2A, center and right panels), reinforcing the hypothesis that the K111/R190 clamp closes upon binding of the substrate, requiring the presence of a high-motility loop from residues 180 to 197.33
Figure 2.
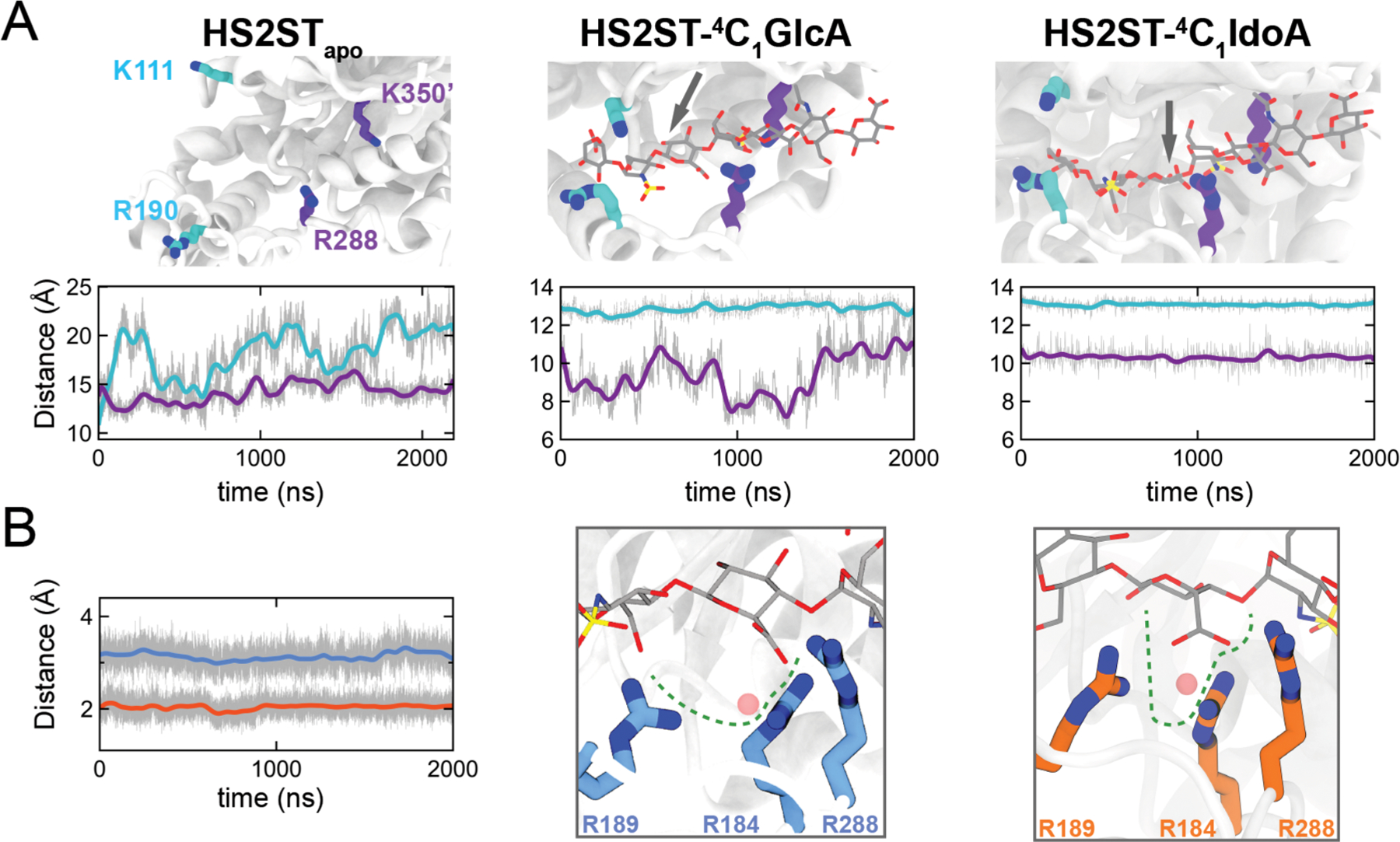
Uronic acid epimers binding to HS2ST. (A) Clamp pairs center of mass distances during simulations with the apoprotein, HS2ST:4C1-GlcA or HS2ST:4C1-IdoA. (B) Center of mass (pink sphere) distance of arginine triad R184/R189/R288 to the C6 atom of either IdoA (in orange) or GlcA (in blue).
The opening of the R288/K350′ clamp varied significantly between the HS2ST:4C1-GlcA and HS2ST:4C1-IdoA simulations (Figure 2A, center and right panels). Specifically, the average opening of R288/K350′ for HS2ST:4C1-IdoA was stable at ∼10.5 Å, while for HS2ST:4C1-GlcA it fluctuated between ∼7 and 12 Å. An analysis of dynamic cross-correlation (DCC) plots, which enables qualitative measurements of the interdependent enzyme dynamic coupling (Figure S3 and Discussion S1), shows that the simple substitution of the acceptor unit from GlcA to IdoA has a substantial effect on HS2ST motility.
Affinity of the HS2ST Sulfotransferase for GlcA and IdoA at Pucker 4C1 as in the Cocrystal Structure.
The substrate binding was affected by enzyme motility, and during our HS2ST:4C1-GlcA MD simulations for two out of three subunits of HS2ST, we observed the dissociation of the oligosaccharide ligand from the enzyme binding pocket (RMSD values are reported in Figure S4). We quantified the binding free energy of the HS2ST:glycan systems by means of MM-GBSA calculations (see the Supporting Information and Methods). These analyses show that the 4C1-IdoA-containing ligand binds to HS2ST with an average ΔG value of −88.5 ± 1.2 kcal/mol, while the formation of the HS2ST:4C1-GlcA complex gains −76.8 ± 2.5 kcal/mol (Table S2).
In order to understand differences in binding affinity, we analyzed the surrounding environment of HS2ST:4C1-GlcA and HS2ST:4C1-IdoA ligand binding pockets during simulations. Consistent with previous experimental data,27,34,35 we identified a positively charged fork formed by three arginine residues, R184, R189, and R288, that anchor the ligand at the sulfate acceptor unit (4C1-GlcA+1 and 4C1-IdoA+1) (Figure 2B). The distance between the C6-acceptor unit of either the HS2ST:4C1-GlcA complex (Figure 2B, with arginines depicted as blue carbons) or the HS2ST:4C1-IdoA complex (Figure 2B, with arginines depicted as orange carbons) and the center of mass of the fork was also tracked over the simulation time. The distance observed was 3.2 Å on average for 4C1-GlcA+1 (blue line) and 2.4 Å on average for 4C1-IdoA+1 (orange line), indicating that the arginine fork provided an efficient electrostatic trap for the negatively charged carboxylated C6 group of the acceptor IdoA residue, while the “in plane” C6OO− of the GlcA acceptor does not fit properly within the fork trap.
To further quantify the contribution of the key amino acids (K111/R190 and R288/K350′ clamps and R184/R189/R288 fork) to the overall binding, an MM-GBSA per-residue energy decomposition analysis was performed on HS2ST:4C1-GlcA and HS2ST:4C1-IdoA (Figure S5 and Table S3). The lysine of the K111/R190 clamp contributes −3.8 ± 0.1 kcal/mol to the binding of the 4C1-IdoA-containing substrate, and −2.3 ± 0.0 kcal/mol for HS2ST:4C1-GlcA, while R190 displays a slight preference toward HS2ST:4C1-IdoA (−5.8 ± 0.8 and −1.9 ± 0.4 kcal/mol, respectively). The contribution of R288 is high for both complexes, being −11.8 ± 0.8 and −12.1 ± 0.6 kcal/mol for HS2ST:4C1-GlcA and HS2ST:4C1-IdoA, respectively. The last residue, K350′, located at the adjacent monomer, contributes −18.7 ± 1.6 kcal/mol in HS2ST:4C1-IdoA and −5.8 ± 2.0 kcal/mol in HS2ST:4C1-GlcA simulations. The three arginine residues that form the fork (R184, R189, and R288) contribute together to a total binding of −20.5 ± 1.5 kcal/mol for HS2ST:4C1-GlcA and −24.0 ± 2.1 kcal/mol for HS2ST:4C1-IdoA. These contributions suggest that the three residues play a fundamental role in binding the substrate, especially R288, which was identified in the two structural motifs responsible for binding the substrate (the R288/K350′ clamp and the R184/R189/R288 fork). It has been reported in the literature that mutating R189 to alanine (R189A) causes a loss of the enzyme preference for IdoA as an acceptor33 and that HS2ST(R189A) is capable of sulfating GlcA to a higher extent in comparison to the wild-type enzyme.27 To date, there are no reports on whether it is the charge and/or steric hindrance of R189 side chains that govern these two observed phenomena.27,33 Therefore we performed an in vitro enzymatic assay on wild-type HS2ST and, in order to evaluate the steric effect of a large but neutral amino acid on the enzyme ability to sulfate the acceptor, we generated a mutant harboring an arginine to leucine mutation (R189L), and a second mutant (R189 K) was generated to evaluate the effect of a different positively charged residue. The R189A mutant was expressed as in Liu et al.34 to coherently enable a comparison of our results. The WT and mutated enzymes were incubated with (GlcNS-GlcA)3 polysaccharide substrates, and the products produced analyzed by strong anion exchange high performance liquid chromatography (HPLC), as as described in Methods in the Supporting Information. The wild-type HS2ST enzyme presented 92% less activity toward [-GlcNS-GlcA-]3 in comparison to [-GlcNS-IdoA-]3 (Figure S6). Similarly, the mutants R189K and R189L exhibited less activity (<10%) toward (GlcNS-GlcA)3, while R189A maintained 100% activity toward (GlcNS-GlcA)3. Thus, our experimental data suggest that there is a balance between the charge and the steric effect of the arginine side chain, reinforcing the idea that the R189 charged guanidinium moiety plays a crucial role in recognizing the hexuronic epimer. We also carried out an in silico investigation of R189A mutation, by performing a 2 μs MD simulation of HS2ST(R189A):4C1-GlcA. In this simulation, A189 did not obstruct the binding of GlcA within the fork as did R189 (Figure S6A,B). Indeed, GlcA can approach the fork and bind to R184 and R288. The contributions of R184 and R288 to ligand binding for HS2ST(R189A):4C1-GlcA are −6.5 ± 0.1 and −2.8 ± 0.5 kcal/mol, respectively, suggesting that, when R189 is abolished, the other two residues (R184 and R288) are not able to “recreate” the electrostatic fork to entrap the hexuronic unit. When they are taken together, the experimental and computational data demonstrate that R189 plays a critical role in defining HS2ST substrate specificity.
On the basis of a per-residue decomposition analysis carried out on all of the amino acids surrounding the binding site, R80 also displays favorable and strong interactions with both HS2ST:4C1-GlcA and HS2ST:4C1-IdoA (−10.8 ± 0.8 and −13.1 ± 0.7 kcal/mol, respectively). While the fork recognizes and fits the carboxylated group of the hexuronic acid, R80 is responsible for recognizing the GlcN2S unit at the +2 position, reinforcing the hypothesis that N-sulfation is a prerequisite for 2-O-sulfation.34 We can also observe distinctive ways in which the NREs of the polysaccharide substrates bind to HS2ST in the HS2ST:4C1-GlcA and HS2ST:4C1-IdoA simulations. The observed contributions of K354′ toward substrate binding for HS2ST:4C1-IdoA (−8.4 ± 2.1 kcal/mol) and HS2ST:4C1-GlcA (∼0 kcal/mol) highlight the idea that contacts between the oligosaccharide NRE and the enzyme are lost when the predominant epimer is GlcA, while when the glycan is IdoA, these contacts are retained.
HS2ST Senses the Conformation of Uronic Acid of Its Substrates.
GlcA and IdoA epimers can adopt different conformations. For the majority of pyranoses, including GlcA, the 4C1 chair conformation is the only relevant conformation, since other conformations require much higher energy.36 However, IdoA shows a higher degree of conformational flexibility. Indeed, all PDB entries (∼100) containing IdoA that were crystallographically solved at an atomic resolution of <1.5 Å up until the time of this study presented the uronic ring in the 1C4 chair conformation. In the recently crystallized HS2ST (PDB ID: 4NDZ), the polysaccharide presents uronic acid in the 4C1 conformation, an unusual pucker for native GAGs. In fact, HS/heparin chains are composed of 30−250 disaccharides, and it has been reported that there is an increase in 1C4-IdoA within polysaccharides above 12 residues,37,38 reinforcing the hypothesis that real substrates comprise iduronic acid in the 1C4 conformation rather than in the 4C1 conformation. Since it is unlikely that HS2ST would induce such a pyranoside ring distortion, the observation of 4C1-IdoA in the HS2ST crystal can be explained by the fact that the oligosaccharide used for crystallization is small.39 In order to explore the free energy landscape for HS2ST:IdoA and HS2ST:GlcA complexes along with the sugar conformations, we carried out well-tempered metadynamics to bias different sugar puckers.
The resulting free energy surface (FES) for HS2ST:GlcA shows three basins associated with the 4C1, 1,4B, and 1C4 glucuronic acid (Figure 3A, top panel) and 4C1, 2S0, and 1C4 iduronic acid (Figure 3A, bottom panel) conformations. These minima are plotted as a Mercator projection of the Cremer–Pople sphere and expressed in terms of θ angle vs the distance between the acceptor oxygen and the PAPS sulfur (Figure 3A). Since Φ (O5–C1–O4–C4) and θ (C1–O4–C4–C5) dihedral angles dictate the orientation of the sugar ring in polysaccharides, the free energy surfaces of Φ/θ projections can be obtained as seen in Figure S7.
Figure 3.
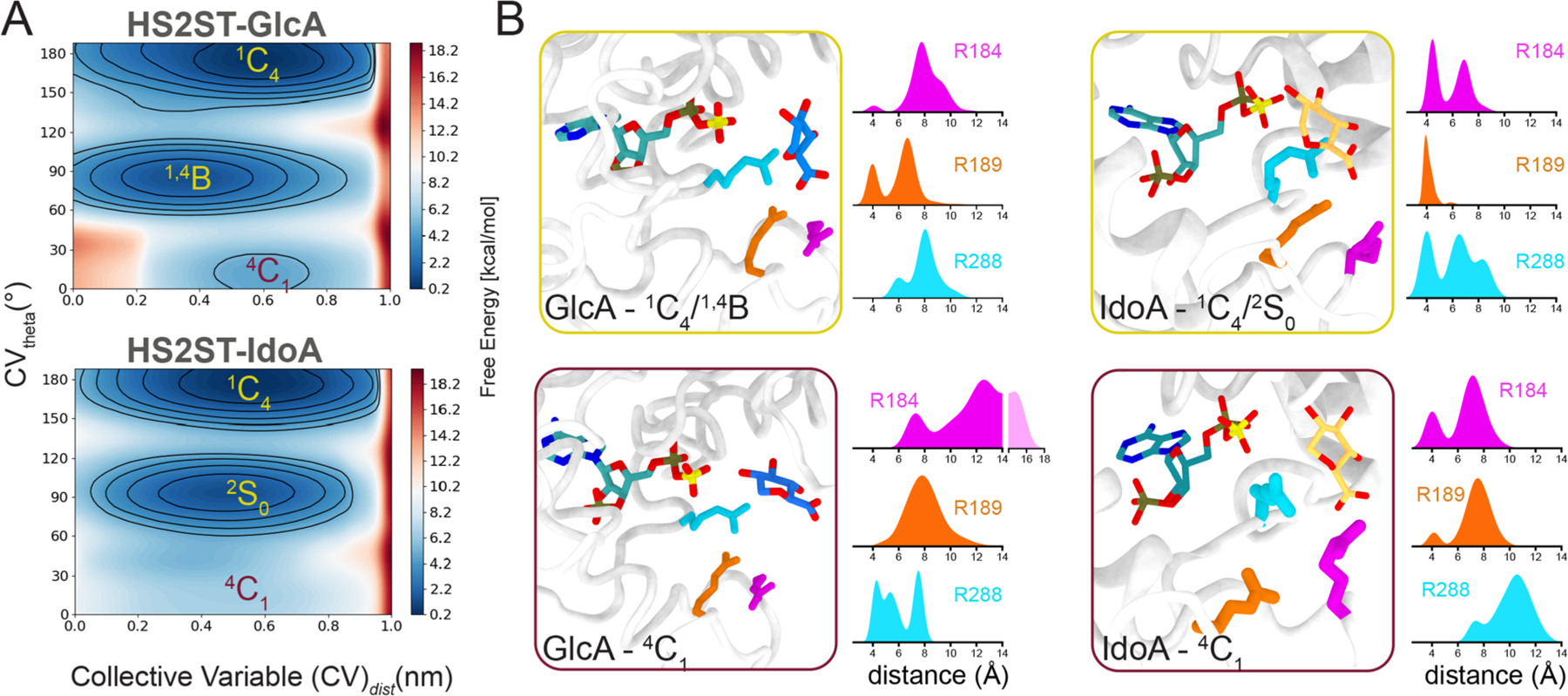
Metadynamics of the HS2ST acceptor sugars. (A) Conformational FES (θ vs distance from PAPS) obtained for HS2ST:GlcA and HS2ST:IdoA. Each contour line of the diagram corresponds to 5 kcal/mol. (B) Distribution of key interactions in terms of the distances from the acceptor hexosamine to the arginines composing the fork in the 1C4/4C1 states (yellow and purple outlines, respectively). The distances of the uronic acceptor heavy atoms to the guanidine groups of R184, R189, and R288 were calculated.
For GlcA, the lowest energy basins are 1,4B and 1C4, while IdoA preferentially adopts a distorted conformation (2S0) along with 1C4 (180°) in the enzyme–substrate complexes. It is known that, in solution, IdoA has three distinguishable minima: the 4C1 chair, the 2S0 skew, and the reverse 1C4 chair. Spiwok et al. have calculated a free energy barrier of ∼7 kcal/mol for the 4C1 to 2S0 transition in water.40 In the HS2ST environment, the pucker minima profiles cover most of the equatorial and northern half of the pseudorotation cycle, with a barrier decreased to 5 kcal/mol between 1,4B and 1C4.
To verify the stability of HS2ST with the differently puckered ligand complexes, we carried out 2 μs MD simulations of HS2ST:1C4-IdoA and HS2ST:1C4-GlcA (where we constrained GlcA to the improbable 1C4 conformation), and then we performed MM-GBSA computations. The total binding free energies, reported in Table S2 and Figure S8, were −81.5 ± 2.9 kcal/mol for HS2ST:1C4-GlcA and −114.7 ± 1.4 kcal/mol for HS2ST:1C4-IdoA. Interestingly, the computed binding affinity is indicative of a tighter binding of 1C4-IdoA with respect to 4C1-IdoA, reinforcing the idea that the enzyme preferentially recognizes the naturally occurring iduronic acid conformation. Thereafter, we quantified the contribution of interface residues on the binding affinity of the substrate. For sake of clarity, the per-residue decomposition results obtained for all of the HS2ST:glycan systems are summarized in Figure S5 and Table S3.
The three arginines (R184, R189, and R288) have a pronounced preference for the 1C4-IdoA unit (Figure S5, orange columns) in comparison to 4C1-IdoA (Figure S5, blue columns), the less probable pucker for iduronic acid. This is also verified for the other epimers, since the fork seems to bind preferably to 4C1-GlcA (Figure S5, green columns) rather than 1C4-GlcA (Figure S5, wine columns), an improbable conformation. The fitting/unfitting of the carboxylated group of the acceptor unit (4C1-IdoA/4C1-GlcA) may provide a plausible mechanistic explanation for the preference of this enzyme for IdoA-over GlcA-containing substrates. We then examined the distribution of the distances of GlcA and IdoA epimers within the arginine triad during the metadynamics simulations (Figure 3B). In the various GlcA conformers, the pyranose ring could be distorted from 4C1 and 1,4B/1S3 to minimize clashes with the recognizing arginine fork (R184, R189, and R288) (Figure 3B, yellow square in the left panel). However, at this pucker the GlcA substrate is not in a position for catalysis, as the GlcA O2 is distanced >5 Å from the PAPS sulfate (Figure 3B, left panel). When the acceptor GlcA is in the 4C1 conformation, it is not within a suitable distance to interact with R184/R189, and arginine fork binding is improbable, despite it being equatorially oriented to receive the sulfate. Interestingly, the 4C1 pucker has a much more drastic effect on the IdoA interaction with R288 within the arginine fork (Figure 3B, purple insert in the left panel). The sampled puckers have little effect on the IdoA acceptor orientation within the PAPS donor, an indication that these two variables are not directly correlated. When they are taken together, our data suggest the steric/hydrogen-bonding effect exerted by R184/R189/R288 over the equatorial exocyclic oxygen atoms is a determinant of the observed HS2ST preference for IdoA over GlcA moieties.
How Does the Uronic Acid Conformation Influence HS2ST Catalysis?
In order to further assess if and how the uronic pucker influences the sulfate transfer by HS2ST, we modeled the reaction mechanism for the following acceptor conformations: the 4C1, 2S0, and 1C4 conformers for IdoA and the 4C1 and 1C4 conformers for GlcA. The starting point for QM/MM calculations corresponding to the NAC (near attack conformation) structure was extracted from our MD simulation (Figure S2). Figure 4 shows a schematic representation of the reaction mechanism and the activation barriers (ΔG⧧) and reaction energies (ΔGr) obtained. When the Rx, TS, and Pd points at the HS2ST:4C1-IdoA, HS2ST:2S0-IdoA, HS2ST:1C4-IdoA, HS2ST:1C4-GlcA, and HS2ST:4C1-GlcA complexes were compared, we found that the network of noncovalent interactions engaged by the active residues with the acceptor does not vary significantly (Figures S9 and S10). The reaction proceeds by migration of the sulfate group from PAPS to the acceptor unit through an asynchronous mechanism,41 where deprotonation of the nucleophile by H142 occurs at a later stage with respect to the nucleophilic attack, while K83 stabilizes the migration of SO3− by protonating the phosphate group on PAPS. While all of the active site residues (K83, S86, and R80) display common activity in all of the investigated profiles, we found that only in HS2ST:1C4-IdoA does R288 protonate the acceptor unit, anchoring the 1C4-IdoA in the active site. While the active site interaction network remains unaltered, the activation barriers (ΔG⧧) and reaction energies (ΔGr) are strongly affected by the puckering of the uronic acid. QM/MM calculations showed that the sulfations of 1C4-, 2S0- and 4C1-IdoA are kinetically allowed paths (ΔG⧧ = 15.4, 18.4, and 9.3 kcal/mol, respectively), but only the sulfation of the most probable conformation 1C4 is exoergonic. With regard to GlcA, the computed ΔG⧧ values suggest that HS2ST prefers to catalyze the sulfation of 4C1-GlcA (15.5 kcal/mol) over 1C4-GlcA (27.9 kcal/mol). HS2ST only recognizes and sulfates those sugar conformers that are naturally occurring and, therefore, physiologically relevant (1C4-IdoA and 4C1-GlcA). The sulfation of improbable puckers (1C4-GlcA) faces a barrier that is insurmountable under physiological conditions. To attain closer insights into HS2ST catalysis, a detailed description of the catalytic mechanism and the role of active site residues is reported in Discussion S2.
Figure 4.
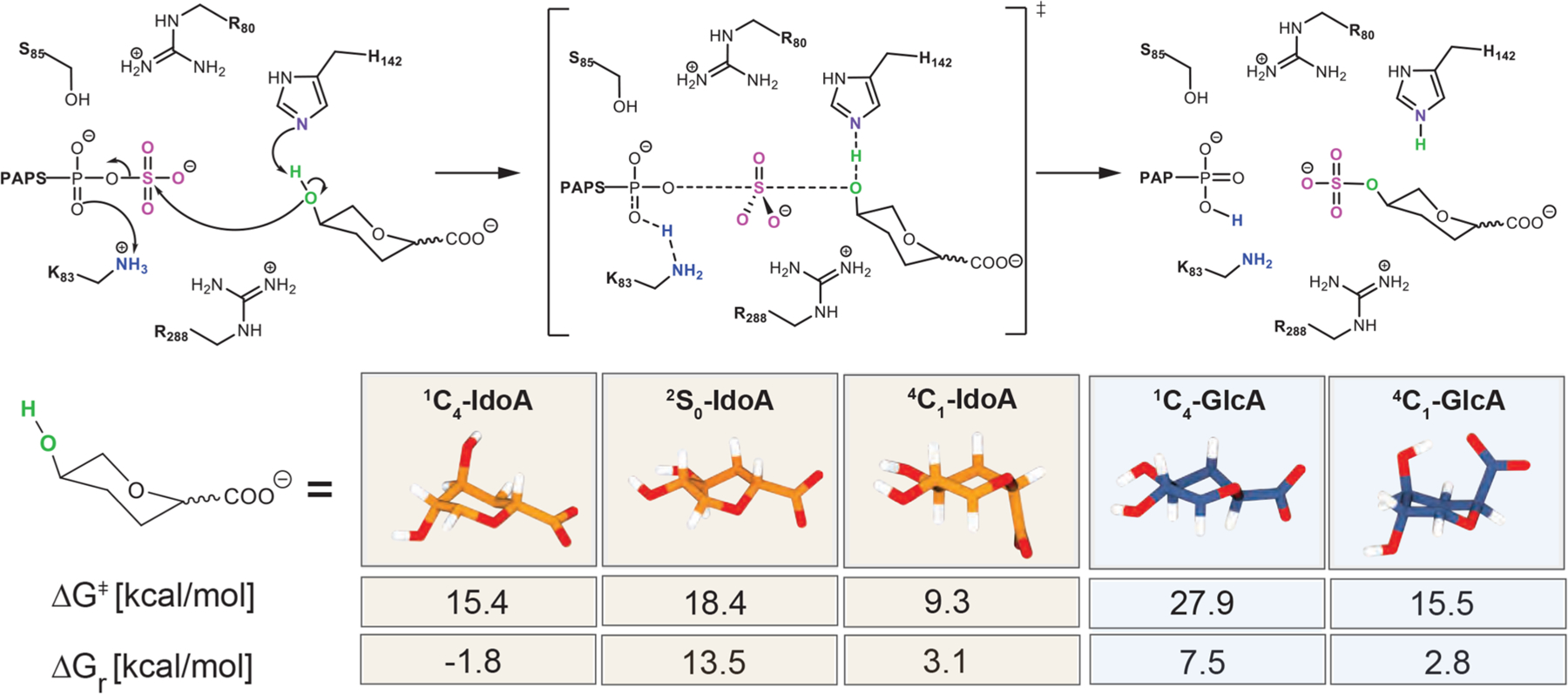
Schematic representation of the critical points computed at the ONIOM[M06–2x/6–31+G*:ff14SB] level of theory for HS2ST catalysis on 4C1-, 2S0-, and 1C4-IdoA and 4C1- and 1C4-GlcA saccharide units. The activation (ΔG⧧) and reaction (ΔGr) Gibbs free energies are reported.
HS6ST Structural Motions with Different Substrates.
In HS/heparin biosynthesis, 2-O-sulfation by HS2ST is followed by 6-O-sulfation by HS6ST. To explore the binding and catalytic processes exerted by HS6ST, we carried out molecular dynamics simulations using the catalytic domain of HS6ST bound to two different substrates (PDB ID 5T03 and 5T05).32 The PDB file 5T03 contains the HS6ST enzyme bound to a polysaccharide composed of GlcNS+1-GlcA+2-GlcNS+3-GlcA+4-GlcNS+5-GlcA+6, where GlcNS+1 is the acceptor sugar unit and GlcA+4 is the sugar unit that is specifically bound by a K131/K132 lysine clamp. This complex is hereafter referred to as HS6ST:GlcA+4. The PDB file 5T05 contains an HS6ST enzyme bound to a polysaccharide that varies from 5T03 only at position +4. 5T05, instead, contains a 2-O-sulfated IdoA2S in a 2S0 skew-boat conformation, GlcNS+1-GlcA+2-GlcNS+3-IdoA2S+4-GlcNS+5-GlcA+6,32 hereafter referred to as HS6ST:IdoA2S+4. The saccharide unit that undergoes sulfation is in position “+1” and is indicated in boldface.
The HS6ST active site contains two tryptophan residues,32 W153 and W210, that are solvent-accessible (orange and yellow sticks in Figure 1C, respectively). The crystal structure 5T05 of HS6ST containing a 2-O-sulfate at position +4 (IdoA2S+4) shows the acceptor N-sulfo moiety GlcNS+1 within hydrogen-bonding distance of the amide backbone of W210. The side chain of W210 points toward the GlcNS+1 acceptor in an unusual perpendicular orientation that may allow CH–π interactions such as typical aromatic–sugar interactions found in many glycan binding proteins.42,43 In order to probe the presence of CH–π contacts between W210 and the acceptor glucosamine GlcNS+1 of both HS6ST:GlcA+4 and HS6ST:Ido2S+4, we analyzed the distance between GlcNS+1 and the center of the indole phenyl (d(CH–π)) and the angle between the CH vector and the phenyl ring plane normal vector (ω(CH–π)).43–45 Aside from the differences between HS6ST:GlcA+4 and HS6ST:IdoA2S+4, our analysis (see Discussion S3) does not meet the criteria for a classical or “T-shaped” CH–π interaction. Instead, W210 and the acceptor glucosamine interact via H-bonds in the HS6ST:Ido2S+4 complex (Figure S11A,B). This is confirmed by the MM-GBSA pairwise decomposition analysis: the binding affinity of W210 to the acceptor glucosamine GlcNS+1 (−11.4 ± 1.5 kcal/mol) is mainly due to electrostatics (−7.3 ± 2.0 kcal/mol), rather than to Van der Waals interactions (−3.1 ± 0.9 kcal/mol) (Figure S12). QM calculations also confirm this behavior (Discussion S3), indicating that H-bonds and nonhydrophobic interactions are dominant in the GlcNS+1–W210 interaction. It is worth noting that the published structure of the N-sulfotransferase domain of the N-deacetylase/N-sulfotransferase (PDB ID 1NST)46 contains a structurally conserved residue with the same topology as W210 (W713), an indication that this tryptophan in heparan sulfate N-deacetylase/N-sulfotransferases might play a similar role in binding GlcNAc. The positioning of W210 also resembles that of other solvent-exposed tryptophan residues found in oligosaccharide lyases, such as W49 of B. fragilis beta-lactamase47 or W31 of R. solanacearum lectin.48 Similar to the tryptophan in these enzymes, W210 allows HS6ST to recognize a greater variety of ligands, conferring a degree of promiscuity to HS6STs.
The loop containing the other tryptophan residue W153 moves away from the active site, opening the active site cleft. We visualized this movement by plotting the distance between H158, the catalytic histidine, and the W153 indole side chain center of mass for the HS6STapo, HS6ST:GlcA+4, and HS6ST:Ido2S+4 simulations in Figure 5A. For the apo enzyme, the distance between the center of mass for H158 and W153 fluctuates with an average amplitude of 5.3 Å; for HS6ST:GlcA+4, the distance between H158 and W153 varies substantially, with an amplitude of 4.4 Å, while for HS6ST:IdoA2S+4 the distance is stable at around 4.1 Å (Figure 5A).
Figure 5.
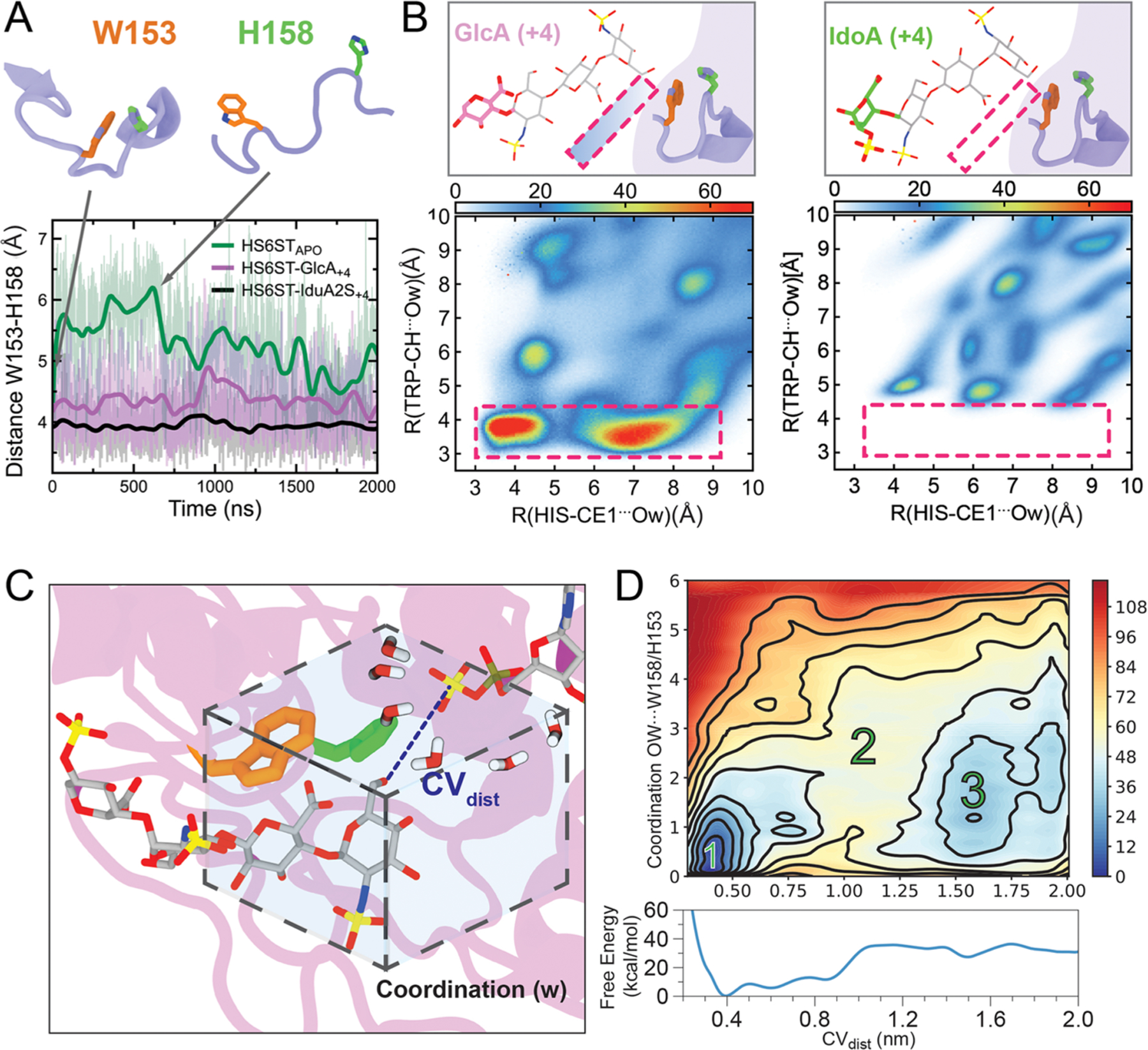
HS6ST substrate at position +4 influences HS6ST motility and active site hydration. (A) Distances between W153 and H158 during HS6ST simulations. (B) 2D-radial distribution functions (2D-RDF) function for the two closest atoms from W153 and H158 for both HS6ST:IdoA2S+4 and HS6ST:GlcA+4 simulations. Pink dashed lines outline the two water clusters observed in HS6ST:GlcA+4 simulations. Atoms are labeled according to AMBER force field nomenclature. (C) Diagrams of coordination parameters defined for free energy calculations of active site hydration and acceptor coupling. The dashed box displays the coordination of waters surrounding the active site collective variable parameter (w). The substrate is shown as gray sticks. (D) Projection of the FES onto CVdist and CVw, as well as onto CVdist alone (bottom). Numbers indicate the most relevant metastable states 1 and 3, separated by a high-energy barrier, 2. Contours are drawn every 12 kcal/mol.
Upon further analysis of the crystal structures, we observed that, while PDB ID 5T03 (HS6ST:GlcA+4) contained four structural water molecules at <4 Å from the catalytic histidine, PDB ID 5T05 (HS6ST:Ido2S+4) only had one. On the basis of the fact that PAPS is highly prone to hydrolysis and that HS6ST presents a shallow cleft architecture in its active site, we suggest that the presence of W153 could create a hydrophobic environment that protects PAPS49,50 within the protein active site from oxidative damage.51 This prompted us to probe the effect of water on substrate binding to the active site of HS6ST:GlcA+4 and HS6ST:IdoA2S+4 complexes, by computing the radial distribution functions (RDF) of water molecules around H158. The weaker binding noted for HS6ST:GlcA+4 was found to affect solvent accessibility around the catalytic histidine, as shown by the radial distribution of water molecules 0.5 g(r) for HS6ST:IdoA2S+4 and 1.0 g(r) for HS6ST:GlcA+4 (Figure S13). Both HS6ST:GlcA+4 and HS6ST:IdoA2S+4 simulations display a typical hydrophilic interaction with a well-defined first hydration shell, showing a density peak at 2.0 Å and a second hydration shell around 3.8 Å (Figure S13A). As the simulations progressed for HS6ST:GlcA+4, interfacing residues became exposed to the solvent, facilitating a glycan–water interaction and causing release of the substrate, and with this, drifting of the substrate was observed. The persistence of structured water molecules between the enzyme and substrate during MD simulations was analyzed through normalized two-dimensional RDF functions (2D-RDF) for HS6ST:IdoA2S+4 and HS6ST:GlcA+4. Essentially, 2D-RDF calculates the pairwise value (Å distance) that reveals the hydrophobicity radius around the selected atom (Figure 5B). No water density was found at a distance below a radius of 4.8 Å from H158CE1 and W153CH (Figure 5B, left and right panels), suggesting the absence of water molecules within the active site of HS6ST:IdoA2S+4. In contrast, for HS6ST:GlcA+4, the calculated pairwise value of water density was ∼3.6 Å, showing two persistent water pockets for HS6ST:GlcA+4 at its binding interface (indicated with a pink rectangle in the left panel in Figure 5B). For HS6ST:GlcA+4, the average residence time for bridging water molecules within the active site was 277 ps, with the most persistent bridging molecule having a residence time of 737 ps, indicating that these water molecules are dynamically exchanged. During HS6ST:IdoA2S+4 simulations, W153 prevents the access of bulk water within a 4.3 Å radius of catalytic H158, while in stark contrast during HS6ST:GlcA+4 simulations, bulk water is able to permeate within 3.6 Å of H158. From the 2D-RDF analysis, we can conclude that W153 repulses water molecules more efficiently when IdoA2S+4 is the substrate (Figure 5B, left and right panels). In order to further explore how water permeation around H158 and W153 precisely affects substrate binding, we calculated the free energy of water coordination of HS6ST:IdoA2S+4 using well-tempered bias-exchange metadynamics52 followed by the corresponding reweighting techniques (Figure S14A).53 The second collective variable was defined as the region within H158 and W153 and their coordinating water sites (w, Figure 5C). After calculating total residence times during the HS6ST:IdoA2S+4 simulations, the water shell shared by both residues averaged six water molecules, which was then defined as the cutoff neighbor list.
The approximate free energy surface (FES) of substrate binding estimated as a function of hydration was obtained by averaging the various unbinding runs. Figure 5C, right panel, shows the free energy landscapes of water coordination at the active site as a function of the substrate binding state. On the basis of the conformational distributions at different water coordinations and CVdist values, the water shell around H158 is negatively correlated with stronger binding to the substrate (Figure 5C, right panel). Specifically, when there are three or more water molecules within W153/H158 (w > 3), the oligosaccharide substrate has a minimum at a bound configuration (O6–S distance ∼4 Å; Figure 5C, right panel, basin 1). Interestingly, no minima exist for w > 3 when the oligosaccharide substrate is in the unbound state (Figure 5C, right panel, basin 3). At high d values (Figure 5C, right panel, basin 3, with the representative structure in Figure S14A, panel 3), approximately two water molecules are coordinated to H158. As d decreases, water molecules are expelled from the active site, showing that during binding dehydration occurs in a stepwise manner. This process of dehydration facilitates catalysis. The water molecules expelled from the vicinity of H158 by W153 are replaced by native ligand contacts with the substrate (Figure 5C, right panel, basin 1). All of these features suggest that, for HS6ST, the solvent plays a crucial role in substrate recognition. Other subsites within HS6ST could influence the enzyme affinity for different substrates and, consequently, its catalysis. In order to evaluate how overall enzyme motions affect substrate affinity, we calculated the dynamic cross-correlation for HS6ST:IdoA2S+4 and HS6ST:GlcA+4 simulations, and the results, reported in the Supporting Information, suggest that the K131/K132 loop works as a lid for the binding cleft (Figure S15). This is in line with the current hypothesis that this loop strongly binds to the negatively charged glycosaminoglycan chain and coordinates the alignment of the glycan within the active site.32 On the basis of the dynamic cross-correlation scores (see Discussion S4), the movement of the lloops containing amino acids W153 and W210 are anti-correlated in the apo structure (green squares, Figure S15, left panel), confirming that the tryptophan residues (W153/W210) flanking the catalytic histidine in HS6STs play a role in promoting the binding of the acceptor. We investigated the effect of the K131/K132 loop for both HS6ST:GlcA+4 and HS6ST:IdoA2S+4 by calculating MM-GBSA values, finding that K131 binds with a weaker binding energy to the GlcA+4 acceptor in comparison to IdoA2S+4, with values of −6.4 ± 1.1 and −11.8 ± 0.4 kcal/mol, respectively (Figure S12). Thus, these tryptophan residues, which are unique to HS6STs, could convey, at least in part, the substrate specificity (albeit with some promiscuity) of this enzyme.
HS6ST Sulfotransferase Hydration and Molecular Recognition.
HS6ST presents unique structural characteristics that differ substantially from the reported binding interfaces for HSNST,46 HS2ST,34 and HS3ST.14,32 In comparison to HS2ST, fewer contacts exist between HS6ST and its substrate;32 only the first four units of a polysaccharide substrate form contacts with HS6ST, versus all units of a heptasaccharide for HS2ST.35 In order to understand the dynamics of the enzyme–substrate interactions in HS6ST:GlcA+4 and HS6ST:IdoA2S+4 complexes, we carried out an interaction fingerprinting analysis. In this type of analysis, the interactions between the individual amino acid residues and the monosaccharide units are classified into water bridge, salt bridge, and hydrogen-bond donor and acceptor. Contacts are expressed on the basis of the frequency (%) with which they appear during the dynamics simulations. While interaction fingerprinting was performed for all oligosaccharide binding residues for both HS6ST:GlcA+4 and HS6ST:IdoA2S+4 conditions, we focused our analysis on W153/W210, which surrounds the acceptor, and the K131/K132 loop. While the HS6ST:IdoA2S+4 K131/K132 pair maintains H-bonds and salt bridges with IdoA2S+4 throughout most of the simulation time (>90%), these interactions are lost for GlcA+4 (∼0.05%, Figure S17A,B, bottom panels). These interactions are shifted from GlcA+4 to GlcNS+1/GlcA+2, indicating that as the chain dissociates from its original binding position it is capable of binding at different locations. The nature of the K131/K132 interactions and contacts with GlcA+4 for the HS6ST:GlcA+4 simulations is not sufficient to keep the reducing end fully bound to the enzyme (Figure S17). GlcA+4 loses water bridges and hydrogen bonds with W153, T151, and Q130. This loss of interactions affects water access to the HS6ST active site: the water shell around GlcNS+1 is 50% higher during HS6ST:GlcA+4 simulations in comparison to HS6ST:IdoA2S+4. These data indicate tighter binding of K131/K132 with IdoA2S+4, in comparison to GlcA+4, preventing water access at the protein acceptor interface. Dislodging a polysaccharide involved in an extensive network of water-mediated hydrogen bonds poses a major challenge for HS6ST. The loss of contacts between the enzyme and substrate observed during the HS6ST:GlcA+4 simulations affect the enzyme not only at the reducing end, as discussed above, but also at the NRE (see Figure S17 and Discussion S5).
Sulfate Transfer to the 6O-Position.
To obtain further information on how HS6ST catalyzes the SO3− transfer to the ligand, we investigated the reaction mechanism of GlcNS sulfation for the HS6ST:GlcA+4 complex. We found that the reaction proceeds via a SN2-like asynchronous mechanism (as shown in Figure 6) and that the sulfation requires 16.1 kcal/mol, in line with the results obtained for HS2ST, suggesting that the activation energy is easily surmountable under physiological conditions. To demonstrate a catalytic role for K104 and H158,35 we exchanged the side chain of K104, replacing the −CH2NH3+ terminal group for a CH3 group, and mutated the H158 as H158A, in order to abolish their potential effects within the active site of HS6ST:GlcA+4. In line with the experimental mutagenesis results obtained by Xu and collegues,32 we found that elimination of the terminal NH3+ group in K104 raises the activation energy to 48.2 kcal/mol, while H158A raises it to 24.9 kcal/mol, confirming that K104 and H158 are crucial for HS6ST catalysis. A detailed representation and description of the critical points obtained for HS6ST catalysis are given in Figure S19 and Discussion S6, respectively.
Figure 6.
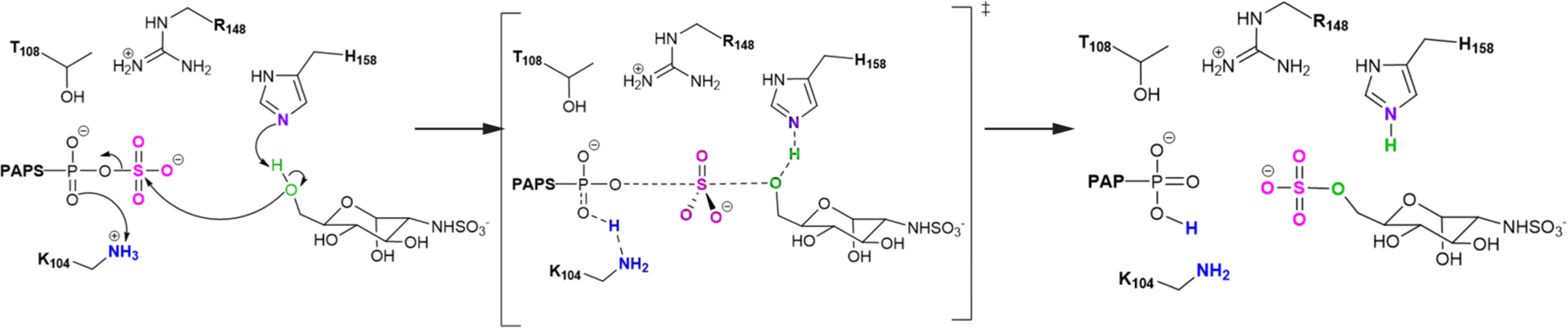
Schematic representation of the critical points computed at the ONIOM[M06–2x/6–31+G*:ff14SB] level of theory for HS6ST catalysis.
HS6ST at the Nonreducing End of the Glycan.
Natural substrates for HS6STs are sugar oligomers that are much longer than the polysaccharide that was cocrystallized with HS6ST. Hence, we systematically evaluated an expansion of the HS6ST ideal substrate, IdoA2S+4, with a polysaccharide to further explore binding at the NRE of HS6ST. The final models were composed of the HS6ST enzyme (PDB ID 5T05) containing its crystal-bound substrate plus a polysaccharide extension with varying degrees of modifications: (GlcNX-GlcAX/IdoAX)x4-GlcNS+1-GlcA+2-GlcNS+3-IdoA2S+4-GlcNS+5-GlcA+6. These additions consisted of (I) GlcNS6S-IdoA2S, (II) GlcNS-IdoA, (III) GlcNS-GlcA2S, and (IV) GlcNAc-GlcA (Figure S20). All models were subjected to molecular dynamics simulations for 1 μs, and each enzyme–monosaccharide interaction was fingerprinted. For extension I, GlcNS6S-IdoA2S, the least probable substrate for HS6ST, was placed at the NRE.
In this case, the substrate (IdoA2S‑3 and IdoA2S‑5) formed water/salt bridges and hydrogen bonds with both R112 and R329 (Figure S20). R116 bound to both GlcNS‑4 and Ido2S‑5, and Ido2S‑5 bound to R112 and R329 with a higher frequency in comparison to R116. Closer to the acceptor GlcNS, GlcA‑1 formed the most contacts with R112/R148/R206 and K104. This network of positively charged residues surrounding GlcA‑1 was persistent across all models assayed, but the nature of interactions shifted from mainly hydrogen bonds and salt bridges to water bridges for the less sulfated substrates. For extensions II and III, which were GlcNS-IdoA and GlcNS-GlcA2S, respectively, contacts at positions −4 to −6 were composed of water bridges, with a notable new hydrogen bond being formed between GlcNS‑2 and H203 (Figure S20). Interestingly, the binding profiles of substrates II and III were similar, despite the absence of 2-O-sulfation on substrate II. This emphasizes the importance of epimerization of the uronic acid for increasing chain flexibility, improving glycan binding. K202, R206, and R329 form hydrogen bonds and water bridges with IdoA/GlcA2S‑3, indicating their probable importance as selectors of sulfated uronic acid at this position. Reinforcing this idea, K202 and R206 did not bind the −3 substrate when GlcNS‑2 was 6-O-sulfated. H203, suggested to have a role in binding the −2 moiety and orienting the acceptor for catalysis,32 formed hydrogen bonds with the GlcNS of substrates II and III, while forming hydrophobic interactions with substrate IV, which only contains GlcNAc-GlcA. This suggests that H203 plays a critical role not as an active site residue but as an extended binding residue orienting the acceptor when the glucosamine is not 6-O-sulfated and binding to the −2 glucosamine in either acetylated or sulfated forms. Tornberg and colleagues previously reported that the mutation R323Q in the HS6ST isoform 1 (HS6ST1) gene is correlated with idiopathic hypogonadotropic hypogonadism (IHH) in humans.54 The corresponding HS6ST residue in zebrafish is R329.32,54 Our extended simulations show that R329 maintained both hydrogen bonds and/or water bridges when the substrate had GlcNS or GlcNAc at −2 (Figure S20, panels (−2)). However, when the substrate at −2 was GlcNS6S, R329 formed fewer (<20% of the dynamics course) hydrogen bonds as an acceptor of C6-O-sulfate, suggesting that this residue plays a role as a processivity gate toward the NRE of the polysaccharide. Beyond its role as a selector for NRE sulfation, R329 appears to play a role in allosterically regulating PAPS access to the active site. When analyzing the HS6STapo simulations, we identified that the loop containing R329 is highly mobile and, in the absence of a substrate, R329 formed a salt bridge with D192 (Video 1 in the Supporting Information). This loop closure occludes the PAPS binding site and reduces the pocket volume from 181 Å3 (open loop) to 6 Å3, as calculated by POVME 3.0.55 Thus, this salt bridge could function as the homotropic regulator controlling PAPS binding site access since, upon substrate binding, the R329/D192 bridge would be dissolved. This idea is strengthened after calculating the pairwise interaction energies using MM-GBSA between the R329-D192 pair during the apo simulations (−12.3 ± 0.2 kcal/mol) with R329 and GlcS+3/Idoa2S+4/GlcNS+5 (−4.9 ± 0.1, 15.7 ± 0.6, and 1.9 ± 0.3 kcal/mol, respectively) (Figure 7A). We hypothesized that the topology of R329/D192 residues influences PAPS binding and, consequently, glycan secondary modifications. To confirm this hypothesis experimentally, we conducted an in vitro site-directed mutagenesis of HS6ST R329 and D192. Mutations retaining the electric charge, D192E and R329K, were designed, as was a double mutant, D192E/R329K. Alanine substitutive mutants were also synthesized and expressed. Wild-type enzymes and mutants were incubated with a hexasaccharide substrate composed of repeating GlcNS-IdoA2S units and the sulfate donor PAPS, and thereafter, the product was analyzed using SAX-HPLC chromatography, as described in Methods in the Supporting Information. Two mutations show reduced activity; specifically, D192E showed a 50% reduction in activity and R329K showed a 43% reduction in activity in comparison to the wild-type enzyme. Substituting the conserved residues R329 and D192 individually to an alanine leads to a 95% reduction of 6-O-sulfation (Figure 7B). Strikingly, when we compared the sequence of HS6STs to the sequence of all verified Golgi sulfotransferases in the NCBI and UniProt databases, we found that these residues are conserved within the whole glycosaminoglycan sulfotransferase family (Figure 7C). This suggests that the mechanism by which this salt bridge is outcompeted by substrate binding allowing PAPS access is conserved among this family of proteins.
Figure 7.
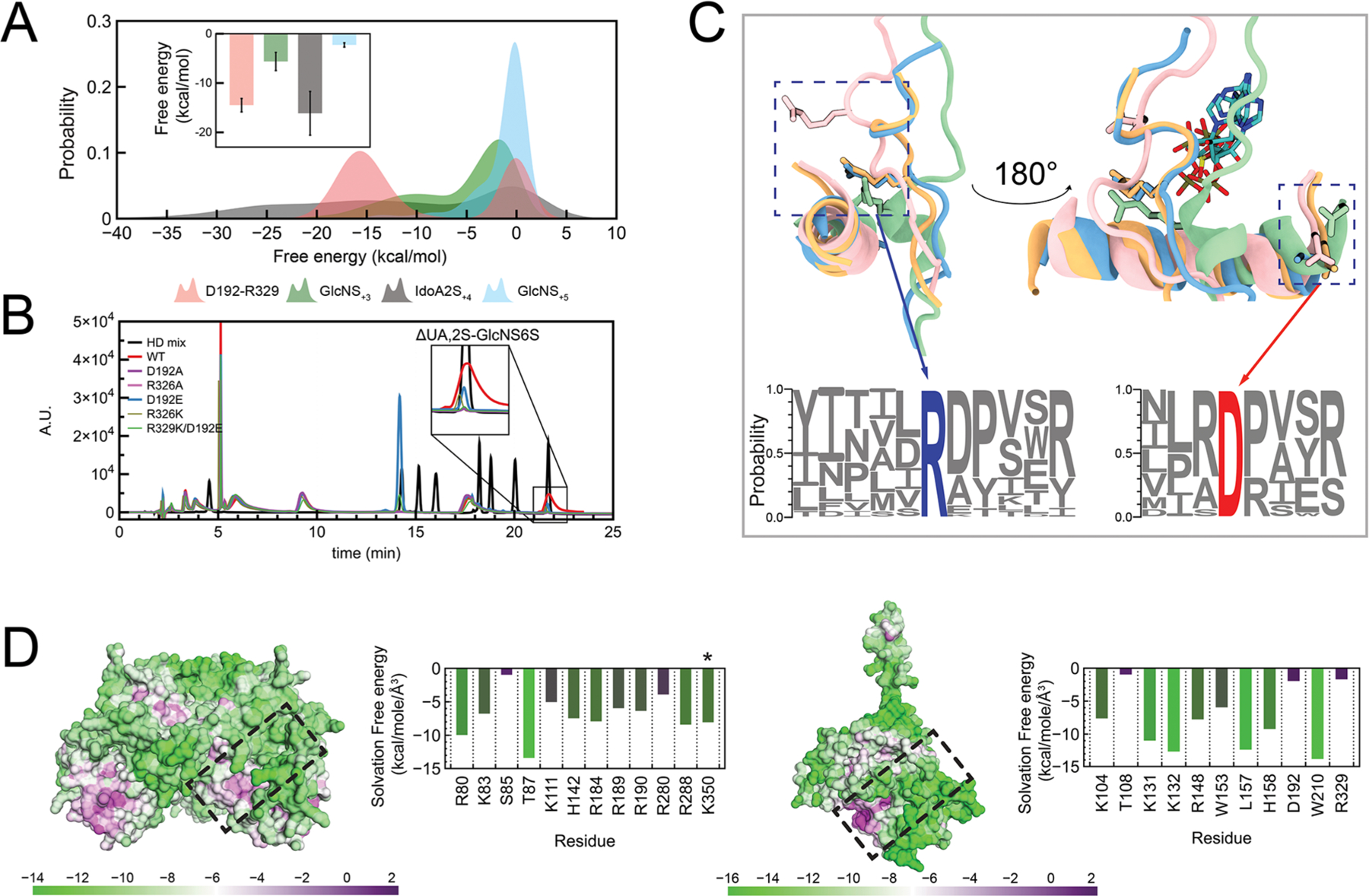
Primer on the shared topologies of heparin sulfotransferases. (A) Probability distribution of calculated pairwise energies during molecular dynamics simulations for (I) the substrate IdoA2S-GlcNS and (II) the D192-R329 salt bridge during HS6ST apoprotein simulations. The inset shows calculated averages. (B) SAX-HPLC analysis of total disaccharide composition after the action of R329/D192 mutants incubated with IdoA2S-GlcNS substrates. The insert highlights the formation of IdoA2S-GlcNS6S moieties. HD mix (black line), heparin disaccharide mix (Iduron, UK). (C) A salt bridge is conserved among glycan sulfotransferases, and its conformation change is triggered by substrate binding. Superposition of HSSTs: light blue, HSNST; pink, HS2ST; orange, HS3ST; green, HS6ST. The bottom logo plot highlights ARG (blue) and ASP (red) conservation among all HSSTs known to date. (D) Calculated solvation free energies on the surface of the binding interfaces of HS2ST (left) and HS6ST (right) as a measure of their hydrophobicity. Tables show the per-residue average hydrophobicity for each apoprotein enzyme. Table colors range from more hydrophobic (purple) to more hydrophilic (green). The asterisk denotes C-terminal “prime” residues.
HS2ST and HS6ST Share Key Molecular Features but Differ Drastically in Their Substrate Recognition.
While our data suggest many similarities in the catalytic mechanisms of HS2ST and HS6ST, we found that the substrate binding modes varied considerably. Since the sulfate transfer requires a hydrophobic environment to protect PAPS from hydrolysis, we calculated the electrostatic surface potentials of both HS2ST and HS6ST using adaptive Poisson–Boltzmann solver (APBS).56 As shown in Figure S21A,B, both enzymes present an extended patch of positive amino acids within their binding cleft. Because the positively charged patch (blue regions) is spatially close to the hydrophobic active site (white patches), we investigated the interplay between these two regions with antagonistic characteristics. We could observe that both sulfotransferase enzymes present a large, discrete patch of positively charged residues, indicating that the macromolecular interface of these enzymes is dominated by electrostatic interactions. This topology is similar to protein–DNA binding systems, where one-dimensional diffusion (sliding) is promoted mainly by electrostatic forces, while specific interactions involving hydrogen bonds and van der Waals forces are absent.57,58
Water molecules that are bound at the ligand binding region of the enzyme can have a crucial effect on the overall ligand binding energy, since they must be displaced first to then allow the ligand to access the binding site.57,59,60 We computed the displacement of perturbed water upon binding by the grid on the basis of the Grid Inhomogeneous Solvation Theory (GIST) for HS2STapo and HS6STapo enzymes.60 The resulting calculated free energy of solvation for each residue within the binding cleft provides quantitative data on hydrophobicity and solvent accessibility, and the solvation free energy grid is mapped onto the protein surfaces in Figure 7D. Hydrophobicity is a metric to calculate the solvation free energy that would need to be replaced by a ligand binding into the enzyme substrate binding region. Green patches show residues that require more energy to be desolvated (higher negative solvation free energy), while water molecules can be more easily displaced from purple regions. A comparison of localized hydrophobicity within both HS2ST and HS6ST sulfotransferase binding interfaces demonstrates the hydrophobic nature of the PAPS binding site. HS2ST S85 and HS6ST T108, known to bind and orient PAPS toward catalysis, are the interface binding site residues displaying higher hydrophobicity. HS2ST ARG/LYS clamps show a similar solvation energy (−6.0 kcal/mol), and T87, a conserved residue among all sulfotransferases, is critical for maintaining the PAPS phosphate equatorially positioned showing the highest hydrophobicity within the protein interface (−0.6 kcal/mol). HS6ST W210 and the loop K131/K132 display the highest solvent exposure (−12.0 kcal/mol), while W153 maintains a hydrophobic environment around the catalytic H158.
We speculated that the strong H-bonding of W210 to the glucosamine acceptor reduces the ability of HS6STs to discriminate among oligosaccharides on the basis of their specific sulfation and epimerization patterns. To counteract this, the K131/K132 loop acts as a “preference” selector. This role is emphasized when we look at the anatomy of the floppy K131/K132 loop, which is N- and C-terminally flanked by two prolines—P123 and P139—which are conserved among all HS6STs. The presence of prolines flanking protein loops has been shown to drastically limit loop conformations61,62 and to reduce loop elasticity,63,64 a phenomenon termed “focused motional freedom”. These studies have shown that rigidity is introduced by the proline pyrrolidine ring, drastically limiting the backbone dihedral angle to 90°. As the polysaccharides are gradually sulfated during the biosynthetic process, negative charges build up and the polymer hydrophilicity increases. Intuitively, this should also increase the binding entropy of substrates to the positively charged binding sites. Because HS6ST acts relatively late within HS biosynthesis, this accumulating negative charge causes a difficult task for the enzyme—HS6ST needs to recognize its substrates with a reasonable degree of flexibility and promiscuity without irreversibly binding them.
The events leading to sulfate transfer are remarkably different for HS2ST. We can assume that to achieve processivity the enzyme should bind moderately to its polymer substrate. Upon binding of the substrate, the swapped C-terminal tail of the HS2ST adjacent monomer bends over and orients the substrate—this compound enzyme interface allows it to completely bury the oligosaccharide, increasing its interaction surface to maintain the large oligosaccharide in a semi-enclosed position, facilitating its processivity. The difference between apo conformations and substrate-bound HS2STs poses indicates the formation of specific and extensive interactions between HS2ST and its cognate substrates, enabling specific discrimination between anomeric or epimeric moieties and size. The influence of motion in cleft/active site recognition has also been described for other glycan binding proteins.65–67
CONCLUSION
Here we analyzed the substrate-binding modes and catalysis of the heparan sulfate biosynthetic sulfotransferases HS2ST and HS6ST. Our QM/MM results highlight common features between the two enzymes—they promote the sulfate transfer from PAPS to the respective acceptor unit in a SN2-like mechanism with a histidine as the catalytic base and a lysine, stabilizing the formation of the 5′-phosphate group on PAPS. Our molecular dynamics simulations, however, revealed important differences between HS2ST and HS6ST during substrate recognition, evidencing the molecular determinants required for each enzyme to selectively recognize and distinguish among a pool of substrates.
For HS2ST, we investigated the affinity of this enzyme toward IdoA or GlcA, starting from the available crystal structure which presents IdoA in the 4C1 conformation; then, we analyzed the effect of different uronic acid puckers within the substrate. Overall, we identified, by in silico methods, three structural determinants for substrate specificity: an electrostatic fork made of R184/R189 and R288, two molecular clamps (K111/R190 and R288/K350′), and R80, which anchors the glucosamine at position +2. The group of three arginines forms a positively charged fork that anchors the ligand by entrapping the C6 carboxyl group of the uronic acid acceptor unit. This arginine fork is able to recognize GlcA or IdoA epimers and their puckers but prefers the carboxyl group of 1C4-IdoA that fits properly within the electrostatic trap. The two molecular clamps identified (K111/R190 and R288/K350′) position the ligand precisely within the active site, while R80 is responsible for recognizing the GlcN2S unit at the +2 position, providing molecular evidence for the N-sulfation as a prerequisite for HS2ST binding and subsequent 2-O-sulfation.34
The role of R189 in the steric hindrance of GlcA was further investigated by in vitro enzymatic assays, suggesting that this residue is a vital component of the fork which determines the increased preference of HS2ST for iduronate. By calculating binding affinities, we confirmed that HS2ST preferably recognizes IdoA over GlcA and, moreover, the enzyme senses the conformation of the sugar ring, binding to the most energetically favorable conformations (1C4/2S0 of IdoA). The preference of HS2ST for ligands with the most plausible sugar conformations was confirmed by the energetic profiles computed for the catalytic mechanism, which suggest that, even if the conformation flexibility of the enzyme allows the binding of different epimers/puckers, HS2ST preferably sulfates 1C4-IdoA over any other puckers and epimers. Overall, we provide evidence, at an atomic level, explaining how HS2ST displays a preference for IdoA over GlcA residues as acceptor units.
The HS6ST binding cleft is unique in comparison to HS2ST and other glycosaminoglycan sulfotransferases. Our data identify a set of exclusive structural determinants: W153, W210, and a R329/D192 salt bridge. The narrow binding cleft presents two solvent-exposed tryptophan residues (W153 and W210) that flank the catalytic histidine at the active site. Our data indicate that W153, with assistance of the K131/K132 loop, maintains active site hydrophobicity by repelling solvent molecules from the catalytic center. Our calculations suggest that water dynamics play an integral role in HS6ST substrate binding and dissociation, since the enzyme must compete with the bulk solvent to bind to a highly sulfated/hydrated substrate which is extremely hydrophilic. W210 helps bind the acceptor substrate by engaging a H-bond with the acceptor unit. Furthermore, we identified a salt bridge formed between R329 and D192 that blocks the cofactor entry of HS6ST. Upon substrate binding, this noncovalent bond breaks, allowing access to the PAPS donor. This bridge is conserved within the heparin/HS sulfotransferase family and, to the best of our knowledge, has not previously been described. This novel structural signature can act as a gate that opens, allowing PAPS to access its binding site, working as a homotropic regulator. This understanding has obvious implications on sulfotransferase design, opening questions for future investigations on how PAPS accesses the binding cleft of these enzymes.
Supplementary Material
ACKNOWLEDGMENTS
The authors acknowledge the use of the Opuntia Cluster and the advanced support from the Center of Advanced Computing and Data Science at the University of Houston and the Texas Advanced Computing Center (TACC) at The University of Texas at Austin for providing high-performance computing resources. URL: http://www.tacc.utexas.edu. The authors thank Francisco Corzana and Søren Balling Engelsen for providing the 2D-RDF source code and Michael Williams for helping with code development. Carbohydrate analysis was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences and Biosciences Division, under award #DE-SC001566 to Paarstoo Azadi.
Funding
This study was supported in part by funds from UH SeFAC (V.J.C.-T.), Mizutani Foundation (V.J.C.-T.), R01 EY029289 (V.J.C.-T.), and the European Union (Marie Skłodowska Curie Actions SUPA-HD 625451) (J.W.M.).
ABBREVIATIONS
- IdoA
α-l-iduronic acid
- GlcA
β-d-glucopyranuronic acid
- HS2ST
heparan sulfate 2-O-sulfotransferase
- HS6ST
heparan sulfate 6-O-sulfotransferase
- PAPS
3′-phosphoadenosine-5′-phosphosulfate
- HPLC
high-performance liquid chromatography
- NRE
nonreducing end
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.1c03088.
Cartesian coordinates for critical points (reactant, transition state, and product) obtained at a QM/MM level (ZIP)
Molecular dynamics of HS6ST shows the formation of a salt bridge between D192 and R329 (MP4)
Methods, a detailed analysis of all enzyme complex conditions investigated, figures and tables (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acscatal.1c03088
The authors declare no competing financial interest.
Contributor Information
Tarsis Ferreira Gesteira, College of Optometry, University of Houston, Houston, Texas 77004, United States.
Tainah Dorina Marforio, Dipartimento di Chimica “Giacomo Ciamician”, Università di Bologna, Bologna 40126, Italy.
Jonathan Wolf Mueller, Institute of Metabolism and Systems Research, University of Birmingham, Birmingham B15 2TT, U.K..
Matteo Calvaresi, Dipartimento di Chimica “Giacomo Ciamician”, Università di Bologna, Bologna 40126, Italy.
Vivien Jane Coulson-Thomas, College of Optometry, University of Houston, Houston, Texas 77004, United States.
REFERENCES
- (1).Nader HB; Dietrich CP; Buonassisi V; Colburn P Heparin sequences in the heparan sulfate chains of an endothelial cell proteoglycan. Proc. Natl. Acad. Sci. U. S. A 1987, 84 (11), 3565–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Gesteira TF; Coulson-Thomas VJ; Ogata FT; Farias EH; Cavalheiro RP; de Lima MA; Cunha GL; Nakayasu ES; Almeida IC; Toma L; Nader HB A novel approach for the characterisation of proteoglycans and biosynthetic enzymes in a snail model. Biochim. Biophys. Acta, Proteins Proteomics 2011, 1814 (12), 1862–9. [DOI] [PubMed] [Google Scholar]
- (3).Attreed M; Saied-Santiago K; Bulow HE Conservation of anatomically restricted glycosaminoglycan structures in divergent nematode species. Glycobiology 2016, 26 (8), 862–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Heparin Market Size, Share & Trends Analysis Report By Type (Low Molecular Weight, Ultra-low Molecule Weight) Grand View Research. https://www.grandviewresearch.com/industry-analysis/heparin-market; accessed December 15, 2019. [Google Scholar]
- (5).Gallagher JT; Walker A Molecular distinctions between heparan sulphate and heparin. Analysis of sulphation patterns indicates that heparan sulphate and heparin are separate families of N-sulphated polysaccharides. Biochem. J 1985, 230 (3), 665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Shriver Z; Capila I; Venkataraman G; Sasisekharan R Heparin and heparan sulfate: analyzing structure and microheterogeneity. Handb. Exp. Pharmacol 2012, 207 (207), 159–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Hileman RE; Fromm JR; Weiler JM; Linhardt RJ Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. BioEssays 1998, 20 (2), 156–67. [DOI] [PubMed] [Google Scholar]
- (8).Tykesson E; Mao Y; Maccarana M; Pu Y; Gao J; Lin C; Zaia J; Westergren-Thorsson G; Ellervik U; Malmstrom L; Malmstrom A Deciphering the Mode of Action of the Processive Polysaccharide Modifying Enzyme Dermatan Sulfate Epimerase 1 by Hydrogen-Deuterium Exchange Mass Spectrometry. Chem. Sci 2016, 7 (2), 1447–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Mandon E; Kempner ES; Ishihara M; Hirschberg CB A monomeric protein in the Golgi membrane catalyzes both N-deacetylation and N-sulfation of heparan sulfate. J. Biol. Chem 1994, 269 (16), 11729–33. [PubMed] [Google Scholar]
- (10).Li J; Hagner-McWhirter A; Kjellen L; Palgi J; Jalkanen M; Lindahl U Biosynthesis of heparin/heparan sulfate. cDNA cloning and expression of D-glucuronyl C5-epimerase from bovine lung. J. Biol. Chem 1997, 272 (44), 28158–63. [DOI] [PubMed] [Google Scholar]
- (11).Sugahara K; Kitagawa H Recent advances in the study of the biosynthesis and functions of sulfated glycosaminoglycans. Curr. Opin. Struct. Biol 2000, 10 (5), 518–27. [DOI] [PubMed] [Google Scholar]
- (12).Nagai N; Habuchi H; Esko JD; Kimata K Stem domains of heparan sulfate 6-O-sulfotransferase are required for Golgi localization, oligomer formation and enzyme activity. J. Cell Sci 2004, 117 (15), 3331–3341. [DOI] [PubMed] [Google Scholar]
- (13).Nader HB; Dietrich CP Effect of heparin sulfate fractions on coagulation and hemostasis. Exp. Biol. Med 1974, 146 (2), 504–8. [DOI] [PubMed] [Google Scholar]
- (14).Moon AF; Edavettal SC; Krahn JM; Munoz EM; Negishi M; Linhardt RJ; Liu J; Pedersen LC Structural analysis of the sulfotransferase (3-o-sulfotransferase isoform 3) involved in the biosynthesis of an entry receptor for herpes simplex virus 1. J. Biol. Chem 2004, 279 (43), 45185–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Nader HB; Buonassisi V; Colburn P; Dietrich CP Heparin stimulates the synthesis and modifies the sulfation pattern of heparan sulfate proteoglycan from endothelial cells. J. Cell. Physiol 1989, 140 (2), 305–10. [DOI] [PubMed] [Google Scholar]
- (16).Coulson-Thomas VJ The role of heparan sulphate in development: the ectodermal story. Int. J. Exp. Pathol 2016, 97 (3), 213–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Coulson-Thomas VJ; Chang SH; Yeh LK; Coulson-Thomas YM; Yamaguchi Y; Esko J; Liu CY; Kao W Loss of corneal epithelial heparan sulfate leads to corneal degeneration and impaired wound healing. Invest. Ophthalmol. Visual Sci 2015, 56 (5), 3004–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Coulson-Thomas VJ; Gesteira TF; Esko J; Kao W Heparan sulfate regulates hair follicle and sebaceous gland morphogenesis and homeostasis. J. Biol. Chem 2014, 289 (36), 25211–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Guibinga GH; Miyanohara A; Esko JD; Friedmann T Cell surface heparan sulfate is a receptor for attachment of envelope protein-free retrovirus-like particles and VSV-G pseudotyped MLV-derived retrovirus vectors to target cells. Mol. Ther 2002, 5 (5), 538–546. [DOI] [PubMed] [Google Scholar]
- (20).Thacker BE; Seamen E; Lawrence R; Parker MW; Xu Y; Liu J; Vander Kooi CW; Esko JD Expanding the 3-O-Sulfate Proteome–Enhanced Binding of Neuropilin-1 to 3-O-Sulfated Heparan Sulfate Modulates Its Activity. ACS Chem. Biol 2016, 11 (4), 971–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Gunal S; Hardman R; Kopriva S; Mueller JW Sulfation pathways from red to green. J. Biol. Chem 2019, 294 (33), 12293–12312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Merry CL; Bullock SL; Swan DC; Backen AC; Lyon M; Beddington RS; Wilson VA; Gallagher JT The molecular phenotype of heparan sulfate in the Hs2st−/− mutant mouse. J. Biol. Chem 2001, 276 (38), 35429–34. [DOI] [PubMed] [Google Scholar]
- (23).Kamimura K; Koyama T; Habuchi H; Ueda R; Masu M; Kimata K; Nakato H Specific and flexible roles of heparan sulfate modifications in Drosophila FGF signaling. J. Cell Biol 2006, 174 (6), 773–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Esko JD; Selleck SB Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu. Rev. Biochem 2002, 71, 435–71. [DOI] [PubMed] [Google Scholar]
- (25).Bullock SL; Fletcher JM; Beddington RS; Wilson VA Renal agenesis in mice homozygous for a gene trap mutation in the gene encoding heparan sulfate 2-sulfotransferase. Genes Dev 1998, 12 (12), 1894–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wilson VA; Gallagher JT; Merry CL Heparan sulfate 2-O-sulfotransferase (Hs2st) and mouse development. Glycoconjugate J 2002, 19 (4–5), 347–54. [DOI] [PubMed] [Google Scholar]
- (27).Bethea HN; Xu D; Liu J; Pedersen LC Redirecting the substrate specificity of heparan sulfate 2-O-sulfotransferase by structurally guided mutagenesis. Proc. Natl. Acad. Sci. U. S. A 2008, 105 (48), 18724–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Atha DH; Stephens AW; Rosenberg RD Evaluation of critical groups required for the binding of heparin to antithrombin. Proc. Natl. Acad. Sci. U. S. A 1984, 81 (4), 1030–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Habuchi H; Tanaka M; Habuchi O; Yoshida K; Suzuki H; Ban K; Kimata K The occurrence of three isoforms of heparan sulfate 6-O-sulfotransferase having different specificities for hexuronic acid adjacent to the targeted N-sulfoglucosamine. J. Biol. Chem 2000, 275 (4), 2859–68. [DOI] [PubMed] [Google Scholar]
- (30).Liu R; Liu J Enzymatic placement of 6-O-sulfo groups in heparan sulfate. Biochemistry 2011, 50 (20), 4382–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Jemth P; Smeds E; Do AT; Habuchi H; Kimata K; Lindahl U; Kusche-Gullberg M Oligosaccharide library-based assessment of heparan sulfate 6-O-sulfotransferase substrate specificity. J. Biol. Chem 2003, 278 (27), 24371–6. [DOI] [PubMed] [Google Scholar]
- (32).Xu Y; Moon AF; Xu S; Krahn JM; Liu J; Pedersen LC Structure Based Substrate Specificity Analysis of Heparan Sulfate 6-O-Sulfotransferases. ACS Chem. Biol 2017, 12 (1), 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Thieker DF; Xu Y; Chapla D; Nora C; Qiu H; Felix T; Wang L; Moremen KW; Liu J; Esko JD; Woods RJ Downstream Products are Potent Inhibitors of the Heparan Sulfate 2-O-Sulfotransferase. Sci. Rep 2018, 8 (1), 11832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Liu C; Sheng J; Krahn JM; Perera L; Xu Y; Hsieh PH; Dou W; Liu J; Pedersen LC Molecular mechanism of substrate specificity for heparan sulfate 2-O-sulfotransferase. J. Biol. Chem 2014, 289 (19), 13407–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Xu D; Song D; Pedersen LC; Liu J Mutational study of heparan sulfate 2-O-sulfotransferase and chondroitin sulfate 2-O-sulfotransferase. J. Biol. Chem 2007, 282 (11), 8356–67. [DOI] [PubMed] [Google Scholar]
- (36).Tvaroška I; Kožár T Theoretical studies on the conformation of saccharides: Part VI. Influence of the pyranoid ring shape on the conformational properties of the glycosidic linkage and on the magnitude of the exoanomeric effect. J. Mol. Struct.: THEOCHEM 1985, 123 (1), 141–154. [Google Scholar]
- (37).Berglund J; Angles d’Ortoli T; Vilaplana F; Widmalm G; Bergenstrahle-Wohlert M; Lawoko M; Henriksson G; Lindstrom M; Wohlert J A molecular dynamics study of the effect of glycosidic linkage type in the hemicellulose backbone on the molecular chain flexibility. Plant J 2016, 88 (1), 56–70. [DOI] [PubMed] [Google Scholar]
- (38).Plazinski W; Lonardi A; Hunenberger PH Revision of the GROMOS 56A6(CARBO) force field: Improving the description of ring-conformational equilibria in hexopyranose-based carbohydrates chains. J. Comput. Chem 2016, 37 (3), 354–65. [DOI] [PubMed] [Google Scholar]
- (39).Hsieh PH; Thieker DF; Guerrini M; Woods RJ; Liu J Uncovering the Relationship between Sulphation Patterns and Conformation of Iduronic Acid in Heparan Sulphate. Sci. Rep 2016, 6, 29602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Spiwok V; Kralova B; Tvaroska I Modelling of beta-D-glucopyranose ring distortion in different force fields: a metadynamics study. Carbohydr. Res 2010, 345 (4), 530–7. [DOI] [PubMed] [Google Scholar]
- (41).Marforio TD; Giacinto P; Bottoni A; Calvaresi M Computational Evidence for the Catalytic Mechanism of Tyrosylprotein Sulfotransferases: A Density Functional Theory Investigation. Biochemistry 2015, 54 (28), 4404–4410. [DOI] [PubMed] [Google Scholar]
- (42).Kadam RU; Garg D; Schwartz J; Visini R; Sattler M; Stocker A; Darbre T; Reymond JL CH-pi ″T-shape″ interaction with histidine explains binding of aromatic galactosides to Pseudomonas aeruginosa lectin LecA. ACS Chem. Biol 2013, 8 (9), 1925–30. [DOI] [PubMed] [Google Scholar]
- (43).Hudson KL; Bartlett GJ; Diehl RC; Agirre J; Gallagher T; Kiessling LL; Woolfson DN Carbohydrate-Aromatic Interactions in Proteins. J. Am. Chem. Soc 2015, 137 (48), 15152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Stankovic IM; Blagojevic Filipovic JP; Zaric SD Carbohydrate - Protein aromatic ring interactions beyond CH/pi interactions: A Protein Data Bank survey and quantum chemical calculations. Int. J. Biol. Macromol 2020, 157, 1–9. [DOI] [PubMed] [Google Scholar]
- (45).Montalvillo-Jimenez L; Santana AG; Corzana F; Jimenez-Oses G; Jimenez-Barbero J; Gomez AM; Asensio JL Impact of Aromatic Stacking on Glycoside Reactivity: Balancing CH/pi and Cation/pi Interactions for the Stabilization of Glycosyl-Oxocarbenium Ions. J. Am. Chem. Soc 2019, 141 (34), 13372–13384. [DOI] [PubMed] [Google Scholar]
- (46).Kakuta Y; Sueyoshi T; Negishi M; Pedersen LC Crystal structure of the sulfotransferase domain of human heparan sulfate N-deacetylase/N-sulfotransferase 1. J. Biol. Chem 1999, 274 (16), 10673–6. [DOI] [PubMed] [Google Scholar]
- (47).Huntley JJ; Fast W; Benkovic SJ; Wright PE; Dyson HJ Role of a solvent-exposed tryptophan in the recognition and binding of antibiotic substrates for a metallo-beta-lactamase. Protein Sci 2003, 12 (7), 1368–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Tobola F; Lelimousin M; Varrot A; Gillon E; Darnhofer B; Blixt O; Birner-Gruenberger R; Imberty A; Wiltschi B Effect of Noncanonical Amino Acids on Protein-Carbohydrate Interactions: Structure, Dynamics, and Carbohydrate Affinity of a Lectin Engineered with Fluorinated Tryptophan Analogs. ACS Chem. Biol 2018, 13 (8), 2211–2219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Mueller JW; Idkowiak J; Gesteira TF; Vallet C; Hardman R; van den Boom J; Dhir V; Knauer SK; Rosta E; Arlt W Human DHEA sulfation requires direct interaction between PAPS synthase 2 and DHEA sulfotransferase SULT2A1. J. Biol. Chem 2018, 293 (25), 9724–9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Foster PA; Mueller JW SULFATION PATHWAYS: Insights into steroid sulfation and desulfation pathways. J. Mol. Endocrinol 2018, 61 (2), T271–t283. [DOI] [PubMed] [Google Scholar]
- (51).Gray HB; Winkler JR Hole hopping through tyrosine/tryptophan chains protects proteins from oxidative damage. Proc. Natl. Acad. Sci. U. S. A 2015, 112 (35), 10920–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Barducci A; Bonomi M; Parrinello M Metadynamics. Wiley Interdiscip. Rev.: Comput. Mol. Sci 2011, 1 (5), 826–843. [Google Scholar]
- (53).Barducci A; Bonomi M; Parrinello M Linking well-tempered metadynamics simulations with experiments. Biophys. J 2010, 98 (9), L44–L46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Tornberg J; Sykiotis GP; Keefe K; Plummer L; Hoang X; Hall JE; Quinton R; Seminara SB; Hughes V; Van Vliet G; Van Uum S; Crowley WF; Habuchi H; Kimata K; Pitteloud N; Bülow HE Heparan sulfate 6-O-sulfotransferase 1, a gene involved in extracellular sugar modifications, is mutated in patients with idiopathic hypogonadotrophic hypogonadism. Proc. Natl. Acad. Sci. U. S. A 2011, 108 (28), 11524–11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Wagner JR; Sorensen J; Hensley N; Wong C; Zhu C; Perison T; Amaro RE POVME 3.0: Software for Mapping Binding Pocket Flexibility. J. Chem. Theory Comput 2017, 13 (9), 4584–4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Jurrus E; Engel D; Star K; Monson K; Brandi J; Felberg LE; Brookes DH; Wilson L; Chen J; Liles K; Chun M; Li P; Gohara DW; Dolinsky T; Konecny R; Koes DR; Nielsen JE; Head-Gordon T; Geng W; Krasny R; Wei GW; Holst MJ; McCammon JA; Baker NA Improvements to the APBS biomolecular solvation software suite. Protein Sci 2018, 27 (1), 112–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Record MT Jr.; Ha JH; Fisher MA Analysis of equilibrium and kinetic measurements to determine thermodynamic origins of stability and specificity and mechanism of formation of site-specific complexes between proteins and helical DNA. Methods Enzymol 1991, 208, 291–343. [DOI] [PubMed] [Google Scholar]
- (58).von Hippel PH From ″simple″ DNA-protein interactions to the macromolecular machines of gene expression. Annu. Rev. Biophys. Biomol. Struct 2007, 36, 79–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Kovacevic B; Baric D; Babic D; Bilic L; Hanzevacki M; Sandala GM; Radom L; Smith DM Computational Tale of Two Enzymes: Glycerol Dehydration With or Without B12. J. Am. Chem. Soc 2018, 140 (27), 8487–8496. [DOI] [PubMed] [Google Scholar]
- (60).Nguyen CN; Young TK; Gilson MK Grid inhomogeneous solvation theory: hydration structure and thermodynamics of the miniature receptor cucurbit[7]uril. J. Chem. Phys 2012, 137 (4), 044101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Van Agthoven JF; Shams H; Cochran FV; Alonso JL; Kintzing JR; Garakani K; Adair BD; Xiong JP; Mofrad MRK; Cochran JR; Arnaout MA Structural Basis of the Differential Binding of Engineered Knottins to Integrins alphaVbeta3 and alpha5beta1. Structure 2019, 27 (9), 1443–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Pavlicek J; Coon SL; Ganguly S; Weller JL; Hassan SA; Sackett DL; Klein DC Evidence that proline focuses movement of the floppy loop of arylalkylamine N-acetyltransferase (EC 2.3.1.87). J. Biol. Chem 2008, 283 (21), 14552–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Yu H; Yan Y; Zhang C; Dalby PA Two strategies to engineer flexible loops for improved enzyme thermostability. Sci. Rep 2017, 7, 41212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Yu H; Zhao Y; Guo C; Gan Y; Huang H The role of proline substitutions within flexible regions on thermostability of luciferase. Biochim. Biophys. Acta, Proteins Proteomics 2015, 1854 (1), 65–72. [DOI] [PubMed] [Google Scholar]
- (65).Cuneo MJ; Changela A; Warren JJ; Beese LS; Hellinga HW The crystal structure of a thermophilic glucose binding protein reveals adaptations that interconvert mono and di-saccharide binding sites. J. Mol. Biol 2006, 362 (2), 259–70. [DOI] [PubMed] [Google Scholar]
- (66).Cuneo MJ; Changela A; Beese LS; Hellinga HW Structural adaptations that modulate monosaccharide, disaccharide, and trisaccharide specificities in periplasmic maltose-binding proteins. J. Mol. Biol 2009, 389 (1), 157–66. [DOI] [PubMed] [Google Scholar]
- (67).Shukla S; Bafna K; Gullett C; Myles DAA; Agarwal PK; Cuneo MJ Differential Substrate Recognition by Maltose Binding Proteins Influenced by Structure and Dynamics. Biochemistry 2018, 57 (40), 5864–5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.