

Abstract
Introduction
Though vascular smooth muscle cells adopt an osteogenic phenotype during pathological vascular calcification, clinical studies note an inverse correlation between bone mineral density and arterial mineral—also known as the calcification paradox. Both processes are mediated by extracellular vesicles (EVs) that sequester calcium and phosphate. Calcifying EV formation in the vasculature requires caveolin-1 (CAV1), a membrane scaffolding protein that resides in membrane invaginations (caveolae). Of note, caveolin-1-deficient mice, however, have increased bone mineral density. We hypothesized that caveolin-1 may play divergent roles in calcifying EV formation from vascular smooth muscle cells (VSMCs) and osteoblasts (HOBs).
Methods
Primary human coronary artery VSMCs and osteoblasts were cultured for up to 28 days in an osteogenic media. CAV1 expression was knocked down using siRNA. Methyl β-cyclodextrin (MβCD) and a calpain inhibitor were used, respectively, to disrupt and stabilize the caveolar domains in VSMCs and HOBs.
Results
CAV1 genetic variation demonstrates significant inverse relationships between bone-mineral density (BMD) and coronary artery calcification (CAC) across two independent epidemiological cohorts. Culture in osteogenic (OS) media increased calcification in HOBs and VSMCs. siRNA knockdown of CAV1 abrogated VSMC calcification with no effect on osteoblast mineralization. MβCD-mediated caveolae disruption led to a 3-fold increase of calcification in VSMCs treated with osteogenic media (p < 0.05) but hindered osteoblast mineralization (p < 0.01). Conversely, stabilizing caveolae by calpain inhibition prevented VSMC calcification (p < 0.05) without affecting osteoblast mineralization. There was no significant difference in CAV1 content between lipid domains from HOBs cultured in OS and control media.
Conclusion
Our data indicate fundamental cellular-level differences in physiological and pathophysiological mineralization mediated by CAV1 dynamics. This is the first study to suggest that divergent mechanisms in calcifying EV formation may play a role in the calcification paradox.
Supplementary Information
The online version contains supplementary material available at 10.1007/s12195-023-00779-7
Keywords: (3-10) Vascular smooth muscle cells, Osteoblasts, Caveolae, Extracellular vesicles, Matrix vesicles, Calpain, Methyl-β-cyclodextrin, Cardioinformatics
Introduction
Cardiovascular diseases represent the global leading cause of morbidity and mortality. Cardiovascular calcification is the most significant predictor of these cardiovascular events [1, 2]. The presence of bone-like mineral in vasculature increases cardiac load required to move blood through the systemic circulation. Furthermore, it may lead to mechanical stress in atherosclerotic plaques, promoting plaque rupture events that result in heart attacks. In pathologies, including atherosclerosis and chronic kidney disease, vascular smooth muscle cells (VSMCs) undergo an osteogenic differentiation [3–5] and release calcifying extracellular vesicles (EVs), imitating physiological bone mineralization by osteoblasts (HOBs).
During bone formation, HOBs release matrix vesicles (MVs), small organelles enriched with osteogenic components, which serve as nucleation sites for early mineral formation within the collagen-based extracellular matrix (ECM) [6, 7]. Crucial components in MVs include free calcium and phosphate and tissue non-specific alkaline phosphatase (TNAP), a well-studied enzyme in calcification [8]. TNAP hydrolyzes pyrophosphate, a potent inhibitor of mineralization, releasing free phosphate for mineral nucleation [3, 9]. Osteogenic VSMCs similarly release calcifying EVs, that initiate calcification in the collagen-rich ECM of the vasculature [8]. Several studies reported similarities between cardiovascular calcification and bone metabolism and the role of osteogenic EVs in regulating the pathological calcification process within cardiovascular tissues [4, 8, 10].
Though bone and vascular mineralization processes share many commonalities, the appearance of cardiovascular calcification correlates highly with a reduction in bone mineral density (BMD) [11], a phenomenon referred to as the "calcification paradox." The mechanisms underlying the calcification paradox remain unclear. Previous studies associate calcification paradox with an imbalanced phosphate/calcium content due to bone loss [2, 12, 13] and/or increased systemic inflammation that promotes simultaneous pathological remodeling in both tissues [14–17]. Interestingly, modulation of a membrane scaffolding protein, caveolin-1 (CAV1), leads to divergent mineralization outcomes in these tissues [18]. Mice with global CAV1 knockout exhibit increased bone mineral density [19], while knockdown of CAV1 in VSMC cultures decreases in-vitro calcification [3]. These effects are likely unrelated to changes in systemic distribution of mineral components or inflammation.
Dynamics of key proteins in membrane domains prior to the liberation of osteogenic vesicles are poorly understood in VSMCs and HOBs. Despite the integral role of CAV1 in VSMC EV biogenesis [2], its role in HOB MV formation has not been studied. CAV1 is anchored to lipid rafts in the caveolar domain, flask-shaped invaginations of the plasma membrane that regulate membrane tension and cellular vesicle trafficking [20]. We recently showed that during mineralization, levels of CAV1 decrease in HOB MVs, whereas CAV1 increases in VSMC calcifying EVs [18]. Here, we further assessed the participation of CAV1 in calcification of VSMCs and HOBs by modulating caveolae stability using methyl-β-cyclodextrin and calpain inhibitor. Methyl-β-cyclodextrin disrupts membrane dynamics by removing cholesterol from the lipid rafts, resulting in the loss of caveolar domains in which CAV1 reside [21]. Conversely, calpain inhibitor strengthens the localization of CAV1 to caveolar domains by preventing cleavage of filament A [22, 23]. Our data indicate that either manipulation of CAV1 trafficking in osteogenic cultures leads to divergent mineralization outcomes in VSMCs and HOBs.
Methods
Human Genetics
To preliminarily assess the role of CAV1 function in the calcification paradox in humans, we investigated the direction of effect that CAV1 genetic variation had on bone-mineral density (BMD) and coronary artery calcification (CAC). Genome-wide association studies (GWAS) summary statistics for BMD for 426,824 individuals in the UK Biobank, spanning 13,705,641 single nucleotide polymorphisms (SNPs), are sourced from Morris et al. 2018 [24]. GWAS summary statistics for CAC are determined from 5771 individuals in the Multi-Ethnic Study of Atherosclerosis (MESA) [25]. The GWAS for CAC is conducted using age, sex, ethnicity of participants, and MESA study site as covariates. GWAS is conducted using plink [26]. For each SNP present in both BMD and CAC GWAS populations, the direction of effect is compared and the number of SNPs with opposite directions of effect for BMD and CAC are considered evidence for differential effects of CAV1 function between the two phenotypes; the probability of observing the number of SNPs with opposite direction of effect is calculated using a binomial distribution. A range of distances between 0 and 1 Mb away from the coding region of the CAV1 gene are included in our analysis to include potential cis-eQTLs that modulate CAC/BMD via CAV1. All heatmaps are generated with shinyheatmap [27].
Osteogenic Stimulation and In-vitro Calcification
Primary human coronary artery vascular smooth muscle cells (VSMCs, ATCC, PCS-100- 021) were cultured using vascular smooth muscle cell media and growth kit (ATCC, PCS- 100-042). VSMCs (passage 4-6) were harvested using 0.05% trypsin-EDTA solution (Caisson Labs, TRL04) and seeded with a density of 26,320 cells.cm-2 and incubated for 72 hours at 37 °C, 5% CO2 with controlled humidity prior to treatment. VSMCs were treated with either control media, consisting of DMEM (HyClone, SH30022.01), 10% v/v bovine calf serum (iron-supplemented, R&D Systems, S11950), and 1% v/v penicillin- streptomycin (Gibco, 15070-063), or with an osteogenic media (OS) optimized to induce calcification [4, 28]. OS media were supplemented with 10 mM β-glycerophosphate (Sigma, 13408-09-8), 0.1 mM L-ascorbic acid (Sigma, 113170-55-1), and 10 nM dexamethasone (Sigma, 50-02-2). To assess the role of various inhibitors, Calpain inhibitor (5 μM, Cayman Chemical Company, 14921) and methyl-β-cyclodextrin (mβCD, 1 mM, Cayman Chemical Company, 21633), were added to the OS media. An equal volume of the vehicle was added to the control and OS groups for each inhibitor. We found that 28 days in OS culture media led to robust calcification by VSMCs; therefore, all cultures (n = 3, independent donors, male and female) were treated for 28 days and media were replaced every three days.
Osteoblasts (from human fetus, hFOB 1.19, ATCC, CRL-11372) were cultured and grown in DMEM containing 10% v/v bovine calf serum and 1% v/v penicillin-streptomycin. Osteoblasts (passage 4-6) were harvested using 0.25% trypsin-EDTA solution (Caisson Labs, TRL01), seeded with a density of 5,200 cell.cm-2, and incubated for 24 hours at 37 °C and 5% CO2 with controlled humidity. The cells were treated in three groups of control, OS, and OS supplemented with inhibitors, Calpain inhibitor (5 μM) and methyl-β-cyclodextrin (1 mM), for 21 days and media were changed every three days.
Alizarin Red S Staining and Quantification
At the end of experiments (28 and 21 days of treatment for VSMCs and HOBs, respectively), media were removed, and the cells were fixed using formalin (10%, Fisher Chemical, SF100) for 15 min. To visualize in-vitro calcification, Alizarin Red S stain (ARS, Ricca, 500-32) was added to the wells and incubated for 30 min at room temperature. The stain was then removed, and the cells were washed three times with milliQ water. To quantify the in-vitro calcification, ARS stain was extracted using acetic acid (1.67 M, Fisher Chemical, A38S) on a shaker. After 30 min, the supernatants were collected, briefly vortexed, and heated at 85 °C for 10 min. The samples were then cooled on ice for 5 min and centrifuged at 20,000 × g for 15 min to remove debris. Sample absorbance of 405 nm light was measured using a multi-mode reader (BioTek, Synergy HTX).
Alkaline Phosphatase Activity Assay
To assess the activity of cellular tissue non-specific alkaline phosphatase (TNAP), a colorimetric assay kit (BioVision, K412) was used. VSMCs (n = 3) and HOBs (n = 3) following 14 days of treatment with control, OS, and OS plus any of the inhibitors, were lysed in 120 μL assay buffer. Each sample (80 uL) was mixed with 50 μL of 5 mM pNPP solution and incubated for 60 min at 25 °C. The colorimetric change resulting from the reaction was detected using a plate reader to measure absorbance at 405 nm. The results were normalized to the total protein for associated samples measured by a BCA protein assay (BioVision, K813).
Lipid Density Gradient
Following 14 days of culture, VSMCs and HOBs were lysed in HEPES lysis buffer, composed of 25 mM HEPES-HCl, 150 mM NaCl, 1 mM EDTA, 100 mM PMSF, protease inhibitor (Thermo Fisher, PIA32965), and 1% Triton X. A volume of 1 ml of lysis buffer to the plate and cells were scraped off using a scraper. Cell lysates were transferred to an ice-cold microcentrifuge tube to incubate for 30 minutes. At this point, cell lysates were either immediately fractioned via density gradient or stored at − 80 °C. The density gradient column was prepared by adding cell lysates and 5 layers of Optiprep (Sigma, D1556) with concentrations of 35, 30, 25, 20, and 0 percent, from the bottom to the top, to the final volume of 1.2 mL. Samples were then centrifuged at ~ 200,000 × g for 4 hours at 4 °C. After centrifugation, seven 160-uL fractions were collected from top to bottom of the gradient column. Samples were stored at − 80 °C for later analysis.
Gel Electrophoresis and Protein Immunoblotting
VSMCs and HOBs were lysed in RIPA lysis and extraction buffer supplemented with protease inhibitor. After adding Laemmli SDS-sample buffer (1:4 v/v, Boston BioProducts, BP-110R) to each lysate, the samples were denatured at 100 °C for 10 min, loaded into 7.5-12% 1-mm SDS-PAGE gel (15 to 20 μg protein per lane), and run at 170 V. The proteins were then transferred to Trans-Blot turbo nitrocellulose membranes (BIO-RAD, 1704158) at 25 V for 7 min. To quantify the total protein, the membranes were stained using 2% w/v Ponceau stain (Alfa Aesar, AAJ6074409) for 20 min, followed by one wash with 5% acetic acid and milliQ water for 5 min. After imaging, the intensity of each lane was measured in ImageJ for total protein normalization. Membranes were blocked with 5% w/v bovine serum albumin (HyClone, SH30574.01) in TBS-Tween (1X) for 1 hour. The membranes were incubated with primary antibodies of interest, including CAV1 (1:200, Abcam, ab2910) and TNAP (1:200, Invitrogen, 702454) overnight at 4 °C. After three washes with TBS-Tween (1X), the membranes were incubated with secondary antibody (1:1000, Li-Cor) for 1 hour, followed by three washes with TBS-Tween (1X). The protein bands were visualized with Odyssey CLx scanner (Li-Cor) and quantified using Image Studio Lite software (Li-Cor).
RNA interference
To assess the role of CAV1 in in-vitro mineralization of HOBs, CAV1 was knocked down in either control or OS cultures. RNA silencing was perfumed using 20 nM validated CAV1 siRNA (Ambion, 4390824) and On-target plus non-targeting pool (negative control, Dharmacon, D-001810-10-20). Following the culture of the HOBs, DharmaFECT (Dharmacon, T-2001-02) was used for CAV1 silencing. The siRNA and media were replaced every three days for 21 days.
Statistics
Data are presented as the mean of independent replications, and error bars represent the standard error of the mean. The reported n values represent independent biological replicates. Statistical significance between groups was calculated using a Mann-Whitney U test or a Kruskal-Wallis ANOVA in GraphPad Prism 8. A p-value less than 0.05 was considered statistically significant.
Results
Previous experimental studies suggest that modulation of CAV1 leads to divergent mineralization responses in bone and vascular tissues. To determine the relevance of CAV1 in the calcification paradox in humans, we started by analyzing SNPs near the CAV1 locus and assessed reported BMD and CAC associated with each SNP (Supplemental Table 1). Genetic variation within CAV1 largely demonstrates an inverse relationship between BMD and CAC (Fig. 1). Table 1 shows the location and BMD/CAC effects of SNPs between the transcription start and stop sites of the CAV1 gene. The direction of effect that SNPs near CAV1 between BMD and CAC is evaluated over a range of p-value thresholds for inclusion of SNPs, as well as a range of distances from the CAV1 coding region. We find that with the exception of analyses including SNPs only within CAV1 with p-value thresholds less than 0.5, all other analyses (36/40) suggest enrichment in SNPs with inverse BMD/CAC effects (p < 0.05). We note that despite the lack of statistical significance at stringent p-value inclusion thresholds and only SNPs within the CAV1 coding region, the p-values are close to significance (p = 0.0625) and a majority of the SNPs have inverse relationships between BMD and CAC, suggesting that the lack of significance is due to the lower number of SNPs rather than the lack of true signal.
Fig. 1.

Row labels are p-values for inclusion of SNPs and column labels are distance upstream and downstream of the CAV1 coding region where SNPs are included. Heatmaps illustrate (A) − log(p-values) of observed number of inversely associated SNPs between BMD and CAC for B percentage of included SNPs with inverted directions between BMD and CAC and C total counts of included SNPs. For ease of viewing, the − log p-value corresponding to a statistical significance level of 0.05 is 1.30. D Locations of all SNPs included in at least one of the analyses above. The region of included SNPs spans 1 MB above and below the coding region of CAV1. SNPs indicated in red are within the transcription start and stop site of CAV1. E Workflow of sources for CAC and BMD data, as well as their analytical methods
Table 1.
All SNPs within the transcription start and stop site of CAV1 and their functional location
| RSID | Functional location | CAC association direction | BMD association direction |
|---|---|---|---|
| rs10256914 | Intron variant | − | + |
| rs1049334 | 3’ UTR | − | + |
| rs11773845 | Intron variant | − | + |
| rs1476833 | Intron variant | − | + |
| rs1997572 | Intron variant | − | + |
| rs3815412 | Intron variant | − | + |
| rs6466587 | Intron variant | − | + |
| rs6466588 | Intron variant | − | − |
| rs729949 | Intron variant | − | + |
| rs7804372 | Intron variant | − | + |
| rs8713 | 3’ UTR | − | + |
| rs917664 | Intron variant | − | − |
| rs959173 | Intron variant | − | + |
| rs9920 | 3’ UTR | + | + |
To compare these population wide observations to differences in cellular responses, we cultured VSMCs and HOBs in osteogenic media (OS) in-vitro. VSMCs (Fig. 2A) and HOBs (Fig. 2B) osteogenic exhibited increased calcification compared to controls cultured in growth media as demonstrated by alizarin red S stain. We have previously shown that knockdown of CAV1 in VSMCs inhibits calcification [3] which is consistent with our current observations (Fig. 2C). We also confirmed that the knockdown persists for 21 days in culture (Supplemental Fig. 1). In this study, we assessed the effects of knocking down CAV1 in HOBs. This had no effect on calcification of HOB in-vitro cultures (Fig. 2D).
Fig. 2.

Alizarin red staining and quantification of VSMCs (A) and HOBs (B) grown in control and OS media after 28 and 21 days, respectively. Alizarin red staining and quantification in control and OS media with siRNA knockdown of CAV1 of VSMCs (C) and HOBs (D). *p < 0.05, **p ≤ 0.01, ***p ≤ 0.001, and ****p ≤ 0.0001. Mann-Whitney U test and Kruskal-Wallis ANOVA were used as non-parametric analyses to test for differences between groups
Methyl-β-cyclodextrin (MβCD) removes cholesterol from the plasma membrane [21], disrupting the caveolar domains in which CAV1 resides [21, 29]. Our results showed that MβCD-supplemented OS media significantly (p < 0.05) increased calcification in VSMCs compared to OS cultures (Fig. 3A). In HOBs, co-treatment with MβCD and OS significantly (p < 0.01) abrogated calcification compared to OS media, with no significant difference from the control group (Fig. 3B). OS media increased cellular CAV1 protein levels in both cell types (Fig. 3C, D). MβCD-supplemented OS media significantly reduced CAV1 protein levels in VSMCs and decreased CAV1 to control levels in HOBs (Fig. 3C, D). Cellular TNAP activity increased in OS cultures of both cell types (Fig. 3E, F). In HOBs, the addition of MβCD reduced activity to control levels (Fig. 3F, p < 0.01 compared to OS alone), while in VSMCs, we observed a trending but non-significant increase in TNAP activity with MβCD compared to OS alone (Fig. 3E). In EVs from VSMCs, TNAP activity increased from control to OS cultures (p < 0.01). MβCD further increased EV TNAP activity in VSMC cultures (Fig. 3G, p < 0.05 compared to OS alone). In MVs, addition of MβCD reduced TNAP activity down to control levels (Fig. 3H, p < 0.05).
Fig. 3.

Alizarin red quantification of VSMCs (A) and HOBs (B) in control, OS, and OS + MβCD media after 28 and 21 days, respectively. Cellular CAV1 in VSMCs (C) and HOBs (D) after 14 days of treatment. Cellular TNAP activity in VSMCs (E) and HOBs (F) after 14 days of culture. VSMC extracellular vesicles’ TNAP activity after 21 days of culture (G). TNAP activity of HOB matrix vesicles released after 14 days of treatments (H). *p < 0.05 and **p ≤ 0.01, Kruskal-Wallis ANOVA was used as a non-parametric analysis to test for differences between groups
Filamin A (FLNA) mediates interactions between CAV1 and the actin cytoskeleton, regulating caveolae trafficking [30]. Calpain is normally responsible for cleaving FLNA at the C-terminal [31]. Calpain inhibitor, therefore, reduces the cleavage of FLNA, stabilizing the interaction between CAV1 and FLNA and altering the trafficking of CAV1. In VSMCs calpain inhibition completely prevented in-vitro calcification (Fig. 4A, p < 0.05). In HOBs, addition of calpain inhibitor led to a trending but non-significant increase in calcification compared to OS cultures alone (Fig. 4B). Cellular levels of CAV1 and TNAP protein increased in VSMC OS cultures (p < 0.05), and exhibited a trending but non-significant increase by the addition of calpain inhibitor (Fig 4C, D). OS media increased the TNAP activity of VSMCs, both within cells (Fig. 4F p < 0.01) and within EVs (p < 0.05). Interestingly, the introduction of calpain inhibitor to OS VSMC media significantly reduced TNAP activity compared to OS cultures (p < 0.05), both within cells (Fig. 4E) and in released EVs (Fig. 4F). EV TNAP activity in cultures containing calpain inhibitor was not significantly different than TNAP activity in control media.
Fig. 4.

Cultures containing Alizarin red staining and quantification in VSMCS (A) and HOB (B) after 28 and 21 days, respectfully. Cellular CAV1 protein (C) and TNAP protein (D) in VSMCs after 14 days of culture. Cellular TNAP activity in VSMCs after 14 days of treatment (E). TNAP activity of EVs released from VSMCs after 14 days of treatment (F). *p < 0.05 and **p ≤ 0.01, Kruskal-Wallis ANOVA was used as a non-parametric analysis to test for differences between groups
Previously, we demonstrated that OS culture conditions resulted in translocation of CAV1 from low density caveolar lipid raft domains to high density non-caveolar domains [18]. In the current study, we found no significant differences in the amount of CAV1 in any layer between control and OS groups (Fig. 5).
Fig. 5.
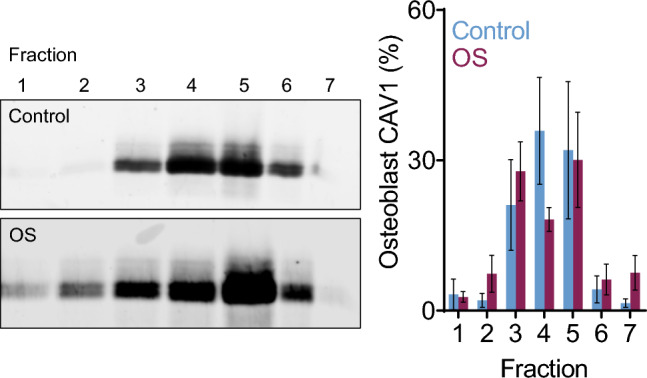
Relative CAV1 content of HOB cell lysate density fractions from control and OS cultures after 14 days
Discussion
The formation of cardiovascular calcification involves VSMC-derived calcifying EVs that serve as nucleating foci for mineralization. This process mimics physiological bone mineralization by HOBs in which MVs are the initializing unit of calcification [8]. Despite their similarities, there seems to be an inverse relation between arterial mineral deposition and bone mineral density - a phenomenon referred to as the “calcification paradox”. This phenomenon has often been observed clinically. Patients with bone disorders, such as osteoporosis are considered high-risk for cardiovascular events [32]. While previous studies have suggested that divergent outcomes in mineralization may be attributed to systemic alterations in inflammation and/or mineral promoting components [2, 14–17], we observe fundamental mechanistic divergence in HOB and VSMC mineralization. Future studies are needed to clarify the relative contributions of systemic pressures and mechanistic differences leading to the calcification paradox.
Previous studies reported that the membrane scaffolding protein, CAV1, actively participates in the biogenesis of calcifying EVs in VSMCs [3]. The removal of CAV1 in VSMCs has been shown to abrogate calcification, indicating that it is an essential part of the calcification pathway in this cell type. In this study, we investigated the outcome of removing CAV1 in HOB cultures. Interestingly, we found that siRNA knockdown of CAV1 had no effect on calcification in HOBs (Fig. 2D). Examining the role of CAV1 further, we can look at the osteogenic vesicles released by each cell type in osteogenic conditions and their CAV1 content. In a previous study we showed that in osteogenic conditions, VSMC EVs contain more CAV1 while the HOB analog MVs contain less CAV1 than control vesicles [18]. Supported by the observation that mice lacking CAV1 exhibit increased BMD [19], these data suggest that CAV1 may play a divergent role in HOB and VSMC mineralization.
To compare effects of CAV1 associated SNPs on BMD and CAC, we used available data from two large cohort studies. We only included SNPs with information available in both cohorts and within a 1 Mb range of the CAV1 locus to include potential cis-eQTLs that modulate CAC/BMD via CAV1. The MESA cohort was used to assess CAC, and the UK Biobank was used to gather information on BMD. Therefore, for each SNP we report information on both CAC and BMD effects. Figure 1D shows all SNPs included in the analyses, and those colored red fall within the open reading frame of CAV1. Supporting a role for CAV1 in the calcification paradox, a majority of the SNPs included in these analyses associate with opposite effects on BMD and CAC. Since the SNPs within the open reading frame all derive from intronic or untranslated regions, it is likely that they effect protein expression rather than function. Future studies will focus on clarifying the functional relevance of these SNPs.
Our next goal was to investigate known CAV1 interactors and their role in modulating VSMC and HOB calcification. Methyl β-cyclodextrin (MβCD), which acts by removing cholesterol from the membrane, disrupts the lipid-rich caveolar domains in which CAV1 resides. The addition of MβCD to osteogenic media increased endpoint calcification in VSMCs, significantly more than OS controls alone (Fig. 3A); however, it completely stopped calcification in HOBs (Fig. 3B). Accordingly, TNAP activity in EVs increased in VSMC cultures supplemented with MβCD, whereas in HOB cultures, the MβCD treatment reduced MV TNAP activity to control levels (Fig 3G, H). These results indicate that the stability of caveolar domains in which CAV1 is housed is apparently essential to the mineralization potential of HOBs. Future studies need to reconcile this finding with observations that CAV1 knockdown in-vitro (Fig. 3B) and knockout in-vivo [19] have no effect and increase bone mineral production, respectively.
Addition of calpain inhibitor reduces the cleavage of FLNA. In endothelial cells, this interference of FLNA cleavage localizes CAV1 to the plasma membrane, preventing its normal cellular movement [30]. In VSMCs, the stabilization of this crosslinking stopped calcification (Fig. 4A). This indicates that FNLA cleavage is an essential step in VSMC calcification. Calpain inhibited VSMCs cultures still had increased CAV1 (Fig. 4C) and TNAP protein (Fig. 4D) in the cell, which is typical of VSMCs in osteogenic conditions; however, the cells did not calcify. We propose that FLNA cleavage is integral to osteogenic EV formation in the initial invagination process and without it, the components of EVs are created but cannot successfully be utilized in the absence of typical CAV1 trafficking. This is further supported by the overall reduction in TNAP activity of EVs being released from calpain inhibited VSMCs (Fig. 4F). The calpain inhibited EV population had a similar TNAP activity to that of control EV populations, indicating that calpain inhibited VSMC cultures do not release calcific EVs with active TNAP. The inhibition of FLNA cleavage had an opposite effect in HOBs, demonstrating a trending increase in mineral formation from osteogenic media alone (Fig. 4B). Due to the fairly low sample size used in this study we did not assume normality and employed stringent non-parametric statistical tests. Therefore, though large differences in magnitude were observed in some analyses, they did not reach statistical significance. Future studies will include larger sample sizes to more rigorously test potential differences between groups. However, the current data indicate that FLNA cleavage may play a regulatory role in HOBs, as its reduction exacerbated mineralization in this cell type. Although the mechanistic role of FNLA in calcification requires further study, these data further suggest that altering CAV1 trafficking affects endpoint calcification differently in VSMCs and HOBs.
To better analyze the movement of CAV1 during calcification, we used an ultracentrifugation lipid raft density gradient assay. The lipid rafts that normally house CAV1 in the membrane have a lighter density profile than other membrane domains, due to their high levels of cholesterol. As such, the cell lysate containing lipid raft components separate into lighter fractions, while other membrane domains accumulate in more dense fractions. Our previous results show that OS culture conditions result in translocation of CAV1 to non-caveolar fractions in VSMCs [18]. The current data demonstrate no significant difference in CAV1 location in osteoblast control and OS cultures (Fig. 5). These data emphasize the fundamental differences in CAV1 trafficking during the calcification process of VSMCs and HOBs.
Overall, our results highlight the likely distinction between the mechanistic pathways of calcification in HOBs and VSMCs. It is important to note that the effect of EVs may go beyond solely serving as nucleating foci for mineral formation. Recent investigations into the function of HOB derived EVs identified a subclass of vesicles, small osteoblast vesicles, which mediate the transition from an osteoblast to osteoclast phenotype [33]. These findings highlight a need to further classify the MVs implicated in mineral formation observed in this study. We have shown that CAV1, a membrane protein present in both cells and known to be involved in VSMC calcification, plays a different role in HOB mineralization. Population-level human genetic evidence robustly and significantly suggests that genetic variations near the CAV1 locus associate with opposite directions of effect on BMD and CAC (Fig. 1). Future studies will need to determine mechanisms through which specific SNPs affect the mineralization response in each tissue. All manipulations of CAV1 trafficking done in this study had opposite outcomes in endpoint calcification, further supporting the idea that these two pathways are individually distinct. The long-term outcomes of this work could further identify the characteristic differences of these two pathways, in order to inform therapeutic development that avoids off-target effects when treating pathologies of either tissue.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This work was supported by Florida International University (Dissertation Year Fellowship to A.B.N); and Florida Heart Research Foundation (Stop Heart Disease Researcher of the Year Award to J.D.H). Research reported in this publication was supported by the National Heart, Lung, and Blood Institute as well as the National Institute of Diabetes and Digestive Kidney Diseases of the National Institutes of Health (1R01DK132090 to B.B.K and 1R01HL160740 to J.D.H.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Declarations
Conflict of interest
The authors (A.B.N., K.K., P.S., B.B.K., and J.D.H.) declare that they have no conflict of interest.
Ethical approval
This study did not involve experiments on live animals. The primary human cells were purchased from the American Type Culture Collection (ATCC). The human genetics analyses used data available from the UK Biobank and MESA cohorts.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations
Amirala Bakhshian Nik and Katherine Kaiser have contributed equally to this work
Contributor Information
Bohdan B. Khomtchouk, Email: bokhomt@iu.edu
Joshua D. Hutcheson, Email: jhutches@fiu.edu
References
- 1.Witteman JC, et al. Aortic calcification as a predictor of cardiovascular mortality. Lancet. 1986;2(8516):1120–1122. doi: 10.1016/S0140-6736(86)90530-1. [DOI] [PubMed] [Google Scholar]
- 2.Bakhshian Nik A, et al. The time-dependent role of bisphosphonates on atherosclerotic plaque calcification. J. Cardiovasc. Dev. Dis. 2022;9(6):168. doi: 10.3390/jcdd9060168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goettsch C, et al. Sortilin mediates vascular calcification via its recruitment into extracellular vesicles. J. Clin. Invest. 2016;126(4):1323–1336. doi: 10.1172/JCI80851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hutcheson JD, et al. Genesis and growth of extracellular-vesicle-derived microcalcification in atherosclerotic plaques. Nat. Mater. 2016;15(3):335–343. doi: 10.1038/nmat4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hutcheson JD, et al. Revisiting cardiovascular calcification: a multifaceted disease requiring a multidisciplinary approach. Semin. Cell Dev. Biol. 2015;46:68–77. doi: 10.1016/j.semcdb.2015.09.004. [DOI] [PubMed] [Google Scholar]
- 6.Iwayama T, et al. Osteoblastic lysosome plays a central role in mineralization. Sci Adv. 2019;5(7):eaax0672. doi: 10.1126/sciadv.aax0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruiz JL, et al. Nanoanalytical analysis of bisphosphonate-driven alterations of microcalcifications using a 3D hydrogel system and in vivo mouse model. Proc. Natl. Acad. Sci. U.S.A. 2021;118(14):e1811725118. doi: 10.1073/pnas.1811725118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bakhshian Nik A, Hutcheson JD, Aikawa E. Extracellular vesicles as mediators of cardiovascular calcification. Front. Cardiovasc. Med. 2017;4:78. doi: 10.3389/fcvm.2017.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui L, et al. Characterisation of matrix vesicles in skeletal and soft tissue mineralisation. Bone. 2016;87:147–158. doi: 10.1016/j.bone.2016.04.007. [DOI] [PubMed] [Google Scholar]
- 10.Reynolds JL, et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J. Am. Soc. Nephrol. 2004;15(11):2857–2867. doi: 10.1097/01.ASN.0000141960.01035.28. [DOI] [PubMed] [Google Scholar]
- 11.Persy V, D'Haese P. Vascular calcification and bone disease: the calcification paradox. Trends Mol. Med. 2009;15(9):405–416. doi: 10.1016/j.molmed.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 12.Klein GL. Is calcium a link between inflammatory bone resorption and heart disease? Elife. 2022;11:e83841. doi: 10.7554/eLife.83841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shanahan CM, et al. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ. Res. 2011;109(6):697–711. doi: 10.1161/CIRCRESAHA.110.234914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hjortnaes J, et al. Arterial and aortic valve calcification inversely correlates with osteoporotic bone remodelling: a role for inflammation. Eur. Heart J. 2010;31(16):1975–1984. doi: 10.1093/eurheartj/ehq237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hutcheson JD, Blaser MC, Aikawa E. Giving calcification its due: recognition of a diverse disease: a first attempt to standardize the field. Circ Res. 2017;120(2):270–273. doi: 10.1161/CIRCRESAHA.116.310060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aikawa M, et al. An HMG-CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation. 2001;103(2):276–283. doi: 10.1161/01.CIR.103.2.276. [DOI] [PubMed] [Google Scholar]
- 17.Deguchi JO, et al. Inflammation in atherosclerosis: visualizing matrix metalloproteinase action in macrophages in vivo. Circulation. 2006;114(1):55–62. doi: 10.1161/CIRCULATIONAHA.106.619056. [DOI] [PubMed] [Google Scholar]
- 18.Bakhshian Nik, A., et al., Epidermal Growth Factor Receptor Inhibition Prevents Caveolin-1-dependent Calcifying Extracellular Vesicle Biogenesis. 2021, bioRxiv. [DOI] [PMC free article] [PubMed]
- 19.Rubin J, et al. Caveolin-1 knockout mice have increased bone size and stiffness. J. Bone Miner. Res. 2007;22(9):1408–1418. doi: 10.1359/jbmr.070601. [DOI] [PubMed] [Google Scholar]
- 20.Insel PA, Patel HH. Membrane rafts and caveolae in cardiovascular signaling. Curr. Opin. Nephrol. Hypertens. 2009;18(1):50–56. doi: 10.1097/MNH.0b013e3283186f82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodal SK, et al. Extraction of cholesterol with methyl-beta-cyclodextrin perturbs formation of clathrin-coated endocytic vesicles. Mol. Biol. Cell. 1999;10(4):961–974. doi: 10.1091/mbc.10.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Muriel O, et al. Phosphorylated filamin A regulates actin-linked caveolae dynamics. J. Cell Sci. 2011;124(Pt 16):2763–2776. doi: 10.1242/jcs.080804. [DOI] [PubMed] [Google Scholar]
- 23.Ravid D, et al. Filamin A is a novel caveolin-1-dependent target in IGF-I-stimulated cancer cell migration. Exp. Cell Res. 2008;314(15):2762–2773. doi: 10.1016/j.yexcr.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 24.Morris JA, et al. An atlas of genetic influences on osteoporosis in humans and mice. Nat. Genet. 2019;51(2):258–266. doi: 10.1038/s41588-018-0302-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bild DE, et al. Multi-Ethnic Study of Atherosclerosis: objectives and design. Am. J. Epidemiol. 2002;156(9):871–881. doi: 10.1093/aje/kwf113. [DOI] [PubMed] [Google Scholar]
- 26.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khomtchouk BB, Hennessy JR, Wahlestedt C. shinyheatmap: Ultra fast low memory heatmap web interface for big data genomics. PLoS ONE. 2017;12(5):e0176334. doi: 10.1371/journal.pone.0176334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goettsch C, et al. miR-125b regulates calcification of vascular smooth muscle cells. Am. J. Pathol. 2011;179(4):1594–1600. doi: 10.1016/j.ajpath.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Graziani A, et al. Cholesterol- and caveolin-rich membrane domains are essential for phospholipase A2-dependent EDHF formation. Cardiovasc. Res. 2004;64(2):234–242. doi: 10.1016/j.cardiores.2004.06.026. [DOI] [PubMed] [Google Scholar]
- 30.Sverdlov M, et al. Filamin A regulates caveolae internalization and trafficking in endothelial cells. Mol. Biol. Cell. 2009;20(21):4531–4540. doi: 10.1091/mbc.e08-10-0997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bandaru S, et al. Filamin A regulates cardiovascular remodeling. Int. J. Mol. Sci. 2021;22(12):6555. doi: 10.3390/ijms22126555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lampropoulos CE, Papaioannou I, D'Cruz DP. Osteoporosis–a risk factor for cardiovascular disease? Nat. Rev. Rheumatol. 2012;8(10):587–598. doi: 10.1038/nrrheum.2012.120. [DOI] [PubMed] [Google Scholar]
- 33.Uenaka M, et al. Osteoblast-derived vesicles induce a switch from bone-formation to bone-resorption in vivo. Nat. Commun. 2022;13(1):1066. doi: 10.1038/s41467-022-28673-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.