

Abstract
Glioblastoma (GBM) is the most prevalent and aggressive primary central nervous system (CNS) malignancy. YM155 is a highly potent broad spectrum anti-cancer drug that was derived from a phenotypic screen for functional inhibitors of survivin expression, but for which the relevant biomolecular target remains unknown. Presumably as a result of its lack of cell type selectivity, YM155 has suffered from tolerability issues in the clinic. Based on its structural similarity to the GBM-selective prodrug RIPGBM, here, we report the design, synthesis, and characterization of a prodrug form of YM155, termed aYM155. aYM155 displays potent cell killing activity against a broad panel of patient-derived GBM cancer stem-like cells (IC50 = 0.7-10 nM), as well as EGFR-amplified and EGFR variant III-expressing (EGFRvIII) cell lines (IC50 = 3.8-36 nM), and becomes activated in a cell type-dependent manner. Mass spectrometry-based analysis indicates that enhanced cell type selectivity results from relative rates of prodrug activation in transformed versus non-transformed cell types. The prodrug strategy also facilitates transport into the brain (brain-to-plasma ratio, aYM155 = 0.56; YM155 = BLQ). In addition, we determine that the survivin-suppressing and apoptosis-inducing activities of YM155 involve its interaction with receptor-interacting protein kinase 2 (RIPK2). In an orthotopic intracranial GBM xenograft model, aYM155 prodrug significantly inhibits brain tumor growth in vivo, which correlates with cell type selective survivin-based pharmacodynamic effects.
Graphical Abstract
INTRODUCTION
Glioblastoma (GBM) is the most common form of brain cancer in adults and corresponds to grade IV astrocytoma by WHO classification.1, 2 The current standard of care treatment for GBM involves maximal surgical resection, followed by daily radiation and concurrent chemotherapy using the antineoplastic DNA alkylating agent Temozolomide (TMZ).3 Despite advances in surgery, the disease remains virtually incurable and is one of the most lethal human cancers, typically causing mortality within one year.4, 5 Much of what makes treatment so difficult is attributed to a population of cells within the tumor termed cancer stem-like cells (CSCs), which possess stem-like characteristics, including invasiveness and therapy resistance, and survive treatment to repopulate bulk tumor.6–9 Thus, there is a pressing need for brain-penetrant drugs that effectively target not only bulk tumor but also the CSC-like population that underlies poor prognosis in GBM.
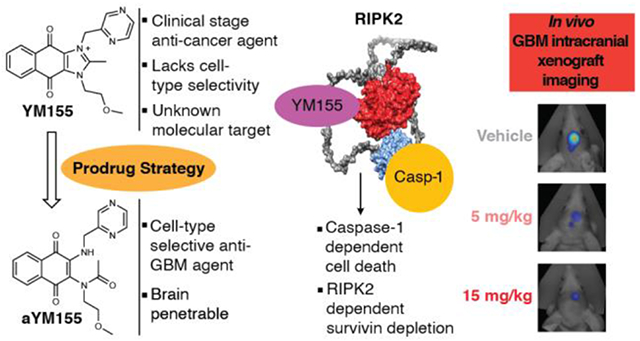
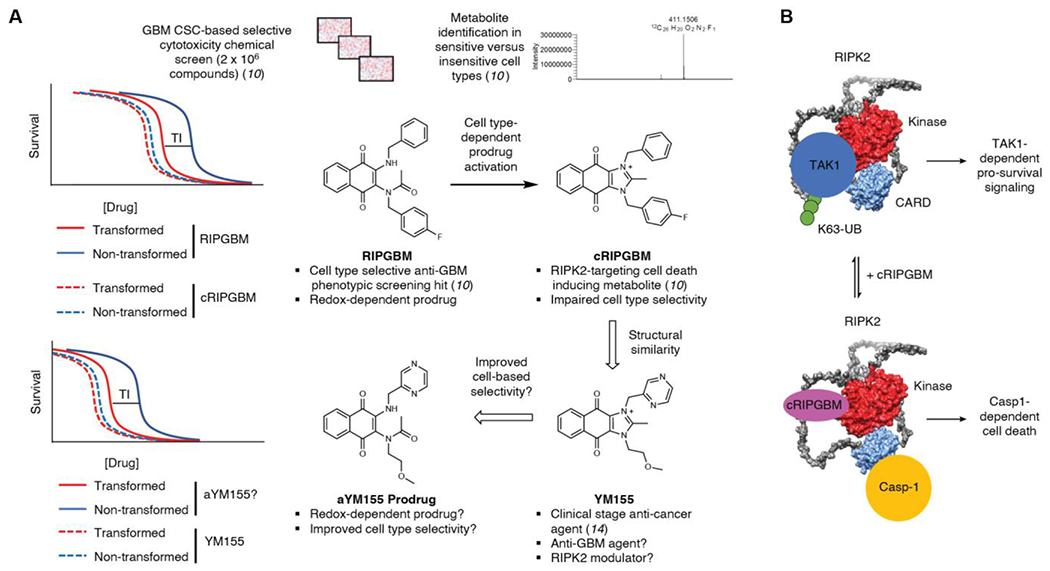
We have previously reported on the discovery of a small molecule termed RIPGBM, which was identified from a large scale cell-based chemical screen (>106 library) involving primary patient-derived GBM CSCs.10 RIPGBM induces cell death selectively in diverse brain tumor cell types, including GBM CSCs, and was found to robustly reduce tumor burden in the context of an orthotopic intracranial GBM xenograft model following oral administration of a well-tolerated dose.10 Metabolite identification studies revealed that RIPGBM undergoes an intramolecular cyclization reaction in GBM cells, to form an imidazolium species termed cRIPGBM.10 Based on observed differences in the rates of cRIPGBM formation in sensitive transformed GBM cells when compared to insensitive non-diseased control cell types, as well as the observed more potent yet less cell type selective cell killing profile of cRIPGBM, it was determined that the cell type selectivity of RIPGBM is derived, at least in part, by its ability to act as a prodrug.10 In addition to the more favorable cell-based selectivity profile of RIPGBM, the prodrug form was found to have excellent brain exposure, whereas the positively charged imidazolium-containing cRIPGBM species is unable to penetrate the blood-brain barrier.10 Using a mass spectrometry-based proteomics approach involving a photoactivatable affinity probe derivative of cRIPGBM, receptor-interacting protein kinase 2 (RIPK2) was identified as a relevant biomolecular target for cRIPGBM in the context of tumor cells.10 Mechanistic studies revealed that cRIPGBM acts as a molecular switch, which upon binding to RIPK2 alters its binding partners, leading to dissociation from Transforming Growth Factor-b (TGF-b)-activated kinase 1 (TAK1) and association with Caspase 1 (Casp1), which leads to the induction of Caspase1 (Casp1)-dependent cell death.10
Here we report on a significant improvement in potency for the RIPGBM scaffold through the characterization of sepantronium bromide (YM155), a clinical stage oncology drug identified based on its ability to suppress the expression of survivin. Additionally, we report on a significant improvement in the cell-based selectivity and therapeutic index profile of YM155 and provide insight into its mechanism of action. Survivin is a member of the inhibitor of apoptosis (IAP) family that plays a critical role in cell cycle progression and cell survival.12 It has been shown to be overexpressed in a number of different carcinomas and has been demonstrated to positively correlate with tumor progression and resistance to chemotherapy.13, 14 YM155 is an imidazolium-containing small molecule that was discovered from a phenotypic high throughput screen for functional inhibitors of survivin expression using a survivin gene promoter reporter-based assay.15 YM155 has been demonstrated to suppress survivin expression and induce apoptosis at pM and low nM concentrations in a variety of preclinical cancer cell models including hormone refractory prostate cancer, non-Hodgkin’s Lymphoma, sarcoma, non-small cell lung cancer, and breast cancer.16, 17 Though its anti-cancer activity has been well studied, the precise mode of action for YM155 remains unclear. Notably, YM155 efficacy has been shown to be contingent upon the expression of the solute carrier SLC35F2, which was demonstrated to be required for YM155 drug import.18 However, the relevant biomolecular target responsible for its survivin-inhibiting and cell killing activity remains to be elucidated.
Despite its remarkable preclinical efficacy and satisfactory results in phase I clinical trials when evaluated at low doses, YM155 has proven to be a disappointment in phase II efficacy trials when evaluated as a single agent.19–23 In general, tolerated human exposure levels are not associated with anti-cancer activity and those associated with moderate efficacy suffer from tolerability and toxicity issues.20, 23 Based on observed cell-based therapeutic indices described below, these observations are predictable and likely result from a lack of cell type selectivity in the killing of tumor versus non-tumor cell types. Based on the similarity of the chemical structures of YM155 and cRIPGBM, we evaluated YM155 in the context of GBM. Potentially due to its complete lack of CNS exposure, YM155 has never been tested in patients with GBM. Our principal findings are that YM155 induces cell death in a broad panel of GBM cell types at low- and sub-nM concentrations and that the cell type selectivity profile and brain exposure properties of YM155 can be significantly enhanced using a prodrug strategy that is analogous to that which we elucidated for RIPGBM (Figure 1A). Additionally, we characterize that YM155-mediated suppression of survivin and its tumor killing activity involves its interaction with RIPK2, thereby shedding mechanistic insight into the mode of action of this clinical stage drug.
Figure 1.
Rationale for a prodrug strategy to improve the cell-based selectivity of YM155 and proposed mechanism of action for RIPK2-dependent induced cell death. (A) Scheme providing a rationale for the YM155 prodrug strategy and design based on comparison of the chemical structures and properties of cRIPGBM to YM155 and of the corresponding RIPGBM and aYM155 prodrugs. (B) Schematic representation of the proposed mechanism of action for cRIPGBM and YM155-induced cell death.10 Predicted full length RIPK2 structure was generated using Alphafold.11
RESULTS
aYM155 prodrug induces cell death selectively in brain cancer cells.
Based on structure-activity relationship (SAR) studies associated with the RIPGBM scaffold and the high degree of structural similarity that exists when comparing the chemical structures of cRIPGBM and YM155 (Figure 1A), we hypothesized that YM155 might serve as an effective modulator of GBM cell survival and act to induce cell death by interacting with RIPK2 via the proposed mechanism for cRIPGBM (Figure 1B).
To explore the relevance of YM155 in the context of GBM, we evaluated the activity of YM155 using the primary patient-derived GBM CSC lines used in the discovery of RIPGBM (i.e., GBM-A and GBM-F), as well as control non-diseased cell types (i.e., primary human astrocytes (HA), WA09 human ES cell-derived neural progenitor cells (NPC), and primary human lung fibroblasts (HLF)).10, 24 YM155 was found to be a highly effective and potent anti-GBM agent (Figures 2A and S1A). The potency of YM155 was observed to be significantly improved relative to cRIPGBM, displaying single digit and sub-nanomolar IC50 values related to cell killing in GBM CSCs (YM155, GBM-A IC50 = 4.5 nM, GBM-F IC50 = 780 pM, Figure 2E; cRIPGBM GBM-A IC50 = 27 nM, GBM-F IC50 = 70 nM, Figures 2B and S1B). However, YM155 was found to suffer from a poor cell type selectivity profile, being similarly cytotoxic to non-transformed cells when evaluated using a panel of control cell types (HA IC50 = 5.1 nM, NPC IC50 = 5.0 nM, HLF IC50 = 92 nM; Figures 2E and S1C).
Figure 2.
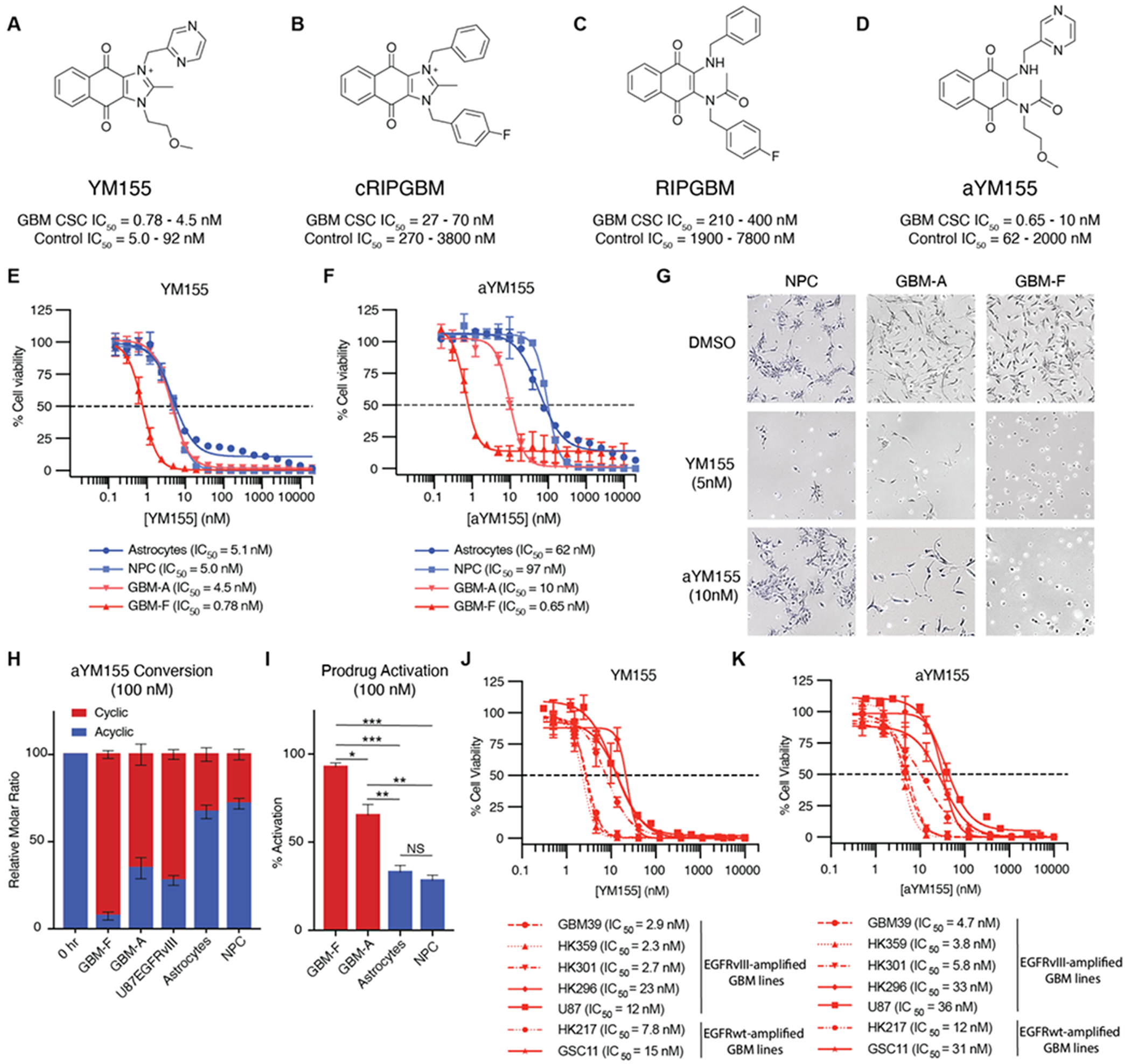
YM155 prodrug induces apoptosis selectively in glioblastoma CSC’s. (A-D) Chemical structure and associated cell-based IC50 profile for cRIPGBM, YM155, RIPGBM, and aYM155. (E) YM155 concentration response curves for two GBM cell lines (GBM-A and GBM-F) and two healthy control cell types (NPC and Astrocytes) after 72 hours. Within each of these assays, individual cell lines were tested in triplicate (biological replicates). (F) aYM155 concentration response curves as in (E). (G) Brightfield images of GBM CSC’s or non-diseased human NPCs treated with either YM155 (5nM) or aYM155 (10nM) for 72 hours. (H) Agilent 6495 triple quadrupole MS-based metabolite identification studies done in GBM CSC’s and non-diseased cells. Cells were treated with 100nM aYM155 for 24 hours prior to extraction. Error bars are from three biological replicates per cell type. (I) Quantification of aYM155 activation in (H) from each cell type tested. (J-K) Concentration response curves for YM155 and aYM155 following a 72-hour incubation across a spectrum of GBM cell types. Student’s t-test was used to assess significance (I). Data are means ± SD. n = 3 independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001. NS = not significant.
Our hypothesis for why YM155 has had challenges in Phase II efficacy-based clinical trials stems from our observations that YM155 lacks cell-based selectivity and is equivalently and potently toxic to non-transformed cell types. We speculate that this narrow therapeutic index has limited efficacy at tolerated doses that have been evaluated in the clinic. Based on our studies with RIPGBM and the associated prodrug model for selective cytotoxicity (i.e., differentiational activation of an acyclic prodrug in target cell types facilitates cell type selectivity), we determined if we could apply an analogous approach to improve the therapeutic index and clinical potential of YM155. The proposed basis for cell type dependent differences in prodrug activation, based on chemical reactivity experiments10, is that altered redox potential or redox-related biotransformation in sensitive cancer cell types leads to reduction of the RIPGBM quinone moiety, which serves to enhance the nucleophilicity of the benzylic amine and facilitates its addition to an adjacent acetamido group. The resulting intramolecular cyclization, followed by dehydration, leads to formation of a cytotoxic imidazolium species (Figure S2). The corresponding putative prodrug acyclic naphthoquinone form of YM155, analogous to the structure of RIPGBM (Figure 2C) and termed aYM155 (Figure 2D), was synthesized and tested using the panels of primary GBM CSC and control cell types described above. aYM155 was found to have potent cytotoxic activity when evaluated in GBM CSC cell types (GBM-A IC50 = 10 nM, GBM-F IC50 = 650 pM; Figures 2F and S1D). Compared to the initial generation RIPGBM molecule (GBM-A IC50 = 200 nM, GBM-F IC50 = 400 nM; Figure S1E), aYM155 demonstrates >100-fold improvement in potency in GBM CSCs. Potentially of more importance, based on its evaluation in control cell types (HA IC50 = 62 nM, NPC IC50 = 97 nM, HLF IC50 = 2.0 μM; Figures 2F and S1C), aYM155 displays a notably improved selectivity profile when compared to YM155 (Figure 2G). Based on comparisons of potency in GBM CSCs and relevant control neural cell types, when compared to YM155, aYM155 is ~equivalent or 2.2-fold less potent in target cell types (i.e., in GBM-F and GBM-A, respectively) and 12- or 19-fold less potent in non-target neural cell types (i.e., HA and NPC, respectively) (Figure 2, E and F).
We hypothesized that the cell type selectivity displayed by aYM155 is derived, at least in part, from our proposed prodrug activation mechanism (Figure S2). To determine relative rates of YM155 formation in target versus control cell types, we used a targeted mass spectrometry-based metabolite identification approach to evaluate relative rates of aYM155 activation, corresponding to a −18 species, in disease and non-diseased cell types. Following 24 hours of aYM155 treatment (100 nM), relative levels of aYM155 and YM155 in normalized extracted cell pellets were quantitatively assessed. Despite being significantly above its IC50 value in GBM CSCs (i.e., <10 nM), 100nM aYM155 was used to overcome instrumentation-based limits of detection on the mass spectrometer, which represented a challenge of working with this new highly potent agent when compared to RIPGBM. At the evaluated concentration and incubation timepoint, aYM155 was converted to the −18 species corresponding to cyclic YM155 molecule in all cell types tested (Figure 2H). Importantly, and consistent with the hypothesis of pro-drug activation corresponding to cell type sensitivity, rates of conversion to the active YM155 cyclic derivative were significantly higher in GBM CSC populations compared to non-transformed control cell types (Figure 2, H and I). Presumably, at lower more relevant concentrations, or shorter periods of in vivo exposure, differences in the relative levels of active drug form in target versus insensitive non-target cell types facilitates cell type-dependent selective cytotoxicity.
We next evaluated aYM155 and YM155 using a broader panel of GBM patient-derived neurosphere cell lines, including both EGFRwt-amplified cells and EGFRvIII-expressing cells, a frequent mutation in GBM which confers increased proliferation and invasiveness to glioma cells.25 Encouragingly, both YM155 and aYM155 were active on all diseased cell types tested (YM155 IC50 = 2.3 nM-12 nM, aYM155 IC50 = 3.8 nM-36 nM; Figure 2, J and K). The observed selectivity of aYM155 prodrug for all GBM cell types compared to control cell types (aYM155 IC50 = 3.8-36 nM versus IC50 = 62-97 nM in control neural cell types; Figure 2, K and F) is significantly improved when compared to YM155 (i.e., 2.3-23 nM versus 5 nM in control neural cell types; Figure 2, J and E). This selectivity is also in stark contrast to that of standard of care drug Temozolomide (TMZ), which has been shown to be selectively toxic to non-diseased cell types and to which GBM CSCs are prone to resistance.6, 7, 10 O6-methylguanine-DNA-methyltransferase (MGMT) promoter methylation status is a prognostic biomarker for the predicted response of GBM patients to TMZ DNA alkylating agent therapy. 26–28 Encouragingly, both YM155 and prodrug aYM155 displayed potent cytotoxic activity when evaluated in either hypo-MGMT methylated (U118MG YM155 IC50 = 3.7 nM, aYM155 IC50 = 11 nM) or hyper-MGMT methylated (LN229 YM155 IC50 = 2.8 nM, aYM155 IC50 = 12 nM) cell lines (Figure S1, F and G).29
YM155 activity is dependent upon its interaction with RIPK2.
Despite having reached a stage of extensive human clinical investigation, following its discovery from a phenotypic screen for inhibitors of survivin expression, the direct biomolecular target and mode of action of YM155 remains unclear. Previously, cRIPGBM was determined to induce caspase-1 (Casp1)-mediated cell death based on its ability to interact with RIPK2 and act as a molecular switch that alters RIPK2 ubiquitination status and an associated change in RIPK2 protein complex formation, ultimately resulting in disruption of TAK1-dependent proliferative signaling and association with and activation of Casp1 (Figure 1B).10 Based on the structural similarity of cRIPGBM and YM155, we hypothesized that RIPK2 is a relevant target of YM155, which could serve to mediate its cell killing function. To explore the mechanistic relevance of RIPK2 in the context of YM155, we first asked if the ability of YM155 to inhibit survivin expression is impacted by loss of RIPK2. shRNA-mediated knockdown of RIPK2 in U87MG cells (Figure S3A), stably expressing EGFRvIII, resulted in a significant rescue of survivin expression following 24 hours of treatment with a high concentration (100 nM) of YM155 (Figure 3A). Further, RIPK2 knockdown was found to significantly ablate the ability of YM155 to induce tumor cell death. Specifically, at a concentration corresponding to the 24-hour IC50 value, loss of RIPK2 resulted in a significant rescue of cell viability in the context of both YM155 (20 nM, Figure 3B) and aYM155 prodrug (160 nM, Figure 3C). We next asked if another member of the RIPK family, receptor-interacting protein kinase 1 (RIPK1), which has been previously implicated in proposed potential therapeutic approaches to the treatment of GBM involving necroptotic-based alternative death pathways,30 might contribute to the mechanism of action of YM155.30 To this end, we assessed the cytotoxicity of YM155 and aYM155 in U87-EGFRvIII cells following shRNA-mediated RIPK1 knockdown. No significant differences in sensitivity to either agent was observed in the context of induced tumor cell death (Figure S3B-D).
Figure 3.
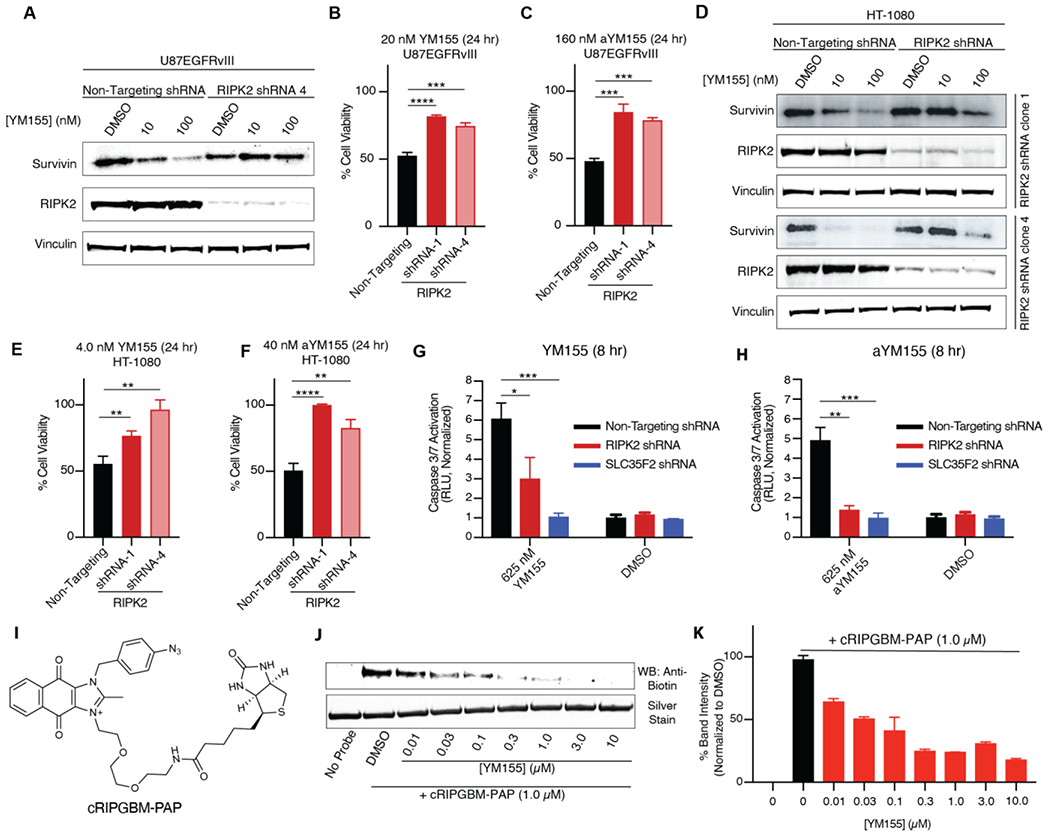
YM155 induces apoptosis in cells by interacting with RIPK2. (A) Western blot analysis of Survivin levels in U87-EGFRvIII cells treated with YM155 for 24 hours following shRNA RIPK2 knockdown. (B and C) Cell-viability values generated from Cell-titer glo assay 24 hours following YM155 and aYM155 treatment, respectively, in U87-EGFRvIII cells. (D) Western blot analysis of Survivin levels in HT-1080 cells treated with YM155 for 24 hours following shRNA RIPK2 knockdown. (E and F) Cell-viability values generated from Cell-titer glo assay 24 hours following YM155 and aYM155 treatment, respectively, in HT-1080 cells. (G and H) YM155 and aYM155, respectively, induced apoptosis in HT-1080 cells measured by Caspase-Glo 3/7 assay following shRNA mediated RIPK2 or SLC35F2 knockdown. (I) Chemical structure of cRIPGBM-PAP. (J) In vitro binding of cRIPGBM-PAP to recombinant human full-length RIPK2 in the absence or presence of competition using varying concentrations of YM155. (K) Band quantification from (D). Values are the mean of two replicates ± SD. Student’s t-test was used to assess significance (C, E-H). Data are means ± SD. n = 3 independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001. NS = not significant.
We next evaluated whether the scope of YM155-RIPK2 dependence expanded beyond brain cancer by conducting further mechanistic studies using HT-1080 fibrosarcoma cells, which are sensitive to YM155 treatment (IC50 = 3.1 nM, Figure S3E), exhibit significant prodrug conversion (Figure S3F), and express RIPK2 to a similar degree as GBM CSCs (Figure S3G). shRNA-mediated knockdown of RIPK2 in HT-1080 cells (Figures S3, H and I) was found to significantly ablate the ability of YM155 to reduce survivin mRNA (Figure S3J) and protein levels (Figures 3D and S3, K-N), following 24 hours of treatment. Indeed, compared to the significant reduction observed with 10 nM YM155 treatment in HT-1080 non-targeting shRNA control cells, a concentration of 100 nM YM155 was required to significantly impact survivin protein levels in RIPK2 shRNA cells (Figures 3D and S3, K-N).
To further examine the potential role of RIPK2 in the mechanism of YM155-mediated cell killing, we measured compound induced cytotoxicity and Casp1 activation in RIPK2 shRNA knockdown and non-targeting shRNA control HT-1080 cells. After 24 hours, loss of RIPK2 resulted in a significant rescue of cell viability in the context of both YM155 (4 nM, Figure 3E) and aYM155 prodrug (40 nM, Figure 3F) treatment. We also assessed Casp-1 and Casp-3/7 activation. Importantly, it has been previously demonstrated that RIPK2 serves as a key mediator of Casp-1-dependent apoptosis in hypoxia induced neuronal cell death, and Casp-1 was demonstrated to facilitate cRIPGBM-mediated RIPK2-dependent GBM cell killing.10, 31, 32 Consistent with the proposed mechanism of action, YM155 was found to induce time dependent activation of Casp-1 in HT-1080 fibrosarcoma cells, as well as both EGFRvIII amplified U87MG cells and GBM CSCs. (Figure S4, A-C). Following 8 hours of treatment, Casp-3/7 activation was observed with either YM155 or aYM155 prodrug, which was significantly reduced in RIPK2 shRNA knockdown cells when compared to non-targeting shRNA control cells (Figure 3, G and H). Interestingly, consistent with the reported requirement of SLC35F2 for transport of YM155 across cell membranes18, SLC35F2 shRNA knockdown ablated the ability of not only YM155 but also aYM155 to facilitate Casp3/7 activation (Figure 3, G and H). Previous studies have shown RIPK2 is a critical downstream mediator of NOD1 and NOD2 signaling and is essential for the activation of IκB kinase (IKK), resulting in the phosphorylation of IκBs.33 To assess the ability of YM155 to modulate RIPK2 function in this context, we analyzed changes in IκBα phosphorylation following stimulation of NOD2 with L-18 MDP. L-18 MDP caused an increase in IκBα phosphorylation in U87EGFRvIII cells, indicative of the expected activation of NFκB pathway signaling (Figure S4D).34 Consistent with the ability of YM155 to modulate RIPK2 function, pretreatment with 100nM YM155 reduced IKK activation, as shown by a decrease in observed IκBα phosphorylation levels (Figure S4D). Together, these data support the ability of YM155 to target RIPK2 and modulate its function.
We next assessed the ability of YM155 to directly interact with RIPK2, via the RIPGBM binding site, using in vitro competitive binding assays involving purified constructs of recombinant full length RIPK2 and our previously reported biotinylated photoactivatable affinity probe termed cRIPGBM-PAP (Figure 3I, apparent Kd = 2.3 μM for RIPK210). YM155 was found to competitively inhibit UV-dependent covalent labeling of RIPK2 by cRIPGBM-PAP (1 μM), by at least 50% at concentrations as low as 30nM (Figure 3, J and K). We next directly compared the ability of YM155 to competitively inhibit UV-dependent covalent labeling, relative to the less potent previous generation molecule (i.e., cRIPGBM). Consistent with their observed relative cell-based potencies, in terms of overall cytotoxicity, YM155 inhibits cRIPGBM-PAP RIPK2 labeling ~10-fold more potently than cRIPGBM (Figure S4, E and F). Collectively, we conclude that YM155 interacts with RIPK2 at the same binding site as cRIPGBM and that enhanced binding affinity directly tracks with observed improvements in cell-based potency. Taken together, these results suggest that RIPK2 is a relevant target of YM155 and that RIPK2 is required for YM155-mediated inhibition of survivin expression and induced cell death.
aYM155 penetrates the brain and inhibits tumor growth in a GBM intracranial xenograft model.
Although YM155 has been tested across a breadth of phase II clinical trials for various indications of cancer, it has never been tested in glioblastoma patients. This could stem from its reported lack of brain exposure35, an issue which significantly hampers the development of therapeutic agents for the treatment of brain cancers. Consistent with previous reports, in a brain tissue distribution pharmacokinetic study using 6-week-old CD-1 mice, we observed that YM155 (10 mg/kg, P.O.) has no brain exposure above limits of quantification (Figure 4A). In contrast, and consistent with what is observed for the relative brain exposure of RIPGBM and cRIPGBM, the aYM155 prodrug was found to possess reasonable brain exposure properties (10 mg/kg, P.O., brain Cmax= 32 nM, brain/plasma concentration ratio = 0.56, Figure 4A). Further, in a two-week mouse tolerability study in naïve animals, oral delivery of aYM155 was well tolerated at 1.0 and 5.0 mg/kg B.I.D. doses, based on weight loss and behavioral observations (Figure S5A). Modulation of the basicity of aromatic amine-containing systems has the potential liability of introducing hERG ion channel inhibitory activity. In a hERG channel inhibition assay, aYM155 prodrug was not found to significantly inhibit the channel at the highest tested concentration (< 20% inhibition at 3 μM, Figure S5B).
Figure 4.
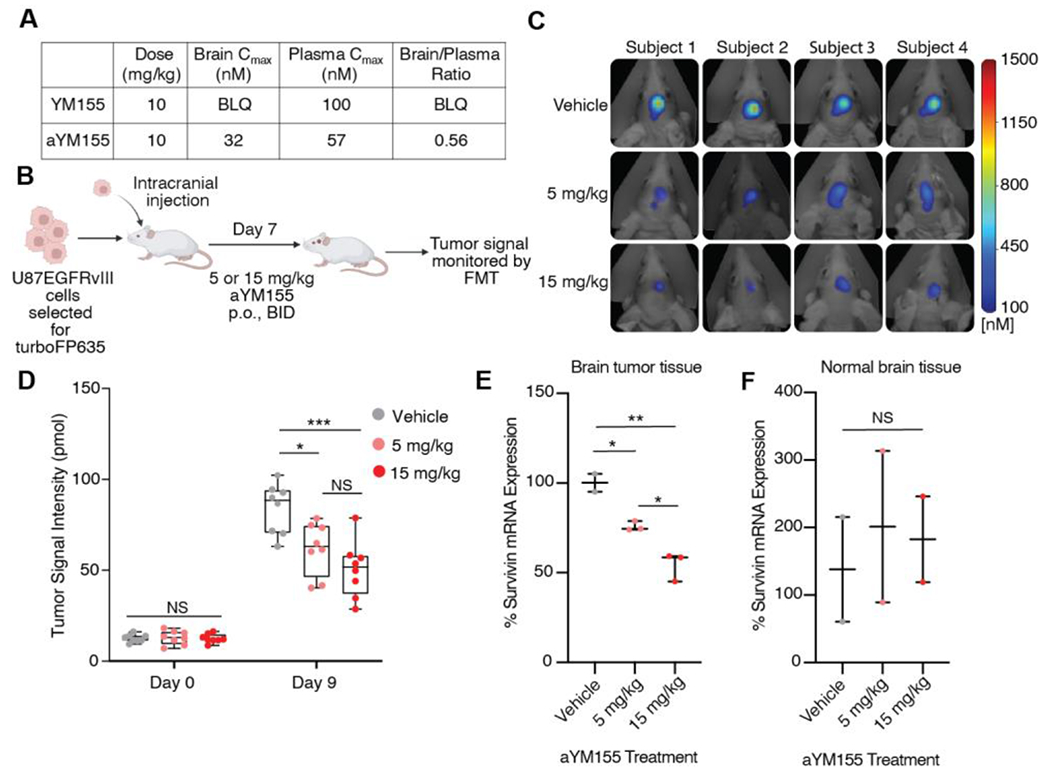
aYM155 penetrates the brain and reduces GBM tumor growth in an intracranial xenograft model. (A) Mouse tissue distribution properties of aYM155 (10 mg/kg, P.O.). (B) Schematic of GBM orthotopic intracranial xenograft model. U87EGFRvIII cells, engineered to carry turboFP635, were intracranially injected into six-week-old female athymic nude mice (n = 8 for each group). aYM155 was administered 7 days post injection. Created with Biorender.com. (C) Representative FMT tumor images of mice at day 9 of aYM155 dosing. (D) Tumor growth curves for mice treated with vehicle, 5 mg/kg, or 15 mg/kg aYM155 administered orally twice daily (n=8 mice per group). (E) qRT-PCR analysis of mouse brain tumor tissue. Student’s t-test was used to assess significance. Each data point represents the mean of 3 technical replicates per mouse (n = 2 mice for vehicle, n = 3 mice for 5 and 15 mg/kg). (F) qRT-PCR analysis of normal mouse brain tissue. Each data point represents the mean of 3 technical replicates per mouse (n = 2 mice). *P < 0.05, **P < 0.01, and ***P < 0.001. NS = not significant.
Given the cell-based selectivity profile of aYM155, its single digit nanomolar IC50 potency with respect to GBM cell killing, including GBM CSC and EGFRvIII-expressing cell types, as well as the observed ability of the prodrug strategy to facilitate a reasonable level of brain exposure, compared to the complete lack of exposure observed for YM155 parent drug, we next sought to assess in vivo efficacy using an orthotopic intracranial GBM mouse xenograft model. U87EGFRvIII cells were engineered to express the turboFP635 protein, to enable non-invasive measurement of tumor growth in the brains of mice using fluorescence molecular tomography (FMT) (Figure 4B). Oral administration of aYM155 (5mg/kg and 15 mg/kg, B.I.D.) to mice bearing U87EGFRvIII intracranial xenografts resulted in a significant inhibition of tumor growth, as monitored by FMT imaging (Figure 4, C and D). To explore the mechanism of action of aYM155 in vivo, we examined the impact of drug treatment on terminal survivin transcript levels, which was demonstrated to be a relevant and robust biomarker of RIPK2 target engagement in our cell-based experiments described above. Human tumor and control mouse brain tissue was harvested 1 hour following the last dose of a 9-day dosing regimen and subjected to RT-qPCR analysis of survivin mRNA transcript levels, using human specific probes designed for analysis of tumor tissue and mouse specific probes designed for analysis of normal brain tissue. At both evaluated doses, aYM155 was found to significantly decrease survivin expression levels specifically in human tumor tissue (Figure 4E) and had no effect in normal brain tissue (Figure 4F), which is consistent with aYM155 being specifically activated in the tumor microenvironment. Collectively, these data demonstrate that the aYM155 prodrug form of YM155 is able to penetrate the brain, reduce survivin specifically in tumor tissue, and significantly reduce orthotopic GBM tumor burden.
CONCLUSION
In summary, based on its structural similarity to cRIPGBM, as well as SAR insights for the chemical scaffold 10, we sought to characterize the clinical stage oncology drug YM155 in the context of GBM and to determine the potential relevance of RIPK2 in its mechanism of action. YM155 was found to be a highly potent anti-GBM agent, with pM to low nM cell-based IC50 cell killing being observed across a broad panel of primary GBM cell lines, including CSC-like and EGFRvIII-expressing lines. Notably, despite its extensive level of evaluation in clinical trials, since its discovery from a phenotypic screen for functional inhibitors of survivin, the relevant target and mechanism of action of YM155 remains unknown. Our findings suggest that the mechanism of YM155-induced suppression of survivin, as well as caspase-dependent cell death, is mediated by its direct interaction with RIPK2. By contributing to an understanding of the mode of action of this highly potent anti-cancer agent, elucidation of RIPK2 as a relevant molecular target of YM155 has implications that may help to facilitate the clinical potential and alternative applications of this drug. These findings also satisfy our objective of finding a more potent analog within the RIPGBM series for development as an anti-GBM agent, and the identified agent is a clinical stage drug.
By applying the same prodrug strategy that facilitates cell-based selectivity within the RIPGBM series, here, we have developed aYM155, which represents a prodrug form of YM155 that retains potent single digit and sub-nanomolar IC50 cell killing values in the context of GBM CSCs, while being significantly less potent on non-transformed control cell types (including neural cell types) when compared to YM155. The improved potency, with respect to RIPGBM, and improved selectivity, with respect to YM155, suggests that aYM155 could represent an excellent starting point for future development efforts in the context of brain cancers. Further, the aYM155 prodrug is also enabling for application of this chemical series in the context of this class of indications, as it facilitates distribution into the CNS, an issue that is frequently limiting and would directly limit YM155 in this context. Indeed, the enabled brain exposure properties of the prodrug allowed us to assess aYM155 activity in vivo using a relevant orthotopic intracranial GBM xenograft model. Based on its ability to significantly reduce brain tumor burden in vivo, its single digit and sub-nM potencies in cell killing assays involving a broad panel of GBM cell types, as well as its improved therapeutic index when compared to YM155, aYM155 represents a promising new potential therapeutic approach to the treatment of GBM.
Supplementary Material
Funding Sources
This work was supported by a grant from the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institute of Health (NIH) to L.L. and P.M. (5R01NS112482-02).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supporting information: Experimental procedures, chemical synthesis and characterization, RIPGBM and cRIPGBM cell toxicity, prodrug activation mechanism, RIPK2 and survivin western blots, RIPK1 knockdown cytotoxicity, Casp-1 activation, IκBα phosphorylation, YM155 binding, aYM155 non-tumor bearing mice toxicity study and hERG inhibition assay (PDF).
The authors declare no competing financial interest.
REFERENCES
- (1).Cloughesy TF; Cavenee WK; Mischel PS, Glioblastoma: from molecular pathology to targeted treatment. Annu Rev Pathol 2014, 9, 1–25. [DOI] [PubMed] [Google Scholar]
- (2).Louis DN; Ohgaki H; Wiestler OD; Cavenee WK; Burger PC; Jouvet A; Scheithauer BW; Kleihues P, The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007, 114 (2), 97–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Davis ME, Glioblastoma: Overview of Disease and Treatment. Clin J Oncol Nurs 2016, 20 (5 Suppl), S2–S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Dunn GP; Rinne ML; Wykosky J; Genovese G; Quayle SN; Dunn IF; Agarwalla PK; Chheda MG; Campos B; Wang A; Brennan C; Ligon KL; Furnari F; Cavenee WK; Depinho RA; Chin L; Hahn WC, Emerging insights into the molecular and cellular basis of glioblastoma. Genes & development 2012, 26 (8), 756–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Chen J; McKay RM; Parada LF, Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell 2012, 149 (1), 36–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Schonberg DL; Lubelski D; Miller TE; Rich JN, Brain tumor stem cells: Molecular characteristics and their impact on therapy. Mol Aspects Med 2014, 39, 82–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Bleau A-M; Hambardzumyan D; Ozawa T; Fomchenko EI; Huse JT; Brennan CW; Holland EC, PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell stem cell 2009, 4 (3), 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hambardzumyan D; Becher OJ; Rosenblum MK; Pandolfi PP; Manova-Todorova K; Holland EC, PI3K pathway regulates survival of cancer stem cells residing in the perivascular niche following radiation in medulloblastoma in vivo. Genes & development 2008, 22 (4), 436–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Bao S; Wu Q; McLendon RE; Hao Y; Shi Q; Hjelmeland AB; Dewhirst MW; Bigner DD; Rich JN, Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444 (7120), 756–60. [DOI] [PubMed] [Google Scholar]
- (10).Lucki NC; Villa GR; Vergani N; Bollong MJ; Beyer BA; Lee JW; Anglin JL; Spangenberg SH; Chin EN; Sharma A; Johnson K; Sander PN; Gordon P; Skirboll SL; Wurdak H; Schultz PG; Mischel PS; Lairson LL, A cell type-selective apoptosis-inducing small molecule for the treatment of brain cancer. Proceedings of the National Academy of Sciences 2019, 116 (13), 6435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Žídek A; Potapenko A; Bridgland A; Meyer C; Kohl SAA; Ballard AJ; Cowie A; Romera-Paredes B; Nikolov S; Jain R; Adler J; Back T; Petersen S; Reiman D; Clancy E; Zielinski M; Steinegger M; Pacholska M; Berghammer T; Bodenstein S; Silver D; Vinyals O; Senior AW; Kavukcuoglu K; Kohli P; Hassabis D, Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596 (7873), 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Altieri DC, Survivin and IAP proteins in cell-death mechanisms. Biochem J 2010, 430 (2), 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Kim YH; Kim SM; Kim YK; Hong SP; Kim MJ; Myoung H, Evaluation of survivin as a prognostic marker in oral squamous cell carcinoma. J Oral Pathol Med 2010, 39 (5), 368–75. [DOI] [PubMed] [Google Scholar]
- (14).Nigam J; Chandra A; Kazmi HR; Parmar D; Singh D; Gupta V; M, N., Expression of survivin mRNA in gallbladder cancer: a diagnostic and prognostic marker? Tumour Biol 2014, 35 (9), 9241–6. [DOI] [PubMed] [Google Scholar]
- (15).Nakahara T; Kita A; Yamanaka K; Mori M; Amino N; Takeuchi M; Tominaga F; Hatakeyama S; Kinoyama I; Matsuhisa A; Kudoh M; Sasamata M, YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res 2007, 67 (17), 8014–21. [DOI] [PubMed] [Google Scholar]
- (16).Nakahara T; Kita A; Yamanaka K; Mori M; Amino N; Takeuchi M; Tominaga F; Kinoyama I; Matsuhisa A; Kudou M; Sasamata M, Broad spectrum and potent antitumor activities of YM155, a novel small-molecule survivin suppressant, in a wide variety of human cancer cell lines and xenograft models. Cancer Sci 2011, 102 (3), 614–21. [DOI] [PubMed] [Google Scholar]
- (17).Kita A; Nakahara T; Yamanaka K; Nakano K; Nakata M; Mori M; Kaneko N; Koutoku H; Izumisawa N; Sasamata M, Antitumor effects of YM155, a novel survivin suppressant, against human aggressive non-Hodgkin lymphoma. Leuk Res 2011, 35 (6), 787–92. [DOI] [PubMed] [Google Scholar]
- (18).Winter GE; Radic B; Mayor-Ruiz C; Blomen VA; Trefzer C; Kandasamy RK; Huber KVM; Gridling M; Chen D; Klampfl T; Kralovics R; Kubicek S; Fernandez-Capetillo O; Brummelkamp TR; Superti-Furga G, The solute carrier SLC35F2 enables YM155-mediated DNA damage toxicity. Nat Chem Biol 2014, 10 (9), 768–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tolcher AW; Mita A; Lewis LD; Garrett CR; Till E; Daud AI; Patnaik A; Papadopoulos K; Takimoto C; Bartels P; Keating A; Antonia S, Phase I and pharmacokinetic study of YM155, a small-molecule inhibitor of survivin. J Clin Oncol 2008, 26 (32), 5198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Satoh T; Okamoto I; Miyazaki M; Morinaga R; Tsuya A; Hasegawa Y; Terashima M; Ueda S; Fukuoka M; Ariyoshi Y; Saito T; Masuda N; Watanabe H; Taguchi T; Kakihara T; Aoyama Y; Hashimoto Y; Nakagawa K, Phase I study of YM155, a novel survivin suppressant, in patients with advanced solid tumors. Clin Cancer Res 2009, 15 (11), 3872–80. [DOI] [PubMed] [Google Scholar]
- (21).Giaccone G; Zatloukal P; Roubec J; Floor K; Musil J; Kuta M; van Klaveren RJ; Chaudhary S; Gunther A; Shamsili S, Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J Clin Oncol 2009, 27 (27), 4481–6. [DOI] [PubMed] [Google Scholar]
- (22).Lewis KD; Samlowski W; Ward J; Catlett J; Cranmer L; Kirkwood J; Lawson D; Whitman E; Gonzalez R, A multi-center phase II evaluation of the small molecule survivin suppressor YM155 in patients with unresectable stage III or IV melanoma. Invest New Drugs 2011, 29 (1), 161–6. [DOI] [PubMed] [Google Scholar]
- (23).Aoyama Y; Kaibara A; Takada A; Nishimura T; Katashima M; Sawamoto T, Population pharmacokinetic modeling of sepantronium bromide (YM155), a small molecule survivin suppressant, in patients with non-small cell lung cancer, hormone refractory prostate cancer, or unresectable stage III or IV melanoma. Investigational new drugs 2013, 31 (2), 443–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wurdak H; Zhu S; Romero A; Lorger M; Watson J; Chiang CY; Zhang J; Natu VS; Lairson LL; Walker JR; Trussell CM; Harsh GR; Vogel H; Felding-Habermann B; Orth AP; Miraglia LJ; Rines DR; Skirboll SL; Schultz PG, An RNAi screen identifies TRRAP as a regulator of brain tumor-initiating cell differentiation. Cell Stem Cell 2010, 6 (1), 37–47. [DOI] [PubMed] [Google Scholar]
- (25).An Z; Aksoy O; Zheng T; Fan Q-W; Weiss WA, Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene 2018, 37 (12), 1561–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Perazzoli G; Prados J; Ortiz R; Caba O; Cabeza L; Berdasco M; Gónzalez B; Melguizo C, Temozolomide Resistance in Glioblastoma Cell Lines: Implication of MGMT, MMR, P-Glycoprotein and CD133 Expression. PLoS One 2015, 10 (10), e0140131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Weller M; Stupp R; Hegi ME; van den Bent M; Tonn JC; Sanson M; Wick W; Reifenberger G, Personalized care in neuro-oncology coming of age: why we need MGMT and 1p/19q testing for malignant glioma patients in clinical practice. Neuro Oncol 2012, 14 Suppl 4 (Suppl 4), iv100–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kaina B; Christmann M, DNA repair in resistance to alkylating anticancer drugs. Int J Clin Pharmacol Ther 2002, 40 (8), 354–67. [DOI] [PubMed] [Google Scholar]
- (29).Switzeny OJ; Christmann M; Renovanz M; Giese A; Sommer C; Kaina B, MGMT promoter methylation determined by HRM in comparison to MSP and pyrosequencing for predicting high-grade glioma response. Clinical Epigenetics 2016, 8 (1), 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tan SK; Jermakowicz A; Mookhtiar AK; Nemeroff CB; Schürer SC; Ayad NG, Drug Repositioning in Glioblastoma: A Pathway Perspective. Front Pharmacol 2018, 9, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Zhang W-H; Wang X; Narayanan M; Zhang Y; Huo C; Reed JC; Friedlander RM, Fundamental role of the Rip2/caspase-1 pathway in hypoxia and ischemia-induced neuronal cell death. Proceedings of the National Academy of Sciences of the United States of America 2003, 100 (26), 16012–16017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wang X; Wang H; Figueroa BE; Zhang WH; Huo C; Guan Y; Zhang Y; Bruey JM; Reed JC; Friedlander RM, Dysregulation of receptor interacting protein-2 and caspase recruitment domain only protein mediates aberrant caspase-1 activation in Huntington’s disease. J Neurosci 2005, 25 (50), 11645–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Hasegawa M; Fujimoto Y; Lucas PC; Nakano H; Fukase K; Núñez G; Inohara N, A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-κB activation. EMBO J 2008, 27 (2), 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Canning P; Ruan Q; Schwerd T; Hrdinka M; Maki JL; Saleh D; Suebsuwong C; Ray S; Brennan PE; Cuny GD; Uhlig HH; Gyrd-Hansen M; Degterev A; Bullock AN, Inflammatory Signaling by NOD-RIPK2 Is Inhibited by Clinically Relevant Type II Kinase Inhibitors. Chem Biol 2015, 22 (9), 1174–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Minematsu T; Sonoda T; Hashimoto T; Iwai M; Oppeneer T; Felder L; Shirai N; Miyashita A; Usui T, Pharmacokinetics, distribution and excretion of YM155 monobromide, a novel small-molecule survivin suppressant, in male and pregnant or lactating female rats. Biopharm Drug Dispos 2012, 33 (3), 160–9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.