

Abstract
The first use of 1,4-pentadiene and 1,5-hexadiene as allylmetal pronucleophiles in regio-, anti-diastereo- and enantioselective carbonyl addition from alcohol proelectrophiles is described. As corroborated by deuterium labelling experiments, primary alcohol dehydrogenation delivers a ruthenium hydride that affects alkene isomerization to furnish a conjugated diene followed by transfer hydrogenative carbonyl addition. Hydrometalation appears to be assisted by formation of a fluxional olefin-chelated homoallylic alkylruthenium complex II, which exists in equilibrium with its pentacoordinate η1 form to enable β-hydride elimination. This effect confers remarkable chemoselectivity: while 1,4-pentadiene and 1,5-hexadiene are competent pronucleophiles, higher 1,n-dienes are not, and olefinic functional groups of the products remain intact under conditions in which the 1,4- and 1,5-dienes isomerize. A survey of halide counterions reveals iodide-bound ruthenium-JOSIPHOS catalysts are uniquely effective in these processes. This method was used to prepare a previously reported C1-C7 substructure of (−)-pironetin in 4 vs 12 steps.
Graphical Abstract

INTRODUCTION
In the course of advancing catalytic methods for hydrogen transfer-mediated carbonyl additions,1 we recently developed novel “chain-walking processes”2 in which carbonyl vinylation or allylation mediated by vinyl halides3a,b and butadiene,3c respectively, is followed by redox isomerization to form saturated ketones (Figure 1).3,4 These rhodium-catalyzed processes rely on the reversible dissociation of monodentate phosphine ligands to facilitate generation of coordinatively unsaturated metal centers that engage in β-hydride elimination from the transient allylic or homoallylic rhodium alkoxides.3 In related ruthenium-catalyzed couplings of primary alcohols and butadiene,5 the homoallylic alcohol products resist redox isomerization due to “double chelation” of the metal by the product and the bis(phosphine) ligand, which leads to full occupancy of all coordination sites and suppression of β-hydride elimination (eq. 1, left). Indeed, if the chelating bis(phosphine) is replaced by a monodentate phosphine ligand, β-hydride elimination is restored (eq. 1, right).6
Figure 1.

Selected examples of merged metal-catalyzed carbonyl C-C coupling-redox isomerization.
 |
(eq. 1) |
The development of enantioselective metal-catalyzed carbonyl addition processes that merge alkene redox-isomerization and carbonyl addition appeared unlikely, as the majority of asymmetric carbonyl reductive couplings to form alcohol products require chiral bis(phosphine) ligands that tend to suppress β-hydride elimination. Reported ruthenium-catalyzed diene isomerizations suggested the feasibility of utilizing “skipped” dienes7 as precursors to conjugated diene pronucleophiles.8 In elegant work by Yin (Figure 1),9a 1,4-pentadienes were used as pronucleophiles for asymmetric ketone (Z)-dienylation, but the diene is activated via deprotonation of the allylic C-H bond. It was unclear whether isomerization via hydrometalation-β-hydride elimination would be possible, as 1,n-dienes such as 1,5-cyclooctadiene (COD) and norbornadiene (NBD) are potent chelators and can suppress catalysis.10 Despite this potential obstacle, we herewith report that iodide-bound ruthenium-JOSIPHOS catalysts recently developed in our laboratory5d,11 promote highly diastereo- and enantioselective redox-neutral C-C couplings of 1,4- or 1,5-diene pronucleophiles with primary alcohol proelectrophiles via hydrogen auto-transfer to form branched products of carbonyl addition as single regioisomers.12
RESULTS AND DISCUSSION
Conditions for merged enantioselective ruthenium-catalyzed redox-isomerization-carbonyl addition were inspired by those utilized for the coupling of 1-sec-alkyl-1-propynes13a or 1-aryl-1-propynes11,13b with primary alcohols, which occur through a dual catalytic process wherein alkyne-to-allene isomerization is followed by allene-carbonyl reductive coupling via hydrogen auto-transfer. In these processes, the dihydride precatalyst RuH2(CO)(PPh3)3 undergoes acid-base reaction14 with an arylsulfonic acid in the presence of Bu4NI and a JOSIPHOS ligand to affect generation of the active iodide-bound ruthenium-JOSIPHOS catalyst (eq. 2). As this catalyst was effective for alkyne-to-allene isomerization, it was posited that it might also promote the coupling of 1,4- pentadiene 1a with 1,3-propanediol tert-butyldiphenylsilyl ether 2a via isomerization of 1a to the conjugated diene. Indeed, after a brief survey of JOSIPHOS ligands,15 arylsulfonic acids and solvents, the catalyst generated from RuH2(CO)(PPh3)3 (5 mol%), SL-J502–2 (5 mol%), 2-NO2PhSO3H (5 mol%) and Bu4NI (10 mol%) in anisole16 (0.5 M) at 80 °C was found to promote formation of the targeted product of C-C coupling 3a in 99% yield as a single regioisomer with excellent control of diastereo- and enantioselectivity (Table 1, entry 1). The use of alternate chiral chelating phosphine ligands did not avail further improvement (Table 1, entries 2–6). Consistent with our prior observations,11 the iodide-bound catalyst performs better than the corresponding chloride or bromide complexes (Table 1, entries 1, 7–9).17 Notably, conditions developed for related couplings of 1,3-butadiene that exploit the precatalyst RuI(CO)(η3-C3H5) gave low yields (<10%),5d demonstrating the unique efficacy of the present catalytic system vis-á-vis olefin isomerization (see Figure S1).
Table 1.
Selected experiments deviating from optimal conditions for the ruthenium-catalyzed coupling of 1,4-pentadiene 1a with alcohol 2a to form homoallylic alcohol 3a.a

|
Yield of material isolated by silica gel chromatography. See Supporting Information for experimental details.
 |
(eq. 2) |
To assess reaction scope, optimal conditions identified for the conversion of 1,4-pentadiene 1a and alcohol 2a to provide homoallylic alcohol 3a were applied to primary alcohols 2b-2r (Table 2). The resulting branched homoallylic alcohols 3b-3r were formed as single regioisomers in good to excellent yields, anti-diastereo- and enantioselectivities. Notably, diverse nitrogen heterocycles were tolerated, including indoles (3b), oxazoles (3c), pyrroles (3d), pyrazoles (3e), pyridines (3f, 3l) phthalimides (3g) and azetidines (3i). Both primary aliphatic alcohols and benzylic alcohols are competent proelectrophiles, as demonstrated by the formation of 3a-3j, 3n-3r and 3k-3m, respectively. Finally, β-stereogenic primary alcohols 2n-2r were exposed to standard reaction conditions using the enantiomeric ruthenium-JOSIPHOS catalysts modified by SL-J502–2 and SL-J502–1. The resulting branched homoallylic alcohols 3n-3r and epi-3n-epi-3r were formed with moderate to good levels of catalyst-directed diastereoselectivity. Most remarkably, while 1,4-pentadiene participates in isomerization, olefinic functional groups of the reactants remain intact, as do the terminal homoallylic olefins of the reaction products. Such chemoselectivity suggests hydrometalation of the skipped diene pronucleophile is chelation-assisted.
Table 2.
Ruthenium-catalyzed coupling of 1,4-pentadiene 1a with alcohols 2a-2r to form homoallylic alcohols 2a-2r.a

|
Yields are of material isolated by silica gel chromatography. Enantioselectivities were determined by HPLC analysis. Diastereoselectivities were determined by 1H NMR analysis of crude reaction mixtures. b7.5 mol% catalyst. cPhOMe (1.0 M). d10 mol% catalyst. e1,4-pentadiene (1000 mol%). f70 °C. g100 °C. h60 °C. See Supporting Information for experimental details.
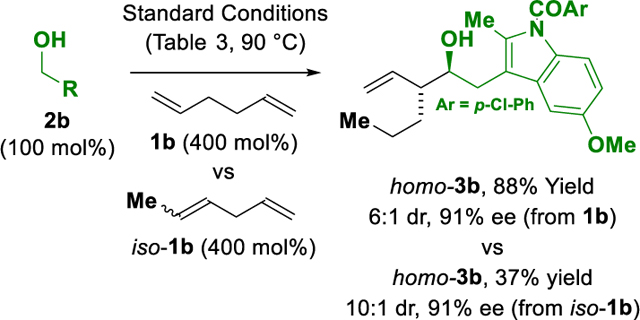
To further explore the scope of this process and probe the influence of chelation on the efficiency of tandem isomerization-hydrogen auto-transfer carbonyl addition, reactions of higher skipped dienes were attempted. 1,5-Hexadiene 1b was found to react with alcohols 2a-2c, 2e, 2q to form the homologous homoallylic alcohols homo-3a-3c, 3e, 3q, epi-3q, although more forcing conditions were required (Table 3). For 1,5-hexadienes, it was also necessary to change the iodide source from Bu4NI to LiI to maintain optimal yields of product. Corresponding reactions of 1,6-heptadiene failed to deliver preparatively useful quantities of the targeted coupling products, and skipped cyclic dienes did not participate in C-C coupling. As illustrated in equation 3, 1,4-hexadiene forms the same product as 1,5-hexadiene, but in significantly lower yield (37% vs 88% yield, respectively). These data again underscore the importance of chelation-initiated isomerization, as 1,5-hexadiene incorporates less substituted olefin moieties, and the stability of late transition metal-olefin π-complex decreases with increasing degree of olefin substitution.18 Finally, 3-methyl-1,4-pentadiene 1c reacts with benzylic alcohol 2k to form Me-3k in good yield (eq. 4). Although absolute stereocontrol was somewhat modest, diastereo- and enantioselective formation of an acyclic quaternary carbon stereocenter bearing methyl, ethyl and vinyl moieties is notable.19
Table 3.
Ruthenium-catalyzed coupling of 1,5-hexadiene 1b with alcohols 2a-2c, 2e, 2q to form the homologous homoallylic alcohols homo-3a-3c, 3e, 3q, epi-3q.a

|
Yields are of material isolated by silica gel chromatography. Enantioselectivities were determined by HPLC analysis. Diastereoselectivities were determined by 1H NMR analysis of crude reaction mixtures. b1,5-hexadiene (1000 mol%). cPhOMe (0.25 M). See Supporting Information for experimental details.
 |
(eq. 3) |
 |
(eq. 4) |
The stereodiads formed via ruthenium-catalyzed coupling of 1,4-pentadiene 1a represent polyketide “butyrate” substructures.20 This motif is evident in (−)-pironetin,20b,21,22 a microtubule destabilizing agent that arrests cell cycle progression at the G2/M phase.22,23 Many FDA approved anticancer drugs perturb microtubule dynamics,24 however, pironetin is unique as it is the only microtubule destabilizing agent that binds α-tubulin.22,25 Consequently, pironetin has attracted the attention of synthetic chemists, resulting in over a dozen total syntheses,26 as well as the synthesis and evaluation of simplified analogues.22 To illustrate the utility of the present ruthenium-catalyzed coupling of 1,4-pentadiene 1a, a known C1-C7 substructure of pironetin was prepared using this method (Scheme 1).26j Thus, propane diol mono-TBS ether 2s was subjected to standard conditions for C-C coupling to form the homoallylic alcohol 3s (not shown), which was exposed to crotonic acid under Mitsunobu conditions27 to furnish the α,β-unsaturated ester 4 with good control of relative and absolute stereochemistry. Ring-closing metathesis27 followed by removal of the TBS ether28 provides the C1-C7 substructure 5 in 4 steps; a compound that previously required a 12-step preparation.26j
Scheme 1.

Synthesis of C1-C7 of pironetin.a
aYields are of material isolated by silica gel chromatography. Enantioselectivities were determined by HPLC analysis. Diastereoselectivities were determined by 1H NMR analysis of crude reaction mixtures. See Supporting Information for experimental details.
A catalytic cycle has been proposed and corroborated by a deuterium labelling experiment (Scheme 2). Hydroruthenation of 1,4-pentadiene 1a by the ruthenium hydride I provides the chelated homoallylic alkylruthenium complex II. Hydrometalation appears to be assisted by formation of the indicated chelate, as 1,4-pentadiene and 1,5-hexadiene are competent pronucleophiles while higher 1,n-dienes are not. This assertion is consistent with a study revealing that 1,4- and 1,5-dienes form chelated ruthenium carbonyl complexes while 1,6-dienes do not.7a Just as a COD or NBD ligand can dissociate from a metal precatalyst, olefin binding in the chelated homoallylic alkylruthenium complex II is fluxional, thus providing an open coordination site for β-hydride elimination of the allylic C-H to form the hydride-containing conjugated η2-diene-ruthenium complex III. Diene hydroruthenation forms the π-allylruthenium complex IV.29 Carbonyl addition from the primary σ-allylruthenium haptomer (not shown) provides the homoallylic ruthenium alkoxide V. Finally, alkoxide exchange with the reactant alcohol 2 releases the product of C-C coupling 3 and forms the primary ruthenium alkoxide VI. β-Hydride elimination from the alkoxide concomitantly forms the aldehyde dehydro-2 and the ruthenium hydride I to close the catalytic cycle. To corroborate this interpretation of the mechanism, the isotopically labeled primary alcohol deuterio-2a was treated with 1,4-pentadiene 1a under standard reaction conditions. The reaction product deuterio-3a incorporated deuterium at every diene-derived carbon atom. Additionally, significant loss of deuterium was observed at the carbinol position. These data demonstrate highly reversible diene hydrometalation-β-hydride elimination and highly reversible hydrogen transfer from the alcohol reactant 2 to 1,4-pentadiene 1a. Re-exposure of the reaction product deuterio-3a to the reaction conditions did not alter the extent of deuterium incorporation at the carbinol position, suggesting chelation of the homoallylic olefin suppresses β-hydride elimination from the homoallylic alkoxide V.
Scheme 2.

Proposed catalytic cycle for successive alkene isomerization-hydrogen auto-transfer carbonyl allylation as corroborated by deuterium labelling experiments.a
aDeuterated materials were characterized by 1H NMR, 2H NMR and HRMS. See Supporting Information for further experimental details.
CONCLUSION
In summary, we report the first use of nonconjugated dienes as allylmetal pronucleophiles in regio-, anti-diastereo- and enantioselective carbonyl additions from alcohol proelectrophiles. As demonstrated by the formation of the C1-C7 substructure of pironetin (4 vs 12 steps), these processes streamline access to butyrate substructures evident in type I polyketide natural products. The collective data, including deuterium labeling studies, corroborate a catalytic mechanism in which hydrometalation of the non-conjugated diene appears to be assisted by formation of the fluxional olefin-chelated homoallylic alkylruthenium complex II, which exists in equilibrium with its pentacoordinate η1 form to enable β-hydride elimination. This effect results in remarkable chemoselectivity: while 1,4-pentadiene and 1,5-hexadiene are competent pronucleophiles higher 1,n-dienes are not, and olefinic functional groups of the products remain intact under conditions in which the 1,4- and 1,5-skipped dienes isomerize.
Supplementary Material
Acknowledgments.
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM069445) are acknowledged for financial support.
Footnotes
The authors declare no competing financial interest.
Supporting Information Placeholder
Supporting Information Available: Experimental procedures and spectral data for all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- (1).For selected reviews on enantioselective carbonyl addition via alcohol-mediated hydrogen auto-transfer, see:Kim SW; Zhang W; Krische MJ Catalytic Enantioselective Carbonyl Allylation and Propargylation via Alcohol-Mediated Hydrogen Transfer: Merging the Chemistry of Grignard and Sabatier. Acc. Chem. Res 2017, 50, 2371−2380.Doerksen RS; Meyer CC; Krische MJ Feedstock Reagents in Metal-Catalyzed Carbonyl Reductive Coupling: Minimizing Preactivation for Efficiency in Target-Oriented Synthesis. Angew. Chem. Int. Ed 2019, 58, 14055–14064.Santana CG; Krische MJ From Hydrogenation to Transfer Hydrogenation to Hydrogen Auto-Transfer in Enantioselective Metal-Catalyzed Carbonyl Reductive Coupling: Past, Present and Future. ACS Catal. 2021, 11, 5572–5585.Ortiz E; Shezaf JZ; Shen W; Krische MJ Historical Perspective on Ruthenium-Catalyzed Hydrogen Transfer and Survey of Enantioselective Hydrogen Auto-Transfer Processes for the Conversion of Lower Alcohols to Higher Alcohols. Chem. Sci 2022, 13, 12625–12633.
- (2).For selected reviews on metal-catalyzed migratory functionalization of olefins, see:Vasseur A; Bruffaerts J; Marek I. Remote Functionalization through Alkene Isomerization. Nat. Chem 2016, 8, 209–219.Sommer H; Julia-Hernandez F; Martin R; Marek I. Walking Metals for Remote Functionalization. ACS Cent. Sci 2018, 4, 153–165.Kochi T; Kanno S; Kakiuchi F. Nondissociative Chain Walking as a Strategy in Catalytic Organic Synthesis. Tetrahedron Lett. 2019, 60, 150938.Li Y; Wu D; Cheng H-G; Yin G. Difunctionalization of Alkenes Involving Metal Migration. Angew. Chem. Int. Ed 2020, 59, 7990–8003.Janssen-Mueller D; Sahoo B; Sun S-Z; Martin R. Tackling Remote sp3 C-H Functionalization via Ni-Catalyzed “Chain-Walking” Reactions. Isr. J. Chem 2020, 60, 195–206.Dhungana RK; Sapkota RR; Niroula D; Giri R. Walking Metals: Catalytic Difunctionalization of Alkenes at Nonclassical Sites. Chem. Sci 2020, 11, 9757–9774.Massad I; Marek I. Alkene Isomerization through Allylmetals as a Strategic Tool in Stereoselective Synthesis. ACS Catal. 2020, 10, 5793–5804.Bonfield HE; Valette D; Lindsay DM; Reid M. Stereoselective Remote Functionalization via Palladium-Catalyzed Redox-Relay Heck Methodologies. Chem. Eur. J 2021, 27, 158–174.Ghosh S; Patel S; Chatterjee I. Chain-Walking Reactions of Transition Metals for Remote C-H Bond Functionalization of Olefinic Substrates. Chem. Commun 2021, 57, 11110–11130.Fiorto D; Scaringi S; Mazet C. Transition Metal-Catalyzed Alkene Isomerization as an Enabling Technology in Tandem, Sequential and Domino Processes. Chem. Soc. Rev 2021, 50, 1391–1406.Zhang M; Ji Y; Zhang C. Transition Metal Catalyzed Enantioselective Migratory Functionalization Reactions of Alkenes through Chain-Walking. Chin. J. Chem 2022, 40, 1608–1622.He Y; Tao R; Zhu S. Nickel-Hydride Catalyzed Remote Hydroarylation of Olefins. Synlett 2022, 33, 224–230.
- (3) <j/>(a).Swyka RA; Shuler WG; Spinello BJ; Zhang W; Lan C; Krische MJ Conversion of Aldehydes to Branched or Linear Ketones via Regiodivergent Rhodium-Catalyzed Vinyl Bromide Reductive Coupling-Redox Isomerization Mediated by Formate. J. Am. Chem. Soc 2019, 141, 6864–6868. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shuler WG; Swyka RA; Schempp TT; Spinello BJ; Krische MJ Vinyl Triflate-Aldehyde Reductive Coupling-Redox Isomerization Mediated by Formate: Rhodium-Catalyzed Ketone Synthesis in the Absence of Stoichiometric Metals. Chem. Eur. J 2019, 25, 12517–12520. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Spinello BJ; Wu J; Cho Y; Krische MJ Conversion of Primary Alcohols and Butadiene to Branched Ketones via Merged Transfer Hydrogenative Carbonyl Addition-Redox Isomerization Catalyzed by Rhodium. J. Am. Chem. Soc 2021, 143, 13507–13512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).For selected reviews on metal-catalyzed alkene isomerization, see:Uma R; Crevisy C; Gree R. Transposition of Allylic Alcohols into Carbonyl Compounds Mediated by Transition Metal Complexes. Chem. Rev 2003, 103, 27–52.Mantilli L; Mazet C. Platinum Metals in the Catalytic Asymmetric Isomerization of Allylic Alcohols. Chem. Lett 2011, 40, 341–344.Larionov E; Li H; Mazet C. Well-Defined Transition Metal Hydrides in Catalytic Isomerizations. Chem. Commun 2014, 50, 9816–9826.Cahard D; Gaillard S; Renaud J-L Asymmetric Isomerization of Allylic Alcohols. Tetrahedron Lett. 2015, 56, 6159–6169.
- (5) <j/>(a).Zbieg JR; Moran J; Krische MJ Diastereo- and Enantioselective Ruthenium Catalyzed Hydrohydroxyalkylation of 2-Silyl-Butadienes: Carbonyl syn-Crotylation from the Alcohol Oxidation Level. J. Am. Chem. Soc 2011, 133, 10582–10586. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zbieg JR; Yamaguchi E; McInturff EL; Krische MJ Enantioselective C-H Crotylation of Primary Alcohols via Hydrohydroxyalkylation of Butadiene. Science 2012, 336, 324–327. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) McInturff EL; Yamaguchi E; Krische MJ Chiral-Anion-Dependent Inversion of Diastereo- and Enantioselectivity in Carbonyl Crotylation via Ruthenium-Catalyzed Butadiene Hydrohydroxyalkylation. J. Am. Chem. Soc 2012, 134, 20628–20631. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ortiz E; Spinello BJ; Cho Y; Wu J; Krische MJ Stereo- and Site-Selective Crotylation of Alcohol Proelectrophiles via Ruthenium-Catalyzed Hydrogen Auto-Transfer Mediated by Methylallene and Butadiene. Angew. Chem. Int. Ed 2022, 61, e202212814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Shibahara F; Bower JF; Krische MJ Diene Hydroacylation from the Alcohol or Aldehyde Oxidation Level via Ruthenium-Catalyzed C-C Bond-Forming Transfer Hydrogenation: Synthesis of β,γ-Unsaturated Ketones. J. Am. Chem. Soc 2008, 130, 14120–14122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).For ruthenium-catalyzed diene isomerization, see:Wuu YM; Zou C; Wrighton MS Thermal Reactions of Tricarbonylbis(ethene)ruthenium with Acrylic, Nonconjugated Dienes and Photochemistry of Ru(CO)4(η2-diene) Complexes. Inorg. Chem 1988, 27, 3039–3044.Wakamatsu H; Nishida M; Adachi N; Mori M. Isomerization Reaction of Olefin Using RuClH(CO)(PPh3)3. J. Org. Chem 2000, 65, 3966–3970.Clark JR; Griffiths JR; Diver ST Ruthenium Hydride-Promoted Dienyl Isomerization: Access to Highly Substituted 1,3-Dienes. J. Am. Chem. Soc 2013, 135, 3327–3330.Scaringi S; Mazet C. Kinetically Controlled Stereoselective Access to Branched 1,3-Dienes by Ru-Catalyzed Remote Conjugative Isomerization. ACS Catal. 2021, 11, 7970–7977.
- (8).For reviews on the use of diene pronucleophiles in metal-catalyzed carbonyl reductive and redox-neutral C-C couplings, see:Holmes M; Schwartz LA; Krische MJ Intermolecular Metal-Catalyzed Reductive Coupling of Dienes, Allenes and Enynes with Carbonyl Compounds and Imines. Chem. Rev 2018, 118, 6026–6052.Xiang M; Pfaffinger DE; Krische MJ Allenes and Dienes as Chiral Allylmetal Pronucleophiles in Catalytic Enantioselective C=X Addition: Historical Perspective and State-of-the-Art Survey. Chem. Eur. J 2021, 27, 13107–13116.
- (9) <j/>(a).Zhong F; Pan Z-Z; Zhou S-W; Zhang H-J; Yin L. Copper(I)-Catalyzed Regioselective Asymmetric Addition of 1,4- Pentadiene to Ketones. J. Am. Chem. Soc 2021, 143, 4556–4562. [DOI] [PubMed] [Google Scholar]; (b) Pan Z-Z; Pan D; Li J-H; Xue X-S; Yin L. Copper(I)-Catalyzed Asymmetric Conjugate Addition of 1,4-Dienes to β‑Substituted Alkenyl Azaarenes. J. Am. Chem. Soc 2023, 145, 1749–1758. [DOI] [PubMed] [Google Scholar]
- (10).Cobley CJ; Lennon IC; McCague R; Ramsden JA; Zanotti-Gerosa A. On the Economic Application of DuPHOS Rhodium(I) Catalysts: A Comparison of COD versus NBD Precatalysts. Tetrahedron Lett. 2001, 42, 7481–7483. [Google Scholar]
- (11).For a study on halide effects in ruthenium-catalyzed carbonyl allylation via hydrogen auto-transfer, see: Ortiz E; Shezaf JZ; Chang Y-H; Gonçalves TP; Huang K-W; Krische MJ. Understanding Halide Counterion Effects in Enantioselective Ruthenium-Catalyzed Carbonyl (α-Aryl)allylation: Alkynes as Latent Allenes and Trifluoroethanol-Enhanced Turnover in The Conversion of Ethanol to Higher Alcohols via Hydrogen Auto-Transfer. J. Am. Chem. Soc 2021, 143, 16709–16717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).For tandem metal-catalyzed vinylboronate-to-allylboronate isomerization-aldehyde allylboration, see:Shimizu H; Igarashi T; Miura T; Murakami M. Rhodium-Catalyzed Reaction of 1-Alkenylboronates with Aldehydes Leading to Allylation Products. Angew. Chem. Int. Ed 2011, 50, 11465–11469.Miura T; Nakahashi J; Murakami M. Enantioselective Synthesis of (E)-δ-Boryl-Substituted anti-Homoallylic Alcohols Using Palladium and a Chiral Phosphoric Acid. Angew. Chem. Int. Ed 2017, 56, 6989–6993.Miura T; Nakahashi J; Zhou W; Shiratori Y; Stewart SG; Murakami M. Enantioselective Synthesis of anti-1,2-Oxaborinan-3-enes from Aldehydes and 1,1-Di(boryl)alk-3-enes Using Ruthenium and Chiral Phosphoric Acid Catalysts. J. Am. Chem. Soc 2017, 139, 10903–10908.Miura T; Nakahashi J; Sasatsu T; Murakami M. Synthesis of γ-Boryl-Substituted Homoallylic Alcohols with anti-Stereochemistry Based on a Double-Bond Transposition. Angew. Chem. Int. Ed 2019, 58, 1138–1142.Miura T; Oku N; Murakami M. Diastereo-and Enantioselective Synthesis of (E)-δ-Boryl-Substituted anti-Homoallylic Alcohols in Two Steps from Terminal Alkynes. Angew. Chem. Int. Ed 2019, 58, 14620–14624.
- (13) <j/>(a).Liang T; Nguyen KD; Zhang W; Krische MJ Enantioselective Ruthenium Catalyzed Carbonyl Allylation via Alkyne-Alcohol C-C Bond Forming Transfer Hydrogenation: Allene Hydrometallation vs. Oxidative Coupling. J. Am. Chem. Soc 2015, 137, 3161–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xiang M; Ghosh A; Krische MJ Diastereo- and Enantioselective Ruthenium-Catalyzed C-C Coupling of 1-Arylpropynes and Alcohols: Alkynes as Chiral Allylmetal Precursors in Carbonyl anti-(α-Aryl)allylation. J. Am. Chem. Soc 2021, 143, 2838–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For acid-base reactions of ruthenium dihydrides with Brønsted acids, see ref. 5b,c and the following study: Dobson A; Robinson SD; Uttley MF Complexes of the Platinum Metals. Part V. Perfluorocarboxylato Derivatives. J. Chem. Soc., Dalton Trans 1975, 370–377. [Google Scholar]
- (15).Blaser H-U; Brieden W; Pugin B; Spindler F; Studer M; Togni A. Solvias Josiphos Ligands: From Discovery to Technical Applications. Top. Catal 2002, 19, 3−16. [Google Scholar]
- (16).Anisole is among the most desirable solvents for industrial chemical synthesis: Byrne FP; Jin S; Paggiola G; Petchey THM; Clark JH; Farmer TJ; Hunt AJ; McElroy CR; Sherwood J. Tools and Techniques for Solvent Selection: Green Solvent Selection Guides. Sustain. Chem. Process 2016, 4, 7/1−7/24. [Google Scholar]
- (17).For selected reviews on halide effects in transition metal catalysis, see:Maitlis PM; Haynes A; James BR; Catellani M; Chiusoli GP Iodide Effects in Transition Metal Catalyzed Reactions. Dalton Trans 2004, 3409–3419.Fagnou K; Lautens M. Halide Effects in Transition Metal Catalysis. Angew. Chem. Int. Ed 2002, 41, 26–47.
- (18) <j/>(a).Cramer R. Olefin Coordination Compounds of Rhodium. V. Relative Stabilities and Rates of Exchange of Olefin Complexes of Rhodium(I). J. Am. Chem. Soc 1967, 89, 4621–4626. [Google Scholar]; (b) Jesse AC; Cordfunke EHP; Ouweltjes W. Calorimetric Investigation of the Reaction of 1,5-Cyclooctadiene with Complexes of the Type (acac)M(olefin)2 [M = Rh(I), Ir(I)]. Thermochim. Acta 1979, 30, 293–302. [Google Scholar]
- (19).For selected reviews on the enantioselective formation of acyclic quaternary carbon stereocenters, see: [Google Scholar]; (a) Das JP; Marek I. Enantioselective Synthesis of All-Carbon Quaternary Stereogenic Centers in Acyclic Systems. Chem. Commun 2011, 47, 4593–4623. [DOI] [PubMed] [Google Scholar]; (b) Marek I; Sklute G. Creation of Quaternary Stereocenters in Carbonyl Allylation Reactions. Chem. Commun 2007, 1683–1691. [DOI] [PubMed] [Google Scholar]; (c) Marek I; Minko Y; Pasco M; Mejuch T; Gilboa N; Chechik H; Das JP All-Carbon Quaternary Stereogenic Centers in Acyclic Systems through the Creation of Several C–C Bonds per Chemical Step. J. Am. Chem. Soc 2014, 136, 2682–2694. [DOI] [PubMed] [Google Scholar]; (d) Minko Y; Marek I. Stereodefined Acyclic Trisubstituted Metal Enolates towards the Asymmetric Formation of Quaternary Carbon Stereocentres. Chem. Commun 2014, 50, 12597–12611. [DOI] [PubMed] [Google Scholar]; (e) Feng J; Holmes M; Krische MJ Acyclic Quaternary Carbon Stereocenters via Enantioselective Transition Metal Catalysis. Chem. Rev 2017, 117, 12564–12580. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Pierrot D; Marek I. Synthesis of Enantioenriched Vicinal Tertiary and Quaternary Carbon Stereogenic Centers within an Acyclic Chain. Angew. Chem. Int. Ed 2020, 59, 36–49. [DOI] [PubMed] [Google Scholar]
- (20) <j/>(a).Vital M; Howe AC; Tiedje JM Revealing the Bacterial Butyrate Synthesis Pathways by Analyzing (Meta)genomic Data. mBio 2014, 5, e00889–14. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kobayashi S; Tsuchiya K; Nishide M; Nishikiori T; Nakagawa T; Shimada N. Pironetin, a Novel Plant Growth Regulator Produced by Streptomyces sp. NK10958. J. Antibiot 1995, 48, 893–895. [PubMed] [Google Scholar]
- (21).Pironetin was isolated independently by two research teams from Streptomyces sp. NK-10958:Kobayashi S; Tsuchiya K; Harada T; Nishide M; Kurokawa T; Nakagawa T; Shimada N; Kobayashi K. Pironetin, A Novel Plant Growth Regulator Produced by Streptomyces sp. NK10958. I. Taxonomy, Production, Isolation, and Preliminary Characterization. J. Antibiot 1994, 47, 697–702.Kobayashi S; Tsuchiya K; Kurokawa T; Nakagawa T; Shimada N; Iitaka Y. Pironetin, A Novel Plant Growth Regulator Produced by Streptomyces sp. NK10958. II. Structural Elucidation. J. Antibiot 1994, 47, 703–707.Tsuchiya K; Kobayashi S; Nishikiori T; Nakagawa T; Tatsuta K. NK10958P, a Novel Plant Growth Regulator Produced by Streptomyces sp. J. Antibiot 1997, 50, 259–260.Yoshida T; Koizumi K; Kawamura Y; Matsumoto K; Itazaki H. Lactone with Immunosuppressive Activity. Japan: Patent Kokai 5–310726, 1993.Yoshida T; Koizumi K; Kawamura Y; Matsumoto K; Itazaki H. Lactone with Immunosuppressive Activity. European: Patent 560389 A1, 1993.
- (22).For a review of the biological properties, total syntheses, and structure activity relationships involving pironetin, see: Coulup SK; Georg GI Revisiting Microtubule Targeting Agents: α-Tubulin and the Pironetin Binding Site as Unexplored Targets for Cancer Therapeu-tics. Bioorg. Med. Chem. Lett 2019, 29, 1865–1873. [DOI] [PubMed] [Google Scholar]
- (23).Usui T; Watanabe H; Nakayama H; Tada Y; Kanoh N; Kondoh M; Asao T; Takio K; Watanabe H; Nishikawa K; Kitahara T; Osada H. The Anticancer Natural Product Pironetin Selectively Targets Lys352 of α-Tubulin. Chem. Biol 2004, 11, 799–806. [DOI] [PubMed] [Google Scholar]
- (24).Reviews:Jordan MA; Wilson L. Microtubules as a Target for Anticancer Drugs. Nat. Rev. Cancer 2004, 4, 253–265.Dumontet C; Jordan MA Microtubule-Binding Agents: A Dynamic Field of Cancer Therapeutics. Nat. Rev. Drug Discov 2010, 9, 790–803.Rohena CC; Mooberry SL Recent Progress with Microtubule Stabilizers: New Compounds, Binding Modes and Cellular Activities Nat. Prod. Rep 2014, 31, 335–355.
- (25).Review: Alpízar-Pedraza D; de la Nuez Veulens; Araujo EC; Piloto-Ferrer J; Sánchez-Lamar Á. Microtubules Destabilizing Agents Binding Sites in Tubulin. J. Mol. Struc 2022, 1259, 132723. [Google Scholar]
- (26).For total syntheses of pironetin, see: [Google Scholar]; (a) Yasui K; Tamura Y; Nakatani T; Kawada K; Ohtani M. Total Synthesis of (−)-PA-48153C, a Novel Immunosuppressive 2-Pyranone Derivative. J. Org. Chem 1995, 60, 7567–7574. [Google Scholar]; (b) Gurjar MK; Henri JT Jr.; Bose DS; Rao AVR Total Synthesis of a Potent Immunosuppressant Pironetin. Tetrahedron Lett. 1996, 37, 6615–6618. [Google Scholar]; (c) Gurjar MK; Chakrabarti A; Rao AVR. A Stereocontrolled Synthesis of Pironetin. Heterocycles 1997, 45, 7–10. [Google Scholar]; (d) Chida N; Yoshinaga M; Tobe T; Ogawa S. Total Synthesis of (−)-PA-48153C (Pironetin) Utilising L-Quebrachitol as a Chiral Building Block. Chem. Commun 1997, 1043–1044. [Google Scholar]; (e) Watanabe H; Watanabe H; Kitahara T. Total Synthesis of (−)-Pironetin. Tetrahedron Lett. 1998, 39, 8313–8316. [Google Scholar]; (f) Watanabe H; Watanabe H; Bando M; Kido M; Kitahara T. An Efficient Synthesis of Pironetins Employing a Useful Chiral Building Block, (1S,5S,6R)-5-Hydroxybicylco[4.1.0]heptan-2-one. Tetrahedron 1999, 55, 9755–9776. [Google Scholar]; (g) Keck GE; Knutson CE; Wiles SA Total Synthesis of the Immunosuppressant (−)-Pironetin (PA48153C). Org. Lett 2001, 3, 707–710. [DOI] [PubMed] [Google Scholar]; (h) Dias LC; de Oliveira LG; de Sousa MA Total Synthesis of (−)-Pironetin. Org. Lett 2003, 5, 265–268. [DOI] [PubMed] [Google Scholar]; (i) Shen X; Wasmuth AS; Zhao J; Zhu C; Nelson SG Catalytic Asymmetric Assembly of Stereodefined Propionate Units: An Enantioselective Total Synthesis of (−)-Pironetin. J. Am. Chem. Soc 2006, 128, 7438–7439. [DOI] [PubMed] [Google Scholar]; (j) Enders D; Dhulut S; Steinbusch D; Herrbach A. Asymmetric Total Synthesis of (−)-Pironetin Employing the SAMP/RAMP Hydrazone Methodology. Chem. Eur. J 2007, 13, 3942–3949. [DOI] [PubMed] [Google Scholar]; (k) Bressy C; Vors J-P; Hillebrand S; Arseniyadis S; Cossy J. Asymmetric Total Synthesis of the Immunosuppressant (−)-Pironetin. Angew. Chem. Int. Ed 2008, 47, 10137–10140. [DOI] [PubMed] [Google Scholar]; (l) Crimmins MT; Dechert A-M Enantioselective Total Synthesis of (−)-Pironetin: Iterative Aldol Reactions of Thiazolidinethiones. Org. Lett 2009, 11, 1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Gu Q; Kong L; Yang L; Zhu L; Hong R. A Stereotetrad-Centered Approach toward Pironetin: Dead Ends, Detour, and Evolution of the Synthetic Strategy. Tetrahedron 2020, 76, 131660. [Google Scholar]; (n) Yang L; Kong L; Gu Q; Shao S; Lin G-Q; Hong R. Stereoselective Access to Polypropionates Expedited by the Double Hydroboration of Allenes: Total Synthesis of Antitumor (−)-Pironetin. CCS Chem. 2020, 3, 769–779. [Google Scholar]
- (27).For a related Mitsunobu reaction and ring-closing metathesis, see: Walleser P; Brückner R. Stereocontrolled Synthesis of a C1–C10 Building Block (“Southwestern Moiety”) for the Unnatural Enantiomers of the Polyene Polyol Antibiotics Filipin III and Pentamycin: A Sultone-Forming Ring-Closing Metathesis for Protection of Homoallylic Alcohols. Eur. J. Org. Chem 2014, 15, 3210–3224. [Google Scholar]
- (28).Böse D; Fernández E; Pietruszka J. Stereoselective Synthesis of Both Enantiomers of Rugulactone. J. Org. Chem 2011, 76, 3463–3469. [DOI] [PubMed] [Google Scholar]
- (29).For stoichiometric reactions of HXRu(CO)(PR3)3 (X = Cl, Br) with allenes or dienes to form discrete π-allylruthenium complexes, see:Hiraki K; Ochi N; Sasada Y; Hayashida H; Fuchita Y; Yamanaka S. Organoruthenium(II) Complexes Formed by Insertion Reactions of Some Vinyl Compounds and Conjugated Dienes into a Hydrido–Ruthenium Bond. J. Chem. Soc., Dalton Trans 1985, 873–877.Hill AF; Ho CT; Wilton-Ely JDET The Coupling of Methylene and Vinyl Ligands at a Ruthenium(II) Centre. Chem. Commun 1997, 2207–2208.Xue P; Bi S; Sung HHY; Williams ID; Lin Z; Jia G. Isomerism of [Ru(η3-allyl)Cl(CO)(PPh3)2] Organometallics 2004, 23, 4735–4743.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.