

Patients with head and neck squamous cell carcinoma that responded to dual PD-1/CTLA4 immune checkpoint blockade exhibited a parallel remodeling of the activated CD4+ regulatory T-cell (Treg) and dysfunctional CD8+ T-cell compartments.
Abstract
To dissect the effect of neoadjuvant PD-1 and CTLA4 blockade on intratumoral T cells in treatment-naive head and neck squamous cell carcinoma, we analyzed primary tumor immune infiltrates from responding and nonresponding patients. At baseline, a higher ratio between active (4-1BB/OX40+) and inactive regulatory CD4+ T cells was associated with immunotherapy response. Furthermore, upon therapy, this active regulatory T-cell (Treg) population showed a profound decrease in responding patients. In an analogous process, intratumoral dysfunctional CD8+ T cells displayed decreased expression of activity and dysfunction-related genes in responding patients, whereas in clinical nonresponders, natural killer cells showed an increased cytotoxic profile early upon treatment. These data reveal immunologic changes in response to dual PD-1/CTLA4 blockade, including a parallel remodeling of presumed tumor-reactive Treg and CD8+ T-cell compartments in responding patients, and indicate that the presence of activated Tregs at baseline may be associated with response.
Significance:
In head and neck squamous cell carcinoma, neoadjuvant PD-1/CTLA4 blockade has shown substantial response rates (20%–35%). As recognition of tumor antigens by T cells appears to be a critical driver of therapy response, a better understanding of alterations in T-cell state that are associated with response and resistance is of importance.
This article is featured in Selected Articles from This Issue, p. 2109
INTRODUCTION
Immune checkpoint blockade (ICB) therapies have shown activity in a wide range of cancers, with response rates to dual anti–PD-1 and anti-CTLA4 therapy of up to 80% when given as neoadjuvant treatment (1, 2). In head and neck squamous cell carcinoma (HNSCC), both neoadjuvant anti–PD-1 monotherapy and neoadjuvant anti–PD-1 and anti-CTLA4 combination therapy have been explored in a total of six clinical trials. Although not designed or powered to compare clinical benefit, the reported data have shown a higher major pathologic response (MPR) rate upon neoadjuvant combined anti–PD-1 and anti-CTLA4 treatment (MPR rate of 20%–35%; Supplementary Table S1; refs. 3, 4) than upon neoadjuvant anti–PD-1 monotherapy (MPR rates ranging from 0% to 14%; Supplementary Table S1; refs. 5–8). At the same time, dual anti–PD-1/anti-CTLA4 therapy is associated with higher rates of immune-related toxicity (3, 4), emphasizing the importance to dissect the mechanisms of response and resistance to such dual therapy.
In tumors responding to neoadjuvant immunotherapy, pathologically complete responses can occur within weeks, demonstrating that effective reinvigoration of the antitumor response can, in at least some cases, lead to rapid cancer cell elimination. The effectiveness of boosting immune activity through ICB is dependent on the immune context (or “immune type”) of a tumor. Specifically, the presence of a tumor-reactive T-cell pool that has the potential to respond to ICB is assumed to be critical to achieve clinical benefit. One T-cell subset that has received ample attention in this regard is formed by the dysfunctional CD8+ T-cell population. Dysfunctional CD8+ T cells are characterized by high expression levels of inhibitory receptors, such as PD-1 and CTLA4, and have been shown to be enriched for tumor reactivity (9–13). In addition, the expression of activation markers, including 4-1BB (encoded by TNFRSF9), and cell cycle–associated genes in a subset of dysfunctional cells suggests that these cells are actively responding to antigen at the tumor site in the absence of therapy (14). Studies that assessed the mechanisms of ICB-induced therapy response have furthermore reported that the dysfunctional CD8+ T-cell compartment expands and shows increased expression of cytotoxic genes upon therapy (15–18). Although these observations explain why many studies have focused on the properties of the intratumoral CD8+ T-cell compartment, there is increasing evidence from mouse and human studies for the capacity of intratumoral regulatory T cells (Treg) to modify clinical response to ICB. First, a high abundance of PD-1+ Tregs relative to PD-1+ CD8+ T cells has been associated with poor response to anti–PD-1 therapy (19), consistent with a suppressive effect of Tregs on the antitumor response as demonstrated in mouse models (20, 21). Furthermore, similar to dysfunctional CD8+ T cells, Tregs in human tumors can be tumor antigen reactive (12) and show signs of local activation, as reflected by their (low-level) clonal expansion and, in particular, the expression of activation markers such as 4-1BB and OX40 (14). In line with this, expression of cell-cycle genes has been observed in 4-1BB+ Tregs at the tumor site (14). Among the tumor types with the most profound regulatory T-cell infiltrate are HNSCCs (22). Moreover, in at least some HNSCC tumors, a high fraction of activated Tregs is observed next to a sizable dysfunctional CD8+ T-cell pool (14), thereby providing a setting in which the dynamics of both cell populations can be studied in parallel.
Here, we describe the effect of neoadjuvant immunotherapy on the CD4+ and CD8+ T-cell compartments in a cohort of patients with treatment-naive HNSCC from the IMCISION trial (17 patients treated with neoadjuvant anti–PD-1 and anti-CTLA4 combination therapy, one patient treated with neoadjuvant anti–PD-1 monotherapy; ref. 4). In this cohort of patients with treatment-naive and mostly human papillomavirus (HPV)–negative HNSCC, dual nivolumab (anti–PD-1) and ipilimumab (anti-CTLA4) therapy induced an MPR at the primary tumor site [i.e., ≥ 90% reduction in viable cancer cells upon therapy (23); see Methods for the exact definition of pathologic response] in 30% of all patients (4). In addition, therapy response in this trial was associated with excellent clinical prospects, as none of the patients with an MPR upon neoadjuvant immunotherapy experienced recurrent disease at a median follow-up of 2 years (4).
To understand how changes in intratumoral T-cell states relate to treatment response, we profiled immune cell (CD45+) populations of primary tumor biopsies harvested prior to therapy (week 0) and 4 weeks after the start of treatment with neoadjuvant ICB (week 5). Analysis of immune cell infiltrates of patients with an MPR and nonresponding patients revealed that clinical response to dual ICB was associated with a higher relative abundance of an activated Treg population compared with an inactive Treg subpopulation at baseline. In addition, upon therapy, the abundance of this activated Treg subpopulation, as well as the abundance of a dysfunctional CD8+ T-cell subpopulation with hallmarks of activation, was reduced in responding tumors. At the same time, a transitional CD8+ T-cell population with lower levels of dysfunction became more abundant in responding patients, potentially due to an influx of cells from the systemic compartment. Natural killer (NK) cells in nonresponding tumors showed increased expression of a cytotoxicity program, suggesting that the immune compartment of nonresponding tumors is not unresponsive to dual ICB. Together, these data demonstrate that the intratumoral CD4+ and CD8+ T-cell compartments undergo a parallel rapid remodeling during the swift and deep HNSCC regressions that are observed upon anti–PD-1 and anti-CTLA4 combination therapy, resulting in reduced levels of T-cell activation and dysfunction in both cell pools.
RESULTS
Intratumoral Immune Cell Types and Cell States in Primary HNSCC
In order to identify characteristics of the intratumoral immune infiltrate that are related to response or resistance to anti–PD-1 and anti-CTLA4 combination therapy, single-cell RNA sequencing (scRNA-seq) and T-cell receptor (TCR) sequencing (TCR-seq) were performed on immune infiltrates of one HPV-positive and 17 HPV-negative HNSCC samples derived from treatment-naive patients who were enrolled in the previously published IMCISION trial (4). In this trial, patients with HNSCC received two courses of anti–PD-1 (nivolumab, 240 mg flat dose in weeks 1 and 3) with or without one course of anti-CTLA4 (ipilimumab, 1 mg/kg in week 1) prior to standard-of-care surgery with curative intent (performed in week 5). Viable immune cells were isolated from pretreatment (week 0) and posttreatment (week 5) primary tumor biopsies of 10 patients responding to anti–PD-1 and anti-CTLA4 combination immunotherapy (one partial pathologic response, nine MPRs) and seven patients nonresponding to anti–PD-1 and anti-CTLA4 combination immunotherapy by fluorescence-activated cell sorting and used for scRNA-seq analysis (Fig. 1A; Supplementary Fig. S1A, Supplementary Table S2). A single patient treated with anti–PD-1 monotherapy (MPR) was included in the dataset, but was excluded from all analyses in which response dynamics were assessed.
Figure 1.
![Figure 1. Intratumoral immune cell state diversity in HNSCC. A, Overview of the analyzed patient cohort. Matched pre- and posttreatment primary tumor biopsies and blood samples [peripheral blood mononuclear cells (PBMC)] were collected from 17 patients (seven NR and 10 RE) who were treated with one cycle of nivolumab (NIVO) and ipilimumab (IPI; week 1), followed by one cycle of nivolumab (week 3) and surgery (week 5). One patient (RE, IMC-04) received two cycles of nivolumab monotherapy, and samples of this patient were left out of all analyses in which response was considered a parameter. Biopsies and blood samples were taken at week 0 and at week 5, at time of surgery. Single-cell RNA and TCR sequencing (10X Genomics) was performed on matched biopsies of all patients. Bulk TCR sequencing was performed on matched blood samples of all responding patients. B, 2D projection of all intratumoral immune cells from all patients, colored by cell state. cDC, conventional dendritic cell; Dysf, dysfunctional; Mono–macro, monocyte–macrophage; pDC, plasmacytoid dendritic cell; Tfh, follicular helper-like T cell. C, Expression of genes that identify the CD4+ and CD8+ T-cell lineages, NK cells, B cells, and myeloid cells across all metacells, projected on the 2D map shown in B. Color scale bar represents log normalized expression, scaled to the minimum and maximum expression levels for each gene. norm., normalized. D, Heat map of the log normalized unique molecular identifier count of marker genes characterizing different subsets of CD4+ and CD8+ T cells, NK cells, B cells, and myeloid cells across all patients, grouped and colored by cell state. Color code corresponds to the color code used in B. Color scale bar represents the log normalized expression of the individual genes.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=10551666_2212fig1.jpg)
Intratumoral immune cell state diversity in HNSCC. A, Overview of the analyzed patient cohort. Matched pre- and posttreatment primary tumor biopsies and blood samples [peripheral blood mononuclear cells (PBMC)] were collected from 17 patients (seven NR and 10 RE) who were treated with one cycle of nivolumab (NIVO) and ipilimumab (IPI; week 1), followed by one cycle of nivolumab (week 3) and surgery (week 5). One patient (RE, IMC-04) received two cycles of nivolumab monotherapy, and samples of this patient were left out of all analyses in which response was considered a parameter. Biopsies and blood samples were taken at week 0 and at week 5, at time of surgery. Single-cell RNA and TCR sequencing (10X Genomics) was performed on matched biopsies of all patients. Bulk TCR sequencing was performed on matched blood samples of all responding patients. B, 2D projection of all intratumoral immune cells from all patients, colored by cell state. cDC, conventional dendritic cell; Dysf, dysfunctional; Mono–macro, monocyte–macrophage; pDC, plasmacytoid dendritic cell; Tfh, follicular helper-like T cell. C, Expression of genes that identify the CD4+ and CD8+ T-cell lineages, NK cells, B cells, and myeloid cells across all metacells, projected on the 2D map shown in B. Color scale bar represents log normalized expression, scaled to the minimum and maximum expression levels for each gene. norm., normalized. D, Heat map of the log normalized unique molecular identifier count of marker genes characterizing different subsets of CD4+ and CD8+ T cells, NK cells, B cells, and myeloid cells across all patients, grouped and colored by cell state. Color code corresponds to the color code used in B. Color scale bar represents the log normalized expression of the individual genes.
Diverse populations of T cells, NK cells, B cells, and myeloid cells were identified across all biopsies (Fig. 1B and C; Supplementary Fig. S1B and S1C; Supplementary Table S3), showing similarities to the cell states that have previously been described in HNSCC tumors (24) and other tumor types, including melanoma (25, 26) and nonsquamous cell lung cancer (27). For the CD8+ T-cell compartment, these included naive-like (expressing IL7R and TCF7), cytotoxic (expressing KLRG1 and CX3CR1), transitional, and dysfunctional CD8+ T cells, which could be further subdivided based on the expression of activation and effector genes (i.e., PDCD1, TNFRSF9, and GZMK; Fig. 1D; Supplementary Fig. S1C). In the CD4+ T-cell compartment, we identified naive-like cells (expressing IL7R and TCF7), follicular helper-like (Tfh) CD4+ T cells (expressing CXCL13 and TOX2), and regulatory CD4+ T cells (Tregs, expressing FOXP3 and IL2RA). Similar to the dysfunctional CD8+ T cells, the latter two populations could be subdivided by expression levels of activation and effector genes. Also in the NK cell compartment, two subpopulations of NK cells were present, which were distinguished by the level of expression of genes related to cytolytic function (e.g., PRF1). Within the population of T cells expressing an αβTCR (Supplementary Fig. S1D), the highest level of clonal expansion was observed within the dysfunctional CD8+ subsets, followed by the transitional CD8+ T-cell and one of the two Tfh (TfhLAG3) populations (Supplementary Fig. S1E).
We identified three myeloid populations, including a mixed population of monocytes and macrophages (expressing CD14 and CD68), a granulocyte population, and a dendritic cell (DC) population. In addition, in the DC population, we identified four subpopulations: a plasmacytoid DC population [pDC; expressing IL3RA (CD123)], a CD1C-expressing conventional DC (cDC) that resembled previously described cDC2 cells, LAMP3-positive cDCs with an activated profile, and CLEC9A-expressing mature cDC1 cells (28–30).
Baseline Differences in Treg and DC Subsets between Responding and Nonresponding Tumors
To dissect the relationship between intratumoral immune cell states and treatment response to dual ICB, we characterized the immune infiltrate of responding and nonresponding patients in pretreatment biopsies. At baseline, immune cell subsets that showed expression of PDCD1 and CTLA4, and thus serving as possible direct targets for checkpoint blockade, included Tregs, nonclassical T cells, dysfunctional CD8+ T cells, and Tfh cells (Supplementary Fig. S2A). Analysis of PD-1 protein levels on the intratumoral T-cell compartment demonstrated higher PD-1 surface levels on T cells of responding tumors than on T cells of nonresponding tumors at baseline (P < 0.1; Supplementary Fig. S2B), providing an incentive to identify specific cell states that were associated with response. Although responding and nonresponding tumors did not show significant differences in the abundance of B cells, CD4+ and CD8+ T cells, myeloid cells, or NK cells at baseline (Supplementary Fig. S2C and S2D), or in the level of clonal expansion or spatial distribution of CD8+ T cells (Supplementary Fig. S2E and S2F), cell states within the intratumoral DC compartment and Treg compartment did show a differential abundance in responding and nonresponding patients (Fig. 2A and B). Specifically, two DC subpopulations, CD1C cDCs and pDC, appeared enriched in biopsies from nonresponding patients as compared with responding patients (both P < 0.1; Fig. 2C). Within the T-cell compartment, a subpopulation of Tregs, characterized by the expression of GIMAP family members (TregGIMAP), was differentially present at baseline. Specifically, TregGIMAP cells seemed more abundant in nonresponders than in responders (P < 0.1; Fig. 2B and C). Notably, the second subset of Tregs, TregTNFRSF9, characterized by the expression of genes of the TNF receptor family, including TNFRSF9 encoding 4-1BB and TNFRSF4 encoding OX40, was slightly enriched in responding patients [not significant (ns), P < 0.5]. Assessment of the ratio between the two Treg subsets in each tumor showed that pretreatment biopsies of responding patients contained a significantly higher ratio of TregTNFRSF9 cells/TregGIMAP cells than nonresponding tumors (P < 0.1; Fig. 2D).
Figure 2.

Pretreatment Treg activation state is associated with response to dual ICB. A, 2D projection of all immune cells from responding (RE) and nonresponding (NR) patients at baseline (n = 17, monotherapy-treated patient IMC-04 excluded). Cells are colored by cell state as in Fig. 1B. Cells from the opposite patient group are colored in light gray. Dysf, dysfunctional; Mono–macro, monocyte–macrophage. B, Predictive coefficient of the abundance of cell fractions for each cell state at baseline in responding vs. nonresponding patient groups. Size of the datapoints corresponds to the P value of the comparison between responding and nonresponding patients of the specific cell state, defined by a two-tailed Mann–Whitney U test. Populations that are differentially abundant (P < 0.1) between responding and nonresponding patients are colored by their cell state. C, Fraction of cDC CD1C and pDC (left) and the two Treg subsets (right) in nonresponding and responding patients at baseline. Datapoints from the one patient with a partial pathologic response (IMC-27) are marked in yellow. Boxes show the median and 25th and 75th percentiles. Whiskers depict 1.5 × interquartile range (IQR), and datapoints represent individual patients, sized by the number of cells that underlie that datapoint. A two-tailed Mann–Whitney U test was performed. D, Left: correlation between fractions of TregTNFRSF9 and TregGIMAP cells per patient at baseline, colored by response. IMC-27 is marked with an asterisk. Right: pretreatment ratio of TregTNFRSF9 over TregGIMAP fraction within total T cells per patient, depicted as the log fold ratio in fraction. Boxes show the median and 25th and 75th percentiles. Whiskers depict 1.5 × IQR, and datapoints represent individual patients, IMC-27 is marked in yellow. A two-tailed Mann–Whitney U test was performed. E, Expression of a Treg activation signature (sig; ref. 32) in TregTNFRSF9 and TregGIMAP cells across all patients at baseline. Log normalized expression of activation-related transcripts is depicted, and a two-tailed Mann–Whitney U test was performed.
Although clinically annotated datasets are at present lacking to validate the differential abundance of these two Treg subsets in patients who do or do not respond to combination ICB, we observed comparable Treg states in 11 treatment-naive HNSCC samples from the cohort published by Kürten and colleagues (ref. 31; no response data available; Supplementary Fig. S3A–S3C). Also in this cohort, a range of TregTNFRSF9 cells/TregGIMAP ratios was observed (Supplementary Fig. S3D). Moreover, the abundance of the TregGIMAP subset was associated with the abundance of the same DC subsets in both cohorts (Supplementary Fig. S3E), suggesting that a shared biology can be observed across HNSCC samples from different cohorts.
Notably, when assessing the expression profiles of TregTNFRSF9 and TregGIMAP cells across patients, the TregTNFRSF9 population showed a significantly higher expression of Treg-specific activation genes (P < 0.001; Fig. 2E; Supplementary Fig. S3F; Supplementary Table S4; ref. 32). The increased ratio between TregTNFRSF9 cells with an activated state and TregGIMAP cells with a less activated state in tumors of responding patients may be reflective of ongoing Treg-mediated immune suppression at the tumor site at baseline. To explore whether the activated Treg state identified in the scRNA-seq data could also be detected by IHC, a method that would be more readily accessible for clinical purposes, we performed IHC for FOXP3 (marking Tregs) and 4-1BB (encoded by TNFRSF9) on the same set of tumor samples. Although the expression of 4-1BB and TNFRSF9 was correlated between IHC and scRNA-seq data (Supplementary Fig. S3G), we discovered that 4-1BB/TNFRSF9 expression marks only part of the activated Treg pool that is identified by scRNA-seq (Supplementary Fig. S3H–S3J). Thus, the development of multiplexed IHC assays will be required to identify and quantify the activated TregTNFRSF9 population that we describe.
Changes in Abundance of Immune Cell Subsets upon Dual ICB
The differences in immune composition between pretreatment biopsies of responding and nonresponding patients raised the question of whether these baseline differences could be associated with a different immunologic response to dual anti–PD-1 and anti-CTLA4 therapy. In order to capture the dynamics in immune cell states in response to treatment, we assessed the changes in abundance of all cell states in matched pretreatment versus posttreatment biopsies (excluding patient IMC-04, who was treated with nivolumab only, and excluding the tumor samples from responder IMC-38 and nonresponder IMC-12 that did not contain sufficient immune cells for matched analysis; Supplementary Fig. S4A and Supplementary Table S3). These analyses demonstrated that the most profound alterations occur in the intratumoral T-cell compartment. Specifically, tumors from responding, but not from nonresponding, patients showed a significant increase in the fraction of transitional CD8+ T cells (P < 0.01). In addition, in primary tumors from responders, a decrease in TregTNFSRF9 (P < 0.05) and naive-like CD8+ T cells (P < 0.05) cells was found upon treatment. Moreover, a considerable but nonsignificant decrease in one of the two dysfunctional CD8+ T-cell subsets, CD8-dysfGZMB, characterized by high expression of dysfunctional and effector markers such as GZMB, PRF1, and PDCD1, was observed (P < 0.1), which was accompanied by an increase in the other dysfunctional CD8+ T-cell subset, CD8-dysfZNF683 (P < 0.1, also observed in nonresponders; Fig. 3A and B; Supplementary Fig. S4B and S4C). Analysis of the ratio of TregTNFSRF9/TregGIMAP and CD8-dysfGZMB/CD8-dysfZNF683 at baseline and upon treatment demonstrated that, in particular, the balance between CD8-dysfGZMB/CD8-dysfZNF683 shifted upon therapy in responding patients (P < 0.01), whereas the balance between TregTNFSRF9 and TregGIMAP cells showed a similar (P < 0.01), though less pronounced, trend upon therapy (Fig. 3B; Supplementary Fig. S4D). In contrast, in nonresponding patients, no such cell state switches occurred. Instead, a decreased abundance of naive-like CD4+ T cells (P < 0.05) and increased abundance of Tfh cells (Tfh LAG3: P < 0.05 and Tfh NR3C1: P < 0.05) was observed upon treatment (Fig. 3A; Supplementary Fig. S4E).
Figure 3.

ICB-induced changes in cell states and clonal dynamics in responding (RE) and nonresponding (NR) patients. A, Median log fold change of cell state abundance in responding (left) and nonresponding (right) patients upon ICB therapy, divided into T cells and non–T cells. Size of the datapoints represents the P value of the paired comparison between pretreatment vs. posttreatment samples. Colors represent the cell states that are differentially abundant (P < 0.1) between pretreatment and posttreatment biopsies. A paired Wilcoxon signed rank test was performed. Samples from IMC-04 (monotherapy treated) and patients for whom matched data were lacking were excluded from the analysis (IMC-38 and IMC-12). Mono−macro, monocyte–macrophage. B, Quantification of the abundance of Treg, CD8+ dysfunctional T-cell, naive-like CD8+ T-cell, and transitional CD8+ T-cell subsets in pretreatment and posttreatment biopsies of responding patients, depicted as fraction of total T cells. Datapoints from IMC-27 are marked in yellow. Boxes show the median and 25th and 75th percentiles. Whiskers depict 1.5 × interquartile range, and datapoints represent individual patients, sized by the number of cells that underlie that datapoint. A paired Wilcoxon signed rank test was performed. C, Expression of cytotoxic (mature) and less cytotoxic (CD56bright) NK cell signatures (sig.) in NK cells from pretreatment and posttreatment biopsies of responding and nonresponding patients (33). A two-tailed Mann–Whitney U test was performed between the pretreatment and posttreatment samples for each patient group. D, TCR sharing between T-cell subsets, as calculated using the STARTRAC transition index (37). E, Expression of a proliferation signature (26) in CD8-dysfGZMB, naive-like, and transitional CD8+ T cells across responding patients, depicted as log normalized expression of cell cycle–related genes. A two-tailed Mann–Whitney U test was performed on cells from pretreatment and posttreatment biopsies per subset. F, Fold change of transitional CD8+ T cells between posttreatment vs. pretreatment samples within individual TCR clones that occurred >2 times in pretreatment and posttreatment samples, in responders and nonresponders. Size of the datapoints corresponds to the summed number of cells in pretreatment and posttreatment biopsies for each TCR clone. G, Stacked bar plot showing the number of nonshared transitional clones (i.e., detected either pretreatment or posttreatment) and their clone size after down-sampling of analyzed immune cells to equal cell numbers. H, Overlap in TCRs that were uniquely detected in transitional CD8+ T cells of either pretreatment or posttreatment tumor samples with TCRs in matched blood samples. Boxes show the median and 25th and 75th percentiles. Whiskers depict 1.5 × interquartile range, and datapoints represent individual patients. A paired Wilcoxon signed rank test was performed.
In addition to these dynamics in the T-cell compartment, we observed a change in the cell states in the NK-cell population in nonresponding patients upon treatment. Although the fraction of the NKKLRC1 subset, characterized by a low cytotoxicity signature (CD56bright; ref. 33), and the NKFGFBP2 subset, expressing a cytotoxic/mature gene signature (33), did not substantially change in nonresponders (Supplementary Fig. S4F and S4G; Supplementary Table S5), we observed a significantly higher expression of the cytotoxic/mature NK signature (33) in NK cells from posttreatment biopsies of patients who did not respond to therapy as compared with NK cells in pretreatment biopsies. Concurrently, the expression of the low cytotoxicity (CD56bright) signature was reduced in those patients upon treatment (Fig. 3C; Supplementary Fig. S4H; Supplementary Tables S6 and S7). These data provide evidence that modulation of immune activity occurs early upon neoadjuvant treatment not only in patients who show a clinical response to dual anti–PD-1 and anti-CTLA4 therapy but also in clinical nonresponding patients.
To further assess the dynamics in cell state abundance that were associated with therapy response, we first investigated whether the increase in transitional CD8+ T cells in responding tumors could be explained by differentiation from other cell states, expansion of a preexisting intratumoral transitional CD8+ T-cell population, or recruitment of a novel T-cell pool from the systemic compartment. The increased abundance of these transitional cells was not explained by therapy-induced differentiation from other cell states at the tumor site, because limited TCR sharing occurred between the transitional population and other CD8+ T-cell populations (Fig. 3D), and those expanded T-cell clones that showed a transitional state upon therapy and that were also detected prior to treatment were already present in the transitional compartment at that time point (Supplementary Fig. S4I). Moreover, analysis of cell-cycle genes (15) in the transitional CD8+ T-cell population of responding patients prior to and after treatment, as a proxy for active on-site proliferation, indicated that the proliferative activity of transitional CD8+ T cells remained low upon treatment in these patients and was comparable with that of naive CD8+ T cells (Fig. 3E). TCR analysis of the top clones furthermore showed that, whereas some transitional clones that were already present before treatment showed an increased prevalence upon treatment (Fig. 3F; Supplementary Fig. S4J), novel T-cell clones were also detected in posttreatment biopsies (Fig. 3G). Notably, the newly occurring T-cell clones in the transitional population were also more prevalent in posttreatment blood than in blood samples taken prior to treatment (Fig. 3H). Taken together, these data are most consistent with cell recruitment from the periphery as an explanation for the increased abundance of transitional CD8+ T cells in response to ICB.
Altered Treg Activation Levels upon Response to Dual ICB
To assess whether the parallel decrease in the abundance of TregTNFRSF9 cells and increase in TregGIMAP cells in responding patients upon treatment could be caused by a shift in cell state, we performed pseudotemporal ordering of all cells within the CD4+ T-cell compartment using Monocle3 (34–36). These analyses suggested that CD4+ T cells were positioned along two gradients of cell states, with naive-like CD4+ T cells on one end of the spectrum and the Tfh and Treg populations on the two other ends of each gradient (Fig. 4A; Supplementary Fig. S5A). The trajectory model of Tfh cells suggested a bifurcation of cell states, whereas the less activated TregGIMAP population was positioned in-between naive-like CD4+ T cells and activated TregTNFRSF9 in the Treg trajectory, suggesting that the TregGIMAP population is more transcriptionally similar to the naive-like CD4+ T-cell population, and that the TregTNFRSF9 cell pool forms the most differentiated Treg population, as also supported by correlation analysis of the most variable genes across the dataset (Supplementary Fig. S5B). In addition, although clonal expansion in the Treg compartment is modest, the observed TCR sharing between the TregGIMAP and TregTNFRSF9 subsets suggests that transitioning between these two cell states may occur (Fig. 3D; Supplementary Fig. S5C). When individual CD4+ T cells were ordered along the TregGIMAP to TregTNFRSF9 axis, cells from pretreatment biopsies of responding patients displayed a more differentiated/activated cell state, while cells from posttreatment biopsies preferentially resided in the less differentiated part of the trajectory (Fig. 4B; Supplementary Table S8). Also at the level of individual Treg clones, a reduction in activation levels was observed specifically in responding patients (Supplementary Fig. S5D). We next identified the gene programs that are associated with this Treg trajectory. In line with the difference in activation state between TregTNFRSF9 and TregGIMAP, gene sets that positively correlated with the trajectory from TregGIMAP to TregTNFRSF9 were related to biological processes reflecting immune cell activity, such as pyruvate metabolism and cytokine responsiveness (Fig. 4C; Supplementary Fig. S5E). Moreover, T-cell activation–related genes, such as TNFRSF4, IL2RA, and TNFRSF1B, were positively correlated with the Treg trajectory (Fig. 4D). Taken together, these results show that the active TregTNFSRF9 subpopulation is reduced upon treatment in responders, whereas the less active TregGIMAP subpopulation increases, potentially due to a therapy-induced cell state transition or replacement.
Figure 4.

Dual ICB induces altered Treg activation levels in responding patients. A, Pseudotemporal ordering of CD4+ T cells from all pretreatment and posttreatment biopsies using Monocle3 (34–36). The trajectory across the two Treg subsets, which forms the basis for B–D, is highlighted in the yellow box, whereas the remainder of the trajectory is depicted in gray. Note that the trajectory represents transcriptional relatedness between cell pools, and hence does not necessarily reflect a direct differentiation path between cell pools. Cells are colored by cell state. UMAP, uniform manifold approximation and projection. B, Heat map of the top 50 genes correlated with the Treg trajectory (in yellow box in A). Genes related to activation are annotated. Cells along the Treg trajectory are grouped into 30 bins based on the pseudotime, and average gene expression is calculated per bin. Bar plots show the fraction (frac.) of cells from pretreatment and posttreatment biopsies in responding (RE; top) and nonresponding (NR; middle) patients and cell state fractions in each of the bins (bottom). Colors of the two top plots represent the patient group and time point of biopsies. Colors of the bottom panel reflect the cell state. C, Single-cell gene set enrichment on the Treg trajectory of five significantly correlated Gene Ontology term gene sets with the Treg trajectory. Cells are divided over 30 bins, and the average gene set score is calculated per bin. The top bar plot shows the fraction of cells from each cell state per bin, as in B. D, Top left: cells along the Treg trajectory, colored by their cell states as in A. Top right/bottom: expression levels of activation-related genes in cells from the Treg trajectory projected on the UMAP shown in the yellow box in A. Colors represent the scaled gene expression of the indicated activation-related genes. norm., normalized.
Reduced Expression of Activation Genes in Dysfunctional CD8+ T Cells upon Response to Treatment
The observation that dual ICB therapy induces changes in cell state in both the Treg and dysfunctional CD8+ T-cell compartments in responding tumors raised the question of whether these alterations could be reflective of a similar process that occurs in both cell subsets. To investigate the transcriptional changes that occurred in the dysfunctional CD8+ T-cell compartment upon therapy, we performed a pseudotemporal cell ordering of all CD8+ αβ T-cell populations including the two dysfunctional CD8+ subsets, naive-like CD8+ T cells, cytotoxic T cells, and transitional CD8+ T cells (Fig. 5A; Supplementary Fig. S6A). The dysfunctional CD8+ T cells were located next to the naive-like CD8+ T cells and transitional CD8+ T cells. CD8-dysfZNF683 cells were located intermediate of the transitional CD8+ T cells/naive-like CD8+ T cells and the CD8-dysfGZMB cells, but the absence of any substantial TCR sharing between the transitional CD8+ T cells and CD8-dysfZNF683 cells argues against a developmental link between these two cell pools (Fig. 3D). In line with the previous analyses (Fig. 3A and B), cells from pretreatment biopsies of responding patients were enriched in the more dysfunctional/activated (CD8-dysfGZMB enriched) part of the dysfunctional trajectory, whereas cells from posttreatment biopsies of responding patients were enriched in the less activated/dysfunctional (CD8-dysfZNF683 enriched) part of the trajectory. Analogous to the transcriptional dynamics that characterized the Treg trajectory, ordering of the dysfunctional CD8+ T cells in pseudotime revealed that the genes that correlated most strongly with the dysfunctional trajectory were related to CD8+ T-cell activity, cytokine expression, and expression of cytotoxicity genes (Supplementary Fig. S6B and S6C). Genes that positively correlated with the CD8-dysfZNF683 to CD8-dysfGZMB trajectory included T-cell dysfunction–associated genes (e.g., LAG3 and CTLA4), T-cell effector genes (GZMB, PRF1, and IFNG), and T-cell activation genes (TNFRSF9, IL2RA, TNFRSF4, and TNFRSF18), which were all markedly lower in the trajectory part in which the CD8-dysfZNF683 population is enriched (Fig. 5B and C; Supplementary Table S9). Of note, following therapy, expression of cell-cycle genes also decreased in the dysfunctional CD8+ T-cell population of responding patients, but not in nonresponding patients (Supplementary Fig. S6D). These data provide further evidence that within weeks after the start of ICB, the dysfunctional CD8+ T-cell population becomes less active in clinically responding patients.
Figure 5.

Dual ICB induces reduced levels of T-cell activation in responding patients. A, Pseudotemporal ordering of CD8+ T cells using Monocle3 (34–36). Trajectory between the dysfunctional CD8+ T-cell subsets is shown in black in the yellow box and forms the basis for analyses in B–E. The remainder of the trajectory between CD8+ T-cell states is depicted in gray. Note that the trajectory represents transcriptional relatedness between cell pools and hence does not necessarily reflect a direct differentiation path between cell pools. Cells are colored by cell states. Dysf, dysfunctional; UMAP, uniform manifold approximation and projection. B, Heat map of the top 50 genes correlated with the dysfunctional trajectory. Genes related to activation are highlighted. Cells are divided over 30 bins, and the average gene expression is calculated per bin. Bar plots show the fraction (frac.) of cells from pretreatment and posttreatment biopsies in responding (RE; top) and nonresponding (NR; middle) patients and cell state fractions in each of the bins (bottom). Colors of the two top plots represent the patient group and time point of biopsies. Colors of the bottom plot reflect the cell state. C, Top left: cells along the dysfunctional CD8+ T-cell trajectory, colored by their cell states as in A. Top right/bottom: expression levels of activation-related genes in cells from the dysfunctional CD8+ T-cell trajectory projected on the UMAP. Colors represent the scaled gene expression of the indicated activation-related genes. norm., normalized. D, Expression of a neoantigen reactivity signature (12) across cells within the CD8-dysfGZMB, CD8-dysfZNF683, naive-like, transitional, and cytotoxic CD8+ T-cell populations. Log-normalized expression of activation-related transcripts is depicted, and a two-tailed Mann–Whitney U test was performed. E, Top 15 largest dysfunctional CD8+ T-cell clones that occurred in >2 cells in pretreatment and posttreatment biopsies, projected onto the dysfunctional trajectory in pseudotime of the responding (left) and nonresponding (right) patients. Cells from pretreatment and posttreatment samples are depicted in light and dark colors, respectively. TCR clones from IMC-27 are marked with an asterisk. Top density plot shows the density of TCR clones of the responding or nonresponding patients, excluding IMC-27, over pseudotime. The bottom scatter plot represents the cell states in pseudotime, colored by cell states as described in A. F, Venn diagram with the overlap in genes between the signatures of the dysfunctional and Treg trajectories, composed of genes that positively correlate with pseudotime. G, Correlation between the signatures of the dysfunctional and Treg trajectories, applied on either Tregs (left) or on dysfunctional CD8+ T cells (right), colored by cell state. A linear regression was performed on both signatures within each of the subsets. Correlation coefficient was calculated with a Pearson correlation. H, Enrichment score of the shared signature in the Tregs and dysfunctional CD8+ T-cell subsets in pretreatment and posttreatment biopsies of responding and nonresponding patients. A two-tailed Mann–Whitney U test was performed between the different patient groups.
The observation that the activity level of dysfunctional CD8+ T cells is lowered upon therapy raised the question of whether the dysfunctional CD8+ T-cell population could contain tumor-reactive T cells that, possibly because of the reduced tumor load that is observed by week 5 (1), display reduced activity due to lack of antigen encounter. To explore the potential presence of tumor-reactive T cells in the dysfunctional CD8+ T-cell compartment, we assessed expression of one of the gene signatures (“neoTCR CD8+ signature”) that has been shown to be enriched in tumor-reactive CD8+ T cells (12, 13). These analyses showed that expression of the tumor reactivity signature was most prominent in the CD8-dysfGZMB population (Fig. 5D). Furthermore, upon therapy, the expression of this signature in the top 25% neoTCR signature–expressing T-cell clones was significantly reduced in responding patients (Supplementary Fig. S6E; P < 0.01), supportive of a model in which reduction in the number of CD8-dysfGZMB cells upon ICB may be driven by reduced antigen exposure.
To further study the dynamics within the dysfunctional CD8+ T-cell population upon treatment in responders, we performed a TCR sharing analysis of cells in the CD8-dysfTNFRSF9 and less dysfunctional/active CD8-dysfZNF683 subpopulations. STARTRAC (37) analysis of the TCR sharing suggested a strong developmental link between the two dysfunctional CD8+ T-cell populations (Fig. 3D). Accordingly, projection of individual T-cell clones on top of the CD8-dysfZNF683 to CD8-dysfTNFRSF9 trajectory showed that multiple expanded T-cell clones in both responding and nonresponding patients consisted of cells from both the CD8-dysfTNFRSF9 and CD8-dysfZNF683 subsets. Notably, within the same clone, cells in pretreatment biopsies of responding patients localized more in the CD8-dysfTNFRSF9–enriched part of the trajectory and posttreatment clones were mostly located in the CD8-dysfZNF683–enriched part, whereas clones from nonresponding patients did not show similar changes (Fig. 5E). Similar dynamics were observed by calculating the change in the fraction of subsets within each TCR clone between pretreatment and posttreatment for responding and nonresponding patients (Supplementary Fig. S6F). Altogether, these findings provide evidence for a model in which the dysfunctional CD8+ T-cell population is characterized by lower levels of dysfunction, but also lower levels of activation, following response to ICB.
Parallel Reprogramming of the Intratumoral Dysfunctional CD8+ and Treg Subsets
As similar therapy-induced alterations in the fractions of activated and nonactivated Treg and dysfunctional CD8+ T-cell states were observed in responding patients, we hypothesized that the response to checkpoint blockade may lead to parallel alterations in the cell state of different intratumoral T-cell pools. To assess whether the potential transition from a TregTNFRSF9 to TregGIMAP state involved similar transcriptional changes as those observed in the proposed CD8-dysfGZMB to CD8-dysfZNF683 transition, we created signatures that were based on genes that showed the strongest positive correlation with either the trajectory from the CD8-dysfZNF683 to CD8-dysfGZMB state (238 genes; Supplementary Table S9) or the trajectory from the TregGIMAP to the TregTNFRSF9 state (372 genes; Supplementary Table S8), and then used these to assess whether dysfunctional CD8+ T cells and Tregs undergo related changes in their transcriptional program upon dual ICB in responding patients. The overlap between the two signatures was 84 genes (35.3% overlap with the dysfunctional signature and 22.8% overlap with the Treg signature), and overlapping genes (hereafter referred to as “shared signature”) included several activation markers (TNFRSF9 and IL2RA) and inhibitory receptors (LAG3 and CTLA4; Fig. 5F; Supplementary Fig. S7A and Supplementary Table S10). Furthermore, expression of the Treg transition and dysfunctional transition signatures in both the dysfunctional CD8+ T-cell and the Treg populations was significantly correlated (P < 0.01; R2 = 0.7 and P < 0.01; R2 = 0.58, respectively; Fig. 5G). Moreover, in line with the shift toward a less differentiated/activated cell state in both the dysfunctional CD8+ T-cell and Treg compartments, expression of the shared signature was decreased in both dysfunctional CD8+ T cells and the Tregs upon treatment in responding tumors (Fig. 5H; Supplementary Fig. S7B). To assess whether this reduction in T-cell activity was observed across additional T-cell subsets, we analyzed the expression of the shared signature in the Tfh pool. Similar to the dynamics observed in Tregs and dysfunctional CD8+ T cells, Tfh cells displayed reduced expression of the shared signature upon treatment (Supplementary Fig. S7B and S7C). In parallel to the reduced activation and differentiation observed across the dysfunctional CD8+ T-cell, Treg, and Tfh pools, response to treatment was associated with a decrease in the expression of IFN-induced genes across multiple immune cell subsets (Supplementary Fig. S7D). In contrast, neither the expression of IFN-induced genes nor expression of the shared signature in Tregs and dysfunctional CD8+ T cells showed such dynamics upon treatment in nonresponding patients (Supplementary Fig. S7B–S7D). Rather, analysis of the expression of inhibitory receptors demonstrated that PDCD1 and CTLA4 were upregulated on Tregs, dysfunctional CD8+ T cells, and Tfh cells upon treatment in nonresponding patients (Supplementary Fig. S7E–S7G), in line with the increased expression of PDCD1 and CTLA4 that was previously observed in bulk RNA-seq data of the same cohort (4). In addition, the inhibitory receptors LAG3 and HAVCR2 (encoding TIM-3) that were not targeted by combination ICB were upregulated in specific T-cell populations (Tregs and Tfh cells, respectively) in nonresponding tumors (Supplementary Fig. S7F and S7G). This observation, together with the increased activity signature observed in NK cells (Fig. 3C), provides evidence that intratumoral immune cells in nonresponding tumors do respond to dual PD-1 and CTLA4 blockade, but that this response is insufficient to achieve clinically effective tumor control.
DISCUSSION
To improve our understanding of the biology underlying response or resistance to anti–PD-1 and anti-CTLA4 (combination) therapies, analysis of baseline characteristics and therapy-induced changes that are associated with response is essential. Analysis of pretreatment (week 0) and posttreatment (week 5) biopsies of a cohort of 18 treatment-naive patients with mostly HPV-negative primary HNSCC revealed cell state changes that were associated with response to dual anti–PD-1 and anti-CTLA4 therapy in multiple immune cell compartments. First, a high ratio of activated TregTNFRSF9 cells/TregGIMAP cells in the tumor microenvironment at baseline appears to be associated with a swift and deep response to neoadjuvant nivolumab and ipilimumab as well as durable favorable clinical outcome in our cohort. Second, analogous transcriptional changes in two Treg subsets and the two dysfunctional CD8+ T-cell subsets were observed in response to immunotherapy. In contrast, a third population of transitional CD8+ T cells expanded after treatment but without evidence for a connected resource pool of cells that could explain the observed population increase. Based on the lack of a consistent increase in the size of preexisting clones, the low expression of cell-cycle genes in this population, and the substantial overlap with TCRs in the blood compartment, we hypothesize that the expansion of the transitional CD8+ T-cell population is due to T-cell recruitment from the periphery, as observed by Liu and colleagues in non–small cell lung cancer (38), and in line with the “clonal replacement” model proposed by Yost and colleagues (39). Of note, as compared with other CD8+ T-cell subsets, the expression of T-cell effector and T-cell activation molecules, as well as expression of a tumor reactivity gene signature, is low in this cell population, and conclusions regarding the role of the transitional CD8+ T-cell pool in tumor elimination should therefore be made with caution.
The cell state changes that were observed in the Treg and dysfunctional CD8+ T-cell population in patients responding to anti–PD-1 and anti-CTLA4 entailed a shift toward a state with reduced expression of inhibitory receptors such as PDCD1, CTLA4, and LAG3 in patients responding to anti–PD-1 and anti-CTLA4. These dynamics in both the dysfunctional CD8+ T-cell and Treg populations would be consistent with four possible models. First, reduced levels of activation and inhibitory receptor expression could be due to reversal of CD8+ T-cell dysfunction and Treg activation. Second, therapy may block the transition of less differentiated cells toward a more activated and more differentiated state. Third, the observed data would be consistent with a loss of cells with high levels of activation, whereas cells with lower activation levels persist. Finally, therapy may drive the increased influx of less differentiated cells, thereby altering the ratio between the two subsets of Tregs and two subsets of dysfunctional CD8+ T cells. Although direct evidence for either model is lacking, in particular, the first two models may deserve further consideration. Specifically, as immune checkpoint expression is driven by antigen encounter, reduced encounter of such antigens due to effective therapy-induced tumor elimination could explain both the reduced levels of T-cell dysfunction and the lower expression of the various T-cell activation signatures observed in patients with a deep response to immunotherapy. The observed decrease in the IFN-responsive signature is likewise in line with a less active tumor microenvironment after tumor clearance in these patients. Evaluation of T-cell states at earlier time points after the start of therapy will be valuable to provide further insights into the primary and secondary effects of ICB on the intratumoral T-cell compartment.
The parallel reduction in dysfunction and activation within the dysfunctional CD8+ T-cell and Treg subsets upon treatment was specific for responders. In contrast, nonresponding tumors showed a trend toward an increased expression in activation and dysfunction signatures upon treatment in some T-cell subsets, along with an increase in the expression of inhibitory receptors PDCD1, CTLA4, and LAG3 across different T-cell subsets. In addition, we observed transcriptional changes, such as increased PRF1, GZMB, and FGFBP2 expression, in the NK-cell population of posttreatment biopsies of nonresponding—but not responding—tumors, suggestive of increased cytotoxic activity in the NK-cell population upon treatment in nonresponders. These data are of particular interest considering prior studies that have suggested a role for NK cells in response to ICB (40, 41). In certain preclinical models, tumor rejection following blockade of the PD-1–PD-L1 axis has been shown to be mediated by NK cells (40, 42). In addition, anti-CTLA4 has been proposed to enhance NK cell function, either through direct binding to CTLA4-expressing NK cells or through inhibition of suppressive immune cell types such as Tregs (41). Whether the increase in NK cell activation is specific for nonresponding patients, or also occurred earlier on during tumor rejection in responding patients, remains to be addressed by analysis of biopsies at earlier time points. Although the cell state alterations across different immune cell subsets are most consistent with a level of treatment-induced immune cell reactivation in clinical nonresponders, these therapy-associated cell state changes evidently did not evoke tumor clearance. As such, the current analyses provide a starting point to further study resistance mechanisms to checkpoint blockade in HNSCC in (preclinical) models that allow perturbation of components of the tumor microenvironment.
Finally, the high ratio of TregTNFRSF9 cells/TregGIMAP cells in responding patients at baseline as compared with nonresponding patients suggests that the activation state of Tregs may reflect the presence of an antitumor response that can be effectively (re)activated by dual ICB. An independent cohort in which response to neoadjuvant dual anti–PD-1 and anti-CTLA4 therapy can be correlated with in-depth (transcriptional) profiling of immune cell subsets for a substantial number of patients with HNSCC will be required to validate the predictive value of Treg activation state with respect to treatment response. Notably, Kumagai and colleagues have previously shown that a high abundance of PD-1+ Tregs is associated with poor response to anti–PD-1 therapy (19), and it may be speculated that the concurrent CTLA4 blockade and/or Treg depletion upon CTLA4 targeting is critical to yield an effective antitumor response in tumors with such an active Treg compartment. In future work, in-depth analyses of single-cell datasets with coupled response information should help address these and other questions, thereby adding to our ability to implement immune checkpoint–blocking therapies in a personalized manner.
METHODS
Processing of Tumor Material
Human tumor tissue was obtained following written informed consent in accordance with national guidelines and after approval by the local Medical Research Ethics Committee of The Netherlands Cancer Institute–Antoni van Leeuwenhoek hospital (MREC AVL; ref. 4). In the original IMCISION cohort, both treatment-naive patients and patients with recurrent disease after radiotherapy were included (4). Patients were classified as responders or nonresponders based on criteria previously defined by Tetzlaff and colleagues (23). Specifically, MPR was defined as ≤10% residual viable tumor cell percentage in the resected tumor bed and a 90% to 100% decrease in viable tumor cells from baseline to after treatment, partial pathologic response as both ≤50% residual viable tumor cells and a 50% to 89% decrease in viable tumor cell percentage from baseline to after treatment, and near-complete clinical response was determined via imaging (RECIST).
The IMCISION trial was designed to investigate the efficacy of dual immunotherapy and not to compare neoadjuvant nivolumab with nivolumab/ipilimumab ICB. The nivolumab-only cohort in the IMCISION trial was a safety run-in of six patients, of whom three had salvage surgery after prior radiotherapy, and only one patient reached an MPR (IMCISION article, ref. 4; Fig. 1A; Supplementary Table S1). As this small and heterogeneous cohort did not allow a systematic analysis of changes in the intratumoral immune compartment between responders and nonresponders in nivolumab-only patients, the current dataset is focused on treatment-naive patients who received combination ICB.
For this study, we selected 18 treatment-naive IMCISION patients, of whom 11 were responders and seven were nonresponders. Of these 18 patients, 17 patients received neoadjuvant combination anti–PD-1 and anti-CTLA4 therapy (2× nivolumab in weeks 1 and 3, and 1× ipilimumab in week 1) prior to surgery with curative intent (in week 5) and one patient (IMC-04) received anti–PD-1 monotherapy (2× nivolumab in weeks 1 and 3) prior to surgery. The eleven responders consisted of nine patients with an MPR (IMC-22, IMC-17, IMC-39, IMC-15, IMC-36, IMC-04, IMC-12, IMC-31, and IMC-29; IMCISION article; Supplementary Table S1; ref. 4), one patient with a clinically near- complete response (IMC-21), and one patient with a partial response of 69% who underwent surgery in week 3 (2 weeks earlier than originally planned, IMC-27). Notably, patient IMC-04 (anti–PD-1 monotherapy) was included for clustering but excluded from all analyses in which response dynamics upon dual ICB were analyzed. Seven nonresponders consisted of six patients with a pathologic response ranging from 0% to 20% (IMC-33, IMC-37, IMC-38, IMC-26, IMC-10, IMC-30; IMCISION article; Supplementary Table S1; ref. 4) and one patient with inoperable disease at time of surgery due to tumor progression (IMC-34). Across the analyzed cohort, one patient had HPV-positive (IMC-22) and 17 patients had HPV-negative HNSCC. Note that the observations described in this work could be reproduced in the homogeneous cohort in which patients with a partial response (IMC-27) and with HPV-positive disease (IMC-22) were excluded (with P < 0.1 for the difference in TregTNFRSF9/TregGIMAP ratio between responders and nonresponders at baseline; P < 0.05 for the difference in TregTNFRSF9 between pretreatment and posttreatment biopsies of responding patients; P < 0.05 for the difference in DysfGZMB between pretreatment and posttreatment biopsies of responding patients).
Posttreatment biopsies were taken at the time of surgery in week 5—that is, 4 weeks after the start of immunotherapy (week 1). Sequential pretreatment and posttreatment biopsies were available for all patients. For nine responding and six nonresponding patients, both biopsies contained sufficient cell numbers for matched analysis (excluding patients IMC-38 and IMC-12, whose samples did not contain sufficient immune cells for matched analysis).
Fresh tumor tissue was dissociated through manual mincing of the material, followed by a 20-minute incubation at 37°C in RPMI 1640 medium (Gibco) with collagenase IV (1 mg/mL, Sigma-Aldrich) and pulmozyme (12.5 μg/mL, Roche), alternated with 3 rounds of dissociation using a gentleMACS dissociator (Miltenyi Biotec). After dissociation, cell suspensions were filtered through a 100-μm filter (Corning) and washed with RPMI 1640 medium with penicillin (100 μg/mL), streptomycin (100 μg/mL), and 10% human serum (Sigma). Subsequently, cell suspensions were frozen at −80°C in 90% fetal calf serum (Sigma) and 10% dimethyl sulfoxide (DMSO, Sigma) until further use.
Sorting of Immune Cells from Tumor Material
Frozen samples were thawed in cold RPMI 1640 medium with 100 μg/mL penicillin, 100 μg/mL streptomycin, 10% human serum, and benzonase (250 U/mL, Novagen). Single-cell suspensions were incubated for 10 minutes at 4°C in cold cell staining buffer (BioLegend) with Human TruStain FcX (1:10, BioLegend), followed by addition of a TotalSeq anti-human Hashtag antibody (numbers 1–10, 10 μg/mL final concentration, BioLegend) to uniquely label cells from each biopsy, and an equal volume of cell staining buffer containing anti–CD45-APC (HI30, BD Biosciences, 1:30), anti–CD3-FITC (SK7, BD Biosciences, 1:30), anti–CD4-BV421 (OKT4, BioLegend, 1:100), anti–CD8-AF700 (3B5, Life Technologies, 1:50), anti–PD-1-PEcy7 (EbioJ105, eBioscience, 1:100), and anti–CD103-BV711 (BerACT8, BD Biosciences, 1:50). After a 30-minute incubation at 4°C and 3 washes with cell staining buffer, samples were resuspended in cold phosphate-buffered saline (PBS) with 0.5% bovine serum albumin (BSA; Sigma) and ethylenediaminetetraacetic acid (EDTA; 2 mmol/L, Life Technologies). Equal aliquots of all samples were collected and, after the addition of AccuCount Blank Particles 13.0 to 17.9 μm (Spherotech) and propidium iodide (PI; Sigma-Aldrich, 0.5 μg/mL), live-cell counts (CD45+ PI−; Supplementary Fig. S1A) per 10,000 counting beads were measured by flow cytometry. Based on the detected cell numbers, equal numbers of viable cells were mixed from all samples and filtered through a 35-μm cell strainer (Falcon tube with cell strainer cap, Corning). After the addition of PI (Sigma-Aldrich, 0.5 μg/mL final concentration), CD45+ PI− cells were sorted using a FACSAria Fusion Flow Cytometer (BD Biosciences) and collected in cooled RPMI 1640 medium supplemented with 100 μg/mL penicillin, 100 μg/mL streptomycin, and 10% human serum. Sorted cells were washed with cold PBS supplemented with 0.04% BSA and resuspended in cold PBS with 0.04% BSA at a concentration of 800 to 1,200 cells/μL for single-cell sequencing using 10X Genomics.
IHC
IHC of the formalin-fixed, paraffin-embedded tumor samples was performed on a Discovery Ultra automated stainer (Ventana Medical Systems). Briefly, paraffin sections were cut at 3 μm, heated at 75°C for 28 minutes, and deparaffinized in the instrument with EZ prep solution (Ventana Medical Systems). Heat-induced antigen retrieval was carried out using Cell Conditioning 1 (CC1; Ventana Medical Systems) for 64 minutes at 95°C.
For the double staining of 4-1BB (Yellow) followed by FOXP3 (Purple), 4-1BB was detected in the first sequence using clone E6Z7F (1/100 dilution, 60 minutes at 37°C, Cell Signaling Technology). 4-1BB–bound antibody was visualized using Anti-Rabbit NP (Ventana Medical systems) for 12 minutes at 37°C followed by Anti-NP AP (Ventana Medical Systems) for 12 minutes at 37°C, followed by the Discovery Yellow detection kit (Ventana Medical Systems). In the second sequence of the double-staining procedure, FOXP3 was detected using clone 236A/E7 (1:100 dilution, 2 hours at 37°C, Abcam). FOXP3 was visualized using Anti-Mouse HQ (Ventana Medical systems) for 12 minutes at 37°C followed by Anti-HQ HRP (Ventana Medical Systems) for 12 minutes at 37°C, followed by the Discovery Purple Detection Kit (Ventana Medical Systems). Slides were counterstained with hematoxylin and bluing reagent (Ventana Medical Systems). A PANNORAMIC 1000 scanner from 3DHISTECH was used to scan the slides at a 40× magnification.
Bulk TCR-seq
Deep sequencing of the TCRβ chain was performed using the Adaptive Biotechnologies ImmunoSEQ platform on genomic DNA extracted from peripheral blood mononuclear cells (PBMC) from all pretreatment and posttreatment samples of responding patients in the cohort. Only productive TCRβ rearrangements were used for downstream analysis. The average number of TCRs per sample was 16,9549 (range, 6,4037–25,1752). To account for differences in sample size between the blood and tumor samples, bootstrapping was applied when calculating TCR overlap.
scRNA-seq and TCR-seq
Sorted immune cells (10,000–20,000) were run over a lane of the 10X Chromium instrument at a concentration of 800 to 1,200 cells/μL for encapsulation in droplets. Single-cell RNA, antibody barcode, and TCR libraries were constructed using the Chromium Next GEM Single-Cell V(D)J Reagent Kits v1.1 for humans (10X Genomics) according to the manufacturer's protocol. Libraries were sequenced on a NextSeq instrument (Illumina) with read lengths of 26/58 for RNA and antibody libraries and 26/91 or 26/130 for TCR libraries, aiming for 30,000 read pairs per cell for RNA libraries and 5,000 reads for both antibody and TCR libraries.
Single-Cell Data Processing
Gene expression, antibody, and TCR-seq reads were respectively mapped to the GRCh38 human reference genome, antibody reference sequences, and IMGT reference using CellRanger Version 3.1 (10X Genomics), followed by the generation of unfiltered unique molecular identifier (UMI) matrices for gene expression and antibody libraries. Demultiplexing of the sample hashtags was done using the HTOdemux function, part of the Seurat package Version 4.0.1, with the positive quantile set to 0.99. Filtering was performed to remove cells with less than 800 UMIs, a fraction of mitochondrial gene expression that exceeded 0.5, or with more than one hashtag (multiplets). Gene sets containing mitochondrial genes, immunoglobulin genes, ribosomal genes, and long noncoding RNA genes, as well as genes that were detected in only one experiment, were removed from the data.
Metacell Modeling and Analysis
For analysis of the scRNA-seq data, we used the MetaCell package (V0.3.5; ref. 43) following the analysis strategy described by Li and colleagues (26). Feature genes were selected based on a scaled variance of 0.08 or higher and a minimum of 100 total UMIs across the dataset. Gene features that were associated with lateral processes, such as cell cycle, type I IFN response, or stress (adapted from Li and colleagues, ref. 26), and genes with an average expression above 4.8 UMIs across all downsampled cells were excluded from metacell formation. Metacell generation was performed on 32,406 cells using 1,690 genes that passed the filtering steps. Cells from all patients were included in metacell formation. K = 100 and 500 bootstrap iteration steps were done, and heterogeneous metacells were split. The confusion matrix (Supplementary Fig. S2B), showing the hierarchical clustering of metacells, was used to annotate groups of metacells that showed similar expression profiles. Four main immune cell types were classified based on the expression of marker genes, including CD19 and MS4A1 for B cells; NCAM1 for NK cells; CD14, CD68, and HLA-DQA1 for myeloid cells; and CD3D and TRBC1 for T cells. Among total immune cells, 20,635 T cells were identified, and within this population, TCR sequences were recovered for 18,504 T cells. All immune cells were further subdivided into distinct cell states based on the confusion matrix and supervised analysis of marker genes, as further described in the main text and below.
Identification of Immune Subsets
T-cell states were annotated using the most variable genes within the dataset, defined based on their variance over all cells divided by the mean. For further analyses, data from posttreatment biopsies of IMC-04 (PD-1 monotherapy treated), IMC-38 (nonresponder, insufficient cell numbers), and IMC-12 (responder, insufficient cell numbers) were left out for analysis. Cells from the patient with a partial pathologic response (IMC-27) were excluded from analyses in which cells from responding and nonresponding patients were compared. To assess Treg activation, the expression of a gene signature derived from Tregs isolated from PBMCs after in vitro stimulation with antigen (32) was plotted as the fraction of activation-related UMIs among total UMIs per cell. Similarly, previously described gene sets associated with CD8+ T-cell activation (adapted from ref. 44), dysfunction (26), and cell-cycle activity (26) were used to infer levels of activation, dysfunction, and proliferation in the CD8+ T-cell population.
Analysis of the Kürten and Colleagues Dataset
For analysis of the scRNA-seq dataset from Kürten and colleagues (31), we used the MetaCell package (V0.3.5; ref. 43) using the same approach as described above. Analysis was restricted to CD45+ cells, and cells with less than 800 UMIs or with a fraction of mitochondrial gene expression >0.4 were removed. Rather than generating new features, we applied the features generated in analysis of the IMCISION cohort on the dataset from Kürten and colleagues. Metacell generation was performed on 23,360 cells using 1,690 genes (from our IMCISION cohort), and annotation was carried out as described above. Patient HN03 was a significant outlier in the analysis and was excluded from further analyses. Similarities between the cell states in the Kürten and colleagues cohort and IMCISION cohort were verified using gene expression markers per cell state, and by projecting signatures of the Tregs and myeloid cells that were generated from the IMCISION cohort on the dataset from Kürten and colleagues.
Cell Proportion Analysis
The coefficient for the difference between the fraction in cell states in the responders versus the nonresponders, the response coefficient, was calculated using a linear model on the fraction of the individual cell states of each patient in responders versus nonresponders, inspired by ref. 17. The coefficient of determination (R2) from the linear model was adjusted to being either negative or positive based on the direction of the slope (m), 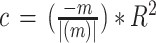 . The P value of each comparison was calculated with a Mann–Whitney U test. In a similar fashion, the treatment coefficient was calculated with a linear model on the pretreatment versus posttreatment samples per patient for individual cell states, with a Mann–Whitney U test for calculation of P values.
. The P value of each comparison was calculated with a Mann–Whitney U test. In a similar fashion, the treatment coefficient was calculated with a linear model on the pretreatment versus posttreatment samples per patient for individual cell states, with a Mann–Whitney U test for calculation of P values.
Trajectory Analysis
The trajectory analysis was performed using Monocle3 (refs. 34–36; v1.0.0) on CD4+ and CD8+ T cells. The dimension reduction in Monocle3 was limited by using the filtered feature gene list used in the MetaCell analysis for generating the metacells. For the preprocessing of data in Monocle3, we used 25 dimensions with the principal component analysis (PCA) method; other parameters were kept on default. Possible batch effects were removed using the build-in function “align_cds.” Dimensions were reduced using uniform manifold approximation and projection as a reduction method and Euclidean distances, with 50 neighbors. The generated trajectory was cropped to the specific subsets—that is, either dysfunctional CD8+ T cells or Tregs—and the pseudotime of these cropped trajectories was extracted from the Monocle3 algorithm.
Signature Generation
The Treg and dysfunctional CD8+ T-cell signatures were generated from the ranked list of correlating genes with the trajectory of all cells in each subset (Treg or dysfunctional CD8+ T cells, respectively) generated with Monocle3 (34–36). Signature genes were selected from the genes that correlated with the trajectory based on a P value <0.01 and a Morgan's test statistic >8. The direction of the correlations was determined by comparing the mean expression of the genes on each side of pseudotime on the trajectory, and genes were filtered on a positive correlation for the signatures. The shared signature was generated by intersecting the Treg and dysfunctional CD8+ T-cell signatures, consisting of positively correlated genes with pseudotime. NK signatures were extracted from (33), and the low cytotoxic NK (CD56bright) signature was derived by combining the differentially expressed genes from the CD56bright blood and bone marrow populations (excluding ribosomal genes). The cytotoxic/mature NK signature was generated from the differentially expressed gene in the active and mature NK populations in blood and bone marrow. The IFN response signature was adapted from ref. 26. The neoTCR CD8+ signature was obtained from ref. 12.
Enrichment Analysis
The enrichment of gene sets and signatures per single cell was calculated using the AUCell (45) package (v1.12.0) unless otherwise stated. By calculating the enrichment of gene sets or signatures, the potential bias caused by highly expressed genes within such gene sets or signatures is minimized. The enrichment analysis in Supplementary Fig. S6D was performed using AUCell on single cells within each of the groups, and the pretreatment versus posttreatment comparison was performed using a linear model and Mann–Whitney U test following the same method as used for the proportion analysis. The gene set enrichment analysis (GSEA) on the correlating genes from the trajectories generated with Monocle3 was performed on the ranked gene list based on the Morans test statistics score. GSEA was performed with the GSEA Java application software (refs. 46, 47; v4.2.3), using the Gene Ontology term gene set list (c5.go.v7.5.1) on default settings.
TCR Repertoire Analysis
For TCR repertoire analysis, TCRβ nucleotide sequences of T cells with a productive TCR rearrangement were used. A total of 19,594 T cells with a productive rearrangement TCR were identified. Clonotypes were assigned based on the nucleotide sequence of the CDR3 region of the TCRβ chain. The STARTRAC package was used to calculate the level of T-cell expansion within a T-cell state for each patient, based on the TCR frequencies in that cell state (37). The extent of TCR sharing between cell states was determined using the STARTRAC transition index. For T-cell clone–based analyses, clonal expansion of T-cell clones was defined by a threshold of >3 occurrences of that TCR per biopsy unless otherwise stated.
Statistical Analysis
All analysis and statics were performed with R in Rstudio (v4.0.2). A Mann–Whitney U test was used for the comparison of the gene scores that were plotted as a fraction of total UMIs. For unbiased identification of differentially expressed genes, differential gene expression analysis was performed on a downsampled UMI matrix. Mann–Whitney U test with false discovery rate correction was performed on the number of transcripts/1,000 UMIs. A Wilcoxon signed rank test was used to test differences in abundance of cell states between matched pretreatment and posttreatment biopsies, whereas a Mann–Whitney U test was used for comparisons between responders and nonresponders unless otherwise stated.
Data and Code Availability
The processed scRNA-seq data and clonotype information have been deposited in the NCBI Gene Expression Omnibus database (GSE232240). The raw scRNA-seq and TCR-seq data have been deposited in the European Genome-phenome Archive (EGA) database (EGAS00001007367 and EGAS00001007368, respectively) and will be made available from the corresponding author upon reasonable request. Data requests will be reviewed by the institutional review board of The Netherlands Cancer Institute and require a data transfer agreement after approval. The code that was used for the analyses in this study is available on GitHub (https://github.com/ZuurLabRep/ms_code_vanderleun_traets_singlecell_IMCISION).
Supplementary Material
Cell state annotations of CD45+ cells and clonality of T cells isolated from HNSCC biopsies.
Cell state fractions and transcriptional differences at baseline.
Validation of the differential abundance of Treg subpopulations at baseline using orthogonal strategies and an independent HNSCC dataset.
ICB-induced changes in immune cell state abundance and gene expression profiles.
Dual ICB-induced changes in activation state of Tregs.
Dual ICB-induced changes in dysfunctional CD8+ T cells of responding patients towards a less dysfunctional state.
Gene expression changes associated with response and resistance to ICB across cell states.
Acknowledgments
We thank Bristol Myers Squibb for scientific input and financial support. This study was funded by the Bristol Myers Squibb International Immuno-Oncology Network and the Riki Foundation, whereas the Netherlands Cancer Institute was the study's sponsor. Both funding sources had no role in the design and execution of the study, data analysis, or writing of the manuscript. We thank staff of the NKI Flow Cytometry facility and the NKI Genomics Core facility for technical support and input, as well as members of the Zuur and Schumacher laboratories for discussions.
The publication costs of this article were defrayed in part by the payment of publication fees. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Footnotes
Note: Supplementary data for this article are available at Cancer Discovery Online (http://cancerdiscovery.aacrjournals.org/).
Authors’ Disclosures
J.B.A.G. Haanen reports grants from Bristol Myers Squibb, MSD, Asher Bio, Sastra Cell Therapy, Novartis, Amgen, and BioNTech outside the submitted work; consultancy roles for Bristol Myers Squibb, CureVac, GSK, Imcyse, Iovance Bio, Instil Bio, Immunocore, Ipsen, Merck Serono, MSD, Molecular Partners, Novartis, Pfizer, Roche/Genentech, Sanofi, Scenic, and Third Rock Ventures; and participated in scientific advisory boards for Achilles Tx, BioNTech, Instil Bio, PokeAcell, T-Knife, Scenic, and Neogene Therapeutics. T.N.M. Schumacher reports personal fees and other support from Third Rock Ventures, Asher Bio, Celsius, Allogene, Merus, and Scenic Biotech, personal fees from Neogene Therapeutics, and other support from Cell Control outside the submitted work. C.L. Zuur reports other support from The Netherlands Cancer Institute during the conduct of the study, as well as personal fees from Sanofi (consultant) outside the submitted work. No disclosures were reported by the other authors.
Authors’ Contributions
A.M. van der Leun: Conceptualization, resources, data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, project administration, writing–review and editing. J.J.H. Traets: Conceptualization, resources, data curation, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. J.L. Vos: Resources, formal analysis, investigation, methodology, writing–review and editing. J.B.W. Elbers: Resources, writing–review and editing. S. Patiwael: Resources, investigation, writing–review and editing. X. Qiao: Supervision, investigation, writing–review and editing. M. Machuca-Ostos: Formal analysis, investigation, writing–review and editing. D.S. Thommen: Formal analysis, supervision, investigation, writing–original draft, writing–review and editing. J.B.A.G. Haanen: Conceptualization, supervision, writing–review and editing. T.N.M. Schumacher: Conceptualization, resources, formal analysis, supervision, funding acquisition, investigation, visualization, methodology, writing–original draft, writing–review and editing. C.L. Zuur: Conceptualization, resources, formal analysis, supervision, funding acquisition, investigation, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Blank CU, Rozeman EA, Fanchi LF, Sikorska K, van de Wiel B, Kvistborg P, et al. Neoadjuvant versus adjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma. Nat Med 2018;24:1655–61. [DOI] [PubMed] [Google Scholar]
- 2. Rozeman EA, Menzies AM, van Akkooi ACJ, Adhikari C, Bierman C, van de Wiel BA, et al. Identification of the optimal combination dosing schedule of neoadjuvant ipilimumab plus nivolumab in macroscopic stage III melanoma (OpACIN-neo): a multicentre, phase 2, randomised, controlled trial. Lancet Oncol 2019;20:948–60. [DOI] [PubMed] [Google Scholar]
- 3. Schoenfeld JD, Hanna GJ, Jo VY, Rawal B, Chen YH, Catalano PS, et al. Neoadjuvant nivolumab or nivolumab plus ipilimumab in untreated oral cavity squamous cell carcinoma: a phase 2 open-label randomized clinical trial. JAMA Oncol 2020;6:1563–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vos JL, Elbers JBW, Krijgsman O, Traets JJH, Qiao X, van der Leun AM, et al. Neoadjuvant immunotherapy with nivolumab and ipilimumab induces major pathological responses in patients with head and neck squamous cell carcinoma. Nat Commun 2021;12:7348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ferris RL, Spanos WC, Leidner R, Goncalves A, Martens UM, Kyi C, et al. Neoadjuvant nivolumab for patients with resectable HPV-positive and HPV-negative squamous cell carcinomas of the head and neck in the CheckMate 358 trial. J Immunother Cancer 2021;9:e002568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Knochelmann HM, Horton JD, Liu S, Armeson K, Kaczmar JM, Wyatt MM, et al. Neoadjuvant presurgical PD-1 inhibition in oral cavity squamous cell carcinoma. Cell Rep Med 2021;2:100426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Uppaluri R, Campbell KM, Egloff AM, Zolkind P, Skidmore ZL, Nussenbaum B, et al. Neoadjuvant and adjuvant pembrolizumab in resectable locally advanced, human papillomavirus-unrelated head and neck cancer: a multicenter, phase II trial. Clin Cancer Res 2020;26:5140–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wise-Draper TM, Gulati S, Palackdharry S, Hinrichs BH, Worden FP, Old MO, et al. Phase II clinical trial of neoadjuvant and adjuvant pembrolizumab in resectable local-regionally advanced head and neck squamous cell carcinoma. Clin Cancer Res 2022;28:1345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, et al. A transcriptionally and functionally distinct PD-1(+) CD8(+) T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med 2018;24:994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gros A, Robbins PF, Yao X, Li YF, Turcotte S, Tran E, et al. PD-1 identifies the patient-specific CD8(+) tumor-reactive repertoire infiltrating human tumors. J Clin Invest 2014;124:2246–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Oliveira G, Stromhaug K, Klaeger S, Kula T, Frederick DT, Le PM, et al. Phenotype, specificity and avidity of antitumour CD8(+) T cells in melanoma. Nature 2021;596:119–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lowery FJ, Krishna S, Yossef R, Parikh NB, Chatani PD, Zacharakis N, et al. Molecular signatures of antitumor neoantigen-reactive T cells from metastatic human cancers. Science 2022;375:877–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Caushi JX, Zhang J, Ji Z, Vaghasia A, Zhang B, Hsiue EH, et al. Transcriptional programs of neoantigen-specific TIL in anti–PD-1-treated lung cancers. Nature 2021;596:126–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zheng L, Qin S, Si W, Wang A, Xing B, Gao R, et al. Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 2021;374:abe6474. [DOI] [PubMed] [Google Scholar]
- 15. Bassez A, Vos H, Van Dyck L, Floris G, Arijs I, Desmedt C, et al. A single-cell map of intratumoral changes during anti–PD-1 treatment of patients with breast cancer. Nat Med 2021;27:820–32. [DOI] [PubMed] [Google Scholar]
- 16. Luoma AM, Suo S, Wang Y, Gunasti L, Porter CBM, Nabilsi N, et al. Tissue-resident memory and circulating T cells are early responders to pre-surgical cancer immunotherapy. Cell 2022;185:2918–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhang Y, Chen H, Mo H, Hu X, Gao R, Zhao Y, et al. Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 2021;39:1578–93. [DOI] [PubMed] [Google Scholar]
- 18. Clarke J, Panwar B, Madrigal A, Singh D, Gujar R, Wood O, et al. Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J Exp Med 2019;216:2128–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol 2020;21:1346–58. [DOI] [PubMed] [Google Scholar]
- 20. Quezada SA, Peggs KS, Simpson TR, Shen Y, Littman DR, Allison JP. Limited tumor infiltration by activated T effector cells restricts the therapeutic activity of regulatory T-cell depletion against established melanoma. J Exp Med 2008;205:2125–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med 2013;210:2435–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mandal R, Senbabaoglu Y, Desrichard A, Havel JJ, Dalin MG, Riaz N, et al. The head and neck cancer immune landscape and its immunotherapeutic implications. JCI Insight 2016;1:e89829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tetzlaff MT, Messina JL, Stein JE, Xu X, Amaria RN, Blank CU, et al. Pathological assessment of resection specimens after neoadjuvant therapy for metastatic melanoma. Ann Oncol 2018;29:1861–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cillo AR, Kurten CHL, Tabib T, Qi Z, Onkar S, Wang T, et al. Immune landscape of viral- and carcinogen-driven head and neck cancer. Immunity 2020;52:183–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell 2019;176:404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell 2020;181:747. [DOI] [PubMed] [Google Scholar]
- 27. Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 2018;24:978–85. [DOI] [PubMed] [Google Scholar]
- 28. Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O'Brien SA, et al. Single-cell analyses inform mechanisms of myeloid-targeted therapies in colon cancer. Cell 2020;181:442–59. [DOI] [PubMed] [Google Scholar]
- 29. Gerhard GM, Bill R, Messemaker M, Klein AM, Pittet MJ. Tumor-infiltrating dendritic cell states are conserved across solid human cancers. J Exp Med 2021;218:e20200264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, et al. A pan-cancer single-cell transcriptional atlas of tumor-infiltrating myeloid cells. Cell 2021;184:792–809. [DOI] [PubMed] [Google Scholar]
- 31. Kurten CHL, Kulkarni A, Cillo AR, Santos PM, Roble AK, Onkar S, et al. Investigating immune and non-immune cell interactions in head and neck tumors by single-cell RNA sequencing. Nat Commun 2021;12:7338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bacher P, Heinrich F, Stervbo U, Nienen M, Vahldieck M, Iwert C, et al. Regulatory T cell specificity directs tolerance versus allergy against aeroantigens in humans. Cell 2016;167:1067–78. [DOI] [PubMed] [Google Scholar]
- 33. Yang C, Siebert JR, Burns R, Gerbec ZJ, Bonacci B, Rymaszewski A, et al. Heterogeneity of human bone marrow and blood natural killer cells defined by single-cell transcriptome. Nat Commun 2019;10:3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cao J, Spielmann M, Qiu X, Huang X, Ibrahim DM, Hill AJ, et al. The single-cell transcriptional landscape of mammalian organogenesis. Nature 2019;566:496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 2017;14:979–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 2014;32:381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 2018;564:268–72. [DOI] [PubMed] [Google Scholar]
- 38. Liu B, Hu X, Feng K, Gao R, Xue Z, Zhang S, et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti–PD-1 therapy in lung cancer. Nat Cancer 2022;3:108–21. [DOI] [PubMed] [Google Scholar]
- 39. Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med 2019;25:1251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hsu J, Hodgins JJ, Marathe M, Nicolai CJ, Bourgeois-Daigneault MC, Trevino TN, et al. Contribution of NK cells to immunotherapy mediated by PD-1/PD-L1 blockade. J Clin Invest 2018;128:4654–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Khan M, Arooj S, Wang H. NK cell-based immune checkpoint inhibition. Front Immunol 2020;11:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dong W, Wu X, Ma S, Wang Y, Nalin AP, Zhu Z, et al. The mechanism of anti-PD-L1 antibody efficacy against PD-L1-negative tumors identifies NK cells expressing PD-L1 as a cytolytic effector. Cancer Discov 2019;9:1422–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Baran Y, Bercovich A, Sebe-Pedros A, Lubling Y, Giladi A, Chomsky E, et al. MetaCell: analysis of single-cell RNA-seq data using K-nn graph partitions. Genome Biol 2019;20:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fuchs YF, Sharma V, Eugster A, Kraus G, Morgenstern R, Dahl A, et al. Gene expression-based identification of antigen-responsive CD8(+) T cells on a single-cell level. Front Immunol 2019;10:2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods 2017;14:1083–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003;34:267–73. [DOI] [PubMed] [Google Scholar]
- 47. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Cell state annotations of CD45+ cells and clonality of T cells isolated from HNSCC biopsies.
Cell state fractions and transcriptional differences at baseline.
Validation of the differential abundance of Treg subpopulations at baseline using orthogonal strategies and an independent HNSCC dataset.
ICB-induced changes in immune cell state abundance and gene expression profiles.
Dual ICB-induced changes in activation state of Tregs.
Dual ICB-induced changes in dysfunctional CD8+ T cells of responding patients towards a less dysfunctional state.
Gene expression changes associated with response and resistance to ICB across cell states.
Data Availability Statement
The processed scRNA-seq data and clonotype information have been deposited in the NCBI Gene Expression Omnibus database (GSE232240). The raw scRNA-seq and TCR-seq data have been deposited in the European Genome-phenome Archive (EGA) database (EGAS00001007367 and EGAS00001007368, respectively) and will be made available from the corresponding author upon reasonable request. Data requests will be reviewed by the institutional review board of The Netherlands Cancer Institute and require a data transfer agreement after approval. The code that was used for the analyses in this study is available on GitHub (https://github.com/ZuurLabRep/ms_code_vanderleun_traets_singlecell_IMCISION).