

Abstract
Damages to the ear are very diverse and can depend on the type of inherited metabolic diseases (IMD). Indeed, IMDs can affect all parts of the auditory system, from the outer ear to the central auditory process. We have identified 219 IMDs associated with various types of ear involvement which we classified into five groups according to the lesion site of the auditory system: congenital external ear abnormalities, acquired external ear abnormalities, middle ear involvement, inner ear or retrocochlear involvement, and unspecified hearing loss. This represents the ninth issue in a series of educational summaries providing a comprehensive and updated list of metabolic differential diagnoses according to system involvement.
Keywords: ear, inborn errors of metabolism, inherited metabolic diseases, hearing loss, tinnitus, sensorineural hearing loss, conductive hearing loss, external ear
1. Introduction
This is the ninth in a series of articles that aim to provide a comprehensive list of inherited metabolic disorders (IMDs) associated with specific signs and symptoms. The first eight issues were dedicated to IMDs associated with movement disorders [1], metabolic liver diseases [2], those with psychiatric presentations [3], metabolic cardiovascular diseases [4], those with cerebral palsy phenotypes [5], metabolic dermatoses [6], ocular phenotypes [7] and neoplasms [8].
The list follows the classification utilized in the knowledgebase of IMDs [9], the Nosology of inborn errors of metabolism (IMDs) [10] and the International Classification of Inherited Metabolic Disorders (ICIMD) [11].
This issue will be dedicated to metabolic ear diseases. Damages to the ear are diverse and can depend on the type of IMD. Indeed, IMDs can affect all parts of the auditory system, from the outer ear to the central auditory process. Data source is the IEMbase (http://www.iembase.org).
2. The auditory system
Knowledge of the anatomo-functional organization of the auditory system is important to better understand auditory damage, especially different types of hearing loss. The organization of the auditory system is summarized in Figure 1. The processing of auditory information is complex and involves both peripheral and central mechanisms. The peripheral auditory system is classically divided into the external, middle and inner ear. Sound corresponds to variations in air pressure that travel to the ear, where it is picked up by the pinna and transmitted into the external auditory meatus (ear canal) where it reaches the tympanic membrane. The tympanic membrane transmits the sound to the inner ear (cochlea) via vibration of the ossicular chain (malleus, incus and stapes), which is located in the middle ear. The inner ear contains the neuro-sensory organ of hearing (organ of Corti), which allows for the mechano-electrical transduction of sound by the sensory cells (inner and outer hair cells), where an action potential will be produced at the level of the cochlear nerve. The auditory information will be then coded by central auditory pathways to the brain in order to understand the auditory information. In case of damage to the external ear (e.g., cerumen impaction) or middle ear (e.g., middle ear effusion in otitis media), the transmission function will be altered, resulting in conductive hearing loss. In case of damage to the inner ear (e.g., loss of hair cells), auditory nerve (e.g., auditory neuropathy spectrum disorder) or central auditory pathway, the perception function will be altered, resulting in sensorineural hearing loss (SNHL). Mixed hearing loss is defined as a combination of conductive hearing loss and SNHL.
Figure 1.

Schematic of auditory system organization with external, middle ear and inner ear views and cross-section of the cochlear canal with the view of the organ of Corti.
Legends: SV: scala vestibuli, ST: scala tympani, SM: scala media
The prevalence of hearing loss in the general population is estimated to be 20.3% [12]. There are more than 1,500 IMDs, each being somewhat rare and characterized by diverse and non-specific signs. As such, hearing health professionals rarely mention IMDs when diagnosing hearing loss. This is understandable, as IMDs are often not obvious, given the limited and poor description of clinical and biological phenotypes provided in the literature. Many IMDs are revealed by a severe decompensation requiring emergent diagnosis. However, some IMDs may be present with mild symptoms and a slow evolution, that can result in a longer and uncertain diagnosis period until the emergence of additional symptoms or worsening of existing symptoms. Consequently, better knowledge of IMDs associated with hearing loss symptoms would have two main benefits: i) best management of IMDs, especially those for which there is an approved treatment, and ii) earlier treatment of hearing loss, thereby improving patients’ comfort and quality of life.
3. Signs and symptoms
No formal guidelines have been published regarding when to suspect IMDs in children with ear or auditory disorders. We have identified 219 IMDs associated with various types of ear involvement which we classified into five groups according to the lesion site of the auditory system (Supplemental Table 1): i) congenital external ear abnormalities, ii) acquired external ear abnormalities, iii) middle ear involvement, iv) inner ear or retrocochlear involvement, and v) unspecified hearing loss.
Inner ear or retrocochlear involvement is the most common abnormality reported for IMDs with ear involvement (111/219; 51%) followed by unspecified hearing loss (75/219; 34%), congenital external ear abnormalities (27/219; 12%), middle ear involvement (14/219; 6%), and acquired external ear abnormalities in (5/219; 2%) (Figure 2). For inner ear or retrocochlear involvement, the most frequently reported disorder is SNHL (40%) (Supplemental Table 2).
Figure 2.
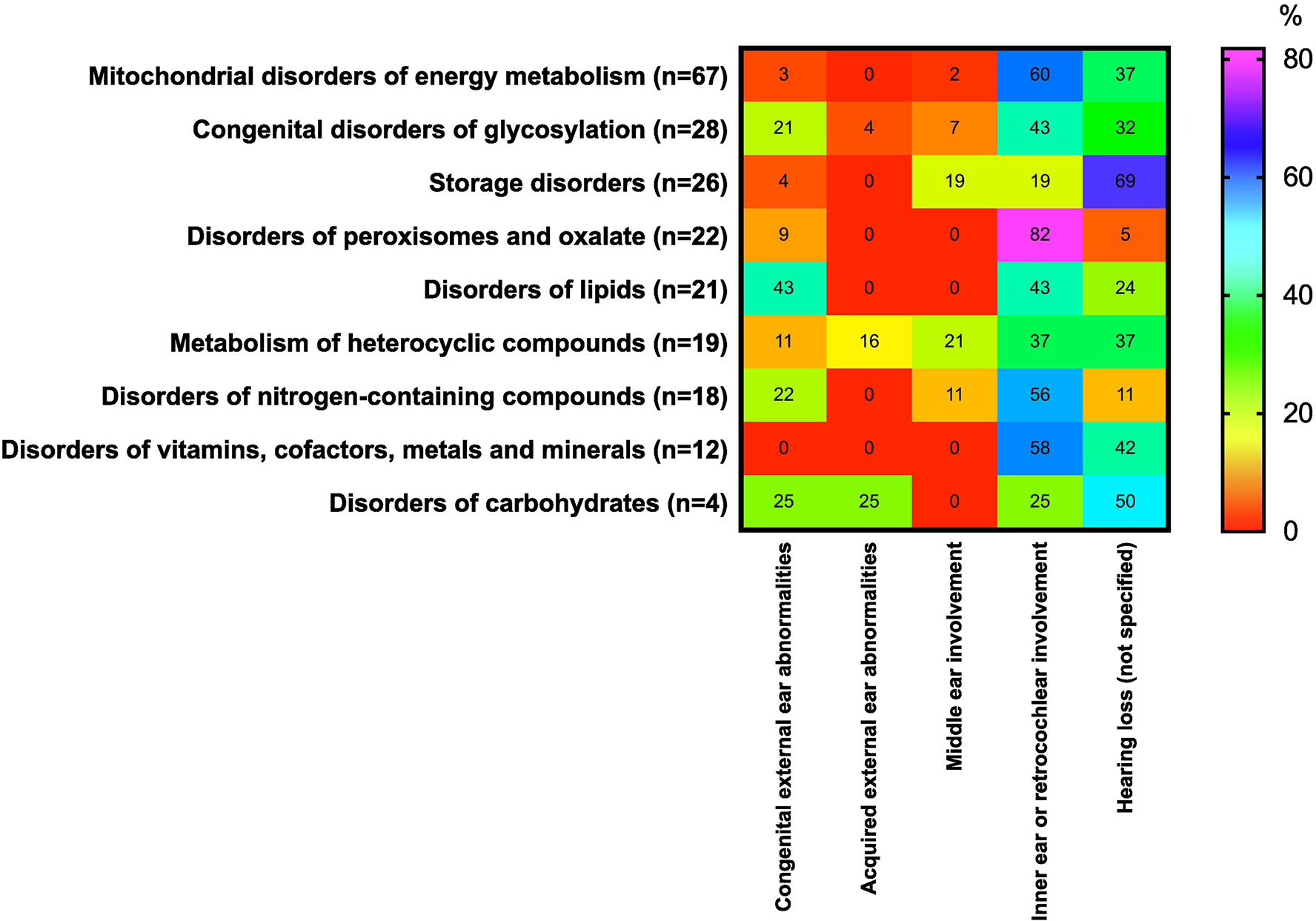
Occurrence (in percent) of ear symptoms associated with the 5 categories of IMDs. The percentages for ear involvement were calculated using the denominator of the total number of IMDs in each category presenting with any ear phenotype. The heat scale ranges from red (0%; diseases with no particular symptom reported) to violet (100%; diseases with particular symptoms reported) within the disorders group. For further information about the 9 categories of disorders affecting auditory system, see Supplemental Table 1. For interpretation of references to color in this figure legend, the reader is referred to the web version of this article.
The most representative group of IMDs associated with ear symptoms is mitochondrial disturbance (n=68). We observed some interesting associations between ear symptoms and groups of metabolic disorders. For example, peroxisomal disorders [e.g., peroxin 1 deficiency (Zellweger), glycerone 3-phosphate acyltransferase deficiency, alkylglycerone 3-phosphate synthase deficiency, X-linked adrenoleukodystrophy and adrenomyeloneuropathy, or peroxisomal straight-chain acyl-CoA oxidase deficiency] were associated with inner ear or retrocochlear involvement in 82% of disorders, with only marginal associations with other types of ear involvement. Similarly, the occurrence of inner ear or retrocochlear involvement was also strongly associated (60%) with mitochondrial disorders of energy metabolism [e.g., pyruvate dehydrogenase kinase isoenzyme 3 superactivity, mitochondrial aconitase deficiency, alpha-ketoglutarate dehydrogenase deficiency, mitochondrial ATP-Mg-phosphate transporter deficiency, BCS1L deficiency-GRACILE syndrome, CEP89 deficiency, mitochondrial ATP synthase F0 subunit 6 deficiency, mitochondrial cytochrome b deficiency, holocytochrome c synthase deficiency, topoisomerase 3α deficiency, mitochondrial ribosomal large subunit 2/3 deficiencies, SERAC1 deficiency-MEGDEL Syndrome, coenzyme Q6 monooxygenase deficiency, mitochondrial tRNA(AA) deficiencies, and more], as well as disorders of vitamins, cofactors, metals, and minerals (58%; e.g., ferredoxin reductase deficiency, frataxin deficiency-Friedreich ataxia, biotinidase deficiency, riboflavin transporter 2/3 deficiencies, vitamin D receptor deficiency, or MEDNIK-like syndrome) and nitrogen-containing compounds (56%; e.g., phosphoribosyl pyrophosphate synthetase 1 deficiency, ribonuclease T2 deficiency, equilibrative nucleoside transporter 3 deficiency, dipeptidase deficiency, aminoacylase 1 deficiency, adenosine kinase deficiency, 17-beta-hydroxysteroid dehydrogenase type 10 deficiency, propionic acidemia due to propionyl-CoA carboxylase subunit alpha/beta deficiency or 3-hydroxyanthranilic acid 3,4-dioxygenase deficiency). Unspecified hearing loss was associated with disorders of vitamins, cofactors, metals, and minerals in 42% of entries (e.g., thiamine transporter 1 deficiency, MEDNIK syndrome, acetyl-CoA transporter deficiency, deficiency of copper chaperone for superoxide dismutase or KCNJ10 deficiency), and with mitochondrial disorders of energy metabolism in 37% [e.g., acyl-CoA Dehydrogenase 9 deficiency, cytochrome c oxidase subunit 6A1 deficiency, mitochondrial DNA polymerase gamma catalytic subunit deficiency, TWINKLE mitochondrial DNA helicase deficiency, single-stranded DNA-binding protein 1 deficiency, OPA1 deficiency, OPA3 deficiency-Costeff syndrome, mitochondrial tRNA(AA) deficiencies, and more]. Since inner ear or retrocochlear involvement is the most frequent symptom in IMDs, the differential diagnosis of IMDs requires specific explorations to distinguish these diagnoses. For example, unspecified hearing loss was associated with 69% of patients with lysosomal storage disease (e.g., acid sphingomyelinase deficiency-Niemann-Pick type A, beta-galactosylceramidase deficiency-Krabbe disease, Krabbe disease-like disorder due to saposin A deficiency, acid ceramidase deficiency, primary neurologic phenotype-Farber disease, alpha-neuraminidase deficiency, beta-mannosidase deficiency, and almost all mucopolysaccharidoses) and 50% of patients with carbohydrate disorders (e.g., ribose-5-phosphate isomerase deficiency; Figure 2). Such distinctions may point to more specific diagnosis of hearing loss.
Interestingly, some IMDs are associated with several ear abnormalities without specificity. For example, 43% of disorders of lipids were associated with congenital external ear abnormalities (e.g., alkaline ceramidase 3 deficiency, ZDHHC9 palmitoyltransferase deficiency, phosphatidylinositol 4-kinase type 2-alpha deficiency, chondrodysplasia punctata 2, recessive-Conradi-Hünermann syndrome, or cytochrome P450 oxidoreductase deficiency-Antley-Bixler syndrome), inner ear or retrocochlear involvement (43%; e.g., very long-chain fatty acid elongase 1 deficiency, phosphatidylserine flippase deficiency, sphingosine-1-phosphate lyase deficiency, Smith-Lemli-Opitz syndrome, or ATP8B1 deficiency-Byler disease), and unspecified hearing loss (24%; e.g., very long-chain fatty acid elongase 5 deficiency, porcupine palmitoyltransferase deficiency-Goltz syndrome, or UDP-glucuronosyltransferase A1 deficiency). Similarly diffuse associations with ear involvement were observed for congenital disorders of glycosylation, disorders of carbohydrates, and metabolism of heterocyclic compounds. Some symptoms (e.g., acquired external ear abnormalities, middle ear involvement) were associated with relatively few IMDs. A more general summary of IMDs associated with 5 categories of symptoms affecting the ear is summarized in Table 2.
Table 2.
Main groups of IMDs associated with 5 categories of symptoms affecting ear.
| Congenital external ear abnormalities | Acquired external ear abnormalities | Middle ear involvement | Inner ear or retrocochlear involvement | Unspecific hearing loss |
|---|---|---|---|---|
|
| ||||
| • Pyrimidine metabolism | • Insulin secretion and signaling | • Pyrimidine metabolism | • Purine metabolism | • Aminoacylase deficiencies |
| • Proline and ornithine metabolism | • O-xylosylation and glycosaminoglyc an synthesis | • Proline and ornithine metabolism | • Nucleotide metabolism | • Branched-chain amino acid metabolism |
| • Asparagine metabolism | • Ribosomal biogenesis | • Mitochond rial carriers | • Glutathione metabolism | • Thiamine metabolism |
| • Insulin secretion and signaling | • Mucolipid oses | • Aminoacylase deficiencies | • Copper metabolism | |
| • Mitochondrial carriers | • Mucopolys accharidoses | • Sulfur amino acid and sulfide metabolism | • Magnesium metabolism | |
| • Mitochondrial protein quality control | • N-linked glycosylation | • Branched-chain amino acid metabolism | • Pentose phosphate pathway and polyol metabolism | |
| • Non-mitochondrial phospholipid metabolism | • Vesicular trafficking | • Tryptophan metabolism | • Insulin secretion and signaling | |
| • Non-lysosomal sphingolipid metabolism | • Ribosomal biogenesis | • Lipoic acid and iron-sulfur metabolism | • Complex I assembly | |
| • Palmitoylation | • Biotin metabolism | • Complex IV subunits | ||
| • Phosphoinositide metabolism | • Riboflavin metabolism | • Complex IV assembly and ancillary proteins | ||
| • Cholesterol biosynthesis | • Vitamin D metabolism | • Mitochondrial DNA depletion, multiple deletion, or intergenomic communication | ||
| • Steroid metabolism | • Copper metabolism | • Mitochondrial ribosomopathies | ||
| • Autophagy | • Glycogen storage diseases | • Mitochondrial translation factors | ||
| • Plasmalogen synthesis | • Pyruvate metabolism | • Mitochondrial tRNA | ||
| • Peroxisomal β-oxidation | • The Krebs cycle | • Mitochondrial tRNA incorporation and recycling | ||
| • N-linked glycosylation | • Thiamine metabolism | • Mitochondrial fission | ||
| • O-xylosylation and glycosaminoglyc an synthesis | • Mitochondrial carriers | • Mitochondrial protein quality control | ||
| • Glycosylphospha tidylinositol biosynthesis | • Complex I assembly | • Primary CoQ10 deficiencies | ||
| • Vesicular trafficking | • Complex III/V subunits | • Fatty acid synthesis and elongation | ||
| • Ribosomal biogenesis | • Complex IV assembly and ancillary proteins | • Non-mitochondrial phospholipid metabolism | ||
| • Mitochondrial cytochrome synthesis and incorporation | • Palmitoylation | |||
| • Mitochondrial transcription and RNA transcript processing | • Phosphoinositide metabolism | |||
| • Mitochondrial ribosomopathies | • Bilirubin metabolism and biliary transport | |||
| • Mitochondrial tRNA | • Autophagy | |||
| • Mitochondrial tRNA incorporation and recycling | • Sphingolipidoses | |||
| • Mitochondrial fission | • Oligosaccharidoses | |||
| • Mitochondrial phospholipid metabolism | • Mucopolysaccharidoses | |||
| • Mitochondrial protein import | • Plasmalogen synthesis | |||
| • Mitochondrial protein quality control | • Peroxisomal β-oxidation | |||
| • Other mitochondrial homeostasis | • N-linked glycosylation | |||
| • Primary CoQ10 deficiencies | • O-xylosylation and glycosaminoglycan synthesis | |||
| • Fatty acid synthesis and elongation | • Glycosylphosphatidylinositol biosynthesis | |||
| • Non-mitochondrial phospholipid metabolism | • Vesicular trafficking | |||
| • Non-lysosomal sphingolipid metabolism | • Non-mitochondrial tRNA processing and aminoacyl-tRNA synthetases | |||
| • Cholesterol biosynthesis | • Ribosomal biogenesis | |||
| • Bilirubin metabolism and biliary transport | • Organelle interplay | |||
| • Autophagy | • Synaptic vesicle cycle | |||
| • Sphingolipidoses | ||||
| • Plasmalogen synthesis | ||||
| • Peroxisomal P-oxidation | ||||
| • Peroxisomal biogénesis | ||||
| • N-linked glycosylation | ||||
| • O-mannosylation | ||||
| • O-xylosylation and glycosaminoglycan synthesis | ||||
| • Glycosylphosphatidylino sitol biosynthesis | ||||
| • Glycolipid glycosylation | ||||
| • Monosaccharide synthesis and interconversion | ||||
| • Nucleotide-sugar synthesis | ||||
| • Non-mitochondrial tRNAprocessing and aminoacyl-tRNA synthetases | ||||
| • Organelle interplay | ||||
To conclude, some IMDs are associated with some specific ear involvement that may greatly help in early diagnosis. Other groups of IMDs have a more variable occurrence of ear involvement, which may depend on how and when the IMDs are revealed. These findings highlight the necessity of rigorous characterization of ear involvement, and complete clinical evaluation according to specific targeted biological investigations and careful family history.
3.1. External ear abnormalities
In case of external ear abnormalities (congenital or acquired), the diagnosis is made after examination of the external ear. Most deformities are present at birth. For example, in case of prominent ears (congenital abnormalities), ears protrude more than 2 cm from the side of the head, with no association with hearing loss.
Cauliflower ears can be associated with diseases affecting the cartilage (e.g., sulfate transporter deficiency) [13]; usually, the deformities affect the external ear at childhood or adolescence.
3.2. Middle ear abnormalities
The most common etiologies are recurrent acute otitis media and otitis media with effusion. In case of acute otitis media, otoscopy is abnormal with bulging and erythema of the tympanic membrane (Figure 3B). In case of recurrent acute otitis media with persistent middle ear effusion, hearing loss can persist. Otoscopy may reveal an opaque tympanic membrane or visible air-fluid level behind the tympanic membrane (Figure 3C). A yellow appearance of the tympanic membrane is often described, and the tympanic membrane can be retracted.
Figure 3.
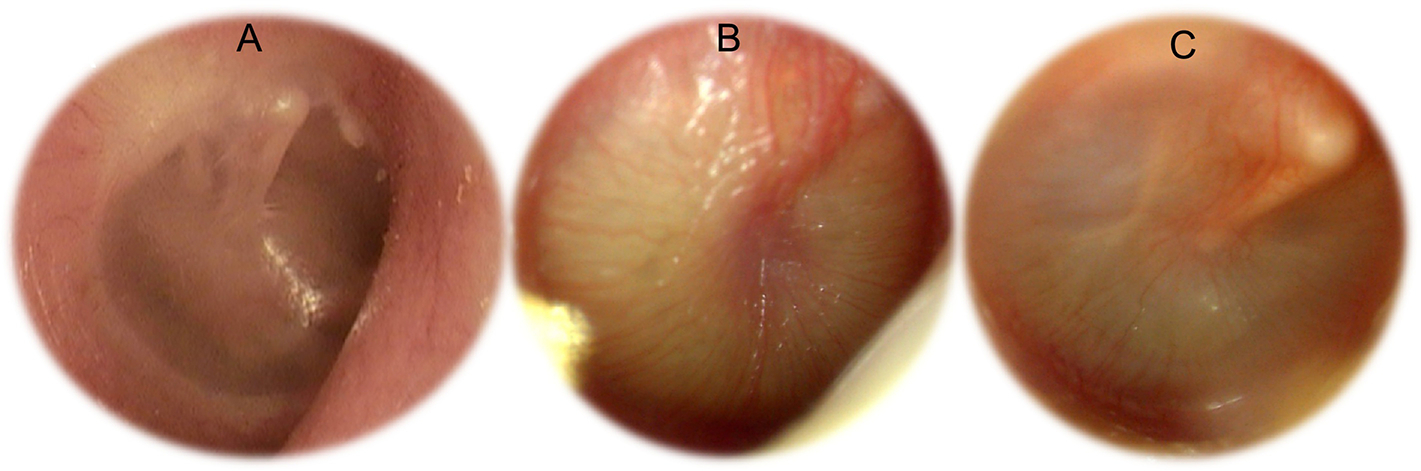
Otoscopies. A: Normal right otoscopy with visualization of the ossicular chain. B: Otitis media. C: Otitis media with effusion
Hearing loss due to middle ear abnormalities (e.g., recurrent media otitis or otitis media with effusion) can be associated with storage disorders such as mucopolysaccharidosis (MPS). Conductive hearing loss due to recurrent otitis has been observed in 80% of patients with MPSII and 25% of patients with MPSVI [14].
3.3. Inner ear or retrochlear abnormalities
In case of SNHL, otoscopy is normal (Figure 3A). Damage mostly occurs in the inner ear (e.g., propionic acidemia) or may have retrocochlear involvement (e.g., auditory nerve demyelination in biotinidase deficiency) [15–16]. In case of SNHL due to inner ear damage, speech intelligibility is related to the degree of hearing loss found with pure-tone audiometry, and auditory brainstem responses exhibit normal latencies. SNHL due to retrocochlear damage (auditory nerve demyelination) is associated with poor speech recognition thresholds and word recognition scored; auditory brainstem responses exhibit abnormally long latencies. Magnetic resonance imaging (MRI) of the brain is used to eliminate tumors such as vestibular schwannoma and to identify inner ear anomalies.
Tinnitus is a symptom that mainly occurs in cases of inner ear or retrocochlear involvement.
Hearing loss in patients affect by IMDs is not uncommon and varies from few patients described as case reports (e.g., Pompe disease) to 100% of patients (e.g., ß-mannosidosis or thiamine-responsive megaloblastic anemia) [14, 17–19].
4. Diagnosis and differential diagnosis
Pure-tone and speech audiometry is used to identify conductive hearing loss, mixed hearing loss, or SNHL, as well as the degree of hearing loss. Tympanometry is used to evaluate the middle ear function and will suggest otitis media effusion in case of a flat tympanogram.
In case of asymmetrical SNHL or poor speech understanding not associated with elevated pure-tone thresholds, MRI of the brain will be used to eliminate retrocochlear involvement (e.g., vestibular schwannoma) and inner ear malformations, and to ensure the presence of the auditory nerve. Objective audiometry (e.g., stapedial reflex testing, otoacoustic emissions, auditory brainstem responses) will be included in the audiologic evaluation, especially if auditory neuropathy is suggested.
Newborn hearing screening allows for identification of hearing loss using objective measures (e.g., automated otoacoustic emissions and auditory brainstem responses). Objective measures such as auditory steady-state responses can be used to determine auditory thresholds in newborns where subjective audiometry cannot be performed. In case of suspected hearing loss, newborns must be referred to a hearing specialist to clarify the diagnosis and evaluate the type and degree of hearing loss.
Certain situations should alert practitioners to the possibility of an IMD in case of association with other sensory disorders, familial history with same symptoms, neurological symptoms, dysmorphic features, or liver failure.
A list of biological investigations to aid in the diagnosis of the various listed IMDs is summarized in Table 1. Some of the investigations (e.g., basic tests in the 1st column of Table 1) are performed by all biological laboratories and results may be obtained in less than 24 hours with few restrictive sampling conditions. Other explorations such as metabolic profiles (e.g., aminoacids, acylcarnitines, organic acids; 2nd column of Table 1) are performed by specific metabolic centers and require biologist expertise in IMDs. Results may be longer to obtain because the tests are not automated (they are mainly based on mass spectrometry) and may require specific conditions for sampling (e.g., fasting, avoiding some drugs, etc.). However, in case of emergency, results are needed in less than 24–48 hours. Thus, these profiles are the standard metabolic explorations that must be available at main hospital centers. Importantly, other metabolic profiles listed in the 2nd column of Table 1 are performed by a limited number of laboratories, as are some specific tests (3rd column of Table 1) that will be performed only in case of highly justified conditions (e.g., SAM & SAH, lysosomal enzymes, etc.). As metabolic testing may take a long time, the explorations to help in the diagnosis must be hierarchized, with priority to treatable disorders. Thus, a close collaboration between the ENT specialist and the metabolic expert is necessary to focus on specific disorders according to hearing loss symptoms, enabling the most appropriate panel, before expanding the diagnosis to include metabolic exploration as a second step. For more details see Supplemental Table 2. Careful examination of all ear structures is required, and the diagnostic approach, as with all IMDs, should prioritize treatable disorders.
Table 1.
Biochemical investigations in metabòlic diseases affecting the ear.
| Basic tests | Profiles | Special tests |
|---|---|---|
|
| ||
| Blood count | Amino acids (P,U) | Copper(S,U) |
| ASAT/ALAT (P) | Organic acids (U) | Ceruloplasmin (S) |
| CK (P) | Acylcarnitines (DBS, P) | Ferritin (S) |
| ALP (P) | Sialotransferins (S) | Manganese (B) |
| Lactate (P) | Sterols (P) | Carnitine (P) |
| Pyruvate (P) | Bile acids (U) | Glycogen (L) |
| Glucose(P) | Oligosaccharides (U) | Lysosomal Enzymes (S) |
| Ammonia (B) | Mucopolysaccharides (U) | Vitamins (S) |
| Bilirubin (P) | Plasmalogens (P) | Flavins (B) |
| Coagulation factors | VLCFA (P) | Interferon-alpha (CSF) |
| Immunoglobulins | Lipid panel (S) | SAM & SAH (P) |
| Polyols (P,U) | CoQ10 (M, P, WBC) | |
ALAT: alanine aminotransferase
ALP: alkaline phosphatase
ASAT: aspartate aminotransferase
CK: creatine kinase
CoQ10: cofactor Q10
SAH: S-adenosyl-homocysteine
SAM: S-adenosyl-methionine
U: urine
VLCFA: very-long chain fatty acids
5. Treatment
Therapy of hearing loss in patients with IMDs involves primary treatment of the underlying condition as enzyme replacement therapy or oral supplementation or restriction, which may increase life expectancy, quality of life and limit disease progression.
In case of conductive hearing loss due to recurrent otitis, myringotomy with tube insertion allows for recovery of normal thresholds and development of oral language, especially in children under 3 years old.
In case of SNHL, hearing aids will be proposed first. If hearing aids are not sufficiently efficacious, cochlear implantation may be proposed to restore hearing.
In any cases of hearing loss associated with delayed language development in childhood, speech therapy will be necessary to improve oral language development.
Treatment for hearing loss needs a multidisciplinary approach that includes pediatricians, internists, ENT practitioners, speech therapists, and audiologists.
6. Conclusion
We have provided a comprehensive list of metabolic diseases associated with ear disease (external ear abnormalities) or hearing loss (middle or inner ear involvement). We have also proposed a battery of standard biological explorations to aid in diagnosis based on the many possible IMDs, with a priority of treatable diseases. This represents the ninth issue in a series of educational summaries providing a comprehensive and updated list of metabolic differential diagnoses according to system involvement. The full list can be freely accessed at www.iembase.org/gamuts, and will be curated and updated on a regular basis.
Supplementary Material
Highlights.
We found 219 inborn errors of metabolism (IMDs) associated with various types of ear involvement and provide a list of specific explorations to aid in their diagnosis.
This is the ninth issue in a series of educational summaries providing a comprehensive list of metabolic differential diagnoses.
This list will be curated and updated on a continual basis in the IEMbase website.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Ferreira CR, Hoffmann GF, Blau N, Clinical and biochemical footprints of inherited metabolic diseases. I. Movement disorders, Mol. Genet. Metab. 127 (2019) 28–30, 10.1016/j.ymgme.2019.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Ferreira CR, Cassiman D, Blau N, Clinical and biochemical footprints of inherited metabolic diseases. II. Metabolic liver diseases, Mol. Genet. Metab. 127 (2019) 117–121, 10.1016/j.ymgme.2019.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Horvath GA, Stowe RM, Ferreira CR, Blau N, Clinical and biochemical footprints of inherited metabolic diseases. III. Psychiatric presentations, Mol. Genet. Metab. 130 (2020) 1–6, 10.1016/j.ymgme.2020.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ferreira CR, Blau N, Clinical and biochemical footprints of inherited metabolic diseases. IV. Metabolic cardiovascular disease, Mol. Genet. Metab. 132 (2021) 112–118, 10.1016/j.ymgme.2020.12.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Horvath GA, Blau N, Ferreira CR, Clinical and biochemical footprints of inherited metabolic disease. V. Cerebral palsy phenotypes, Mol. Genet. Metab. (2021) 10.1016/j.ymgme.2021.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ferreira CR, Martinelli D, Blau N. Clinical and biochemical footprints of inherited metabolic diseases. VI. Metabolic dermatoses. Mol Genet Metab. 2021. Sep-Oct;134(1–2):87–95. doi: 10.1016/j.ymgme.2021.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Garanto A, Ferreira CR, Boon CJF, van Karnebeek CDM, Blau N. Clinical and biochemical footprints of inherited metabolic disorders. VII. Ocular phenotypes. Mol Genet Metab. 2022. Apr;135(4):311–319. doi: 10.1016/j.ymgme.2022.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jerves T, Blau N, Ferreira CR. Clinical and biochemical footprints of inherited metabolic diseases. VIII. Neoplasias. Mol Genet Metab. 2022. Mar 28:S1096-7192(22)00281-5. doi: 10.1016/j.ymgme.2022.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lee JJY, Wasserman WW, Hoffmann GF, van Karnebeek CDM, Blau N, Knowledge base and mini-expert platform for the diagnosis of inborn errors of metabolism, Genet. Med. Off. J. Am. Coll. Med. Genet. 20 (2018) 151–158, 10.1038/gim.2017.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ferreira CR, van Karnebeek CDM, Vockley J, Blau N, A proposed nosology of inborn errors of metabolism, Genet. Med. Off. J. Am. Coll. Med. Genet. 21 (2019) 102–106, 10.1038/s41436-018-0022-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ferreira CR, Rahman S, Keller M, Zschocke J, Group Icimd advisory, an international classification of inherited metabolic disorders (ICIMD), J. Inherit. Metab. Dis. 44 (2021) 164–177. doi: 10.1002/jimd.12348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].GBD 2019 Hearing Loss Collaborators. Hearing loss prevalence and years lived with disability, 1990–2019: findings from the Global Burden of Disease Study 2019. Lancet. 397 (2021) 996–1009. doi: 10.1016/S0140-6736(21)00516-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Macias-Gomez NM, Megarbane A, Leal-Ugarte E, Rodriguez-Rojas LX, Barros-Nunez P Diastrophic dysplasia and atelosteogenesis type II as expression of compound heterozygosis: first report of a Mexican patient and genotype-phenotype correlation. Am. J. Med. Genet. 129 (2004) 190–192. doi: 10.1002/ajmg.a.30149. [DOI] [PubMed] [Google Scholar]
- [14].Trinh TT, Blasco H, Maillot F, Bakhos D. Hearing loss in inherited metabolic disorders: A systematic review. Metabolism. 122 (2021) 154841.doi: 10.1016/j.metabol.2021.154841. [DOI] [PubMed] [Google Scholar]
- [15].Heller AJ, Stanley C, Shaia WT, Sismanis A, Spencer RF, Wolf B. Localization of biotinidase in the brain: implications for its role in hearing loss in biotinidase deficiency. Hear Res 173 (2002) 62–68. doi: 10.1016/s0378-5955(02)00609-3. [DOI] [PubMed] [Google Scholar]
- [16].Grünert SC, Bodi I, Odening KE Possible mechanisms for sensorineural hearing loss and deafness in patients with propionic acidemia Orphanet J Rare Dis, 12 (2017. 13) 30. doi: 10.1186/s13023-017-0585-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Labay V, Raz T, Baron D, Mandel H, Williams H, Barrett T, et al. Mutations in SLC19A2 cause thiamine-responsive megaloblastic anaemia associated with diabetes mellitus and deafness. Nat Genet, 22 (1999) 300–304. doi: 10.1038/10372. [DOI] [PubMed] [Google Scholar]
- [18].Sedel F, Friderici K, Nummy K, Caillaud C, Chabli A, Dürr A, et al. Atypical Gilles de la Tourette Syndrome with beta-mannosidase deficiency. Arch Neurol, 63 (2006) 129–131. doi: 10.1001/archneur.63.1.129 [DOI] [PubMed] [Google Scholar]
- [19].Hanisch F, Rahne T, Plontke SK. Prevalence of hearing loss in patients with late-onset Pompe disease: audiological and otological consequences. Int J Audiol 52 (2013) 816–823. doi: 10.3109/14992027.2013.840932. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.