

Abstract
Anthracene core-based hole-transporting material containing TIPs (triisopropylsilylacetylene) has been spotlighted as potential donors for perovskite solar cells (SCs) due to their appropriate energy levels, efficient hole transport capacity, high stability, and high power conversion efficiency. Herein, we have efficiently designed seven new highly conjugated A–B–D–C–D molecules (AS1–AS7) containing an anthracene core. We used end-capped modifications of donor units with acceptor units on one side and then theoretically characterized them for their appropriate use for SC applications. Modern quantum chemistry techniques have theoretically described the R (reference molecule) and developed (AS1–AS7) molecules. Moreover, the proposed (AS1–AS7) molecules are explored with density functional theory (DFT) and time-dependent density functional theory (TD-DFT) employing B3LYP/6-31G(d,p), and numerous parameters like photovoltaic, optical and electronic characteristics, frontier molecular orbital, excitation, binding and reorganization (λe and λh) energies, open circuit voltage, light harvesting efficiency, transition density matrix, fill factor, and the density of states have been studied. End-capped modification causes a smaller band gap between the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO), higher UV–vis absorption maxima, tuned energy levels, lower binding and reorganizational (λe and λh) energies, and larger Voc values in proposed (AS1–AS7) molecules than R. AS5 has a remarkable absorption maximum of 495.94 nm and a narrow optimal energy gap (Eg) of 1.46 eV. Furthermore, a complex study of AS5:PC61BM has revealed extraordinary charge shifting at the HOMO (AS5)–LUMO (PC61BM) interface. Our results suggested that newly developed anthracene core-based compounds (AS1–AS7) would be effective candidates with excellent photovoltaic and optoelectronic properties and could be employed in future organic and perovskite SC applications.
1. Introduction
Perovskite types of organic–inorganic hybrid halides have exceptional characteristics, such as their significant absorption constant,1 considerable charge-carrier diffusion length,2 minimum trap density,3 and low excitation binding energy.4 With efficiencies of 25.5%, they have become one of the most promising future photovoltaic materials. One of the most crucial variables in getting excellent performance in perovskite solar cells (PSCs) is the presence of a hole-transporting material. Hole-transporting materials (HTMs) are significant in perovskite types of organic solar cells (OSCs) by transporting the cations from the perovskite to the counter electrode.5–9 2,2′,7,7′-tetrakis(N,N-di-p-methoxyphenylamine)-9,9′-spirobifluorene (Spiro-OMeTAD) is a popular HTM that has found widespread use in PSCs. However, for PSCs to be helpful in the future, new HTMs must be developed to compensate for their poor stability and excellent pricing.
For its exceptional hole-carrying capacity, dimethoxy-arylamine has been extensively employed as a donor component by many studies. Furthermore, donor (arylamine) π spacer-donor (arylamine) materials have been designed to synthesize novel HTMs by straightforward modifications. Thiophene compounds,10,11 fluorene-thiophene,12 and carbazole compounds,13,14 bound by donors (amine groups), are extensively explored in PSCs to substitute the costly and unstable Spiro-OMeTAD. From all these compounds, anthracene-based compounds stand out for their promising features and widespread use in these devices, such as organic transistors with thin films, light-producing diodes, and perovskite-type OSCs.15–19 Using easily synthesized anthracene HTMs, Liu recently achieved a PEC of 17.27%.20 A102-based SCs function better than A101-based cells due to superior vertical charge transfer from regulated intermolecular interactions. However, A101 devices still have trouble dissolving in water. The power conversion efficiencies of PSCs using organic small molecules as HTMs have attracted a lot of interest. Siddique et al. developed four tiny molecule donors for SCs using a triphenylamine (TPA) donor moiety. The proposed D1-D4 molecules had unique end-capped units, and their structure–property connections were theoretically estimated. The D3 molecule was found to be the best candidate for future SCs, with a small excitation energy, an energy gap, and the smallest reorganization energies. The theoretically modeled simulation supports the possibility of adjusting end-capped unit configurations for optoelectronic characteristics.21–24 Recently, Paek successfully synthesized and designed donor–spacer–donor type HTM (i.e., PEH-16), incorporating anthracene containing triisopropylsilylacetylene (TIPs) derivative (π-conjugation part) and dimethoxy TPA (donor part) for PSCs. Anthracene containing TIP molecules displayed high stability and had an outstanding efficiency of power conversion than Spiro-OMeTAD.25
In this work, we have efficiently designed seven anthracene core-based (AS1–AS7) molecules by side-chain modification of the reference molecule (R) for future photovoltaic applications. In these designed systems, the end-cap donor part of the reference molecule on one side is replaced with the bridge (furan) and robust alternative electron-withdrawing groups, forming a backbone such as A–B–D–C–D. These alternative electron-withdrawing groups (AL1–AL7) have great potential to boost the absorption range, and the same furan bridge is utilized for all molecules (AS1–AS7). Furthermore, it worked as a transporter to transfer the charge from the electron-donating part to the acceptor region. These acceptor groups (AL1–AL7) are electron-deficient, allowing for better charge separation and reducing recombination loss, and thus are helpful in improving the photovoltaic performances of the optoelectronic devices. Moreover, these groups also have higher conjugation and electron affinity, which is greatly beneficial in separating HOMO and LUMO in designed molecules.26–28 After this, all designed (AS1–AS7) compounds are explored with DFT and time-dependent theory and compared with the R (reference molecule); parameters like photovoltaic, optical and electronic properties, open circuit voltage, frontier molecular orbital (FMO), binding, reorganizational (λe and λh) and excitation energies, light harvesting efficiency, transition density matrix (TDM), and density of states (DOS) have been studied. Furthermore, a detailed study is carried out to examine the charge mobilities from the donor part toward the acceptor unit to suggest that the proposed molecules might be good options for highly efficient OSCs. Finally, we will recommend a unique setup for future inventions in the field of OSCs.
2. Computational Details
DFT, or density functional theory, is a very effective computational tool in the scientific community. This technique has been shown to be effective in assessing a wide range of quantum mechanical properties with the highest reliability and precision.29,30 The quantum chemical computations were performed using Gaussian 09W,31 and the R molecule and newly proposed molecule (AS1–AS7) configurations were seen using Gauss View 5.0.32 Initially, the optimization of the R (reference molecule) was carried out with five alternative DFT-based functionals such as B3LYP,33 MPW1PW91,34 CAM-B3LYP,35 ωB97XD,36 and M062X37 at 6-31G (d,p). The selected functionals were then utilized to carry out further studies. To determine the absorption maxima (λmax) of the standard R (reference molecule), DFT was performed at these abovementioned functional levels in both the solvent (tetrahydrofuran) and gaseous phase. Using a modestly common technique, we compared the theoretically predicted UV–vis value of the R at these levels to the observed UV–vis range to determine the optimal functional level for this investigation. We discovered that the UV–vis λmax of the R molecule measured at 6-31G (d,p)/B3LYP was the most closely matched with the actual absorption maxima value25 as is typical for the most acceptable theoretical functional. Hence, the fundamental optical and photovoltaic characteristics of AS1–AS7 were computed using this functional level (i.e., B3LYP). For R25 and AS1–AS5 donors, we carried out maximum absorption (λmax), geometric optimization, the DOS, TDM, molecular electrostatic potential (MEP), reorganizational (λe and λh) energy, open-circuit voltage, FMO, and charge transfer analysis at the selected DFT level. Data from a Gaussian evaluation was used to create a map of absorption spectra using Origin 8.0.38 The DOS of the R and AS1-AS7 molecules were studied using PyMOlyze-1.139 and Origin 8.0 to determine the role of individual atoms and groups of atoms in allocating electronic masses among the possible states. TDM data were converted into maps showing excitation mobility and interaction among the atoms using Multiwfn software.40 The reorganizational energy of a molecule is the most significant feature in determining the electron and hole mobilities of that molecule. RE is categorized into external (RE) and internal reorganizational energy. The first is associated with perturbations in the external environment and the effects of polarity on charge transport generally, which is often disregarded, while the second is related to modification of the interior geometry of the molecule. The reorganizational (λe and λh) energies were estimated by eqs 1 and 2(41,42)
| 1 |
| 2 |
Herein, E– and E+ are the anionic and cationic energies derived using optimized anionic and cationic molecules, respectively. E0 denotes the ground state energy of the optimized neutral molecule, whereas E+0 and E–0 are the anion and cation energies computed from the molecular geometry of the cationic and anionic structures in the neutral condition, respectively. Moreover, E0+ and E0– show the energy of the neutral molecule, which is obtained through optimized cationic and anionic geometry.43
3. Results and Discussion
The primary goal of this investigation was to theoretically improve a new anthracene core containing TIP donor molecules with latent acceptor units and calculate their optical and electronic properties. The experimentally synthesized donor–π–donor anthracene core-based PEH-16 derivative (selected as a reference in this investigation) can be used to design new molecules. The reference molecule has two components: (i) dimethoxy TPA (a donor unit) and (ii) a core that contains a TIPs-anthracene derivative. The end-capped modification of the donor unit is critical to getting better optoelectronic properties. The dimethoxy component of the donor group of the R on one side was replaced with a bridge and alternative acceptor groups (AL1–AL7), while the core was preserved to produce seven new (AS1–AS7) organic materials having remarkable photovoltaic characteristics. The different acceptor groups (AL1–AL7) are shown in Figure 2.
Figure 2.

Structural illustration of the acceptor groups (AL1–AL7).
Initially, the optimization of R (reference molecule) was performed at the 6-31G (d,p) level of DFT in five different functionals (CAM-B3LYP, B3LYP, ωB97XD, M062X, and MPW1PW91). After optimization with the functionals as mentioned above, the reference molecule was analyzed by UV–vis spectroscopy in tetrahydrofuran using the CPCM solvation model. Reference molecule absorption maxima in tetrahydrofuran solvent were measured to be 304.35, 367.38, 300.39, 313.80, and 353.68 nm at functionals CAM-B3LYP, B3LYP, ωB97XD, M062X, and MPW1PW91, respectively. Figure 1 and Table 1 show the reported and calculated λmax of R at various basis sets. Absorption peaks at 380 nm were observed experimentally.21 Based on the results, B3LYP and 6-31G (d,p) have opted for further investigation since they were observed near the experimentally measured absorption maximum value.
Figure 1.
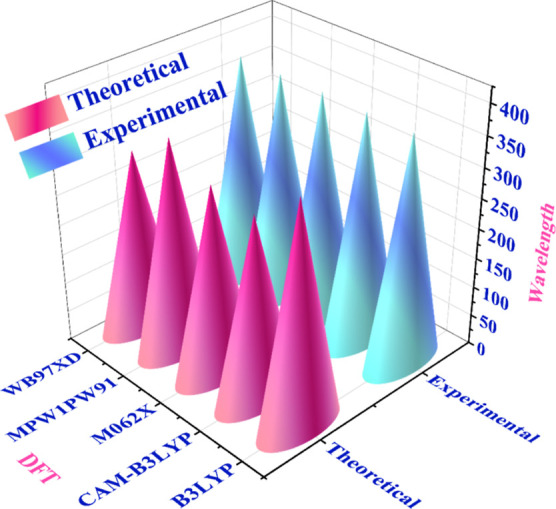
3D bar chart of the R (reference molecule) with five distinct functionals in a tetrahydrofuran solvent.
Table 1. Reported and Calculated Value of λmax of the R at Various Basis Sets.
| DFT functionals | DFT calculated values λmax (nm) | reported value λmax (nm) |
|---|---|---|
| R | 380 | |
| B3LYP | 367.38 | |
| CAM-B3LYP | 304.35 | |
| M062X | 313.39 | |
| MPW1PW91 | 353.68 | |
| ωB97XD | 300.39 |
After selecting the DFT functional, the R and all designed molecules (AS1–AS7) were optimized at their ground state. Figure 3 displays the designed structures of the modeled (AS1–AS7) compounds. The molecules in planar geometry are more stable, with less steric hindrance and more charge carrier mobilities.44Figure 4 shows that all molecules have a planar shape as determined by analyzing the optimal geometries of the R and AS1–AS7.
Figure 3.

Designed structures of all modeled molecules (AS1–AS7) and R.
Figure 4.

Optimized geometries of R along with the AS1–AS7 molecules.
The dihedral angles of all developed molecules and the R were estimated on both sides of the molecules with the help of these optimized structures to investigate the effect of the bridge (furan) and acceptor units, as illustrated in Table 2. As a result, the dihedral angles (Φ1 and Φ2) of all developed (AS1–AS7) molecules show deviation in the range of 35.48–35.84° (Φ1) and −36.06 to −37.11° (Φ2) than the dihedral angles of the reference molecule, which has Φ1 = 35.63° and Φ2 = −35.95° value. These Φ1 and Φ2 values of the proposed structures (AS1–AS7) indicate that end-capped modification of the donor group with bridge and acceptor moieties has enhanced conjugation, structural planarity, and optical and electronic properties. Additionally, it substantially contributes to determining charge transport capabilities and the reorganizational energies of materials.
Table 2. Dihedral Angle (Φ1 and Φ2) for R and AS1–AS7.
| compounds | dihedral angle Φ1 | dihedral angle Φ2 |
|---|---|---|
| R | 35.63 | –35.95 |
| AS1 | 35.84 | –36.45 |
| AS2 | 35.80 | –36.65 |
| AS3 | 35.76 | –36.53 |
| AS4 | 35.48 | –36.67 |
| AS5 | 35.76 | –36.06 |
| AS6 | 35.51 | –37.11 |
| AS7 | 35.74 | –36.56 |
3.1. Frontier Molecular Orbitals
The R molecule and molecular design structures (AS1–AS7) were optimized using the B3LYP/6-31G (d,p) DFT functional. The HOMO and LUMO patterns are used to calculate electrical and optical characteristics.45–48 For example, in a photovoltaic device, “the location and energy of both bands”49–52 determine the device’s efficiency. Therefore, the LUMO and HOMO positions are critical in enhancing the device’s efficacy. Figure 5 displays the FMOs distribution pattern computed at 6-31G (d,p)/B3LYP and their corresponding bar chart, along with the band gap in Figure 6.
Figure 5.
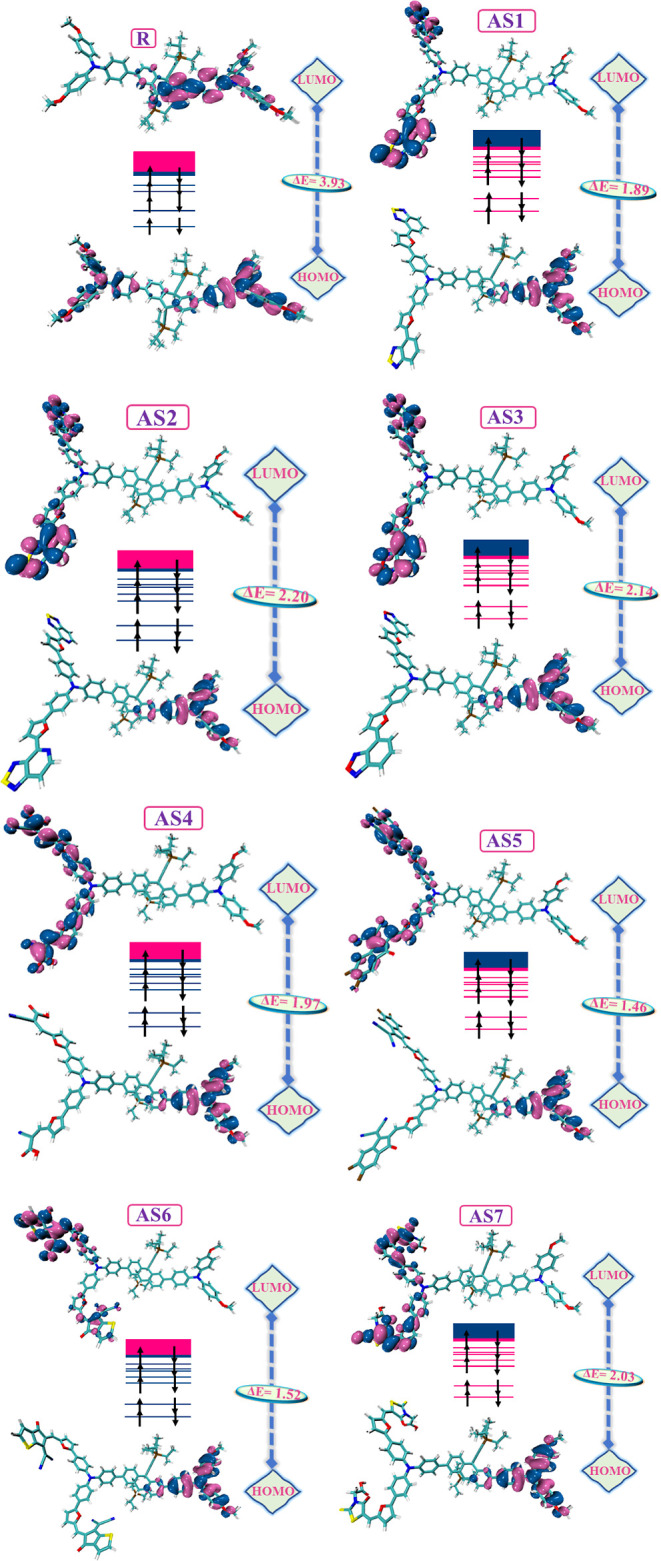
FMO distribution patterns of R and AS1–AS7.
Figure 6.
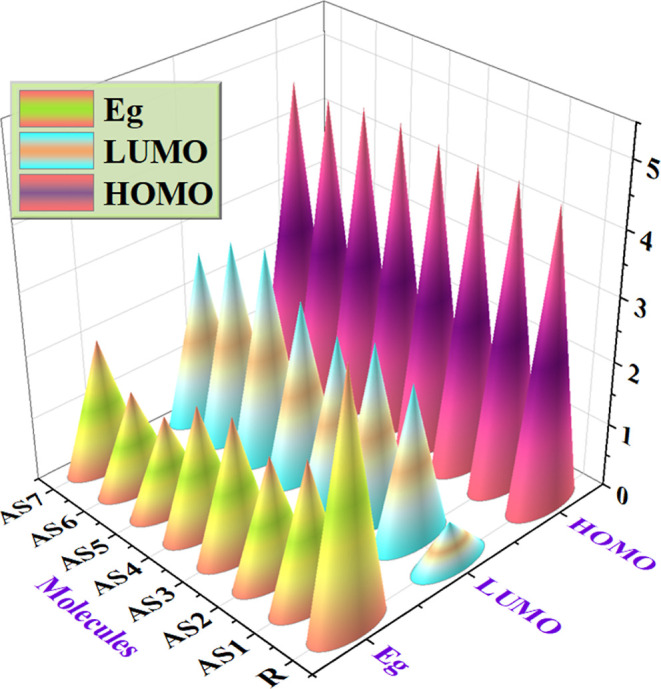
3D plot of EHOMO, ELUMO, and band gap of R along with AS1–AS7 molecules.
The valence band FMOs52–55 are also known as HOMOs, whereas the conduction band FMOs are called LUMOs. The band gap is the difference in the energy gaps between these two orbitals. The photovoltaic efficiency of OSCs heavily depends on the band gap of the orbitals. The band gap represents the separation of these two orbitals and is also called the HOMO–LUMO energy gap. There is a one-to-one relationship between charge transfer and the band gap. The energy gap determines the charge transfer efficiency; a high gap means a weaker transfer. By enabling a rapid transfer of newly produced charge carriers to the electrodes with lower recombination losses, the increased carrier mobility enhances efficiency. As a result, OSCs with a smaller energy gap perform better in photovoltaic systems, while OSCs with a wider gap perform poorly. Due to the inverse relationship between the band gap and the photovoltaic performance of OSCs, molecules with smaller energy gaps have greater PCE, and vice versa.56,57 Therefore, the small energy gap and strong charge-transferring capacity are responsible for attaining an excellent PCE and photophysical capacity. The conductivity of the standard molecule R and the molecules with specific modifications (AS1–AS7) may be calculated from this approach. Table 3 lists the HOMO and LUMO energies and the band gap that has been computed. The reference molecule has a −4.58 eV value of HOMO, a −0.64 eV value for LUMO orbitals, and a band gap value of 3.93 eV. The newly designed (AS1–AS7) molecules had the HOMO values of −4.65, −4.63, −4.68, −4.74, −4.74, −4.63, and −4.68 eV. Energy gaps of 2.20, 1.89, 2.14, 1.97, 1.46, 1.52, and 2.03 eV correspond to the LUMO values of −2.45, −2.75, and −2.54, respectively, −2.77, −3.28, −3.11, and −2.65 eV for (AS1–AS7). Based on the data presented above, it is observed that the band gap of all the newly created molecules (AS1–AS7) is less than that of R. Possibly due to (Z)-2-(2-ethylidene-5,6-difluoro-3-oxo-2,3-dihydro-1H-in den-1-ethylidene)malononitrile, the AS5 molecule has a narrower energy gap of 1.46 eV than other designed molecules. After comparing the developed molecules to the R (reference molecule), it becomes clear that AS5 has superior photovoltaic performance. AS6 has a second narrow band gap, possibly attributable to the (Z)-2-(5-ethylidene-6-oxo-5,6-dihydro-4H-cyclopenta[b]thiophen-4-ylidene)malononitrile designed group. The energy band gap is in increasing order as follows: AS5 < AS6 < AS2 < AS4 < AS7 < AS3 < AS1 < R. Based on the evidence presented above, it seems that the conduction bands of these newly created molecules are narrower than R, making them exciting aspirants for future use in SCs.
Table 3. Computed Values of HOMO–LUMO Energies and Energy Gap of the AS1–AS7 and the R.
| compounds | EHOMO (eV) | ELUMO (eV) | Eg (eV) |
|---|---|---|---|
| R | –4.58 | –0.64 | 3.93 |
| AS1 | –4.65 | –2.45 | 2.20 |
| AS2 | –4.63 | –2.75 | 1.89 |
| AS3 | –4.68 | –2.54 | 2.14 |
| AS4 | –4.74 | –2.77 | 1.97 |
| AS5 | –4.74 | –3.28 | 1.46 |
| AS6 | –4.63 | –3.11 | 1.52 |
| AS7 | –4.68 | –2.65 | 2.03 |
FMOs are further assisted through the DOS graphs.58 Diagrams showing the DOS provide insight into how the LUMO–HOMO populations are allocated throughout a molecule. B3LYP/6-31G(d,p) functional of DFT is operated for the DOS estimation. In the DOS graph, the fragment of studied molecules (AS1–AS7), such as core, donors, bridge, and acceptors, is characterized by purple, orange, pink, and green peaks, respectively, whereas the fragment of the R has only purple and orange lines that indicate the core and donors. These lines specify the extent of the contribution of different components in improving FMOs. A gray line illustrates the whole DOS. The density of the HOMO is positioned on the donor component, and the minor HOMO density is on the anthracene core, whereas the population of the LUMO is distributed chiefly on the anthracene core and donor part in the reference molecule, as shown in Figure 7. HOMO and LUMO density distribution patterns change because of the addition of acceptor units to the proposed (AS1–AS7) molecules. The HOMO density is mainly found on the donor component, with just a small amount on the anthracene core, and the LUMO population is primarily situated on the acceptor units and furan bridge that connect with the donor atoms. In all of the developed molecules, the acceptor units and bridge atoms carry a significant share of the LUMO density, whereas the donor atoms have a much smaller percentage. DOS spectra provide a complete overview of the energy levels. A high DOS value indicates a greater concentration of electronic states at the energy level. This indicates that the electrons at this energy level can access various possible energy states. All of the compounds in our investigation had their Fermi levels determined by summing the energies of their FMOs (Figure 7). Fermi levels are used to determine whether an electron is available in the conduction or valence band. When the Fermi energy is close to the conduction band, the charge may flow more efficiently because electrons can hop from the valence band to the conducting band. The AS5 molecule is more stable, and the lower valence band (i.e., HOMO) may account for its lower Fermi level, showing that it has improved photovoltaic performance as a donor molecule among optoelectronic devices.59–61 According to the above discussion, it is concluded that when the acceptor unit is attached to the reference molecule, it significantly increases the electron-donating potential of donor units toward the acceptor part in all newly proposed compounds (AS1–AS7), making them excellent candidates for highly efficient SC devices.
Figure 7.

DOS spectra and computed Fermi levels of R and designed molecules (AS1–AS7).
3.2. Optical Properties
UV–vis analysis at the B3LYP/6-31G (d,p) DFT level in the organic solvent tetrahydrofuran and the gaseous phase was utilized to estimate the optical and photophysical characteristics of the R molecule and the newly modeled (AS1–AS7) compounds. Tables 4 and 5 provide the solvent and gaseous phase data for the oscillator frequency (fos), the maximum absorption wavelength (λmax), the assignments that describe the type of transitions in proposed (AS1–AS7) molecules, R (reference molecule), and the transition energy (Ex).
Table 4. Actual and Theoretically Computed λmax, Excitation Energy, Oscillating Frequency, and Major Molecular Transitions for R and All Studied Molecules (AS1–AS7) in Tetrahydrofuran Solvent.
| molecules | DFT calculated λmax (nm) | reported λmax (nm) | Ex (eV) | fos | major MO assignment |
|---|---|---|---|---|---|
| R | 367.38 | 380.00 | 3.37 | 1.41 | HOMO → LUMO (62%) |
| AS1 | 407.13 | 3.05 | 1.06 | HOMO → LUMO (96%) | |
| AS2 | 679.29 | 1.83 | 0.76 | HOMO → LUMO (93%) | |
| AS3 | 386.12 | 3.21 | 0.81 | HOMO → LUMO (95%) | |
| AS4 | 400.35 | 3.10 | 1.09 | HOMO → LUMO (96%) | |
| AS5 | 495.94 | 2.50 | 1.07 | HOMO → LUMO (88%) | |
| AS6 | 549.06 | 2.26 | 0.53 | HOMO → LUMO (96%) | |
| AS7 | 626.72 | 1.98 | 0.91 | HOMO → LUMO (99%) |
Table 5. Actual and Theoretically Computed λmax, Excitation Energy, Oscillating Frequency, and Major Molecular Transitions for R and All Studied Molecules (AS1–AS7) in the Gaseous Phase.
| molecules | DFT calculated λmax (nm) | reported λmax (nm) | Ex (eV) | fos | major MO assignment |
|---|---|---|---|---|---|
| R | 356.88 | 380.00 | 3.47 | 1.40 | HOMO → LUMO (65%) |
| AS1 | 399.67 | 3.10 | 0.91 | HOMO → LUMO (96%) | |
| AS2 | 661.64 | 1.87 | 0.68 | HOMO → LUMO (99%) | |
| AS3 | 377.34 | 3.29 | 0.73 | HOMO → LUMO (86%) | |
| AS4 | 386.73 | 3.21 | 1.10 | HOMO → LUMO (86%) | |
| AS5 | 467.86 | 2.65 | 1.08 | HOMO → LUMO (80%) | |
| AS6 | 527.97 | 2.35 | 0.44 | HOMO → LUMO (94%) | |
| AS7 | 589.56 | 2.10 | 0.80 | HOMO → LUMO (99%) |
Absorption maxima of the reference molecule were measured experimentally to be 380 nm,25 but DFT calculations show that they are 367.38 and 356.88 nm in the tetrahydrofuran and the gaseous phases, respectively. Typically, increasing the PCE of a SC is accomplished by moving the absorption spectrum toward the red end of the visible-light spectrum. The absorption spectrum of AS1–AS7 compounds is red-shifted because of the substitution of a side-chain donor unit (bis(4-methoxyphenyl)aniline) by a variety of end-capped acceptor units in the R atom (reference molecule), as presented in Figure 8. This investigation found that the maximum absorption (λmax) of all seven studied molecules (AS1–AS7) was greater than that of the standard reference (R). AS2 has the highest absorption value, determined to be 661.64 nm in gas and 679.29 nm in tetrahydrofuran among all designed molecules due to the end-cap ([1,2,5]thiadiazolo[3,4-c]pyridine) unit, which causes the spectrum to be red-shifted and enhances the PCE of SCs. Maximum absorption wavelengths of designed molecules are 407.13 (AS1), 679.29 (AS2), 386.12 (AS3), 400.35 (AS4), 495.94 (AS5), 549.06 (AS6), and 626.72 nm (AS7) in a tetrahydrofuran solvent. On the other hand, the λmax of designed molecules in the gas phase are 399.67 nm (AS1), 661.64 nm (AS2), 377.34 nm (AS3), 386.73 nm (AS4), 467.86 nm (AS5), 527.97 nm (AS6), and 589.56 nm (AS7). These absorption λmax values illustrate the effectiveness of acceptor units for AS1–AS7 modeled molecules. The decreasing order of λmax values of all compounds is noted as AS2 > AS7 > AS6 > AS5 > AS1 > AS4 > AS3 > R in the tetrahydrofuran solvent and the gas phase.
Figure 8.

UV–vis plot of R and all proposed molecules (AS1–AS7) in the tetrahydrofuran solvent and gas phase.
3.3. Open-Circuit Voltage
OSC performance may also be assessed by determining the open-circuit voltage of the device. Voc is a critical measure for evaluating the performance of PV systems. The voltage or current magnitude allows solar devices to reach the zero-current state.62 The effectiveness of OSCs is greatly improved by increasing their fill factor (FF), which rises when the Voc rises. Since Voc is a positive factor in the recombination capability of photovoltaic systems, it influences both the photogenerated current and the saturation voltage. Many factors, such as light intensity, type of material, the heat of the SC-based device, and the available energy, all influence the Voc.54,63 The Voc is proportional to the LUMO–HOMO energy gap of the acceptor and donor molecules. For Voc calculations, PC61BM was selected for effective electron injection from donor molecules to the acceptor (PC61BM). Research has shown that electron injection efficiency from the HOMO orbital of the donor molecule to the LUMO of the acceptor is maximized when the ELL values are between 0.2 and 1 eV.64 All of the proposed molecules had ELL in the highly competitive range of 0.42–0.95 eV except for a bit of divergence in the energies of AS1, AS3, and AS7, and their values are 1.25, 1.16, and 1.05 eV. The minute deviation of these values from the standard value suggests further investigation of the donor–acceptor-based interface. The interfacial charge transfer energy (ECT) was determined to advance the research and ensure the effectiveness of the PC61BM (acceptor)-based interface. The values of ECT and ELL are tabulated in Table 6. The ECT values are found in the range of 0.93–1.04 eV, which is lower than the energy gap of the designed molecules. This result suggests that this interface is more effective than the proposed molecules’ internal charge transfer capacities (AS1–AS7).
Table 6. Calculated Values of ELL, ECT, and Energy Loss Incurred during Charge Creation and Charge Recombination for R, along with AS1–AS7.
| molecules | ELL (eV) | ECT (eV) | energy loss incurred during charge generation | energy loss incurred during charge recombination |
|---|---|---|---|---|
| R | 3.06 | 0.88 | 3.06 | 0.30 |
| AS1 | 1.25 | 0.95 | 1.25 | 0.30 |
| AS2 | 0.95 | 0.93 | 0.95 | 0.30 |
| AS3 | 1.16 | 0.98 | 1.16 | 0.30 |
| AS4 | 0.93 | 1.04 | 0.93 | 0.30 |
| AS5 | 0.42 | 1.04 | 0.42 | 0.30 |
| AS6 | 0.59 | 0.93 | 0.59 | 0.30 |
| AS7 | 1.05 | 0.98 | 1.05 | 0.30 |
In the next step, we estimate the Voc of all molecules (R along with AS1–AS7) using the equation Scharber and co-workers developed.65
| 3 |
The Voc is approximated by subtracting the donor’s EHOMO from the acceptor’s ELUMO. In addition, Voc is linked to this HOMO–LUMO energy gap. The Voc for the investigated (AS1–AS7) molecules and the R was thus theoretically calculated at B3LYP/6-31G (d,p). The primary focus of Voc is to synchronize the HOMO of the donor molecules (AS1–AS7) with the LUMO of the acceptor polymer material PC61BM. It is found that the LUMO and HOMO of PC61BM are situated at 3.7 and 6.1 eV. Figure 9 summarizes the relationship between the HOMOdonor-LUMOPC61BM energy gaps and the Voc values derived from eq 3. R and AS1–AS7 have respective Voc values of 1.18, 1.25, 1.23, 1.28, 1.34, 1.34, 1.23, and 1.28 V. The seven proposed compounds (AS1–AS7) have Voc values higher than those of the reference molecule. Among all developed molecules, AS4 and AS5 represent the highest Voc values. Voc values for both designed molecules and the reference increase as follows: R < AS2 = AS6 < AS1< AS3 = AS7 < AS4 = AS5. The LUMO and HOMO levels of the acceptor (PC61BM) and donor molecules are often used to compute the Voc. The photophysical and optoelectronic characteristics of OSCs are enhanced since the HOMO value of all molecules (AS1–AS7) is increased, leading to a more excellent Voc. The higher HOMO of the proposed molecules (AS1–AS7) is responsible for enhancing electron mobility from the HOMO orbital of donors (AS1–AS7) to the LUMO orbital of acceptor PC61BM, which in turn improves the optoelectronic capabilities of OSCs.
Figure 9.

Voc of the R and all modeled (AS1–AS7) molecules concerning the PC61BM acceptor polymer.
3.4. Transition Density Matrix
The TDM evaluation is often required to estimate the extent of transitions and their modes in the molecules under study. The TDM illustrations were computed at the B3LYP/6-31G (d,p) level of theory. The TDM representations have refined the estimated localization of electrons, holes, and electron–hole interactions inside the molecules. In addition, they reveal the existence of multiple charge transfers between excited states.66,67 The studied molecules (AS1–AS7) were categorized into donor and core groups to assess TDM characterizations more accurately.46
Moreover, the hydrogen impact was disregarded in these studies since hydrogen does not take part in carrier transitions. The type of transition in each molecule (designed AS1–AS7 and reference molecules) is shown in the TDM diagrams. Figure 10 shows the TDM outcomes for all examined compounds (R and AS1–AS7). It was determined through these analyses that the electrical charge mobility was distributed evenly among the core and end-capped groups. The donor units of the reference R molecule contain most of the charge coherence and a small amount of charge on the core. A large electronic charge is available on the core, while a minor amount is available on the donor units in the proposed AS1 molecule. The remaining molecules, AS2–AS7 specifically engineered, showed the constant movement of electronic charge density from the donor units to the core. The behavior of electron coherence in the R and proposed (AS1–AS7) molecules indicates that charge coherence successfully shifts from donor to core unit, then diagonally toward the bridge, acting as a facilitator for allocating electrons without tricking them, and that electron density has finally arrived at the acceptor part. This suggests that while all designed compounds may have lower electron coupling than the reference molecule, their exciton dissociation in the excited state may be more significant and simpler. Compared to R, we observed that all of our created molecules exhibited successful spectrum transitions, revealing their hidden potential as a photoactive material for creating efficient devices in the future.
Figure 10.

TDM outcomes for R and all proposed molecules (AS1–AS7).
3.5. Binding Energy
Predicting the optoelectronic properties of materials using the binding energy (Eb) estimate is an exciting new direction. The Eb affects the SC efficacy because it affects the dissociation potential. The Eb is proportional to the inverse of the charge mobility. Therefore, higher binding energy molecules tend to have smaller current intensities and charge movements. The Coulombic force between electrons and holes in a compound was analyzed using this method. Exciton breakdown in an excited state is proportional to the Eb, which, in turn, depends inversely on the strength of the interaction between holes and electrons. Therefore, an excited electron and hole in a photoactive film of a SC may easily dissociate from a molecule with a small Eb. Hence, in the photoactive film of an OSC, a molecule with a small Eb will demonstrate an excellent PCE. Equation 4 was used to compute the Eb of the R and the developed compounds (AS1–AS7)68,69
| 4 |
The energy difference between the HOMO and LUMO levels and the energy of the first excited singlet state (Eopt) are used to derive Eb. Table 7 shows that the Eb value for R is 0.46 eV, whereas the binding energy for AS1–AS7 ranges from 0.16 eV (AS1), 0.13 eV (AS2, AS3, AS4, AS6, and AS7), and 0.12 eV (AS5). Figure 11 shows that the Eb values of the developed (AS1–AS7) materials were lower than those of the R (reference molecule) because of the electron-accepting nature of the terminal units. From the smaller Eb values of the designed (AS1–AS7) molecules, it is estimated that they can dispose of a more significant charge concentration than R (reference molecule). The smallest Eb is observed for AS5, which may have been caused by the presence of a side-chain acceptor unit ((Z)-2-(2-ethylidene-5,6-difluoro-3-oxo-2,3-dihydro-1H-in den-1-ethylidene)malononitrile). The observed pattern suggests that developed molecules (AS1–AS7), especially AS5, would be superior for developing OSCs.
Table 7. Calculated Values of Eg, Eopt, and Eb of R and AS1–AS7.
| compounds | Eg | Eopt (eV) | Eb (eV) |
|---|---|---|---|
| R | 3.93 | 3.47 | 0.46 |
| AS1 | 2.20 | 2.04 | 0.16 |
| AS2 | 1.89 | 1.76 | 0.13 |
| AS3 | 2.14 | 2.01 | 0.13 |
| S4 | 1.97 | 1.85 | 0.13 |
| AS5 | 1.46 | 1.34 | 0.12 |
| AS6 | 1.52 | 1.40 | 0.13 |
| AS7 | 2.03 | 1.90 | 0.13 |
Figure 11.
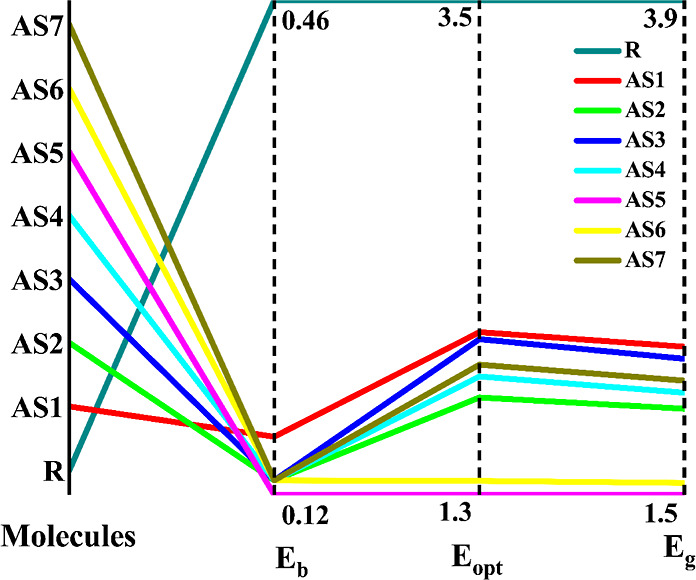
Comparison between Eopt, band gap (Eg), and binding energy (Eb) of R along with AS1–AS7.
3.6. Molecular Electrostatic Potential
The MEP investigation also plays a significant role in elucidating the existence of dynamic charging sites within the molecules and the varying charge separations and positions within the molecules. Overall, the molecule has several charge sites or combinations of charging sites, and each of these sites is represented by its own set of data describing the molecule’s unique features. Molecular efficiency is also profoundly affected by the components.70 MEP plots of developing (AS1–AS7) and R molecules were computed using the B3LYP/6-31G (d,p) level of theory. Figure 12 shows the results of the calculations of the MEP plots. Individual properties of the molecule may be seen in the MEP plots when the color patterns and their combinations are broken down and analyzed. For instance, a neutral part of the molecule is shown in light green, whereas blue indicates an electropositive region.
Figure 12.

MEP surfaces of modeled (AS1–AS7) molecules and R calculated at B3LYP/6-31G(d,p).
On the other hand, a negative area inside the molecule is highlighted by the red color. Our developed molecules (AS1–AS7) have followed the same color distribution patterns as the R molecule. The presence of many charging sites also discloses the distinct properties of that molecule, which might be used to estimate its potential usefulness in SC devices. As a result, these MEP plots may help reveal the full potential of the materials. Furthermore, there is a uniform allocation of charges over the MEP surfaces of designed molecules, making them far more relevant to the R. So, it is possible to increase the PCE and consistency of OSCs by using practical molecular side-chain enhancements to construct better and more proficient molecules for future use in OSC applications. Our MEP studies also revealed that owing to their comparable charge separation patterns, our end-capped engineered molecules might be an active and efficient alternative to R.
3.7. Excitation Energy
Excitation energy is a significant additional component that is crucial for improving the optoelectronic characteristics of OSCs. The excitation energy of a SC is another indicator of its effectiveness.47,62 The excitation energy level is typically low when a high PCE is obtained. Additionally, considerable charge transfer and outstanding optical properties are possible at low excitation energies and oscillator strengths. Figure 13 displays a comparison between the oscillating strength and excitation energy of all the proposed (AS1–AS7) molecules (AS1–AS7) and the R. All the studied (AS1–AS7) molecules have smaller Ex and fos in comparison to the R because of side-chain acceptor units. The excitation energy of the R is 3.37 eV and for all proposed molecules are 3.05 eV (AS1), 1.83 eV (AS2), 3.21 eV (AS3), 3.10 eV (AS4), 2.50 eV (AS5), 2.26 eV (AS6), and 1.98 eV (AS7). Excitation energies for all proposed molecules decrease from highest to lowest as follows: R > AS3 > AS4 > AS1 > AS5 > AS6 > AS7 > AS2. The smallest Ex and the most apparent red shift in the UV–vis spectrum of the AS2 molecule are observed as the number of electrons decreases in the excited state. These molecules had the largest density allocation from the HOMO to the LUMO (99%), as shown by their related molecular assignments in the solvent, which was calculated for all the AS1–AS7 compounds. Other newly developed compounds have also demonstrated lower Ex values and more excellent absorption, promising their proper use in developing effective SC devices.
Figure 13.
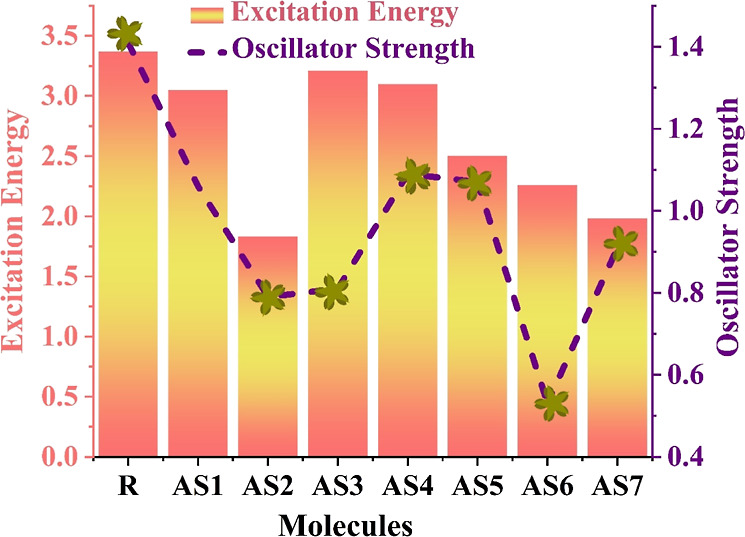
Comparison between the excitation energy and oscillating strength of the developed (AS1–AS7) molecules and the R.
3.8. Light Harvesting Energy
The quantity of incoming light that is absorbed in a given wavelength is measured using a metric known as light-harvesting energy, which is also one of the crucial variables that are inversely related to the incident photon to the current efficiency of the photosensitizing molecule. High LHE values help to improve the short-circuit current of the devices and, thus, enhance the efficiency of the devices.71 Any optoelectronic material can generate charge carriers during the light-harvesting process.65,72 It is also calculated using eq 5 and is believed to be directly related to the Jsc values of materials created following the fabrication of devices73
| 5 |
The efficiency of charge collection is represented by ηcollect, whereas that of electron injection is represented by φinj. The LHE phenomenon was investigated using eq 6,48,73 and the corresponding values are illustrated in Table 8
| 6 |
Table 8. LHE with Corresponding Oscillator Frequency (fos) of R and All Investigated Molecules (AS1–AS7) in Both the Tetrahydrofuran and Gas Phase.
| molecules | solvent |
gas
phase |
||
|---|---|---|---|---|
| fos | LHE | fos | LHE | |
| R | 1.41 | 0.9611 | 1.40 | 0.9602 |
| AS1 | 1.06 | 0.9129 | 0.91 | 0.8770 |
| AS2 | 0.79 | 0.8378 | 0.68 | 0.7911 |
| AS3 | 0.81 | 0.8451 | 0.73 | 0.8138 |
| AS4 | 1.09 | 0.9187 | 1.10 | 0.9206 |
| AS5 | 1.07 | 0.9149 | 1.08 | 0.9168 |
| AS6 | 0.53 | 0.7049 | 0.44 | 0.6369 |
| AS7 | 0.91 | 0.8770 | 0.80 | 0.8415 |
Newly created molecules (AS1–AS7) and values for R that correlate to the oscillator strength are shown in Figure 14. The importance of fos in the gas phase and tetrahydrofuran are used to estimate the LHE. Although the LHE values of AS1, AS4, and AS5 were more significant compared to the other created molecules, those of AS2, AS3, AS6, and AS7 were lower, indicating that these molecules could be able to produce even more Jsc in SC devices. Furthermore, the created molecule AS4 has the highest LHE (91.87%), which suggests outstanding Jsc performance. It shows our efficient design strategy for photovoltaic OSC devices.
Figure 14.
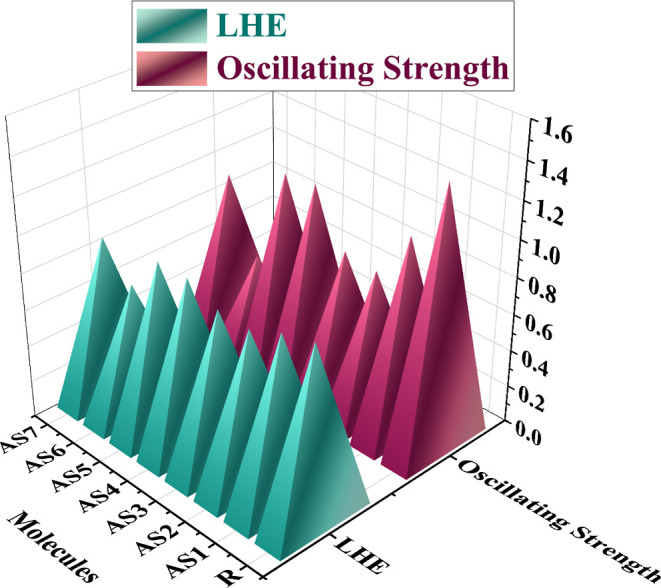
3D plot of oscillating strength and LHE of the R and all investigated (AS1–AS7) molecules in tetrahydrofuran solvent.
3.9. Heat Map
The overlap between electrons and holes in the developed molecules (AS1–AS7) has also been theoretically calculated and the results of this analysis are expressed in Figure 15. The parameters demonstrate that the narrow energy gap and strong mobility of hole–electrons in the (AS1–AS7) molecules under study have resulted in high-intensity overlap for all seven proposed compounds. In addition, AS5 has a high-intensity overlap among all proposed molecules due to a narrow band gap and high hole/electron mobility. These findings provide convincing evidence that the molecules under study are outstanding candidates for the photoactive film of OSCs, as they exhibit effective overlapping and charge coherence.
Figure 15.

Hole–electron overlap surfaces of R along with AS1–AS7.
3.10. Fill Factor
One of the most significant metrics for evaluating SC performance is the FF. The FF is a measure of a SC’s efficiency in converting incoming light into usable electricity. When the FF of a SC is high, its electrical output exceeds its theoretical maximum. This points to an internal recombination of charge carriers and decreased resistance losses inside the device. Voc, Jsc, and FF are the three major factors used to calculate the potential output of a given photovoltaic device. Voc and FF for the designed molecules (AS1–AS7) and R were also calculated. The FF for R and the proposed molecules were calculated using eq 7
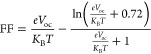 |
7 |
Thus, 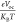 = Voc stands
for normalized Voc, with “e” for the elementary charge, which equals 1. Equation 7 is used to get the
molecular Voc, where KB is the Boltzmann constant, its value is 8.61733034 ×
10–5 eV/K, and T is the temperature
at 300 K.74,75 Compared to the reference and other developed
molecules, the AS4 and AS5 molecules had a higher value of FF (0.9066).
The high FF percentage of all proposed molecules (AS1–AS7)
compared to that of R demonstrated the efficiency of our design methodology
for creating efficient photovoltaic materials suitable for use in
SCs.
= Voc stands
for normalized Voc, with “e” for the elementary charge, which equals 1. Equation 7 is used to get the
molecular Voc, where KB is the Boltzmann constant, its value is 8.61733034 ×
10–5 eV/K, and T is the temperature
at 300 K.74,75 Compared to the reference and other developed
molecules, the AS4 and AS5 molecules had a higher value of FF (0.9066).
The high FF percentage of all proposed molecules (AS1–AS7)
compared to that of R demonstrated the efficiency of our design methodology
for creating efficient photovoltaic materials suitable for use in
SCs.
Moreover, using eq 8, we can develop a reasonable approximation of PCE for all newly developed molecules (AS1–AS7)73
| 8 |
Herein, the power from the external light source is denoted by Pinput. It is well-known that charge mobility rates, LHE, and the band gap of materials are crucial to the Jsc values. All these parameters were calculated theoretically and revealed that our newly proposed molecules (AS1–AS7) had shown better PCE values than the R molecule. Figure 16 represents the calculated PCE of all molecules (R and AS1–AS7). These materials (AS1–AS7) have comparable Voc and FF, which has inspired us to suggest them for developing high-performance SC devices. Table 9 displays the results of the calculations for Voc, FF, and PCE. Voc and FF values for all newly developed molecules were significantly higher than those for R, ensuring their suitability for future use in SC devices.
Figure 16.
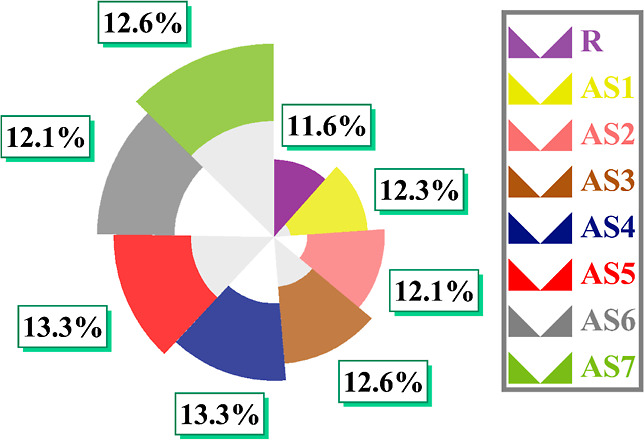
Pie graph for approximation of PCE of all molecules (R and AS1–AS7).
Table 9. Calculated Voc, % FF, and PCE of All Modeled Molecules (AS1–AS7) and R.
| compounds | Voc | % age FF | PCE |
|---|---|---|---|
| R | 1.18 | 89.68 | 23.48 |
| AS1 | 1.25 | 90.14 | 25 |
| AS2 | 1.23 | 90.01 | 24.57 |
| AS3 | 1.28 | 90.32 | 25.65 |
| AS4 | 1.34 | 90.66 | 26.96 |
| AS5 | 1.34 | 90.66 | 26.96 |
| AS6 | 1.23 | 90.01 | 24.57 |
| AS7 | 1.28 | 90.32 | 25.65 |
3.11. Reorganizational Energy
Reorganization energy is an important parameter for photovoltaic materials to get an estimation of their nature, i.e., hole transfer (λh) or electron transfer (λe). RE is a critical parameter influencing charge mobility between the donor and acceptor regions of materials. Donor and acceptor molecules, their structures, and their surroundings all have a role in determining the extent of the reorganizational energy required. Reorganizational energy helps in better charge separation and recombination.76,77 There is a reverse relation between the reorganization energy and charge transfer (the transfer of electrons or holes). Higher charge mobilities are often associated with low reorganizational energies, and vice versa.78–80 Reorganizational energy also affects the photovoltaic performance of the devices. The lower values of reorganizational energy result in better photovoltaic efficiencies.81–83 For charged particles such as electrons and holes, the energy required to rearrange their configuration equals the inverse of their mobility. Internal (λint) and external (λext) reorganization energies are the two most common forms of RE. According to Marcus’s theory,84 the former is a qualitative indication of the charge transfer speed (the more effective the λ, the lower the charge transfer rate). It is formed by geometrical changes when an electron is withdrawn or supplied to a molecule. The latter is associated with polarization-induced changes in the ambient medium, which is discarded because λext is difficult to calculate correctly. The outer region of the polarized force field is calculated to be substantially weaker than the inner region.85 The reorganization energy was computed using DFT at B3LYP/6-31G (d,p).
Table 10 shows that the R and proposed molecule’s electron RE (λe) values range from 0.0024 to 0.0078 eV. All newly developed SCs have low electron reorganization energy because of the efficient side-chain acceptor moieties, demonstrating the superior electron mobility in these materials (AS1–AS7). Among all developed molecules, AS6 has the most extensive electron transfer capability from the donor component to the acceptor part because it has the lowest value of λe. While the hole RE (λh) values range from 0.0016 to 0.0024 eV and are also listed in Table 10. AS1, AS2, and AS3 molecules have the same λh value (0.0016). It means that their capacity to carry holes is roughly the same. The value of λh suggests that the hole mobility rate is higher than the electron mobility rate for all designed compounds. According to the reorganizational energy analysis results, our developed (AS1–AS7) compounds are promising aspirants for highly efficient OSCs.
Table 10. Computed Reorganizational, T(hole), and T(electron) Energies of All Proposed (AS1–AS7) Molecules and R.
| molecules | λe | λh | T(hole) | T(electron) |
|---|---|---|---|---|
| R | 0.0078 | 0.0023 | 0.01 | 0.03 |
| AS1 | 0.0025 | 0.0016 | 0.07 | 0.02 |
| AS2 | 0.0026 | 0.0016 | 0.12 | 0.02 |
| AS3 | 0.0028 | 0.0016 | 0.13 | 0.02 |
| AS4 | 0.0042 | 0.0023 | 0.32 | 0.07 |
| AS5 | 0.0025 | 0.0024 | 0.35 | 0.05 |
| AS6 | 0.0024 | 0.0022 | 0.28 | 0.04 |
| AS7 | 0.0031 | 0.0019 | 0.18 | 0.05 |
Similarly, T(hole) and T(electron) calculations of R and all proposed (AS1–AS7) molecules were performed and are also shown in Table 10. The T(hole) values of the created molecules are higher than those of the R. In contrast, T(electron) values are comparable to the R, indicating that side-chain modification of molecules with electron-withdrawing groups enhances the photovoltaic characteristics of molecules for SC application. The decreasing order of T(hole) values of all (AS1–AS7) molecules is observed as AS5 > AS4> AS6> AS7 > AS3 > AS2 > AS1 > R and T(electron) values are as follows: AS4 > AS5 = AS7> AS6> R > AS1 = AS2 = AS3. The detailed comparison between T(hole) and T(electron) energies and between λe and λh energies is shown in Figure 17.
Figure 17.

Line graph of T(hole) and T(electron) and 3D plot of reorganizational (λe and λh) energies for modeled molecules (AS1–AS7) and the R.
3.12. Charge Transfer Analysis
CT analysis is conducted to determine whether the developed compounds might be helpful as SC materials. The real incentive behind this calculation is comprehending the donating properties of our recently developed (AS1–AS7) molecules in their acceptor (PC61BM) presence. These donor and acceptor materials have been tuned individually and together using B3LYP/6-31G (d,p). To comprehend the CT behavior of the complex, we chose one of the highest-performing modeled (AS5) molecules to produce the complex (AS5:PC61BM). The AS5 molecule was selected because it has appropriate reorganizational (λe and λh) energy, has reduced Eb, Ex, and Eg, and a high value for FF and Voc, and exhibits red-shifting absorption. Acceptor PC61BM in the AS5:PC61BM complex system has been optimized at B3LYP/6-31G (d,p) to a parallel arrangement with donor AS5 and their optimized structure is shown in Figure 18.
Figure 18.

Optimized complex of AS5/PC61BM at B3LYP/6-31G (d,p); FMO distribution patterns of the HOMO on AS5 (donor) and the LUMO on PC61BM (acceptor).
In addition, we calculated the FMOs of AS5 (donor) and PC61BM (acceptor) and the HOMO and LUMO dispersion patterns of AS5: PC61BM are also shown in Figure 18. Donor HOMO and acceptor LUMO have a significant impact on this CT. FMO diagrams showed that the population of the HOMO charge is concentrated on the donor part of the AS5 molecule, whereas the LUMO charge is uniformly distributed across the acceptor material (PC61BM). From the HOMO–LUMO distribution patterns, it has been seen that the bulk of the charge exchange occurred at the donor–acceptor contact (AS5:PC61BM). Table 11 displays the PC61BM (acceptor) and AS5 (donor) orbitals engaged in charge transfer.
Table 11. Calculated Values of NBO Charge Transfer from the Donor AS5 Molecule to the Acceptor PC61BM Polymer and Their Related Stabilization Energies.
| donor NBO (AS5) | acceptor NBO (PC61BM) | E2 kcal/mol |
|---|---|---|
| π C1–C6 | π* C247–C258 | 0.11 |
| π C25–C26 | σ* C286–H299 | 0.81 |
| σ C44–H48 | σ* O218–C289 | 0.05 |
| σ C70–H159 | π* C271–C273 | 0.08 |
| π C187–C188 | σ* C290–H302 | 0.21 |
| σ C188–H191 | σ* O217–C290 | 0.06 |
| σ C188–H191 | π* O218–C289 | 0.08 |
| σ C188–H191 | σ* C290–H302 | 0.11 |
The donor NBO (bonded orbitals) is responsible for signaling charge density, while the acceptor NBO (antibonding orbitals) accepts it. The E2 kcal/mol represents the energy necessary for this charge transfer process at the donor (AS5): acceptor (PC61BM) contact. The π bond (C25–C26) of AS5 (donor) provides electron density to the σ* bond (C286–H299) of PC61BM acceptor through a 0.81 kcal/mol energy transition, allowing for efficient charge transfer between the complex AS5:PC61BM. The 0.21 kcal/mol change in energy is caused by the transfer of electron density from the π bond (C187–C188) of the AS5 donor to σ* (C290–H302) of the PC61BM acceptor.
Moreover, all of the carbon and hydrogen atoms of the AS5 donor molecule are significantly closer to the carbon and hydrogen of the PC61BM acceptor molecule, permitting an efficient interfacial charge transfer process. The CT analysis demonstrates that the charge was successfully transferred from the donor part of the molecule to the end-capped acceptors, resulting in high-performance SCs. Moreover, the energy lost during charge creation (Eg – ECT) and charge recombination (ECT – eVoc) was evaluated to understand charge transport properties clearly. The energy lost in producing a charge is distinct from the energy lost during the recombination of charge, which is almost identical at 0.3 eV for all the developed molecules. Among all created molecules, the AS5/PC61BM interface has shown the minimum power loss of 0.42 eV during charge conduction. All designed donor–acceptor interfaces are highly efficient, but the AS5/PC61BM interface is promising for making highly efficient SCs.86
4. Conclusions
In summary, seven unique highly conjugated A–B–D–C–D molecules (AS1–AS7) containing an anthracene core were developed by end-capped modification on one side, and their behavior in charge transfer, optical, electronic, and photovoltaic properties, and structural activity were studied using modern quantum chemistry calculations. The UV–vis plots revealed that all the developed compounds have higher absorption maxima values in the 377.34–661.64 nm range in the gaseous phase and 386.12–679.27 nm in the tetrahydrofuran than the R molecule that has 356 nm in the gas phase and 367 nm in the tetrahydrofuran. The AS2 molecule had the highest maximum absorption value of 679.29 nm in tetrahydrofuran and 661.64 nm in the gaseous phase, showing that it has exhibited outstanding photophysical properties. All the proposed molecules (AS1–AS7) have shown narrow HOMO–LUMO band gap (Eg) values (1.46–2.20 eV) in comparison to the R (3.93 eV), demonstrating the extensive conjugation that is suggestive of the outstanding electron-accepting capacity of side-chain acceptor units. The Voc values of developed molecules (AS1–AS7) were calculated to be 1.23–1.34 V using the polymer PC61BM (acceptor), proving that newly proposed molecules have a large Voc and are effective optoelectronic candidates for SC applications. It was demonstrated that AS6 has the highest electron transport capability among all designed molecules from the electron-donating part to the acceptor unit because it has the smallest value of λe. In contrast, the values of λh indicate that the hole density transfer speed is higher than the electron transfer speed for all of the designed compounds. Moreover, the excitation and binding energy of all proposed (AS1–AS7) molecules are lower than the R, causing enlarged exciton dissociation in the excited state. AS1–AS7 has remarkable optical and electronic characteristics compared to the R due to end-capped acceptor units having a more robust electron-withdrawing capacity. Hence, our results suggest that the proposed molecules, notably AS5, can be employed as hole and electron transporters in OSCs with exceptional photophysical properties and can be used in future efficient SC devices.
Acknowledgments
The authors extend their appreciation to the Ministry of Education in KSA for funding this research work through the Project number KKU-IFP2-DB-7.
Data Availability Statement
Data will be made available upon reasonable request to the corresponding author.
Author Contributions
# A.S. and M.A. contributed equally to the first authorship. A.S. performed the research work and wrote the initial draft. M.A., R.H., and Z.I. analyzed the research data, provided supervision, used software, and edited the manuscript. U.F. helped in generating and compiling the dataset.
The authors declare no competing financial interest.
References
- Stranks S. D.; Eperon G. E.; Grancini G.; Menelaou C.; Alcocer M. J. P.; Leijtens T.; Herz L. M.; Petrozza A.; Snaith H. J. Electron-Hole Diffusion Lengths Exceeding 1 Micrometer in an Organometal Trihalide Perovskite Absorber. Science 2013, 342 (6156), 341–344. 10.1126/science.1243982. [DOI] [PubMed] [Google Scholar]
- Irshad Z.; Adnan M.; Lee J. K. Sequentially dip-coated processed MA y FA1- y PbI3- x Br x perovskite layers from an aqueous halide-free lead precursor for efficient perovskite solar cells. J. Mater. Sci.: Mater. Electron. 2022, 33 (18), 15132–15142. 10.1007/s10854-022-08432-1. [DOI] [Google Scholar]
- Shi D.; Adinolfi V.; Comin R.; Yuan M.; Alarousu E.; Buin A.; Chen Y.; Hoogland S.; Rothenberger A.; Katsiev K.; et al. Low trap-state density and long carrier diffusion in organolead trihalide perovskite single crystals. Science 2015, 347 (6221), 519–522. 10.1126/science.aaa2725. [DOI] [PubMed] [Google Scholar]
- De Wolf S.; Holovsky J.; Moon S.-J.; Löper P.; Niesen B.; Ledinsky M.; Haug F.-J.; Yum J.-H.; Ballif C. Organometallic Halide Perovskites: Sharp Optical Absorption Edge and Its Relation to Photovoltaic Performance. J. Phys. Chem. Lett. 2014, 5 (6), 1035–1039. 10.1021/jz500279b. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Xu X.; Cai N.; Qian S.; Luo R.; Huo Y.; Tsang S.-W. Rational Design of Dopant-Free Coplanar D-π-D Hole-Transporting Materials for High-Performance Perovskite Solar Cells with Fill Factor Exceeding 80%. Adv. Energy Mater. 2019, 9 (39), 1901268. 10.1002/aenm.201901268. [DOI] [Google Scholar]
- Vegiraju S.; Ke W.; Priyanka P.; Ni J.-S.; Wu Y.-C.; Spanopoulos I.; Yau S. L.; Marks T. J.; Chen M.-C.; Kanatzidis M. G. Benzodithiophene Hole-Transporting Materials for Efficient Tin-Based Perovskite Solar Cells. Adv. Funct. Mater. 2019, 29 (45), 1905393. 10.1002/adfm.201905393. [DOI] [Google Scholar]
- Zimmermann I.; Aghazada S.; Nazeeruddin M. K. J. A. C. Lead and HTM Free Stable Two-Dimensional Tin Perovskites with Suitable Band Gap for Solar Cell Applications. Angew. Chem. 2019, 131 (4), 1084–1088. 10.1002/ange.201811497. [DOI] [PubMed] [Google Scholar]
- Ke W.; Priyanka P.; Vegiraju S.; Stoumpos C. C.; Spanopoulos I.; Soe C. M. M.; Marks T. J.; Chen M.-C.; Kanatzidis M. G. Dopant-Free Tetrakis-Triphenylamine Hole Transporting Material for Efficient Tin-Based Perovskite Solar Cells. J. Am. Chem. Soc. 2018, 140 (1), 388–393. 10.1021/jacs.7b10898. [DOI] [PubMed] [Google Scholar]
- Liu X.; Zhang F.; Liu Z.; Xiao Y.; Wang S.; Li X. Dopant-free and low-cost molecular “bee” hole-transporting materials for efficient and stable perovskite solar cells. J. Mater. Chem. C 2017, 5 (44), 11429–11435. 10.1039/C7TC03931J. [DOI] [Google Scholar]
- Li H.; Fu K.; Hagfeldt A.; Grätzel M.; Mhaisalkar S. G.; Grimsdale A. C. A simple 3, 4-ethylenedioxythiophene based hole-transporting material for perovskite solar cells. Angew. Chem. 2014, 126 (16), 4169–4172. 10.1002/ange.201310877. [DOI] [PubMed] [Google Scholar]
- Adnan M.; Lee J. k. All sequential dip-coating processed perovskite layers from an aqueous lead precursor for high efficiency perovskite solar cells. Sci. Rep. 2018, 8 (1), 2168. 10.1038/s41598-018-20296-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba M.; Orlandi S.; Matsui T.; Aghazada S.; Cavazzini M.; Correa-Baena J.-P.; Gao P.; Scopelliti R.; Mosconi E.; Dahmen K.-H.; et al. A molecularly engineered hole-transporting material for efficient perovskite solar cells. Nat. Energy 2016, 1 (2), 15017. 10.1038/nenergy.2015.17. [DOI] [Google Scholar]
- Rakstys K.; Paek S.; Drevilkauskaite A.; Kanda H.; Daskeviciute S.; Shibayama N.; Daskeviciene M.; Gruodis A.; Kamarauskas E.; Jankauskas V.; et al. Carbazole-Terminated Isomeric Hole-Transporting Materials for Perovskite Solar Cells. ACS Appl. Mater. Interfaces 2020, 12 (17), 19710–19717. 10.1021/acsami.9b23495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luizys P.; Xia J.; Daskeviciene M.; Kantminiene K.; Kasparavicius E.; Kanda H.; Zhang Y.; Jankauskas V.; Rakstys K.; Getautis V.; et al. Branched Methoxydiphenylamine-Substituted Carbazole Derivatives for Efficient Perovskite Solar Cells: Bigger Is Not Always Better. Chem. Mater. 2021, 33 (17), 7017–7027. 10.1021/acs.chemmater.1c02114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawar A. M.; Yahia I. S. Fabrication and characterization of anthracene thin films for wide-scale organic optoelectronic applications based on linear/nonlinear analyzed optical dispersion parameters. Opt. Mater. 2017, 70, 1–10. 10.1016/j.optmat.2017.05.006. [DOI] [Google Scholar]
- Matsuo Y.; Okada H.; Kondo Y.; Jeon I.; Wang H.; Yu Y.; Matsushita T.; Yanai M.; Ikuta T. Anthracene-Based Organic Small-Molecule Electron-Injecting Material for Inverted Organic Light-Emitting Diodes. ACS Appl. Mater. Interfaces 2018, 10 (14), 11810–11817. 10.1021/acsami.8b00603. [DOI] [PubMed] [Google Scholar]
- Inoue Y.; Tokito S.; Ito K.; Suzuki T. Organic thin-film transistors based on anthracene oligomers. J. Appl. Phys. 2004, 95 (10), 5795–5799. 10.1063/1.1707206. [DOI] [Google Scholar]
- Choi H.; Ko H. M.; Cho N.; Song K.; Lee J. K.; Ko J. Electron-Rich Anthracene Semiconductors Containing Triarylamine for Solution-Processed Small-Molecule Organic Solar Cells. ChemSusChem 2012, 5 (10), 2045–2052. 10.1002/cssc.201200242. [DOI] [PubMed] [Google Scholar]
- Qamar Kayani K.; Yaqoob U.; Jabeen S.; Iqbal S.; Yaseen M.; Khalid M.; Salim Akhter M.; Iqbal J. Tris-isopropyl-sily-ethynyl anthracene based small molecules for organic solar cells with efficient photovoltaic parameters. Comput. Theor. Chem. 2021, 1202, 113305. 10.1016/j.comptc.2021.113305. [DOI] [Google Scholar]
- Liu X.; Kong F.; Ghadari R.; Jin S.; Yu T.; Chen W.; Liu G.; Tan Z. a.; Chen J.; Dai S. Anthracene-arylamine hole transporting materials for perovskite solar cells. Chem. Commun. 2017, 53 (69), 9558–9561. 10.1039/C7CC03444J. [DOI] [PubMed] [Google Scholar]
- Adnan M.; Lee J. k. Highly efficient planar heterojunction perovskite solar cells with sequentially dip-coated deposited perovskite layers from a non-halide aqueous lead precursor. RSC Adv. 2020, 10 (9), 5454–5461. 10.1039/C9RA09607H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddique S. A.; Siddique M. B. A.; Hussain R.; Liu X.; Mehboob M. Y.; Irshad Z.; Adnan M. Efficient tuning of triphenylamine-based donor materials for high-efficiency organic solar cells. Comput. Theor. Chem. 2020, 1191, 113045. 10.1016/j.comptc.2020.113045. [DOI] [Google Scholar]
- Akram M.; Siddique S. A.; Iqbal J.; Hussain R.; Mehboob M. Y.; Siddique M. B. A.; Naveed S.; Ali B.; Hanif A.; Sajid M.; et al. End-capped engineering of bipolar diketopyrrolopyrrole based small electron acceptor molecules for high performance organic solar cells. Comput. Theor. Chem. 2021, 1201, 113242. 10.1016/j.comptc.2021.113242. [DOI] [Google Scholar]
- Siddique S. A.; Arshad M.; Naveed S.; Mehboob M. Y.; Adnan M.; Hussain R.; Ali B.; Siddique M. B. A.; Liu X. Efficient tuning of zinc phthalocyanine-based dyes for dye-sensitized solar cells: a detailed DFT study. RSC Adv. 2021, 11 (44), 27570–27582. 10.1039/D1RA04529F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paek S. Novel Anthracene HTM Containing TIPs for Perovskite Solar Cells. Processes 2021, 9 (12), 2249. 10.3390/pr9122249. [DOI] [Google Scholar]
- Sun Z.-Z.; Long R. Thia[5]helicene-Based D-π-A-type Molecular Semiconductors for Stable and Efficient Perovskite Solar Cells: A Theoretical Study. J. Phys. Chem. C 2023, 127 (19), 8953–8962. 10.1021/acs.jpcc.3c01963. [DOI] [Google Scholar]
- Sun Z.-Z.; Yang J.; Ding W.-L.; Liu J.-L.; Xu X.-L. Structural Engineering of FDT toward Promising Spiro-Typed Hole-Transporting Materials: Promoting the Hole Transport and Stabilizing the HOMO Levels. J. Phys. Chem. C 2022, 126 (28), 11529–11536. 10.1021/acs.jpcc.2c03878. [DOI] [Google Scholar]
- Sun Z.-Z.; Long R. Interfacial and Molecular Engineering of a Helicene-Based Molecular Semiconductor for Stable and Efficient Perovskite Solar Cells. J. Phys. Chem. C 2023, 127, 12913–12922. 10.1021/acs.jpcc.3c02776. [DOI] [Google Scholar]
- Plazinski W.; Drach M. Calcium-α-l-Guluronate Complexes: Ca2+ Binding Modes from DFT-MD Simulations. J. Phys. Chem. B 2013, 117 (40), 12105–12112. 10.1021/jp405638k. [DOI] [PubMed] [Google Scholar]
- Parr R. G.Density functional theory of atoms and molecules. In Horizons of Quantum Chemistry: Proceedings of the Third International Congress of Quantum Chemistry Held at Kyoto, Japan, October 29-November 3, 1979; Springer, 1980; pp 5–15.
- Frisch M. J.Gaussian 09, 2009.
- Dennington R.; Keith T.; Millam J. J. I.. GaussView 5.0; Gaussian: Wallingford, 2008; Vol. 20.
- Civalleri B.; Zicovich-Wilson C. M.; Valenzano L.; Ugliengo P. B3LYP augmented with an empirical dispersion term (B3LYP-D*) as applied to molecular crystals. CrystEngComm 2008, 10 (4), 405–410. 10.1039/B715018K. [DOI] [Google Scholar]
- Adamo C.; Barone V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664–675. 10.1063/1.475428. [DOI] [Google Scholar]
- Yanai T.; Tew D. P.; Handy N. C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393 (1–3), 51–57. 10.1016/j.cplett.2004.06.011. [DOI] [Google Scholar]
- Chai J. D.; Head-Gordon M. Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615. 10.1039/b810189b. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Verma P.; Zhang L.; Li Y.; Liu Z.; Truhlar D. G.; He X. M06-SX screened-exchange density functional for chemistry and solid-state physics. Proc. Natl. Acad. Sci. U.S.A. 2020, 117 (5), 2294–2301. 10.1073/pnas.1913699117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Origin(Pro), Version 2021. OriginLab Corporation, Northampton, MA, USA.
- O’Boyle N. M.; Tenderholt A. L.; Langner K. M. cclib: A library for package-independent computational chemistry algorithms. J. Comput. Chem. 2008, 29 (5), 839–845. 10.1002/jcc.20823. [DOI] [PubMed] [Google Scholar]
- Lu T.; Chen F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- Adnan M.; Irshad Z.; Lee J. K. Facile all-dip-coating deposition of highly efficient (CH 3) 3 NPbI 3- x Cl x perovskite materials from aqueous non-halide lead precursor. RSC Adv. 2020, 10 (48), 29010–29017. 10.1039/D0RA06074G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adnan M.; Irshad Z.; Hussain R.; Lee W.; Yup Yang J.; Lim J. Influence of end-capped engineering on 3-dimenional star-shaped triphenylamine-based donor materials for efficient organic solar cells. Arabian J. Chem. 2023, 16 (6), 104709. 10.1016/j.arabjc.2023.104709. [DOI] [Google Scholar]
- Tang S.; Zhang J. Design of donors with broad absorption regions and suitable frontier molecular orbitals to match typical acceptors via substitution on oligo(thienylenevinylene) toward solar cells. J. Comput. Chem. 2012, 33 (15), 1353–1363. 10.1002/jcc.22966. [DOI] [PubMed] [Google Scholar]
- Yoosefian M.; Raissi H.; Nadim E. S.; Farzad F.; Fazli M.; Karimzade E.; Nowroozi A. Substituent effect on structure, electron density, and intramolecular hydrogen bonding in nitroso-oxime methane. Int. J. Quantum Chem. 2011, 111 (14), 3505–3516. 10.1002/qua.22793. [DOI] [Google Scholar]
- Irshad Z.; Adnan M.; Lee J. K. Efficient planar heterojunction inverted perovskite solar cells with perovskite materials deposited using an aqueous non-halide lead precursor. Bull. Korean Chem. Soc. 2020, 41 (9), 937–942. 10.1002/bkcs.12092. [DOI] [Google Scholar]
- Adnan M.; Mehboob M. Y.; Hussain R.; Irshad Z. In silico designing of efficient C-shape non-fullerene acceptor molecules having quinoid structure with remarkable photovoltaic properties for high-performance organic solar cells. Optik 2021, 241, 166839. 10.1016/j.ijleo.2021.166839. [DOI] [Google Scholar]
- Atiq K.; Adnan M.; Muhammad S.; Hussain R.; Irshad Z.; Khan M. U. Fused ring pyrrolo[3,2-b]pyrrole-based tilde-shaped acceptor molecules for highly efficient organic solar cells. J. Phys. Chem. Solids 2023, 176, 111228. 10.1016/j.jpcs.2023.111228. [DOI] [Google Scholar]
- Hussain R.; Adnan M.; Nawab S.; Khan M. U.; Khalid M.; Irshad Z.; Ayub K.; Lim J. Role of novel carbon-oxygen-bridged Z-shaped non-fullerene acceptors for high efficiency organic solar cells. Synth. Met. 2022, 290, 117159. 10.1016/j.synthmet.2022.117159. [DOI] [Google Scholar]
- Hussain R.; Khan M. U.; Mehboob M. Y.; Khalid M.; Iqbal J.; Ayub K.; Adnan M.; Ahmed M.; Atiq K.; Mahmood K. Enhancement in Photovoltaic Properties of N,N-Diethylaniline Based Donor Materials by Bridging Core Modifications for Efficient Solar Cells. ChemistrySelect 2020, 5, 5022–5034. 10.1002/slct.202000096. [DOI] [Google Scholar]
- Mehboob M. Y.; Hussain R.; Irshad Z.; Adnan M. Enhancement in the Photovoltaic Properties of Hole Transport Materials by End-Capped Donor Modifications for Solar Cell Applications. Bull. Korean Chem. Soc. 2021, 42 (4), 597–610. 10.1002/bkcs.12238. [DOI] [Google Scholar]
- Mehboob M. Y.; Hussain R.; Khan M. U.; Adnan M.; Umar A.; Alvi M. U.; Ahmed M.; Khalid M.; Iqbal J.; Akhtar M. N.; et al. Designing N-phenylaniline-triazol configured donor materials with promising optoelectronic properties for high-efficiency solar cells. Comput. Theor. Chem. 2020, 1186, 112908. 10.1016/j.comptc.2020.112908. [DOI] [Google Scholar]
- Hassan T.; Hussain R.; Khan M. U.; Habiba U.; Irshad Z.; Adnan M.; Lim J. Development of non-fused acceptor materials with 3D-Interpenetrated structure for stable and efficient organic solar cells. Mater. Sci. Semicond. Process. 2022, 151, 107010. 10.1016/j.mssp.2022.107010. [DOI] [Google Scholar]
- Adnan M.; Kim H. S.; Jeong H.; Ko H. M.; Woo S. K.; Lee J. K. Efficient synthesis and characterization of solvatochromic fluorophore. Bull. Korean Chem. Soc. 2017, 38 (9), 1052–1057. 10.1002/bkcs.11219. [DOI] [Google Scholar]
- Rafique A.; Hussain R.; Irshad Z.; Adnan M.; Lim J. Over 1000 nm photoresponse with cyclopentadithiophene-based non-fullerene acceptors for efficient organic solar cells. Comput. Theor. Chem. 2022, 1216, 113852. 10.1016/j.comptc.2022.113852. [DOI] [Google Scholar]
- Irshad Z.; Adnan M.; Lee J. K. Designing novel Zn-decorated inorganic B12P12 nanoclusters with promising electronic properties: a step forward toward efficient CO2 sensing materials. Simple preparation of highly efficient MA x FA1- x PbI3 perovskite films from an aqueous halide-free lead precursor by all dip-coating approach and application in high-performance perovskite solar cells. J. Mater. Sci. 2022, 57 (3), 1936–1946. 10.1007/s10853-022-06867-9. [DOI] [Google Scholar]
- Chi W.-J.; Li Q.-S.; Li Z.-S. Exploring the electrochemical properties of hole transport materials with spiro-cores for efficient perovskite solar cells from first-principles. Nanoscale 2016, 8 (11), 6146–6154. 10.1039/C6NR00235H. [DOI] [PubMed] [Google Scholar]
- Chi W.-J.; Sun P.-P.; Li Z.-S. How to regulate energy levels and hole mobility of spiro-type hole transport materials in perovskite solar cells. Phys. Chem. Chem. Phys. 2016, 18 (39), 27073–27077. 10.1039/C6CP03316D. [DOI] [PubMed] [Google Scholar]
- Birowosuto M.; Skipetrov S.; Vos W. L.; Mosk A. P. Observation of spatial fluctuations of the local density of states in random photonic media. Phys. Rev. Lett. 2010, 105 (1), 013904. 10.1103/physrevlett.105.013904. [DOI] [PubMed] [Google Scholar]
- Shehzad R. A.; Muhammad S.; Iqbal J.; Al-Sehemi A. G.; Yaseen M.; Aloui Z.; Khalid M. Exploring the optoelectronic and third-order nonlinear optical susceptibility of cross-shaped molecules: insights from molecule to material level. J. Mol. Model. 2021, 27 (1), 12. 10.1007/s00894-020-04619-7. [DOI] [PubMed] [Google Scholar]
- Abbas M.; Ali U.; Faizan M.; Siddique M. B. A. Spirofluorene based small molecules as an alternative to traditional non-fullerene acceptors for organic solar cells. Opt. Quantum Electron. 2021, 53 (5), 246. 10.1007/s11082-020-02672-3. [DOI] [Google Scholar]
- Mehboob M. Y.; Khan M. U.; Hussain R.; Khalid M.; Yaqoob J.; Rehman R.; Siddique M. B. A.; Alam M. M.; Imran M.; Ayub K. First example of vinylbenzene based small photovoltaic molecules: Towards the development of efficient D-π-A configured optoelectronic materials for bulk heterojunction solar cells. Phys. B 2022, 633, 413769. 10.1016/j.physb.2022.413769. [DOI] [Google Scholar]
- Irshad Z.; Adnan M.; Lee J. K. Controlling phase and morphology of all-dip-coating processed HC (NH2) 2PbI3 perovskite layers from an aqueous halide-free lead precursor. J. Phys. Chem. Solids 2022, 160, 110374. 10.1016/j.jpcs.2021.110374. [DOI] [Google Scholar]
- Hussain R.; Adnan M.; Atiq K.; Usman Khan M.; H. Farooqi Z.; Iqbal J.; Begum R. Designing of silolothiophene-linked triphenylamine-based hole transporting materials for perovskites and donors for organic solar cells-A DFT study. Sol. Energy 2023, 253, 187–198. 10.1016/j.solener.2023.02.016. [DOI] [Google Scholar]
- Hussain R.; Adnan M.; Irshad Z.; Muhammad S.; Khan M. U.; Lim J. Environmentally compatible 3-dimensional star-shaped donor materials for efficient organic solar cells. Int. J. Energy Res. 2022, 46 (15), 22145–22161. 10.1002/er.8617. [DOI] [Google Scholar]
- Scharber M. C.; Mühlbacher D.; Koppe M.; Denk P.; Waldauf C.; Heeger A. J.; Brabec C. J. Design Rules for Donors in Bulk-Heterojunction Solar Cells—Towards 10% Energy-Conversion Efficiency. Adv. Mater. 2006, 18 (6), 789–794. 10.1002/adma.200501717. [DOI] [Google Scholar]
- Adnan M.; Mehboob M. Y.; Hussain R.; Irshad Z. Banana-Shaped Nonfullerene Acceptor Molecules for Highly Stable and Efficient Organic Solar Cells. Energy Fuels 2021, 35 (14), 11496–11506. 10.1021/acs.energyfuels.1c01431. [DOI] [Google Scholar]
- Riaz S.; Hussain R.; Adnan M.; Khan M. U.; Muhammad S.; Yaqoob J.; Alvi M. U.; Khalid M.; Irshad Z.; Ayub K. Ab Initio Study of Two-Dimensional Cross-Shaped Non-Fullerene Acceptors for Efficient Organic Solar Cells. ACS Omega 2022, 7 (12), 10638–10648. 10.1021/acsomega.2c00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adnan M.; Iqbal J.; BiBi S.; Hussain R.; Akhtar M. N.; Rashid M. A.; Eliasson B.; Ayub K. J. Fine tuning the optoelectronic properties of triphenylamine based donor molecules for organic solar cells. Z. Phys. Chem. 2017, 231 (6), 1127–1139. 10.1515/zpch-2016-0790. [DOI] [Google Scholar]
- Adnan M.; Irshad Z.; Hussain R.; Lee W.; Kim M.; Lim J. Efficient ternary active layer materials for organic photovoltaics. Sol. Energy 2023, 257, 324–343. 10.1016/j.solener.2023.04.012. [DOI] [Google Scholar]
- Hussain R.; Mehboob M. Y.; Khan M. U.; Khalid M.; Irshad Z.; Fatima R.; Anwar A.; Nawab S.; Adnan M. Efficient designing of triphenylamine-based hole transport materials with outstanding photovoltaic characteristics for organic solar cells. J. Mater. Sci. 2021, 56, 5113–5131. 10.1007/s10853-020-05567-6. [DOI] [Google Scholar]
- Zahid S.; Rasool A.; Ans M.; Yaseen M.; Iqbal J. Quantum Chemical Approach of Donor-π-Acceptor Based Arylborane-Arylamine Macrocycles with Outstanding Photovoltaic Properties Toward High-Performance Organic Solar Cells. Energy Fuels 2021, 35 (18), 15018–15032. 10.1021/acs.energyfuels.1c02260. [DOI] [Google Scholar]
- Irshad Z.; Lee W.; Adnan M.; Choi Y.; Park T.; Lim J. Elucidating Charge Carrier Dynamics in Perovskite-Based Tandem Solar Cells. Small Methods 2023, 2300238. 10.1002/smtd.202300238. [DOI] [PubMed] [Google Scholar]
- Rasool A.; Zahid S.; Ans M.; Iqbal J.; Adnan M.; Sherif E.-S. M.; Al-Buriahi M. S. Synergistic engineering of end-capped acceptor and bridge on arylborane-arylamine macrocycles to boost the photovoltaic properties of organic solar cells. Opt. Mater. 2022, 123, 111907. 10.1016/j.optmat.2021.111907. [DOI] [Google Scholar]
- Parr R. G.Density Functional Theory of Atoms and Molecules. In Horizons of Quantum Chemistry; Fukui K., Pullman B., Eds.; Springer Netherlands: Dordrecht, 1980; pp 5–15. [Google Scholar]
- Lu S.; Yin Z.; Liao S.; Yang B.; Liu S.; Liu M.; Yin L.; Zheng W. An asymmetric encoder-decoder model for Zn-ion battery lifetime prediction. Energy Rep. 2022, 8, 33–50. 10.1016/j.egyr.2022.09.211. [DOI] [Google Scholar]
- Zahid S.; Rasool A.; Ans M.; Salim Akhter M.; Iqbal J.; Al-Buriahi M. S.; Alomairy S.; Alrowaili Z. A. Environmentally compatible and highly improved hole transport materials (HTMs) based on benzotrithiophene (BTT) skeleton for perovskite as well as narrow bandgap donors for organic solar cells. Sol. Energy 2022, 231, 793–808. 10.1016/j.solener.2021.12.010. [DOI] [Google Scholar]
- Ans M.; Iqbal J.; Eliasson B.; Saif M. J.; Javed H. M. A.; Ayub K. Designing of non-fullerene 3D star-shaped acceptors for organic solar cells. J. Mol. Model. 2019, 25 (5), 129. 10.1007/s00894-019-3992-9. [DOI] [PubMed] [Google Scholar]
- Yasir Mehboob M.; Zaier R.; Hussain R.; Adnan M.; Muhammad Asif Iqbal M.; Irshad Z.; Bilal I.; Ramzan Saeed Ashraf Janjua M. In silico modelling of acceptor materials by End-capped and π-linker modifications for High-Performance organic solar Cells: Estimated PCE > 18%. Comput. Theor. Chem. 2022, 1208, 113555. 10.1016/j.comptc.2021.113555. [DOI] [Google Scholar]
- Mehboob M. Y.; Hussain R.; Adnan M.; Irshad Z.; Khalid M. Impact of π-linker modifications on the photovoltaic performance of rainbow-shaped acceptor molecules for high performance organic solar cell applications. Phys. B 2022, 625, 413465. 10.1016/j.physb.2021.413465. [DOI] [Google Scholar]
- Mehboob M. Y.; Hussain R.; Khan M. U.; Adnan M.; Alvi M. U.; Yaqoob J.; Khalid M. Efficient designing of half-moon-shaped chalcogen heterocycles as non-fullerene acceptors for organic solar cells. J. Mol. Model. 2022, 28 (5), 125. 10.1007/s00894-022-05116-9. [DOI] [PubMed] [Google Scholar]
- Siddique S. A.; Altaf S.; Ahmed E.; Naveed S.; Siddique M. B. A.; Hussain R.; Liu X.; Rauf A.; Arshad M. Discovery of versatile bat-shaped acceptor materials for high-performance organic solar cells-a DFT approach. Int. J. Energy Res. 2022, 46 (10), 13393–13408. 10.1002/er.8049. [DOI] [Google Scholar]
- Abbas F.; Ali U.; Tallat A.; Ahmad H. M. R.; Siddique S. A.; Zeb Z.; Siddique M. B. A. An optoelectronic study to design better benzodithiophene (BDT) donor unit based non-fullerene organic solar cells (OSCs): the DFT approaches. Chem. Pap. 2022, 76 (8), 4977–4987. 10.1007/s11696-022-02224-x. [DOI] [Google Scholar]
- Siddique S. A.; Naveed S.; Alvi M. U.; Mehboob M. Y.; Ali B.; Rauf A.; Siddique M. B. A.; Hussain R.; Arshad M.; Liu X. Deciphering the role of end-capped acceptor units for amplifying the photovoltaic properties of donor materials for high-performance organic solar cell applications. Comput. Theor. Chem. 2021, 1205, 113454. 10.1016/j.comptc.2021.113454. [DOI] [Google Scholar]
- Nelsen S. F.; Trieber D. A.; Ismagilov R. F.; Teki Y. Solvent Effects on Charge Transfer Bands of Nitrogen-Centered Intervalence Compounds. J. Am. Chem. Soc. 2001, 123 (24), 5684–5694. 10.1021/ja003436n. [DOI] [PubMed] [Google Scholar]
- Norton J. E.; Brédas J. L. Polarization Energies in Oligoacene Semiconductor Crystals. J. Am. Chem. Soc. 2008, 130 (37), 12377–12384. 10.1021/ja8017797. [DOI] [PubMed] [Google Scholar]
- Arkhipov V. I.; Heremans P.; Bässler H. Why is exciton dissociation so efficient at the interface between a conjugated polymer and an electron acceptor?. Appl. Phys. Lett. 2003, 82 (25), 4605–4607. 10.1063/1.1586456. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data will be made available upon reasonable request to the corresponding author.