

Abstract
Defining clinically relevant MET amplification levels in non-small cell lung cancer remains a challenge. Using results of Guardant360 assay, we described the relationship between MET copy number, overlapping oncogenes and amplicon size, to explore means of enrichment for the patients that will most likely derive benefit from MET targeted therapy.
Introduction:
Defining clinically relevant MET amplification levels in non-small cell lung cancer (NSCLC) remains challenging. We hypothesize that oncogene overlap and MET amplicon size decline with increase in MET plasma copy number (pCN), thus enriching for MET-dependent states.
Patients and Methods:
We interrogated cell-free DNA NGS results of 16,782 patients with newly diagnosed advanced NSCLC to identify those with MET amplification as reported using Guardant360. Co-occurring genomic mutations and copy number alterations within each sample were evaluated. An exploratory method of adjusting for tumor fraction was also performed and amplicon size for MET was analyzed when available.
Results:
MET amplification was detected in 207 (1.2%) of samples. pCN ranged from 2.1 to 52.9. Of these, 43 (20.8%) had an overlapping oncogenic driver, including 23 (11.1%) METex14 skipping or other MET mutations. The degree of (non-MET) oncogene overlap decreased with increases in pCN. Patients with MET pCN ≥ 2.7 had lower rates of overlapping drivers compared to those with MET pCN < 2.7 (6.1% vs. 16.3%, P = .033). None of the 7 patients with pCN > 6.7 had an overlapping driver. After adjusting for tumor fraction, adjusted pCN (ApCN) was also lower for those with overlapping drivers than those without (median ApCN 4.9 vs. 7.3, P = .024). There was an inverse relationship between amplicon size and pCN.
Conclusions:
We propose that a high MET pCN and/or ApCN, together with the absence of overlapping oncogenic drivers and small MET amplicon size, will enrich for patients most likely to derive benefit from MET targeted therapy.
Keywords: Amplification, Cell-free DNA, Next-generation sequencing, Non-small cell lung cancer, c-MET
Introduction
The MET proto-oncogene receptor tyrosine kinase (MET) has emerged as a key target in the treatment landscape of non-small cell lung cancer (NSCLC). Activation of MET that renders the associated cancer cell susceptible to MET inhibition can occur in several ways. Recently, both capmatinib and tepotinib were approved by the FDA for metastatic NSCLC harboring MET exon 14 (METex14) skipping alterations.1,2 There is also evidence of efficacy of these drugs in patients with primary MET amplification.1,3 However, the precise definition of MET amplification associated with benefit from MET inhibition is still unclear.
Unlike the binary nature of genomic events leading to METex14 skipping, MET amplification represents a continuous variable.4 In addition, depending on the assay used, MET copy number gain may be quantified relative to multiple different variables. Eg, using fluorescence in situ hybridization (FISH) assays, which involve direct cell visualization, MET may be quantified relative to the tumor cell count (mean MET/cell) or relative to the centromere of chromosome 7 (MET/CEP7 ratio).4 For next generation sequencing (NGS) assays, copy number is defined relative to some approximation of disomy based on other sequence reads within the underlying bioinformatics platform. Depending on the details of the comparator used, focal (true) amplification of a gene may or may not be able to be differentiated from gene copy numbers which are elevated due to high polysomy.
Prior efforts introduced the concept of oncogene overlap analysis to enrich for clinically-relevant levels of MET amplification. Because most true oncogenic drivers are mutually exclusive5–7, as MET copy number increased a threshold was hypothesized to exist above which other oncogenic drivers would disappear. At such a level, MET-dependence should be highly likely. Among 1164 patients with adenocarcinoma of the lung who had received broad panel molecular analyses, including MET FISH testing, overlapping oncogenic drivers disappeared with MET/CEP7ratios ≥ 5.8 At ratios, 1.8 to ≤ 2.2 and >2.2 but <5, oncogene overlap occurred in 52% and 50% of cases, respectively.
Using mean MET/cell quantification, oncogene overlap was still present in 41% of the cases with mean MET copies per cell ≥ 7, the highest category explored in the study.8 Subsequent studies demonstrated similar results.9 The specific categories of MET/CEP7 ratios explored in the oncogene overlap analysis were chosen because the same initial categories had been used to subdivide patients treated within a separate crizotinib trial. Consistent with the oncogene analysis data, the highest objective response rates were associated with the highest category of MET/CEP7 ratio.10
NGS assays conducted on circulating cell-free DNA (cfDNA) have become commonplace as a rapid and convenient means for molecularly profiling solid tumors, distinct from tissue-based assays. While comparable oncogene overlap analyses are underway based on tissue NGS (D.R. Camidge, personal communication), cfDNA assays offer additional questions with regard to determining the cutoff for defining clinically relevant gene amplification levels. Here, we used oncogene overlap analysis to explore the appropriate criteria for defining MET amplification using the broadly used commercial Guardant360 cfDNA assay by examining the relationship between MET copy number and overlapping oncogenic drivers in patients with newly diagnosed NSCLC. In addition, we examined the relationship between MET amplification segment (amplicon) size and MET copy number to explore the value of incorporating amplicon size within the determination of a clinically relevant definition of MET amplification.
Materials and Methods
Study Design and Patient Selection
We performed a de-identified, retrospective analysis of 16,782 patients who were newly diagnosed with advanced lung adenocarcinoma or NSCLC and underwent Guardant360 analysis between December 2019 and July 2021. The standard Guardant360 assays reports on levels of MET amplification calculated to represent a plasma Copy Number (pCN) of 2.1 to 2.6 [referred to as medium or (++)] or a pCN ≥2.7 [referred to as high or (+++)]. Patients reported to have medium or high-level MET amplification were then assessed for the presence of other overlapping oncogenic drivers. Overlapping oncogenic drivers were defined as genetic aberrations listed in OncoKB as FDA Level 1 and/or 2 evidence for NSCLC (Supplementary table 1). The generation of de-identified data sets by Guardant Health for research purposes was approved by the Quorum Institutional Review Board. The study was also approved by the Colorado Multiple Institutional Review Board.
Molecular Analysis
Two 10mL STRECK tubes were collected from each patient. cfDNA for the Guardant360 assay, a New York State Department of Health-approved test, was isolated from plasma, and NGS was performed as previously described at Guardant Health Inc, a CLIA-certified, CAP- accredited laboratory. Two versions of Guardant360 assay were used in this study and analyzed single nucleotide variants (SNVs) and indels in 74 or 83 genes, copy number amplifications (CNAs) in 18 or 19 genes, and fusions in up to 11 genes, depending on the panel version performed Detailed descriptions of the assay and its validation were published previously.11,12 Copy number alterations (CNAs) for all samples were determined using the methods previously published.11,12 For 33/207 samples with MET amplification, a new methodology was used which is able to determine segment size of the amplicon.
CNA Analysis
Gene CNAs was detected from probe-level molecule coverage normalized to coverage profile learned for a set of training clinical samples, probe GC content and signal saturation. Normalized coverage is robustly summarized at the gene level, and CNA detection is based on training set–established decision thresholds for both absolute copy number deviation from per-sample diploid baseline and deviation from the baseline variation of probe-level normalized signal (z-score) in the context of background variation within each sample’s own diploid baseline.11,12
Additionally, off-target genomic regions are binned into 100kb regions and the observed molecule coverage, similar to probe coverage, is normalized in order to remove systematic biases. The whole genome coverage resulting from on-target (probe-level) and off-target coverage is partitioned into regions of consistent copy number utilizing Circular Binary Segmentation.13 The resulting genomic segments are used to infer the size of the amplified regions.
Statistical Analysis
Adjusted plasma copy number (ApCN) was calculated using the exploratory formula ApCN = [Observed pCN − 2 *(1−T%)]/T% where T% = 2 X VAFmax/100.14 The maximum variant allele fraction (VAFmax) is defined as the VAF of the variant with maximum somatic allele frequency of SNVs, Indels, and Fusions excluding synonymous variants. For variants that occur on an amplified gene, VAFmax was normalized CN normalized VAF = VAF/log2(CN). Because the formula is linear and likely overestimates ApCN at low VAFmax, patients with VAFmax < 1% were excluded from analysis. The Mann-Whitney U Test and Fisher Exact Test were used to examine nominal and categorical relationships when applicable. Spearman’s correlation was used to examine relationships between continuous variables.
Results
Patient Data Set
Data were collected on 16,782 unique newly diagnosed NSCLC patient samples tested on the Guardant360 cfDNA assay. Of these samples, 15,496 (92.3%) had at least one somatic alteration detected (supplementary figure 1). Among these patients, 207 (1.3%) had medium or high levels of MET amplification as reported by the Guardant360 algorithm. Of the 207 samples, 183 (88.4%) were labelled as “lung adenocarcinoma” in histology, while the remaining samples were labelled as “non-small cell lung cancer” in histology. Reported genetic aberrations for individual patients were listed for those with a non-MET-related overlapping oncogenic driver (supplementary table 2), for those without an overlapping oncogenic driver (supplementary table 3), and for those with an overlapping MET-related mutation, separate from MET amplification (supplementary table 4).
Characteristics of Overlapping Oncogenic Drivers
Of the 207 patients with medium or high-level MET amplification reported per the Guardant360 algorithm, 43 (20.8%) had an overlapping oncogenic driver. Two patients had EGFR T790M mutations in the absence of an EGFR activating mutation or other actionable driver oncogene being present. As both of these were quantified as minor subclones compared to other abnormalities in the blood [VAF (variant allele frequency)EGFR T790M/VAFmax (somatic variant with highest VAF) <0.05], these patients were included within the group described as being without an overlapping oncogenic driver (n = 164).
Among the 43 patients with an overlapping oncogenic driver, 22 patients had METex14 skipping and 1 patient had a MET D1010Y mutation. Together these 23 cases were considered within the group described as having overlapping MET-related mutations. Among the other 20 patients with overlapping oncogenic drivers, 1 had an ALK-rearrangement, 11 had EGFR mutations (4 L858R, 3 exon 19 deletion, 1 exon 20 insertion and 3 G719X mutations), 2 had ERBB2 mutations (1 exon 20 insertion and 1 R678Q mutation), 5 had KRAS G12C mutations, and 1 had a RET rearrangement (supplementary table 2).
Observed MET Copy Number Among Those With and Without Overlapping Oncogenic Drivers
Among the patients defined as having medium or high MET amplification per the Guardant360 algorithm, the range of MET pCN for patients with (N = 43) and without (N = 164) overlapping oncogenic drivers (including the MET-related oncogenic mutations) were 2.1 to 6.7 and 2.1 to 52.9, respectively. The median pCN for those with and without overlapping oncogenic drivers were 2.5 and 2.8, respectively. The mean pCN for those with and without overlapping oncogenic drivers were 2.9 and 3.8, respectively. These differences were statistically significant (Figure 1A, Mann-Whitney U Test: P = .033).
Figure 1.
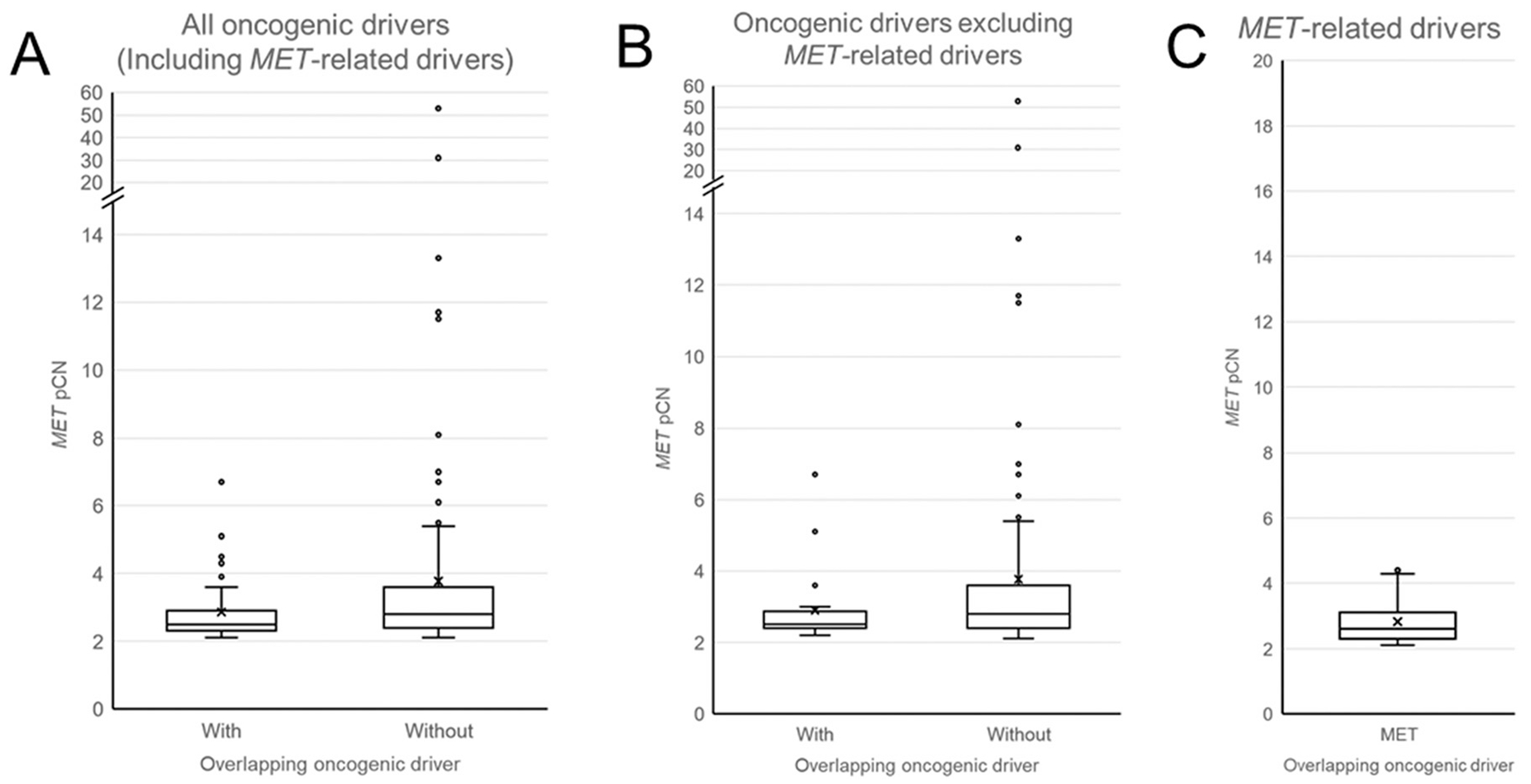
Box plots of MET plasma copy number (pCN) by overlapping oncogenic driver. MET pCN for patients with and without overlapping oncogenic drivers by OncoKB database including MET-related overlapping drivers (A) and excluding MET-related overlapping drivers (B). Panel C represents MET pCN for only those patients with MET-related overlapping drivers.
The range of MET pCN for the patients with overlapping MET-related mutations was 2.1 to 4.5. The median and mean pCN were 2.6 and 2.8, respectively (Figure 1C). Because it is unclear to what degree the oncogenic activity of MET amplification and METex14 skipping mutations or D1010Y are independent for those patients in whom the two aberrations co-exist, all subsequent oncogene overlap analyses below were performed excluding patients with overlapping MET mutations.
When patients with overlapping MET-related mutations (n = 23) were excluded, 20 of 184 patients (10.9%) had an overlapping oncogenic driver. The range of MET pCN for patients with (N = 20) and without (N = 164) overlapping (non-MET) oncogenic drivers were 2.2 to 6.7 and 2.1 to 52.9, respectively. The median pCN for those with and without overlapping (non-MET) oncogenic drivers were 2.5 and 2.8, respectively. The mean pCN for those with and without overlapping (non-MET) oncogenic drivers were 2.9 and 3.8, respectively. These differences were not statistically significant (Figure 1B, Mann-Whitney U Test: P = .116). These results were summarized in Table 1.
Table 1.
Summary of pCN for patients with and without overlapping oncogenic Drivers and Respective Statistics
| Including MET-related Overlapping Oncogenic Driver | Excluding MET-Related Overlapping Oncogenic Driver | MET-Related Overlapping Oncogenic Drivers Only | |||
|---|---|---|---|---|---|
|
| |||||
| With Overlapping Oncogenic Driver | Without Overlapping Oncogenic Driver | With Overlapping Oncogenic Driver | Without Overlapping Oncogenic Driver | ||
|
| |||||
| N | 43 | 146 | 20 | 146 | 23 |
| Median pCN | 2.5 | 2.8 | 2.5 | 2.8 | 2.6 |
|
| |||||
| Mean pCN | 2.9 | 3.8 | 2.9 | 3.8 | 2.8 |
|
| |||||
| pCN range | 2.1-6.7 | 2.1-52.9 | 2.2-6.7 | 2.1-52.9 | 2.1-4.5 |
|
| |||||
| Mann-Whitney U test P value | .033 | 0.116 | |||
Percentage of Patients With Overlapping Oncogenic Drivers at Various Observed MET Copy Number Cutoffs
The percentage of patients with overlapping (non-MET) oncogenic drivers at each interval of pCN are shown in Figure 2A. Overlap occurred at all levels below 6.7 pCN. However, the degree of overlap appeared related to the pCN.
Figure 2.
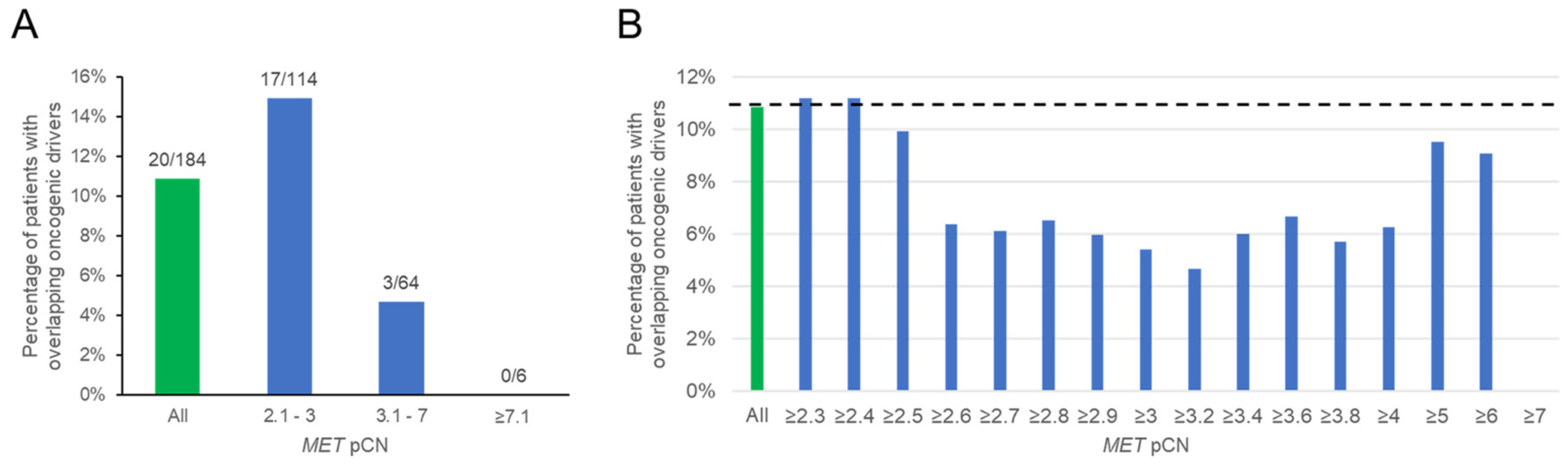
(A) Percentage of patients with overlapping oncogenic drivers by OncoKB in the entire MET-amplified cohort (green bar) and stated pCN intervals (blue bars). Numbers above each bar denotes the number of patients with overlapping drivers out of those with pCN levels within each category. (B) Percentage of patients with overlapping oncogenic drivers by OncoKB at or above a pCN cutoff (blue bars). Green bar represents entire MET-amplified cohort with dotted line representing its associated percentage of oncogene overlap.
Figure 2B illustrates the percentage of with overlapping oncogenic drivers at or above stated pCN cutoffs. The first cutoff level with a subjectively noticeable reduction in oncogene overlap was at pCN ≥2.6. The ~ 6% overlapping oncogenic driver appeared consistent from pCN ≥ 2.7 - 6.7, although the overlapping percentage varies for pCN >5 or pCN > 6 due to the limited sample size in these categories. This suggests that a pCN 2.7 would be a reasonable cutoff for minimal oncogene overlap and 6.7 for defining a non-overlap cutoff.
Based on Guardant Health’s medium and high pCN definitions, 14 of 86 (16.3%) patients with low-level MET amplification (pCN 2.1-2.6) had an overlapping oncogenic driver, whereas 6 of 98 (6.1%) patients with high-level MET amplification (pCN ≥ 2.7) had an overlapping oncogenic driver (Fisher Exact Test: P = .033).
Adjusted MET Copy Number Among Those With and Without Overlapping Oncogenic Drivers
The observed pCN may be impacted by tumor copy number as well as tumor burden, tumor location and the intrinsic rate of DNA shed from the tumor with respect to cfDNA. The ApCN is an exploratory means of modifying the observed pCN, adjusting for the fraction of tumor DNA in the blood. Using all available nonsynonymous tumor derived DNA alterations, VAFmax is used as a surrogate for tumor fraction.
Five patients were excluded from this analysis because ApCN could not be calculated as they either had no co-mutation or only had synonymous mutations. Another 7 patients were excluded because the VAFmax in the sample was < 1%, as ApCN may be overestimated at low VAFmax. There was a correlation between observed between pCN and ApCN (Figure 3A; Spearman correlation = 0.524, P < .001).
Figure 3.
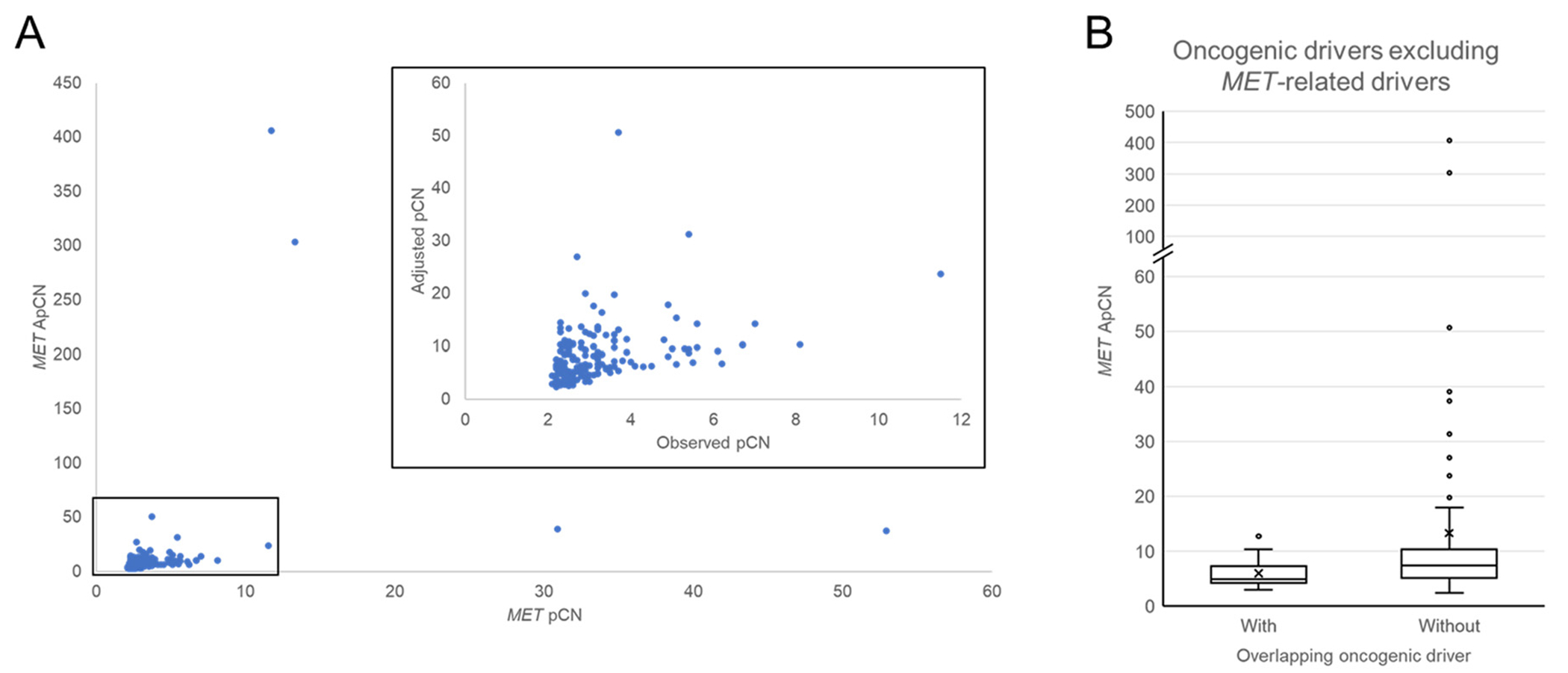
(A) Correlation between MET pCN and ApCN. (B) Adjusted pCN (ApCN) for patients with and without overlapping putative drivers by OncoKB database.
The range of ApCN for patients with (N = 18) and without (N = 154) overlapping (non-MET) oncogenic drivers were 3.0 to 12.7 and 2.4 to 406.2, respectively. The median ApCN for those with and without (non-MET) overlapping oncogenic drivers were 4.9 and 7.3, respectively. The mean ApCN for those with and without overlapping (non-MET) oncogenic drivers were 6.0 and 13.3, respectively. These differences were statistically significant (Figure 3B, Mann-Whitney U Test: P = .024).
Percentage of Patients With Overlapping Oncogenic Drivers at Various Adjusted MET Copy Number Cutoffs
The percentage of patients with overlapping (non-MET) oncogenic drivers at each interval of ApCN are shown in Figure 4A. Overlap occurred at all levels below 12.7 ApCN. The degree of overlap appeared related to the ApCN.
Figure 4.
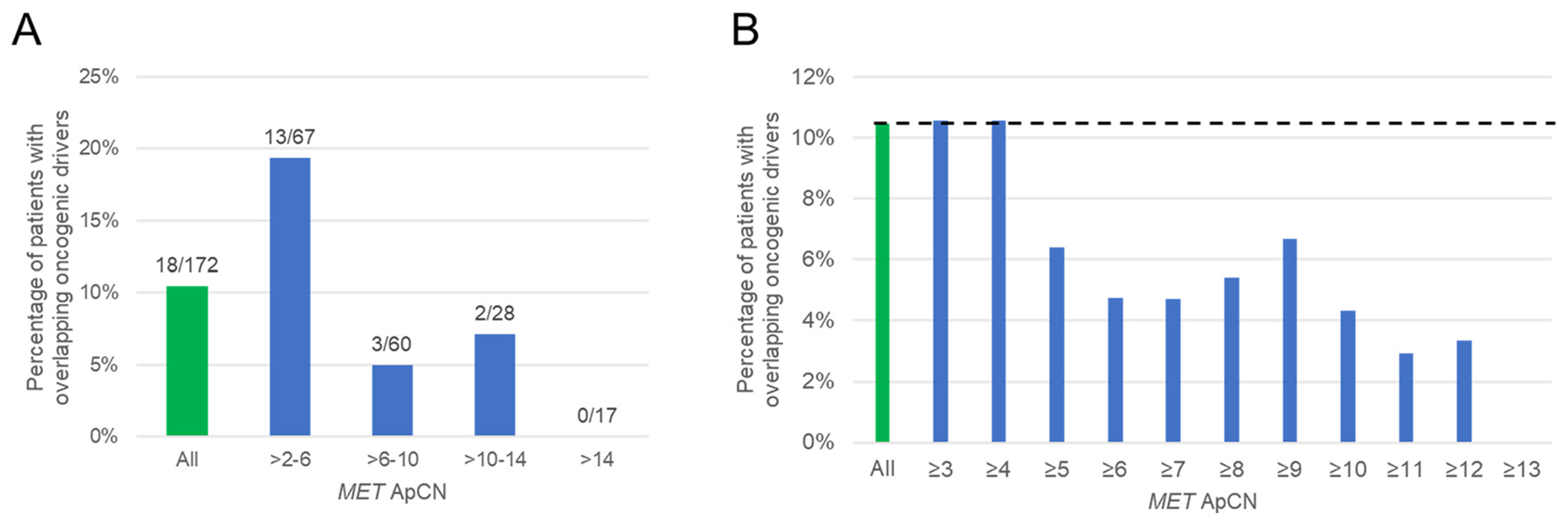
(A) Percentage of patients with overlapping oncogenic drivers by OncoKB in the entire MET-amplified cohort (green bar) and stated ApCN intervals (blue bars). Numbers above each bar denotes the number of patients with overlapping drivers out of those with pCN levels within each category. (B) Percentage of patients with overlapping oncogenic drivers by OncoKB at or above a ApCN cutoff (blue bars). Green bar represents entire MET-amplified cohort with dotted line representing its associated percentage of oncogene overlap.
Figure 4B illustrates the percentage of patients with overlapping (non-MET) oncogenic drivers at or above stated ApCN cutoffs. The first cutoff level with a subjectively noticeable reduction in oncogene overlap was at ApCN ≥ 5.
Relationship Between MET Amplicon Sizes and MET Copy Number Gain
Data on amplicon size were available for 33 of the 207 patients with MET amplification. Among these patients, 7 had an overlapping oncogenic driver including 6 with METex14 skipping mutations and 1 with an EGFR exon 19 deletion. Two of the 33 patients had subclonal EGFR T790M mutations without a sensitizing EGFR mutation, and therefore were considered to be among those without an overlapping oncogenic driver as discussed above.
Figure 5A illustrates a trend towards decreasing MET pCN as amplicon size increased. Using a previously described cutoff of 20 Mbps to categorize focal (<20 Mbps) vs. non-focal (≥ 20 Mbps) MET amplification15, pCN was significantly higher for those with focal MET amplification compared to those with non-focal amplification (Figure 5B, Mann-Whitney U Test: P = .001). However, a statistical difference would also exist for amplicon cutoffs ranging from 5 Mbps to 100 Mbps (Table 2).
Figures 5.
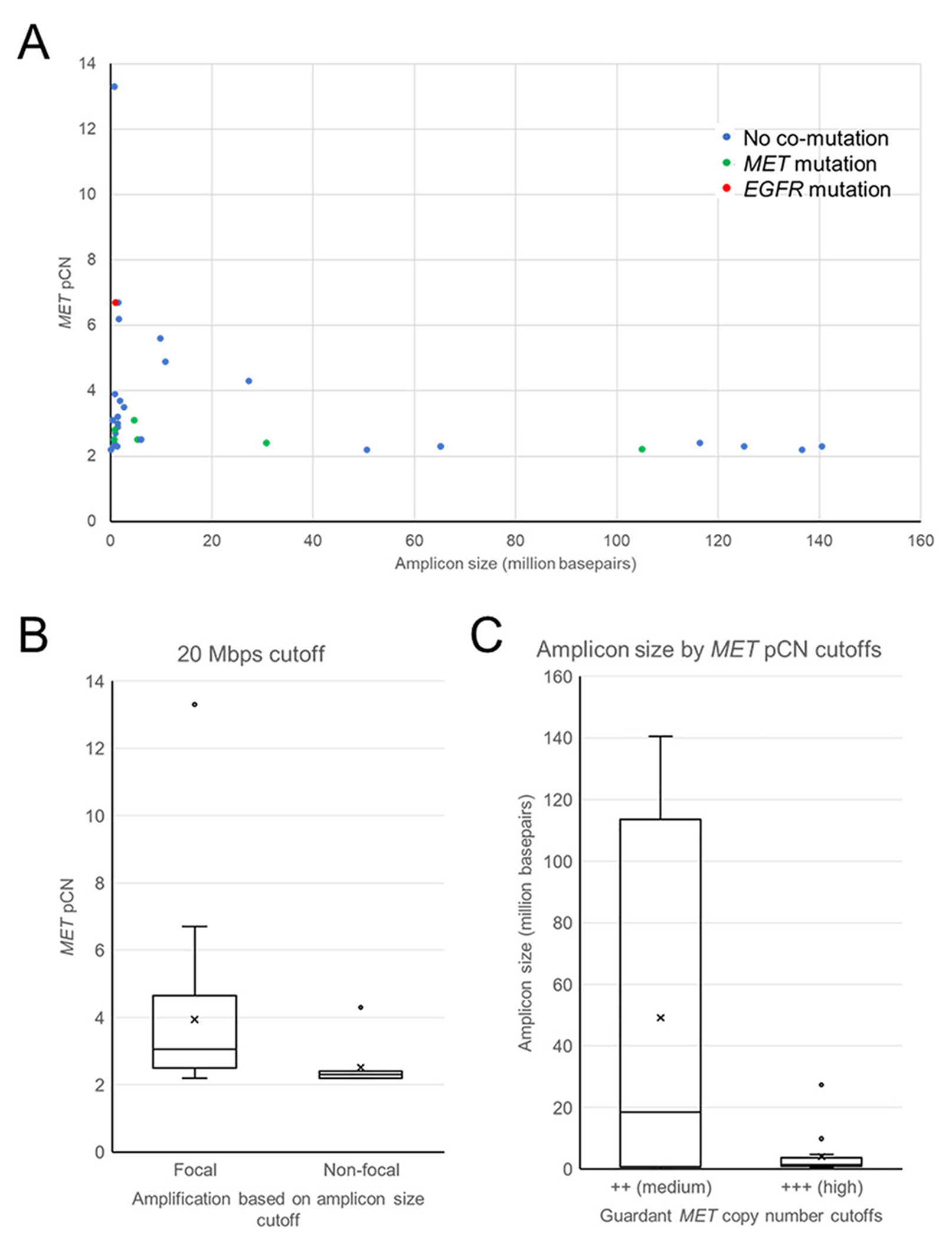
(A) Correlation between amplicon size and MET pCN. (B) pCN for patients with focal (amplicon size < 20 Mbps) and non-focal (amplicon size ≥ 20 Mbps) MET amplification. (C) amplicons size for patients with medium (pCN 2.1-2.6) MET amplification (pCN ≥ 2.7).
Table 2.
Median and Mean pCN for Samples with MET Amplicon Sizes at Various Amplicon Size Cutoff
| Amplicon Size Cutoff (Million Basepairs) | Below Cutoff | At or Above Cutoff | Mann-Whitney P value | ||
|---|---|---|---|---|---|
|
| |||||
| Median pCN | Mean pCN | Median pCN | Mean pCN | ||
|
| |||||
| 1 | 2.6 | 3.8 | 2.9 | 3.5 | .862 |
| 5 | 3.1 | 4.0 | 2.4 | 2.9 | .033 |
|
| |||||
| 10 | 3.0 | 3.9 | 2.3 | 2.8 | .01 |
|
| |||||
| 20 | 3.1 | 3.9 | 2.3 | 2.5 | .001 |
|
| |||||
| 50 | 3.1 | 3.9 | 2.3 | 2.3 | <.001 |
|
| |||||
| 100 | 3.0 | 3.8 | 2.3 | 2.3 | .003 |
Using the Guardant360 high and medium pCN cutoff definitions, there was a trend towards smaller amplicon sizes occurring in association with pCN ≥ 2.7 compared to those with lower pCN levels (Figure 5C, Mann-Whitney U Test: P = .157).
The presence of only 1 patient with MET amplification and a non-MET mutant overlapping oncogene prevented meaningful oncogene overlap analyses to be explored based on amplicon size. However, amplicon sizes of the different cases are listed in supplementary table 5.
Discussion
While it is believed that MET amplification alone can create a true MET-dependent, oncogene-addicted state in some NSCLCs, attempts to categorically define criteria related to MET amplification to define populations with and without significant chances of benefit from MET inhibition continue. Unlike with common activating EGFR mutations, or ALK and ROS1 gene rearrangements, which are associated with 80% to 90% response rates to targeted therapy16–21, the response rate to MET inhibitors in variously defined MET-amplified NSCLC, is commonly reported to be approximately 40%.1,3 These data suggest that in some cases, the apparent MET amplification ‘fails to launch’ as a true driver oncogene, or that it is operating only as a co-driver of a more complex oncogenic state. Oncogene overlap analysis offers a means to both better understand the MET copy number level within a given assay most likely to be exclusively MET driven, and to enrich for tumors more likely to be MET-driven at any MET copy number level if overlapping drivers are excluded. Using tumoral FISH assays across a limited number of MET/CEP7 ratio categories, ratios ≥ 5 identified a population of patients without overlapping oncogenes8,9, in whom crizotinib was associated with a 67% response rate.22 Using a ratio of ≥ 4, which was the level taken forward in the final expansion of the study, the objective response rate was 40%.10 Rare responses were seen with crizotinib among patients with even lower MET/CEP7 ratios. However, as crizotinib is a multi-targeted drug, it was not clear whether every response seen was truly MET-related. Nevertheless, while higher response rates to highly specific MET inhibitors have also been associated with higher MET copy number levels, responses to these drugs have also been observed at low MET copy number levels, albeit rarely. Eg, in a phase I study (NCT01324479), 7 of 15 patients (47%) with MET gene copy number (GCN) ≥ 6 by FISH responded to capmatinib, compared to 2 of 12 (17%) with MET GCN ≥ 4 and < 6, and 0 of 17 (0%) with MET GCN <4.23 Illustrating the potential for complexity within the MET field, of note, 3 of the 7 responders in the MET GCN ≥ 6 group also harbored METex14 skipping alterations.
NGS assays applied to both tissue and blood have revolutionized the routine molecular profiling of NSCLC. Gene copy number, such as for MET, is potentially calculated differently by bioinformatics algorithms within each associated NGS assay and there is a potential need to explore the biological relevance of the level of amplification for each assay. Our results showed that among patients with newly diagnosed, advanced NSCLC who have MET amplification called using the Guardant360 cfDNA assay, those with overlapping oncogenes tend to have lower MET copy number. None of the 7 patients with MET pCN >6.7 had an overlapping oncogenic driver. Based on our prior data, a pCN of >6.7 could be argued to be the amplification level that would be most enriched for patients with true MET addiction. As with the prior FISH data, which calculated the incidence of patients with a MET/CEP7 ratio ≥ 5 at 0.34% of the NSCLC population, this population is expected to be extremely rare. Using the overall cfDNA dataset the incidence is estimated to be no more than 7 of 16,782, although this does not factor in the known false negative rate of the cfDNA analysis, which may be highly dependent on the tumor burden, its location and the inherent DNA shed from the tumor. However, given the responses to MET inhibitors seen across a range of different MET copy number levels, MET amplification may need to be viewed similarly to the way in which PD-L1 tumor proportion scores (TPS) are used as predictive biomarkers for PD-(L)1 inhibition. Specifically, no PD-L1 TPS is associated with a 100% chance of a response to immunotherapy and no level is associated with a zero percent chance. Yet, as the chance of benefit is enriched as the TPS increases, the level can still be used to determine the acceptability of immune monotherapy, or of alternative strategies in the first or later lines of therapy. Therefore, if MET copy number gain were viewed the same way, multiple different factors, including but not limited to the MET copy number, could be used to determine when MET inhibition is proposed as monotherapy and in what line of therapy, given the alternatives.
Our results showed that oncogene overlap begins to decline at around pCN ≥2.6 (Figure 2). While we were unable to statistically analyze every level, the Guardant360 ‘high’ cutoff of pCN ≥2.7 was associated with statistically less overlapping oncogenic drivers and a trend towards smaller amplicon size, compared to those with pCN <2.7. As such, we postulate that a pCN of ≥ 2.6 to 2.7 is a level at which significant enrichment for a MET-addicted state may begin. Consistent with this, the VISION study showed a response rate of 41.7% to tepotinib in patients with MET copy number ≥ 2.5 by the Guardant360 cfDNA assay.3
Our data are limited by not having access to any MET inhibitor outcomes in the patients with differing MET copy number levels by cfDNA. However, it is intriguing to ask whether any subgroups within the VISION trial would be associated with any higher response if, in addition to a MET copy number ≥ 2.5, the patients were also separated out by the presence or absence of a non-MET mutant overlapping oncogene and by the size of their MET amplicon. Strikingly, the objective response rate to capmatinib among those with a tumoral MET gene copy number of ≥ 6 appeared similar to that seen with tepotinib using a cfDNA based MET copy number of ≥ 2.5. Putting aside any potential drug-drug differences, these data initially seem to refute the idea that MET gene copy number is correlated with response to MET inhibition. However, it is far more likely to be due to the fact that the observed pCN of a gene in plasma-derived cfDNA samples is a function of not just the tissue copy number of the gene, but also of tumor burden, location and the propensity of the tumor to shed DNA.
When the observed copy number in our analyses was adjusted for tumoral contribution to the cfDNA, to calculate the ApCN, the copy number values generated appear far closer to the values associated with tumor tissue-based assays used in the capamatinib studies, which were associated with a comparable response rate to MET inhibition. This method of adjusting for tumor fraction is exploratory. While it has been mostly explored in GI malignancies to date, we propose it should also be considered for further investigation in NSCLC in the future.14,24,25
We excluded patients with co-occurring METex14 skipping mutations in most of our oncogene overlap analyses due to the close association between MET amplification and METex14 skipping mutations. About 7.6% to 40.5% of patients with METex14 skipping mutations also harbor MET amplification.1,26–28 While many studies have shown the prevalence of MET amplification among patients with METex14 skipping mutations, few studies have shown the prevalence of METex14 skipping mutations among those with de novo MET amplification. Our data show that, using Guardant360’s algorithm, 10.7% (22/207) patients with MET amplification harbor METex14 skipping mutations. The MET pCN tends to be relatively low in the 2.1 to 4.5 range in the METex14 group, consistent with other studies.27 Early evidence, albeit with small sample size, suggest that patients with overlapping METex14 skipping mutations and high MET amplification had a very high response rate (~80%) to MET TKIs.1,2
There are multiple limitations in our study. First, the assumption of newly-diagnosed NSCLC depends on the accuracy of the database and it is possible that in some cases, MET amplification is an acquired resistance mechanism, rather than a primary oncogenic driver. Second, the decision on whether a specific variant is an oncogenic driver is based on whether it is listed in the OncoKB database with FDA level 1 and 2 evidence, which may exclude some putative drivers with lower levels of evidence. Third, as noted above, we do not have access to relevant clinical correlates related to the impact of MET inhibition among our analyzed patients.
Our study described the relationship between MET pCN, overlapping oncogenic drivers and amplicon size. The assumptions that a level of MET amplification will lead to increased levels of MET protein, MET signaling and true MET addiction in a tumor are, arguably, higher risk assumptions than are associated with assuming that a common EGFR mutation or ALK or ROS1 gene rearrangement will lead to comparable oncogene addicted states. As such, MET amplification by level, amplicon size and oncogene overlap, may need to be viewed in a manner more akin to how PD-L1 TPS is used to determine when and how PD(L)-1 inhibitors are employed.29
The goal of this effort was to explore means of enrichment for the patient population with de novo MET amplification that will most likely derive benefit from MET tyrosine kinase inhibitors. Whether the same or different criteria will be required for other MET-targetable states, eg, with MET-directed antibodies or antibody drug conjugates will need to be explored in the future.30 Equally, the approaches described here might also help pave the way for exploring clinically relevant criteria for other gene amplifications in oncology and possibly shed light on the criteria required for determining clinically-relevant gene amplification criteria manifesting in the acquired resistance setting.
Supplementary Material
Clinical Practice Points.
Defining clinically relevant MET amplification levels in non-small cell lung cancer remains a challenge.
FISH analysis in the past showed that oncogene overlap disappears at high MET/CEP7 ratio (≥ 5), at which MET-dependence is likely based on the principle that oncogenic drivers tend to be mutually exclusive.
Using cell-free DNA next-generation sequencing results, our study showed that the degree of oncogene overlap subjectively decreased with increase in observed MET plasma copy number (pCN) as well as adjusted MET pCN (ApCN) after adjusting for tumor fraction.
An inverse relationship was observed between MET amplicon size and MET pCN.
These results pave the way for means of enriching for patients most likely to derive benefit from MET targeted therapy by pCN and/or ApCN, presence or absence of overlapping oncogenes and amplicon size.
Acknowledgments
D Gao is partially supported by The Cancer Center Support Grant (P30CA046934). The work of DRC was partially supported by the University of Colorado Cancer Center’s Thoracic Oncology Research Initiative and the Joyce Zeff Chair in Lung Cancer Research.
Footnotes
Disclosure
David C.C. Tsui: none. Leylah M. Drusbosky: employee and stockholder of Guardant Health. Sara Wienke: employee and stockholder of Guardant Health. Dexiang Gao: none. Adrian Bubie: employee and stockholder of Guardant Health. Catalin Barbacioru: employee and stockholder of Guardant Health. D. Ross Camidge: consulting or advisory role with Inivata Ltd, Regeneron Pharmaceuticals Inc, AbbVie Inc and EMD Serono Inc.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cllc.2022.07.002.
References
- 1.Wolf J, Seto T, Han JY, et al. Capmatinib in MET Exon 14-mutated or MET-amplified non-small-cell lung cancer. N Engl J Med. 2020;383:944–957. [DOI] [PubMed] [Google Scholar]
- 2.Paik PK, Felip E, Veillon R, et al. Tepotinib in non-small-cell lung cancer with MET Exon 14 skipping mutations. N Engl J Med. 2020;383:931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Le X, Paz-Ares LG, Van Meerbeeck J, et al. Tepotinib in patients (pts) with advanced non-small cell lung cancer (NSCLC) with MET amplification (METamp). Journal of Clinical Oncology. 2021;39:9021–9021. [Google Scholar]
- 4.Drilon A, Cappuzzo F, Ou SI, Camidge DR. Targeting MET in lung cancer: will expectations finally be MET? J Thorac Oncol. 2017;12:15–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gainor JF, Varghese AM, Ou SH, et al. ALK rearrangements are mutually exclusive with mutations in EGFR or KRAS: an analysis of 1,683 patients with non-small cell lung cancer. Clin Cancer Res. 2013;19:4273–4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takeuchi K, Soda M, Togashi Y, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012;18:378–381. [DOI] [PubMed] [Google Scholar]
- 7.Farago AF, Taylor MS, Doebele RC, et al. Clinicopathologic features of non-small-cell lung cancer harboring an NTRK gene fusion. JCO Precis Oncol. 2018;2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Noonan SA, Berry L, Lu X, et al. Identifying the appropriate FISH criteria for defining MET copy number-driven lung adenocarcinoma through oncogene overlap analysis. J Thorac Oncol. 2016;11:1293–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tong JH, Yeung SF, Chan AWH, et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of non–small cell lung carcinoma with poor prognosis. Clin Cancer Res. 2016;22:3048–3056. [DOI] [PubMed] [Google Scholar]
- 10.Camidge DR, Otterson GA, Clark JW, et al. Crizotinib in patients with MET-amplified NSCLC. J Thorac Oncol. 2021;16:1017–1029. [DOI] [PubMed] [Google Scholar]
- 11.Lanman RB, Mortimer SA, Zill OA, et al. Analytical and clinical validation of a digital sequencing panel for quantitative, highly accurate evaluation of cell-free circulating tumor DNA. PLoS One. 2015;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Odegaard JI, Vincent JJ, Mortimer S, et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin Cancer Res. 2018;24:3539–3549. [DOI] [PubMed] [Google Scholar]
- 13.Olshen AB, Bengtsson H, Neuvial P, Spellman PT, Olshen RA, Seshan VE. Parent-specific copy number in paired tumor-normal studies using circular binary segmentation. Bioinformatics. 2011;27:2038–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siravegna G, Sartore-Bianchi A, Nagy RJ, et al. Plasma HER2 (ERBB2) copy number predicts response to HER2-targeted therapy in metastatic colorectal cancer. Clin Cancer Res. 2019;25:3046–3053. [DOI] [PubMed] [Google Scholar]
- 15.Ou S, Pavlick D, Stephens PJ, et al. OA 12.08 Genomic Analysis of Non-Small Cell Lung Cancer (NSCLC) cases with focal and non-focal MET amplification. J Thor Oncol. 2017;12:S1778–S1779. [Google Scholar]
- 16.Soria JC, Ohe Y, Vansteenkiste J, et al. Osimertinib in untreated EGFR-mutated advanced non-small-cell lung cancer. N Engl J Med. 2018;378:113–125. [DOI] [PubMed] [Google Scholar]
- 17.Peters S, Camidge DR, Shaw AT, et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N Engl J Med. 2017;377:829–838. [DOI] [PubMed] [Google Scholar]
- 18.Camidge DR, Kim HR, Ahn MJ, et al. Brigatinib versus Crizotinib in ALK-positive non-small-cell lung cancer. N Engl J Med. 2018;379:2027–2039. [DOI] [PubMed] [Google Scholar]
- 19.Shaw AT, Bauer TM, de Marinis F, et al. First-line lorlatinib or crizotinib in advanced ALK-positive lung cancer. N Engl J Med. 2020;383:2018–2029. [DOI] [PubMed] [Google Scholar]
- 20.Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371:1963–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drilon A, Siena S, Dziadziuszko R, et al. Entrectinib in ROS1 fusion-positive non-small-cell lung cancer: integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020;21:261–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Camidge DR, Ou S-HI, Shapiro G, et al. Efficacy and safety of crizotinib in patients with advanced c-MET-amplified non-small cell lung cancer (NSCLC). Journal of Clinical Oncology. 2014;32:8001–8001. [Google Scholar]
- 23.Schuler M, Berardi R, Lim WT, et al. Molecular correlates of response to capmatinib in advanced non-small-cell lung cancer: clinical and biomarker results from a phase I trial. Ann Oncol. 2020;31:789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janjigian YY, Maron SB, Chatila WK, et al. First-line pembrolizumab and trastuzumab in HER2-positive oesophageal, gastric, or gastro-oesophageal junction cancer: an open-label, single-arm, phase 2 trial. Lancet Oncol. 2020;21:821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakamura Y, Okamoto W, Kato T, et al. Circulating tumor DNA-guided treatment with pertuzumab plus trastuzumab for HER2-amplified metastatic colorectal cancer: a phase 2 trial. Nat Med. 2021;27:1899–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Awad MM, Oxnard GR, Jackman DM, et al. MET Exon 14 mutations in non-small-cell lung cancer are associated with advanced age and stage-dependent MET genomic amplification and c-met overexpression. J Clin Oncol. 2016;34:721–730. [DOI] [PubMed] [Google Scholar]
- 27.Le X, Hong L, Hensel C, et al. Landscape and clonal dominance of co-occurring genomic alterations in non–small-cell lung cancer harboring MET Exon 14 skipping. JCO Precision Oncology. 2021;5:1802–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schrock AB, Frampton GM, Suh J, et al. Characterization of 298 patients with lung cancer harboring MET Exon 14 skipping alterations. J Thorac Oncol. 2016;11:1493–1502. [DOI] [PubMed] [Google Scholar]
- 29.Camidge DR, Doebele RC, Kerr KM. Comparing and contrasting predictive biomarkers for immunotherapy and targeted therapy of NSCLC. Nat Rev Clin Oncol. 2019;16:341–355. [DOI] [PubMed] [Google Scholar]
- 30.Camidge DR, Morgensztern D, Heist RS, et al. Phase I study of 2- or 3-week dosing of telisotuzumab vedotin, an antibody-drug conjugate targeting c-met, monotherapy in patients with advanced non-small cell lung carcinoma. Clin Cancer Res. 2021;27:5781–5792. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.