

Abstract
Transforming growth factor–β1 (TGF-β1) is inextricably linked to regulatory T cell (Treg) biology. However, precisely untangling the role for TGF-β1 in Treg differentiation and function is complicated by the pleiotropic and context-dependent activity of this cytokine and the multifaceted biology of Tregs. Among CD4+ T cells, Tregs are the major producers of latent TGF-β1 and are uniquely able to activate this cytokine via expression of cell surface docking receptor glycoprotein A repetitions predominant (GARP) and αv integrins. Although a preponderance of evidence indicates no essential roles for Treg-derived TGF-β1 in Treg immunosuppression, TGF-β1 signaling is crucial for Treg development in the thymus and periphery. Furthermore, active TGF-β1 instructs the differentiation of other T cell subsets, including TH17 cells. Here, we will review TGF-β1 signaling in Treg development and function and discuss knowledge gaps, future research, and the TGF-β1/Treg axis in the context of cancer immunotherapy and fibrosis.
INTRODUCTION
Transforming growth factor–β (TGF-β) was discovered biochemically in the 1970s in an attempt to isolate soluble factors that induce anchorage-independent growth or transform normal cells (1, 2). The high-affinity receptors of TGF-β1 (TGF-βRI/RII) were discovered shortly thereafter (3, 4). Human TGF-β cDNA was cloned in 1985 (5), followed immediately by cloning of mouse TGF-β cDNA (6). The successful cloning of TGF-β cDNA facilitated a boon to genetic studies and rapid unraveling of its roles in biology, including cancer, fibrosis, and immune tolerance. In the early 1990s, using loss-of-function genetic approaches, two groups independently reported that TGF-β1 deletion in mice resulted in extensive multiorgan inflammation and early death (7–9). The similarity of this phenotype to mice with major defects in regulatory T cells (Tregs) sparked an intense and ongoing interest in the intersection between Treg and TGF-β1 biology. Figure 1 uses a timeline to outline key findings that helped establish the critical roles of TGF-β1 in immune tolerance and Treg biology.
Fig. 1. Timeline of notable discoveries on the role of TGF-β1 and TGF-β1 signaling in the biology of T cells and Tregs.
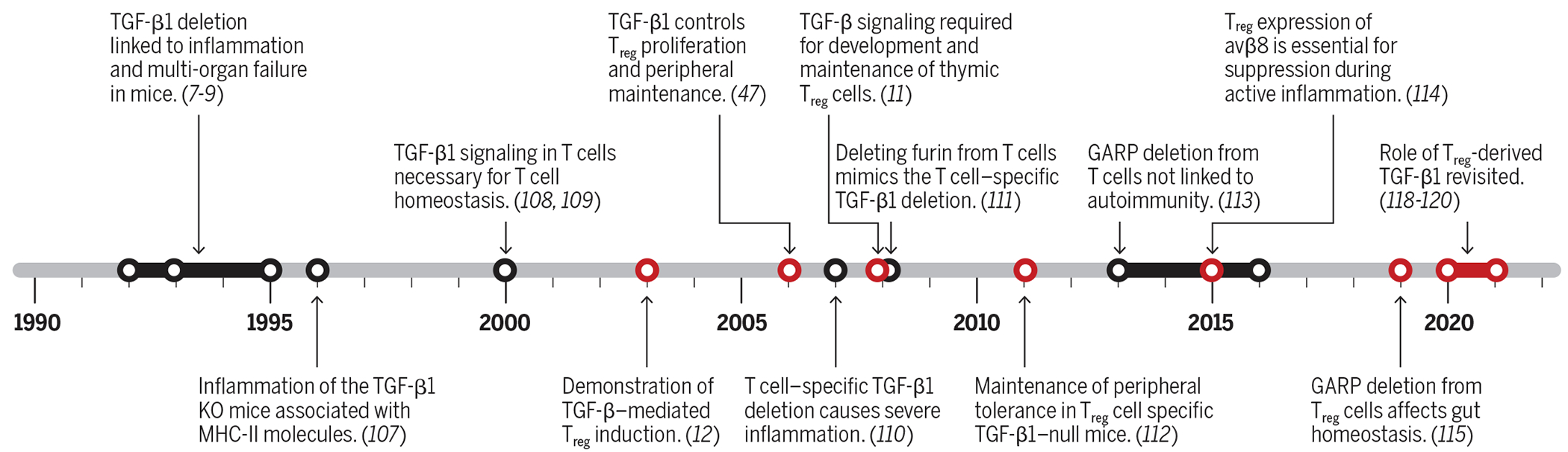
Work on the roles of TGF-β in Treg biology is highlighted in red.
The biogenesis, activation, and signaling of TGF-β1 are complex [reviewed by (10)]. Nearly all cells can produce latent TGF-β1 (LTGF-β1), which can be secreted but requires activation before it can act in an autocrine or paracrine manner. Furthermore, because of the ubiquitous expression pattern of TGF-β receptors and their signaling modules, most cells can respond to active TGF-β1. Proper development of thymus-derived Tregs (tTregs) (11) and induced Tregs (12) in the periphery requires active TGF-β1 signaling. In addition, TGF-β1 instructs the differentiation of other T cell subsets [e.g., T helper 17 (TH17) cells] (13). Intriguingly, among CD4+ T cells, Tregs are the major producers of LTGF-β1 and are uniquely able to activate LTGF-β1 via the expression of cell surface docking receptor glycoprotein A repetitions predominant (GARP) (14–16) and αv integrins (17). These observations suggest that TGF-β1 plays important roles in Treg biology. Recent discoveries underscore the importance of TGF-β1 signaling in Treg biology and emphasize that the function of this cytokine is highly context dependent. Here, we will review our current understanding of TGF-β1 biology as it specifically relates to Tregs. We will discuss key roles that extrinsic TGF-β1 signaling plays in regulating immune responses and Tregs and will also discuss how Treg-derived TGF-β1 modulates the environment in contexts such as cancer and fibrosis. We will address current controversies in this field and outline future directions and potential therapeutic implications of targeting this pathway for the treatment of human diseases.
MECHANISM OF ACTIVATION AND SIGNALING OF TGF-β SUPERFAMILY
TGF-β1 biogenesis
The TGF-β family of ligands consists of three isoforms (TGF-β1, TGF-β2, and TGF-β3) that signal via TGF-β receptors (RI and RII) and co-receptors (such as betaglycan). These TGF-β isoforms are part of a larger superfamily, including bone morphogenetic proteins, activins, and growth differentiation factors (18). In this Review, we will focus our discussion on the TGF-β1 isoform. TGF-β1 is synthesized as a propeptide in the rough endoplasmic reticulum and is composed of the N-terminal latency-associated peptide (LAP) and the C-terminal TGF-β1 protein (Fig. 2). After initial biogenesis, two TGF-β1 propeptides combine to form a homodimer by intermolecular disulfide bonds and translocate to the Golgi, where furin performs a critical proteolytic cleavage step that separates TGF-β1 from LAP (19). Furin cleavage depends on a unique R-H-R-R sequence (20), but the two cleaved products remain noncovalently bound to each other. The resulting small latent complex (SLC) is the LTGF-β1 and is inactive because the mature TGF-β1 is held deep inside the structure to prevent its exposure and activation. TGF-β1 can be secreted extracellularly as the SLC or bound to LTGF-β–binding proteins (LTBPs) to form the large latent complex (LLC) (21). Downstream from biogenesis, the bioavailability and activity of TGF-β are controlled by multiple accessory molecules collectively called “TGF-β milieu molecules” (22), including LTBP (to anchor LTGF-β to the extracellular complex), GARP (16, 23, 24), and LRRC33 (to dock TGF-β onto the cell surface) (22) and αvβ6 and αvβ8 integrins (for activation of LTGF-β) (25).
Fig. 2. TGF-β1 biogenesis, activation, and signaling.
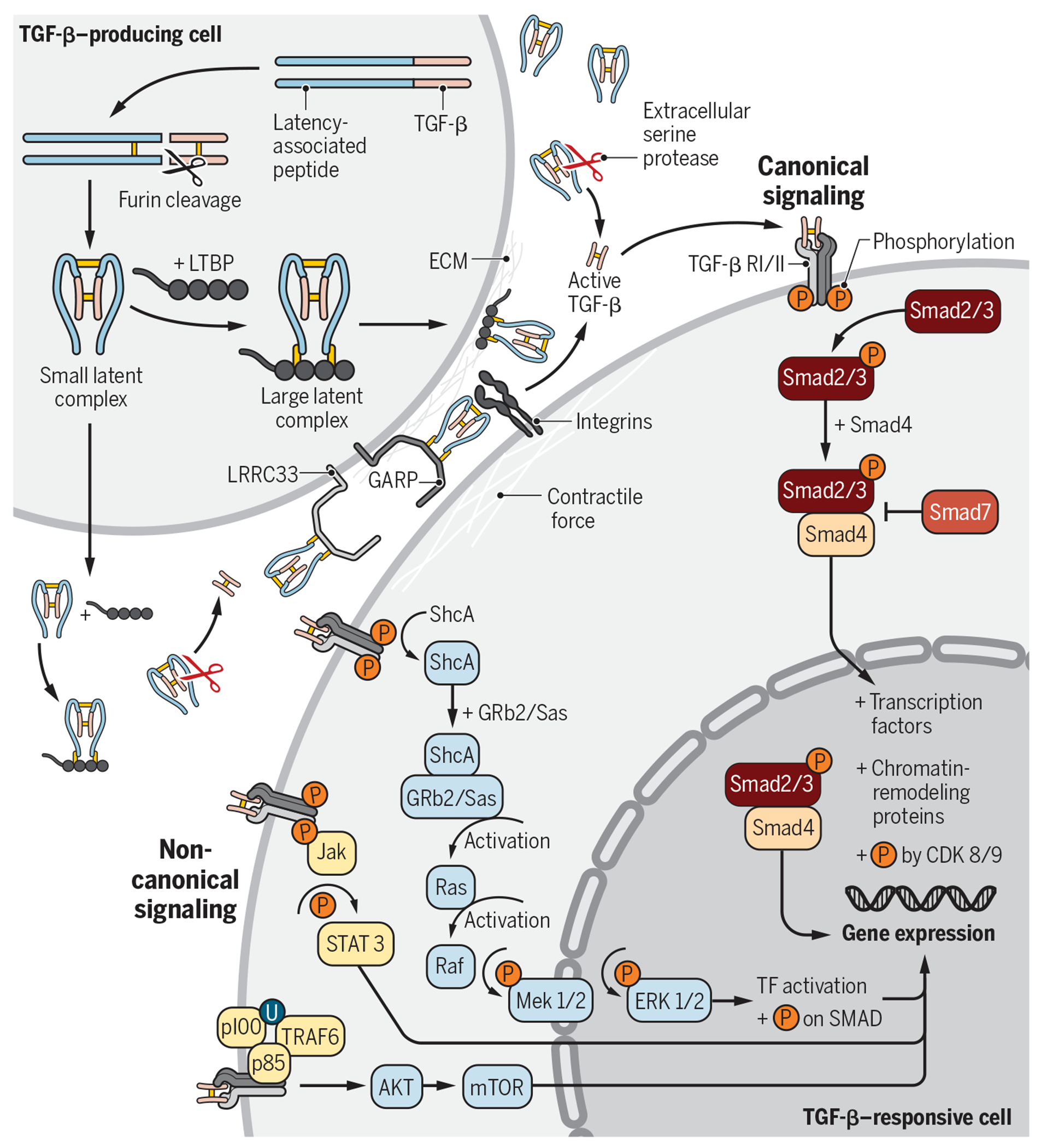
TGF-β1 can be produced and secreted by a number of cell types, including Tregs. LTGF-β1 can be secreted in the SLC or LLC form. Upon secretion, the LLC can bind to fibronectin or fibrillin in the ECM. The SLC can bind to cell surface GARP (e.g., on Tregs, platelets, and endothelial cells) or LRRC33 (e.g., on macrophages and microglia). Activation of LTGF-β1 requires cleavage or conformational change of the SLC or LLC, which releases the mature TGF-β1 from the complex or exposes its active binding motif. Active TGF-β1 binds to TGF-βRII on target cells, which recruits and activates TGF-βRI intracellular domain. Phosphorylation of the TGF-β RI cytoplasmic tails leads to activation and signaling via the canonical (Smad-dependent) or noncanonical pathways. All signaling pathways regulate downstream gene expression. TF, transcription factor.
Upon secretion, the LLC can bind to the extracellular matrix (ECM) via covalent interactions between LTBP and ECM proteins, such as fibronectin or fibrillin. Alternatively, the SLC can bind to GARP [e.g., on Tregs, platelets, and endothelial cells; (14–16, 26)] or LRRC33 [e.g., on macrophages and microglia; (22)]. GARP and its homolog LRRC33 are type I transmembrane cell surface docking receptors for LTGF-β, which modulate its bioavailability and downstream signaling. The binding of LTGF-β to LTBP, GARP, or LRRC33 requires disulfide bond formation with a key cysteine residue in the LAP (27, 28). The association of these complexes ultimately participates in the activation and release of TGF-β1 (29, 30). Unbound extracellular SLC may also exist, and it is sensitive to cleavage and subsequent activation by serine proteases such as matrix metalloproteinases, plasmin, and cathepsin D.
Release of mature and active TGF-β1 requires cleavage or conformational change of the SLC or LLC. Integrin-mediated activation of LTGF-β1 occurs via binding of αvβ6 or αvβ8 integrin heterodimers, expressed on specialized cells including Tregs, to tripeptide Arg-Gly-Asp motifs in the LAP. Such an interaction triggers a contractile force that unfastens the “straitjacket” conformation of LTGF-β1 liberating the active form (25, 31, 32). αvβ8 integrin was recently shown by cryo–electron microscopy to activate LTGF-β1 through a mechanism that does not require release of the mature TGF-β1 for subsequent signaling (33). One study also suggests that thrombin-mediated cleavage of GARP can activate LTGF-β1 from the cell surface GARP–LTGF-β1 complex (34).
TGF-β1 signaling cascade
Active TGF-β1 binds to TGF-β receptor II (TGF-βRII) on target cells, which recruits TGF-β receptor I (TGF-βRI) to create a heterotetrameric receptor complex. Binding and activation of this complex leads to phosphorylation of the cytoplasmic tails, which can lead to signaling via the canonical (Smad) or noncanonical pathways. Within the canonical signaling pathway, Smad2/3 phosphorylation results in the binding to Smad4 to create a signaling complex that translocates to the nucleus and induces downstream changes in gene expression. Upon nuclear translocation, the Smad complex binds to other transcription factors (e.g., NFAT and activating protein 1 in immune cells) or chromatin-remodeling proteins (e.g., histone acetylases) to regulate gene expression [e.g., Foxp3 in Tregs; (35)]. TGF-β1 can also signal via a plethora of noncanonical Smad-independent pathways [reviewed in (36)], including via the mitogen-activated protein (MAP) kinase pathway, which signals via extracellular signal–regulated kinase 1/2 or TGF-β–activated kinase 1 (TAK1).
TGF-β1 SIGNALING IN TREG DEVELOPMENT AND FUNCTION
The majority of Tregs develop in the thymus (tTregs) from CD4 single-positive T cell progenitors (37). However, some immunoregulatory T cells arise via conversion of mature CD4+ conventional T cells in peripheral tissues [peripherally derived Tregs (pTregs)] (38, 39). While the importance of cell-intrinsic TGF-β1 signaling in tTreg development and homeostasis is not fully resolved, this cytokine is required for pTreg induction (39). The underlying molecular mechanisms of this pathway have long been exploited in vitro to convert conventional T cells into a suppressive population [in vitro induced Tregs (iTregs)], which resemble in vivo Tregs. Manipulation of these cells presents a unique opportunity for developing immunoregulatory adoptive cell therapies (12, 40). Below, we discuss the relative contribution of TGF-β1 signaling to the biology of both tTregs and peripheral Tregs (described in Fig. 3A).
Fig. 3. An integrated view of TGF-β1 in Treg biology.
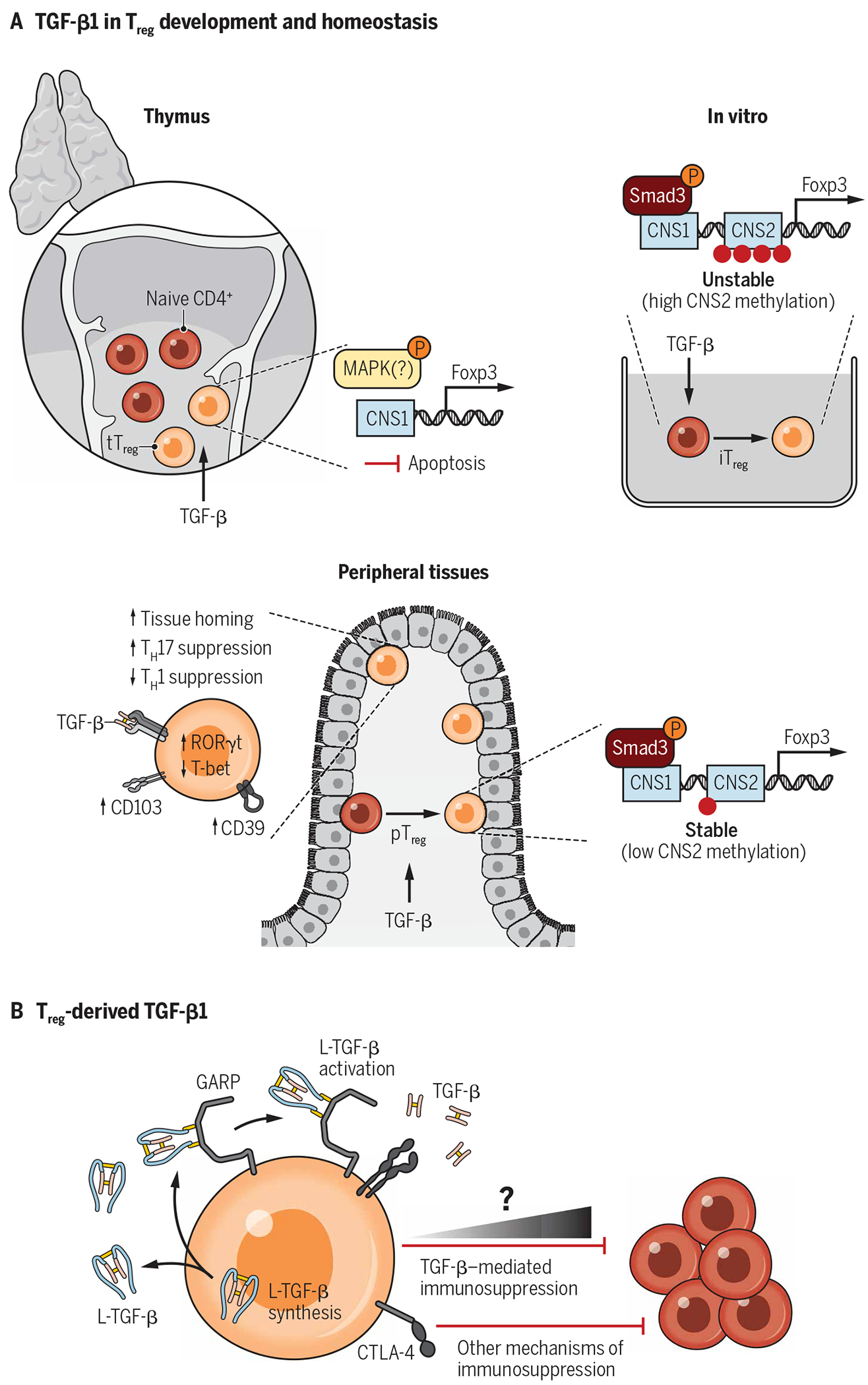
TGF-β1 is uniquely poised to be a focal point in Treg biology because Tregs highly express the TGF-β receptor, are major producers of latent TGF-β1, and have their activating machinery. (A) Signaling through receptor dimers of TGF-βRI and TGF-βRII is a requirement for both thymic Treg development and induced Treg differentiation. (B) Treg-derived TGF-β1 has long been hypothesized to have a major role in Treg function as these cells are a source of latent TGF-β1 and express both GARP and αvβ8 integrin, which work in tandem to activate TGF-β1. Accumulating evidence suggests that Treg-produced TGF-β1 contributes to immunoregulation but is one of several mechanisms in the Treg arsenal. Although not critical to homeostatic function, Treg generation of bioactive TGF-β1 is likely to have tissue- and context-specific immunomodulatory activity.
TGF-β1 is a cryptic driver of thymic Treg development
Foxp3 is a master Treg transcriptional regulator whose expression first appears in CD4 single-positive thymocytes localized to the thymic medulla (41, 42). These T cell progenitors receive signals required for Treg development, including T cell receptor (TCR)–mediated recognition of peptide/major histocompatibility complex II complexes, CD28 costimulation, and a variety of soluble cytokine cues, especially interleukin-2 (IL-2), IL-7, and IL-15. Active TGF-β1 is highly enriched in the medulla, and its expression temporally correlates with neonatal waves of thymocyte negative selection and Treg differentiation. It is thought to be released in response to thymocyte apoptosis (43, 44). While accumulating evidence supports a role for TGF-β1 signaling in Treg development, the molecular details remain poorly defined. Notably, TGF-β1 signaling negatively regulates medullary thymic epithelial cells (mTECs), which instruct T cell development (42, 45). Ablation of TGF-βRII in mTECs led to their expansion and enhanced function and ultimately increased production of tTregs (45). This inhibitory role for TGF-β1 signaling in the thymic medulla illustrates the nuanced, complicated, and sometimes opposing pathways regulated by this highly pleiotropic cytokine.
Early in vivo experiments using animals with global TGF-β1 knockout (KO) or T cell–specific TGF-βRII deletion described the differential requirements for TGF-β1 by peripheral and thymic Tregs (46, 47). The loss of either TGF-β1 or TGF-βRII from T cells precipitated a substantial reduction of Tregs in the peripheral lymphoid organs of mice older than 8 days but induced no defect in tTregs, with equivalent or higher numbers produced by KO mice (46–48). Supporting these experiments, T cell–specific deletion of TGF-β1 signaling proteins Smad2 and Smad3 results in mild expansion of tTregs, whereas pTregs are reduced (49, 50). These findings implied that TGF-β1 signaling is dispensable for tTreg generation; however, subsequent analysis of neonate mice with T cells lacking TGF-βRI found a profound tTreg deficiency. Thymic Treg numbers recovered as the mice aged but correlated with increased levels of thymic IL-2 and proliferation of the remaining Tregs, suggesting that increased IL-2 production rescued the tTreg phenotype. Co-deletion of IL-2 and TGF-βRI prevented tTreg rebound (11). A later study confirmed the early loss of tTregs in a TGF-βRII deletion model, which appeared to be due to a cell survival benefit that TGF-β1 sensing bestows on developing Tregs. TGF-βRII–deficient tTregs underwent increased apoptosis, which correlated with reduced expression of the anti-apoptotic protein B-cell lymphoma 2. Double deletion of TGF-βRII and pro-apoptotic Bim rescued the defect in tTreg generation (51). On the basis of these results, the authors proposed a model where TGF-β1 protects the tTreg lineage from thymic negative selection and promotes survival and maturation (51, 52). However, TGF-β1 broadly protects thymocytes and other lymphocytes from apoptosis, indicating that this pathway is not specific to Treg development (52–55).
Strikingly, tTregs from Foxp3-Cre Tgfbr1fl/fl mice, which lose TGF-βRI expression only after Foxp3 induction, do not exhibit any defects at the neonatal or adult stage (43). Thus, TGF-β1 signaling appears to have a direct impact on commitment to the tTreg lineage, which may be more important than its role in attenuating thymic negative selection. Intrathymic transfer of TCR-transgenic Foxp3− CD4 single-positive uncommitted thymocytes into wild-type mice resulted in Foxp3 expression (43, 56). Blocking TGF-β1 signaling or using TGF-βRI–deficient cells prevented induction of Foxp3. Polyclonal double-negative thymocytes transferred into wild-type mice did not commit to tTreg lineage if donor cells were TGF-βRI deficient (43). In vitro differentiation experiments also support a direct role for TGF-β1 in tTreg development and Foxp3 expression because costimulation with TGF-β1 and common γ chain cytokines synergistically induced Foxp3+ cells (43).
Although these data suggest that TGF-β1 induces tTreg development via Foxp3 signaling, the underlying molecular pathways remain undetermined. Foxp3 expression in CD4+ T cells is driven by conserved noncoding DNA sequence elements (CNS0 to CNS3) at the Foxp3 locus (57). CNS1 contains binding sites for TGF-β1–responsive transcription factors that induce Foxp3, including NFAT and Smad3 (38, 58–60). However, although CNS1 is required for the generation of peripheral and induced Tregs (38, 58–60), in vivo competition experiments demonstrated equivalent numbers of CNS1-sufficient and CNS1-deficient Foxp3+ neonatal thymocytes (38). Therefore, TGF-β1–induced Foxp3 expression in tTregs may occur through a noncanonical pathway, which does not require direct Smad binding to the Foxp3 locus and may occur independently of Smad signaling altogether (36, 52). For example, TGF-β1 can activate MAP kinase signaling, including TAK1, which has been associated with thymic T cell development (36, 61, 62). In addition, TGF-β1 may repress negative regulators of Treg fate, such as was recently hinted in deletion studies of the transcription factors MAZR and hematopoietically-expressed homeobox (63, 64).
TGF-β1 signaling is required for Treg induction
Unlike its complicated role in tTreg development, TGF-β1 is known to synergize with other signals to induce Foxp3 expression in mature CD4+ T cells. This signaling cascade induces CD4+ T cells to display immunoregulatory properties reminiscent, although not identical, to those of tTregs (39, 65). The ability of TGF-β1 to endow naive T cells with suppressive function was first demonstrated in vitro using CD4+ T cells derived from human blood that were stimulated with irradiated allogeneic peripheral blood mononuclear cells in the presence or absence of TGF-β1. The suppressive cell fraction highly expressed CD25; however, the authors concluded that these suppressor cells were derived from existing CD25+ cells rather than converted from CD25−CD4+ T cells (40). These findings were extended in a landmark paper that identified this phenomenon in vivo and determined that it was modulated via Foxp3 expression (12). This aspect of TGF-β1 biology is of particular clinical interest because it opens the possibility of large-scale in vitro iTreg generation for cell therapy applications (66, 67). In addition, pTregs tend to be enriched and functionally active in peripheral tissues, such as the gastrointestinal tract, where their augmentation could provide a strategy for antigen-specific targeting of inflammatory disease (38, 39, 66, 68, 69).
Induction of pTregs is dependent on TGF-β1–mediated activation of Smad3. Binding of Smad3 at CNS1 in the Foxp3 locus promotes Foxp3 transcription, leading to acquisition of a Treg phenotype (58–60). Intriguingly, CNS1 is only present in placental mammals; during pregnancy, pTregs accumulate in the placenta, suggesting that this pathway evolved as a means to maintain maternal-fetal tolerance (59). TGF-β1 may also promote Foxp3 expression through CNS1-independent pathways. TGF-β1–activated STAT5 can demethylate CNS2, accessibility of which is critical for stable Foxp3 gene expression (39, 70). Conversely, TGF-β1 signaling represses methylation by sequestering Uhrf1 from the nucleus (71). Recently, a posttranslational mechanism was also identified wherein TGF-β1 activation of TAK1 causes Nemo-like kinase to phosphorylate Foxp3 and protect it from proteasomal degradation (72). In addition, other factors such as moesin in CD4+ T cells in the tumor microenvironment (TME) augment pTreg induction through up-regulation of TGF-βRII (73).
Although TGF-β1 synergizes with other components of the in vivo milieu to generate pTregs, these signaling factors do not guarantee that a T cell will commit to a regulatory lineage. By inducing Foxp3, TGF-β1 may allow integration of TCR signaling and effectively broaden the range of cognate ligand affinities favorable for conversion to a regulatory state but not necessarily enforce it (74). Interestingly, mechanisms do exist to prevent peripheral T cells from converting to pTregs. Although naïve T cells readily express Foxp3 in response to TGF-β1, differentiated effector and memory populations resist conversion (75). TH cytokines produced by these cells actively interfere with the potency of TGF-β1 (75). Poly(rC) binding protein 1, an RNA binding protein involved in posttranslational repression of TGF-β1 signaling, attenuates pTreg induction (76). Nonetheless, secondary signals may be able to overcome such resistance, such as programmed cell death protein 1 (PD-1) ligation on TH1 cells or combined rapamycin/retinoic acid stimulation of TH2 cells (77, 78). As TGF-β1 mediates differentiation toward both the TH17 and Treg fate, experiments comparatively dissecting how this choice is made are instructive toward understanding how these elements coalesce in vivo. For example, metabolic requirements that drive differentiation and bias against glycolysis and mitochondrial respiration support Treg differentiation (79). Similarly, bile acid metabolites enriched within the gastrointestinal tract promote Treg differentiation. The metabolite isoallolithocholic acid directly increased Treg induction in a TGF-β1–dependent mechanism involving CNS3, whereas another metabolite (3-oxo-5β-cholanoic acid) inhibited TH17 differentiation (80).
The cellular and tissue context required for pTreg generation and a precise molecular picture of how TGF-β1 signaling transactivates the Foxp3 promoter remain to be elucidated. A caveat in harnessing iTregs for cell therapy is that TGF-β–induced Foxp3 expression in human T cells does not necessarily correlate with suppressive function (81). Although both in vitro and in vivo humanized mouse experiments demonstrate that Foxp3-expressing cells can be generated, confirming that suppressive activity will be essential for the development of clinically useful products (82, 83). A notable drawback to iTregs is that they lack the functional stability of their in vivo counterparts (84). This is a critical issue for the therapeutic potential of these cells as reversion to an effector phenotype after adoptive transfer has been reported (85). A specific difference between natural and in vitro induced subsets is the level of methylation at the CNS2 region (84). The demethylated state exhibited by tTregs and pTregs allows for efficient recruitment of transcription factors to the Foxp3 locus and suggests activity of TGF-β1 synergizing factors (39, 86, 87). For example, signaling induced by IL-2, vitamin C, and rapamycin all lead to CNS2 demethylation and can augment iTreg stability, although less potently in human T cells (88–91). Other molecules, such as retinoic acid, also enhance TGF-β1–induced Treg stability but, apparently, through methylation-independent mechanisms (92). Nonetheless, given the substantial evidence linking pTreg generation to peripheral organs (38, 39, 66, 68, 69), deciphering how tissue-specific signals interact with TGF-β1 signaling will be fundamental for developing strategies to modulate Tregs during inflammatory disease.
An unassuming role for TGF-β1 signaling in Treg homeostasis and function
Although TGF-β1 signaling is required for Treg induction, accumulated evidence does not suggest a central role for this cytokine in Treg maintenance or function. Instead, TGF-β1 signaling likely fine-tunes Treg function by modulating specific molecular pathways contextual to local tissue and inflammatory environments. One-year-old Foxp3-CreTgfbr1fl/fl mice do not exhibit overt inflammation or any defect in Treg abundance in lymphoid organs, lung, and colon lamina propria (93). These observations are largely in agreement with earlier studies using a tamoxifen-inducible Cre-ERT2 system to delete Tgfbr2 in peripheral CD4+ T cells. After tamoxifen administration, increased Treg proliferation and accumulation were noted in the spleen and lungs but not in the colon lamina propria or Peyer’s patches, suggesting tissue-specific modulation of the Treg compartment. Studies using mixed bone marrow chimeras indicated that Treg expansion was a cell-intrinsic response and Tgfbr2−/− Tregs proliferated more robustly after anti-CD3 stimulation in vitro (94). Therefore, TGF-β does not appear to control Treg maintenance under homeostatic conditions but may restrain Treg proliferation in response to TCR signaling.
Although Foxp3-CreTgfbr1fl/fl mice do not exhibit overt inflammation over time, proinflammatory TH17 cells accumulate within the colon lamina propria and skin. More TH17 but fewer TH1 cells infiltrated the central nervous system during experimental autoimmune encephalomyelitis, and Tregs from Foxp3-CreTgfbr1fl/fl were unable to control inflammation in a T cell transfer model of colitis (93). Tgfbr1-deficient Tregs demonstrated enhanced suppression of TH1 cells in vitro but were less effective at restraining TH17s (93). Mechanistically, the loss of TGF-β1 responsiveness led to increased expression of the TH1-defining transcription factor T-bet, which facilitates Treg suppression of type 1 inflammation (93, 95). In agreement with this imbalance in TH1 and TH17 control, TGF-β1 has been shown to induce expression of the transcription factor c-Maf (69). Generation of Rorγt+ Tregs requires c-Maf, and these Tregs play a key role in maintaining homeostasis of the intestinal microbiota through TH17 regulation (69, 96). In addition, Foxp3-CreTgfb1fl/fl animals with TGF-β1–deficient Tregs have a high frequency of interferon-γ–producing Foxp3+ cells. In vitro culture assays indicated that autocrine TGF-β1 down-regulates IL-12 receptor genes to prevent IL-12–driven acquisition of a TH1 phenotype (97). TGF-β may also modify Treg activity via metabolic regulation because signaling through this pathway has been associated with reduced expression of genes involved in glycolysis (98, 99).
TGF-β1 has important nonimmune functions in wound healing and barrier tissue homeostasis (100, 101). Given this tissue-centric biology, the gastrointestinal, skin, and central nervous system effects associated with loss of TGF-β signaling in Tregs hint at a broader role in tuning Treg function within peripheral organs. Notably, Treg-specific deletion of TGF-βRI altered expression of several molecules known to regulate immune cell trafficking and retention in gut tissues, including G protein–coupled receptor 15 and the integrin CD103 (93). Accordingly, Tgfbr1-deficient Tregs were unable to accumulate within the colon or control intestinal inflammation in a T cell transfer model of colitis (93). As CD103 also contributes to the persistence and function of CD8+ tissue-resident memory T cells (Trm) in the skin, it is possible that TGF-β1 signaling broadly licenses Treg activity at epithelial sites through modulation of integrin expression (102–104). However, when we deleted Tgfbr2 in Tregs, these mice developed no immediate quantitative or qualitative defects in the skin (105). Long-term experiments will be necessary to determine whether TGF-β1 controls Treg tissue homeostasis and function in the same manner as CD8+ Trm (103, 106).
TREG-DERIVED TGF-β1 IN TREG FUNCTION
A lasting debate: Evidence for and against Treg-derived TGF-β1 as an important mediator of immune tolerance
Our understanding of the immunological roles of TGF-β1 signaling started almost 30 years ago with the discovery that ubiquitous depletion of TGF-β1 from mice causes a severe inflammatory phenotype with fatal multiorgan insufficiency (7–9). Since then, there have been many studies to uncover the significance of TGF-β1 and its signaling cascade in conventional T cell and Treg biology using different genetic mouse models (11, 46, 47, 49, 107–120) (Fig. 1). Unlike the well-defined roles of TGF-β1 and its signaling cascade in the establishment of pTregs, the roles of endogenous TGF-β1 in the biology and function of committed Tregs remain controversial. TGF-β1 is produced in large quantities by Tregs (121), which has been speculated to be responsible for Treg-suppressive function, a hypothesis reinforced by the discovery that Tregs can potently activate TGF-β1 via cell surface GARP and αv integrins. (Fig. 3B). However, despite the rationale of this premise, conflicting reports exist regarding the relative contribution of Treg-produced TGF-β1 to their suppressive function. Here, we discuss the current controversy and provide clarity surrounding the roles of Treg-derived TGF-β1 in immunosuppression. In essence, Treg-intrinsic TGF-β1 is not required for the homeostatic suppressive function of Tregs.
The support for TGF-β1 to mediate Treg immune suppression initially came from transfer colitis experiments. Shortly after Tregs were identified as the CD4+CD25+-expressing cell subpopulation with crucial immune suppressive properties, it was reported that their ability to prevent colitis induced by the transfer of naive CD45RBhigh CD4+ T cells into immunodeficient mice was TGF-β1 mediated. This was based on the observation that the protective effect that they conferred over colitis was lost when TGF-β1 was neutralized with anti–TGF-β1 monoclonal antibodies (122, 123). In a follow-up study using mice whose T cells are unresponsive to TGF-β1 signaling due to expressing a dominant-negative TGF-βRII (dnTβRII), Tregs fail to rescue the colitis phenotype (124). Incongruously, CD4+CD25+ Tregs purified from TGF-β1−/− mice showed no loss in their suppressive potential compared with Tregs from wild-type mice despite the lack of endogenous TGF-β1. However, the authors hypothesized that the TGF-β1−/− Tregs relied on exogenous TGF-β1 to induce their suppressive function. In line with the earlier reports, Nakamura et al. (125) showed that Tregs purified from TGF-β1−/− mice failed to rescue colitis in vivo, whereas they demonstrated normal capacity to block T cell proliferation in vitro. TGF-β1 has also been reported to be indispensable for Treg-mediated immunosuppression in the context of cancer because Tregs suppressed wild-type but not dnTβRII tumor-specific CD8+ T cells (126). When Li et al. (110) generated the first T cell–specific TGF-β1 KO mice, they showed that Tregs from these mice had decreased capacity to rescue T cell–induced colitis, although in vitro suppressive activity remained intact. To examine the role of Treg produced TGF-β1 in their suppressive capacity, Pesu et al. (111) generated a mouse with T cell–specific deletion of furin, which is involved in the biogenesis of TGF-β1. Tregs from these mice were inferior at suppressing colitis compared with wild-type Tregs.
However, the literature on the roles of Treg-derived TGF-β1 in their suppressive function has not always been supportive of its relevance. Various reports from experiments using similar settings to those described above, from anti–TGF-β1 neutralizing antibodies in vitro to various loss-of-function genetic mouse mutants, yield contradictory results. Early reports indicated that Tregs maintain their suppressive activity in vitro, even in the presence of anti–TGF-β1 neutralizing antibodies or genetic deletion (TGF-β1−/−) (127–129). Similarly, piccirillo et al. (129) showed that neutralizing TGF-β1 in vitro was not sufficient to alter their suppressive activity, as was the case with Tregs isolated from Tgfb1−/− mice. They further reinforced this finding by using target T cells from either Smad3−/− or dnTβRII mice, both unable to respond to TGF-β1, which, however, were subject to suppression by Tregs as efficiently as the wild-type T cells. When the same Smad3−/− or dnTβRII T cells were used to induce colitis in vivo, the disease was still able to be rescued by Tregs, although the T cells were unable to respond to TGF-β1 (129, 130). Akin, TGF-β1–nonproducing Tregs, purified from Tgfb1−/− mice, were able to prevent T cell–induced colitis in vivo, indicating that they sustain their suppressive activity despite the lack of autocrine TGF-β1 (130, 131).
In most of the aforementioned studies, the role of Treg-derived TGF-β1 in suppressive function was assessed either indirectly using target T cells that could not respond to TGF-β1 or directly using TGF-β1–nonproducing Tregs from Tgfb1−/− mice (110, 124, 125, 129–131). Gutcher et al. (112) generated the first Treg-specific TGF-β1 KO mouse model, wherein T cells lose TGF-β1 production only after they commit to the Treg fate. These mice remained healthy with no signs of autoimmune disease until late adulthood, providing the first concrete evidence that self-tolerance can be established and maintained in the absence of Treg-produced TGF-β1. However, the technique by which they conditionally delete TGF-β1 from Tregs raises some concerns regarding the efficiency of TGF-β1 deletion from the progeny. Because of the importance of understanding the essence of the debate, some technical details regarding how the TGF-β1 KO mice were generated are provided here. Briefly, their mutant mice have both exon 1 of Tgfb1 and exon 4 of the adjacent gene B9d2 flanked with loxP (110). Although they rescued B9D2 expression by introducing a Tgfb1-null allele with the intact B9d2 locus, their Treg-specific TGF-β1 KO mice are still B9d2 heterozygous because of the fact that the other KO allele contains a 5′ loxP site upstream of exon 4 of B9d2. When these B9d2exon4Tgfb1exon1 floxed mice were later used to study the biological roles of B9D2, Town et al. (132) found that TGF-β1 was still produced upon Cre recombination. This expression resulted from an in-frame chimeric mRNA transcript that formed after excision of B9d2 exon4 and Tgfb1 exon1. A subsequent mouse model generated with a loxP-flanked Tgfb1 exon6 allele was predicted to delete the active form of TGF-β1 only in Cre-expressing cells while sparing adjacent genes (133). However, the two loxP sites in these mice, deposited to the Jackson Laboratory, were later found to have reverse orientation, which was confirmed by our independent study (119). Although it is unclear whether this genetic anomaly occurred in the founder mice or represents genetic drift later during the propagation of this colony, such an anomaly makes these mice unreliable for the conditional deletion of Tgfb1. Therefore, until recently, as described below, it was unclear whether these models accurately represent TGF-β1 deletion from Tregs, and better genetic tools for the conditional deletion of TGF-β were needed.
Turner et al. (118) recently revisited the roles of Treg-produced TGF-β1 in Treg biology using the same Tgfb1exon6 mice mentioned above. Mice with two loxP-flanked Tgfb1exon6 alleles developed severe autoimmunity when crossed with mice that expressed Cre recombinase from the Treg cell–specific Foxp3 locus (Foxp3YFP-cre), which led to their conclusion that Treg-derived TGF-β1 is indispensable for self-tolerance (118). This conclusion was based on the presumption that Tgfb1 is deleted from the Cre-expressing Foxp3+ Tregs in the Foxp3YFP-creTgfb1exon6fl/fl progeny. However, a study from our group demonstrated that these mice develop autoimmunity because of a germline genetic lesion and not because their Tregs do not produce TGF-β1 (119). Briefly, we provided evidence that an inverted 3′-loxP site in the mice used by Turner et al. leads to chromosomal 7 abnormalities during Cre recombination, which ultimately render the Tregs highly apoptotic. Therefore, these mice develop severe autoimmunity because of Treg depletion, similar to the phenotype seen in Scurfy mice (134–136). This was further supported by the fact that the phenotype of these mice was not rescued when crossed with Tgfb1 knock-in mice, which have Tregs with restored TGF-β1 (119). We also showed that an alternative Treg-specific TGF-β1 KO mouse strain generated by targeting exon2 of Tgfb1 remained healthy with no signs of autoimmunity despite the complete absence of TGF-β1 from Tregs (119). Choi et al. (120) similarly provided evidence against the importance of Treg-derived TGF-β1 in Treg-mediated immunosuppression. By comparing Tregs from the Tgfb1exon1 floxed mice described earlier with wild-type Tregs (110), they performed in vitro and in vivo experiments to assess the functionality of Tregs in the absence of endogenous TGF-β1. They found no evidence for compromised Treg function despite reliable deletion of TGF-β1 (120).
Together, this evidence favors the notion that Treg-mediated immunosuppression appears largely impervious to the loss of endogenous TGF-β1 under baseline conditions. despite the initial premise and seemingly conflicting reports in the literature, we believe that the current debate has been settled that Treg-derived TGF-β1 is dispensable for Treg-mediated immune suppression and that self-tolerance can be maintained in the absence of Treg-produced TGF-β1. This could simply be due to the unique ability of Tregs to activate LTGF-β1 in a paracrine fashion via GARP and αv integrin (discussed later), a hypothesis that we are actively studying. However, the potential significance of Treg-derived TGF-β1 under pathological conditions such as infection, autoimmunity, and cancer remains to be fully elucidated.
Role of LTGF-β1 bound to the surface of Tregs
Activated Tregs can store LTGF-β on their surface bound to GARP (14, 16, 23, 24, 121). The release of mature TGF-β1 from the LTGF-β/GARP complex on Treg surface is primarily mediated by αv integrins, allowing the binding of active TGF-β1 to its receptor. In the gut, αv integrins expressed by a subset of CD103+ dendritic cells have been reported to mediate TGF-β1 activation and iTreg induction (137). Recently, it was reported that GARP-bound TGF-β1 can activate its receptor even without being dissociated from the complex (33). GARP can also be proteolytically cleaved by thrombin to release active TGF-β1 (34), a process for TGF-β1 activation that might be preferentially used by platelets compared with Tregs (26). Nonetheless, the role of the surface-bound LTGF-β in Treg function has not been fully elucidated. Consistent with Treg-specific deletion of TGF-β1, mice with conditional GARP ablation from Tregs successfully establish self-tolerance. However, these Tregs show reduced accumulation in the colon, which ultimately leads to enhanced T cell immunity and better control of azoxymethane/dextran sodium sulfate–induced colon cancer development (115).
Tregs themselves also express machinery for activating LTGF-β and may use this pathway to modulate immune function during active inflammation, tissue injury, and cancer. Both mouse and human Tregs have been reported to express the integrin αvβ8, which interacts with GARP to release mature TGF-β1 (17, 114). Deletion of αvβ8 on Tregs did not cause spontaneous autoimmunity but did interfere with the ability of these cells to ameliorate a T cell transfer model of colitis (114). Likewise, monoclonal antibodies that prevent TGF-β1 activation by targeting the GARP:TGF-β1 complex inhibited Treg-mediated immunosuppression in a xenogeneic graft-versus-host disease model (138) and stimulated anticancer immunity (139, 140). We recently observed that specific deletion of αvβ8 on Tregs impaired skin responses to epithelial injury by repressing keratinocyte-driven innate inflammation. Disruption of this circuit exposed mice to uncontrolled bacterial infection through a compromised skin barrier (105). Therefore, although the ability of Tregs to produce LTGF-β per se is not a major contributor to self-tolerance during homeostasis, the regulatory mechanism of Tregs to active LTGF-β produced by neighboring cells in a paracrine fashion is likely important under specific disease and tissue conditions. Although speculative, it remains possible that it is the ability of Tregs to produce and activate LTGF-β that matters in immune tolerance. The proof of this hypothesis has to come from future creative strategies such as loss-of-function studies to examine the impact of simultaneous deletion of LTGF-β and its activating machinery from Tregs.
CLINICAL CONTEXT OF TGF-β1 SIGNALING IN TREG BIOLOGY
Because of their ability to be induced by, produce, activate, and respond to TGF-β1, Tregs have long been implicated as key players in a wide spectrum of human diseases. Here, we will discuss the current knowledge on the role of Treg/TGF-β1 axis in two major pathological states, related to both immunological and nonimmunological roles of Tregs: fibrosis and cancer.
Fibrosis
Fibrosis is characterized by dysregulation of ECM deposition leading to gross disruption in tissue architecture and ultimately organ dysfunction. As an insidious component of a large number of diseases, fibrosis results in an enormous clinical burden. TGF-β1 is strongly implicated in the pathogenesis of fibrotic disorders because it can activate fibroblasts and drives their conversion into myofibroblasts. TGF-β1 also acts on many other cell types, including immune cells to establish a profibrogenic local environment (141). Therapeutic blockade of TGF-β1 in patients with systemic sclerosis decreased fibrotic biomarkers, highlighting the clinical relevance of this cytokine (142).
The role of Tregs in fibrosis has been controversial because they have been reported to both promote and control fibrosis across a variety of tissues and disease models. Experimental Treg ablation worsened skin, lung, and liver fibrosis (143–145). Moreover, Treg infiltration into the livers of chronically infected patients with hepatitis C correlated with reduced fibrotic pathology (146). In murine skin, Tregs are uniquely poised to control TH2 immune responses (143, 147). Recently, it was shown that this function of skin Tregs results in an overall regulation of fibroblast activation and tissue fibrosis induced by TH2 cytokines, both in the steady state and after treatment with bleomycin (143). It remains to be determined whether this is a unique feature of skin Tregs or whether this occurs in other tissues. Furthermore, it is currently unknown whether TGF-β1 biology plays any role in this process. In all the studies mentioned above, Tregs were found to restrain fibrotic disease by suppressing pathologic tissue inflammation, including through direct dampening of TH1, TH2, and TH17 activity (143–145). Conversely, other studies observed improved fibrosis after Treg depletion (148–150). This inconsistency may be due to the quality of Treg activity in fibrotic tissues. Tregs exhibiting a proinflammatory and profibrotic phenotype reportedly accumulate in heart tissue after myocardial infarction. Treg ablation and subsequent reconstitution generated cells with normal suppressive capability (150). Similarly, Treg-specific deficiency of the costimulatory molecule CD226 exacerbated renal fibrosis secondary to their acquisition of a TH2 phenotype (151). Therefore, local features within some fibrotic lesions may polarize Tregs toward a profibrotic phenotype.
It remains unclear how TGF-β1 contributes to the balance between pro- and antifibrotic Treg function. However, pTregs accumulate in and attenuate bleomycin-induced lung fibrosis after in vivo TGF-β1 stimulation (152). Therefore, secondary signals in the milieu of certain fibrotic lesions may co-opt pTreg induction to generate unstable or pathogenic Tregs. Although TGF-β1 produced by Tregs could also contribute to fibrogenesis, the typically potent ability of these cells to suppress other profibrotic immune cells is likely dominant. In addition, Tregs in fibrotic tissues may act as TGF-β sinks and sequester this cytokine through either the TGF-β receptor or GARP (143). Together, it is likely that augmentation of bona fide Treg activity will have therapeutic benefit in fibrotic disorders.
Cancer
The contribution of TGF-β1 to immune evasion during cancer is well known and has been recently reviewed elsewhere (153). It is also known that TGF-β1–rich stroma reduces the efficacy of immune checkpoint inhibitors (CPIs) due to restrained intratumoral T cell trafficking (154, 155). Importantly, rich infiltration of the TME by Tregs is the rule rather than the exception, which is a key roadblock for effective cancer immunotherapy (156). On the one hand, understanding how tumor-associated iTregs are generated and maintained by TGF-β1 and how Tregs adopt to the TME and tolerize the tumor-specific immune responses holds promise for the design and development of next-generation immunotherapeutics. On the other hand, although Tregs are not the sole source of TGF-β1 within the TME, they are critical drivers of the TGF-β1 pathway. It was recently shown that αvβ8 integrin–expressing Tregs activate LTGF-β1 produced by tumor cells, promoting cancer immune evasion (157). Tumor cells and platelets positively regulate the local TGF-β1 levels and function via GARP (26, 158). Intratumoral TGF-β1 converts conventional T cells into pTregs, which increases the Treg pool in the TME and ultimately increases local TGF-β1 production through secretion and activation of LTGF-β via surface GARP (153, 158). As described in Fig. 4, despite some favorable roles of TGF-β1 in immunity (159), TGF-β1 signaling and Treg activation for the most part negatively influence many aspects of adaptive and innate immunity, contributing to immune evasion, immunotherapy resistance, and tumor progression. For example, the TGF-β1/Treg axis blunts the cytotoxic activity of CD8+ cells (160), blocks TH1 cells (161), and promotes tolerogenic function of dendritic cells (162). Moreover, TGF-β1 signaling potently suppresses the functions of natural killer cells by repressing the mammalian target of rapamycin pathway (163). It also polarizes macrophages toward a tumor-promoting M2 phenotype (164, 165). Last, the TGF-β1/Treg axis has also been implicated in promoting oncogenesis and resistance to CPI therapy via enhancing the activity of cancer-associated fibroblasts (166).
Fig. 4. Contributions of TGF-β1 to immune evasion and immunity in cancer.
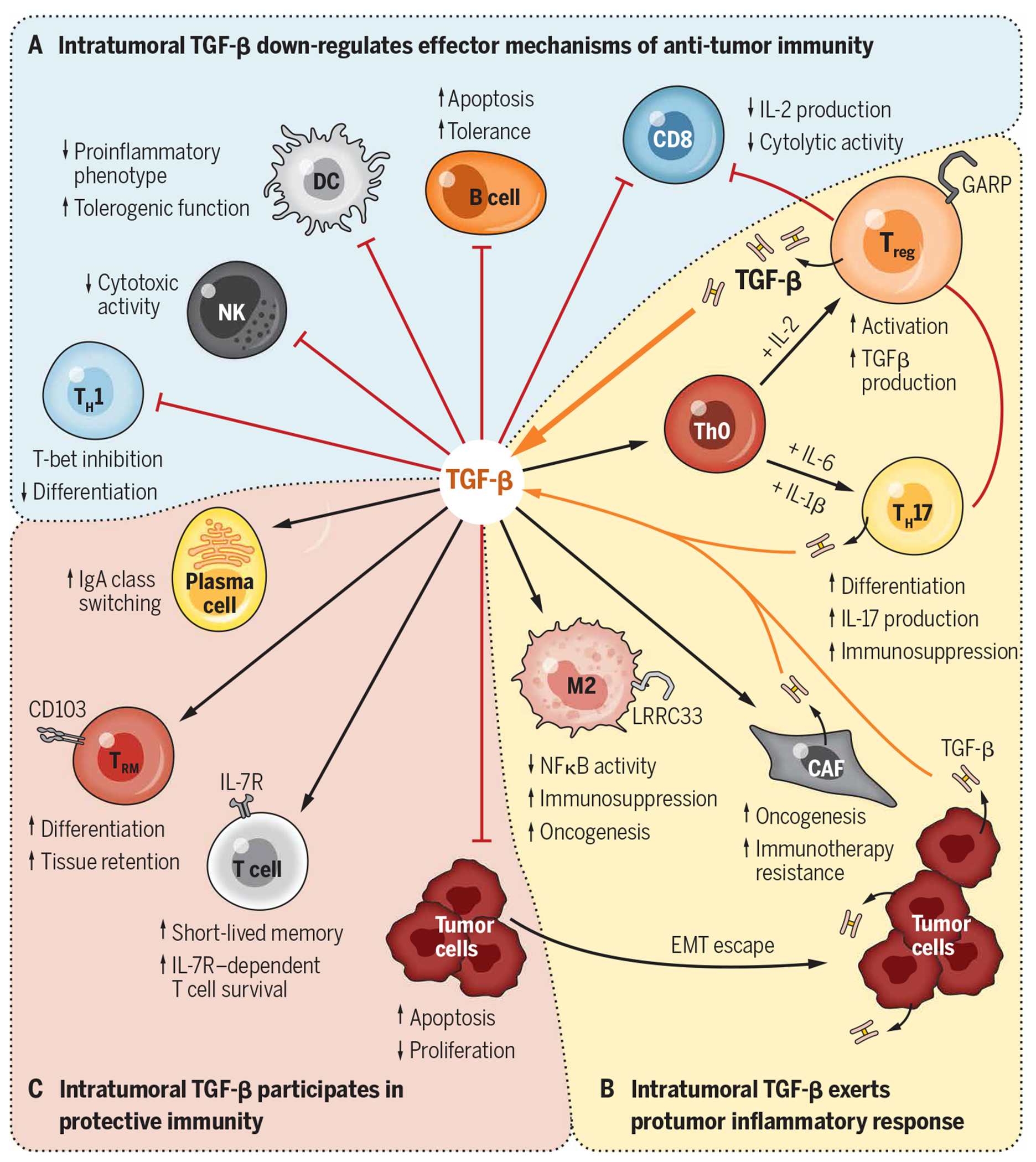
The pleiotropic roles of TGF-β1 within the TME include (A) dampening the antitumor immune responses, (B) enhancing protumorigenic responses, and (C) promoting other elements of protective immunity through interactions with various cell types. Key functional aspects of these interactions are depicted. DC, dendritic cell; NK, natural killer; IgA, immunoglobulin A; NFκB, nuclear factor κB.
However, targeting TGF-β1 therapeutically has proven to be challenging, partly because it is daunting to design a strategy to block TGF-β1 function in the cell type–specific fashion at the right place and the right time. The discovery of the GARP:TGF-β1 axis potentially provides a unique therapeutic approach to fine-tune TGF-β1 activation locally, avoiding the systemic side effects of global TGF-β1 deletion. Monoclonal antibodies that prevent active TGF-β1 release by blocking the GARP:TGF-β complex on Tregs have been shown to inhibit immunosuppression in a xenogeneic graft-versus-host disease model (138). The same anti-GARP targeting strategy seems to also stimulate anticancer immunity by eliminating the Treg-mediated TGF-β1 activation in the TME, which contributes to immunotherapy resistance (139, 140), and is now being tested in a phase 1 clinical trial for the treatment of various cancers (NCT03821935). Similarly, other anti-GARP antibodies, which block the interaction of free Treg GARP with LTGF-β1, could be also used to control local TGF-β1 levels by inhibiting the presentation and, ultimately, activation of LTGF-β1 by Tregs derived from other cellular sources (158). Furthermore, the expression of GARP on activated Tregs creates an opportunity to target GARP using chimeric antigen receptor–expressing T cells for not only GARP-expressing tumors but also the immunosuppressive Tregs in the TME. Future research is expected to define the exact place of GARP:TGF-β targeting strategies within the therapeutic arsenal against human diseases.
Other cancer therapeutic strategies targeting TGF-β1 pathway either alone or in combination with existing modalities include but are not limited to blocking integrins (139, 167), depleting LTGF-β pool systemically (168), CD4+ T cell–specific TGF-β targeting using a bifunctional anti-CD4 antibody–TGF-β trap (4T-Trap) (169, 170) and others [reviewed in (171)]. Notably, bifunctional therapies combining anti–PD-L1 monoclonal antibodies and a TGF-β1 “trap” showed promising results in preclinical models and early-phase clinical trials (172, 173). However, this strategy suffered several setbacks including a failed phase 3 trial to demonstrate superiority over PD-1 blockade alone against non–small cell lung cancer. It is the high hope that continuing fundamental research coupled with innovative drug development strategies will eventually make the TGF-β pathway druggable for cancer.
CONCLUSIONS AND FUTURE PERSPECTIVES
The discovery of Tregs led to a paradigm shift in our understanding of how the immune system functions and maintains homeostasis. We now know that the immune system is always “on,” reacting to both self and foreign antigens. It is effectively dampened by regulatory cells including Tregs that are also always on, preventing most individuals from developing chronic inflammatory and/or autoimmune diseases. Cancer diverts these regulatory mechanisms to prevent antitumor immunity, and pathologic fibrosing conditions may result from defects in regulatory cells that normally suppress profibrotic immune responses and subsequent fibroblast activation. Treg biology and TGF-β1 biology are inextricably linked. iTregs require TGF-β1 for their induction from naïve CD4+ T cells, are capable of secreting large amounts of LTGF-β1, and highly express the molecular machinery to activate LTGF-β1 to act back on themselves or to signal to other neighboring cells. These tenets are widely accepted and supported by numerous experiments. Everything else regarding Treg and TGF-β1 biology is not entirely clear. Given the multitude of cells that can express and/or activate this cytokine and the even larger number of cells that can respond to it, dissecting the functional consequences of any individual cell type activating or responding to TGF-β1 in a complex tissue environment has been difficult. Tregs have been no exception. Emerging data suggest that these cells use TGF-β1 to mediate their functions, but this is highly context dependent and is influenced by the tissue, the type of inflammation, and possibly the type of Tregs. In addition, it may be only in rare circumstances that Treg utilization of TGF-β1 is not redundant with other cells using this cytokine in the local tissue environment. Teleologically, this redundancy makes sense, given the overall importance of the TGF-β1 pathway in preventing systemic inflammation and mortality. New transcriptome profiling platforms at the single-cell level have recently advanced our perception of Treg diversity and functional heterogeneity, especially in the nonlymphoid sites. As cutting-edge omics technologies continue to evolve, it is very likely that distinct TGF-β1 signatures identified within specific Treg subpopulations will help define the tissue and disease contexts where the Treg–TGF-β1 axis plays a dominant role. The goal of future research will undoubtedly be to develop therapeutic strategies to selectively augment or inhibit this axis. Despite its complexity, this is an extremely exciting area of translational investigation, with the first hints of clinical efficacy just beginning to emerge.
Acknowledgments:
We would like to thank the patients who enrolled in clinical trials, as their commitment to research is key in developing new therapeutics, including those related to TGF-β1 We would like to thank I. Boothby for key discussions relating to topics discussed in this Review.
Funding:
This publication was supported by the Ohio State University Comprehensive Cancer Center (OSUCCC) and the NIH under grant P30CA016058. This research was made possible through resources, expertise, and support provided by the Pelotonia Institute for Immuno-Oncology (PIIO), which is funded by the Pelotonia community and the OSUCCC. Z.L. was supported by NIH grants R01AI077283 and R01CA262069. J.M.M. was supported by the Cancer Research Society (grant 26005).
Footnotes
Competing interests: M.D.R. is a consultant and cofounder of TRex Bio Inc. and Sitryx Therapeutics. He is also a consultant for Mozart Therapeutics. Z.L. sits on scientific advisory boards or the board of directors for the following companies: Heat Biologics Inc., Alphamab Oncology (chair), Hengenix Biotech Inc., Ikonisys, Shanghai Henlius Biotech, and the University of Science and Technology of China. Z.L. provides consulting services for Yumed and Houston Methodist Hospital Research Institute. The other authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.de Larco JE, Todaro GJ, Growth factors from murine sarcoma virus-transformed cells. Proc. Natl. Acad. Sci. U.S.A 75, 4001–4005 (1978). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts AB, Anzano MA, Lamb LC, Smith JM, Sporn MB, New class of transforming growth factors potentiated by epidermal growth factor: Isolation from non-neoplastic tissues. Proc. Natl. Acad. Sci. U.S.A 78, 5339–5343 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Massague J, Czech MP, Iwata K, DeLarco JE, Todaro GJ, Affinity labeling of a transforming growth factor receptor that does not interact with epidermal growth factor. Proc. Natl. Acad. Sci. U.S.A 79, 6822–6826 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Massague J, Like B, Cellular receptors for type β transforming growth factor: Ligand binding and affinity labeling in human and rodent cell lines. J. Biol. Chem 260, 2636–2645 (1985). [PubMed] [Google Scholar]
- 5.Derynck R, Jarrett JA, Chen EY, Eaton DH, Bell JR, Assoian RK, Roberts AB, Sporn MB, Goeddel DV, Human transforming growth factor-β complementary DNA sequence and expression in normal and transformed cells. Nature 316, 701–705 (1985). [DOI] [PubMed] [Google Scholar]
- 6.Derynck R, Jarrett JA, Chen EY, Goeddel DV, The murine transforming growth factor-β precursor. J. Biol. Chem 261,4377–4379 (1986). [PubMed] [Google Scholar]
- 7.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, Annunziata N, Doetschman T, Targeted disruption of the mouse transforming growth factor-β 1 gene results in multifocal inflammatory disease. Nature 359, 693–699 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S, Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. U.S.A 90, 770–774 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Diebold RJ, Eis MJ, Yin M, Ormsby I, Boivin GP, Darrow BJ, Saffitz JE, Doetschman T, Early-onset multifocal inflammation in the transforming growth factor β 1-null mouse is lymphocyte mediated. Proc. Natl. Acad. Sci. U.S.A 92, 12215–12219 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hinck AP, Mueller TD, Springer TA, Structural biology and evolution of the TGF-β family. Cold Spring Harb. Perspect. Biol 8, a022103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, Zhang P, Li J, Kulkarni AB, Perruche S, Chen WJ, A critical function for TGF-β signaling in the development of natural CD4+CD25+Foxp3+ regulatory T cells. Nat. Immunol 9, 632–640 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM, Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J. Exp. Med 198, 1875–1886 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B, TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24, 179–189 (2006). [DOI] [PubMed] [Google Scholar]
- 14.Wang R, Wan Q, Kozhaya L, Fujii H, Unutmaz D, Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces Foxp3 expression. PLOS ONE 3, e2705 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang R, Kozhaya L, Mercer F, Khaitan A, Fujii H, Unutmaz D, Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proc. Natl. Acad. Sci. U.S.A 106, 13439–13444 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM, GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc. Natl. Acad. Sci. U.S.A 106, 13445–13450 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edwards JP, Thornton AM, Shevach EM, Release of active TGF-β1 from the latent TGF-β1/GARP complex on T regulatory cells is mediated by integrin β8. J. Immunol 193, 2843–2849 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Massague J, TGF-β signal transduction. Annu. Rev. Biochem 67, 753–791 (1998). [DOI] [PubMed] [Google Scholar]
- 19.Dubois CM, Laprise MH, Blanchette F, Gentry LE, Leduc R, Processing of transforming growth factor β1 precursor by human furin convertase. J. Biol. Chem 270, 10618–10624 (1995). [DOI] [PubMed] [Google Scholar]
- 20.Dubois CM, Blanchette F, Laprise MH, Leduc R, Grondin F, Seidah NG, Evidence that furin is an authentic transforming growth factor-beta1-converting enzyme. Am. J. Pathol 158, 305–316 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanzaki T, Olofsson A, Morén A, Wernstedt C, Hellman U, Miyazono K, Claesson-Welsh L, Heldin CH, TGF-beta 1 binding protein: A component of the large latent complex of TGF-beta 1 with multiple repeat sequences. Cell 61, 1051–1061 (1990). [DOI] [PubMed] [Google Scholar]
- 22.Qin Y, Garrison BS, Ma W, Wang R, Jiang A, Li J, Mistry M, Bronson RT, Santoro D, Franco C, Robinton DA, Stevens B, Rossi DJ, Lu C, Springer TA, A milieu molecule for TGF-β required for microglia function in the nervous system. Cell 174, 156–171.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stockis J, Colau D, Coulie PG, Lucas S, Membrane protein GARP is a receptor for latent TGF-beta on the surface of activated human Treg. Eur. J. Immunol 39, 3315–3322 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Wu BX, Metelli A, Thaxton JE, Hong F, Rachidi S, Ansa-Addo E, Sun S, Vasu C, Yang Y, Liu B, Li Z, GP96 is a GARP chaperone and controls regulatory T cell functions. J. Clin. Invest 125, 859–869 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dong X, Zhao B, Iacob RE, Zhu J, Koksal AC, Lu C, Engen JR, Springer TA, Force interacts with macromolecular structure in activation of TGF-β. Nature 542, 55–59 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rachidi S, Metelli A, Riesenberg B, Wu BX, Nelson MH, Wallace C, Paulos CM, Rubinstein MP, Garrett-Mayer E, Hennig M, Bearden DW, Yang Y, Liu B, Li Z, Platelets subvert T cell immunity against cancer via GARP-TGFβ axis. Sci. Immunol 2, eaai7911 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saharinen J, Taipale J, Keski-Oja J, Association of the small latent transforming growth factor-beta with an eight cysteine repeat of its binding protein LTBP-1. EMBO J. 15, 245–253 (1996). [PMC free article] [PubMed] [Google Scholar]
- 28.Gleizes PE, Beavis RC, Mazzieri R, Shen B, Rifkin DB, Identification and characterization of an eight-cysteine repeat of the latent transforming growth factor-β binding protein-1 that mediates bonding to the latent transforming growth factor-β1. J. Biol. Chem 271, 29891–29896 (1996). [DOI] [PubMed] [Google Scholar]
- 29.Liénart S, Merceron R, Vanderaa C, Lambert F, Colau D, Stockis J, van der Woning B,De Haard G, Saunders M, Coulie PG, Savvides SN, Lucas S, Structural basis of latent TGF-β1 presentation and activation by GARP on human regulatory T cells. Science 362, 952–956 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Wang R, Zhu J, 5Dong X, Shi M, Lu C, Springer TA, GARP regulates the bioavailability and activation of TGFβ. Mol. Biol. Cell 23, 1129–1139 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Munger JS, Huang X, Kawakatsu H, Griffiths MJD, Dalton SL, Wu J, Pittet JF, Kaminski N, Garat C, Matthay MA, Rifkin DB, Sheppard D, The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 96, 319–328 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA, Latent TGF-β structure and activation. Nature 474, 343–349 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Campbell MG, Cormier A, Ito S, Seed RI, Bondesson AJ, Lou J, Marks JD, Baron JL, Cheng Y, Nishimura SL, Cryo-EM reveals integrin-mediated TGF-β activation without release from latent TGF-β. Cell 180, 490–501.e16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Metelli A, Wu BX, Riesenberg B, Guglietta S, Huck JD, Mills C, Li A, Rachidi S, Krieg C, Rubinstein MP, Gewirth DT, Sun S, Lilly MB, Wahlquist AH, Carbone DP, Yang Y,Liu B, Li Z, Thrombin contributes to cancer immune evasion via proteolysis of platelet-bound GARP to activate LTGF-β. Sci. Transl. Med 12, eaay4860 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mullen AC, Orlando DA, Newman JJ, Lovén J, Kumar RM, Bilodeau S, Reddy J, Guenther MG, DeKoter RP, Young RA, Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell 147, 565–576 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Derynck R, Budi EH, Specificity, versatility, and control of TGF-β family signaling. Sci. Signal 12, eaav5183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klein L, Robey EA, Hsieh CS, Central CD4+ T cell tolerance: Deletion versus regulatory T cell differentiation. Nat. Rev. Immunol 19, 7–18 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, Umetsu DT,Rudensky AY, Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature 482, 395–399 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanamori M, Nakatsukasa H, Okada M, Lu Q, Yoshimura A, Induced regulatory T cells: Their development, stability, and applications. Trends Immunol. 37, 803–811 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Yamagiwa S, Gray JD, Hashimoto S, Horwitz DA, A role for TGF-beta in the generation and expansion of CD4+CD25+ regulatory T cells from human peripheral blood. J. Immunol 166, 7282–7289 (2001). [DOI] [PubMed] [Google Scholar]
- 41.Fontenot JD, Dooley JL, Farr AG, Rudensky AY, Developmental regulation of Foxp3 expression during ontogeny. J. Exp. Med 202, 901–906 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hinterberger M, Aichinger M, Prazeres da Costa O, Voehringer D, Hoffmann R, Klein L, Autonomous role of medullary thymic epithelial cells in central CD4+ T cell tolerance. Nat. Immunol 11, 512–519 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Konkel JE, Jin W, Abbatiello B, Grainger JR, Chen W, Thymocyte apoptosis drives the intrathymic generation of regulatory T cells. Proc. Natl. Acad. Sci. U.S.A 111, E465–E473 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen W, Frank ME, Jin W, Wahl SM, TGF-beta released by apoptotic T cells contributes to an immunosuppressive milieu. Immunity 14, 715–725 (2001). [DOI] [PubMed] [Google Scholar]
- 45.Hauri-Hohl M, Zuklys S, Hollander GA, Ziegler SF, A regulatory role for TGF-β signaling in the establishment and function of the thymic medulla. Nat. Immunol 15, 554–561 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Marie JC, Liggitt D, Rudensky AY, Cellular mechanisms of fatal early-onset autoimmunity in mice with the T cell-specific targeting of transforming growth factor-beta receptor. Immunity 25, 441–454 (2006). [DOI] [PubMed] [Google Scholar]
- 47.Li MO, Sanjabi S, Flavell RA, Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 25, 455–471 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Marie JC, Letterio JJ, Gavin M, Rudensky AY, TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J. Exp. Med 201, 1061–1067 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takimoto T, Wakabayashi Y, Sekiya T, Inoue N, Morita R, Ichiyama K, Takahashi R, Asakawa M, Muto G, Mori T, Hasegawa E, Shizuya S, Hara T, Nomura M, Yoshimura A, Smad2 and Smad3 are redundantly essential for the TGF-beta-mediated regulation of regulatory T plasticity and Th1 development. J. Immunol 185, 842–855 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Gu AD, Wang Y, Lin L, Zhang SS, Wan YY, Requirements of transcription factor Smad-dependent and -independent TGF-β signaling to control discrete T-cell functions. Proc. Natl. Acad. Sci. U.S.A 109, 905–910 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ouyang W, Beckett O, Ma Q, Li MO, Transforming growth factor-beta signaling curbs thymic negative selection promoting regulatory T cell development. Immunity 32, 642–653 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen W, Konkel JE, Development of thymic Foxp3+ regulatory T cells: TGF-β matters. Eur. J. Immunol 45, 958–965 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McKarns SC, Schwartz RH, Distinct effects of TGF-beta 1 on CD4+ and CD8+ T cell survival, division, and IL-2 production: A role for T cell intrinsic Smad3. J. Immunol 174, 2071–2083 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Konkel JE, Maruyama T, Carpenter AC, Xiong Y, Zamarron BF, Hall BE,Kulkarni AB, Zhang P, Bosselut R, Chen WJ, Control of the development of CD8αα+ intestinal intraepithelial lymphocytes by TGF-β. Nat. Immunol 12, 312–319 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen W, Jin W, Tian H, Sicurello P, Frank M, Orenstein JM, Wahl SM, Requirement for transforming growth factor beta1 in controlling T cell apoptosis. J. Exp. Med 194, 439–454 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wirnsberger G, Mair F, Klein L, Regulatory T cell differentiation of thymocytes does not require a dedicated antigen-presenting cell but is under T cell-intrinsic developmental control. Proc. Natl. Acad. Sci. U.S.A 106, 10278–10283 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Colamatteo A, Carbone F, Bruzzaniti S, Galgani M, Fusco C, Maniscalco GT, Rella FD, de Candia P, De Rosa V, Molecular mechanisms controlling Foxp3 expression in health and autoimmunity: From epigenetic to post-translational regulation. Front. Immunol 10, 3136 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tone Y, Furuuchi K, Kojima Y, Tykocinski ML, Greene MI, Tone M, Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat. Immunol 9, 194–202 (2008). [DOI] [PubMed] [Google Scholar]
- 59.Xu L, Kitani A, Stuelten C, McGrady G, Fuss I, Strober W, Positive and negative transcriptional regulation of the Foxp3 gene is mediated by access and binding of the Smad3 protein to enhancer I. Immunity 33, 313–325 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zheng Y, Josefowicz S, Chaudhry A, Peng XP, Forbush K, Rudensky AY, Role of conserved non-coding DNA elements in the Foxp3 gene in regulatory T-cell fate. Nature 463, 808–812 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wan YY, Chi H, Xie M, Schneider MD, Flavell RA, The kinase TAK1 integrates antigen and cytokine receptor signaling for T cell development, survival and function. Nat. Immunol 7, 851–858 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Xing Y, Wang X, Jameson SC, Hogquist KA, Late stages of T cell maturation in the thymus involve NF-κB and tonic type I interferon signaling. Nat. Immunol 17, 565–573 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Andersen L, Gülich AF, Alteneder M, Preglej T, Orola MJ, Dhele N, Stolz V,Schebesta A, Hamminger P, Hladik A, Floess S, Krausgruber T, Faux T, Andrabi SBA, Huehn J, Knapp S, Sparwasser T, Bock C, Laiho A, Elo LL, Rasool O, Lahesmaa R, Sakaguchi S, Ellmeier W, The transcription factor MAZR/PATZ1 regulates the development of FOXP3+ regulatory T cells. Cell Rep. 29, 4447–4459.e6 (2019). [DOI] [PubMed] [Google Scholar]
- 64.Jang SW, Hwang SS, Kim HS, Kim MK, Lee WH, Hwang SU, Gwak J, Yew SK, Flavell RA, Lee GR, Homeobox protein Hhex negatively regulates Treg cells by inhibiting Foxp3 expression and function. Proc. Natl. Acad. Sci. U.S.A 116, 25790–25799 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, Ziegelbauer J, Yassai M, Li SH, Relland LM, Wise PM, Chen A, Zheng YQ, Simpson PM, Gorski J, Salzman NH, Hessner MJ, Chatila TA, Williams CB, A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity 35, 109–122 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sharabi A, Tsokos MG, Ding Y, Malek TR, Klatzmann D, Tsokos GC, Regulatory T cells in the treatment of disease. Nat. Rev. Drug Discov 17, 823–844 (2018). [DOI] [PubMed] [Google Scholar]
- 67.MacMillan ML, Hippen KL, McKenna DH, Kadidlo D, Sumstad D, DeFor TE, Brunstein CG, Holtan SG, Miller JS, Warlick ED, Weisdorf DJ, Wagner JE, Blazar BR, First-in-human phase 1 trial of induced regulatory T cells for graft-versus-host disease prophylaxis in HLA-matched siblings. Blood Adv. 5, 1425–1436 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hadis U, Wahl B, Schulz O, Hardtke-Wolenski M, Schippers A, Wagner N, Müller W, Sparwasser T, Förster R, Pabst O, Intestinal tolerance requires gut homing and expansion of FoxP3+ regulatory T cells in the lamina propria. Immunity 34, 237–246 (2011). [DOI] [PubMed] [Google Scholar]
- 69.Xu M, Pokrovskii M, Ding Y, Yi R, Au C, Harrison OJ, Galan C, Belkaid Y, Bonneau R,Littman DR, c-MAF-dependent regulatory T cells mediate immunological tolerance to a gut pathobiont. Nature 554, 373–377 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ogawa C, Tone Y, Tsuda M, Peter C, Waldmann H, Tone M, TGF-β–mediated Foxp3 gene expression is cooperatively regulated by Stat5, Creb, and AP-1 through CNS2. J. Immunol 192, 475–483 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sun X, Cui Y, Feng H, Liu H, Liu X, TGF-β signaling controls Foxp3 methylation and T reg cell differentiation by modulating Uhrf1 activity. J. Exp. Med 216, 2819–2837 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fleskens V, Minutti CM, Wu X, Wei P, Pals CEGM, Crae JM, Hemmers S, Groenewold V, Vos H-J, Rudensky A, Pan F, Li H, Zaiss DM, Coffer PJ, Nemo-like kinase drives Foxp3 stability and is critical for maintenance of immune tolerance by regulatory T cells. Cell Rep. 26, 3600–3612.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ansa-Addo EA, Zhang Y, Yang Y, Hussey GS, Howley BV, Salem M, Riesenberg B, Sun S,Rockey DC, Karvar S, Howe PH, Liu B, Li Z, Membrane-organizing protein moesin controls Treg differentiation and antitumor immunity via TGF-β signaling. J. Clin. Invest 127, 1321–1337 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gottschalk RA, Corse E, Allison JP, TCR ligand density and affinity determine peripheral induction of Foxp3 in vivo. J. Exp. Med 207, 1701–1711 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hill JA, Hall JA, Sun CM, Cai Q, Ghyselinck N, Chambon P, Belkaid Y, Mathis D, Benoist C, Retinoic acid enhances Foxp3 induction indirectly by relieving inhibition from CD4+CD44hi Cells. Immunity 29, 758–770 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ansa-Addo EA, Huang H-C, Riesenberg B, lamsawat S, Borucki D, Nelson MH, Nam JH, Chung D, Paulos CM, Liu B, Yu X-Z, Philpott C, Howe PH, Li Z, RNA binding protein PCBP1 is an intracellular immune checkpoint for shaping T cell responses in cancer immunity. Sci. Adv 6, eaaz3865 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kim BS, Kim IK, Park YJ, Kim YS, Kim YJ, Chang WS, Lee YS, Kweon MN, Chung Y, Kang CY, Conversion of Th2 memory cells into Foxp3+ regulatory T cells suppressing Th2-mediated allergic asthma. Proc. Natl. Acad. Sci. U.S.A 107, 8742–8747 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Amarnath S, Mangus CW, Wang JCM, Wei F, He A, Kapoor V, Foley JE, Massey PR, Felizardo TC, Riley JL, Levine BL, June CH, Medin JA, Fowler DH, The PDL1-PD1 axis converts human TH1 cells into regulatory T cells. Sci. Transl. Med 3, 111ra120 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, Chi H, HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med 208, 1367–1376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hang S, Paik D, Yao L, Kim E, Trinath J, Lu J, Ha S, Nelson BN, Kelly SP, Wu L, Zheng Y, Longman RS, Rastinejad F, Devlin AS, Krout MR, Fischbach MA, Littman DR, Huh JR, Bile acid metabolites control TH17 and Treg cell differentiation. Nature 576, 143–148 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tran DQ, Ramsey H, Shevach EM, Induction of FOXP3 expression in naive human CD4+FOXP3 T cells by T-cell receptor stimulation is transforming growth factor-beta dependent but does not confer a regulatory phenotype. Blood 110, 2983–2990 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Riquelme P, Haarer J, Kammler A, Walter L, Tomiuk S, Ahrens N, Wege AK, Goecze I,Zecher D, Banas B, Spang R, Fändrich F, Lutz MB, Sawitzki B, Schlitt HJ, Ochando J,Geissler EK, Hutchinson JA, TIGIT+ iTregs elicited by human regulatory macrophages control T cell immunity. Nat. Commun 9, 2858 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Alvarez-Salazar EK, Cortes-Hernandez A, Arteaga-Cruz S, Alberu-Gomez J, Soldevila G, Large-scale generation of human allospecific induced tregs with functional stability for use in immunotherapy in transplantation. Front. Immunol 11,375 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, Klein-Hessling S, Serfling E, Hamann A, Huehn J, Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 5, e38 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koenecke C, Czeloth N, Bubke A, Schmitz S, Kissenpfennig A, Malissen B, Huehn J,Ganser A, Forster R, Prinz I, Alloantigen-specific de novo-induced Foxp3+ Treg revert in vivo and do not protect from experimental GVHD. Eur. J. Immunol 39, 3091–3096 (2009). [DOI] [PubMed] [Google Scholar]
- 86.Feng Y, Arvey A, Chinen T, van der Veeken J, Gasteiger G, Rudensky AY, Control of the inheritance of regulatory T cell identity by a cis element in the Foxp3 locus. Cell 158, 749–763 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li X, Liang Y, LeBlanc M, Benner C, Zheng Y, Function of a Foxp3 cis-element in protecting regulatory T cell identity. Cell 158, 734–748 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yue X, Trifari S, Äijö T, Tsagaratou A, Pastor WA, Zepeda-Martínez JA, Lio CWJ, Li X, Huang Y, Vijayanand P, Lähdesmäki H, Rao A, Control of Foxp3 stability through modulation of TET activity. J. Exp. Med 213, 377–397 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen Q, Kim YC, Laurence A, Punkosdy GA, Shevach EM, IL-2 controls the stability of Foxp3 expression in TGF-beta-induced Foxp3+ T cells in vivo. J. Immunol 186, 6329–6337 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kim J, Hope CM, Perkins GB, Stead SO, Scaffidi JC, Kette FD, Carroll RP, Barry SC, Coates PT, Rapamycin and abundant TCR stimulation are required for the generation of stable human induced regulatory T cells. Clin. Transl. Immunol 9, e1223 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nikolouli E, Hardtke-Wolenski M, Hapke M, Beckstette M, Geffers R, Floess S, Jaeckel E, Huehn J, Alloantigen-induced regulatory T cells generated in presence of Vitamin C display enhanced stability of foxp3 expression and promote skin allograft acceptance. Front. Immunol 8, 748 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lu L, Ma J, Li Z, Lan Q, Chen M, Liu Y, Xia Z, Wang J, Han Y, Shi W, Quesniaux V,Ryffel B, Brand D, Li B, Liu Z, Zheng SG, All-trans retinoic acid promotes TGF-β-Induced Tregs via histone modification but not DNA demethylation on Foxp3 gene locus. PLOS ONE 6, e24590 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Konkel JE, Zhang D, Zanvit P, Chia C, Zangarle-Murray T, Jin W, Wang S, Chen WJ, Transforming growth factor-β signaling in regulatory T cells controls T helper-17 cells and tissue-specific immune responses. Immunity 46, 660–674 (2017). [DOI] [PubMed] [Google Scholar]
- 94.Śledzińska A, Hemmers S, Mair F, Gorka O, Ruland J, Fairbairn L, Nissler A, MQller W,Waisman A, Becher B, Buch T, TGF-β signalling is required for CD4+ T cell homeostasis but dispensable for regulatory T cell function. PLoS Biol. 11, e1001674 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, Campbell DJ, The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol 10, 595–602 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Neumann C, Blume J, Roy U, Teh PP, Vasanthakumar A, Beller A, Liao Y, Heinrich F, Arenzana TL, Hackney JA, Eidenschenk C, Gálvez EJC, Stehle C, Heinz GA, Maschmeyer P, Sidwell T, Hu Y, Amsen D, Romagnani C, Chang H-D, Kruglov A, Mashreghi M-F, Shi W, Strowig T, Rutz S, Kallies A, Scheffold A, c-Maf-dependent Treg cell control of intestinal TH17 cells and IgA establishes host-microbiota homeostasis. Nat. Immunol 20, 471–481 (2019). [DOI] [PubMed] [Google Scholar]
- 97.Choi G, Na H, Kuen DS, Kim BS, Chung Y, Autocrine TGF-β 1 maintains the stability of Foxp3+ regulatory T cells via IL-12Rβ2 downregulation. Biomolecules 10, 819 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Priyadharshini B, Loschi M, Newton RH, Zhang JW, Finn KK, Gerriets VA, Huynh A, Rathmell JC, Blazar BR, Turka LA, Cutting edge: TGF-β and phosphatidylinositol 3-kinase signals modulate distinct metabolism of regulatory T cell subsets. J. Immunol 201 , 2215–2219 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen X, Feng L, Li S, Long D, Shan J, Li Y, TGF-β1 maintains Foxp3 expression and inhibits glycolysis in natural regulatory T cells via PP2A-mediated suppression of mTOR signaling. Immunol. Lett 226, 31–37 (2020). [DOI] [PubMed] [Google Scholar]
- 100.Xu X, Zheng L, Yuan Q, Zhen G, Crane JL, Zhou X, Cao X, Transforming growth factor-β in stem cells and tissue homeostasis. Bone Res. 6, 2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pakyari M, Farrokhi A, Maharlooei MK, Ghahary A, Critical role of transforming growth factor beta in different phases of wound healing. Adv. Wound Care 2, 215–224 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moreau JM, Gouirand V, Rosenblum MD, T cell adhesion in healthy and inflamed skin. JID Innovations 1, 100014 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mackay LK, Rahimpour A, Ma JZ, Collins N, Stock AT, Hafon ML, Vega-Ramos J, Lauzurica P, Mueller SN, Stefanovic T, Tscharke DC, Heath WR, Inouye M,Carbone FR, Gebhardt T, The developmental pathway for CD103+CD8+ tissue-resident memory T cells of skin. Nat. Immunol 14, 1294–1301 (2013). [DOI] [PubMed] [Google Scholar]
- 104.Hirai T, Yang Y, Zenke Y, Li H, Chaudhri VK, De La Cruz Diaz JS, Zhou PY,Nguyen BA-T, Bartholin L, Workman CJ, Griggs DW, Vignali DAA, Singh H,Masopust D, Kaplan DH, Competition for active TGFβ cytokine allows for selective retention of antigen-specific tissue-resident memory T cells in the epidermal niche. Immunity 54, 84–98.e5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Moreau JM, Dhariwala MO, Gouirand V, Boda DP, Boothby IC, Lowe MM, Cohen JN, Macon CE, Leech JM, Kalekar LA, Scharschmidt TC, Rosenblum MD, Regulatory T cells promote innate inflammation after skin barrier breach via TGF-β activation. Sci. Immunol 6, eabg2329 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ferreira C, Barros L, Baptista M, Blankenhaus B, Barros A, Figueiredo-Campos P, Konjar š., Lainé A, Kamenjarin N, Stojanovic A, Cerwenka A, Probst HC, Marie JC, Veldhoen M, Type 1 Treg cells promote the generation of CD8+ tissue-resident memory T cells. Nat. Immunol 21,766–776 (2020). [DOI] [PubMed] [Google Scholar]
- 107.Letterio JJ, Geiser AG, Kulkarni AB, Dang H, Kong L, Nakabayashi T, Mackall CL, Gress RE, Roberts AB, Autoimmunity associated with TGF-beta1-deficiency in mice is dependent on MHC class II antigen expression. J. Clin. Invest 98, 2109–2119 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lucas PJ, Kim SJ, Melby SJ, Gress RE, Disruption of T cell homeostasis in mice expressing a T cell-specific dominant negative transforming growth factor beta II receptor. J. Exp. Med 191,1187–1196 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gorelik L, Flavell RA, Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity 12, 171–181 (2000). [DOI] [PubMed] [Google Scholar]
- 110.Li MO, Wan YY, Flavell RA, T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 26, 579–591 (2007). [DOI] [PubMed] [Google Scholar]
- 111.Pesu M, Watford WT, Wei L, Xu L, Fuss I, Strober W, Andersson J, Shevach EM, Quezado M, Bouladoux N, Roebroek A, Belkaid Y, Creemers J, O’Shea JJ, T-cell-expressed proprotein convertase furin is essential for maintenance of peripheral immune tolerance. Nature 455, 246–250 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO, Autocrine transforming growth factor-β1 promotes in vivo Th17 cell differentiation. Immunity 34, 396–408 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Edwards JP, Fujii H, Zhou AX, Creemers J, Unutmaz D, Shevach EM, Regulation of the expression of GARP/latent TGF-β1 complexes on mouse T cells and their role in regulatory T cell and Th17 differentiation. J. Immunol 190, 5506–5515 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Worthington JJ, Kelly A, Smedley C, Bauché D, Campbell S, Marie JC, Travis MA, Integrin αvβ8-mediated TGF-β activation by effector regulatory T cells is essential for suppression of T-cell-mediated inflammation. Immunity 42, 903–915 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Salem M, Wallace C, Velegraki M, Li A, Ansa-Addo E, Metelli A, Kwon H, Riesenberg B,Wu B, Zhang Y, Guglietta S, Sun S, Liu B, Li Z, GARP dampens cancer immunity by sustaining function and accumulation of regulatory T cells in the colon. Cancer Res. 79, 1178–1190 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhang N, Bevan MJ, TGF-β signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat. Immunol 13, 667–673 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang N, Bevan MJ, Transforming growth factor-β signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 39, 687–696 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Turner JA, Stephen-Victor E, Wang S, Rivas MN, Abdel-Gadir A, Harb H, Cui Y, Fanny M, Charbonnier L-M, Fong JJH, Benamar M, Wang L, Burton OT, Bansal K, Bry L, Zhu C, Li Q-Z, Clement RL, Oettgen HC, Crestani E, Rachid R, Sage PT, Chatila TA, Regulatory T cell-derived TGF-β1 controls multiple checkpoints governing allergy and autoimmunity. Immunity 53, 1202–1214.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Velegraki M, Salem M, Ansa-Addo EA, Wu BX, Li Z, Autocrine transforming growth factor β1 in regulatory T cell biology-gone but not missed. Immunity 54, 395–396 (2021). [DOI] [PubMed] [Google Scholar]
- 120.Choi G, Kim BS, Chang JH, Chung Y, Defining the role of transforming growth factor β1 in Foxp3+ T regulatory cells. Immunity 54, 393–394 (2021). [DOI] [PubMed] [Google Scholar]
- 121.Nakamura K, Kitani A, Strober W, Cell contact-dependent immunosuppression by CD4+CD25+ regulatory T cells is mediated by cell surface-bound transforming growth factor beta. J. Exp. Med 194, 629–644 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Powrie F, Carlino J, Leach MW, Mauze S, Coffman RL, A critical role for transforming growth factor-beta but not interleukin 4 in the suppression of T helper type 1-mediated colitis by CD45RB(low) CD4+ T cells. J. Exp. Med 183, 2669–2674 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Read S, Malmstrom V, Powrie F, Cytotoxic T lymphocyte-associated antigen 4 plays an essential role in the function of CD25+CD4+ regulatory cells that control intestinal inflammation. J. Exp. Med 192, 295–302 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fahlén L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, Powrie F, T cells that cannot respond to TGF-beta escape control by CD4+CD25+ regulatory T cells. J. Exp. Med 201, 737–746 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Nakamura K, Kitani A, Fuss I, Pedersen A, Harada N, Nawata H, Strober W, TGF-β 1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J. Immunol 172, 834–842 (2004). [DOI] [PubMed] [Google Scholar]
- 126.Chen ML, Pittet MJ, Gorelik L, Flavell RA, Weissleder R, von Boehmer H, Khazaie K, Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-β signals in vivo. Proc. Natl. Acad. Sci. U.S.A 102, 419–424 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Takahashi T, Kuniyasu Y, Toda M, Sakaguchi N, Itoh M, Iwata M, Shimizu J, Sakaguchi S, Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: Induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol 10, 1969–1980 (1998). [DOI] [PubMed] [Google Scholar]
- 128.Thornton AM, Shevach EM, CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J. Exp. Med 188, 287–296 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Piccirillo CA, Letterio JJ, Thornton AM, McHugh RS, Mamura M, Mizuhara H, Shevach EM, CD4+CD25+ regulatory T cells can mediate suppressor function in the absence of transforming growth factor β1 production and responsiveness. J. Exp. Med 196, 237–246 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kullberg MC, Hay V, Cheever AW, Mamura M, Sher A, Letterio JJ, Shevach EM,Piccirillo CA, TGFβ1 production by CD4+CD25+ regulatory T cells is not essential for suppression of intestinal inflammation. Eur. J. Immunol 35, 2886–2895 (2005). [DOI] [PubMed] [Google Scholar]
- 131.Mamura M, Lee WK, Sullivan TJ, Felici A, Sowers AL, Allison JP, Letterio JJ, CD28 disruption exacerbates inflammation in Tgf-β1−/− mice: In vivo suppression by CD4+CD25+ regulatory T cells independent of autocrine TGF-β1. Blood 103, 4594–4601 (2004). [DOI] [PubMed] [Google Scholar]
- 132.Town T, Breunig JJ, Sarkisian MR, Spilianakis C, Ayoub AE, Liu X, Ferrandino AF, Gallagher AR, Li MO, Rakic P, Flavell RA, The stumpy gene is required for mammalian ciliogenesis. Proc. Natl. Acad. Sci. U.S.A 105, 2853–2858 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Azhar M, Yin M, Bommireddy R, Duffy JJ, Yang J, Pawlowski SA, Boivin GP, Engle SJ, Sanford LP, Grisham C, Singh RR, Babcock GF, Doetschman T, Generation of mice with a conditional allele for transforming growth factor beta 1 gene. Genesis 47, 423–431 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Godfrey VL, Wilkinson JE, Rinchik EM, Russell LB, Fatal lymphoreticular disease in the scurfy (sf) mouse requires T cells that mature in a sf thymic environment: potential model for thymic education. Proc. Natl. Acad. Sci. U.S.A 88, 5528–5532 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bennett CL, Christie J, Ramsdell F, Brunkow ME, Ferguson PJ, Whitesell L, Kelly TE,Saulsbury FT, Chance PF, Ochs HD, The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet 27, 20–21 (2001). [DOI] [PubMed] [Google Scholar]
- 136.Brunkow ME, Jeffery EW, Hjerrild KA, Paeper B, Clark LB, Yasayko SA, Wilkinson JE, Galas D, Ziegler SF, Ramsdell F, Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet 27, 68–73 (2001). [DOI] [PubMed] [Google Scholar]
- 137.Worthington JJ, Czajkowska BI, Melton AC, Travis MA, Intestinal dendritic cells specialize to activate transforming growth factor-β and induce Foxp3+ regulatory T cells via integrin αVβ8. Gastroenterology 141,1802–1812 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cuende J, Liénart S, Dedobbeleer O, van der Woning B, De Boeck G, Stockis J,Huygens C, Colau D, Somja J, Delvenne P, Hannon M, Baron F, Dumoutier L, Renauld J-C, De Haard H, Saunders M, Coulie PG, Lucas S, Monoclonal antibodies against GARP/TGF-β1 complexes inhibit the immunosuppressive activity of human regulatory T cells in vivo. Sci. Transl. Med 7, 284ra256 (2015). [DOI] [PubMed] [Google Scholar]
- 139.Stockis J, Liénart S, Colau D, Collignon A, Nishimura SL, Sheppard D, Coulie PG, Lucas S, Blocking immunosuppression by human Tregs in vivo with antibodies targeting integrin αVβ8. Proc. Natl. Acad. Sci. U.S.A 114, E10161–E10168 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.de Streel G, Bertrand C, Chalon N, Liénart S, Bricard O, Lecomte S, Devreux J, Gaignage M, de Boeck G, Mariën L, van de Walle I, van der Woning B, Saunders M, de Haard H, Vermeersch E, Maes W, Deckmyn H, Coulie PG, van Baren N, Lucas S, Selective inhibition of TGF-β1 produced by GARP-expressing Tregs overcomes resistance to PD-1/PD-L1 blockade in cancer. Nat. Commun 11 , 4545 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Frangogiannis N, Transforming growth factor–β in tissue fibrosis. J. Exp. Med 217, e20190103 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rice LM, Padilla CM, McLaughlin SR, Mathes A, Ziemek J, Goummih S, Nakerakanti S, York M, Farina G, Whitfield ML, Spiera RF, Christmann RB, Gordon JK, Weinberg J, Simms RW, Lafyatis R, Fresolimumab treatment decreases biomarkers and improves clinical symptoms in systemic sclerosis patients. J. Clin. Invest 125, 2795–2807 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kalekar LA, Cohen JN, Prevel N, Sandoval PM, Mathur AN, Moreau JM, Lowe MM, Nosbaum A, Wolters PJ, Haemel A, Boin F, Rosenblum MD, Regulatory T cells in skin are uniquely poised to suppress profibrotic immune responses. Sci. Immunol 4, eaaw2910 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Moyé S, Bormann T, Maus R, Sparwasser T, Sandrock I, Prinz I, Warnecke G, Welte T, Gauldie J, Kolb M, Maus UA, Regulatory T cells limit pneumococcus-induced exacerbation of lung fibrosis in mice. J. Immunol 204, 2429–2438 (2020). [DOI] [PubMed] [Google Scholar]
- 145.Taylor AE, Carey AN, Kudira R, Lages CS, Shi T, Lam S, Karns R, Simmons J, Shanmukhappa K, Almanan M, Chougnet CA, Miethke AG, Interleukin 2 promotes hepatic regulatory T cell responses and protects from biliary fibrosis in murine sclerosing cholangitis. Hepatology 68, 1905–1921 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Claassen MA, de Knegt RJ, Tilanus HW, Janssen HL, Boonstra A, Abundant numbers of regulatory T cells localize to the liver of chronic hepatitis C infected patients and limit the extent of fibrosis. J. Hepatol 52, 315–321 (2010). [DOI] [PubMed] [Google Scholar]
- 147.Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, Hall JA, Yagi R, Naik S, Bhairavabhotla R, Paul WE, Bosselut R, Wei G, Zhao K, Oukka M, Zhu J, Belkaid Y, GATA3 controls Foxp3+ regulatory T cell fate during inflammation in mice. J. Clin. Invest 121,4503–4515 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Xiong S, Guo R, Yang Z, Xu L, du L, Li R, Xiao F, Wang Q, Zhu M, Pan X, Treg depletion attenuates irradiation-induced pulmonary fibrosis by reducing fibrocyte accumulation, inducing Th17 response, and shifting IFN-γ, IL-12/IL-4, IL-5 balance. Immunobiology 220, 1284–1291 (2015). [DOI] [PubMed] [Google Scholar]
- 149.Liu F, Liu J, Weng D, Chen Y, Song L, He Q, Chen J, CD4+CD25+Foxp3+ regulatory T cells depletion may attenuate the development of silica-induced lung fibrosis in mice. PLOS ONE 5, e15404 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bansal SS, Ismahil MA, Goel M, Zhou G, Rokosh G, Hamid T, Prabhu SD, Dysfunctional and proinflammatory regulatory T-lymphocytes are essential for adverse cardiac remodeling in ischemic cardiomyopathy. Circulation 139, 206–221 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mu Y, Zhang J, Liu Y, Ma J, Jiang D, Zhang X, Yi X, Cheng K, Shen S, Yang Y, Zhuang R, Zhang Y, CD226 deficiency on regulatory T cells aggravates renal fibrosis via up-regulation of Th2 cytokines through miR-340. J. Leukoc. Biol 107, 573–587 (2020). [DOI] [PubMed] [Google Scholar]
- 152.Kitani A, Fuss I, Nakamura K, Kumaki F, Usui T, Strober W, Transforming growth factor (TGF)-β1-producing regulatory T cells induce Smad-mediated interleukin 10 secretion that facilitates coordinated immunoregulatory activity and amelioration of TGF-β1-mediated fibrosis. J. Exp. Med 198, 1179–1188 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Batlle E, Massague J, Transforming growth factor-β signaling in immunity and cancer. Immunity 50, 924–940 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel III EE, Koeppen H, Astarita JL, Cubas R, Jhunjhunwala S, Banchereau R, Yang Y, Guan Y,Chalouni C, Ziai J, §enbabaoğlu Y, Santoro S, Sheinson D, Hung J, Giltnane JM, Pierce AA, Mesh K, Lianoglou S, Riegler J, Carano RAD, Eriksson P, Höglund M, Somarriba L, Halligan DL, van der Heijden MS, Loriot Y, Rosenberg JE, Fong L, Mellman I, Chen DS, Green M, Derleth C, Fine GD, Hegde PS, Bourgon R, Powles T, TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Cañellas A, Hernando-Momblona X, Byrom D,Matarin JA, Calon A, Rivas EI, Nebreda AR, Riera A, Attolini CSO, Batlle E, TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 554, 538–543 (2018). [DOI] [PubMed] [Google Scholar]
- 156.Zheng L, Qin S, Si W, Wang A, Xing B, Gao R, Ren X, Wang L, Wu X, Zhang J, Wu N, Zhang N, Zheng H, Ouyang H, Chen K, Bu Z, Hu X, Ji J, Zhang Z, Pan-cancer single-cell landscape of tumor-infiltrating T cells. Science 374, abe6474 (2021). [DOI] [PubMed] [Google Scholar]
- 157.Lainé A, Labiad O, Hernandez-Vargas H, This S, Sanlaville A, Léon S, Dalle S,Sheppard D, Travis MA, Paidassi H, Marie JC, Regulatory T cells promote cancer immune-escape through integrin αvβ8-mediated TGF-β activation. Nat. Commun 12, 6228 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Metelli A, Wu BX, Fugle CW, Rachidi S, Sun S, Zhang Y, Wu J, Tomlinson S, Howe PH, Yang Y, Garrett-Mayer E, Liu B, Li Z, Surface expression of TGFβ docking receptor GARP promotes oncogenesis and immune tolerance in breast cancer. Cancer Res. 76, 7106–7117 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Travis MA, Sheppard D, TGF-β activation and function in immunity. Annu. Rev. Immunol 32, 51–82 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Thomas DA, Massague J, TGF-β directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell 8, 369–380 (2005). [DOI] [PubMed] [Google Scholar]
- 161.Gorelik L, Constant S, Flavell RA, Mechanism of transforming growth factor β-induced inhibition of T helper type 1 differentiation. J. Exp. Med 195, 1499–1505 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ramalingam R, Larmonier CB, Thurston RD, Midura-Kiela MT, Zheng SG, Ghishan FK, Kiela PR, Dendritic cell-specific disruption of TGF-β receptor II leads to altered regulatory T cell phenotype and spontaneous multiorgan autoimmunity. J. Immunol 189, 3878–3893 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Viel S, Marçais A, Guimaraes FS-F, Loftus R, Rabilloud J, Grau M, Degouve S, Djebali S, Sanlaville A, Charrier E, Bienvenu J, Marie JC, Caux C, Marvel J, Town L, Huntington ND, Bartholin L, Finlay D, Smyth MJ, Walzer T, TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci. Signal 9, ra19 (2016). [DOI] [PubMed] [Google Scholar]
- 164.Gong D, Shi W, Yi SJ, Chen H, Groffen J, Heisterkamp N, TGFβ signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol. 13, 31 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhang M, Pan X, Fujiwara K, Jurcak N, Muth S, Zhou J, Xiao Q, Li A, Che X, Li Z, Zheng L, Pancreatic cancer cells render tumor-associated macrophages metabolically reprogrammed by a GARP and DNA methylation-mediated mechanism. Signal Transduct. Target. Ther 6, 366 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chakravarthy A, Khan L, Bensler NP, Bose P, De Carvalho DD, TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun 9, 4692 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Seed RI, Kobayashi K, Ito S, Takasaka N, Cormier A, Jespersen JM, Publicover J, Trilok S, Combes AJ, Chew NW, Chapman J, Krummel MF, Lou J, Marks J, Cheng Y, Baron JL, Nishimura SL, A tumor-specific mechanism of Treg enrichment mediated by the integrin αvβ8. Sci. Immunol 6, eabf0558 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gabriely G, da Cunha AP, Rezende RM, Kenyon B, Madi A, Vandeventer T, Skillin N, Rubino S, Garo L, Mazzola MA, Kolypetri P, Lanser AJ, Moreira T, Faria AMC, Lassmann H, Kuchroo V, Murugaiyan G, Weiner HL, Targeting latency-associated peptide promotes antitumor immunity. Sci. Immunol 2, eaaj1738 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Li S, Liu M, Do MH, Chou C, Stamatiades EG, Nixon BG, Shi W, Zhang X, Li P, Gao S,Capistrano KJ, Xu H, Cheung NKV, Li MO, Cancer immunotherapy via targeted TGF-β signalling blockade in TH cells. Nature 587, 121–125 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Liu M, Kuo F, Capistrano KJ, Kang D, Nixon BG, Shi W, Chou C, Do MH, Stamatiades EG, Gao S, Li S, Chen Y, Hsieh JJ, Hakimi AA, Taniuchi I, Chan TA, Li MO, TGF-β suppresses type 2 immunity to cancer. Nature 587, 115–120 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kim BG, Malek E, Choi SH, Ignatz-Hoover JJ, Driscoll JJ, Novel therapies emerging in oncology to target the TGF-β pathway. J. Hematol. Oncol 14, 55 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Strauss J, Gatti-Mays ME, Cho BC, Hill A, Salas S, McClay E, Redman JM, Sater HA, Donahue RN, Jochems C, Lamping E, Burmeister A, Marté JL, Cordes LM, Bilusic M,Karzai F, Ojalvo LS, Jehl G, Rolfe PA, Hinrichs CS, Madan RA, Schlom J, Gulley JL, Bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in patients with human papillomavirus-associated malignancies. J. Immunother. Cancer 8, e001395 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lan Y, Zhang D, Xu C, Hance KW, Marelli B, Qi J, Yu H, Qin G, Sircar A, Hernández VM, Jenkins MH, Fontana RE, Deshpande A, Locke G, Sabzevari H,Radvanyi L, Lo KM, Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci. Transl. Med 10, eaan5488 (2018). [DOI] [PubMed] [Google Scholar]