

Abstract
Despite the success of Sonogashira coupling for the synthesis of arylalkynes and conjugated enynes, the engagement of unactivated alkyl halides in such reactions remains historically challenging. We report herein a strategy that merges Cu-catalyzed alkyne transfer with the aryl radical activation of carbon–halide bonds to enable a general approach for the coupling of alkyl iodides with terminal alkynes. This unprecedented Sonogashira-type cross-coupling reaction tolerates a broad range of functional groups and has been applied to the late-stage cross-coupling of densely functionalized pharmaceutical agents as well as the synthesis of positron emission tomography tracers.
Keywords: copper, alkyne, Sonogashira coupling, aryl radical, halogen abstraction
Graphical Abstract

INTRODUCTION
As one of the most valuable functional groups in organic chemistry, alkynes are ubiquitous in a diverse range of natural products, pharmaceuticals, agrochemicals, and organic materials.1 In addition, alkynes have been widely used in Cu-catalyzed and strain-promoted azide–alkyne cycloaddition reactions, that is, click chemistry, for the conjugation of bioactive molecules.2 Moreover, alkynes are vital synthetic precursors for the construction of a wide variety of functionalities, including alkenes, aldehydes, acids, and heterocycles.3 Recent work has shown that alkyne-tagged small molecules are promising agents for Raman imaging, as the Raman bands of alkynyl groups do not overlap with Raman scattering from endogenous molecules in live cells.4 Due to the importance of alkyne-containing molecules, the development of new methods that can efficiently install alkynyl groups into functionalized organic molecules has been the subject of numerous studies.5,6
Sonogashira coupling reactions,7 in which aryl and vinyl (pseudo)halides react with terminal alkynes under Pd-catalyzed, Cu-cocatalyzed conditions, are among the most efficient and modular approaches for the synthesis of alkynes.8 Despite the success of Sonogashira coupling for constructing arylalkynes and conjugated enynes, the engagement of unactivated alkyl halides in such reactions remains a formidable challenge.9 This limitation is largely due to the sluggish oxidative addition of alkyl halides and the tendency of alkylpalladium intermediates to undergo the competing β-hydrogen elimination reactions.10 The classic reaction of acetylide ions with alkyl halides suffers from poor functional group tolerance. Few methods allow for the coupling of unactivated and β-hydrogen bearing alkyl halides with terminal alkynes (Figure 1a). Seminal work by Fu11 and Glorius12 showed that the use of sterically bulky N-heterocyclic carbene ligands facilitated the Pd-catalyzed Sonogashira coupling of unactivated alkyl halides. Hu13 and Liu14 have later reported that the analogous reactions could be catalyzed by Ni complexes using pincer and pyridine bisoxazoline ligands, respectively. Very recently, based on the pioneering work of Fu and Peters,15 and Hwang,16 the Lalic group reported a photoinduced, Cu-catalyzed approach for coupling unactivated alkyl iodides with terminal alkynes.17 Nonetheless, the generality of these elegant precedents remain rather limited, due in part to the requirement for strong bases, specially designed ligands, or the high reactivity of photoexcited metal species. Indeed, few of these methods have been applied to the cross-coupling of densely functionalized molecules.
Figure 1.

Development of a new approach for the cross-coupling of terminal alkynes with unactivated alkyl halides.
Our group has recently shown that merging the unique reactivity of aryl radicals with copper catalysis could enable the cross coupling of hitherto challenging alkyl electrophiles with organozinc reagents.18 This approach complements the elegant work by MacMillan19 and Leonori,20 who have demonstrated that silyl radicals and α-amino alkyl radicals, respectively, could activate alkyl halides for Cu catalysis. More specifically, our strategy is inspired by the underexplored reactions between aryl radicals with alkyl iodides to generate alkyl radicals at rates approaching the diffusion-controlled limit (k = 109 M−1 s−1).21 We reasoned that combining the unique ability of Cu catalysts to generate aryl radicals with arenediazonium salts, as in the Sandmeyer reactions,22 with the ability of aryl radicals to activate alkyl iodides23 would provide a versatile platform for Cu-catalyzed cross-coupling reactions. This previously unexplored approach has promoted a Cu-catalyzed Negishi-type difluoromethylation reaction (Figure 1b).18b We recently questioned whether this aryl radical activation strategy could be translated to the Sonogashira-type cross-coupling, thus enabling a general approach to the construction of C(sp3)–C(sp) moieties. We herein disclose the successful execution of these ideas and present a broadly applicable protocol for the cross-coupling of unactivated alkyl iodides with terminal alkynes under mild conditions (Figure 1c).
RESULTS AND DISCUSSION
Mechanistically, we reason that CuI catalyst 1 reacts with terminal alkyne 2, in the presence of base 3, to form [CuI-acetylide] species 4 (Figure 2).24 Subsequent single electron transfer (SET) from 4 to diazonium salt 5, followed by the extrusion of N2, produces aryl radical 6 and [CuII-acetylide] intermediate 7.25 The feasibility of this SET step was supported by the observation of an irreversible reduction peak at −0.26 V [vs SCE in dimethyl sulfoxide (DMSO)] for a diazonium salt and a peak at −0.96 V (vs SCE in DMSO) for an in situ generated CuI-acetylide species (Figure S1). Fast iodine abstraction from alkyl iodide 8 by the aryl radical 6 forms aryl iodide 9 and alkyl radical 10. The latter reacts with the [CuII-acetylide] species 7 to generate a formal organocopper(III) species 11,26 which reductively eliminates to give the desired alkyne coupling product 12.27 The reductive elimination reaction also regenerates the CuI catalyst, thus closing the catalytic cycle. We realized that a few challenges could be associated with this hypothesis. First, the formation rate of CuI-acetylide species 4 should be sufficiently high to match the fast reaction of a CuI complex with a diazonium salt. Additionally, the formation of dimerized alkynes under oxidative conditions, that is, Glaser coupling,28 might compete with the cross-coupling pathway. Moreover, the known cross-coupling of the diazonium salts with the alkynes must be minimized.29 We postulated that these challenges could be addressed by tuning the steric and electronic properties of the diazonium salts.
Figure 2.

Reaction design of aryl-radical-enabled, Cu-catalyzed Sonogashira-type cross-coupling.
Reaction Optimization.
A survey of a various combinations of different Cu catalysts, ligands, bases, and arenediazonium salts identified the optimized conditions, as shown in Table 1, entry 1 (see Supporting Information for other conditions studied). Thus, in the presence of [Cu(CH3CN)4]-BF4 as the catalyst, 2,2′; 6′,2″-terpyridine (terpyridine) as the ligand, and potassium carbonate as the base, diazonium salt 14 facilitated the cross-coupling of alkyl iodide 13 with phenylacetylene to afford the desired alkyne product 15 in 91% yield at 50 °C. Consistent with our previous work on the aryl-radical-enabled difluoromethylation reaction,18b an electron-rich and sterically hindered diazonium salt 14 was the optimal promoter for this alkynylation reaction, whereas electron-deficient or less sterically hindered ones were less effective (entry 2–3). We hypothesize that the steric hindrance of the thus-produced aryl radical prevented its direct coupling with the alkyne. Solvent screening revealed that this transformation was most efficient in DMSO, whereas the use of other standard organic solvents decreased the yield of the desired products (entries 4 and 5). Cu salts with weakly coordinating ligands were preferred for this coupling reaction, presumably by facilitating the formation of the copper acetylide species (entries 6 and 7). The commercially available terpyridine ligand was found to be the most effective ligand, while other bidentate ligands, including bipyridine and phenanthroline, had deleterious effects on the coupling reactions (entries 8 and 9). Previous work by Lalic suggested that the use of tridentate ligands could help prevent the undesired alkyne polymerization pathway.17 The base also played an important role in this reaction, with potassium carbonate being uniquely effective when compared with other commonly employed organic and inorganic bases (entries 10–12). Notably, although Cu-catalyzed Glaser coupling is known to occur under aerobic conditions, this aryl-radical-enabled protocol could afford the product in synthetic useful yield under an air atmosphere (entry 13). The desired coupling product could be formed in good yield with lower catalyst loading. (entries 14 and 15). As expected, no coupling products were obtained when the reaction was conducted without a copper catalyst, diazonium salt, or base (entries 16–18). The alkyl iodide remained intact in the absence of the diazonium salt, consistent with the Cu catalyst alone not being reactive toward an unactivated alkyl iodide.
Table 1.
Reaction Optimization for Aryl-Radical-Enabled Sonogashira Coupling of Alkyl Iodidesa

| |||||
|---|---|---|---|---|---|
| entry | variations | yield (%)b | entry | variations | yield (%)b |
| 1 | none | 91 | 10 | Na2CO3 as base | N.D. |
| 2 | 4-methoxy-arenediazonium salt | 30 | 11 | K3PO4 as base | 19 |
| 3 | 4-bromo-arenediazonium salt | N.D. | 12 | Et3N as base | N.D. |
| 4 | acetonitrile as solvent | 69 | 13 | under air | 60 |
| 5 | THF as solvent | 15 | 14 | 20 mol % Cu catalyst and ligand | 79 |
| 6 | CuBr as catalyst | 43 | 15 | 15 mol % Cu catalyst and ligand | 65 |
| 7 | CuI as catalyst | 48 | 16 | no Cu catalyst | N.D. |
| 8 | 2,2’-bipyridine as ligand | 12 | 17 | no diazonium salt | N.D. |
| 9 | 1,10-phenanthroline as ligand | 8 | 18 | no ligand | N.D. |
Reactions were conducted using 13 (0.050 mmol, 1.0 equiv), diazonium salt (0.10 mmol, 2.0 equiv), phenylacetylene (0.055 mmol, 1.1 equiv), potassium carbonate (0.15 mmol, 3 equiv), [Cu(CH3CN)4]BF4 (25 mol %), and ligand (25 mol %) in DMSO at 50 °C.
Yields were determined by GC with 1-ethylnaphthalene as the internal standard. N.D., not detected.
Substrate Scope.
With these optimized conditions in hand, we then explored the scope of this aryl-radical-enabled Sonogashira-type cross-coupling reaction. As shown in Table 2, this approach was broadly applicable to the coupling of a wide array of alkyl iodides with terminal alkynes. Secondary alkyl iodides appended to various four- to seven-membered rings efficiently reacted with a terminal alkyne, affording the corresponding coupling products in good to excellent yield (16 to 27, 52–85% yield). Functional groups including acetals, carbamates, esters, sulfonamides, and amides were well tolerated. Notably, a substrate possessing a tertiary amine, a traditionally challenging functional group for transition metal catalysis, delivered the alkynylated product in good yield (17, 73% yield). Heterocycles commonly encountered in medicinal chemistry, such as furan, thiophene, piperidine, azepane, and pyrrolidone, were compatible with this protocol. The coupling reaction is also tolerant of conjugated alkenes (21), which are known to rapidly react with aryl radicals,30 highlighting the high selectivity of aryl radicals toward the activation of alkyl iodide bonds. Several medicinally relevant bicyclic and tricyclic compounds formed the desired products with good efficiency (24–27, 58–85% yield). Moreover, secondary acyclic alkyl iodides (28–31) and primary alkyl iodides bearing diverse functional groups, including alkyl bromides, silyl ethers, and hydroxyl groups, could be converted into the corresponding alkynes in good to high yield (32–37, 47–82% yield). A tertiary alkyl iodide derived from adamantane also participated in this transformation, albeit with moderate efficiency (38, 40% yield). Nonetheless, coupling of an acyclic tertiary alkyl iodide, tert-butyl iodide, failed to form the desired product (39), presumably due to the steric hindrance of the tert-butyl radical intermediate.
Table 2.
Substrate Scope of the Aryl Radical-Activated, Copper-Catalyzed Sonogashira-type Coupling of Alkyl Iodidesa

|
Reactions were run with 0.20 mmol of alkyl iodides (1.0 equiv), 0.22 mmol of alkyne (1.1 equiv), 0.40 mmol of diazonium salt 14 (2.0 equiv), terpyridine (25 mol %), and [Cu(CH3CN)4]BF4 (25 mol %) in of DMSO at 50 °C under Ar. Isolated yields were reported.
dr (diastereomeric ratio) of the starting alkyl iodides >20:1.
The compatibility of different alkynes with this Cu-catalyzed reaction was then studied. We were pleased to find that the efficacy of this protocol showed little dependence on the electronic properties of aryl substituents, as various phenyl acetylenes with electron-withdrawing, -neutral, and -donating groups reacted with an alkyl iodide to afford the corresponding alkynes in good yield (40–50, 62–85% yield). Interestingly, despite the known reactions between ferrocene and aryldiazonium salts,31 ethynylferrocene readily participated in this coupling reaction (47, 82% yield). Other conjugated alkynes, including an ethynylthiophene (48), an enyne (49), and a propiolate (50), could react smoothly, furnishing the conjugated alkyne products in good yield (58–75%). Moreover, coupling reactions proceeded with nonconjugated alkynes bearing various functional groups (hydroxyl, halide, sulfone, nitrile, nitro, acetal, amide, and sily; 51–61, 35–78% yield). Finally, the scalability of the reaction was evidenced by the preparation of 23 on a half gram scale (75% yield).
Late-Stage Cross-Coupling of Bioactive Molecules.
In an effort to demonstrate the applicability of our protocol to the synthesis of drug-like molecules, we tested the cross-coupling of a wide range of bioactive molecules using this Sonogashira-type transformation (Table 3). Alkyl iodides derived from various monosaccharides (glucose, xylose, ribose, fructose, and galactose) were smoothly converted to the corresponding alkyne derivatives in moderate to good yield (66–71, 40–75% yield). Notably, a single diastereomer was formed for the alkynylation of a glucose derivative (66), whereas the previous synthesis of an analogous alkyne molecule required a five-step synthesis.32 In addition, a variety of steroid derivatives successfully underwent coupling reactions (72–75, 41–70% yield). Three alkynes derived from bioactive molecules—norethindrone (76), caffeine (77), and clodinafop (78)—were successfully coupled under the standard conditions. A benzoquinone-containing drug agent (idebenone), a xanthine derivative (proxyphylline), and an estrogen receptor modulator (ospemifene) were all smoothly converted to the desired products (79–81).
Table 3.
Late-Stage Cross-Coupling of Bioactive Moleculesa

|
See Supporting Information for detailed conditions.
(diastereomeric ratio) of the starting alkyl iodides >20:1.
Empaglifloizin, a glucoside compound and one of the top-selling small molecule pharmaceuticals in 2021,33 formed the corresponding alkynylated analogue in 50% yield (82). The compatibility of unprotected triol groups further highlights the mild conditions of this protocol. Densely functionalized pharmaceutical agents including tedizolid (83), which consists of a tetrazole, a pyridine, and an oxazolidone ring, as well as ticagrelor (84) which incorporates a thioether, a diol, and a triazolo[4,5,-d]pyrimidine scaffold, were converted to their alkynylated analogues in 66% and 45% yield, respectively. Considering the value of unnatural amino acids in chemical biology and drug discovery, we also applied this protocol to the synthesis of two alkyne-containing analogues of methionine and alanine (85 and 86). Moreover, we developed a one-pot protocol for the synthesis of terminal alkynes, given their wide range of applications in click chemistry. Thus, the coupling of trimethylsilylacetylene with the alkyl iodide derived from proxyphylline under the standard conditions, followed by the in situ deprotection of the silyl group, afforded the alkyne derivative (87) in overall 65% yield. Finally, to demonstrate the potential of this method for the conjugation of functionalized alkynes and alkyl iodides, an alkyl iodide derived from empaglifloizin successfully coupled with the alkyne derived from clodinafop, affording the desired product (88) in 62% yield. It is worthwhile mentioning that this coupling product was not obtained under the previously reported photoinduced or Ni-catalyzed conditions, further highlighting the potential utility of this aryl-radical-enabled protocol for the synthesis of previously inaccessible alkyne molecules.
Mechanistic Studies.
Mechanistic studies shed light on this aryl-radical-enabled, Cu-catalyzed protocol (Figure 3). The addition of 2 equiv of TEMPO, a radical-trapping agent, completely suppressed the formation of the coupled product, whereas the TEMPO trapping product (89) was isolated in 55% yield (Figure 3a). Additionally, the coupling of a cyclopropyl-containing alkyl iodide 90 afforded mainly the ring-opened product 91, confirming the intermediacy of a cyclopropylcarbinyl radical, which is known to undergo rapid ring opening to the homoallylic radical (rate constant k = 2.7 × 1011 s−1) (Figure 3b).34 The alkynylation of alkene-containing alkyl iodide 92 afforded the cyclized product 93 in 52% yield, consistent with a 5-exo-trig radical cyclization pathway (Figure 3c). These results together support the formation of alkyl radicals in these coupling reactions.
Figure 3.

Mechanistic studies support the proposed aryl radical activation mechanism. (a) TEMPO trapping experiment; (b) radical clock experiment; (c) ring closure experiment; and (d) reactivity of Cu-acetylide species.
Finally, the involvement of Cu–acetylide species in this reaction was studied (Figure 3d). Using a stoichiometric amount of Cu-acetylide complex 94, no reactions occurred with alkyl iodide 13, regardless of the presence of terpyridine as the ligand. In addition, although the diazonium salt 14 could undergo SET with 94, as indicated by the immediate effervescence of the reaction mixture, the coupling product 15 was not formed in the absence of terpyridine. On the contrary, 94 smoothly transferred the alkyne group to the alkyl iodide 13 in the presence of both the diazonium salt and terpyridine to afford the alkyne product 15 in 43% yield. These findings are consistent with the involvement of a ligand-bound Cu-acetylide species that transfers the alkyne group via the aryl radical activation pathway.
Synthetic Application for Positron Emission Tomography.
Positron emission tomography (PET) is a non-invasive imaging technique that allows tracking radiolabeled drug molecules inside living subjects.35 Central to this technology is the development of novel PET tracers—targeted molecules that consist of positron-emitting isotopes.36 Due to the short half-lives of such isotopes, including 18F (t1/2 = 109.8 min), synthesis of PET tracers requires the reactions to be completed within a short period and conducted at the late stage of the overall synthesis process. Given the fast reaction kinetics of our alkynylation reaction (<20 min) and its broad functional group tolerance, we hypothesize that this protocol could offer a unique opportunity to the rapid conjugation of radio-labeled molecules for the development of PET tracers.
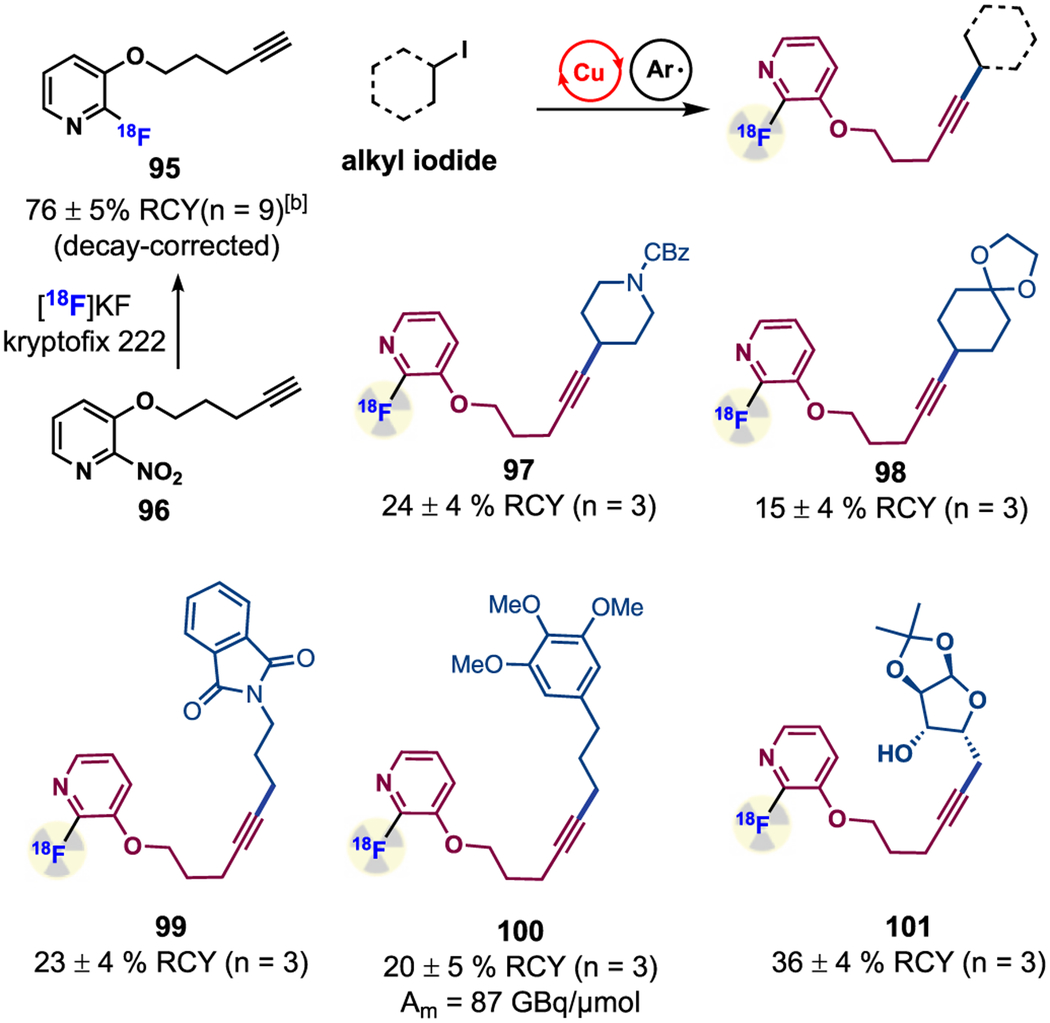
Thus, we aimed to develop a radio-conjugation approach that can couple 18F-labeled alkynes with alkyl iodides. Such an indirect method for 18F-radiolabeling reactions could circumvent a harsh step to carry out direct 18F-fluorination on complex molecules, which is incompatible with many sensitive functional groups.35 In addition, the use of a communal 18F-labeled prosthetic group could allow for rapid assembly of a variety of target molecules and their corresponding derivatives without the need of ad hoc and lengthy syntheses of complex precursor molecules in the absence of their PET imaging utility. The major challenge for the radio-conjugation is to form the reactive Cu-18 F-acetylide intermediates using 18F-alkynes, the concentration of which is typically in the range of picomolar to nanomolar. In comparison, high concentration of alkynes (~0.2 M) is employed under catalytic conditions to facilitate the formation of such intermediates. In addition, the trace Cu-18 F-acetylide complex needs to rapidly participate in the ensuing SET and alkyne transfer steps to form the 18F-labeled products.
18F–alkyne 95 was chosen as the model substrate due to its ease of preparation; treatment of its NO2 precursor (96) with 18F-fluoride in DMSO afforded 95 with a high radiochemical yield (76%, see Supporting Information for details). Under conditions similar to the catalytic reactions, 95 successfully coupled with primary and secondary alkyl iodides to afford the 18F-labeled alkyne products with moderate to good radiochemical yields (Table 4, 97–101, 15–36%). In addition, as high molar activity (Am) radiotracers are often needed for imaging low abundance of receptors in vivo, we measured the Am of compound 100, which was synthesized in >99% radiochemical purity. Gratifyingly, tracer 100 exhibited Am activity of 87 GBq/μmol (2.36 Ci/μmol), which is sufficient for imaging purposes. Given these promising proof-of-concept results, we anticipate that this aryl radical strategy could be applied to the rapid installation of other radioisotopes for the synthesis of novel radiotracers. These studies are currently carried out in our laboratories.
Table 4.
Synthesis of 18F-Labeled Alkynesa
|
Reaction conditions: alkyl iodide (0.01 mmol), 18F-alknye (0.5–1 mCi), [Cu(CH3CN)4]BF4 (1.6 mg), terpyridine (1.2 mg), K2CO3 (2.8 mg), 2,4,6-trimethylbenzenediazonium tetrafluoroborate (4.7 mg), DMSO (0.5 mL), 60 °C, 20 min; Radiochemical yield (RCY) and product identity were determined by radio-high-performance liquid chromatography (HPLC).
Reaction conditions: alkyne (2 mg), K2CO3/kryptofix 222 (1/5 mg), DMSO (0.4 mL), 150 °C, 10 min; Radiochemical yield of the isolated product was reported as decay-corrected, and the isolation was performed by radio-HPLC.
CONCLUSIONS
We report herein a widely applicable approach for the Cu-catalyzed Sonogashira-type cross-coupling of unactivated alkyl iodides with alkynes. This method harnessed the iodine abstraction ability of aryl radicals to allow the participation of unactivated alkyl iodides in Cu-catalyzed Sonogashira coupling reactions. This aryl-radical-enabled coupling reaction demonstrated high functional group tolerance and empowered the late-stage coupling of densely functionalized molecules. Mechanistic studies were consistent with the transfer of alkyne groups from Cu-acetylide species to alkyl radicals, which were generated via an aryl radical activation pathway. Moreover, given the fast kinetics of the aryl radical-enabled coupling reaction, this protocol has also been applied to the conjugation of [18F]-labeled molecules for the synthesis of novel PET tracers. Given the rich medicinal potential of functionalized alkynes as well as the broad availability of alkyl halides and terminal alkynes, we anticipate that this protocol will find wide application in drug development. From a broader perspective, we envision that this aryl radical activation strategy will be of great utility in both modern synthetic chemistry and pharmaceutical research.
Supplementary Material
ACKNOWLEDGMENTS
W.L. acknowledges financial support from the ACS Herman Frasch Foundation Grant and National Institute of General Medical Sciences (R35GM146765). NMR experiments were performed using a Bruker AVANCE NEO 400 MHz NMR spectrometer, funded by NSF-MRI grant CHE-1726092. S.H.L. acknowledges financial support from National Institute of Health (MH128705, AG070060, AG073428, AG075444, and AG079956), and members of Emory PET Imaging Center & Radiopharmaceutical Discovery Program, Department of Radiology and Imaging Sciences, and Emory University School of Medicine.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.2c05901.
Experimental details, characterizations of new compounds, and copies of NMR spectra for new compounds (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acscatal.2c05901
The authors declare no competing financial interest.
Contributor Information
Xiaojun Zeng, School of Chemistry and Chemical Engineering, Nanchang University, Nanchang, Jiangxi 330031, China.
Chao Wang, Department of Chemistry, University of Cincinnati, Cincinnati, Ohio 45221, United States.
Wenhao Yan, Department of Chemistry, University of Cincinnati, Cincinnati, Ohio 45221, United States.
Jian Rong, Department of Radiology and Imaging Sciences, Emory University, Atlanta, Georgia 30322, United States.
Yanshan Song, School of Chemistry and Chemical Engineering, Nanchang University, Nanchang, Jiangxi 330031, China.
Zhiwei Xiao, Department of Radiology and Imaging Sciences, Emory University, Atlanta, Georgia 30322, United States.
Aijie Cai, Department of Chemistry, University of Cincinnati, Cincinnati, Ohio 45221, United States.
Steven H. Liang, Department of Radiology and Imaging Sciences, Emory University, Atlanta, Georgia 30322, United States
Wei Liu, Department of Chemistry, University of Cincinnati, Cincinnati, Ohio 45221, United States.
REFERENCES
- (1).(a) Talele TT Acetylene Group, Friend or Foe in Medicinal Chemistry. J. Med. Chem 2020, 63, 5625–5663. [DOI] [PubMed] [Google Scholar]; (b) Trost BM; Li C-J Modern Alkyne Chemistry: Catalytic and Atom-Economic Transformations; John Wiley & Sons, 2015, pp 1–424. [Google Scholar]; (c) Diederich F; Stang PJ; Tykwinski RR Acetylene Chemistry: Chemistry, Biology and Material Science; John Wiley & Sons, 2006, pp 1–507. [Google Scholar]; (d) Lam J Chemistry and Biology of Naturally-Occurring Acetylenes and Related Compounds (NOARC); Elsevier Science Pub. Co., 1988. [Google Scholar]; (e) Patai S; Rappoport Z The Chemistry of Triple-Bonded Functional Groups; Wiley-Blackwell, 1994; Vol. 22, Supplement C2, pp 1–800. [Google Scholar]
- (2).For selected reviews on click chemistry:; (a) Devaraj NK; Finn MG Introduction: Click Chemistry. Chem. Rev 2021, 121, 6697–6698. [DOI] [PubMed] [Google Scholar]; (b) Thirumurugan P; Matosiuk D; Jozwiak K Click Chemistry for Drug Development and Diverse Chemical-Biology Applications. Chem. Rev 2013, 113, 4905–4979. [DOI] [PubMed] [Google Scholar]; (c) Fantoni NZ; El-Sagheer AH; Brown T A Hitchhiker’s Guide to Click-Chemistry with Nucleic Acids. Chem. Rev 2021, 121, 7122–7154. [DOI] [PubMed] [Google Scholar]; (d) Kolb HC; Finn M; Sharpless KB Click chemistry: diverse chemical function from a few good reactions. Angew. Chem., Int. Ed 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]; (e) Baskin JM; Prescher JA; Laughlin ST; Agard NJ; Chang PV; Miller IA; Lo A; Codelli JA; Bertozzi CR Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. U.S.A 2007, 104, 16793–16797. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Jewett JC; Bertozzi CR Cu-free click cycloaddition reactions in chemical biology. Chem. Soc. Rev 2010, 39, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Stang PJ; Diederich F Modern Acetylene Chemistry; VCH Weinheim, 1995, pp 1–606. [Google Scholar]
- (4).(a) Yamakoshi H; Dodo K; Palonpon A; Ando J; Fujita K; Kawata S; Sodeoka M Alkyne-Tag Raman Imaging for Visualization of Mobile Small Molecules in Live Cells. J. Am. Chem. Soc 2012, 134, 20681–20689. [DOI] [PubMed] [Google Scholar]; (b) Bakthavatsalam S; Dodo K; Sodeoka M A decade of alkyne-tag Raman imaging (ATRI): applications in biological systems. RSC Chem. Biol 2021, 2, 1415–1429. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wei L; Hu F; Shen Y; Chen Z; Yu Y; Lin C-C; Wang MC; Min W Live-cell imaging of alkyne-tagged small biomolecules by stimulated Raman scattering. Nat. Methods 2014, 11, 410–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).For selected reviews:; (a) Shaw R; Elagamy A; Althagafi I; Pratap R Synthesis of alkynes from non-alkyne sources. Org. Biomol. Chem 2020, 18, 3797–3817. [DOI] [PubMed] [Google Scholar]; (b) Habrant D; Rauhala V; Koskinen AM Conversion of carbonyl compounds to alkynes: general overview and recent developments. Chem. Soc. Rev 2010, 39, 2007–2017. [DOI] [PubMed] [Google Scholar]
- (6).For selected recent examples on alkyne synthesis:; (a) Smith JM; Qin T; Merchant RR; Edwards JT; Malins LR; Liu Z; Che G; Shen Z; Shaw SA; Eastgate MD; Baran PS Decarboxylative Alkynylation. Angew. Chem., Int. Ed 2017, 56, 11906–11910. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bi HP; Zhao L; Liang YM; Li CJ The Copper-Catalyzed Decarboxylative Coupling of the sp3-Hybridized Carbon Atoms of α-Amino Acids. Angew. Chem., Int. Ed 2009, 48, 792–795. [DOI] [PubMed] [Google Scholar]; (c) Liu X; Wang Z; Cheng X; Li C Silver-catalyzed decarboxylative alkynylation of aliphatic carboxylic acids in aqueous solution. J. Am. Chem. Soc 2012, 134, 14330–14333. [DOI] [PubMed] [Google Scholar]; (d) Zhou QQ; Guo W; Ding W; Wu X; Chen X; Lu LQ; Xiao WJ Decarboxylative Alkynylation and Carbonylative Alkynylation of Carboxylic Acids Enabled by Visible-Light Photoredox Catalysis. Angew. Chem., Int. Ed 2015, 54, 11196–11199. [DOI] [PubMed] [Google Scholar]; (e) Le Vaillant F; Courant T; Waser J Room-Temperature Decarboxylative Alkynylation of Carboxylic Acids Using Photoredox Catalysis and EBX Reagents. Angew. Chem., Int. Ed 2015, 127, 11352–11356. [DOI] [PubMed] [Google Scholar]; (f) Hatakeyama T; Okada Y; Yoshimoto Y; Nakamura M Tuning Chemoselectivity in Iron-Catalyzed Sonogashira-Type Reactions Using a Bisphosphine Ligand with Peripheral Steric Bulk: Selective Alkynylation of Nonactivated Alkyl Halides. Angew. Chem., Int. Ed 2011, 50, 10973–10976. [DOI] [PubMed] [Google Scholar]; (g) Fu L; Zhang Z; Chen P; Lin Z; Liu G Enantioselective Copper-Catalyzed Alkynylation of Benzylic C-H Bonds via Radical Relay. J. Am. Chem. Soc 2020, 142, 12493–12500. [DOI] [PubMed] [Google Scholar]; (h) Xia H-D; Li Z-L; Gu Q-S; Dong X-Y; Fang J-H; Du X-Y; Wang L-L; Liu X-Y Photoinduced Copper-Catalyzed Asymmetric Decarboxylative Alkynylation with Terminal Alkynes. Angew. Chem., Int. Ed 2020, 59, 16926–16932. [DOI] [PubMed] [Google Scholar]; (i) Ni S; Li C-X; Mao Y; Han J; Wang Y; Yan H; Pan Y Ni-catalyzed deaminative cross-electrophile coupling of Katritzky salts with halides via C–N bond activation. Sci. Adv 2019, 5, No. eaaw9516. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Zhang X; Qi D; Jiao C; Liu X; Zhang G Nickel-catalyzed deaminative Sonogashira coupling of alkylpyridinium salts enabled by NN2 pincer ligand. Nat. Commun 2021, 12, 4904. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Zhang Z-H; Dong X-Y; Du X-Y; Gu Q-S; Li Z-L; Liu X-Y Copper-catalyzed enantioselective Sonogashira-type oxidative cross-coupling of unactivated C(sp3)–H bonds with alkynes. Nat. Commun 2019, 10, 5689. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Liu W; Li L; Li C-J Empowering a transition-metal-free coupling between alkyne and alkyl iodide with light in water. Nat. Commun 2015, 6, 6526. [DOI] [PubMed] [Google Scholar]; (m) Huang L; Olivares AM; Weix DJ Reductive Decarboxylative Alkynylation of N -Hydroxyphthalimide Esters with Bromoalkynes. Angew. Chem., Int. Ed 2017, 56, 11901–11905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).For early report on Sonogashira coupling:; (a) Sonogashira K; Tohda Y; Hagihara N A convenient synthesis of acetylenes: catalytic substitutions of acetylenic hydrogen with bromoalkenes, iodoarenes and bromopyridines. Tetrahedron Lett. 1975, 16, 4467–4470. [Google Scholar]; (b) Dieck a. H.; Heck F Palladium catalyzed synthesis of aryl, heterocyclic and vinylic acetylene derivatives. J. Organometallic. Chem 1975, 93, 259–263. [Google Scholar]; (c) Cassar L Synthesis of aryl- and vinyl-substituted acetylene derivatives by the use of nickel and palladium complexes. J. Organometallic. Chem 1975, 93, 253–257. [Google Scholar]
- (8).For selected reviews:; (a) Chinchilla R; Nájera C The Sonogashira Reaction: A Booming Methodology in Synthetic Organic Chemistry. Chem. Rev 2007, 107, 874–922. [DOI] [PubMed] [Google Scholar]; (b) Chinchilla R; Nájera C Recent advances in Sonogashira reactions. Chem. Soc. Rev 2011, 40, 5084–5121. [DOI] [PubMed] [Google Scholar]; (c) Bakherad M Recent progress and current applications of Sonogashira coupling reaction in water. Appl. Organomet. Chem 2013, 27, 125–140. [Google Scholar]; (d) Wang D; Gao S Sonogashira coupling in natural product synthesis. Org. Chem. Front 2014, 1, 556–566. [Google Scholar]; (e) Thomas AM; Sujatha A; Anilkumar G Recent advances and perspectives in copper-catalyzed Sonogashira coupling reactions. RSC Adv. 2014, 4, 21688–21698. [Google Scholar]
- (9).For Cu-catalyzed Sonogashira coupling of activated alkyl electrophiles:; (a) Cao Y-X; Dong X-Y; Yang J; Jiang S-P; Zhou S; Li Z-L; Chen G-Q; Liu X-Y A Copper-Catalyzed Sonogashira Coupling Reaction of Diverse Activated Alkyl Halides with Terminal Alkynes Under Ambient Conditions. Adv. Syn. & Catal 2020, 362, 2280–2284. [Google Scholar]; (b) Dong X-Y; Zhang Y-F; Ma C-L; Gu Q-S; Wang F-L; Li Z-L; Jiang S-P; Liu X-Y A general asymmetric copper-catalysed Sonogashira C(sp3)-C(sp) coupling. Nat. Chem 2019, 11, 1158–1166. [DOI] [PubMed] [Google Scholar]; (c) Luo F-X; Xu X; Wang D; Cao Z-C; Zhang Y-F; Shi Z-J Cu-Catalyzed Alkynylation of Unactivated C(sp3)-X Bonds with Terminal Alkynes through Directing Strategy. Org. Lett 2016, 18, 2040–2043. [DOI] [PubMed] [Google Scholar]; (d) Zhang H; Sun N; Hu B; Shen Z; Hu X; Jin L Copper-catalyzed direct couplings of terminal alkynes with primary and secondary benzyl bromides. Org. Chem. Front 2019, 6, 1983–1988. [Google Scholar]
- (10).Choi J; Fu GC Transition metal-catalyzed alkyl-alkyl bond formation: Another dimension in cross-coupling chemistry. Science 2017, 356, No. eaaf7230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Eckhardt M; Fu GC The First Applications of Carbene Ligands in Cross-Couplings of Alkyl Electrophiles: Sonogashira Reactions of Unactivated Alkyl Bromides and Iodides. J. Am. Chem. Soc 2003, 125, 13642–13643. [DOI] [PubMed] [Google Scholar]
- (12).Altenhoff G; Würtz S; Glorius F The first palladium-catalyzed Sonogashira coupling of unactivated secondary alkyl bromides. Tetrahedron Lett. 2006, 47, 2925–2928. [Google Scholar]
- (13).Vechorkin O; Barmaz D; Proust V; Hu X Ni-Catalyzed Sonogashira Coupling of Nonactivated Alkyl Halides: Orthogonal Functionalization of Alkyl Iodides, Bromides, and Chlorides. J. Am. Chem. Soc 2009, 131, 12078–12079. [DOI] [PubMed] [Google Scholar]
- (14).Yi J; Lu X; Sun Y-Y; Xiao B; Liu L Nickel-Catalyzed Sonogashira Reactions of Non-activated Secondary Alkyl Bromides and Iodides. Angew. Chem., Int. Ed 2013, 52, 12409–12413. [DOI] [PubMed] [Google Scholar]
- (15).(a) Chen C; Peters JC; Fu GC Photoinduced copper-catalysed asymmetric amidation via ligand cooperativity. Nature 2021, 596, 250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Creutz SE; Lotito KJ; Fu GC; Peters JC Photoinduced Ullmann C-N Coupling: Demonstrating the Viability of a Radical Pathway. Science 2012, 338, 647. [DOI] [PubMed] [Google Scholar]; (c) Bissember AC; Lundgren RJ; Creutz SE; Peters JC; Fu GC Transition-Metal-Catalyzed Alkylations of Amines with Alkyl Halides: Photo-induced, Copper-Catalyzed Couplings of Carbazoles. Angew. Chem., Int. Ed 2013, 52, 5129–5133. [DOI] [PubMed] [Google Scholar]; (d) Kainz QM; Matier CD; Bartoszewicz A; Zultanski SL; Peters JC; Fu GC Asymmetric copper-catalyzed C-N cross-couplings induced by visible light. Science 2016, 351, 681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sagadevan A; Hwang KC Photo-Induced Sonogashira C-C Coupling Reaction Catalyzed by Simple Copper(I) Chloride Salt at Room Temperature. Adv. Syn. & Catal 2012, 354, 3421–3427. [Google Scholar]
- (17).Hazra A; Lee MT; Chiu JF; Lalic G Photoinduced Copper-Catalyzed Coupling of Terminal Alkynes and Alkyl Iodides. Angew. Chem., Int. Ed 2018, 57, 5492–5496. [DOI] [PubMed] [Google Scholar]
- (18).(a) Cai A; Yan W; Liu W Aryl Radical Activation of C-O Bonds: Copper-Catalyzed Deoxygenative Difluoromethylation of Alcohols. J. Am. Chem. Soc 2021, 143, 9952–9960. [DOI] [PubMed] [Google Scholar]; (b) Cai A; Yan W; Wang C; Liu W Copper-Catalyzed Difluoromethylation of Alkyl Iodides Enabled by Aryl Radical Activation of Carbon-Iodine Bonds. Angew. Chem., Int. Ed 2021, 60, 27070–27077. [DOI] [PubMed] [Google Scholar]
- (19).(a) Le C; Chen TQ; Liang T; Zhang P; MacMillan DWC A radical approach to the copper oxidative addition problem: Trifluoromethylation of bromoarenes. Science 2018, 360, 1010. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kornfilt DJP; MacMillan DWC Copper-Catalyzed Trifluoromethylation of Alkyl Bromides. J. Am. Chem. Soc 2019, 141, 6853–6858. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhao X; MacMillan DWC Metallaphotoredox Perfluoroalkylation of Organobromides. J. Am. Chem. Soc 2020, 142, 19480–19486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).(a) Górski B; Barthelemy A-L; Douglas JJ; Juliá F; Leonori D Copper-catalysed amination of alkyl iodides enabled by halogen-atom transfer. Nat. Catal 2021, 4, 623–630. [Google Scholar]; (b) Constantin T; Zanini M; Regni A; Sheikh NS; Juliá F; Leonori D Aminoalkyl radicals as halogen-atom transfer agents for activation of alkyl and aryl halides. Science 2020, 367, 1021. [DOI] [PubMed] [Google Scholar]; (c) Zhang Z; Górski B; Leonori D Merging Halogen-Atom Transfer (XAT) and Copper Catalysis for the Modular Suzuki-Miyaura-Type Cross-Coupling of Alkyl Iodides and Organoborons. J. Am. Chem. Soc 2022, 144, 1986–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Galli C Radical reactions of arenediazonium ions: An easy entry into the chemistry of the aryl radical. Chem. Rev 1988, 88, 765–792. [Google Scholar]
- (22).Mo F; Qiu D; Zhang L; Wang J Recent Development of Aryl Diazonium Chemistry for the Derivatization of Aromatic Compounds. Chem. Rev 2021, 121, 5741–5829. [DOI] [PubMed] [Google Scholar]
- (23).(a) Tatunashvili E; McErlean CSP Generation and reaction of alkyl radicals in open reaction vessels. Org. Biomol. Chem 2020, 18, 7818–7821. [DOI] [PubMed] [Google Scholar]; (b) Chaambi A; Kurtay G; Abderrahim R; Robert F; Landais Y Aryl Radical-Mediated Alkenylation of Alkyl Halides. Helv. Chim. Acta 2019, 102, No. e1900140. [Google Scholar]; (c) Kurandina D; Yadagiri D; Rivas M; Kavun A; Chuentragool P; Hayama K; Gevorgyan V Transition-Metal- and Light-Free Directed Amination of Remote Unactivated C(sp3)-H Bonds of Alcohols. J. Am. Chem. Soc 2019, 141, 8104–8109. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Clerici A; Cannella R; Panzeri W; Pastori N; Regolini E; Porta O TiCl3/PhN2+-mediated radical addition of ethers to aldimines generated in situ under aqueous conditions. Tetrahedron Lett. 2005, 46, 8351–8354. [Google Scholar]; (e) Cao L; Li C p-MeOC6H4/TiCl3: a novel initiator for halogen atom-transfer radical reactions in aqueous media. Tetrahedron Lett. 2008, 49, 7380–7382. [Google Scholar]
- (24).Lang H; Jakob A; Milde B Copper(I) Alkyne and Alkynide Complexes. Organometallics 2012, 31, 7661–7693. [Google Scholar]
- (25).(a) Bakhoda A; Okoromoba OE; Greene C; Boroujeni MR; Bertke JA; Warren TH Three-Coordinate Copper(II) Alkynyl Complex in C-C Bond Formation: The Sesquicentennial of the Glaser Coupling. J. Am. Chem. Soc 2020, 142, 18483–18490. [DOI] [PubMed] [Google Scholar]; (b) Ziegler MS; Lakshmi K; Tilley TD Dicopper Cu(I)Cu(I) and Cu(I)Cu(II) Complexes in Copper-Catalyzed Azide-Alkyne Cycloaddition. J. Am. Chem. Soc 2017, 139, 5378–5386. [DOI] [PubMed] [Google Scholar]
- (26).(a) Hickman AJ; Sanford MS High-valent organometallic copper and palladium in catalysis. Nature 2012, 484, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) DiMucci IM; Lukens JT; Chatterjee S; Carsch KM; Titus CJ; Lee SJ; Nordlund D; Betley TA; MacMillan SN; Lancaster KM The Myth of d8 Copper(III). J. Am. Chem. Soc 2019, 141, 18508–18520. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Keown W; Gary JB; Stack TDP High-valent copper in biomimetic and biological oxidations. JBIC J. Biol. Inorg. Chem 2017, 22, 289–305. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Casitas A; Ribas X The Role of Organometallic Copper(III) Complexes in Homogeneous Catalysis. Chem. Sci 2013, 4, 2301–2318. [Google Scholar]; (e) Liu L; Xi Z Organocopper(III) Compounds with Well-defined Structures Undergo Reductive Elimination to Form C-C or C-Heteroatom Bonds. Chin. J. Chem 2018, 36, 1213–1221. [Google Scholar]; (f) Liu H; Shen Q Well-defined organometallic Copper(III) complexes: Preparation, characterization and reactivity. Coord. Chem. Rev 2021, 442, 213923. [Google Scholar]
- (27).(a) Rovira M; Font M; Acuña-Parés F; Parella T; Luis JM; Lloret-Fillol J; Ribas X Aryl-Copper(III)-Acetylides as Key Intermediates in C sp 2-CspModel Couplings under Mild Conditions. Chem.—Eur. J 2014, 20, 10005–10010. [DOI] [PubMed] [Google Scholar]; (b) Lu Z; Liu H; Liu S; Leng X; Lan Y; Shen Q A Key Intermediate in Copper-Mediated Arene Trifluoromethylation, [n Bu 4 N][Cu(Ar)(CF 3) 3]: Synthesis, Characterization, and C(sp 2)–CF 3 Reductive Elimination. Angew. Chem., Int. Ed 2019, 58, 8510–8514. [DOI] [PubMed] [Google Scholar]; (c) Liu S; Liu H; Liu S; Lu Z; Lu C; Leng X; Lan Y; Shen Q C(sp3)-CF3 Reductive Elimination from a Five-Coordinate Neutral Copper(III) Complex. J. Am. Chem. Soc 2020, 142, 9785–9791. [DOI] [PubMed] [Google Scholar]; (d) Paeth M; Tyndall SB; Chen L-Y; Hong J-C; Carson WP; Liu X; Sun X; Liu J; Yang K; Hale EM; Tierney DL; Liu B; Cao Z; Cheng M-J; Goddard WA; Liu W Csp3-Csp3 Bond-Forming Reductive Elimination from Well-Defined Copper(III) Complexes. J. Am. Chem. Soc 2019, 141, 3153–3159. [DOI] [PubMed] [Google Scholar]
- (28).Glaser C Beiträge zur Kenntniss des Acetenylbenzols. Ber. Dtsch. Chem. Ges 1869, 2, 422–424. [Google Scholar]
- (29).Roglans A; Pla-Quintana A; Moreno-Mañas M Diazonium Salts as Substrates in Palladium-Catalyzed Cross-Coupling Reactions. Chem. Rev 2006, 106, 4622–4643. [DOI] [PubMed] [Google Scholar]
- (30).Kindt S; Heinrich MR Recent Advances in Meerwein Arylation Chemistry. Synthesis 2016, 48, 1597–1606. [Google Scholar]
- (31).Rosenblum M; Howells WG; Banerjee A; Bennett C The Structure and Chemistry of Ferrocene. VI. Mechanism of the Arylation Reaction. J. Am. Chem. Soc 1962, 84, 2726–2732. [Google Scholar]
- (32).Betkekar VV; Panda S; Kaliappan KP A Tandem Enyne/Ring Closing Metathesis Approach to 4-Methylene-2-cyclohexenols: An Efficient Entry to Otteliones and Loloanolides. Org. Lett 2012, 14, 198–201. [DOI] [PubMed] [Google Scholar]
- (33).McGrath NA; Brichacek M; Njardarson JT A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ 2010, 87, 1348–1349. [Google Scholar]
- (34).Newcomb M; Johnson CC; Manek MB; Varick TR Picosecond radical kinetics. Ring openings of phenyl-substituted cyclopropylcarbinyl radicals. J. Am. Chem. Soc 1992, 114, 10915–10921. [Google Scholar]
- (35).(a) Deng X; Rong J; Wang L; Vasdev N; Zhang L; Josephson L; Liang SH Chemistry for Positron Emission Tomography: Recent Advances in 11 C-, 18 F-, 13 N-, and 15 O-Labeling Reactions. Angew. Chem., Int. Ed 2019, 58, 2580–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Krishnan HS; Ma L; Vasdev N; Liang SH 18F-Labeling of Sensitive Biomolecules for Positron Emission Tomography. Chem. Eur. J 2017, 23, 15553–15577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).(a) Rong J; Haider H; Liang SH Precision Radiochemistry for Fluorine-18 Labeling of PET Tracers. In Organofluorine Chemistry: Synthesis, Modeling, and Applications; Wiley-VCH GmbH: Boschstr, 2021, pp 397–425. [Google Scholar]; (b) Rong J; Chen Z; Vasdev N; Liang SH Aryl-18F bond formation from nucleophilic [18F]fluoride. In Frontiers of Organofluorine Chemistry; World Scientific: Singapore, 2020, pp 617–648. DOI: 10.1142/9781786347336_0013 [DOI] [Google Scholar]; (c) Rong J; Liang SH Aliphatic [18F]Fluorination Chemistry for Positron Emission Tomography. In fluoriation; Springer: New York, 2018, pp 1–14. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.