

Abstract
Alzheimer’s disease (AD) is one of the most common neurodegenerative diseases. However, the AD mechanism has not yet been fully elucidated to date, hindering the development of effective therapies. In our work, we perform a brain imaging genomics study to link genetics, single-cell gene expression data, tissue-specific gene expression data, brain imaging-derived volumetric endophenotypes, and disease diagnosis to discover potential underlying neurobiological pathways for AD. To do so, we perform brain-wide genome-wide colocalization analyses to integrate multidimensional imaging genomic biobank data. Specifically, we use (1) the individual-level imputed genotyping data and magnetic resonance imaging (MRI) data from the UK Biobank, (2) the summary statistics of the genome-wide association study (GWAS) from multiple European ancestry cohorts, and (3) the tissue-specific cis-expression quantitative trait loci (cis-eQTL) summary statistics from the GTEx project. We apply a Bayes factor colocalization framework and mediation analysis to these multi-modal imaging genomic data. As a result, we derive the brain regional level GWAS summary statistics for 145 brain regions with 482,831 single nucleotide polymorphisms (SNPs) followed by posthoc functional annotations. Our analysis yields the discovery of a potential AD causal pathway from a systems biology perspective: the SNP chr10:124165615:G>A (rs6585827) mutation upregulates the expression of BTBD16 gene in oligodendrocytes, a specialized glial cells, in the brain cortex, leading to a reduced risk of volumetric loss in the entorhinal cortex, resulting in the protective effect on AD. We substantiate our findings with multiple evidence from existing imaging, genetic and genomic studies in AD literature. Our study connects genetics, molecular and cellular signatures, regional brain morphologic endophenotypes, and AD diagnosis, providing new insights into the mechanistic understanding of the disease. Our findings can provide valuable guidance for subsequent therapeutic target identification and drug discovery in AD.
Keywords: Alzheimer’s disease, Imaging-genetics, Multi-modality, Colocalization, Mediation analysis
1. Introduction
Alzheimer’s disease (AD) is a common neurodegenerative disorder (Scandurra et al., 2019) that plagues 29 million people worldwide (Vos et al., 2016); the number of cases is expected to increase to 152 million by 2050 (Christina 2018). However, the mechanism of AD is not yet fully uncovered, hindering the development of effective therapies. Recent advances in brain imaging genomics have provided unprecedented opportunities to fully tape the disease pathogenesis of AD.
Understanding the genetic architecture of AD is critical because genetics is, in nature, close to the underlying etiology of AD. Previous twin studies estimated 58%–79% heritability for AD (Gatz et al., 2006) and identified several protective/risk genetic loci. For example, a recent large-scale genome-wide association study (GWAS) on an AD case-control design identified 75 risk loci (Bellenguez et al., 2022). Until 2022, the NHGRI-EBI GWAS Catalog documented 1741 associations from over 135 studies (Buniello et al., 2019). These large-scale GWAS analyses can guide future therapeutic target identification, drug discoveries, and personalized medicine.
Brain volumetric endophenotypes or quantitative traits (QTs), extracted from magnetic resonance imaging (MRI), have been used for decades to capture in vivo imaging biomarkers of AD (Jack Jr et al. 2011; McKhann et al., 2011; Albert et al., 2011). For example, brain atrophy has been robustly shown in the hippocampus and medial temporal regions in AD patients (Baron et al., 2001; Busatto et al., 2003; Hampel et al., 2008). Brain volumetric variations are heritable, with more than 80% of estimated heritability from twin studies (Kremen et al., 2010; Blokland et al., 2012; Den Braber et al. 2013; Eyler et al., 2014; Jansen et al., 2015; Wen et al., 2016). The regional-level volumetric QTs are shown to have a high degree of genetic correlation with AD (Zhao et al., 2019). In addition, recent studies showed strong and robust associations between brain volumetric QTs and genetic determinants [e.g., single nucleotide polymorphisms (SNP)]. In 2018, the first large-scale region-based whole brain imaging-genetics GWAS study identified 148 genome-wide significant associations (Elliott et al., 2018). Later, larger-scale GWAS analyses on brain regions of interest (ROIs) identified 365 variants significantly associated with 101 brain volumetric QTs (Zhao et al., 2019), and 109 genome loci significantly associated with specific tracts of white matter (Zhao et al., 2021). Recently, patterns of structural covariance in brain morphology were discovered to be significantly associated with 915 genomic loci, which correlates with the reelin signaling, apoptosis, neurogenesis, and appendage development pathways (Wen et al., 2022).
An open question in AD GWAS is that association does not imply causation. This gap has hindered the identification of causal genes for the development of gene-guided therapies. Recent GWAS improved the biological interpretation of association findings by cis-expression quantitative traits loci (cis-eQTL) colocalizations. This enables the identification of potential causal variants for GWAS and cis-eQTL signals in a given genome region (Hormozdiari et al., 2016). Colocalization has been widely used, and many studies have confirmed its effectiveness. For example, Kunkle et al. found evidence of a shared causal variant affecting gene expression and AD risk in 66 genes over 20 loci (Kunkle et al., 2019); Bellenguez et al. found that the TMEM106B and GRN signals in frontotemporal lobar degeneration with TAR DNA-binding protein inclusions shared causal variants with AD-related dementias (Bellenguez et al., 2022).
The success of colocalization advances molecular and pathological insights into AD. However, it is underexplored in brain imaging genomics studies. The full landscape of AD causal pathways, from underlying genetic variants to gene expression, brain morphological changes, and finally, the clinical manifestation of AD, has not been comprehensively established. To bridge this gap, we hypothesize that genetic variants caused the tissue-specific differential gene expression, led to volumetric brain changes, and resulted in the manifestation of AD. Herein, we perform a comprehensive brain imaging genomics study through analyzing large-scale brain MRI and genetic data using brain-wide genome-wide colocalization and mediation analyses to test this hypothesis and establish a novel AD putative causal pathway.
2. Materials and methods
Our brain-wide genome-wide colocalization framework integrates the analyses of multidimensional biobank data including genetics, tissue-specific gene expression data, brain imaging-derived volumetric endophenotypes, and AD diagnosis (Fig. 1). It provides an efficient and explainable bioinformatics strategy to discover potential underlying neurobiological pathways for AD. In this framework, we first performed a brain-wide genome-wide imaging genomics association study (Fig. 1 Block 4) using UKBB MRI data (Fig. 1 Block 2) and imputed genotyping data (Fig. 1 Block 3). After that, we conducted a colocalization analysis to integrate summary statistics from our imaging genomics study with the existing AD GWAS summary statistics (Bellenguez et al., 2022) (Fig. 1 Block 1) to identify SNPs associated with both imaging QTs (iQTs) and AD diagnosis (Fig. 1 Block 5). For each identified (SNP, iQT, diagnosis) triplet, we further performed mediation analyses to discover significant mediation relationships from SNP to iQT to diagnosis (Fig. 1 Block 6). For each significant SNP→iQT→diagnosis mediation relationship, we performed another colocalization analysis using the GWAS summary data for the GTEx v7 tissue-specific gene expression QTs (eQTs) (Fig. 1 Block 7) to identify SNPs related to both tissue-specific gene expression QT (eQT) and imaging-based brain volumetric QT (iQT) (Fig. 1 Block 8). Integrating all the above findings, we discovered a putative AD pathway from genetics (SNP) to tissue-specific gene eQT, to imaging-based brain volumetric iQTs, and to AD diagnosis. Finally, we performed tissue-specific and cell-type-specific differential expression analyses using brain samples to characterize our imaging genomic findings (Fig. 1 Block 9 and Block 10).
Fig. 1.
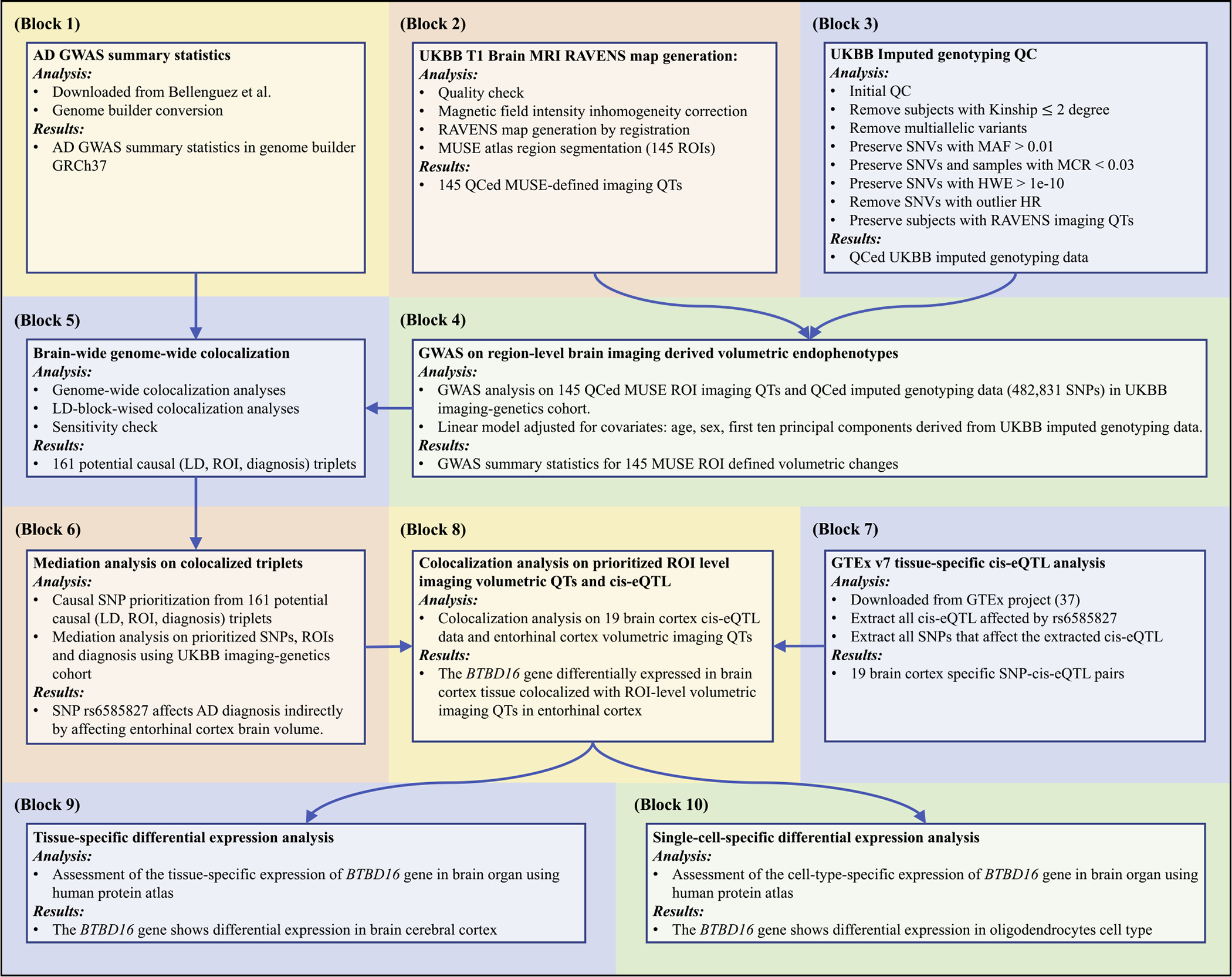
The brian-wide genome-wide colocalization framework. Block 1) AD GWAS summary statistics. Block 2) UKBB T1-weighted MRI preprocessing and RAVENS map generation using MUSE atlas (Materials and methods: UKBB imaging genomics data preprocessing). Block 3) UKBB imputed genotyping QC (Materials and methods: UKBB imaging genomics data preprocessing). Block 4) GWAS on 145 MUSE ROIs using imputed genotyping data (Materials and methods: GWAS of brain imaging quantitative traits). Block 5) LD-block-wised brain-volumetric-QTs-diagnosis colocalization analyses (Materials and methods: Brain-wide genome-wide colocalization analysis). Block 6) Mediation analyses on prioritized SNP-ROI pairs (Materials and methods: Mediation analysis). Block 7) GTEx v7 tissue-specific cis-eQTL analysis (Materials and methods: Colocalization on mediation analysis prioritized SNP-ROI pairs). Block 8) Colocalization analysis on prioritized ROIs and cis-eQTL on potential causal SNPs (Materials and methods: Colocalization on mediation analysis prioritized SNP-ROI pairs). Block 9) Tissue-specific differential expression analysis (Materials and methods: Tissue-specific differential expression analysis and single-cell-specific differential expression analysis). Block 10) Single-cell-specific differential expression analysis (Materials and methods: Tissue-specific differential expression analysis and single-cell-specific differential expression analysis). Abbreviations: AD: Alzheimer’s disease; GWAS: genome-wide association study; GRCh37: Genome Reference Consortium Human Build 37; UKBB: UK biobank; MRI: magnetic resonance imaging; RAVENS: voxel-wise regional volumetric maps; MUSE: multi-atlas region segmentation; ROI: region of interest; QC: quality control; QT: quantitative trait; SNV: single nucleotide variation; MAF: minor allele frequency; MCR: missing call rate; HWE: Hardy-Weinberg equilibrium; HR: heterozygosity rate; LD: linkage disequilibrium; SNP: single nucleotide polymorphism; cis-eQTL: cis-expression-quantitative-trait-loci; GTEx: Genotype-Tissue Expression project.
2.1. AD case-control GWAS summary statistics
Our brain-wide genome-wide colocalization study investigates the GWAS summary statistics derived from multiple European ancestry cohorts including EADB, GR@ACE, EADI, GERAD/PERADES, DemGene, Bonn, the Rotterdam study, the CCHS study, NxC, and the UKBB (Bellenguez et al., 2022) and the individual-level data from the UK biobank (Miller et al., 2016). We downloaded summary statistics from Bellenguez et al. (Bellenguez et al., 2022) via NHGRI-EBI GWAS Catalog (Buniello et al., 2019) under accession No. GCST90027158 (Fig. 1 Block 1). The study included 487,511 European ancestry samples with 39,106 clinically diagnosed cases, 46,828 proxy cases, and 401,577 healthy controls (CN). In total, 21,101,114 SNP-AD associations were performed, where 5637 associations reached the genome-wide significance threshold of 5e-8. To match the genome builder between the downloaded AD GWAS summary statistics and our brain volumetric imaging QTs (detailed below), we converted the genome builder from GRCh38 to GRCh37 using ANNOVAR (Wang et al., 2010). We excluded all ambiguous mapping SNPs. This preserved 21,057,932 SNPs with 5625 genome-wide significant SNP-AD associations.
2.2. UKBB imaging genomics data preprocessing
The UKBB dataset recruits 500,000 UK adults sampled via population-based registries (http://www.ukbiobank.ac.uk). The UK Biobank received ethical approval from the National Research Ethics Service Committee North West–Haydock (reference 11/NW/0382). All participants provided informed consent and were aged from approximately 40 to 69 years of age at the time of enrollment (http://biobank.ctsu.ox.ac.uk/crystal/field.cgi?id=200). Participants were recruited from the United Kingdom, and initial enrollment was carried out from 2006 to 2010. Participants provided socio-demographic, cognitive, and medical data via questionnaires and physical assessments. Starting in 2014, a subset of the original sample underwent brain MRI (UK Biobank Brain Imaging Documentation, http://www.ukbiobank.ac.uk). The MRI data used in the current study were acquired between 2014 and 2019. To sum up, the image protocols of T1-weighted MRI are as follows: a 3T Siemens Skyra machine (MPRAGE) was used; image resolution: 1 × 1 × 1 mm and time to echo (TE): 2000 ms (Alfaro-Almagro et al., 2018). More details can be found at http://biobank.ctsu.ox.ac.uk/crystal/crystal/docs/brain_mri.pdf.
We created an imaging-genetics cohort from UKBB by preserving all subjects with both T1-weighted MRI data and imputed genotyping data. The UKBB imaging-genetics cohort includes 38,195 subjects with 9788 AD-by-proxy cases and 28,407 controls. AD-by-proxy is defined based on the presence of self-reported parental AD diagnosis and is widely used when the direct diagnosis of AD is not feasible (Marioni et al., 2018). The validity of the use of AD-by-proxy in AD research is supported by a few prior studies. For example, Marioni et al. (2018) found that the genetic correlation between maternal and paternal AD was not significantly different from unity, and both traits had a high genetic correlation with the case-control summary output from the International Genomics of Alzheimer’s Project (IGAP) study. This suggests that the self-reported measure of parental AD is an accurate proxy for clinical diagnosis, validating the use of AD-by-proxy (Marioni et al., 2018). Moreover, Jansen et al. conducted a large genome-wide association study of clinically diagnosed AD and AD-by-proxy and identified 29 risk loci (Jansen et al., 2019). Furthermore, a separate study conducted a comprehensive transcriptome-wide association study (TWAS) using meta-analysis results of both clinical AD and AD-by-proxy GWAS, yielding largely consistent associations that further supported the use of AD-by-proxy in AD research (Sun et al., 2021).
T1-weighted MRIs were downloaded from the UK biobank (UKBB) study (Fig. 1 Block 2) (Miller et al., 2016). Raw 3D T1-weighted MRI scans were first quality checked (QCed) for motion, image artifacts, or restricted field-of-view. Another QC was performed as follows: First, the images were examined by manually evaluating for pipeline failures (e.g., poor brain extraction, tissue segmentation, and registration errors). Furthermore, a second-step automated procedure automatically flagged images based on outlying values of quantified metrics (i.e., ROI values), and those flagged images were re-evaluated.
The quality-controlled MRI scans were first corrected for magnetic field intensity inhomogeneity (Tustison et al., 2010). Voxel-wise regional volumetric maps (RAVENS) for each tissue volume (Davatzikos et al., 2001) were generated by spatially aligning the skull-stripped images to a template residing in the MNI space (Ou et al., 2011). A multi-atlas parcellation method (MUSE) (Doshi et al., 2016) was then used to extract 145 regions of interest (ROI) from gray matter and white matter tissue maps (Supplementary eTable 1).
Raw genetic data (Version 3) were downloaded from the UKBB website (https://www.ukbiobank.ac.uk/enable-your-research/about-our-data/genetic-data) on July 2021. The imputation was performed by the original UKBB genetics study (Bycroft et al., 2018). We first conducted an initial QC according to the UKBB QC protocol (UKBB Resource 1955 and UKBB Category 100313). Then, we removed subjects with Kinship smaller than or equal to the second degree. Next, we filtered out the 1) multiallelic variants, 2) variants with missing call rates greater than 0.03, 3) variants with minor allele frequencies smaller than 0.01, and 4) variants with Hardy-Weinberg equilibrium exact test p-value below the 1e-10 threshold. Next, we filtered out the subjects 1) with missing call rate exceeding 0.03, 2) with heterozygosity rate outside 5 standard deviations of the population heterozygosity rate. Finally, we matched the QCed imputed genotyping cohort with the QCed imaging cohort. All the QC steps were completed using the PLINKv1.91 (Chang et al., 2015), PLINKv2.02 (Chang et al., 2015), and R. Our QCed UKBB imputed genetic data set consisted of 482,831 SNPs and 38,195 subjects (Fig. 1 Block 3), which was used later in our GWAS and colocalization analysis. We further derived the first 50 genetic principal components (PCs) using the FlashPCA.3
2.3. GWAS of brain imaging quantitative traits
We performed GWAS on volumetric imaging QTs of 145 MUSE ROIs using the preprocessed genotyping data (Fig. 1 Block 4). Specifically, we fit a linear regression model for each ROI-SNP pair by treating imaging volumetric QT as a dependent variable and common-variant autosomal individual SNP as an independent variable. Our model was adjusted for age, sex, and the first 10 principal components (PCs). The genome-wide significant threshold was set as 5e-8. All GWAS analyses were performed using PLINK v1.9 (Chang et al., 2015). For each ROI volumetric imaging QT, we performed the post-GWAS analysis using FUMA4 (Watanabe et al., 2017; Watanabe et al., 2019). FUMA is a web-based platform using information from multiple biological resources to facilitate functional annotation of GWAS results. We adopted the FUMA analysis protocol from our previous publication Wen et al. (Wen et al., 2022). FUMA constructed linkage disequilibrium (LD) blocks by tagging all variants with minor allele frequency greater than or equal to 0.0005 and with at least one of the independent significant variants. Of note, the LD blocks are constructed from the 1000 Genomes as reference panels, which may not be overlapped with the variants in the current study. Finally, FUMA merges the LD blocks of independent significant variants into a single genomic locus if they are within 250 kilobases from the closest boundary variants of LD blocks. We used the default parameters settings on FUMA online platform for the other unmentioned parameters. The independent significant SNPs are determined inside the borders of genomic loci defined using LD r2 < 0.6, and the leading SNPs are further clumped from the independent SNPs with the borders of genomic loci defined using LD r2 < 0.1.
2.4. Brain-wide genome-wide colocalization analysis
Genetic colocalization is a method investigating if two potentially related phenotypic traits share the common genetic causal variants in a given genome region. Intuitively, given a genome region, two traits are assumed to follow one of the five following hypotheses (Giambartolomei et al., 2014):
H0: Neither of the traits is significantly associated with any of the SNPs in the given genome region.
H1: Only trait 1 has a significant genetic association in the given genome region.
H2: Only trait 2 has a significant genetic association in the given genome region.
H3: Both traits have genetic associations in the given genome region, but with different causal variants.
H4: Both traits have genetic associations in the given genome region, and they share a single causal variant.
Giambartolomei et al. proposed a Bayesian approach to estimate the posterior probability for each of the hypotheses (Giambartolomei et al., 2014). They assumed that two traits were measured in two distinct datasets using unrelated individuals where samples were drawn from the same ethnic group. Given a region of interest (ROI) in the genome, GWAS summary statistics were obtained by treating each trait as a dependent variable and each SNP as an independent variable. They defined “configuration” for each trait by one binary vector of (0, 1) values of length , where is the number of shared variants in the given genome region of interest by both traits. The value of 1 denotes the corresponding SNP is causally associated with the trait whereas 0 denotes it is not. To calculate the posterior probability, intuitively, they group the configurations into five sets representing the assignments of all SNPs in the genome region of interest to the functional role of the five hypothesis H0, H1, H2, H3, H4. Then, the posterior probabilities can be calculated by
where is the prior probability of configuration; is the probability of the observed data given a configuration . They calculated the posterior probabilities by Approximate Bayes Factor (Wakefield 2009) with appropriate priors (Wallace 2020). In our analyses, we performed the sensitivity check for each high posterior probability H4 signal and preserved those robust signals. All colocalization analyses, sensitivity checks, and the calculation of the posterior probability of the causal SNP in the given LD block are performed via the “coloc” R package.5
In our study, we performed brain-wide genome-wide colocalization analyses on 145 MUSE ROIs and AD/CN diagnoses using the GWAS summary statistics (Fig. 1 Block 5). We first merged SNPs shared by brain imaging QTs GWAS (482,831 SNPs) and AD/CN GWAS summary statistics (21,101,114 SNPs) (Bellenguez et al., 2022). This resulted in 476,286 SNPs for the colocalization analysis. Assuming that, at most, one shared causal SNP in the given genome region of interest, we calculated the posterior probabilities for five different hypotheses adapted from the previous paragraph:
H0: Neither the AD diagnosis nor brain ROI is significantly associated with any SNPs in the given genome region.
H1: Only AD diagnosis has a significant genetic association in the given genome region.
H2: Only brain ROI has a significant genetic association in the given genome region.
H3: Both AD diagnosis and brain ROI have genetic associations in the given genome region but with different causal variants.
H4: Both AD diagnosis and brain ROI have genetic associations in the given genome region, and they share a single putative causal variant.
We applied the approximately independent linkage disequilibrium (LD) blocks derived from European populations6 (Berisa and Pickrell 2016). In their paper, they heuristically chose LD block segment boundaries given a mean segment size of 10,000 SNPs. They denoted to be number of genetic variants on a chromosome and they applied the following steps for segmentation:
Estimate the covariance matrix using shrinkage estimator from Wen and Stephens (Wen and Stephens 2010);
Convert the covariance matrix into a squared Pearson product moment correlation coefficient matrix ;
- Convert the correlation coefficient matrix to a vector using
where ; Apply low-pass filters and local search to adjust the segment boundaries. (Detailed description can be found in Wen and Stephens (Wen and Stephens 2010).)
As a result of the above LD blocks, we partitioned the genome (476,286 SNPs common SNPs) into 1703 LD blocks. The distribution of the number of SNPs within LD blocks is presented in Supplementary eFigure 1. We performed the colocalization analyses on 145 MUSE ROIs and AD diagnosis on each LD block (Fig. 1 Block 5). Triplets (LD vs. ROI vs. AD diagnosis) with posterior probability for H4 ≥ 0.8 were identified. To ensure robust colocalization results, we then performed the sensitivity check for the prioritized triplets. A sensitivity analysis is a post-hoc approach used to identify the range of prior probabilities that still support a given conclusion. This analysis involves adjusting the prior probability values and assessing their impact on the posterior probabilities for hypotheses H0 - H4. In our study, we varied the prior probability from 10−8 to10−4. If all different prior probabilities resulted in a posterior probability of “H4>0.8”, we concluded that the targeted signals had successfully passed the sensitivity check.
2.5. Mediation analysis
From the previous section, we calculated the posterior probability for each SNP within the LD being causal in the prioritized triplets. The SNP with the highest posterior probability was considered a potential causal SNP candidate for the corresponding LD in prioritized triplets. In the present section, we applied the mediation analysis7 (Baron and Kenny 1986; Tingley et al., 2014) to the prioritized potential causal SNP candidate of the corresponding triplets to distinguish the pleiotropy and causality (Fig. 1 Block 6). Specifically, after getting the posterior probability for potential causal SNP candidate for each (LD, ROI, diagnosis) triplet, we excluded all the triplets with posterior probability for potential causal SNP candidate < 0.8. We then applied the mediation analysis (Baron and Kenny 1986) on prioritized triplets using the UKBB imaging-genetics cohort. We treated the AD-by-proxy as the dependent variable , the additive recoded imputed genotyping data as independent variables , and prioritized MUSE ROI brain volumetric imaging QTs as mediators . In the first step, we regress the on using logistic regression to confirm that is a significant predictor of . In the second step, we regress the on to confirm that is a significant predictor of . In the third step, we regress the on both and to confirm that affects through the mediation of . The mediation analyses in this study are performed using the R package “mediation”8 (Tingley et al., 2014). We prioritized the triplets where the SNP mutations significantly affect the AD diagnosis indirectly by affecting the regional brain volumes.
2.6. Colocalization on mediation analysis prioritized SNP-ROI pairs
We further investigated potential molecular causal pathways for those SNPs influencing AD by affecting the regional level brain volumes (Fig. 1 Block 7, Block 8). We downloaded the Genotype-Tissue Expression (GTEx) project (Lonsdale et al., 2013) cis-eQTL data for all SNP-gene pairs of the brain tissues for the mediation analysis prioritized ROIs. In our analysis, all the mediation analyses prioritized brain ROIs were found to be located in brain cortex tissue; hence, we only focus on the brain cortex-specific cis-eQTL analysis. In GTEx cis-eQTL v7 analysis, 205 samples from healthy donors are included in their study with 64 females and 141 males. A detailed description of the project, tissue collection, and data can be found online https://www.gtexportal.org/home/.
In the GTEx cis-eQTL v7 brain cortex specific SNP-gene associations, there are in total 172,489,882 SNP-gene associations where 23,959 unique genes and 10,227,789 unique variants are included. Among all SNP-gene associations, 210,939 associations reach the genome-wide significant threshold 5e-8. For each triplet prioritized by our mediation analysis, we first selected all the genes associated with the potential causal SNP and then extracted all SNP-gene associations. We performed the colocalization analyses for each gene and each ROI (left or right entorhinal cortex separately) on their shared SNPs. We prioritized the gene-ROI pairs with H4 posterior probability > 0.5 and extracted the SNP with the highest posterior probability to be causal, given that H4 is true.
2.7. Tissue-specific differential expression analysis and single-cell-specific differential expression analysis
To characterize our imaging genomic findings, we queried the tissue-specific differential gene expression analysis and single-cell-specific differential expression analysis using the human protein atlas9 (Uhlén et al., 2015; Sjöstedt et al., 2020; Karlsson et al., al.) where they aim to map the human proteins in cells, tissues, and organs via the integration of various omics technologies (https://www.proteinatlas.org/) (Uhlén et al., 2015; Sjöstedt et al., 2020; Karlsson et al., al.). In our analysis, we focused on the results specific to brain samples and utilized their tissue-based RNA expression mapping function and single-cell-based RNA expression mapping function. (Fig. 1 Block 9, Block 10).
3. Results
3.1. Human brain volumetric changes are polygenic
Our GWAS identified 6376 genome-wide significant associations (p-value < 5e–8) with 2824 independent significant SNPs (LD r2 < 0.6) and 1824 leading SNPs (LD r2 < 0.1) (Fig. 2). The cerebellum regions were the most polygenic, with the highest independent significant SNP and leading SNP counts. Compared to other autosomal chromosomes, chromosome 12 had the largest number of lead SNP associations weighted by chromosome length. The SNP-level and gene-level Manhattan plots and QQ plots summarized by FUMA are displayed in Supplementary eFigure 2, 3, 4, and 5. All SNP-QT associations are provided in Supplementary eTable 2.
Fig. 2.
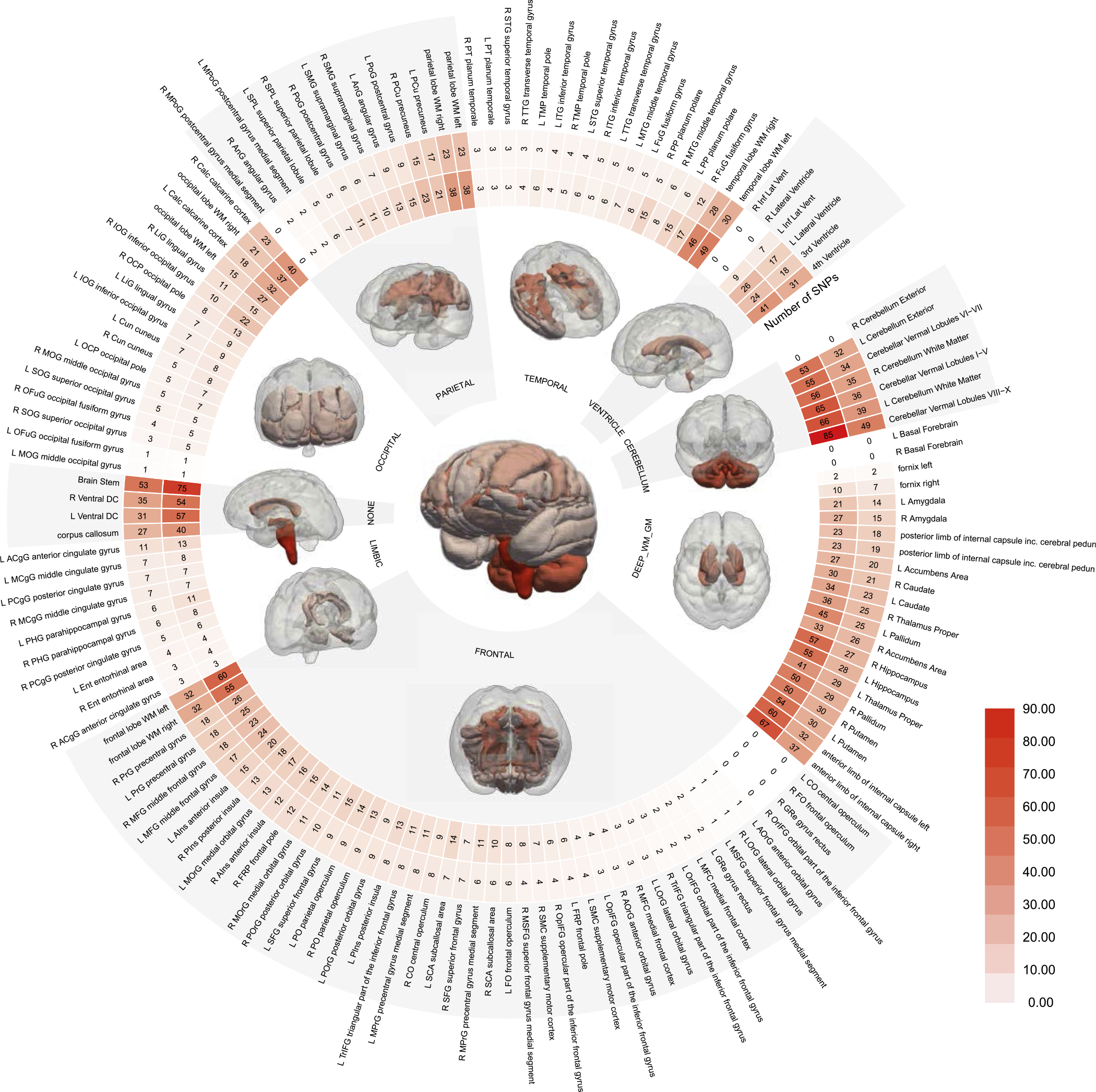
Genome-wide association study (GWAS) between brain volumes and single nucleotide polymorphisms (SNPs). Human brain volumes are highly polygenic, in particular, the cerebellum and brain stem. The circular plot shows the number of independent significant SNPs (the inner circle) and leading SNP counts (the outer circle) (Supplementary eMethod 4), respectively. The statistical brain maps present the number of lead SNPs per anatomical region (e.g., frontal cortex) and the entire brain (i.e., the central figure). Refer to Supplementary eTable 1 for details. WM: white matter; GM: gray matter.
3.2. LD-block-wised colocalization identifies 161 statistical causal LD for ROI-diagnosis pairs
The LD-block-wised colocalization produced 246,790 colocalized (LD, ROI, diagnosis) triplets. A posterior probability for each triplet indicated that the imaging QT and AD diagnosis shared a single putative causal SNP within the given LD genome region (Supplementary eTables 3 and 4) (Fig. 3). For each QT, we presented the posterior probabilities for LD blocks with the top 1% highest posterior probability for H4 (Supplementary eFigure 6). Among the 246,790 colocalized triplets, 397 had the highest posterior probability for H4 (Table 1). From the 397 triplets, we further prioritized 205 triplets with posterior probability for H4 > 0.8. Finally, we performed the sensitivity check for the prioritized 205 colocalization triplets, 161 of which passed the sensitivity check (Supplementary eTable 5, eFigure 7).
Fig. 3.
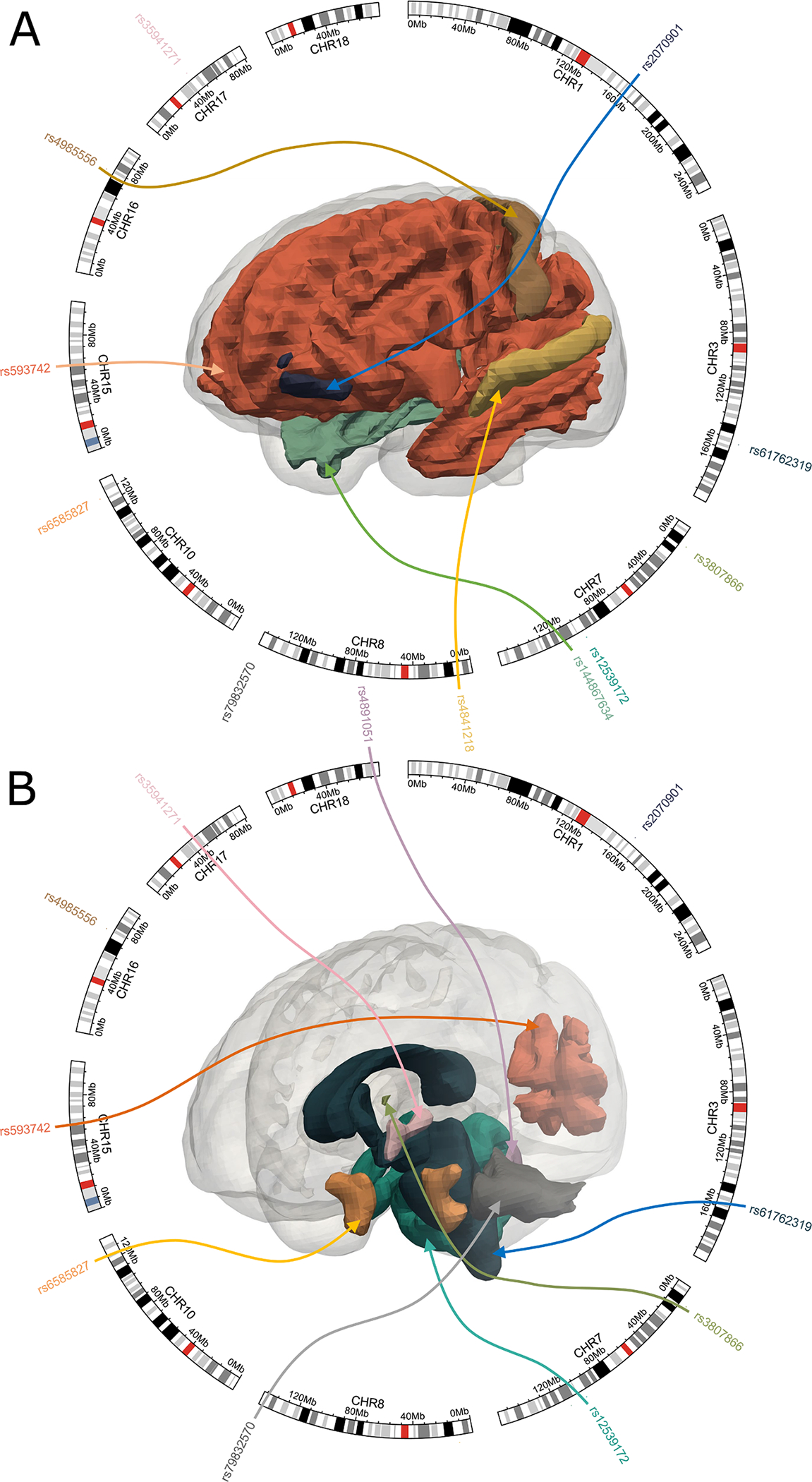
LD-block-wised colocalization analysis on AD/CN and brain volumetric quantitative traits (QTs). We performed colocalization analyses for the 145 brain volumetric QTs (i.e., MUSE ROIs) and AD/CN on the 476,286 SNPs. We prioritized 205 colocalization triplets (i.e., SNP vs. ROI vs. AD/CN) with posterior probability for H4 > 0.8. We then prioritized 161 triplets that passed the sensitivity check. We plotted the top colocalization signals for visualization with sensitivity check posterior probability > 0.99. The colored arrow represents each colocalization triplet (AD/CN is not shown). Refer to Supplementary eTable 1 for details. CN: healthy control.
Table 1.
Number of LD-ROI pairs with the highest posterior probability for H0-H4.
| Hypothesis | H0 | H1 | H2 | H3 | H4 |
|---|---|---|---|---|---|
|
| |||||
| Number of pairs | 235,486 | 7482 | 3181 | 244 | 397 |
3.3. SNP chr10:124165615:G>A influences AD by affecting the volumetric changes of the entorhinal cortex
Among the 161 triplets from the colocalization analyses, we further prioritized 27 triplets with a probability for the top causal SNP > 0.8, given that H4 was true. We performed the mediation analyses on prioritized triplets using individual-level UKBB AD-by-proxy and the imputed genotyping data. As a result, we found two triplets where the SNPs significantly affected AD outcomes indirectly mediated by imaging volumetric QTs (Table 2). Specifically, we found that chr10:124165615: G>A influences AD by affecting the volume of the entorhinal cortex in both the left and right hemispheres (in the brain cortex tissue).
Table 2.
Number of LD-ROI pairs with the highest posterior probability for H0-H4. “Img” indicates the unique identifier of the MUSE-defined ROI corresponding to Supplementary eTable 1. “SNP” denotes the SNP with the highest probability to be causal given that H4 is true in its corresponding approximate independent LD block. “AveCausalMedEffect” is the indirect effect of SNP on diagnosis mediated by corresponding ROI-level brain imaging volumetric QT. “ACME_P” is the p-value for the indirect effect of SNP on the diagnosis. “AveDirectEffect” is the direct effect of SNP on the diagnosis. “ADE_P” is the p-value for the direct effect of SNP on the diagnosis. “TotalEffect” is the total effect of SNP on diagnosis, which is equal to the sum of “AveCausalMedEffect” and “AveDirectEffect”. “TE_P” is the p-value for the total effect.
| Image | SNP | AveCausalMedEffect | ACME_P | AveDirectEffect | ADE_P | TotalEffect | TE_P |
|---|---|---|---|---|---|---|---|
|
| |||||||
| ROI105 | rs2070901 | − 0.00018 | 0.014 | − 0.00032 | 0.896 | − 0.0005 | 0.864 |
| ROI116 | rs6585827 | −0.00018 | 0.026 | −0.00897 | 0.002 | −0.00914 | 0.002 |
| ROI117 | rs6585827 | −0.0002 | 0.012 | −0.0089 | 0.006 | −0.0091 | 0.006 |
| ROI170 | rs12539172 | − 0.00012 | 0.052 | − 0.00941 | 0.002 | − 0.00954 | 0.002 |
| ROI177 | rs4985556 | 0.000279 | 0.038 | 0.003723 | 0.42 | 0.004002 | 0.382 |
| ROI201 | rs4841218 | − 3.84E-05 | 0.678 | − 0.00125 | 0.726 | − 0.00129 | 0.718 |
| ROI35 | rs61762319 | − 6.69E-05 | 0.73 | 0.006254 | 0.506 | 0.006188 | 0.51 |
| ROI38 | rs12539172 | 1.88E-05 | 0.692 | − 0.00918 | 0.01 | − 0.00916 | 0.01 |
| ROI41 | rs79832570 | 1.20E-05 | 0.942 | 0.019392 | 0 | 0.019404 | 0 |
| ROI47 | rs12539172 | − 4.29E-05 | 0.514 | − 0.00926 | 0.01 | − 0.0093 | 0.01 |
| ROI4 | rs35941271 | 0.000287 | 0.026 | 0.003151 | 0.558 | 0.003438 | 0.53 |
| ROI61 | rs61762319 | − 0.00027 | 0.068 | 0.006448 | 0.472 | 0.006174 | 0.492 |
| ROI71 | rs12539172 | 3.30E-05 | 0.572 | − 0.00923 | 0.004 | − 0.0092 | 0.006 |
| ROI72 | rs4891051 | − 4.14E-05 | 0.72 | − 0.0047 | 0.238 | − 0.00475 | 0.236 |
| ROI73 | rs17125924 | 0.000131 | 0.266 | 0.010905 | 0.056 | 0.011036 | 0.048 |
| ROI81 | rs593742 | − 5.76E-05 | 0.318 | − 0.01418 | 0 | − 0.01424 | 0 |
| ROI82 | rs593742 | − 5.03E-05 | 0.374 | − 0.01401 | 0 | − 0.01406 | 0 |
| ROI84 | rs593742 | 2.01E-06 | 0.986 | − 0.0142 | 0 | − 0.0142 | 0 |
| ROI85 | rs593742 | − 1.43E-05 | 0.77 | − 0.01423 | 0 | − 0.01424 | 0 |
| ROI86 | rs593742 | − 3.85E-05 | 0.426 | − 0.01418 | 0 | − 0.01422 | 0 |
| ROI87 | rs144867634 | − 4.44E-05 | 0.824 | − 0.00572 | 0.568 | − 0.00577 | 0.576 |
| ROI87 | rs593742 | − 7.59E-06 | 0.886 | − 0.01445 | 0 | − 0.01446 | 0 |
| ROI88 | rs593742 | − 1.75E-05 | 0.764 | − 0.01422 | 0 | − 0.01423 | 0 |
| ROI89 | rs3807866 | 5.02E-05 | 0.468 | − 0.00624 | 0.06 | − 0.00619 | 0.06 |
| ROI91 | rs61762319 | − 0.00024 | 0.146 | 0.006099 | 0.514 | 0.005861 | 0.534 |
| ROI95 | rs61762319 | − 0.00027 | 0.092 | 0.006542 | 0.496 | 0.006271 | 0.516 |
| ROI95 | rs3807866 | − 0.00011 | 0.092 | − 0.0063 | 0.036 | − 0.00641 | 0.032 |
3.4. SNP chr10:124165615:G>A affects the volumetric changes of the entorhinal cortex through the up-regulation of the BTBD16 gene
In GTEx v7 cis-eQTL analysis, 19 unique genes were associated with SNP chr10:124165615:G>A in the brain cortex. Our ROI-cis-eQTL colocalization analysis identified two triplets with posterior probability for H4 > 0.5: (LD1087, BTBD16, right entorhinal area) triplet and (LD1087, BTBD16, left entorhinal area) triplet (Table 3). Surprisingly, given H4 is true, the SNP with the highest probability to be causal is chr10:124165615:G>A, which aligns with our findings from colocalization and mediation analyses on imaging volumetric QTs and AD.
Table 3.
Colocalization between ROI-level imaging volumetric changes and brain-cortex-specific cis-eQTL on mediation analysis prioritized SNP-ROI pairs. “ROI” indicates the unique identifier of the MUSE-defined ROI corresponding to Supplementary eTable 1. “Gene” indicates the ensemble gene ID from GTEx cis-eQTL analysis v7. H0-H4 represents the posterior probability for H0-H4. “topSNP” indicates the SNP with the highest posterior probability to be causal given that H4 is true, where “top-SNPprob” is the corresponding posterior probability.
| ROI | Gene | H0 | H1 | H2 | H3 | H4 | topSNP | topSNPprob |
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| ROI116 | ENSG00000066468.16 | 0.1226 | 0.7637 | 0.0058 | 0.0360 | 0.0720 | rs6585827 | 0.9819 |
| ROI116 | ENSG00000226864.1 | 0.0000 | 0.0002 | 0.1382 | 0.8609 | 0.0008 | rs11200194 | 0.5931 |
| ROI116 | ENSG00000107669.13 | 0.1262 | 0.7865 | 0.0057 | 0.0357 | 0.0458 | rs6585827 | 0.9710 |
| ROI116 | ENSG00000107672.10 | 0.1255 | 0.7820 | 0.0059 | 0.0368 | 0.0497 | rs6585827 | 0.9646 |
| ROI116 | ENSG00000138162.13 | 0.1216 | 0.7578 | 0.0053 | 0.0329 | 0.0824 | rs6585827 | 0.9851 |
| ROI116 | ENSG00000138152.7 | 0.0538 | 0.3353 | 0.0030 | 0.0181 | 0.5899 | rs6585827 | 0.9965 |
| ROI116 | ENSG00000107679.10 | 0.0734 | 0.4571 | 0.0031 | 0.0191 | 0.4473 | rs6585827 | 0.9875 |
| ROI116 | ENSG00000254636.1 | 0.1259 | 0.7841 | 0.0064 | 0.0397 | 0.0439 | rs6585827 | 0.9626 |
| ROI116 | ENSG00000166033.7 | 0.1268 | 0.7901 | 0.0053 | 0.0328 | 0.0450 | rs6585827 | 0.9413 |
| ROI116 | ENSG00000187908.11 | 0.1256 | 0.7823 | 0.0057 | 0.0357 | 0.0507 | rs6585827 | 0.9739 |
| ROI116 | ENSG00000272135.1 | 0.1195 | 0.7439 | 0.0061 | 0.0381 | 0.0924 | rs6585827 | 0.9868 |
| ROI116 | ENSG00000138161.8 | 0.1227 | 0.7641 | 0.0098 | 0.0610 | 0.0423 | rs6585827 | 0.9707 |
| ROI116 | ENSG00000213185.2 | 0.1249 | 0.7777 | 0.0068 | 0.0426 | 0.0480 | rs6585827 | 0.9642 |
| ROI116 | ENSG00000255624.1 | 0.1242 | 0.7732 | 0.0053 | 0.0332 | 0.0641 | rs6585827 | 0.9788 |
| ROI116 | ENSG00000119965.8 | 0.1222 | 0.7607 | 0.0057 | 0.0351 | 0.0764 | rs6585827 | 0.9782 |
| ROI116 | ENSG00000179988.9 | 0.1205 | 0.7505 | 0.0068 | 0.0422 | 0.0800 | rs6585827 | 0.9815 |
| ROI116 | ENSG00000095574.7 | 0.1245 | 0.7754 | 0.0060 | 0.0371 | 0.0569 | rs6585827 | 0.9778 |
| ROI116 | ENSG00000196177.8 | 0.1181 | 0.7354 | 0.0063 | 0.0392 | 0.1009 | rs6585827 | 0.9461 |
| ROI116 | ENSG00000154473.13 | 0.1268 | 0.7894 | 0.0055 | 0.0342 | 0.0440 | rs6585827 | 0.9669 |
| ROI117 | ENSG00000066468.16 | 0.1107 | 0.7742 | 0.0052 | 0.0365 | 0.0734 | rs6585827 | 0.9969 |
| ROI117 | ENSG00000226864.1 | 0.0000 | 0.0002 | 0.1250 | 0.8741 | 0.0007 | rs11200194 | 0.5889 |
| ROI117 | ENSG00000107669.13 | 0.1141 | 0.7982 | 0.0052 | 0.0363 | 0.0463 | rs6585827 | 0.9950 |
| ROI117 | ENSG00000107672.10 | 0.1135 | 0.7938 | 0.0053 | 0.0374 | 0.0500 | rs6585827 | 0.9944 |
| ROI117 | ENSG00000138162.13 | 0.1098 | 0.7679 | 0.0048 | 0.0333 | 0.0842 | rs6585827 | 0.9975 |
| ROI117 | ENSG00000138152.7 | 0.0477 | 0.3338 | 0.0027 | 0.0180 | 0.5979 | rs6585827 | 0.9996 |
| ROI117 | ENSG00000107679.10 | 0.0656 | 0.4589 | 0.0028 | 0.0191 | 0.4535 | rs6585827 | 0.9986 |
| ROI117 | ENSG00000254636.1 | 0.1138 | 0.7960 | 0.0058 | 0.0404 | 0.0441 | rs6585827 | 0.9931 |
| ROI117 | ENSG00000166033.7 | 0.1148 | 0.8028 | 0.0048 | 0.0333 | 0.0444 | rs6585827 | 0.9910 |
| ROI117 | ENSG00000187908.11 | 0.1135 | 0.7938 | 0.0052 | 0.0362 | 0.0514 | rs6585827 | 0.9953 |
| ROI117 | ENSG00000272135.1 | 0.1077 | 0.7537 | 0.0055 | 0.0386 | 0.0945 | rs6585827 | 0.9975 |
| ROI117 | ENSG00000138161.8 | 0.1108 | 0.7754 | 0.0089 | 0.0619 | 0.0429 | rs6585827 | 0.9909 |
| ROI117 | ENSG00000213185.2 | 0.1129 | 0.7895 | 0.0062 | 0.0432 | 0.0483 | rs6585827 | 0.9938 |
| ROI117 | ENSG00000255624.1 | 0.1121 | 0.7842 | 0.0048 | 0.0337 | 0.0652 | rs6585827 | 0.9964 |
| ROI117 | ENSG00000119965.8 | 0.1103 | 0.7714 | 0.0051 | 0.0356 | 0.0776 | rs6585827 | 0.9964 |
| ROI117 | ENSG00000179988.9 | 0.1088 | 0.7608 | 0.0061 | 0.0428 | 0.0815 | rs6585827 | 0.9963 |
| ROI117 | ENSG00000095574.7 | 0.1124 | 0.7866 | 0.0054 | 0.0377 | 0.0579 | rs6585827 | 0.9959 |
| ROI117 | ENSG00000196177.8 | 0.1069 | 0.7481 | 0.0057 | 0.0399 | 0.0994 | rs6585827 | 0.9973 |
| ROI117 | ENSG00000154473.13 | 0.1146 | 0.8014 | 0.0050 | 0.0347 | 0.0443 | rs6585827 | 0.9947 |
3.5. Differential expression of the BTBD16 gene locates oligodendrocytes cell type in the brain cortex
The tissue-specific differential gene expression annotation to BTBD16 highlighted the cerebral cortex with 2.3 normalized protein-coding transcripts per million (nTPM)10 (Fig. 4). The single-cell-specific differential gene expression annotation on the brain highlighted oligodendrocytes cell type with normalized RNA expression with 10.9 nTPM11 (Fig. 5), approximately 10 folds larger than the other cell types.
Fig. 4.
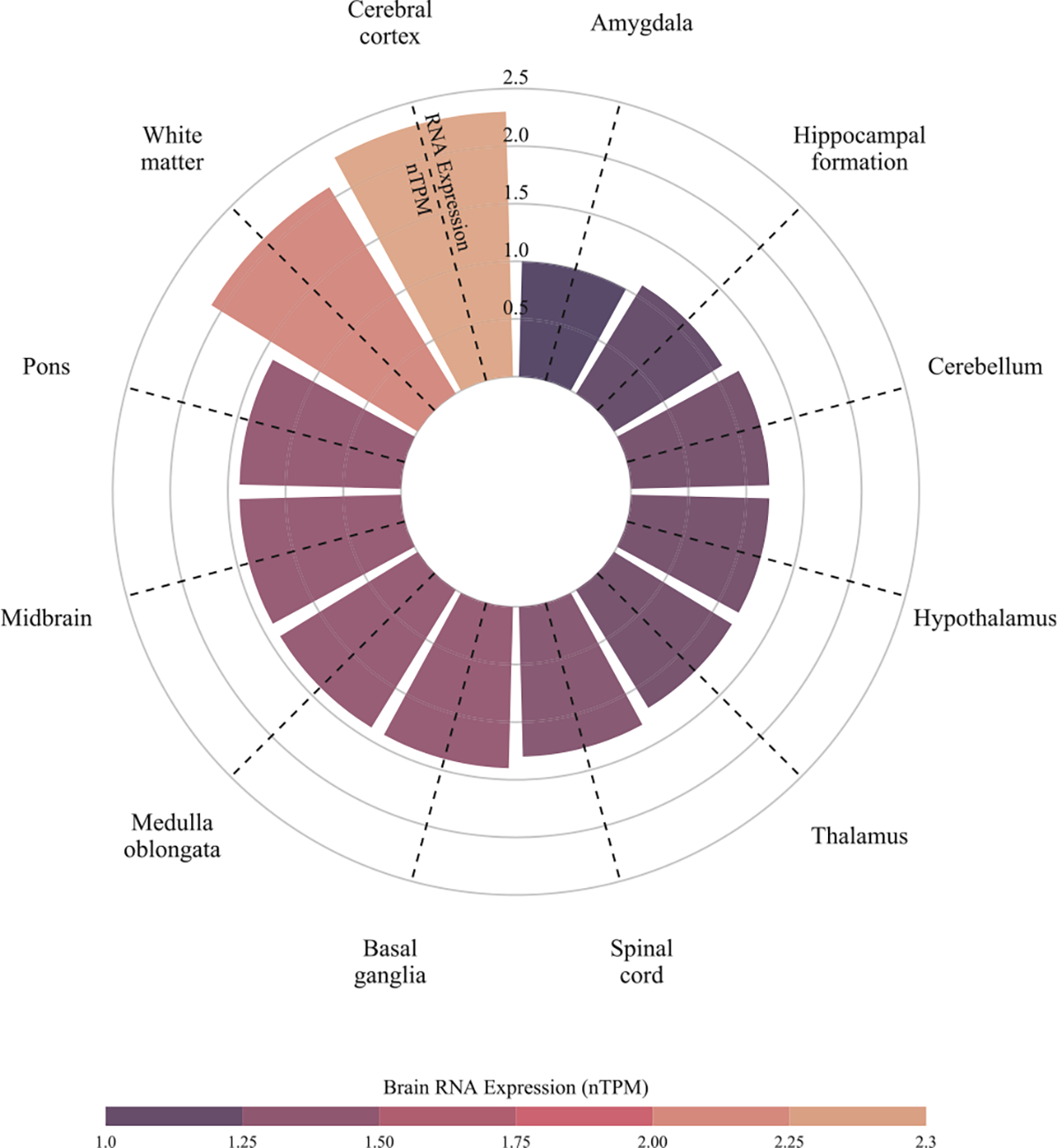
Tissue-specific differential expression of the BTBD16 gene in different brain tissues/regions. Among different brain tissues and regions, the cerebral cortex showed the highest brain RNA expression quantified by normalized protein-coding transcripts per million (nTPM). Data were downloaded from the human protein atlas (Uhlén et al., 2015; Sjöstedt et al., 2020; Karlsson et al., al.). The expression level is quantified by nTPM.
Fig. 5.
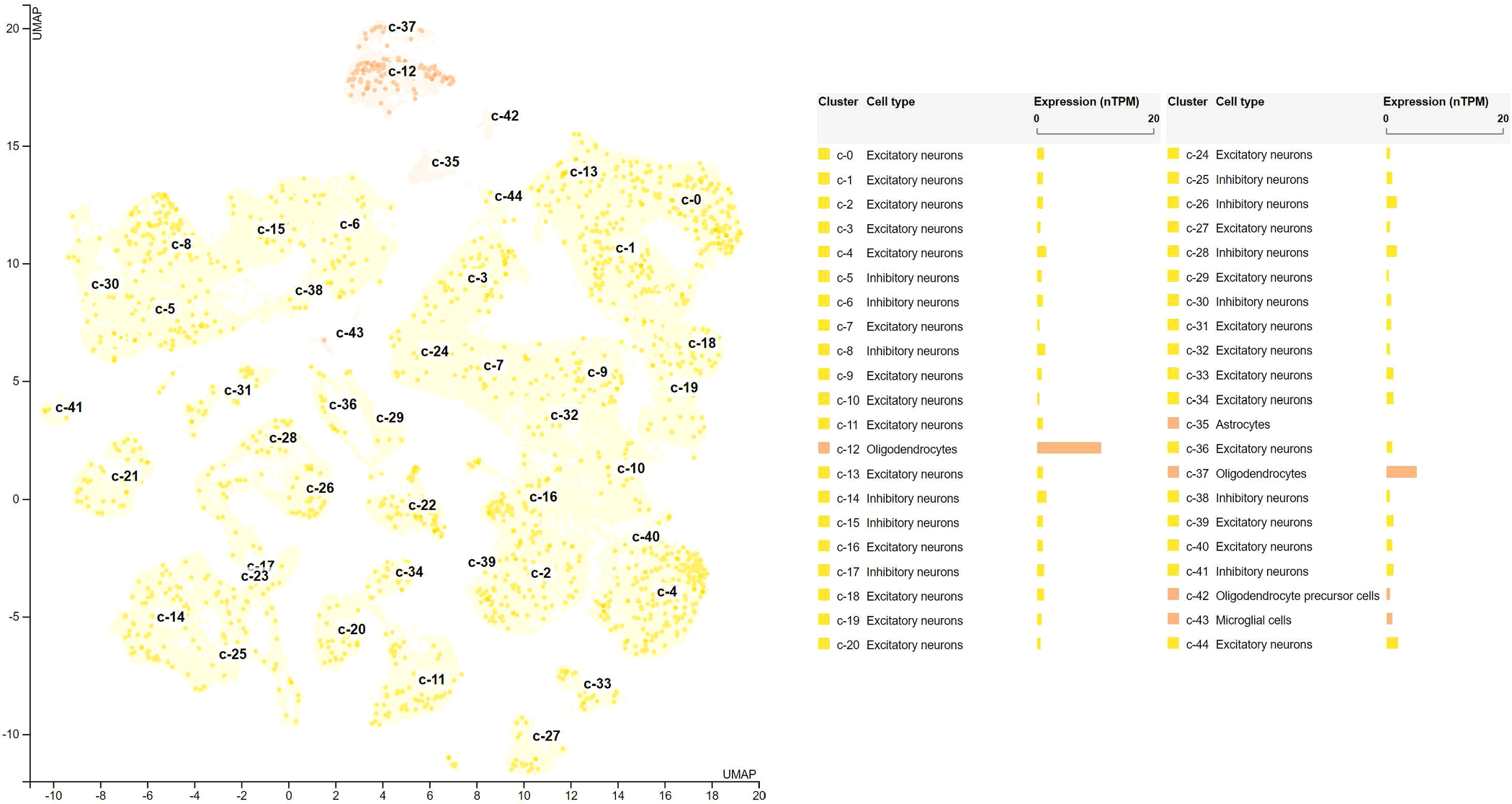
Cell-specific differential expression of BTBD16 in brain organ. The single-cell differential expression of BTBD16 highlights the oligodendrocytes cell type with normalized RNA expression with 10.9 normalized protein-coding transcripts per million (nTPM), which is more than 10 folds higher than the other cell types. The single-cell differential expression profile of BTBD16 supports our findings in the statistical causal pathway. That is, the SNP rs6585827 variant upregulates the expression of the BTBD16 gene in oligodendrocytes, specialized glial cells, in the brain cortex, and affects the diagnosis of AD/CN by affecting the regional brain volumetric variations. Data were downloaded from the human protein atlas (Uhlén et al., 2015; Sjöstedt et al., 2020; Karlsson et al., al.). The expression level is quantified by nTPM.
4. Discussion
In the current study, we conceptualized a putative AD causal pathway: the SNP chr10:124165615:G>A (rs6585827) allele upregulates the expression of the BTBD16 gene in oligodendrocytes, delays brain atrophy in the entorhinal cortex, and finally results in a protective effect of the manifestation of clinical AD (Fig. 6).
Fig. 6.
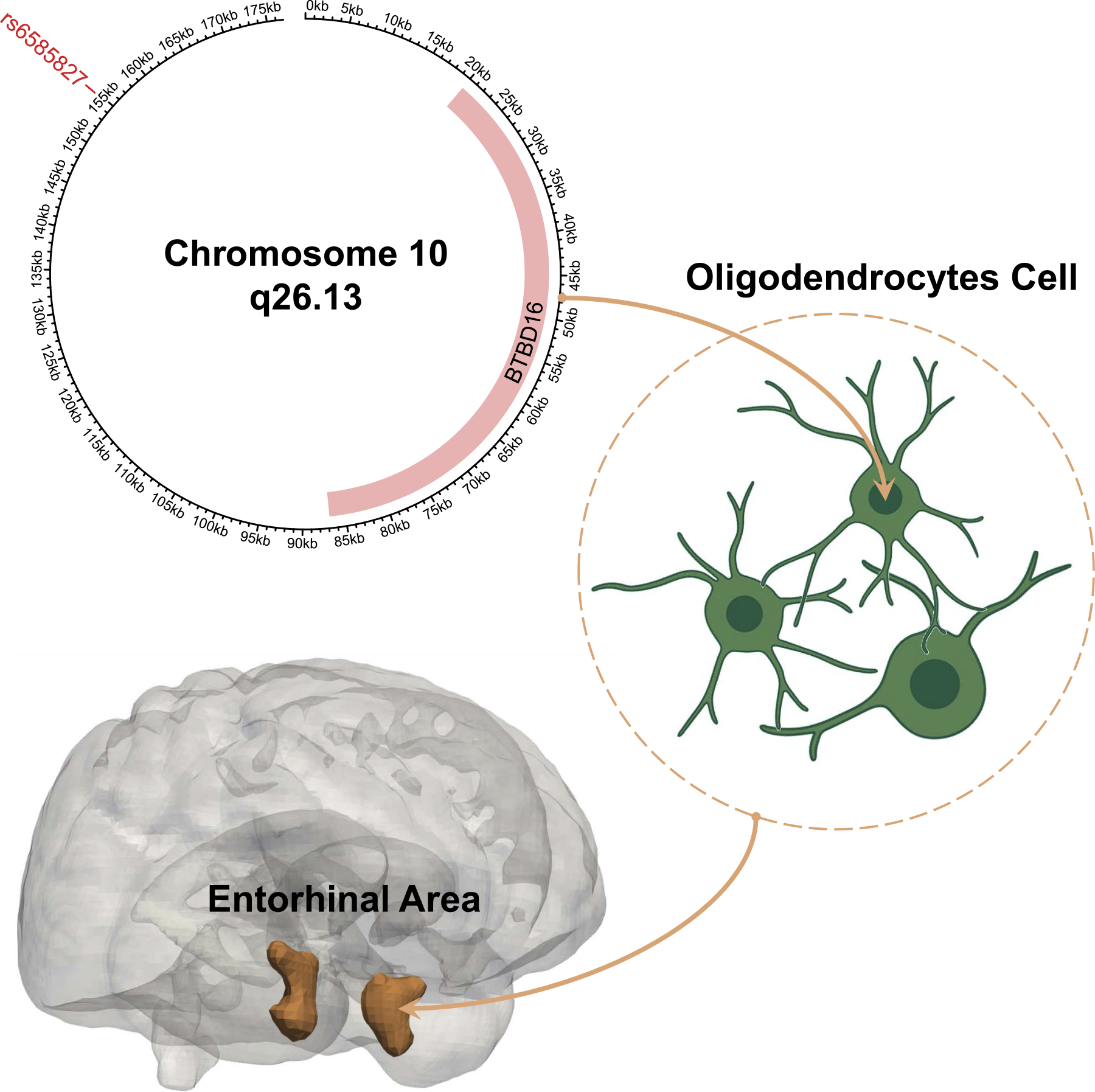
A putative AD causal pathway from genetics to the entorhinal cortex.121 Our study discovers a putative AD causal pathway: the SNP chr10:124165615:G>A (rs6585827) allele upregulates the expression of the BTBD16 gene in oligodendrocytes, leads to larger brain volumes of the entorhinal cortex, and exerts a protective effect against AD. Our evidence is summarized below: 1.SNP rs6585827 is associated with age-related macular degeneration (AMD), where the amyloid ) exists in both the AD brain and AMD drusen; they may share a common pathogenic mechanism. 2.The BTBD16 gene is a protein-coding gene that encodes a protein with a BTB/POZ domain associated with AD-related disorders. 3.The oligodendrocyte is a specialized glial cell in the central nervous system that can induce myelin breakdown. The loss of the myelin sheath may initiate AD before the appearance of amyloid and tau pathology. 4.The entorhinal cortex regions function as a widespread network hub for memory, navigation, and time suggested by rat experiments. These brain functions are tested in the clinical cognitive assessment and serve as direct evidence for AD diagnosis.
From the genomics perspective, SNP chr10:124165615:G>A was reported to have a significant association with age-related macular degeneration (AMD) (Woo et al., 2016; Anderson et al., 2010), where the amyloid ) is found to be existing in both the AD brain and AMD drusen, and they might share a common pathogenic mechanism (Ohno-Matsui 2011). A case-control study also inferred a cause-effect relation linking both diseases (Roca-Santiago et al., 2006). Kaarniranta et al. also reported the retina and brain tissue to share a similar organelle and signaling association in cellular aging processes (Kaarniranta et al., 2011).
From the molecular signature perspective, several findings in prior studies at the gene level support our AD pathway discovery. The BTBD16 gene is a protein-coding gene that encodes a protein with a BTB/POZ domain. Several studies indicated a strong association between BTBD16 and AD-related disorders. Grupe et al. found a functionally significant association between BTBD16 and late-onset AD (Grupe et al., 2006). Multiple association signals have been found between BTBD16 and AMD (Naj et al., 2013). Moreover, BTBD16 is also significantly associated with bipolar disorder (Smith et al., 2009), which is clinically and epidemiologically related to AD (Drange et al., 2019; Nunes et al., 2007; Diniz et al., 2017). A longitudinal study also showed that abnormalities of amyloid during an acute bipolar disease episode have a similar pattern as cerebrospinal fluid Aβ42 in AD patients (Knorr et al., 2022). From the genomics point of view, evidence also showed that AD and bipolar disorder might have overlap generic origin (Drange et al., 2019).
From the cell-type perspective, an oligodendrocyte is a specialized glial cell in the central nervous system (Gordon and Woodruff 2017). The oligodendrocytes in AD pathology are vulnerable and can induce myelin breakdown. Losing the myelin sheath may initiate AD before the appearance of amyloid and tau pathology (Gordon and Woodruff 2017; Cai and Xiao 2016). The formation of oligodendrocytes in the adult brain is associated with glial-restricted progenitor cells (Menn et al., 2006). The mouse models showed that the disruption of glial-restricted progenitor cells is an early pathological sign in AD and potentially accounts for accelerated myelin loss and cognitive decline (Vanzulli et al., 2020).
From the brain tissue and neuroimaging perspective, rat models suggested that the entorhinal cortex regions function as a widespread network hub for memory, navigation, and time (Tsao et al., 2018). These brain functions are tested in the clinical cognitive assessment, with results treated as direct evidence for AD diagnosis (Jack Jr et al. 2011; McKhann et al., 2011; Albert et al., 2011). The functional MRI study on humans and mice showed that the lateral entorhinal cortex dysfunction was affected in preclinical AD disease, where it spread to the parietal cortex and amyloid precursor protein expression potentiated tau toxicity in driving lateral entorhinal cortex dysfunction (Khan et al., 2014). More evidence directly associates AD with volumetric changes in the entorhinal cortex (López et al., 2014). For example, atrophy in the hippocampal and entorhinal cortex is observed in mild cognitive impairment (MCI) patients progressing to AD (deToledo-Morrell et al., 2004; Devanand et al., 2007). An anatomical study even showed that the entorhinal cortex is more involved in AD progression than the parahippocampal cortex among progressive MCI patients (Devanand et al., 2007).
Our brain imaging genomics discovery integrates individual signals across various levels, including genetics, genomics, cellular omics, brain morphometry, and disease phenotype, into a holistic neurobiological pathway. While these individual findings could be studied separately in each component of the systems biology (genome mutations, molecular signatures, imaging biomarkers), the whole is greater than the sum of the parts, and thus it is more important to connect these “dots” together to form a unified and coherent framework. Given that AD is a highly complex pathological system involving various biological processes and interactions, the discovery of our brain-wide genome-wide colocalization analysis yields new insights into the mechanistic understanding of the disease, and provides valuable guidance for subsequent therapeutic target identification and drug discovery.
Besides our major pathway findings, our ROI-level GWAS results highlight the brain stem, cerebellum, and some sub-cortical regions such as the hippocampus and putamen. Chromosome 12 has the largest number of leading SNP associations after weighting by chromosome length among all SNP-ROI-volume associations, which matches the findings from Zhao et al. (Zhao et al., 2019).
There are some limitations in our work. For example, given many stringent thresholds used in our analysis and the limited sample size in GTEx tissue-specific cis-eQTL data, some potential causal signals might be missed in our analysis. In addition, although our potential AD causal pathway underwent rigorous statistical analyses, experimental validation is still required to conclude the causality.
In summary, our brain-wide genome-wide colocalization study has identified a putative neurobiological pathway from genetics, molecular and cellular signatures, regional brain morphologic endophenotypes, to AD diagnosis. It yields new insights into the mechanistic understanding of AD, and provides valuable information for subsequent therapeutic target identification and drug discovery.
Supplementary Material
Acknowledgments
This study was partly supported by NIH grants S10 OD023495, U01 AG068057, U01 AG066833, R01 AG 071470, R01 LM013463, R01 AG058854, RF1 AG068191, RF1 AG063481, R01 AG071174. This research has been conducted using data from UK Biobank, a major biomedical database.
Footnotes
Declaration of Competing Interest
The authors declare no conflicts of interest.
CRediT authorship contribution statement
Jingxuan Bao: Conceptualization, Data curation, Formal analysis, Methodology, Visualization, Writing – original draft, Writing – review & editing. Junhao Wen: Data curation, Writing – original draft, Writing – review & editing. Zixuan Wen: Writing – review & editing. Shu Yang: Writing – review & editing. Yuhan Cui: Data curation, Writing – review & editing. Zhijian Yang: Data curation, Writing – review & editing. Guray Erus: Data curation, Writing – review & editing. Andrew J. Saykin: Writing – review & editing. Qi Long: Funding acquisition, Writing – review & editing. Christos Davatzikos: Data curation, Writing – review & editing. Li Shen: Conceptualization, Data curation, Funding acquisition, Methodology, Supervision, Writing – review & editing.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.neuroimage.2023.120346.
The oligodendrocyte cell plot is created by BioRender.com.
Data availability
The data used for the preparation of this article is from the UK Biobank (UKBB), which is publicly available at https://www.ukbiobank.ac.uk/.
References
- Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH, 2011. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheim. Dement. 7, 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfaro-Almagro F, Jenkinson M, Bangerter NK, Andersson JLR, Griffanti L, Douaud G, Sotiropoulos SN, Jbabdi S, Hernandez-Fernandez M, Vallee E, Vidaurre D, Webster M, McCarthy P, Rorden C, Daducci A, Alexander DC, Zhang H, Dragonu I, Matthews PM, Miller KL, Smith SM, 2018. Image processing and Quality Control for the first 10,000 brain imaging datasets from UK Biobank. Neuroimage 166, 400–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, Hancox LS, Hu J, Ebright JN, Malek G, Hauser MA, Bowes Rickman C, Bok D, Hageman GS, Johnson LV, 2010. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog. Retin. Eye Res. 29, 95–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron J-C, Chételat G, Desgranges B, Perchey G, Landeau B, de L, Sayette V, Eustache F, 2001. In vivo mapping of gray matter loss with voxel-based morphometry in mild Alzheimer’s disease. Neuroimage 14, 298–309. [DOI] [PubMed] [Google Scholar]
- Baron RM, Kenny DA, 1986. The moderator–mediator variable distinction in social psychological research: conceptual, strategic, and statistical considerations. J. Pers. Soc. Psychol. 51, 1173. [DOI] [PubMed] [Google Scholar]
- Bellenguez C, Küçükali F, Jansen IE, Kleineidam L, Moreno-Grau S, Amin N, Naj AC, Campos-Martin R, Grenier-Boley B, Andrade V, Holmans PA, Boland A, Damotte V, van der Lee SJ, Costa MR, Kuulasmaa T, Yang Q, de Rojas I, Bis JC, Yaqub A, Prokic I, Chapuis J, Ahmad S, Giedraitis V, Aarsland D, Garcia-Gonzalez P, Abdelnour C, Alarcón-Martín E, Alcolea D, Alegret M, Alvarez I, Álvarez V, Armstrong NJ, Tsolaki A, Antúnez C, Appollonio I, Arcaro M, Archetti S, Pastor AA, Arosio B, Athanasiu L, Bailly H, Banaj N, Baquero M, Barral S, Beiser A, Pastor AB, Below JE, Benchek P, Benussi L, Berr C, Besse C, Bessi V, Binetti G, Bizarro A, Blesa R, Boada M, Boerwinkle E, Borroni B, Boschi S, Bossù P, Bråthen G, Bressler J, Bresner C, Brodaty H, Brookes KJ, Brusco LI, Buiza-Rueda D, Bûrger K, Burholt V, Bush WS, Calero M, Cantwell LB, Chene G, Chung J, Cuccaro ML, Carracedo Á, Cecchetti R, Cervera-Carles L, Charbonnier C, Chen H-H, Chillotti C, Ciccone S, Claassen JAHR, Clark C, Conti E, Corma-Gómez A, Costantini E, Custodero C, Daian D, Dalmasso MC, Daniele A, Dardiotis E, Dartigues J-F, de Deyn PP, de P, Lopes K, de Witte LD, Debette S, Deckert J, del Ser T, Denning N, DeStefano A, Dichgans M, Diehl-Schmid J, Diez-Fairen M, Rossi PD, Djurovic S, Duron E, Düzel E, Dufouil C, Eiriksdottir G, Engelborghs S, Escott-Price V, Espinosa A, Ewers M, Faber KM, Fabrizio T, Nielsen SF, Fardo DW, Farotti L, Fenoglio C, Fernández-Fuertes M, Ferrari R, Ferreira CB, Ferri E, Fin B, Fischer P, Fladby T, Fließbach K, Fongang B, Fornage M, Fortea J, Foroud TM, Fostinelli S, Fox NC, Franco-Macías E, Bullido MJ, Frank-García A, Froelich L, Fulton-Howard B, Galimberti D, García-Alberca JM, García-González P, Garcia-Madrona S, Garcia-Ribas G, Ghidoni R, Giegling I, Giorgio G, Goate AM, Goldhardt O, Gomez-Fonseca D, González-Pérez A, Graff C, Grande G, Green E, Grimmer T, Grünblatt E, Grunin M, Gudnason V, Guetta-Baranes T, Haapasalo A, Hadjigeorgiou G, Haines JL, Hamilton-Nelson KL, Hampel H, Hanon O, Hardy J, Hartmann AM, Hausner L, Harwood J, Heilmann-Heimbach S, Helisalmi S, Heneka MT, Hernández I, Herrmann MJ, Hoffmann P, Holmes C, Holstege H, Vilas RH, Hulsman M, Humphrey J, Biessels GJ, Jian X, Johansson C, Jun GR, Kastumata Y, Kauwe J, Kehoe PG, Kilander L, Ståhlbom AK, Kivipelto M, Koivisto A, Kornhuber J, Kosmidis MH, Kukull WA, Kuksa PP, Kunkle BW, Kuzma AB, Lage C, Laukka EJ, Launer L, Lauria A, Lee C-Y, Lehtisalo J, Lerch O, Lleó A, Longstreth W, Lopez O, de Munain AL, Love S, Löwemark M, Luckcuck L, Lunetta KL, Ma Y, Macías J, MacLeod CA, Maier W, Mangialasche F, Spallazzi M, Marquié M, Marshall R, Martin ER, Montes AM, Rodríguez CM, Masullo C, Mayeux R, Mead S, Mecocci P, Medina M, Meggy A, Mehrabian S, Mendoza S, Menéndez-González M, Mir P, Moebus S, Mol M, Molina-Porcel L, Montrreal L, Morelli L, Moreno F, Morgan K, Mosley T, Nöthen MM, Muchnik C, 2022. New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berisa T, Pickrell JK, 2016. Approximately independent linkage disequilibrium blocks in human populations. Bioinformatics 32, 283–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokland GAM, de Zubicaray GI, McMahon KL, Wright MJ, 2012. Genetic and environmental influences on neuroimaging phenotypes: a meta-analytical perspective on twin imaging studies. Twin Res. Hum. Genet. 15, 351–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buniello A, MacArthur JAL, Cerezo M, Harris LW, Hayhurst J, Malangone C, McMahon A, Morales J, Mountjoy E, Sollis E, 2019. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucl. Acid. Res. 47. D1005–D12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busatto GF, Garrido GEJ, Almeida OP, Castro CC, Camargo CHP, Cid CG, Buchpiguel CA, Furuie S, Bottino CM, 2003. A voxel-based morphometry study of temporal lobe gray matter reductions in Alzheimer’s disease. Neurobiol. Aging 24, 221–231. [DOI] [PubMed] [Google Scholar]
- Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J, Cortes A, Welsh S, Young A, Effingham M, McVean G, Leslie S, Allen N, Donnelly P, Marchini J, 2018. The UK Biobank resource with deep phenotyping and genomic data. Nature 562, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Xiao M, 2016. Oligodendrocytes and Alzheimer’s disease’. Int. J. Neurosci. 126, 97–104. [DOI] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, Lee JJ, 2015. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4 s13742–015-0047–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christina P, 2018. ‘World Alzheimer report 2018. [Google Scholar]
- Davatzikos C, Genc A, Xu D, Resnick SM, 2001. Voxel-based morphometry using the RAVENS maps: methods and validation using simulated longitudinal atrophy. Neuroimage 14, 1361–1369. [DOI] [PubMed] [Google Scholar]
- Den Braber A, Bohlken MM, Brouwer RM, Ent D.van’t, Kanai R, Kahn RS, de Geus EJC, Pol HEH, Boomsma DI, 2013. Heritability of subcortical brain measures: a perspective for future genome-wide association studies. Neuroimage 83, 98–102. [DOI] [PubMed] [Google Scholar]
- deToledo-Morrell L, Stoub TR, Bulgakova M, Wilson RS, Bennett DA, Leurgans S, Wuu J, Turner DA, 2004. MRI-derived entorhinal volume is a good predictor of conversion from MCI to AD. Neurobiol. Aging 25, 1197–1203. [DOI] [PubMed] [Google Scholar]
- Devanand DP, Pradhaban G, Liu X, Khandji A, De Santi S, Segal S, Rusinek H, Pelton GH, Honig LS, Mayeux R, 2007. Hippocampal and entorhinal atrophy in mild cognitive impairment: prediction of Alzheimer disease. Neurology 68, 828–836. [DOI] [PubMed] [Google Scholar]
- Diniz BS, Teixeira AL, Cao F, Gildengers A, Soares JC, Butters MA, Reynolds III CF, 2017. History of bipolar disorder and the risk of dementia: a systematic review and meta-analysis. Am. J. Geriatr. Psychiatry 25, 357–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doshi J, Erus G, Ou Y, Resnick SM, Gur RC, Gur RE, Satterthwaite TD, Furth S, Davatzikos C, 2016. MUSE: mUlti-atlas region Segmentation utilizing Ensembles of registration algorithms and parameters, and locally optimal atlas selection. Neuroimage 127, 186–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drange OK, Smeland OB, Shadrin AA, Finseth PI, Witoelar A, Frei O, Group Psychiatric Genomics Consortium Bipolar Disorder Working, Yunpeng Wang, Sahar Hassani, and Srdjan Djurovic, 2019. Genetic overlap between Alzheimer’s disease and bipolar disorder implicates the MARK2 and VAC14 genes. In: Front. Neurosci, 13, p. 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott LT, Sharp K, Alfaro-Almagro F, Shi S, Miller KL, Douaud G, Marchini J, Smith SM, 2018. Genome-wide association studies of brain imaging phenotypes in UK Biobank. Nature 562, 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyler LT, Vuoksimaa E, Panizzon MS, Fennema-Notestine C, Neale MC, Chen C-H, Jak A, Franz CE, Lyons MJ, Thompson WK, 2014. Conceptual and data-based investigation of genetic influences and brain asymmetry: a twin study of multiple structural phenotypes. J. Cogn. Neurosci. 26, 1100–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatz M, Reynolds CA, Fratiglioni L, Johansson B, Mortimer JA, Berg S, Fiske A, Pedersen NL, 2006. Role of genes and environments for explaining Alzheimer disease. Arch. Gen. Psychiatry 63, 168–174. [DOI] [PubMed] [Google Scholar]
- Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, Plagnol V, 2014. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon R, Woodruff TM, 2017. Chapter 3 - Neuroinflammation as a therapeutic target in neurodegenerative diseases. In: Baekelandt Veerle, Lobbestael Evy (Eds.), Disease-Modifying Targets in Neurodegenerative Disorders. Academic Press. [Google Scholar]
- Grupe A, Li Y, Rowland C, Nowotny P, Hinrichs AL, Smemo S, Kauwe JSK, Maxwell TJ, Cherny S, Doil L, Tacey K, Luchene R, van Myers, A., Vrièze FW-D, Kaleem M, Hollingworth P, Jehu L, Foy C, Archer N, Hamilton G, Holmans P, Morris CM, Catanese J, Sninsky J, White TJ, Powell J, Hardy J, O’Donovan M, Lovestone S, Jones L, Morris JC, Thal L, Owen M, Williams J, Goate A, 2006. A scan of chromosome 10 identifies a novel locus showing strong association with late-onset Alzheimer’s disease. Am. Hum. Genet. 78, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel H, Bürger K, Teipel SJ, Bokde ALW, Zetterberg H, Blennow K, 2008. Core candidate neurochemical and imaging biomarkers of Alzheimer’s disease. Alzheimer. Dementia 4, 38–48. [DOI] [PubMed] [Google Scholar]
- Hormozdiari F, van de Bunt M, Segrè AV, Li X, Joo JWJ, Bilow M, Sul JH, Sankararaman S, Pasaniuc B, Eskin E, 2016. Colocalization of GWAS and eQTL signals detects target genes. Am. Hum. Genet. 99, 1245–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR Jr, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC, Thies B, Phelps CH, 2011. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia 7, 257–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen AG, Mous SE, White T, Posthuma D, Polderman TJC, 2015. What twin studies tell us about the heritability of brain development, morphology, and function: a review. Neuropsychol. Rev. 25, 27–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, Sealock J, Karlsson IK, Hägg S, Athanasiu L, Voyle N, Proitsi P, Witoelar A, Stringer S, Aarsland D, Almdahl IS, Andersen F, Bergh S, Bettella F, Bjornsson S, Brækhus A, Bråthen G, de Leeuw C, Desikan RS, Djurovic S, Dumitrescu L, Fladby T, Hohman TJ, Jonsson PV, Kiddle SJ, Rongve A, Saltvedt I, Sando SB, Selbæk G, Shoai M, Skene NG, Snaedal J, Stordal E, Ulstein ID, Wang Y, White LR, Hardy J, Hjerling-Leffler J, Sullivan PF, van der Flier WM, Dobson R, Davis LK, Stefansson H, Stefansson K, Pedersen NL, Ripke S, Andreassen OA, Posthuma D, 2019. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 51, 404–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaarniranta K, Salminen A, Haapasalo A, Soininen H, Hiltunen M, 2011. Age-related macular degeneration (AMD): alzheimer’s disease in the eye? J. Alzheimers Dis. 24, 615–631. [DOI] [PubMed] [Google Scholar]
- Karlsson Max, Cheng Zhang, Loren Méar, Wen Zhong, Andreas Digre, Borbala Katona, Evelina Sjöstedt, Lynn Butler, Jacob Odeberg, Philip Dusart, Fredrik Edfors, Per Oksvold, Kalle von Feilitzen, Martin Zwahlen, Muhammad Arif, Ozlem Altay, Xiangyu Li, Mehmet Ozcan, Adil Mardinoglu, Linn Fagerberg, Jan Mulder, Yonglun Luo, Fredrik Ponten, Mathias Uhlén, and Cecilia Lindskog. ‘A single–cell type transcriptomics map of human tissues’, Science Advances, 7: eabh2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan UA, Liu L, Provenzano FA, Berman DE, Profaci CP, Sloan R, Mayeux R, Duff KE, Small SA, 2014. Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease’. Nat. Neurosci. 17, 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knorr U, Simonsen AH, Jensen CS, Zetterberg H, Blennow K, Akhøj M, Forman J, Hasselbalch SG, Vedel Kessing L, 2022. Alzheimer’s disease related biomarkers in bipolar disorder – A longitudinal one-year case-control study’. J. Affect. Disord. 297, 623–633. [DOI] [PubMed] [Google Scholar]
- Kremen WS, Prom-Wormley E, Panizzon MS, Eyler LT, Fischl B, Neale MC, Franz CE, Lyons MJ, Pacheco J, Perry ME, 2010. Genetic and environmental influences on the size of specific brain regions in midlife: the VETSA MRI study. Neuroimage 49, 1213–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunkle BW, Grenier-Boley B, Sims R, Bis JC, Damotte V, Naj AC, Boland A, Vronskaya M, van der Lee SJ, Amlie-Wolf A, Bellenguez C, Frizatti A, Chouraki V, Martin ER, Sleegers K, Badarinarayan N, Jakobsdottir J, Hamilton-Nelson KL, Moreno-Grau S, Olaso R, Raybould R, Chen Y, Kuzma AB, Hiltunen M, Morgan T, Ahmad S, Vardarajan BN, Epelbaum J, Hoffmann P, Boada M, Beecham GW, Garnier J-G, Harold D, Fitzpatrick AL, Valladares O, Moutet M-L, Gerrish A, Smith AV, Qu L, Bacq D, Denning N, Jian X, Zhao Y, Zompo MD, Fox NC, Choi S-H, Mateo I, Hughes JT, Adams HH, Malamon J, Sanchez-Garcia F, Patel Y, Brody JA, Dombroski BA, Naranjo MCD, Daniilidou M, Eiriksdottir G, Mukherjee S, Wallon D, Uphill J, Aspelund T, Cantwell LB, Garzia F, Galimberti D, Hofer E, Butkiewicz M, Fin B, Scarpini E, Sarnowski C, Bush WS, Meslage S, Kornhuber J, White CC, Song Y, Barber RC, Engelborghs S, Sordon S, Voijnovic D, Adams PM, Vandenberghe R, Mayhaus M, Adrienne Cupples L, Albert MS, Deyn PPD, Gu W, Himali JJ, Beekly D, Squassina A, Hartmann AM, Orellana A, Blacker D, Rodriguez-Rodriguez E, Lovestone S, Garcia ME, Doody RS, Munoz-Fernadez C, Sussams R, Lin H, Fairchild TJ, Benito YA, Holmes C, Karamujić-Čomić H, Frosch MP, Thonberg H, Maier W, Roshchupkin G, Ghetti B, Giedraitis V, Kawalia A, Li S, Huebinger RM, Kilander L, Moebus S, Hernández I, Ilyas Kamboh M, Brundin RM, Turton J, Yang Q, Katz MJ, Concari L, Lord J, Beiser AS, Dirk Keene C, Helisalmi S, Kloszewska I, Kukull WA, Koivisto AM, Lynch A, Tarraga L, Larson EB, Haapasalo A, Lawlor B, Mosley TH, Lipton RB, Solfrizzi V, Gill M, Longstreth WT, Montine TJ, Frisardi V, Diez-Fairen M, Rivadeneira F, Petersen RC, Deramecourt V, Alvarez I, Salani F, Ciaramella A, Boerwinkle E, Reiman EM, Fievet N, Rotter JI, Reisch JS, Hanon O, Cupidi C, Andre Uitterlinden AG, Royall DR, Dufouil C, Maletta RG, de Rojas I, Sano M, Brice A, Cecchetti R, George-Hyslop PS, Ritchie K, Tsolaki M, Tsuang DW, Dubois B, Craig D, Wu C-K, Soininen H, Avramidou D, Albin RL, Fratiglioni L, Germanou A, Apostolova LG, Keller L, Koutroumani M, Arnold SE, Panza F, Gkatzima O, Asthana S, Hannequin D, Whitehead P, Atwood CS, Caffarra P, Hampel H, Quintela I, Carracedo Á, Lannfelt L, Rubinsztein DC, Barnes LL, Pasquier F, Frölich L, Barral S, McGuinness B, Beach TG, Johnston JA, Becker JT, Passmore P, Bigio EH, Schott JM, Bird TD, Warren JD, Boeve BF, Lupton MK, Bowen JD, Proitsi P, Boxer A, Powell JF, Burke JR, Kauwe JSK, Burns JM, Mancuso M, Buxbaum JD, Bonuccelli U, Cairns NJ, McQuillin A, Cao C, Livingston G, Carlson CS, Bass NJ, Carlsson CM, Hardy J, Carney RM, Bras J, Carrasquillo MM, Guerreiro R, Allen M, Chui HC, Fisher E, Masullo C, Crocco EA, DeCarli C, Bisceglio G, Dick M, Ma L, Duara R, Graff-Radford NR, Evans DA, Hodges A, Faber KM, Scherer M, Fallon KB, Riemenschneider M, Fardo DW, Heun R, Farlow MR, Kölsch H, Ferris S, Leber M, Foroud TM, Heuser I, Galasko DR, Giegling I, Gearing M, Hüll M, Geschwind DH, Gilbert JR, Morris J, Green RC, Mayo K, Growdon JH, Feulner T, Hamilton RL, Harrell LE, Drichel D, Honig LS, Cushion TD, Huentelman MJ, Hollingworth P, Hulette CM, Hyman BT, Marshall R, Jarvik GP, Meggy A, Abner E, Menzies GE, Jin L-W, Leonenko G, Real LM, Jun GR, Baldwin CT, Grozeva D, Karydas A, Russo G, Kaye JA, Kim R, Jessen F, Kowall NW, Vellas B, Kramer JH, Vardy E, LaFerla FM, Jöckel K-H, Lah JJ, Dichgans M, Leverenz JB, Mann D, Levey AI, Pickering-Brown S, Lieberman AP, 2019. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale J, Thomas J, Salvatore M, 2013. ‘The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López ME, Bruña R, Aurtenetxe S, Pineda-Pardo JÁ, Marcos A, Arrazola J, Reinoso AI, Montejo P, Bajo R, Maestú F, 2014. Alpha-band hypersynchronization in progressive mild cognitive impairment: a magnetoencephalography study. J. Neurosci. 34, 14551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marioni RE, Harris SE, Zhang Q, McRae AF, Hagenaars SP, David Hill W, Davies G, Ritchie CW, Gale CR, Starr JM, 2018. GWAS on family history of Alzheimer’s disease. Transl. Psychiatry 8, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jr CRJ, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH, 2011. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer. Dementia 7, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menn B, Garcia-Verdugo JM, Yaschine C, Gonzalez-Perez O, Rowitch D, Alvarez-Buylla A, 2006. Origin of oligodendrocytes in the subventricular zone of the adult brain. J. Neurosci. 26, 7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KL, Alfaro-Almagro F, Bangerter NK, Thomas DL, Yacoub E, Xu J, Bartsch AJ, Jbabdi S, Sotiropoulos SN, Andersson JLR, Griffanti L, Douaud G, Okell TW, Weale P, Dragonu I, Garratt S, Hudson S, Collins R, Jenkinson M, Matthews PM, Smith SM, 2016. Multimodal population brain imaging in the UK Biobank prospective epidemiological study. Nat. Neurosci. 19, 1523–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naj AC, Scott WK, Courtenay MD, Cade WH, Schwartz SG, Kovach JL, Agarwal A, Wang G, Haines JL, Pericak-Vance MA, 2013. Genetic factors in nonsmokers with age-related macular degeneration revealed through genome-wide gene-environment interaction analysis. Ann. Hum. Genet. 77, 215–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes PV, Forlenza OV, Gattaz WF, 2007. Lithium and risk for Alzheimer’s disease in elderly patients with bipolar disorder. Br. J. Psychiatry 190, 359–360. [DOI] [PubMed] [Google Scholar]
- Ohno-Matsui K, 2011. Parallel findings in age-related macular degeneration and Alzheimer’s disease. Prog. Retin. Eye Res. 30, 217–238. [DOI] [PubMed] [Google Scholar]
- Ou Y, Sotiras A, Paragios N, Davatzikos C, 2011. DRAMMS: deformable registration via attribute matching and mutual-saliency weighting. Med. Image Anal. 15, 622–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca-Santiago HM, Lago-Bouza JR, Millán-Calenti JC, Gómez-Ulla-Irazazábal F, 2006. [Alzheimer’s disease and age-related macular degeneration]. Arch Soc Esp Oftalmol 81, 73–78. [DOI] [PubMed] [Google Scholar]
- Scandurra V, Gialloreti LE, Barbanera F, Scordo MR, Pierini A, Canitano R, 2019. Neurodevelopmental disorders and adaptive functions: a study of children with autism spectrum disorders (ASD) and/or attention deficit and hyperactivity disorder (ADHD). Front. Psychiatry 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjöstedt E, Zhong W, Fagerberg L, Karlsson M, Mitsios N, Adori C, Oksvold P, Edfors F, Limiszewska A, Hikmet F, Huang J, Du Y, Lin L, Dong Z, Yang L, Liu X, Jiang H, Xu X, Wang J, Yang H, Bolund L, Mardinoglu A, Zhang C, von Feilitzen K, Lindskog C, Pontén F, Luo Y, Hökfelt T, Uhlén M, Mulder J, 2020. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 367, eaay5947. [DOI] [PubMed] [Google Scholar]
- Smith EN, Bloss CS, Badner JA, Barrett T, Belmonte PL, Berrettini W, Byerley W, Coryell W, Craig D, Edenberg HJ, Eskin E, Foroud T, Gershon E, Greenwood TA, Hipolito M, Koller DL, Lawson WB, Liu C, Lohoff F, McInnis MG, McMahon FJ, Mirel DB, Murray SS, Nievergelt C, Nurnberger J, Nwulia EA, Paschall J, Potash JB, Rice J, Schulze TG, Scheftner W, Panganiban C, Zaitlen N, Zandi PP, Zöllner S, Schork NJ, Kelsoe JR, 2009. ‘Genome-wide association study of bipolar disorder in European American and African American individuals. Mol. Psychiatry 14, 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Zhu J, Zhou D, Canchi S, Wu C, Cox NJ, Rissman RA, Gamazon ER, Wu L, 2021. A transcriptome-wide association study of Alzheimer’s disease using prediction models of relevant tissues identifies novel candidate susceptibility genes. Genome Med 13, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tingley D, Yamamoto T, Hirose K, Keele L, Imai K, 2014. mediation: R Package for Causal Mediation Analysis. J. Stat. Softw. 59 (5), 1–38. 10.18637/jss.v059.i05.26917999 [DOI] [Google Scholar]
- Tsao A, Sugar J, Lu L, Wang C, Knierim JJ, Moser M-B, Moser EI, 2018. Integrating time from experience in the lateral entorhinal cortex. Nature 561, 57–62. [DOI] [PubMed] [Google Scholar]
- Tustison NJ, Avants BB, Cook PA, Zheng Y, Egan A, Yushkevich PA, Gee JC, 2010. N4ITK: improved N3 Bias Correction. IEEE Trans. Med. Imaging 29, 1310–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström Björn M, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson IM, Edlund K,Lundberg E, Navani S, Szigyarto Cristina A-K, Odeberg J, Djureinovic D, Takanen Jenny O, Hober S, Alm T, Edqvist P-H, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk Jochen M, Hamsten M, Feilitzen K, von, Forsberg M, Persson L, Johansson F, Zwahlen M, Heijne G.von, Nielsen J, Pontén F, 2015. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Vanzulli I, Papanikolaou M, Chacon De-La-Rocha I, Pieropan F, Rivera AD, Gomez-Nicola D, Verkhratsky A, Rodríguez JJ, Butt AM, 2020. Disruption of oligodendrocyte progenitor cells is an early sign of pathology in the triple transgenic mouse model of Alzheimer’s disease. Neurobiol. Aging 94, 130–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos T, Allen C, Arora M, 2016. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet North Am. Ed. 388, 1545–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakefield J, 2009. Bayes factors for genome-wide association studies: comparison with P-values. Genet. Epidemiol.: Off. Public. Int. Genet. Epidemiol. Soc. 33, 79–86. [DOI] [PubMed] [Google Scholar]
- Wallace C, 2020. Eliciting priors and relaxing the single causal variant assumption in colocalisation analyses. PLoS Genet. 16, e1008720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H, 2010. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic. Acid. Res. 38 e164–e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Taskesen E, van Bochoven A, Posthuma D, 2017. Functional mapping and annotation of genetic associations with FUMA’. Nat. Commun. 8, 1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Umićević Mirkov M, de Leeuw CA, van den Heuvel MP, Posthuma D, 2019. Genetic mapping of cell type specificity for complex traits. Nat. Commun. 10, 3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Fu CHY, Tosun D, Veturi Y, Yang Z, Abdulkadir A, Mamourian E, Srinivasan D, Skampardoni I, Singh A, Nawani H, Bao J, Erus G, Shou H, Habes M, Doshi J, Varol E, Scott Mackin R, Sotiras A, Fan Y, Saykin AJ, Sheline YI, Shen L, Ritchie MD, Wolk DA, Albert M, Resnick SM, Davatzikos C, 2022a. Characterizing Heterogeneity in Neuroimaging, Cognition, Clinical Symptoms, and Genetics Among Patients With Late-Life Depression. JAMA Psychiatry 79, 464–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen J, Nasrallah IM, Abdulkadir A, 2022b. Novel genomic loci and pathways influence patterns of structural covariance in the human brain. medRxiv, 2022.07.20.22277727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen W, Thalamuthu A, Mather KA, Zhu W, Jiang J, Lafaye de Micheaux P, Wright MJ, Ames D, Sachdev PS, 2016. Distinct genetic influences on cortical and subcortical brain structures. Sci. Rep. 6, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Stephens M, 2010. Using linear predictors to impute allele frequencies from summary or pooled genotype data. Ann. Appl. Stat. 4, 1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo HJ, Yu C, Kumar K, Gold B, Reifman J, 2016. Genotype distribution-based inference of collective effects in genome-wide association studies: insights to age-related macular degeneration disease mechanism. Bmc Genom. [Electron. Resour.] 17, 695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Li T, Yang Y, Wang X, Luo T, Shan Y, Zhu Z, Xiong D, Hauberg Mads E, Bendl J, Fullard John F, Roussos P, Li Y, Stein Jason L, Zhu H, 2021. Common genetic variation influencing human white matter microstructure. Science 372, eabf3736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Luo T, Li T, Li Y, Zhang J, Shan Y, Wang X, Yang L, Zhou F, Zhu Z, Zhu H, 2019. Genome-wide association analysis of 19,629 individuals identifies variants influencing regional brain volumes and refines their genetic co-architecture with cognitive and mental health traits. Nat. Genet. 51, 1637–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used for the preparation of this article is from the UK Biobank (UKBB), which is publicly available at https://www.ukbiobank.ac.uk/.