

Abstract
Once-daily (o.d.) administration of 20 mg of amikacin per kg of body weight to neutropenic patients has been validated by clinical studies, but amikacin pharmacokinetics have been documented only for the 7.5-mg/kg twice-daily (b.i.d.) regimen in this population. In order to determine in neutropenic patients (i) the influence of the dosing regimen on the kinetics of amikacin, (ii) the linearity of kinetics of amikacin in the range of 7.5 to 20 mg/kg, and (iii) the influence of patient characteristics on the disposition of amikacin and (iv) to provide a rationale for dosing recommendations, we evaluated the population pharmacokinetics of amikacin administered to 57 febrile neutropenic adults (neutrophil count, <500/mm3) being treated for a hematological disorder and receiving amikacin at 7.5 mg/kg b.i.d. (n = 29) or 20 mg/kg o.d. (n = 28) and administered intravenously over 0.5 h. A total of 278 blood samples were obtained (1 to 14 samples per patient) during one or several administration intervals (1 to 47). Serum amikacin levels were measured by the enzyme-multiplied immunoassay technique. A mixed-effect modeling approach was used to fit a bicompartmental model to the data (NONMEM software). The influences of the dosing regimen and the demographic and biological indices on the pharmacokinetic parameters of amikacin were evaluated by the maximum-likelihood ratio test on the population model. The dosing regimen had no influence on amikacin pharmacokinetic parameters, i.e., the kinetics of amikacin were linear over the range of 7.5 to 20 mg/kg. Amikacin elimination clearance (CL) was only correlated with creatinine clearance or its covariates, namely, sex, age, body weight, and serum creatinine level. The interindividual variability of CL was 21%, while those of the central volume of distribution, the distribution clearance, and the tissue volume of distribution were 15, 30, and 25%, respectively. On the basis of the expected distribution of amikacin concentrations in this population, dosing recommendations as a function of creatinine clearance (CLCR) are proposed: for patients with normal renal function (CLCR of 80 to 130 ml/min), 20 mg/kg o.d. is recommended, whereas for patients with severe renal impairment (CLCR, 10 to 20 ml/min), a dosage of 17 mg/kg every 48 h is recommended.
Infection remains the primary cause of morbidity and mortality in neutropenic patients (6). The use of broad-spectrum antibiotics has been shown to improve significantly the prognosis of bacterial infections in these patients (30). Aminoglycosides in association with a β-lactam antibiotic are still commonly prescribed as the first-line combination during prolonged, febrile, severe neutropenia because of their broad-spectrum, peak-dependent bactericidal activities, their marked postantibiotic effects, and their ability to prevent the emergence of resistant mutants (20). The rationale for once-daily (o.d.) dosing of aminoglycosides is well established (10), and several recent studies have documented the clinical and microbiological efficacies of o.d. dosing of amikacin in combination with a β-lactamine during febrile neutropenia (9, 16). Although clinically interesting and probably cost-beneficial, these studies did not include any pharmacokinetic data apart from peak and trough concentrations, thus giving no pharmacokinetic rationale for the optimal amikacin dosage with o.d. dosing during febrile neutropenia. This lack of information is particularly important to a population in which considerable changes in pharmacokinetic parameters have been reported. These modifications concerned the aminoglycosides (11, 15, 17, 25, 40), the glycopeptides (7, 21), and, to a lesser extent, the β-lactams, and mainly consist of increased volume of distribution and/or clearance leading to low concentrations of the drugs in serum. Low serum aminoglycoside concentrations are associated with a higher risk of clinical failure (27, 28) and the selection of resistant strains (10). So far, modifications of aminoglycoside kinetics in febrile neutropenic patients have been reported for conventional dosages administered twice daily (b.i.d.) or three times daily (t.i.d.) (11, 15, 17, 25, 40). No specific study documented the pharmacokinetics of high-dose amikacin given o.d. to neutropenic patients. The use of a high dose of amikacin also raises the question of the linearity of the kinetics; i.e., do circulating amikacin concentrations remain proportional to the dose in febrile neutropenic patients? Reports on this point are in favor of proportionality in nonneutropenic patients (36, 37), although there was a tendency to a lower than expected peak in one study. The optimal peak concentration of amikacin in febrile neutropenic patients is unknown, but in nonneutropenic patients, peak concentrations in serum (measured 1 h after the start of the infusion) of <20 mg/liter in patients treated t.i.d. (28) and <40 mg/liter in intensive care unit patients treated o.d. (3) were associated with a less favorable prognosis. Hence, a less than proportional increase in the peak amikacin level could affect efficacy. Therefore, it appeared pertinent to determine potential modifications of the pharmacokinetics of amikacin administered o.d. to febrile neutropenic adults and to correlate them to the demographic and biological parameters for these patients. The most useful method for such an analysis is the population approach (33), which we recently used to study the pharmacokinetics of teicoplanin in the same population (21). Knowledge of population pharmacokinetic parameters allows individualization of the antibiotic dosage either before or after drug administration by using Bayesian methods (19).
The aims of our study were to determine for a population of febrile, severely neutropenic adults with hematological malignancies the pharmacokinetic parameters of amikacin administered o.d. or b.i.d. and the demographic and biological parameters that influence the variability of these pharmacokinetic parameters in this population and to propose adapted regimens that can be used to obtain the desired peak and trough levels in the serum of most patients as a function of creatinine clearance (CLCR).
MATERIALS AND METHODS
Patients and treatments.
Febrile neutropenic patients of both sexes (ages >18 years) with an expected duration of neutropenia of >7 days and hospitalized in single rooms of the Hematology Unit of Avicenne Hospital to receive treatment for a primary hematological disorder were included in this prospective trial. Two distinct periods were defined in the study: from January 1993 to December 1994, the patients received amikacin at 7.5 mg/kg of body weight b.i.d. combined with piperacillin (4 g t.i.d.); from January 1995 to December 1995, the patients received amikacin at 20 mg/kg (a dose which had been used in two recent large clinical studies [9, 16]) in combination with piperacillin (4 g)–tazobactam (0.5 g) t.i.d. Amikacin was administered through a short catheter by gravity flow. All of the patients had a central venous catheter, and all gave their informed consent to participate in the study. Pregnant women and human immunodeficiency virus-infected patients were not included.
Neutropenia was defined as a neutrophil count of <500/mm3, and fever was defined as a body temperature of >38.0°C measured twice within 3 h or by an episode of body temperature of >38.5°C. On the first day of neutropenia, all of these patients received partial digestive decontamination consisting of nifuroxazide (400 mg t.i.d.) and amphotericin B (500 mg t.i.d.). Systematic microbiological investigations consisted of at least three cultures of peripheral blood, a culture of blood drawn from the catheter, and urinalysis; a chest X ray was also taken. On the first day that a neutropenic patient became febrile, amikacin was injected into a peripheral vein over 30 min while the β-lactamine was given in another peripheral vein. In patients whose CLCR (estimated by the method of Cockcroft and Gault [8]) was <20 ml/min, the β-lactamine was administered at the same dose but b.i.d. The amikacin dosage was adjusted to obtain 1-h peak and 24-h trough serum amikacin levels of >40 and <5 mg/liter, respectively. These thresholds were set on the basis of the results of two studies on the efficacy of o.d. dosing of amikacin in intensive care unit patients (3, 26) and one study on the efficacy and tolerance of netilmicin with o.d. dosing (34). During the neutropenic phase a physical examination was performed at least daily for all of these patients. Teicoplanin (6 mg/kg given at 0, 12, and 24 h and then o.d.) was administered at 48 h when fever persisted or initially when infection with a gram-positive organism was suspected or documented. Patients who did not respond to this combination were given amphotericin B (1 mg/kg/day) intravenously over 6 h or any other antibiotic regimen as a function of bacteriological test results.
Measurements.
Blood samples (6 ml) were collected by a research nurse from the central venous catheter at 1 h (time of the peak concentration; measured 0.5 h after the end of the infusion), 12 h, or 24 h (time of the trough concentration) after the beginning of the first infusion and then every 3 days during the neutropenic episode for peak and trough amikacin concentration determinations. Additional samples, normally taken for the determination of biological or hematological parameters, were also obtained from most patients at 2 and 8 h during the first dosing interval and were stored for subsequent determination of serum amikacin levels. Dosing and sampling times were recorded by a research nurse. The accuracies of the records were further assessed by a pharmacist participating in the study. All the serum samples were stored and kept frozen (−20°C) until analysis. Amikacin levels were measured by the enzyme-multiplied immunoassay (Cobas, Roche, France). The limit of quantification of the assay was 2.5 mg/liter, and the precision was better than 6% over the entire calibration range (2.5 to 50 mg/liter). When concentrations were found to be greater than 50 mg/liter, the samples were diluted in order to be in the calibration range. Concentrations below the quantification limit were recorded as measured; i.e., they were neither recorded as zero nor dropped from the analysis. The following variables were recorded to evaluate their respective influences on amikacin pharmacokinetics: weight, age, sex, serum creatinine and albumin levels, and hematological parameters (in particular, leukocyte and neutrophil counts).
Pharmacokinetic modeling.
Since the sparse sampling schedule did not enable individual pharmacokinetic parameters to be estimated by usual methods for most patients, a population pharmacokinetic method based on a nonlinear mixed-effect modeling approach was used (33). Basically, an open two-compartment pharmacokinetic model with zero-order input was fitted to concentration-versus-time data for amikacin in serum. The four parameters were the elimination clearance (CL), the volume of distribution in the central compartment V1, the distribution clearance describing amikacin exchange between the central and the peripheral compartments (CLD), and the volume of the peripheral compartment (Vt). The model enabled the computation of the amikacin concentration at any time for any given dosing regimen (38).
Two levels of variability were considered. Interindividual variability was taken into account by assuming that individual pharmacokinetic parameters arise from a log-normal distribution. The value of a given parameter in subject j, Pj, represents the typical value of that parameter in the population, P̄, by Pj = P̄ exp (ηj), where ηj is a random effect normally distributed with a mean of zero and variance to be estimated in the analysis.
The second level of random variability implemented in the model was residual variability. This variability is a normally distributed random effect (ɛ) with a mean of zero and variance to be estimated. ɛ accounts for the deviation of the observed amikacin concentration (Cij) from the predicted concentration at time ti (Ĉij), Cij = Ĉij + ɛiĈijb, where Ĉij is calculated given Pj. The exponent b of the power variance model is also to be estimated.
Model building.
Assumptions about the population model (e.g., one- versus two-compartment model) were evaluated according to the likelihood ratio test (39), which was the main criterion of selection. Other criteria were the Akaike criterion (38), the aspect of the residual plots, and the values of the random-effects variance. Possible correlations between the demographic and biological indices and the parameters of the model (CL, V1, CLD, Vt) were explored by the approach proposed recently (22, 24). First, the structural model (without any covariates) was fitted to the data to obtain the population parameters (the mean and variance of each parameter). Individual pharmacokinetic parameters were obtained by using a Bayesian maximum a posteriori estimator. Second, individual parameters were regressed on the potential covariates by using a multivariate linear model after visual examination of the parameter-versus-covariate plots. Third, the relationships found in the second step were incorporated into the structural model, with the initial values of the parameters of the covariate model being set at the values found in step 2. Population and individual parameters were then reestimated as in step 1. Covariates were finally retained when the correlations were significant at the 0.05 level according to the likelihood ratio test (39).
Assessment of goodness of fit.
The population model was validated according to several criteria (1): (i) visual examination of the goodness of fit of each individual concentration-versus-time curve compared to the experimental data; (ii) visual comparison of the distribution of the standardized residuals to that of the normal distribution (N); (iii) visual examination of the scatter plot of observed versus predicted amikacin concentrations; and (iv) visual comparison of the distribution of the a posteriori estimates of the pharmacokinetic parameters with the log-normal distribution (LN).
Simulations.
The population model of amikacin in neutropenic patients was used to generate simulations of the mean ± standard deviation (SD) concentrations for 1,000 individuals by randomly choosing values of the random effects (η’s) only according to their covariance matrix. Amikacin (20 mg/kg given o.d.) was assumed to be administered intravenously over 0.5 h for 8 days. The concentrations at 1 and 23.9 h after the start of each infusion were calculated. Relevant statistics were then based on the distribution of the concentrations at each “sampling” time.
Also, in order to derive all the pharmacokinetic parameters of interest, the distribution of CL, the elimination half-life (t1/2β), and the volume of distribution at steady-state (VSS) were obtained by simulation. The values of the relevant covariates and V1, CLD, and Vt were randomly chosen for 1,000 individuals, and the corresponding values of t1/2β and VSS were calculated for each individual by using the relationships existing between these parameters (38).
Finally, in order to derive dosing recommendations, the expected distributions of the 1-h peak (after the first administration) and predose levels at steady-state amikacin concentrations were calculated by simulation for a population of 500 fictitious individuals. Body weight (required for dose simulation) was assumed to be normally distributed with mean ± SD of 67 ± 13 kg, and CLCR was assumed to be uniformly distributed within different bounds.
Programs.
Fitting of the population model and individual Bayesian estimations were made by using the NONMEM IV software (2). The first-order conditional estimation (FOCE) method was used (keyword, METHOD = COND). With the final model, the η-ɛ interaction was taken into account (keyword, INTERACTION). Simulations were performed with our POPSIM software, which has been described elsewhere (appendix of reference 35). Analysis of covariate models, statistical tests, and relevant graphs were computed by using SPSS for Windows (release 6.1; SPSS France, Boulogne, France).
RESULTS
Patients.
A total of 57 patients were enrolled in the study: 29 in the b.i.d. group and 28 in the o.d. group. Hematological disorders were similar in both groups and consisted of acute myeloblastic leukemia (n = 18), acute lymphoblastic leukemia (n = 8), non-Hodgkin’s lymphoma (n = 21), Hodgkin’s lymphoma (n = 1), myeloma (n = 7), agranulocytosis (n = 1), and aplastic anemia (n = 1). As indicated in Table 1, both groups were similar with respect to age and weight. The mean estimated CLCR value in the b.i.d. group was 12.5% lower than that in the o.d. group, but the difference was not statistically significant (the difference in the median values was only 6%).
TABLE 1.
Demographic data for the 57 febrile, severely neutropenic patients receiving amikacin o.d. or b.i.d.
| Group | No. of patients | Wt (kg)
|
Age (yr)
|
No. of males/no. of females | CLCR (ml/min)
|
|||
|---|---|---|---|---|---|---|---|---|
| Mean (range) | Median (range) | Mean (SD) | Median (range) | Mean (SD) | Median (range) | |||
| o.d. | 28 | 65.8 (13.0) | 64 (44–95) | 50.2 (16.8) | 51 (19–85) | 16/12 | 104 (39) | 107 (25–213) |
| b.i.d. | 29 | 68.1 (13.1) | 66 (49–106) | 51.3 (16.0) | 50 (18–74) | 19/10 | 91 (36) | 101 (20–150) |
Amikacin levels.
A total of 278 samples, including 93 samples containing peak concentrations, 117 samples containing trough concentrations, and 68 samples containing intermediate concentrations, were analyzed. The patients received 1 to 47 amikacin administrations, and the median number of samples per patient was 4 (range, 1 to 14). Table 2 presents the experimental concentrations of amikacin measured in both groups of patients. The interindividual variability was very broad, with the ratios between the extreme concentrations within each group being ca. 6 and 20 for peaks and trough levels, respectively. The mean peak values (normalized to the dose) reached in the o.d. and b.i.d. groups differed significantly (P < 0.0001).
TABLE 2.
Experimental concentrations of amikacin given o.d. or b.i.d. to 57 neutropenic adults
| Dosage | Timea | No. of samples | Concn (mg/liter)
|
|||
|---|---|---|---|---|---|---|
| Mean | Median | SD | Range | |||
| First day of administration | ||||||
| 20 mg/kg o.d. | P | 23 | 75.4 | 66.7 | 28.9 | 43–170 |
| T (24) | 24 | 2.1 | 1.0 | 2.8 | 0.5–14.5 | |
| 7.5 mg/kg b.i.d. | P | 23 | 36.0 | 30.4 | 14.8 | 19.4–66.0 |
| T (12) | 29 | 4.7 | 3.8 | 3.9 | 1.0–17.0 | |
| All administrations | ||||||
| 20 mg/kg o.d. | P | 43 | 74.1 | 75.5 | 26.4 | 29–170 |
| T (24) | 53 | 1.7 | 1.0 | 2.0 | 0.5–14.5 | |
| 7.5 mg/kg b.i.d. | P | 50 | 39.3 | 36.6 | 17.0 | 13.7–86.0 |
| T (12) | 64 | 5.0 | 3.7 | 4.3 | 1.0–20.7 | |
P, one-hour peak levels (i.e., 0.5 h after the end of the infusion); T (24), 24-h trough levels; T (12), 12-h trough levels.
Model building.
The main models and hypotheses tested are described in Table 3. The likelihood ratio test and the Akaike criterion showed that a two-compartment model was more adequate than a one-compartment model. Figure 1 shows the plots of predicted concentrations for a typical subject obtained with the one- and two-compartment models, corresponding to step 1 and step 2 in Table 3, respectively. The one-compartment model was unable to fit adequately peaks and 12- or 24-h trough concentrations. In the earlier steps of model building, the only significant covariate was CLCR, which explained in part the interindividual variability in amikacin clearance. The other demographic and biological indices (in particular, leukocyte and neutrophil counts) did not correlate significantly with the variations in the amikacin pharmacokinetic parameters. Therefore, the final model was first written as CLj = 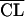 exp(ηCLj),
exp(ηCLj), 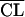 = a CLCR + b(a = 0.367, b = 1.40, and CLCR is in liters per hour), V1j =
= a CLCR + b(a = 0.367, b = 1.40, and CLCR is in liters per hour), V1j = 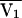 exp(ηV1j) (where
exp(ηV1j) (where 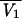 is the typical value of V1), CLDj =
is the typical value of V1), CLDj = 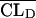 exp(ηCLDj) (where
exp(ηCLDj) (where 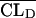 is the typical value of CLD), and Vtj =
is the typical value of CLD), and Vtj = 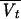 exp(ηVtj) (where
exp(ηVtj) (where 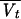 is the typical value of Vt).
is the typical value of Vt).
TABLE 3.
Main steps in population model buildinga
| Step | Model description | OBFVb | Comments |
|---|---|---|---|
| 1 | One-compartment model, CL = θ1CLCR | 1,476 | ςɛ2 = 16.8 |
| 2 | Two-compartment model, CL = θ1CLCR | 1,325 | ςɛ2 = 0.441; much better than step 1 |
| 3 | Infusion duration implemented as a random variable | 1,325 | Not better than step 2 |
| 4 |
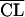 = θ1CLCR + θ2 = θ1CLCR + θ2
|
1,290 | Better than step 2 |
| 5 |
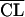 = θ1 × 6 × [θ3 − (age/100)]/(bw/SCR) + θ4, with i = 1 for males and 2 for females = θ1 × 6 × [θ3 − (age/100)]/(bw/SCR) + θ4, with i = 1 for males and 2 for females |
1,281 | Better than step 4c |
| 6 | Similar to step 5, but θ1 = θ2 | 1,285 | Sex is a significant covariatec |
| 7 to 10 | Typical values of either CL, V1, CLD, or Vt are allowed to differ according to o.d. or b.i.d. regimen | 1,282 to 1,287 | No influence of dosing regimen |
| 11 to 14 | Similar to steps 7 to 10 but with separated η’s according to o.d. or b.i.d. regimen | 1,282 to 1,289 | No influence of dosing regimen |
| 15 |
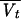 = θ7 + [(θ8 − θ7) × time]/(θ9 + time) = θ7 + [(θ8 − θ7) × time]/(θ9 + time) |
1,281 | θ8 tends to θ7; no influence of time on Vt |
| 16 | Similar to step 15, but for V1 | 1,281 | No influence of length of therapy |
| 17 |
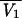 = θ5 × (bw/65) + θ6 = θ5 × (bw/65) + θ6
|
1,280 | Not significantc |
| 18 |
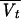 = θ7 (bw/65) + θ8 = θ7 (bw/65) + θ8
|
1,281 | Not significantc |
| 19 | Similar to step 5, but with FOCE η-ɛ interaction method | 1,248 | ςɛ2 = 0.189 |
| 20 | Similar to step 19, but C = Ĉ + ɛ1Ĉb + ɛ2 with var (ɛ2) fixed to 0.25 | 1,258 | Not better than step 19 |
After step 2 the two-compartment model was always used. After step 6 the clearance model described in step 5 was always used.
OBFV, objective function value.
Additional criteria (see Materials and Methods section) were also considered for the decision.
FIG. 1.

Simulation of amikacin kinetics for a typical patient (CLCR = 100 ml/min) using a one- or two-compartment model.
However, a significant reduction in the objective function value was obtained by replacing the estimated CLCR by a covariate model including its covariates, namely, sex, age, body weight (bw), and serum creatinine concentration (SCR) in a formula similar to that of Cockcroft and Gault (8):
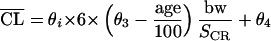 |
with i equal to 1 for men and 2 for women and the rest of the model remaining unchanged. θ is the population parameter to be estimated. Although the difference between θ1 and θ2 is small, removal of the covariate sex in the model presented above resulted in a significantly poorer fit. Allowing for covariance between η’s did not improve the fit.
The influence of the length of therapy on parameter values was first assessed by visual examination of the plots of the parameter values versus time of the last sample. Although no particular trend emerged, the influence of time was formally assessed by testing a hyperbolic relationship between V1 or Vt and time. The rationale for this relationship is that accumulation of amikacin in the deep compartment is expected to increase the apparent volume of distribution from an initial value (θ7 in step 18) to a final higher value (θ8 in step 18), with the rate of increase being controlled by θ9, the time at which half of the maximal increase is reached. However, none of the decision criteria supported this model, and the parameter values do not change during treatment.
Inclusion in the population model of a categorical covariate describing the mode of administration (o.d. or b.i.d.) in order to assess a hypothetical difference in the values of the pharmacokinetic parameters for amikacin between the two patient groups (steps 7 to 14 in Table 3) did not result in an improved fit according to the likelihood ratio test. Therefore, the values of the pharmacokinetic parameters for amikacin do not change, regardless of whether the dose is 7.5 or 20 mg/kg, i.e., amikacin pharmacokinetics are linear with respect to dose in the range of 7.5 to 20 mg/kg. The values of the parameters of the final model, based on the data for 57 patients, are summarized in Table 4.
TABLE 4.
Values of population pharmacokinetic parameters for amikacin estimated for 57 febrile, severely neutropenic patients
| Parameter | Population mean
|
% Interindividual variability
|
||
|---|---|---|---|---|
| Estimate | SE | Estimatea | SEb | |
| θ1c | 0.797 | 0.181 | ||
| θ2c | 0.640 | 0.162 | ||
| θ3c (y/100) | 0.985 | 0.106 | ||
| θ4c (liter/h) | 1.66 | 0.34 | ||
| CL (liter/h) | 21 | 8 | ||
| V1 (liter) | 8.92 | 1.17 | 15 | 6 |
| CLD (liter/h) | 4.43 | 0.76 | 30 | 6 |
| Vt (liter) | 11.4 | 1.3 | 25 | 7 |
| θ8d | 0.939 | 0.060 | ||
| ςɛ2e | 0.189 | 0.083 | ||
Estimate of variability expressed as a coefficient of variation.
Standard error (SE) of the coefficient of variation, taken as [SE (var)/var] × 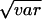 .
.
Parameters expressing clearance as a function of covariates (see text).
Exponent b of the residual-error model (see text).
Variance of the residual-error model (see text).
The graph of the predicted concentrations (more precisely, the individual predictions based on the population estimates of the values of the pharmacokinetic parameters for amikacin according to the covariates of each individual) versus observed concentrations is presented in Fig. 2. In this plot, the residuals (i.e., the difference between the observed and the predicted concentrations) are randomly distributed around the identity line, possibly with the exception of observed concentrations of >150 mg/liter, but there were only three of these. The plot of weighted residuals (i.e., the residuals divided by their SDs) versus time (Fig. 3) does not show any systematic deviation from the reference line. With this model, the population parameters have been obtained with reasonable precision, as shown by the standard errors of the estimates (Table 4).
FIG. 2.

Scatter plot of predicted versus observed amikacin concentrations. Predicted concentrations were calculated by using the population model, the covariates of each patient, and the patient’s dosing history.
FIG. 3.

Weighted residuals (i.e., the difference between the observed and the predicted concentrations normalized to their SDs) versus time. Each points represents one observation.
The correlation between amikacin clearance (a posteriori estimates) and estimated CLCR (calculated by the formula of Cockcroft and Gault [8]) is illustrated in Fig. 4. The variability in CLCR can explain 57% of the variability in amikacin clearance, and the residual interindividual variability of amikacin clearance is 21% once CLCR has been taken into account. In order to allow comparison of the amikacin kinetics reported in other publications, the characteristics of the distribution of CL, t1/2β, and VSS were derived by simulation and are summarized in Table 5. The simulation was based on data for 1,000 fictitious individuals with covariate distributions similar to those of our patients: age and body weight were assumed to be normally distributed with means ± SD of 51 ± 16 years and 67 ± 13 kg, respectively, while the serum creatinine level was assumed to be log-normally distributed, with a mean ± SD of 85 ± 37 μmol. The simulation was done separately for men and women, with the values of the population parameters for amikacin given in Table 4. The difference between men and women with respect to amikacin CL and t1/2β were small (ca. 10%). The mean predicted curve with the 95% confidence interval for male and female patients after the administration of a 20-mg/kg dose is presented in Fig. 5.
FIG. 4.

Scatter plot of amikacin clearance versus estimated CLCR. Amikacin clearance was estimated by the Bayesian method. CLCR was estimated as described by Cockcroft and Gault (8).
TABLE 5.
Derived values of population pharmacokinetic parameters for amikacin in neutropenic patientsa
| Value | CL (liters/h)
|
t1/2β (h)
|
VSS (liter) | ||
|---|---|---|---|---|---|
| Men | Women | Men | Women | ||
| Mean | 3.82 | 3.40 | 5.6 | 6.0 | 20.6 |
| SD | 1.52 | 1.28 | 2.0 | 2.2 | 3.4 |
| Median | 3.60 | 3.25 | 5.2 | 5.6 | 20.1 |
| 5 and 95b | 2.0 and 6.5 | 1.8 and 5.7 | 2.9 and 9.2 | 3.2 and 10.0 | 15.8 and 26.4 |
Based on data for 1,000 simulated individuals with the following covariate distributions: age = 51 ± 16 (N); body weight = 67 ± 13 kg (N); SCR = 85 ± 37 μmol (LN).
5 and 95, 5th and 95th percentiles of the distribution, respectively.
FIG. 5.

Simulation of amikacin kinetics at steady state for male and female patients following administration of a 20-mg/kg dose. The middle pair of curves is the mean profile based on data for 500 fictitious individuals. The upper and lower pairs of curves are ±2 SD around the mean. For each pair of curves, the upper curve is for females and the lower curve is for males.
DISCUSSION
In the studies performed earlier with neutropenic patients to determine the values of the pharmacokinetic parameters for amikacin, the drug was given at 7.5 mg/kg b.i.d. and its pharmacokinetics were determined after the administration of the first dose or at steady state. By contrast, in our study amikacin was given o.d. or b.i.d. at doses of 20 or 7.5 mg/kg to two groups of patients with severe and prolonged neutropenia. The patients also required prolonged antibiotic treatment. Amikacin concentrations were measured at several points during treatment and were analyzed by a population approach. These particular conditions gave us the opportunity to study the influences of dose, length of therapy, and demographic and biological indices on the pharmacokinetics of amikacin. Among the earlier studies, those with sufficient details about the patients, the methods, and the results are presented in Table 6. The values of the parameters have been expressed in a homogeneous system of units to allow comparison. Compared to these results, we found amikacin clearance to be lower than those in the earlier studies by about half and to have a volume of distribution similar to those in the earlier studies, which resulted in an almost doubled half-life. One possible explanation would be that NONMEM provided biased parameter estimates. However, bias in parameter estimates with NONMEM has been demonstrated only in the case of estimation by the so-called first-order method. It was shown recently that the more sophisticated FOCE method is much more accurate and yields negligible bias, at least in the examples studied (4). In our study, we used the FOCE method taking into account the η-ɛ interaction, which is a priori even more accurate than the simple FOCE method. Therefore, a large bias in our parameter estimates is unlikely. Part of the discrepancy with other studies of the pharmacokinetics of amikacin can be explained by differences in the methods applied. Indeed, the terminal half-life might have been underestimated in most studies because they were based on a two-sample (a peak and a trough) design, with the values of the pharmacokinetic parameters for amikacin being estimated by the method of Sawchuk and Zaske (31), i.e., with the implicit assumption of a one-compartment model. The study by Hary et al. (14) was based on a two-compartment model, but the samples were only taken during the 8 h following the administration of the first dose, so that it is difficult to estimate a half-life longer than 3 or 4 h. Blaser et al. (5), in a study of netilmicin administered t.i.d. or o.d. to patients with serious infections, found that the half-life estimates determined between 8 and 24 h were much longer (mean, 5 to 7 h) than those calculated between 1 and 8 h (mean, 3 h). In contrast, in our study samples were obtained at different dosing intervals and also included nonpeak and nontrough values. Thus, the discrepancies between our results and those of previous investigators in terms of amikacin clearance might be explained in part by methodological considerations.
TABLE 6.
Studies of the pharmacokinetics of amikacin given at a dosage of 7.5 mg/kg b.i.d. to neutropenic patients and healthy volunteers
| Reference | No. of subjects | CLCR (ml/min) | Subject characteristic | Sampling designa | Modelb | CL (liters/h) | VSS (liter/kg) | t1/2β (h) |
|---|---|---|---|---|---|---|---|---|
| 14 | 8 | 105 ± 11 | Nonfebrile | 10 samples (0–8 h) | Two | 9.2 | 0.45 | 2.1 |
| 17 | 10 | 108 ± 38 | Febrile | P, T | One | 7.6 | 0.40 | 2.9 |
| 11 | 28 | 86 ± 30 | Febrile | P, T | One | 5.7 | 0.38 | 3.8 |
| 40 | 27 | 103 ± 28 | Febrile | P and T at steady state | One | 7.2 | 0.40 | 2.3 |
| 14 | 8 | 106 ± 5 | Healthy volunteers | 10 samples (0–8 h) | Two | 6.7 | 0.34 | 2.3 |
| 13 | 6 | <125c | Healthy volunteers | 11 samples (0–24 h) | Two | 7.6 | 0.30 | 1.9 |
P, peak; T, trough.
One and two, one-compartment model and two-compartment model, respectively.
The value is the SCR (in micromolar).
Table 6 also presents the results of two studies of amikacin kinetics in healthy volunteers in which a two-compartment model was fitted to the data. Compared to healthy subjects, our patients had lower CL and a higher or equal volume of distribution of amikacin which resulted in a longer t1/2β.
A high proportion (40%) of amikacin clearance was not associated with CLCR in our study, which is surprising owing to the almost complete elimination of aminoglycosides by the renal route. However, similar findings were made in other population studies involving aminoglycosides (1, 26, 35). In those studies, CLCR was estimated from the serum creatinine level, usually by the formula of Cockcroft and Gault (8). Although this estimation method is one of the most precise, about one-third of the patients are not well evaluated (29), which will confound the relationship between amikacin clearance and CLCR. Other possible explanations are the tubular secretion of creatinine, which results in overestimation of the glomerular filtration rate in patients with severe renal impairment, and the possible fluctuation of CLCR over the dosing interval, which was not accounted for (only one measurement of the serum creatinine level was obtained each day).
One major goal of our study was to assess the linearity of amikacin kinetics with respect to the dose, because some reports on aminoglycoside kinetics have suggested that nonlinearity may exist. With regard to amikacin, the peak concentrations were proportional to the dose in the range of 7.5 to 15 mg/kg in one study (36), but they were less than proportional in another one (37). In the latter study, although the difference was not significant, the peaks at the higher dose were 21% lower than expected compared to the concentrations measured after administration of the lower dose. Owing to the high amikacin dose used to treat neutropenic patients (20 mg/kg), the consequences of nonlinear kinetics could have been more pronounced. However, statistical analysis based on the population model did not confirm the nonlinearity in amikacin kinetics since the values of the population parameters were not significantly different for the o.d. group and the b.i.d. group. Therefore, it can be concluded that amikacin kinetics in neutropenic patients are linear in the range of 7.5 to 20 mg/kg.
A comparison of the pharmacokinetics of amikacin given o.d. versus those of amikacin given b.i.d. has been performed for other populations, but with a smaller dose range. Maller (23) studied 45 elderly patients and found that mean peak values (measured at the end of the infusion) were 55 mg/liter after the administration of 15 mg/kg and 33 mg/liter after the administration of 7.5 mg/kg, while t1/2β (estimated after fitting a two-compartment model to the data) was 4.4 to 5.2 h. Marik (26) studied 100 critically ill patients; for a subgroup of 40 adults with CLCR above 50 ml/min/1.73 m2, they found a mean t1/2β of 3.45 h (range, 1.09 to 6.47 h). The mean ± SD 1-h peak concentrations for the 100 patients receiving drug either o.d. or b.i.d. were 33.7 ± 4.8 and 19.4 ± 3.1 mg/liter, respectively. Tulkens (36) compared amikacin given at a dosage of 14.5 mg/kg o.d. to amikacin given at a dosage of 7.7 mg/kg b.i.d. with 40 young women suffering from pelvic inflammatory disease. No difference in the values of the pharmacokinetic parameters estimated from the data of each arm was found. Therefore, our results regarding the linearity of amikacin kinetics are in agreement with those data, although we assessed a larger dose range (7.5 to 20 mg/kg). It appears that critically ill patients have lower peak concentrations than other populations, including neutropenic patients.
When the study was designed, clinical experience with amikacin given o.d. to neutropenic patients was limited, and no recommendation was available for the peak and trough serum amikacin levels. We chose to adjust the amikacin dosing to obtain 1-h peak and 24-h trough serum amikacin levels of >40 and <5 mg/liter, respectively. These breakpoints were based on (i) the study of Beaucaire et al. (3), who observed a higher mortality rate in intensive care unit patients when the first peak serum amikacin level was <40 mg/liter, and (ii) the study of Ter Braak et al. (34), who noted a 24-h trough netilmicin level of 2.8 mg/liter for patients who developed nephrotoxicity versus a level of 1.1 mg/liter for other patients. Since the recommended amikacin levels are ca. 2 times higher than those of netilmicin with the administration of multiple daily doses, we hypothesized that patients with a 24-h trough amikacin level of >5 mg/liter could be at a higher risk for nephrotoxicity. Since the present study was designed, two reports by the International Antimicrobial Therapy Cooperative Group of the European Organization for Research and Treatment of Cancer on o.d. amikacin administration for neutropenic patients have been published, and those reports support the breakpoint regarding the trough value. Those studies demonstrated that 24-h trough levels of <10 mg/liter ensure a very low incidence of nephrotoxicity. In the first study (16), nephrotoxicity occurred in 12 of 351 patients (3%) in the o.d. amikacin group, but toxicity did not develop until other nephrotoxic drugs (amphotericin B, glycopeptide antibiotics, furosemide) were used for 11 of the 12 patients. Auditory toxicity was found in 6 of 70 patients who underwent audiometric testing, but neither the peak nor the trough serum amikacin concentrations were higher in patients with ototoxicity than in those without it. In the second study (9), nephrotoxicity developed in 5 of 854 episodes (0.6%). However, in those studies, almost all patients had a trough level of <5 mg/liter. Therefore, at least when the treatment duration does not exceed 10 days, the maximal 24-h trough level of 5 mg/liter is supported by clinical data.
With regard to the peak value, it is known that a peak concentration/MIC ratio of >6 is required to obtain the highest probability of a favorable outcome in immunocompetent patients (27). Although we did not assess the relationship between peak amikacin concentration and short-term outcome, it has been reported that neutropenic patients with gram-negative bacterial infections require higher peak bactericidal concentrations than nonneutropenic patients to improve the outcome (32). Moreover, the postantibiotic effect of aminoglycosides is dependent on the peak concentration and time of exposure, but it is markedly reduced in neutropenic animals (10, 12). Finally, adaptive resistance to aminoglycosides (i.e., the increase in the MIC after the first exposure to the antibiotic) is decreased by a factor of 2 to 3 when the peak concentration/MIC ratio increases from 8 to 24 (18). Therefore, the value of 40 mg/liter that holds for intensive care unit patients might be too low for neutropenic patients. Since the MICs at which 90% of strains susceptible to amikacin are inhibited are <8 mg/liter, a peak amikacin level of >60 mg/liter seems to be a reasonable goal for avoiding inefficacy in severely neutropenic patients.
It has been shown that the peak serum amikacin level obtained after the administration of the first dose is the most important factor for a favorable outcome (3, 28). Therefore, the population model was used to propose dosing recommendations for amikacin in febrile neutropenic patients, individualized on the basis of their biological and demographic characteristics. For the sake of simplicity, the population model involving only the estimated CLCR was used. The goal was to adjust the dose and its administration interval so that 90% of the patients would have a peak serum amikacin level of >60 mg/liter (1 h after the start of the first administration) and 95% of the patients would have a trough serum amikacin level of <5 mg/liter (predose level at steady state). Our proposals for obtaining these objectives are summarized in Table 7. These proposals should now be prospectively correlated with clinical and microbiological outcomes to determine their relevance.
TABLE 7.
Proposed amikacin dosing regimens to achieve first 1-h-peak level of >60 mg/liter in 90% of patients and trough (predose) level at steady state of <5 mg/liter in 95% of patients
| CLCR (ml/min) | Dose (mg/kg) | Interval (h) |
|---|---|---|
| 80–130 | 20 | 24 |
| 60–80 | 22 | 36 |
| 40–60 | 20 | 36 |
| 20–40 | 20 | 48 |
| 10–20 | 17 | 48 |
With regard to the consequences for therapeutic drug monitoring in clinical practice, two samples (one with a peak concentration and one with a trough concentration) should be obtained at 1 and 12 h (regardless of the dosing interval) after administration of the first dose. Sampling at 12 h ensures that the amikacin concentration will be measurable. If necessary, the dosing schedule should be individualized by using the Bayesian method based on the population model described in this study. Serum amikacin levels (1-h peak and predose trough levels) should be controlled after the third dose has been given and should be further monitored if the patient’s renal function is unstable.
REFERENCES
- 1.Aarons L, Vozeh S, Wenk M, Weiss P, Follath F. Population pharmacokinetics of tobramycin. Br J Clin Pharmacol. 1989;28:305–314. doi: 10.1111/j.1365-2125.1989.tb05431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beal S L, Boeckman A, Sheiner L B. NONMEM user’s guides, version IV. San Francisco: NONMEM Project Group, University of California; 1992. [Google Scholar]
- 3.Beaucaire, G., O. Leroy, C. Beuscart, P. Karp, C. Chidiac, and M. Caillaux. 1991. Clinical and bacteriological efficacy, and practical aspects of amikacin given once daily for severe infections. J. Antimicrob. Chemother. 27(Suppl. C):91–103. [DOI] [PubMed]
- 4.Bennett J E, Wakefield J C. A comparison of a Bayesian population method with two methods as implemented in commercially available software. J Pharmacokinet Biopharm. 1996;24:403–432. doi: 10.1007/BF02353520. [DOI] [PubMed] [Google Scholar]
- 5.Blaser J, Simmen H P, Thurnheer U, König C, Lüthy R. Nephrotoxicity, high frequency ototoxicity, efficacy and serum kinetics of once daily versus thrice daily dosing of netilmicin in patients with serious infections. J Antimicrob Chemother. 1995;36:803–814. doi: 10.1093/jac/36.5.803. [DOI] [PubMed] [Google Scholar]
- 6.Brown A E. Neutropenia, fever and infection. Am J Med. 1984;76:421–428. doi: 10.1016/0002-9343(84)90661-2. [DOI] [PubMed] [Google Scholar]
- 7.Chang D, Liem L, Malagolowkin M. A prospective study of vancomycin pharmacokinetics and dosage requirements in pediatric cancer patients. Pediatr Infect Dis J. 1994;13:969–974. doi: 10.1097/00006454-199411000-00007. [DOI] [PubMed] [Google Scholar]
- 8.Cockcroft D W, Gault M H. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–47. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 9.Cometta A, Zinner S, de Bock R, Calandra T, Gaya H, Klastersky J, Langenaeken J, Paesmans M, Viscoli C, Glauser M P the International Antimicrobial Therapy Cooperative Group of the European Organization for Research and Treatment of Cancer. Piperacillin-tazobactam plus amikacin versus ceftazidime plus amikacin as empiric therapy for fever in granulocytopenic patients with cancer. Antimicrob Agents Chemother. 1995;39:445–452. doi: 10.1128/aac.39.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Craig, W. A. 1995. Once-daily versus multiple-daily dosing of aminoglycosides. J. Chemother. 7(Suppl. 2):47–52. [PubMed]
- 11.Davis R L, Lehman D, Stidley C A, Neidhart J. Amikacin pharmacokinetics in patients receiving high-dose cancer chemotherapy. Antimicrob Agents Chemother. 1991;35:944–947. doi: 10.1128/aac.35.5.944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fantin B, Elbert S, Leggett J, Vogelman B, Craig W A. Factors affecting duration of in vivo post-antibiotic effect for aminoglycosides against gram-negative bacilli. J Antimicrob Chemother. 1991;27:829–836. doi: 10.1093/jac/27.6.829. [DOI] [PubMed] [Google Scholar]
- 13.Garraffo R, Drugeon H B, Dellamonica P, Bernard E, Lapalus P. Determination of optimal dosage regimen for amikacin in healthy volunteers by study of pharmacokinetics and bactericidal activity. Antimicrob Agents Chemother. 1990;34:614–621. doi: 10.1128/aac.34.4.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hary L, Andrejak M, Bernaert F, Desablens B. Pharmacokinetics of amikacin in neutropenic patients. Curr Ther Res. 1989;46:821–827. [Google Scholar]
- 15.Higa G M, Murray W E. Alterations in aminoglycoside pharmacokinetics in patients with cancer. Clin Pharm. 1987;6:963–966. [PubMed] [Google Scholar]
- 16.The International Antimicrobial Therapy Cooperative Group of the European Organization for Research and Treatment of Cancer. Efficacy and toxicity of single daily doses of amikacin and ceftriaxone versus multiple daily doses of amikacin and ceftazidime for infection in patients with cancer and granulocytopenia. Ann Intern Med. 1993;119:584–593. doi: 10.7326/0003-4819-119-7_Part_1-199310010-00006. [DOI] [PubMed] [Google Scholar]
- 17.Kaojarern S, Maoleekoonpairoj S, Atichartakarn V. Pharmacokinetics of amikacin in hematologic malignancies. Antimicrob Agents Chemother. 1989;33:1406–1408. doi: 10.1128/aac.33.8.1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karlowsky J A, Zhanel G G, Davidson R J, Hoban D J. Post antibiotic effect in Pseudomonas aeruginosa following single and multiple aminoglycoside exposures in vitro. J Antimicrob Chemother. 1994;33:937–947. doi: 10.1093/jac/33.5.937. [DOI] [PubMed] [Google Scholar]
- 19.Kosirog J L, Rospond R M, Destache C, Hall P. Aminoglycoside forecasting in neutropenic patients with cancer. Clin Pharmacokinet. 1993;24:79–87. doi: 10.2165/00003088-199324010-00007. [DOI] [PubMed] [Google Scholar]
- 20.Lortholary O, Tod M, Cohen Y, Petitjean O. Aminoglycosides. Med Clin N Am. 1995;79:761–787. doi: 10.1016/s0025-7125(16)30038-4. [DOI] [PubMed] [Google Scholar]
- 21.Lortholary O, Tod M, Rizzo N, Padoin C, Biard O, Casassus P, Guillevin L, Petitjean O. Population pharmacokinetic study of teicoplanin in severely neutropenic patients. Antimicrob Agents Chemother. 1996;40:1242–1247. doi: 10.1128/aac.40.5.1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maitre P O, Bührer M, Thomson D, Stanski D R. A three step approach combining Bayesian regression and NONMEM population analysis: application to midazolam. J Pharmacokinet Biopharm. 1991;19:377–384. doi: 10.1007/BF01061662. [DOI] [PubMed] [Google Scholar]
- 23.Maller R, Isaksson B, Nilsson L, Soren L. A study of amikacin given once versus twice daily in serious infections. J Antimicrob Chemother. 1988;22:75–79. doi: 10.1093/jac/22.1.75. [DOI] [PubMed] [Google Scholar]
- 24.Mandema J W, Verotta D, Sheiner L B. Building population pharmacokinetic-pharmacodynamic models: models for covariate effects. J Pharmacokinet Biopharm. 1992;20:511–528. doi: 10.1007/BF01061469. [DOI] [PubMed] [Google Scholar]
- 25.Manny R P, Hutson P R. Aminoglycoside volume of distribution in hematology-oncology patients. Clin Pharm. 1986;5:629–632. [PubMed] [Google Scholar]
- 26.Marik, P. E., I. Havlik, F. S. E. Monteagudo, and J. Lipman. 1991. The pharmacokinetics of amikacin in critically ill adult and pediatric patients: comparison of once-versus twice-daily dosing regimens. J. Antimicrob. Chemother. 27(Suppl. C):81–89. [DOI] [PubMed]
- 27.Moore R D, Lietman P S, Smith C R. Clinical response to aminoglycoside therapy: importance of the ratio of peak concentration to minimal inhibitory concentration. J Infect Dis. 1987;155:93–99. doi: 10.1093/infdis/155.1.93. [DOI] [PubMed] [Google Scholar]
- 28.Moore R D, Smith C R, Lietman P S. Association of aminoglycoside plasma levels with therapeutic outcome in gram-negative pneumonia. Am J Med. 1984;77:657–662. doi: 10.1016/0002-9343(84)90358-9. [DOI] [PubMed] [Google Scholar]
- 29.O’Connell M B, Dwinell A M, Bannick-Mohrland S D. Predictive performance of equations to estimate creatinine clearance in hospitalized elderly patients. Am Pharmacother. 1992;26:627–632. doi: 10.1177/106002809202600503. [DOI] [PubMed] [Google Scholar]
- 30.Pizzo P A, Hathorn J W, Hiemenz J, Browne M, Commers J, Cotton D, Gress J, Longo D, Marshall D, McKnight J, Rubin M, Skelton J, Thaler M, Wesley R. A randomized trial comparing ceftazidime alone with combination antibiotic therapy in cancer patients with fever and neutropenia. N Engl J Med. 1986;315:552–558. doi: 10.1056/NEJM198608283150905. [DOI] [PubMed] [Google Scholar]
- 31.Sawchuck R J, Zaske D E. Pharmacokinetics of dosing regimens which utilize multiple intravenous infusions: gentamicin in burn patients. J Pharmacokinet Biopharm. 1976;4:183–195. doi: 10.1007/BF01086153. [DOI] [PubMed] [Google Scholar]
- 32.Sculier J P, Klastersky J. Significance of serum bactericidal activity in gram-negative bacillary bacteremia in patients with and without granulocytopenia. Am J Med. 1984;76:429–435. doi: 10.1016/0002-9343(84)90662-4. [DOI] [PubMed] [Google Scholar]
- 33.Sheiner L B, Ludden T M. Population pharmacokinetics/dynamics. Annu Rev Pharmacol Toxicol. 1992;32:185–209. doi: 10.1146/annurev.pa.32.040192.001153. [DOI] [PubMed] [Google Scholar]
- 34.Ter Braak E W, De vries P J, Bouter K P, Van der Vegt S G, Dorrestein G C, Nortier J W. Once-daily dosing regimen for aminoglycoside plus beta-lactam combination therapy of serious bacterial infections: comparative trial with netilmicin plus ceftriaxone. Am J Med. 1990;89:58–66. doi: 10.1016/0002-9343(90)90099-y. [DOI] [PubMed] [Google Scholar]
- 35.Tod M, Padoin C, Minozzi C, Cougnard J, Petitjean O. Population pharmacokinetic study of isepamicin in intensive care unit patients. Antimicrob Agents Chemother. 1996;40:983–987. doi: 10.1128/aac.40.4.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tulkens, P. M. 1991. Pharmacokinetic and toxicological evaluation of a once-daily regimen versus conventional schedules of netilmicin and amikacin. J. Antimicrob. Chemother. 27(Suppl. C):49–61. [DOI] [PubMed]
- 37.Van der Auwera P, Klastersky J. Serum bactericidal activity and post antibiotic effect in serum of patients with urinary tract infection receiving high dose amikacin. Antimicrob Agents Chemother. 1987;31:1061–1068. doi: 10.1128/aac.31.7.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wagner J G. Pharmacokinetics for the pharmaceutical scientist. Lancaster, Pa: Technomic Publishing Co.; 1993. [Google Scholar]
- 39.White D B, Walawander C A, Liu D Y, Grasela T H. Evaluation of hypothesis testing for comparing two populations using NONMEM analysis. J Pharmacokinet Biopharm. 1992;20:295–313. doi: 10.1007/BF01062529. [DOI] [PubMed] [Google Scholar]
- 40.Zeitany R G, El Saghir N S, Santhosh-Kumar C R, Sigmon M A. Increased aminoglycoside dosage requirements in hematologic malignancy. Antimicrob Agents Chemother. 1990;34:702–708. doi: 10.1128/aac.34.5.702. [DOI] [PMC free article] [PubMed] [Google Scholar]