

Abstract
Mesenchymal stem cells (MSCs) exert beneficial therapeutic effects in acute kidney injury (AKI), while the detailed repair mechanism remains unclear. Herein, we probed the underlying mechanisms of MSC therapy in AKI by performing unbiased single-cell RNA sequencing in IRI model with/without MSC treatment. Our analyses uncovered the tubular epithelial cells (TECs) and immune cells transcriptomic diversity and highlighted a repair trajectory involving renal stem/progenitor cell differentiation. Our findings also suggested that profibrotic TECs expressing pro-fibrotic factors such as Zeb2 and Pdgfb promoted the recruitment of inflammatory monocytes and Th17 cells to injured kidney tissue, inducing TGF-β1 secretion and renal fibrosis. Finally, in addition to activating the repair properties of renal progenitor/stem cells, we uncovered a role for MSC-derived miR-26a-5p in mediating the therapeutic effects of MSCs by inhibiting Zeb2 expression and suppressing pro-fibrotic TECs and its subsequent recruitment of immune cell subpopulations. These findings may help to optimize future AKI treatment strategies.
Keywords: acute kidney injury, mesenchymal stem cells, single-cell sequencing, intercellular crosstalk, trajectory analysis
Graphical abstract
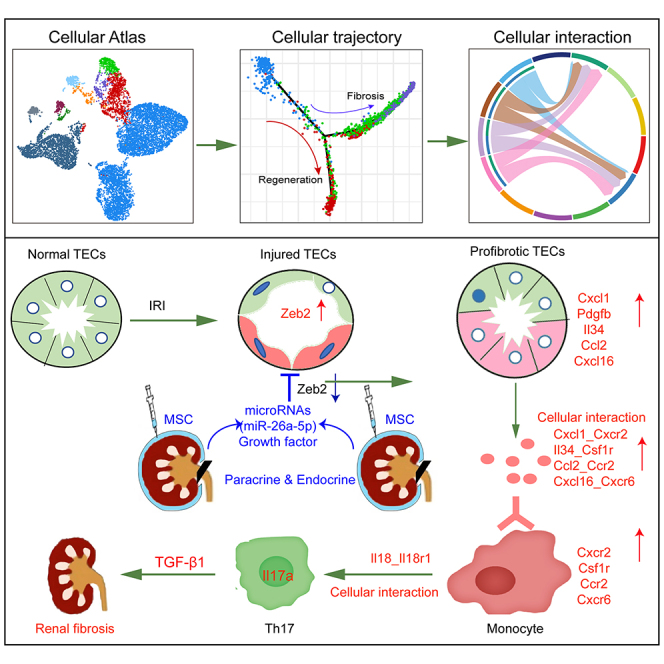
Cai and colleagues found that MSCs exhibit promising therapeutic effects on acute kidney injury by inducing proliferation and differentiation of Prom1+Sox9+Pcna+ progenitor epithelium and secreting miR-26a to inhibit Zeb2 expression and the infiltration of myeloid cells and Th17 cells, providing a theoretical basis for the clinical application of MSCs.
Introduction
Acute kidney injury (AKI) is a clinical syndrome characterized by a sharp decline in renal function.1 AKI occurs in approximately 10%–15% of inpatients, but its incidence is more than 50% in patients receiving treatment in intensive care.2 To date, there are no effective clinical treatments capable of reducing tissue damage, promoting repair, or preventing the development of chronic fibrosis following AKI.
The clinical application of mesenchymal stem cells (MSCs) in regenerative medicine has drawn increasing attention in recent years. MSCs from various sources have also been found to promote tissue repair and modulate immune responses in renal disease.3 They are known to exert many of their functions via the secretion of extracellular vesicles (EVs), which mainly mediate intercellular interaction by delivering a variety of bioactive substances such as nucleic acids (including coding and non-coding RNAs) and proteins to recipient cells.4,5 The molecules delivered in this way promote cell proliferation and suppress both apoptosis and inflammation, thereby stimulating tissue repair and regeneration following injury.6,7
Despite the benefits of MSC treatment, the cellular landscape in the MSC-treated AKI microenvironment and the mechanisms and targets of MSC therapy-induced repair and regeneration in AKI remain incompletely understood. The current lack of mechanistic understanding of the effects of MSC therapy in AKI affects renal repair after IRI by modulating the immune response. To address this, we carried out an unbiased single-cell transcriptomic analysis of kidney tissues in a murine model of AKI with or without umbilical cord MSC treatment. Using this approach, we hoped to elucidate the mechanism by which MSCs regulated the tubular epithelial cells (TECs) repair and immune inflammation underlying the MSC-treated AKI microenvironment and determine a theoretical basis for stem cell treatment in AKI.
Results
Cellular characteristics of the IRI-AKI microenvironment and response to MSC therapy
To comprehensively dissect the cellular and molecular characteristics of the MSC-treated renal microenvironment in AKI, we generated a murine ischemia/reperfusion injury (IRI)-AKI model with or without human umbilical cord MSCs, which were sacrificed for analysis at 1 and 3 days post-IRI (from now on referred to as sham, IRI-1 d, IRI-3 d, and MSC group) (Figure 1A). Results of renal function, compared with the sham group, showed that IRI mice exhibited significantly elevated serum concentrations of serum creatinine (Scr) and blood urea nitrogen (BUN) at 1 and 3 days post-IRI, which was mostly significantly decreased in the MSC therapy group (Figure 1B). In parallel, acute tubular necrosis (ATN) scores showed a similar trend (Figure 1C). Serious renal damage was observed in the IRI-1 d group, including vacuolar degeneration of TECs, partial detachment of the brush border, disordered cellular arrangement, and many formations of casts (Figure 1D). The severity of the above pathological lesions was somewhat lower in the IRI-3 d group, while the MSC-treated group showed the lowest number of pathological lesions (Figure 1D). Hematoxylin and eosin (H&E) staining and immunofluorescence staining of CD45 in kidney tissues also showed that the renal interstitial immune cell infiltration was higher in the IRI-1 d and IRI-3 d groups than that in the sham group, which was reduced in MSC-treated mice (Figure 1D).
Figure 1.

Overview of single-cell transcriptome profiling conducted in a murine model of acute kidney injury and MSC therapy
(A) Summary of the IRI-AKI model, single-cell RNA sequencing, and functional experiments. (B) Biochemical detection of representative renal function indicators (Scr, serum creatinine; BUN, blood urea nitrogen) in IRI-AKI- and MSC-treated kidneys. (C) Acute tubular necrosis (ATN) scores. (D) PAS staining, H&E staining, and immunofluorescence staining of CD45 in mouse IRI-AKI- and MSC-treated kidneys. (E and F) Distribution of 102,162 high-quality kidney cells by cell type (E) and treatment group (F). (G) Heatmap showing the expression level of top differentially expressed genes (DEGs) in each cell type. (H) Violin plot showing the expression of canonical markers in each cell type. (I) Proportions of each cell type in distinct sample groups. Data are expressed as mean ± SD (n = 8); one-way ANOVA was used for comparisons of three or more groups. ∗∗p < 0.01.
Next, single-cell RNA sequencing (scRNA-seq) was conducted to fully analyze the underlying mechanisms. After cell filtering, a total of 102,162 cells from 13 samples across four pathology groups passed quality control (Figures 1F and S1). Unbiased clustering and cell annotation revealed nine cell subtypes, including TECs, distal tubular cells, intercalated cells/principal cells, loop of Henle, endothelial cells, and mesangial cells, and immune cell types such as T cells, B cells, and myeloid cells (Figures 1E and 1G). Expression of identified genes and the typical markers of each cell types are shown in Figures 1G and 1H. Comparison of the different groups showed that the proportions of TECs and immune cells changed markedly during IRI: the TEC population was reduced in the IRI-1 d and IRI-3 d groups, increasing in the MSC-treated group, while myeloid cells showed the opposite trend (Figure 1I). These findings indicate that AKI induces immune cell infiltration and TEC dysfunction or death, while MSC therapy triggers the regeneration of TECs and blocks immune infiltration, thus alleviating kidney injury.
Molecular diversity of the tubular epithelium during IRI-induced AKI
Having observed that TEC numbers in injured kidneys were modulated by MSCs, we further analyzed the differing transcriptomic phenotypes of the TECs induced by MSC therapy. Here, subclustering of the TECs revealed four main subtypes (Figure 2A): normal TECs, characterized by high levels of Slc34a1 expression; injured TECs, which expressed Havcr1 (encoding the kidney injury molecule 1, KIM-1); pro-fibrotic TECs, which expressed Pdgfb and Zeb2 (fibrotic markers) and Nfkb1, C3, Cxcl1, and Il34 (inflammatory markers); and renal stem/progenitor epithelial cells, characterized by high levels of Pcna (a proliferative marker), Prom1 (encoding the stem cell marker CD133), and Epcam (encoding epithelial cell adhension molecule) (Figures 2B and 2C). In addition, immunostaining experiments revealed that MSC treatment promoted the expressions of the renal progenitor cell marker SOX9 and proliferation marker Ki67 and reduced the expression of ZEB2 in kidney tissue (Figure S2); MSC treatment also resulted in lower levels of apoptosis (Figure S2). Using the scRNA-seq data to compare TEC subtypes in sham and IRI kidneys showed that normal TECs were depleted at 1 and 3 days post-IRI, while the proportions of injured TECs and pro-fibrotic TECs were increased. Furthermore, relative to the IRI-1 d and IRI-3 d groups, the MSC therapy group exhibited higher proportions of renal stem/progenitor cells and lower proportions of injured and pro-fibrotic TECs (Figure 2D). Gene set variation analysis (GSVA) further revealed that the pro-fibrotic TECs and injured TECs exhibited enrichment for inflammatory and fibrotic pathways, including TNF-α signaling, hypoxia, TGF-β signaling, IL-6-JAK-STAT3, and fibrosis, consistent with their pro-fibrotic and pro-inflammatory identity (Figure 2E).
Figure 2.

MSC treatment inhibited TEC inflammation and transdifferentiation
(A) Subclustering of TECs from sham-, IRI-AKI-, and MSC-treated kidneys. (B) Expression of the top 15 DEGs in the TEC subclusters. (C) Expression of representative marker genes in the TEC subclusters. (D) Relative cell percentage distribution and enrichment in the treatment groups. (E) Violin plots showing pathway enrichment in the TEC subclusters, estimated by GSVA. (F) Western blotting showing the levels of KIM-1, TGF-β1, pi-NF-κB, caspase-3, and p53 proteins in IRI-AKI- and MSC-treated kidneys. (G) Multiplex immunostaining in kidney sections showing the expression of fibrotic markers α-SMA (green) and vimentin (red) in IRI-AKI (d 3)- and MSC-treated kidneys. (H) Representative electron microscopic images showing changes in mitochondrial morphology in sham-, IRI-AKI (d 3)-, and MSC-treated kidneys.
Western blotting was applied to verify the protective role of MSC therapy in kidney injury and showed that KIM-1 levels increased by approximately 25.9-fold following IRI and were significantly reduced by about 30% after MSC treatment in comparison with the IRI-3 d group. Similar trends were observed for TGF-β1, phosphorylated NF-κB, caspase-3, and p53 (Figures 2F and S3). Furthermore, immunostaining demonstrated decreased levels of vimentin and α-SMA in MSC-treated kidney tissue compared with the IRI-3 d kidney tissue, indicating that the fibrotic phenotype was reduced in the renal epithelium (Figure 2G). Observation of mitochondrial structure by transmission electron microscopy (TEM) showed reduced numbers of mitochondria, accompanied by mitochondrial fission, swelling, and deformation and crest fracture in the IRI-3 d group, which were markedly attenuated by MSC treatment (Figure 2H). Taken together, this indicated that MSC therapy alleviates kidney injury by inhibiting the expression of pro-inflammatory and pro-fibrotic chemokines and reducing fibrosis.
Transcriptional evolution of TECs during MSC-induced repair
To investigate the MSC-induced repair process, we performed single-cell trajectory analysis via Monocle. This showed that differentiation of the renal stem/progenitor epithelium was followed by two alternative trajectories: the repair trajectory, producing normal TECs, or the fibrotic trajectory, producing pro-fibrotic TECs (Figure 3A). We also conducted trajectory analysis of the gene expression changes occurring along the renal stem/progenitor epithelium differentiation trajectory to explore dynamic cellular changes during injury/recovery. We identified four different patterns (modules 1–4, Figure 3B), representing cell fates and/or points along the trajectory. Moreover, several genes, including Tacstd2, Cldn7, Sox9, Prom1, and Pcna, exhibited differential expression during MSC therapy in IRI model, suggesting that they might have a role in determining the epithelial differentiation trajectory (Figures 3B and 3C). Gene ontology (GO) term analysis of the top 50 genes in modules 1–4 revealed differential enrichment of specific pathways/GO terms. For example, renal epithelium ATP metabolism was enriched in module 1, representing normal TECs, while in module 2, kidney development, renal tubule development, and tube formation were enriched, representing renal stem/progenitor differentiation. For module 3, response to wounding showed upregulation, representing the injured condition, and in module 4, angiogenesis and regulation of epithelial cell apoptosis were upregulated, representing the fibrotic condition (Figure 3D). Analysis of the scRNA-seq data also revealed significant upregulations of Pcna and Prom1 in the MSC-treated group (Figure 3E), which was validated by western blotting analysis (Figure 3F). These findings suggest that MSC therapy alleviates kidney injury by promoting renal stem/progenitor proliferation and differentiation.
Figure 3.

MSC treatment promoted tissue repair by enhancing renal stem/progenitor epithelium differentiation
(A) Potential renal stem/progenitor epithelial cell differentiation routes revealed by trajectory analysis. (B) Heatmap showing the scaled expression of dynamic genes along the two branches of the renal stem/progenitor epithelial trajectory. (C) Trajectory analysis showing the dynamic expression of representative stem cell and fibrosis genes. (D) Enriched GO terms for modules 1–4. (E) mRNA levels of Pcna, and Prom1 (Cd133) in sham-, IRI-AKI-, and MSC-treated kidneys. (F) Western blotting and semi-quantitative analysis of PCNA and CD133 protein levels in sham-, IRI-AKI-, and MSC-treated kidneys. Data are expressed as mean ± SD (n = 4); one-way ANOVA was used for comparisons of three or more groups. ∗p < 0.05, ∗∗p < 0.01.
Infiltration of myeloid cells and intercellular crosstalk with pro-fibrotic TECs
Given the results that MSC significantly reduced the proportion of myeloid cells, we identified nine myeloid cell subtypes (Figure 4A) based on the top 8 differentially expressed genes (DEGs) (Figure 4B): three dendritic cell subtypes, Ear2+ neutrophiles, Ly6c2+ monocytes, Cxcr2+ neutrophiles, Arg1+ macrophages, Trem2+ macrophages, and CD81+ resident macrophages (Figures 4B and S4A). GSVA (Figure S4B) revealed that Ly6c2+ monocytes and Cxcr2+ neutrophiles were highly enriched for inflammatory pathways (including TNF-α, IL-6-JAK-STAT3, and IL2-STAT5 signaling pathways), while Trem2+ and CD81+ macrophages exhibited anti-inflammatory signatures. Comparison of the injury/treatment groups showed that the proportions of neutrophiles and monocytes (Cxcr2+ and Ly6c2+ subsets) were increased by 2- to 4-fold in the IRI-1 d and IRI-3 d groups (relative to the sham controls) and then decreased following MSC therapy (Figure 4C). The scRNA-seq findings also revealed an increase in the frequency of anti-inflammatory Trem2+ macrophages in the MSC-treated group compared with the IRI-3 d group (Figure 4C). Flow cytometric analysis confirmed that the proportion of Ly6c2+CD11b+ monocytes was increased in the IRI-1 d group, relative to the sham group (Figure 4D). Furthermore, the percentage of total macrophages/monocytes was decreased in the group receiving MSC therapy relative to IRI-3 d group (Figure 4E). Trajectory analysis indicated the differentiation of infiltrating inflammatory monocytes into anti-inflammatory macrophages (Figures S4C and S4D), further suggesting that MSCs might inhibit the inflammatory microenvironment induced by IRI by promoting monocytic differentiation.
Figure 4.

MSC treatment inhibited the infiltration of inflammatory monocytes
(A) Subclustering of myeloid cells from sham-, IRI-AKI-, and MSC-treated kidneys. (B) Expression of the top 8 DEGs in the 9 myeloid cell subclusters. (C) Relative contribution of each myeloid cell type in the sham-, IRI-AKI-, and MSC-treated groups. (D) Flow cytometric analysis of Ly6c2+CD11b+ monocyte frequencies in the IRI-1 d and sham groups. (E) Flow cytometric analysis of CD11b+F/480+ macrophages frequencies in the IRI-3 d and MSC therapy groups. (F) Heatmap showing intercellular crosstalk between myeloid cells and TECs (ligand-receptor interaction pairs). (G) Circos plots showing the myeloid-TEC immune interaction networks.
Interestingly, we found that the abundance of pro-fibrotic and injured TECs was positively linked to the abundance of Cxcr2+ neutrophiles (Figure S4E). This finding prompted us to further analyze ligand-receptor interactions in TECs and myeloid cell sub-populations (Figure 4F). For example, pro-fibrotic and injured TECs expressed high levels of mRNAs encoding inflammatory cytokines such as Il34, Osm, C3, and Cxcl1, while monocytes and neutrophiles expressed mRNAs encoding the corresponding receptors, namely Csf1r, Lifr and Il6st, C3ar1, and Cxcr2 (Figures 4G and S5). Moreover, MSC therapy resulted in reduced expressions of Il34 and Cxcl1 (Figures S6A and S6B). Taken together, these analyses illuminate the potentially intercellular crosstalk mechanisms underlying inflammatory monocyte recruitment/infiltration in IRI kidneys and highlight possible interactions targeted by MSC therapy.
Reduced renal fibrosis in MSC-treated mice via blockade of Th17 cells infiltration
We also performed subclustering analysis of lymphocyte cells, and generated seven cell subtypes (Figure 5A) showing differing frequencies in post-AKI- and MSC-treated mice (Figure 5B). As defined by the top DEGs, these cell subtypes were identified as NK cells, proliferative Mki67+ CD8 T cells, Gzmk+ CD8 T cells, Tcf7+ CD4 T cells, Th17 cells, Cd28+ CD4 T cells, and B cells (Figure 5C). In particular, Th17 cells were increased by 2- to 3.4-fold in the IRI-1 d and IRI-3 d groups relative to the sham control groups and decreased in the MSC therapy group (Figure 5B). Notably, the abundance of Th17 cells was positively linked to the frequencies of injured and pro-fibrotic TECs while negatively linked to the frequencies of normal TECs, although the p values were not significant because the number of sequenced samples was relatively small (Figure 5D). These findings suggested that Th17 cells might influence the function of inflammatory and pro-fibrotic TECs.
Figure 5.

MSC treatment suppressed the activation of Th17 cells and secretion of TGF-β1
(A) Subclustering of lymphocytes from sham-, IRI-AKI-, and MSC-treated kidneys. (B) Relative contribution of each lymphocyte subtype in the sham-, IRI-AKI-, and MSC-treated groups. (C) The expression of the top 7 differentially expressed genes in each lymphocyte subtype. (D) Spearman’s correlation analysis showing the relationships between Th17 cell abundance and TEC subtype abundance. (E) Potential CD4 T cell differentiation routes revealed by trajectory analysis. (F) Heatmap showing the scaled expression of dynamic genes along the CD4 T cell trajectories. (G) Interaction analysis for immune cell subsets and TEC subsets showing significant receptor-ligand pairs. (H) The expression of ligand-receptor pairs genes in TCEs and immune cell subsets. (I) Representative immunofluorescence staining of the Th17 cell marker IL17A (red) in kidney sections from IRI-AKI (d 3)- and MSC-treated mice. (J) Flow cytometry plots showing the frequencies of CD45+CD3+CD8-IL17A+Th17 cells in IRI-3 d- and MSC-treated kidneys. (K) Levels of TGF-β1 in IRI-3 d- and MSC-treated kidneys, determined by ELISA. Data are expressed as mean ± SD (n = 3–6); Student’s t test was used for comparisons of two groups. ∗p < 0.05.
Pseudotime analysis indicated a differentiation trajectory from naive Tcf7+ CD4 T cells to Th17 cells, stimulated by IRI-AKI (Figures 5E and 5F). Moreover, several ligand-receptor pairs (IL18-IL18r1, Ccl2-Ccr2, and Cxcl16-Cxcr6) might contribute to the recruitment of Th17 cells by inflammatory monocytes (Figure 5G). Th17 cells also expressed high levels of Tgfb1/3 and Il17a RNAs, suggestive of intercellular crosstalk with pro-fibrotic TECs, which expressed Tgfβr1, aVb6 complex, and Il17ra/c (Figure 5G). The typical expression of ligand-receptor pair genes in TCEs and immune cell subsets is presented in Figure 5H. Immunostaining showed that MSC therapy reduced IL17A secretion (Figure 5I). Cytometric analysis revealed that the proportion of Th17 cells was reduced by about 50% in the group receiving MSC therapy relative to the untreated group (Figure 5J). Finally, enzyme-linked immunosorbent assay (ELISA) analysis showed that TGF-β1 levels were also significantly suppressed in the MSC group (Figure 5K). The above results suggest that MSC therapy reduces renal fibrosis by blockading Th17 cell infiltration.
Silencing of Zeb2 expression by MSC-derived miR-26a-5p
To validate our scRNA-seq findings, we carried out bulk RNA-seq in sham controls and mice at 1, 3, and 7 days post-IRI. These analyses showed that, relative to the sham groups, the three IRI groups expressed higher levels of RNAs encoding the pro-fibrotic markers Zeb2, Egfr, Pdgfb, and Twist1, and the inflammatory markers Cxcl1, Il34, and Nfκb1 (Figure 6A). As our results so far suggested that MSCs modulated the inflammatory and pro-fibrotic microenvironment, we wished to determine whether EVs contributed to the suppression of fibrosis by MSCs. We purified and characterized the EVs secreted by the MSCs used in our experiments. Structural analysis by TEM revealed typical bowl-shaped vesicles (Figure 6B), and western blotting showed that MSC-EVs expressed EV markers, including CD9, TSG101, and CD63, but not the cell marker calnexin (Figure 6C). Nanoparticle tracking analysis (NTA) indicated a median diameter ranging between 50 and 200 nm (Figure S7). Furthermore, fluorescence microscopy showed efficient internalization of 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (Dil)-labeled MSC-EVs by a TEC cell line (HK2 cells) (Figure 6E).
Figure 6.

miR-26a-5p derived from MSC-EVs inhibited pro-inflammatory/fibrotic gene expression in vitro
(A) Heatmap showing the expression of pro-fibrotic and inflammatory markers in sham, IRI-1 d, IRI-3 d, and IRI-7 d kidneys, determined by bulk sequencing. (B and C) Characterization of MSC-EVs by electron microscopy (B) and western blotting (C). (D) Expression of miRNA-26a-5p in HK2 cells, MSCs, and MSC-EVs, determined by PCR. (E) Internalization of Dil-labeled EVs (red) by HK2 cells. (F) Western blotting and semiquantitative analysis showing the levels of ZEB2 protein in H/R-HK2 cells +/− administration of MSC-EVs. (G) Detection of internalized Cy3-miR-26a-5p (red) in MSC-EV-treated HK2 cells. (H) Results of luciferase reporter assays following co-transfection of miR-26a-5p or negative control (NC) with a Zeb2 3′ UTR reporter (H27161) or control reporter (H306) in 293T cells. (I) Transfection efficiency of miR-26a-5p mimic in HK2 cells was determined by PCR. (J) Detection of transfected Cy3-labeled miR-26a-5p mimic (red) in HK2 cells. (K) Western blotting showing ZEB2 and pi-NF-κB protein levels in HK2 cells undergoing H/R +/− exogenous miR-26a-5p. Data are expressed as mean ± SD (n = 3); Student’s t test was used for comparisons of two groups; one-way ANOVA was used for comparisons of three or more groups. ∗p < 0.05, ∗∗p < 0.01.
As our results of scRNA-seq and bulk RNA-seq showed, profibrotic marker Zeb2 expression increased after IRI, while MSCs downregulated its expression. Therefore, we speculated whether MSCs regulated Zeb2 expression through EVs. Western blotting showed that hypoxia-reoxygenation (H/R) injury resulted in a 4-fold increase in the level of ZEB2 protein (p < 0.5), which was significantly reduced by treating the cells with 20 μg/mL MSC-EVs during the reoxygenation process (Figure 6F). According to previous microRNA sequencing results of MSC-EVs, miR-26a-5p is highly enriched in MSC-EVs.8 Bioinformatics analysis based on four databases, including tarbase, mirtarbase, diana_microt, and elmmo, indicated that miR-26a-5p might be capable of inhibiting Zeb2 expression. qPCR analysis showed that the level of miR-26a-5p was 4-fold higher in MSCs than in HK2 cells and was further significantly enriched in MSC-EVs (Figure 6D). To determine miR-26a-5p transfer between cells was mediated by EVs, we then transfected a Cy3-labeled miR-26a-5p mimic into MSCs and extracted the EVs from the culture supernatant. Following incubation of the EVs with HK2 cells, internalized Cy3-labeled miR-26a-5p was detected in the cells (Figure 6G). Next, luciferase reporter assays showed that miR-26a-5p mimic reduced Zeb2 luciferase reporter activity by 28.84% relative to the NC group (Figure 6H). To confirm these findings at the protein level, a miR-26a-5p mimic was directly transfected into HK2 cells before H/R (Figures 6I and 6J). Western blotting of the transfected cell extracts showed that H/R upregulated ZEB2 levels, but administration of the miR-26a-5p mimic led to a 60% reduction relative to basal levels (p < 0.01); a similar trend was observed for pi-NF-κB (Figures 6K and S8). Overall, these data indicate that miR-26a-5p suppresses the levels of the pro-fibrotic marker ZEB2 and suggest that miR-26a-5p derived from MSC-EVs may be able to protect TECs from fibrosis induced by H/R.
Weakened protective effects of MSC-EVs on IRI mice caused by inhibition of miR-26a-5p
Finally, we investigated the contribution of MSCs, which were transfected with an inhibitor of miR-26a-5p (miR-26a-5pIN-MSCs) or negative control (miR-26a-5pNC-MSCs), to tubular repair in our IRI-AKI model on days 3 and 7. Histological staining of 3-day post-IRI kidney tissue revealed higher levels of renal damage in mice treated with miR-26a-5pIN-MSCs rather than miR-26a-5pNC-MSCs (Figures 7A and S9A). Moreover, immunohistochemical staining showed that miR-26a-5pIN-MSC treatment also resulted in higher levels of ZEB2 and TGF-β1 (Figure 7A). To determine the effects of miR-26a-5pIN-MSCs on subsequent immune cell activation, we conducted flow cytometric analysis, which showed that the proportion of Th17 cells was significantly increased in the miR-26a-5pIN-MSC group (Figures 7B and S10).
Figure 7.

miR-26a-5p derived from MSC-EVs inhibited kidney inflammation and fibrosis in vivo
(A) PAS staining and immunohistochemical staining (ZEB2, TGF-β1) of IRI-3 d kidney tissue +/− miR-26a-5p inhibitor treatment. (B) Flow cytometry plots showing the proportion of Th17 cells in IRI-3 d kidney tissue +/− miR-26a-5p inhibitor treatment. (C) PAS, Masson’s trichome, and Sirius red staining indicating fibrosis levels in IRI-7 d kidney tissue +/− miR-26a-5p inhibitor treatment. (D) Representative images of immunohistochemical staining showing the levels of ZEB2, TGF-β1, and E-cadherin in IRI-7 d kidney tissue +/− miR-26a-5p inhibitor treatment.
Analysis on day 7 then allowed us to evaluate the contribution of miR-26a-5pIN-MSCs to AKI-chronic kidney disease (CKD) progression. Compared with the miR-26a-5pNC-MSC group, TEC shedding, formation of casts, and TEC dilation (as assessed by ATN scores) were significantly increased in the kidneys of mice treated with miR-26a-5pIN-MSCs (Figures 7C and S9B). Masson’s trichrome and Sirius red staining of kidney tissues also indicated a larger area of renal fibrosis in the miR-26a-5pIN-MSC group (Figure 7C). Finally, immunohistochemical staining revealed higher levels of ZEB2 and TGF-β1 in the TECs of miR-26a-5pIN-MSC-treated mice; expression of the epithelial marker E-cadherin was decreased in this group (Figure 7D). Taken together, these results suggest that MSC-EVs exert their therapeutic effects by targeting the miR-26a-5p/ZEB2 pathway in vivo, leading to suppression of fibrosis.
Discussion
In this study, we aimed to uncover the dynamic changes in cellular transcriptomic diversity occurring during AKI- and MSC-induced tissue repair. To do so, we have generated a valuable resource to elucidate the MSC-treated kidney microenvironment and aid the discovery of potential therapeutic targets for AKI.
Here, we characterized diverse transcriptomic phenotypes of TECs at a molecular level and revealed the expression of repair molecules such as Tacstd2, Tmsb10, Phgdh, Foxc1, Akap12, Plet1, and Tinagl1 not previously known to involved in MSC-induced tissue repair during AKI (Figure 3B). Our results indicated that the possible intercellular communication between renal TECs and immune cells inferred at the gene level might played an important role in AKI, with Zeb2- and Pdgfb-expressing pro-fibrotic TECs potentially acting to promote inflammation and fibrosis by recruiting inflammatory monocytes via several ligand-receptor pairs. Our analyses highlighted a role for infiltrating monocytes in the activation and expansion of the Th17 cells in the acute phase of IRI, potentially further contributing to the development of renal fibrosis, which were consistent with previous scRNA-seq findings.9
Furthermore, pro-fibrotic TECs activation and Th17 cells infiltration was suppressed by MSC treatment, thus reducing the subsequent cascading activation of the immune response and progression of fibrosis (Figure S11). These findings are consistent with the anti-inflammatory and immunomodulatory functions of MSCs10,11,12 and their ability to promote tissue regeneration via the secretions of various nutrients and growth factors.13,14,15 Previous studies showed the injured TECs recruited immune cells through classical Ccl2-Ccr216 and Cxcl1-Cxcr217 pairs. We found that pro-fibrotic TECs also could recruit mononuclear macrophages through Il34-Csf1r and Cxcl16-Cxcr6 pairs. Further, we demonstrated that exogenous MSCs could reduce the expression of Il34 and thus reduced their interaction with mononuclear phagocytes, enriching the mechanism by which MSCs reduced renal inflammation.
ZEB2 possesses C-terminal zinc finger cluster, thereby allowing it to bind to regulatory DNA sequences in their target promoters, involving in different biological events, such as embryogenesis, renal tubulointerstitial fibrosis, and tumor progression.18,19 Our findings are consistent with a recent study showing the importance of ZEB2 in AKI and fibrosis, whereby Zeb2 knockout in TECs reduced pathological damage in IRI and partially blocked the progression of AKI to CKD.19 Many studies showed Zeb2 were regulated by miR-124, miR-145, miR-30a, miR-3653, miR-138-5p, miR-101, miR-873-5p, miR-505, miR-203, miR-218, miR-205-5p, miR-377, miR-653, miR-153, and miR-653 during tumor proliferation, metabolism, and metastasis.20 However, studies on MSC-derived microRNAs inhibiting Zeb2 expression in the progression of AKI-CKD are lacking. In this study, analysis of an MSC-EV miRNA dataset8 and verification using several online databases (including Tar Base, and Mirta Base, which is verified by degradome sequencing21) identified miR-26a-5p as a potential candidate capable of inhibiting Zeb2. In agreement with this finding, miR-26a-5p significantly suppressed ZEB2 and NF-κB protein levels in TECs in vitro, indicating an important protective role for miR-26a-5p in inflammation and renal fibrosis. Moreover, consistent with previous studies, we observed concomitant upregulation of E-cadherin, indicative of partial repression of fibrosis.22 Importantly, inhibition of miR-26a-5p in MSCs promoted the activation and expansion of Th17 cells, accompanied by higher ZEB2 and TGF-β1 levels and lower E-cadherin levels, and promoted renal fibrosis. These findings suggested that miR-26a-5p derived from MSC-EVs blocked inflammation and renal fibrosis by suppressing Zeb2 expression in ischemic AKI.
As an active cell therapy, MSCs play a key role in renal fibrosis of later IRI as well as TEC injury of early IRI, such as significantly reducing the expression of KIM-1 (Figure 2F), a classic specific marker of TEC injury. In this study, MSCs also promoted the proliferation of resident renal stem/progenitor epithelium, as observed previously.23 Interestingly, the renal stem/progenitor epithelium exhibited two differentiation trajectories, leading to the production of either normal or pro-fibrotic TECs. Furthermore, we identified the pro-fibrotic factors Zeb2 and Pdgfb and the inflammatory cytokines Cxcl1 and Il34 as likely key players determining renal fibrosis. Transient activation of Sox9, Egr1, Prom1, and Pcna is more likely to promote the choice of repair trajectory of TECs (Figure 3). Among them, the trends of both Egr1 and Sox9 were consistent in the pseudo-time analysis (Figure 3C). Mechanically, we have found that EGR1 increased Sox9 expression in renal TECs by directly binding to the promoter of the Sox9 gene, thus promoting Sox9+ cell proliferation to repair injured TECs.24 But how MSCs upregulate EGR1 expression is not clear yet. Therefore, it will be the focus of future research to clarify the mechanism of how MSCs alleviate TEC injury in the early stage of IRI, thus alleviating subsequent cascade immune inflammation and renal fibrosis.
Here, we also present a safe MSC delivery method for the targeted treatment of AKI. The main disadvantage of the traditional intravenous infusion method of MSC delivery is poor targeting—most MSCs are intercepted in the lung and spleen, and a relatively small proportion manage to reach the injured kidney.25 Increasing the number of MSC transfusions increases the risk of pulmonary embolism.26 Another problem that cannot be ignored is that intravenous infusion of human-derived MSCs in mice may cause xenografting rejection,27,28 in which NK/T cells play a key role.29 In response to the above problems, we developed a method that used subcapsular injection of MSCs mixed with collagen to improve their targeting and level of adhesion, reduce xenografting rejection, and prolong cell survival time.30 However, as this method may increase the pressure in the capsule, potentially resulting in bleeding, strict control of injection volumes (≤80 μL) is necessary. In our other studies, we also optimized the targeted delivery of MSCs. One technique is to use ultrasound to guide the local delivery of MSCs and/or MSC-EVs to the submentum surrounding the kidney.31 Alternatively, artificial kidney capsules constructed by 3D printing and loaded with MSCs in hydrogel could be used to repeatedly carry out localized therapy in the kidney.32 The development of such strategies offers promising alternatives for the future treatment of CKD.
Our study has some limitations: (1) day 7 after IRI is still considered a short period for a CKD model. The role of MSCs in anti-fibrosis over a longer period should also be included, (2) specific targeted binding sites between miR-26a-5p and Zeb2 remain to be further verified by luciferase binding experiments, and (3) due to the nature of the microRNA hairpin structure, a single microRNA can target multiple genes, and a single gene can be regulated by multiple microRNAs simultaneously. Therefore, we cannot exclude the possibility of protective effects mediated by other miRNAs potentially also targeting Zeb2 or by the targeting of other genes by miR-26a-5p. Further investigation is required to explore these possibilities.
Taken together, we demonstrated the potential repair trajectories involving renal stem/progenitor epithelial differentiation and the inhibition of the miR-26a-5p/Zeb2 pathway on fibrosis and immune cells infiltration in response to MSC therapy during AKI-CKD progression. Overall, these findings provide a theoretical basis for the clinical application of MSCs that, we hope, will aid the development of AKI treatment strategies in the future.
Materials and methods
Animal models and cell-based therapy
The animal care and experimental procedures were approved by the ethics committees for animal experimentation at the Chinese People’s Liberation Army General Hospital (no. 2022-X18-30). Wild male C57BL/6 mice (8–9 weeks old, 22–24 g body weight) were supplied by SPF Biotechnology (Beijing, China). Sample size was determined by the resource balance method.33 In this study, a single-blind method was used for experimental design, and a random number table was used for experimental grouping. Bilateral IRI injury was induced in mice by renal pedicle clamping for 30 min, as described previously.34,35 In brief, following anesthetization of the mice, the dorsal hair above the kidney was shaved and the bilateral renal pedicle vessels were then exposed and clamped using a nontraumatic vascular clamp (Harvard Apparatus, Holliston, MA) for 30 min; during this process, the body temperature of the mice was strictly maintained at 37°C. In sham control mice, the kidneys were exposed but the renal pedicle vessels were not clamped. In the MSC treatment model, 2 × 106 MSCs were re-suspended in 80 μL rat tail collagen I (Thermo Fisher Scientific, Waltham, MA) and slowly injected under the renal capsule (subcapsular transplantation) using an insulin needle at random, as described previously.30 To investigate whether miR-26a-5p contributed to MSC-EV-mediated tubular repair in vivo, MSCs were transfected with miR-26a-5p inhibitors or NC and then administered by subcapsular transplantation, as above. Mice were euthanized at 1 day (IRI-1 d), 3 day (IRI-3 d), and 7 day (IRI-7 d) post-IRI. If there are obvious lesions such as renal cysts or subcapsular bleeding, these samples should be excluded.
Cell culture
Human umbilical cord-derived MSCs were donated by the Wuhan Optics Valley Vcanbio Pharmaceutical (China) and maintained in MSC medium (Cyagen Biosciences, Santa Clara, CA) supplemented with 10% fetal bovine serum (FBS) (Gibco, Thermo Fisher Scientific) or EV-depleted FBS (System Biosciences, Palo Alto, CA). MSCs of the sixth to eighth generation were selected for follow-up animal therapy and EVs (described below) extraction. After the MSCs were cultured for 48 h, the cell culture supernatants were collected and used for the extraction of EVs. HK2 cells (human proximal tubular cells) were cultured in DMEM/F12 medium supplemented with 10% FBS. 293T cells (human embryonic kidney cells) were cultured in DMEM medium supplemented with 10% FBS. The above cells were authenticated without mycoplasma infection. They were grown under standard conditions (37°C, 5% CO2) and the media were routinely replaced every other day.
Scr and BUN tests
Blood samples were collected from the vena cava at the indicated time points. The serum was obtained by centrifugation at 3,000 rpm for 15 min at 4°C, and then used to measure serum Scr and BUN levels through picric acid method and enzymatic method, respectively.
Histopathological examination and immunological staining
Kidney tissues were fixed in 4% formaldehyde for 48 h, embedded in paraffin, sectioned at a thickness of 2 μm, and then stained with periodic acid-Schiff (PAS) or H&E using standard protocols. ATN scores were assessed on PAS-stained sections in a blinded manner, as follows. Ten nonoverlapping fields (200×) were randomly assessed by two professional pathologists and scored from 0 to 4, whereby 0 = normal, 1 = 1%–25% of the field showing damage, 2 = 26%–50% of the field showing damage, 3 = 51%–75% of the field showing damage, and 4 ≥ 75% of the field showing damage. Immunohistochemical and immunofluorescence staining were performed as described previously.36 The primary antibodies used were as follows: anti-Ki67 (1:200, ab16667), anti-Sox9 (1:500, ab185966), anti-TGF-β1 (1:500, ab215715), anti-vimentin (1:400, ab92547), anti-E-cadherin (1:500, ab231303), and anti-α-SMA (1:400, ab92547) from Abcam (Cambridge, UK), anti-CD45 (1:300, GB113886) from Servicebio (Wuhan, China), and anti-ZEB2 (1:1000, NBP1-82991) from Novus Biologicals (Centennial, CO).
Establishment of in vitro hypoxia model and MSC-EV treatment
To mimic IRI in vitro, HK2 cells were maintained in a humidified H/R chamber under hypoxic conditions (5% CO2, 1% O2, and 94% N2) for 12 h, followed by reoxygenation (5% CO2 and 21% O2) for an additional 24 or 48 h34; during this time the cells were incubated with MSC-EVs with/without miR-26a-5p mimic.
Transfection of miRNA mimic/inhibitor
MSCs or HK2 cells were grown to 50% confluence before being transfected with 30 nmol/L synthetic miR-26a-5p mimic, inhibitor, or negative control (NC) (Gene Pharma, Nanjing, China) using Lipofectamine 3000 in Opti-MEM (Invitrogen, Carlsbad, CA). Following transfection for 12 h, the cells were maintained in complete medium containing EV-free FBS for 36 h. EVs were extracted from the conditioned medium of miR-26a-5p mimic- or NC-transfected cells. The sequences were as follows: miR-26a-5p mimic, 5′-UUCAAGUAAUCCAGGAUAGGCU-3′; miR-26a-5p mimic NC, 5′-UUCUCCGAACGUGUCACGUTT-3′; miR-26a-5p inhibitor, 5′- AGCCUAUCCUGGAUUACUUGAA-3′, miR-26a-5p inhibitor NC, 5′-UCUACUCUUUCUAGGAGGUUGUGA-3′.
Luciferase reporter assays
293T cells were co-transfected with 3′ UTR luciferase reporter constructs (pMIR-REPORT Luciferase vector H306, a control reporter, and pMIR-REPORT Luciferase-Zeb2 3′ UTR vector H27161), miRNA (miRNA-NC or miR-26a-5p), and Renilla luciferase using GP-transfect-Mate (OBIO, Shanghai, China) for 48 h. Luciferase activity was measured using a Dual Luciferase Reporter Assay Kit (Promega, Madison, WI) and microplate reader (Tecan, Mannedorf, Switzerland).
Isolation and characterization of MSC-EVs
To obtain high-purity EVs, MSC-EVs were isolated by differential ultracentrifugation and size-exclusion chromatography as described previously.37,38 The collected cell supernatants were centrifuged at 300 × g, 4°C, for 10 min to remove dead cells and then at 3,000 × g, 4°C, for 10 min to remove cell debris, followed by filtration using a 0.22 μm filter. The supernatants were then ultracentrifuged using a P50A72-986 rotor (CP100NX; Hitachi, Tokyo, Japan) at 100,000 × g, 4°C, for 2 h to pellet the EVs. Subsequently, the EV pellets were resuspended in phosphate-buffered saline (PBS) and recentrifuged at 100,000 × g, 4°C for 2 h, washed with PBS, diluted 1.5-fold in PBS, and further purified using Exosupur columns (EchoBiotech, Beijing, China). The samples were eluted in 0.01 M PBS, and 2 mL eluate fractions were collected according to the manufacturer’s instructions. The fractions were concentrated to 200 μL using 100 kDa molecular weight cut-off Amicon Ultra spin filters (Merck, NJ) and stored at −80°C until further use. The protein content of the EVs was quantified using a Pierce BCA assay kit (Thermo Fisher Scientific). Western blotting, NTA, and TEM were performed to characterize the MSC-EVs.39
Internalization of EVs
MSC-EVs labeled with Dil (Yeasen Biotechnology, Shanghai, China) were prepared as described previously.34 To evaluate EV internalization, MSC-EVs (20 μg/mL) were added to the medium of HK2 cells for 8 h; after washing three times with PBS, the internalized EVs were detected using laser scanning confocal microscopy.
Western blotting
Kidneys, cells, and MSC-EVs were lysed in radioimmune precipitation assay lysis buffer (Thermo Fisher Scientific) containing phenylmethylsulphonyl fluoride and phosphatase inhibitors (Beyotime, Shanghai, China); protein concentration was assayed using a BCA protein assay kit (Thermo Fisher Scientific). Protein samples (20–30 μg/lane) were electrophoresed on 7.5%, 10%, or 12% sodium dodecyl sulfate-polyacrylamide gels and then transferred to nitrocellulose filter membranes. Membranes were blocked with 5% defatted milk at room temperature for 2 h and then incubated with the following primary antibodies at 4°C overnight: anti-CD133 (1:1,000, Abcam, Cambridge, UK, ab284389), anti-TGF-β1 (1:1,000, Abcam, ab215715), anti-TSG101 (1:1,000, Abcam, ab125011), anti-caspase-3 (1:5,000, Abcam, ab32351UK), anti-pi-NF-κB (1:1,000, CST, Danvers, MA, 3033), anti-p53 (1:1,000, CST, 2524S), anti-PCNA (1:1,000, CST, 13110), anti-GAPDH (1:2,000, CST, 14C10), anti-KIM-1(1:5,000, R&D Systems, Minneapolis, MN, AF1817), anti-CD9 (1:1,000, Proteintech, San Diego CA, 60232), anti-calnexin (1:1,000, Proteintech, 10427), anti-β-actin (1:10,000, Proteintech, 60008), anti-CD63 (1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA, sc-5275), anti-β-tubulin (1:20,000, Proteintech, 66240), and anti-ZEB2 (1:1,000, Novus Biologicals, NBP1-82991). Secondary antibodies (1:1,000, Beyotime) were used for detection by chemiluminescence and autoradiography (Bio-Rad, Hercules, CA). Image J was used for intensity analysis of the relative protein expression normalized to β-actin, β-tubulin, or GAPDH.
Detection of mRNA and miRNA
Total RNA from kidney tissues, MSCs, HK2 cells, or EVs was extracted using TRIzol reagent (Invitrogen). mRNAs were quantified using PrimeScript reverse transcription reagent and a real-time PCR kit with SYBR Green (Takara Bio, Osaka, Japan). Mature miRNAs were quantified using a real-time PCR detection kit and miRNA-specific primers (BGI, Shenzhen, China). Primer sequences are shown in Table 1.
ELISA
TGF-β1 was detected using ELISA (Multiscience, Hangzhou, China). After being activated with hydrochloric acid, the supernatant of the tissue homogenate was added to the enzyme-labeled plate (soaked in advance); the detection antibody was then added and incubated at room temperature for 1.5 h. After the plate was washed in PBS, horseradish peroxidase-labeled streptavidin was added and incubated at room temperature for 30 min, after which the chromogenic substrate was added. After 20 min, the termination solution was added and optical densities were determined for each well.
Flow cytometry
Single-cell suspensions of kidney tissue (100 μL) were incubated with the appropriate antibodies at room temperature for 30 min, protected from light. For detection of IL-17A, the cells were induced using Cell Stimulation Cocktail (Tonbo Biosciences, San Diego, CA), which was mixed with the single-cell suspension at 1:500. The suspension was then cultured at 37°C for 6 h followed by immunostaining, performed according to the procedure outlined above. The antibodies were purchased from BioLegend (San Diego, CA): PerCP/Cyanine5.5 anti-CD3, APC anti-CD8a, PE anti-IL-17A, APC/Cyanine7 anti- CD11b, PE/Cyanine7 anti-F4/80, APC anti-Ly6c2, and FITC anti-CD45. The data were analyzed using Kaluza analysis software.
Analysis of apoptosis
Apoptosis was detected in mouse kidney paraffin sections using terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling assays (Beyotime).
Sample processing and single-cell mRNA library preparation and sequencing
Fresh surgically resected IRI AKI and MSC-treated kidney samples were minced and enzymatically digested to obtain single-cell suspensions as reported previously.40 Then, the single-cell suspensions were incubated with 7-AAD (BioLegend), and 7-AAD-negative live cells were sorted and CD45+ immune cells were enriched using a BD FACSAria II (BD Biosciences, Franklin Lakes, NJ), and then were pooled together. The single cells suspensions were loaded onto a microfluidic chip at 700–1,000 cells/μL and a complementary deoxyribonucleic acid library was generated using the commercial 10x Genomics platform (10X Genomics, Pleasanton, CA). Single-cell transcriptome amplification and library preparation were performed by BerryGenomics (Beijing, China) and CapitalBio Technology Corporation (Beijing, China) using the Single-Cell 3′ Library Kit v.3 (10X Genomics) according to the manufacturer’s instructions. Then, the libraries were pooled and sequenced over six lanes on an Illumina NovaSeq 6000 System (Illumina, San Diego, CA).
Pre-processing of scRNA-seq data
Gene expression matrixes for the sequenced samples were produced using the Cell Ranger (10X Genomics, v.5) count function by aligning raw sequencing FASTQ files to the mm10 reference genome via the STAR algorithm. Seurat41 R package (v.4.0.0) was employed to perform subsequent data analysis including normalization, scaling, principal-component analysis (PCA), Uniform Manifold Approximation and Projection (UMAP) dimension reduction, and visualization of gene expression. We filtered low-quality cells that had >6,000 or <301 expressed genes, or >30% of UMIs derived from the mitochondrial genome. We removed potential cell doublets using the DoubletFinder42 R package as the dropout effect of 10× data and employed the SeuratWrappers package to integrate single-cell transcriptome expression using the RunFastMNN function. Then, we selected highly variable genes for PCA; the top 30 significant principal components were selected for UMAP dimension reduction and visualization of gene expression.
Cell type determination and abundance estimation
Cell types were annotated according to known canonical marker genes and DEGs, calculated by the FindAllMarker function using default parameters provided by Seurat. The ratio (R) of observed (O) to random expected (E) cell number was used to adjust cell sampling bias for each sample, indicating cluster enrichment in a particular sample. The R(O/E) for each cluster in distinct samples was calculated using the chi-squared test; R(O/E) > 1 indicated enrichment of the cell cluster in the sample, as reported previously.43
Trajectory analysis
We employed the Monocle algorithm44 to infer the cell differentiation trajectories followed by TECs during renal repair, using the default parameters after dimension reduction and cell ordering as reported previously.40
Pathway analysis
We employed GSVA45 to assess pathway enrichment in distinct TEC and myeloid subsets using the hallmark gene sets provided by the Molecular Signatures Database; this analysis was performed via a linear model offered by the limma package.
Intercellular crosstalk analysis
To explore potential intercellular crosstalk networks, we assessed ligand-receptor distribution and expression levels in infiltrating immune cells and TEC subpopulations via a standard pipeline implemented in R using the CellChat46 R package, as reported previously.40 We selected receptors and ligands expressed in >10% of the cells in a specific cluster for subsequent analysis. Interaction pairs whose ligands belonged to the Il, Ccl, Tgfb, complement, and Cxcl families were selected for the evaluation of intercellular crosstalk between distinct TEC subpopulations and infiltrating immune cells.
Statistical analysis
Data were analyzed using the Student’s t test for two groups and one-way ANOVA with Tukey’s multiple comparisons test for three or more groups. Pearson’s correlation analysis was used for the relationship between the two variables. Statistical analyses were performed using GraphPad Prism 5.0 and data are expressed as mean ± SD. p < 0.05 was considered statistically significant.
Data and code availability
The scRNA-seq data required to reproduce the analysis and figures have been deposited on the OMIX dataset: OMIX004421.
Acknowledgments
This study was supported by grants from the National Natural Science Foundation of China (no. 82170686), grant for G.C. (no. 22KJLJ001), the National Key R&D Program of China (no. 2018YFA0108803), the Young Elite Scientists Sponsorship Program from the China Association for Science and Technology (no. 2022QNRC001), and Young Talent Project of Chinese PLA General Hospital (no. 20230403). We particularly acknowledge Wuhan Optics Valley VCANBIO Pharmaceutical Co. Ltd for providing MSCs. We thank BerryGenomics and Capitalbio Technology Corporation for conducting the scRNA-seq experiment.
Author contributions
G.C., X.C., W.W., and M.Z. conceived and designed the experiments. W.W. and X.R. conducted the experiments. M.Z. performed the data analysis. Q.H. and Z.F. provided valuable advice and were responsible for research supervision, coordination, and strategy. Y.S., Y.X., K.Z., T.X., X.G., and S.W. provided technical assistance. W.W. and M.Z. drafted the manuscript. G.C. and X.C. reviewed and edited the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.07.024.
Contributor Information
Xiangmei Chen, Email: xmchen301@126.com.
Guangyan Cai, Email: caiguangyan@sina.com.
Supplemental information
References
- 1.Ali T., Khan I., Simpson W., Prescott G., Townend J., Smith W., Macleod A. Incidence and outcomes in acute kidney injury: a comprehensive population-based study. J. Am. Soc. Nephrol. 2007;18:1292–1298. doi: 10.1681/ASN.2006070756. [DOI] [PubMed] [Google Scholar]
- 2.Ronco C., Bellomo R., Kellum J.A. Acute kidney injury. Lancet (London, England) 2019;394:1949–1964. doi: 10.1016/s0140-6736(19)32563-2. [DOI] [PubMed] [Google Scholar]
- 3.Pavyde E., Usas A., Maciulaitis R. Regenerative pharmacology for the treatment of acute kidney injury: Skeletal muscle stem/progenitor cells for renal regeneration? Pharmacol. Res. 2016;113:802–807. doi: 10.1016/j.phrs.2016.03.014. [DOI] [PubMed] [Google Scholar]
- 4.Brennan M.Á., Layrolle P., Mooney D.J. Biomaterials functionalized with MSC secreted extracellular vesicles and soluble factors for tissue regeneration. Adv. Funct. Mater. 2020;30 doi: 10.1002/adfm.201909125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hade M.D., Suire C.N., Mossell J., Suo Z. Extracellular vesicles: Emerging frontiers in wound healing. Med. Res. Rev. 2022;42:2102–2125. doi: 10.1002/med.21918. [DOI] [PubMed] [Google Scholar]
- 6.Tetta C., Ghigo E., Silengo L., Deregibus M.C., Camussi G. Extracellular vesicles as an emerging mechanism of cell-to-cell communication. Endocrine. 2013;44:11–19. doi: 10.1007/s12020-012-9839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Birtwistle L., Chen X.M., Pollock C. Mesenchymal Stem Cell-Derived Extracellular Vesicles to the Rescue of Renal Injury. Int. J. Mol. Sci. 2021;22 doi: 10.3390/ijms22126596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao J.Y., Wang B., Tang T.T., Wen Y., Li Z.L., Feng S.T., Wu M., Liu D., Yin D., Ma K.L., et al. Exosomal miR-125b-5p deriving from mesenchymal stem cells promotes tubular repair by suppression of p53 in ischemic acute kidney injury. Theranostics. 2021;11:5248–5266. doi: 10.7150/thno.54550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doke T., Abedini A., Aldridge D.L., Yang Y.W., Park J., Hernandez C.M., Balzer M.S., Shrestra R., Coppock G., Rico J.M.I., et al. Single-cell analysis identifies the interaction of altered renal tubules with basophils orchestrating kidney fibrosis. Nat. Immunol. 2022;23:947–959. doi: 10.1038/s41590-022-01200-7. [DOI] [PubMed] [Google Scholar]
- 10.Fazekas B., Griffin M.D. Mesenchymal stromal cell-based therapies for acute kidney injury: progress in the last decade. Kidney Int. 2020;97:1130–1140. doi: 10.1186/s13287-021-02289-710.1016/j.kint.2019.12.019. [DOI] [PubMed] [Google Scholar]
- 11.Huang Y., Yang L. Mesenchymal stem cells and extracellular vesicles in therapy against kidney diseases. Int. J. Mol. Sci. 2021;12:219. doi: 10.3390/ijms2007161910.1186/s13287-021-02289-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yun C.W., Lee S.H. Potential and Therapeutic Efficacy of Cell-based Therapy Using Mesenchymal Stem Cells for Acute/chronic Kidney Disease. 2019;20 doi: 10.3390/ijms20071619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu J., Wang X., Chen J., Chen S., Li Z., Liu H., Bai Y., Zhi F. Embryonic stem cell-derived mesenchymal stem cells promote colon epithelial integrity and regeneration by elevating circulating IGF-1 in colitis mice. Theranostics. 2020;10:12204–12222. doi: 10.7150/thno.47683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y., Pan Y., Liu Y., Li X., Tang L., Duan M., Li J., Zhang G. Exosomes derived from human umbilical cord blood mesenchymal stem cells stimulate regenerative wound healing via transforming growth factor-β receptor inhibition. Stem Cell Res. Ther. 2021;12:434. doi: 10.1186/s13287-021-02517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xie Q., Liu R., Jiang J., Peng J., Yang C., Zhang W., Wang S., Song J. What is the impact of human umbilical cord mesenchymal stem cell transplantation on clinical treatment? Stem Cell Res. Ther. 2020;11:519. doi: 10.1186/s13287-020-02011-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raghu H., Lepus C.M., Wang Q., Wong H.H., Lingampalli N., Oliviero F., Punzi L., Giori N.J., Goodman S.B., Chu C.R., et al. CCL2/CCR2, but not CCL5/CCR5, mediates monocyte recruitment, inflammation and cartilage destruction in osteoarthritis. Ann. Rheum. Dis. 2017;76:914–922. doi: 10.1136/annrheumdis-2016-210426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang L., Zhang Y.L., Lin Q.Y., Liu Y., Guan X.M., Ma X.L., Cao H.J., Liu Y., Bai J., Xia Y.L., et al. CXCL1-CXCR2 axis mediates angiotensin II-induced cardiac hypertrophy and remodelling through regulation of monocyte infiltration. Eur. Heart J. 2018;39:1818–1831. doi: 10.1093/eurheartj/ehy085. [DOI] [PubMed] [Google Scholar]
- 18.Verschueren K., Remacle J.E., Collart C., Kraft H., Baker B.S., Tylzanowski P., Nelles L., Wuytens G., Su M.T., Bodmer R., et al. SIP1, a novel zinc finger/homeodomain repressor, interacts with Smad proteins and binds to 5'-CACCT sequences in candidate target genes. J. Biol. Chem. 1999;274:20489–20498. doi: 10.1074/jbc.274.29.20489. [DOI] [PubMed] [Google Scholar]
- 19.Inotani S., Taniguchi Y., Nakamura K., Nishikawa H., Matsumoto T., Horino T., Fujimoto S., Sano S., Yanagita M., Terada Y. Knockout of Zeb2 ameliorates progression of renal tubulointerstitial fibrosis in a mouse model of renal ischemia-reperfusion injury. Nephrol. Dial. Transpl. 2022;37:454–468. doi: 10.1093/ndt/gfab311. [DOI] [PubMed] [Google Scholar]
- 20.Ashrafizadeh M., Ang H.L., Moghadam E.R. MicroRNAs and Their Influence on the ZEB Family: Mechanistic Aspects and Therapeutic Applications in Cancer Therapy. 2020;10 doi: 10.3390/biom10071040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang X., Zuo X., Yang B., Li Z., Xue Y., Zhou Y., Huang J., Zhao X., Zhou J., Yan Y., et al. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell. 2014;158:607–619. doi: 10.1016/j.cell.2014.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Long J., Zuo D., Park M. Pc2-mediated sumoylation of Smad-interacting protein 1 attenuates transcriptional repression of E-cadherin. J. Biol. Chem. 2005;280:35477–35489. doi: 10.1074/jbc.M504477200. [DOI] [PubMed] [Google Scholar]
- 23.Xia H., Liang C., Luo P., Huang J., He J., Wang Z., Cao X., Peng C., Wu S. Tissue repair and regeneration with endogenous stem cells. Stem Cell Res. Ther. 2018;9:174–193. doi: 10.1038/s41578-018-0027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen J.W., Huang M.J., Chen X.N., Wu L.L., Li Q.G., Hong Q., Wu J., Li F., Chen L.M., Dong Y., et al. Transient upregulation of EGR1 signaling enhances kidney repair by activating SOX9(+) renal tubular cells. Theranostics. 2022;12:5434–5450. doi: 10.7150/thno.73426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zonta S., De Martino M., Bedino G., Piotti G., Rampino T., Gregorini M., Frassoni F., Dal Canton A., Dionigi P., Alessiani M. Which is the most suitable and effective route of administration for mesenchymal stem cell-based immunomodulation therapy in experimental kidney transplantation: endovenous or arterial? Transpl. Proc. 2010;42:1336–1340. doi: 10.1016/j.transproceed.2010.03.081. [DOI] [PubMed] [Google Scholar]
- 26.Chen C., Hou J. Mesenchymal stem cell-based therapy in kidney transplantation. Stem Cell Res. Ther. 2016;7:16. doi: 10.1186/s13287-016-0283-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rossignol J., Boyer C., Thinard R., Remy S., Dugast A.S., Dubayle D., Dey N.D., Boeffard F., Delecrin J., Heymann D., et al. Mesenchymal stem cells induce a weak immune response in the rat striatum after allo or xenotransplantation. J. Cell. Mol. Med. 2009;13:2547–2558. doi: 10.1111/j.1582-4934.2009.00657.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tee B.C., Sun Z. Xenogeneic mesenchymal stem cell transplantation for mandibular defect regeneration. Xenotransplantation. 2020;27 doi: 10.1111/xen.12625. [DOI] [PubMed] [Google Scholar]
- 29.Moadsiri A., Polchert D., Genrich K., Napoles P., Reina E., Turian J., Smith B., Bartholomew A. Mesenchymal stem cells enhance xenochimerism in NK-depleted hosts. Surgery. 2006;140:315–321. doi: 10.1016/j.surg.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 30.Huang M., Li D., Chen J., Ji Y., Su T., Chen Y., Zhang Y., Wang Y., Li F., Chen S., et al. Comparison of the treatment efficacy of umbilical mesenchymal stem cell transplantation via renal subcapsular and parenchymal routes in AKI-CKD mice. Stem Cell Res. Ther. 2022;13:128. doi: 10.1186/s13287-022-02805-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Y., Geng X., Chi K., Liu C., Liu R., Chen X., Hong Q., Cai G. Ultrasound enhances the therapeutic potential of mesenchymal stem cells wrapped in greater omentum for aristolochic acid nephropathy. Stem Cell Res. Ther. 2021;12:261. doi: 10.1186/s13287-021-02243-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fu Z., Chu Y., Geng X., Ma Y., Chi K., Song C., Liao S., Hong Q., Wu D., Wang Y. Artificial Kidney Capsule Packed with Mesenchymal Stem Cell-Laden Hydrogel for the Treatment of Rhabdomyolysis-Induced Acute Kidney Injury. ACS Biomater. Sci. Eng. 2022;8:1726–1734. doi: 10.1021/acsbiomaterials.1c01595. [DOI] [PubMed] [Google Scholar]
- 33.Dell R.B., Holleran S., Ramakrishnan R. Sample size determination. ILAR J. 2002;43:207–213. doi: 10.1093/ilar.43.4.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao Z., Zhang C., Peng F., Chen Q., Zhao Y., Chen L., Wang X., Chen X. Hypoxic mesenchymal stem cell-derived extracellular vesicles ameliorate renal fibrosis after ischemia-reperfusion injure by restoring CPT1A mediated fatty acid oxidation. Stem Cell Res. Ther. 2022;13:191. doi: 10.1186/s13287-022-02861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao L., Zhou D., Tan R.J., Fu H., Zhou L., Hou F.F., Liu Y. Sustained Activation of Wnt/β-Catenin Signaling Drives AKI to CKD Progression. J. Am. Soc. Nephrol. 2016;27:1727–1740. doi: 10.1681/asn.2015040449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bai M., Zhang L., Fu B., Bai J., Zhang Y., Cai G., Bai X., Feng Z., Sun S., Chen X. IL-17A improves the efficacy of mesenchymal stem cells in ischemic-reperfusion renal injury by increasing Treg percentages by the COX-2/PGE2 pathway. Kidney Int. 2018;93:814–825. doi: 10.1016/j.kint.2017.08.030. [DOI] [PubMed] [Google Scholar]
- 37.Théry C., Amigorena S., Raposo G., Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cel. Biol. 2006;Chapter 3 doi: 10.1002/0471143030.cb0322s30. Unit 3.22. [DOI] [PubMed] [Google Scholar]
- 38.Böing A.N., van der Pol E., Grootemaat A.E., Coumans F.A.W., Sturk A., Nieuwland R. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J. Extracell. Vesicles. 2014;3:23430. doi: 10.3402/jev.v3.23430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang C., Shang Y., Chen X., Midgley A.C., Wang Z., Zhu D., Wu J., Chen P., Wu L., Wang X., et al. Supramolecular Nanofibers Containing Arginine-Glycine-Aspartate (RGD) Peptides Boost Therapeutic Efficacy of Extracellular Vesicles in Kidney Repair. ACS nano. 2020;14:12133–12147. doi: 10.1021/acsnano.0c05681. [DOI] [PubMed] [Google Scholar]
- 40.Zhang M., Wu L., Deng Y., Peng F., Wang T., Zhao Y., Chen P., Liu J., Cai G., Wang L., et al. Single Cell Dissection of Epithelial-Immune Cellular Interplay in Acute Kidney Injury Microenvironment. Front. Immunol. 2022;13 doi: 10.3389/fimmu.2022.857025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stuart T., Butler A., Hoffman P., Hafemeister C., Papalexi E., Mauck W.M., 3rd, Hao Y., Stoeckius M., Smibert P., Satija R. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. doi: 10.1016/j.cell.2019.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McGinnis C.S., Murrow L.M., Gartner Z.J. DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst. 2019;8:329–337.e4. doi: 10.1016/j.cels.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guo X., Zhang Y., Zheng L., Zheng C., Song J., Zhang Q., Kang B., Liu Z., Jin L., Xing R., et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat. Med. 2018;24:978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 44.Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., Lennon N.J., Livak K.J., Mikkelsen T.S., Rinn J.L. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hänzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-Seq data. BMC bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jin S., Guerrero-Juarez C.F., Zhang L., Chang I., Ramos R., Kuan C.H., Myung P., Plikus M.V., Nie Q. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 2021;12:1088. doi: 10.1038/s41467-021-21246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The scRNA-seq data required to reproduce the analysis and figures have been deposited on the OMIX dataset: OMIX004421.