

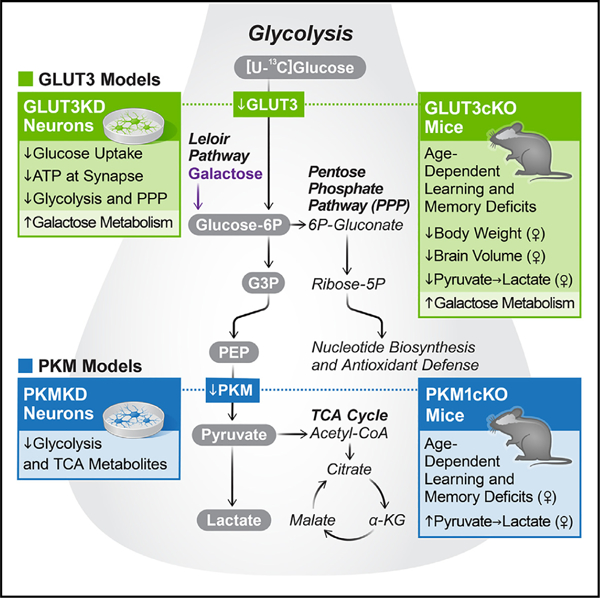
In brief
Whether neurons can and must take up glucose and metabolize it by glycolysis was unclear, especially in vivo. Li et al. combine metabolomics and live imaging of human neurons with mouse models disrupting key steps in glucose metabolism to demonstrate that neurons require glucose uptake and glycolysis for normal function.
SUMMARY
Neurons require large amounts of energy, but whether they can perform glycolysis or require glycolysis to maintain energy remains unclear. Using metabolomics, we show that human neurons do metabolize glucose through glycolysis and can rely on glycolysis to supply tricarboxylic acid (TCA) cycle metabolites. To investigate the requirement for glycolysis, we generated mice with postnatal deletion of either the dominant neuronal glucose transporter (GLUT3cKO) or the neuronal-enriched pyruvate kinase isoform (PKM1cKO) in CA1 and other hippocampal neurons. GLUT3cKO and PKM1cKO mice show age-dependent learning and memory deficits. Hyperpolarized magnetic resonance spectroscopic (MRS) imaging shows that female PKM1cKO mice have increased pyruvate-to-lactate conversion, whereas female GLUT3cKO mice have decreased conversion, body weight, and brain volume. GLUT3KO neurons also have decreased cytosolic glucose and ATP at nerve terminals, with spatial genomics and metabolomics revealing compensatory changes in mitochondrial bioenergetics and galactose metabolism. Therefore, neurons metabolize glucose through glycolysis in vivo and require glycolysis for normal function.
Graphical abstract
INTRODUCTION
The brain requires large amounts of glucose, but the extent and requirement for neuronal glucose metabolism remains unknown. Glucose may be primarily metabolized by glia into lactate, which is then exported to neurons to serve as the primary fuel supporting aerobic respiration (lactate shuttle1). However, more recent studies using glucose analogs2,3 and electrophysiology support both direct glucose uptake by neurons and their inability to rely solely on the lactate shuttle.4 Moreover, the primary neuronal glucose transporter (GLUT3) is not expressed by glia,5 while another glucose transporter, GLUT4, is translocated to the presynaptic plasma membrane of cultured neurons in response to activity, and knockdown (KD) or deletion of both of these GLUTs compromises neuronal function.6,7 These findings suggest that glucose uptake by neurons is critical, although its impact on neuronal glucose and energy metabolism has not been defined.
Even if neurons require direct glucose uptake, we lack definitive evidence that the glucose is metabolized by glycolysis, and it remains unknown if neurons actually require glycolysis, especially in vivo. Indeed, neurons are proposed to perform very little glycolysis under physiologic conditions, and pharmacologically inhibiting neuronal glycolysis may protect against ischemia and neurodegeneration.8–12 Nonetheless, findings using an NADH/NAD+ biosensor as a surrogate for glycolytic flux do support that neuronal glycolysis is upregulated during neuronal activity in hippocampal slices.13 However, uncertainty remains due to the inability of current metabolomic and metabolic imaging approaches, such as fluorine-18 fluorodeoxyglucose positron emission tomography ([18F]FDG-PET), to distinguish between the contributions of neurons and glia to glucose uptake and metabolism. In contrast, hyperpolarized (HP) 13C magnetic resonance spectroscopic imaging (MRSI) enables real-time monitoring of metabolic reactions in vivo, using an exogenous increase in the MR signal of 13C compounds.14–16 This approach has been widely used to study the conversion of HP pyruvate to lactate in cancer, both in preclinical models and in patients.17–19 HP 13C MRSI has the potential to inform on downstream glucose metabolism and complement [18F]FDG-PET findings.
To dissect the contribution of neurons to glucose uptake and metabolism, we generated cellular and mouse models with neuronal disruption of either the primary glucose transporter or pyruvate kinase, which catalyzes the final step in glycolysis. Applying metabolomics, HP 13C MRSI, and behavioral analysis, we investigated the requirement for glucose uptake and glycolysis in neuronal function in vitro and in vivo.
RESULTS
Human neurons metabolize glucose through glycolysis
One challenge to determining the capacity of neurons to metabolize glucose by glycolysis is that both brain tissue and primary neuronal cultures contain substantial glial components, and, until recently, the field has lacked paradigms to generate pure neuronal populations for metabolomic studies. To study glucose metabolism in pure neuronal cultures lacking glia, we used near-homogenous induced pluripotent stem cell (iPSC)-derived human excitatory neurons.20,21 Specifically, we generated human iPSC lines co-expressing neurogenin 2 (Ngn2), an inducible dCas9-KRAB for CRISPR inhibition (CRISPRi),22 and either a non-targeting (NTG) control single guide RNA (sgRNA) or an sgRNA that targets pyruvate kinase (PKM; both the M1 and M2 isoforms), the enzyme catalyzing the final step in glycolysis. iPSC lines were differentiated into neurons by inducing Ngn2 with doxycycline and then cultured for 12 days22 (Figure S1A). Neurons were then incubated with [U-13C]glucose for up to 24 h prior to processing for metabolomics.23
We performed two complementary experiments to quantify glycolytic, pentose phosphate pathway (PPP), and tricarboxylic acid (TCA) cycle metabolites, the first using ion chromatography-mass spectrometry (IC-MS) to resolve pyruvate and glucose-6P and analyze metabolite levels in the media, and the second using ultra-performance liquid chromatography-mass spectrometry (UPLC-MS) to resolve six-carbon sugars, including glucose and fructose. Quantification of metabolites containing [U-13C]glucose-derived carbons in control neurons revealed utilization of glucose-derived metabolites in both glycolysis and the downstream PPP and TCA cycle pathways (Figures 1A and S1B). Considering that these cultures contain essentially no contaminating glia (Figure S1A),21 these data prove that neurons can metabolize glucose through glycolysis for downstream metabolic pathways including the TCA cycle.
Figure 1. Human neurons metabolize glucose through glycolysis.
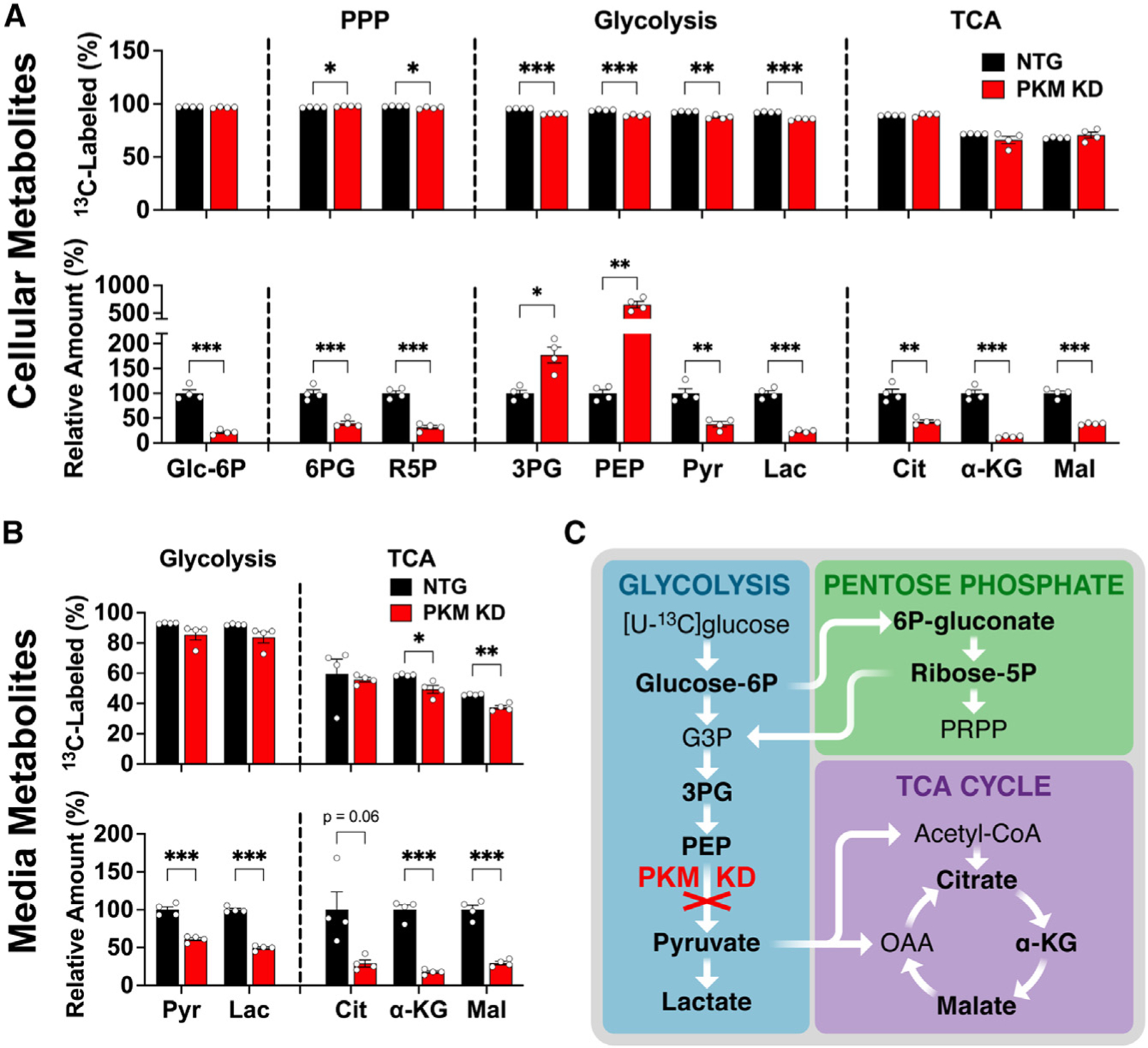
(A and B) (A) Cellular and (B) media-derived metabolites from neurons expressing either an NTG guide (control) or a CRISPRi sgRNA targeting pyruvate kinase (PKM KD). Groups were compared for fractional [U-13C]glucose labeling (top) and total relative metabolite levels (bottom). See Figure S1D for corresponding isotopologue data. Data are means ± SEM. n = 4 samples/group.
(C) Schematic of PKM KD impact on [U-13C] glucose metabolism.
*p < 0.05, **p < 0.01, and ***p < 0.001 by Welch’s t tests.
Notably, the fractional labeling of glycolytic metabolites after 24-h incubation with [U-13C]glucose was only slightly decreased by downregulation of PKM KD, despite 73% gene KD of PKM in the first experiment (Figures 1A and S1C–S1E) and greater than 99% KD in the second (Figures S1B and S1C). However, the total cellular levels of the upstream glycolytic metabolites 3-phosphoglycerate (3PG) and phosphoenolpyruvate (PEP) were markedly increased in both experiments (Figures 1A and S1B). PKM KD also decreased the levels of pyruvate (Pyr) and downstream pyruvate metabolites including lactate (Lac) and malate (Mal) (Figures 1A and S1B).
The media levels of metabolites downstream of PKM also markedly decreased upon PKM KD at 4 and 24 h, again with little or no change in fractional labeling (Figures 1B, 1C, and S1F), consistent with decreased production and release of these metabolites. This decrease in cellular and media metabolites downstream of PKM supports that decreased neuronal glycolysis decreases production of downstream TCA metabolites necessary for bioenergetic production. These data show that neurons can derive the majority of pyruvate from glycolysis, and do so, at least in the absence of glia.
Neurons require glucose uptake in vivo
Although the above data show that neurons are capable of metabolizing glucose through glycolysis, it remains possible that they either do not or do not need to take up glucose in the presence of glia in vivo. To test the neuronal requirement for glucose uptake in vivo, we selectively deleted GLUT3 from CA1 hippocampal neurons (Figure 2A). We bred floxed GLUT3 mice24 with CamKIIα (CamKCre) mice, which express Cre recombinase from p19 in a subset of hippocampal neurons, including nearly all CA1 neurons, and in cortical and other neurons scattered throughout the forebrain.25,26 GLUT3cKO (GLUT3lox/lox;CamKCre) mice were the progeny of GLUT3lox/lox and GLUT3wt/lox;CamKCre. GLUT3WT included control mice (GLUT3wt/lox and GLUT3lox/lox) lacking the Cre transgene. GLUT3cKO mice were born in Mendelian proportions (control 50.0%, GLUT3 heterozygotes [GLUT3cHET] 25.2%, GLUT3cKO 24.8%, n = 238), and there were no differences in survival through more than 1 year (Figure S2A). GLUT3cKO male mice had similar body weights compared with controls through 12 months of age (Figure 2B). GLUT3cKO and control females had similar weight at 3 months, but both GLUT3cKO and GLUT3cHET females had progressively decreased weight at 7 and 12 months.
Figure 2. Neurons have an age-dependent requirement for neuronal glucose uptake in vivo.
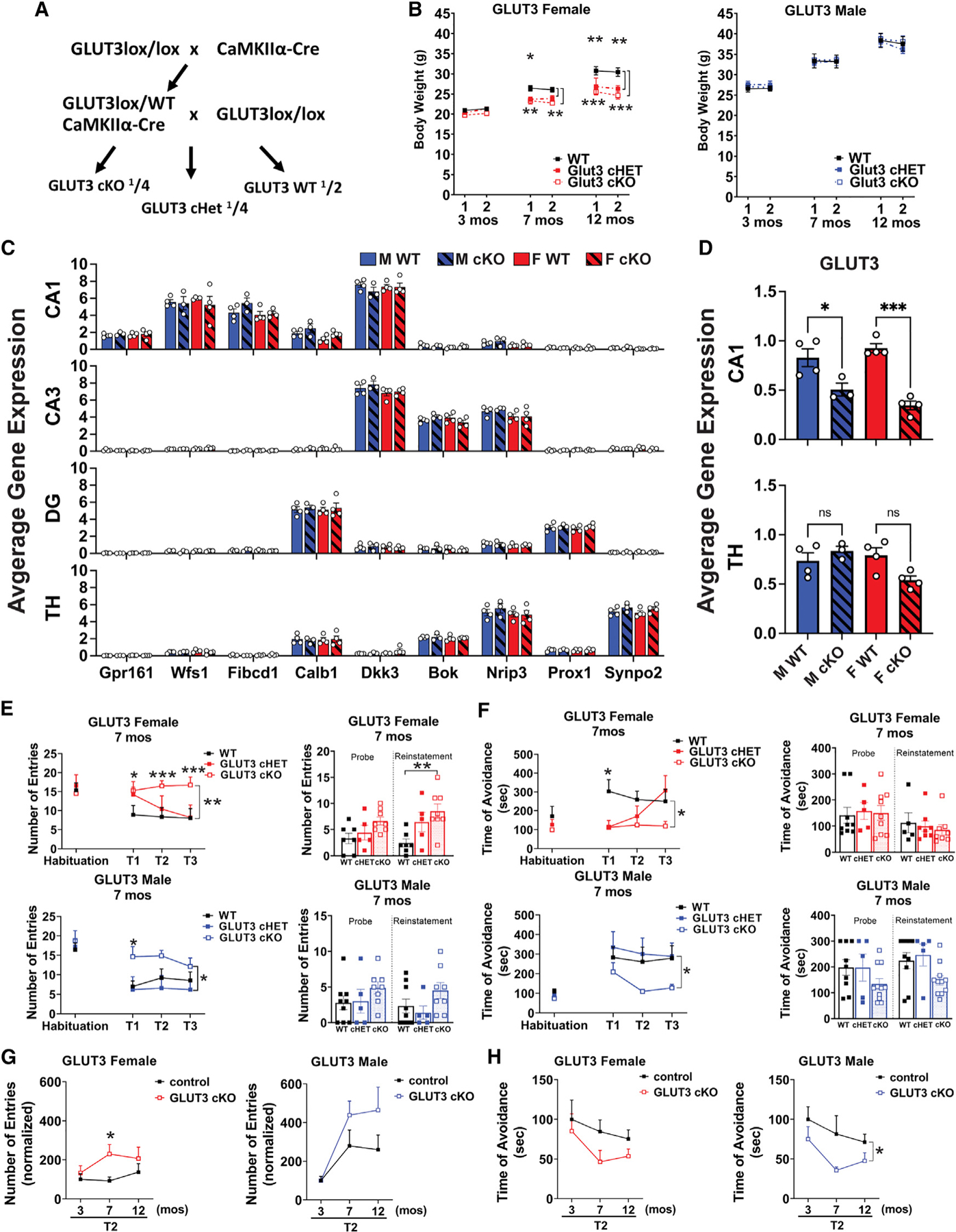
(A) Breeding scheme for GLUT3cKO mice.
(B) Weight measurements at age 3, 7, and 12 months. Data are means ± SEM; n = 9 GLUT3 WT, 5 Het, and 8 KO females, and n = 8–9 WT, 5 Het, and 8 KO males at each time point.
(C) Graphs show regional signatures unique to neurons in CA1 (20–36 capture areas/mouse), CA3 (11–30 capture areas/mouse), dentate gyrus (DG; 13–34 capture areas/mouse), and thalamus (TH; 65–170 capture areas/mouse). n = 3–4 mice/group.
(D) GLUT3 expression in CA1 and thalamic neurons. n = 3–4 mice/group.
(E and F) GLUT3cKO mice develop age-dependent spatial learning and memory deficits as shown by active place avoidance. At 7 months, GLUT3cKO female and male mice have increased entrances into the aversive zone (E) and decreased maximal time of avoidance of this zone (F).
(G and H) Longitudinal analysis shows change in second time point (T2) of active place avoidance, with each mouse normalized to the mean control value at 3 months. Although GLUT3cKO mice are equivalent to controls at 3 months, 7- and 12-month-old GLUT3cKO mice of both sexes enter the aversive zone more frequently than controls (G; see Figure S2 for full data from 3 and 12 months) and avoid it for less time (H). n = 9 WT, 5 Het, 8 KO females, and n=8–9 WT, 5 Het and 8 KO males at each time point, compiled from three cohorts.
ns, not significant.
*p % 0.05; **p < 0.01; ***p < 0.001 by Welch ANOVA with Dunnett’s T3 multiple comparisons test (B, E, F), two-way ANOVA with Tukey’s multiple comparison test (C), one-way ANOVA with Sidak’s multiple comparison test (D), and Welch’s t test (G and H). Brackets in graphs (E–H) show significance of linear mixed modeling for genotype (E and F) or the interaction of genotype and age (H).
To confirm loss of GLUT3 expression in CA1 neurons, we used Visium Spatial Gene Expression from 10X Genomics. We validated our capacity to detect gene expression in CA1 neurons, where Cre is expressed in essentially all neurons.27 CA1 neurons showed high expression of expected marker genes (Calb1, Wfs1, Fibcd1, Gpr161, and Dkk3), which enabled their clear differentiation from neurons in adjacent regions such as dentate gyrus (Calb1 and Prox1), CA3 (Dkk3, Bok, and Nrip3), and thalamus (Nrip3 and Synpo2) (Figure 2C).28 In both male and female GLUT3cKO mice, GLUT3 expression was decreased specifically in CA1, but not in the thalamus where Cre is not expressed (Figure 2D).
To determine if GLUT3 is required for normal hippocampal function, we used the active place avoidance task to examine how GLUT3cKO affects visual-spatial learning and memory.29 Both male and female GLUT3cKO mice displayed normal learning and memory at 3 months (Figures S2B and S2C), but developed severe deficits by 7 months (Figures 2E–2H). GLUT3cKO mice had an increase in the total number of entrances and a decrease in maximum time of avoidance to the aversive zone compared with controls. These deficits were also apparent, although less prominent, at 12 months of age (Figures S2D and S2E). In contrast, GLUT3cHET mice performed similarly to controls (Figures 2E, 2F, and S2B–S2E).
GLUT3cKO mice displayed no abnormalities in elevated plus maze testing at any age, indicating no gross changes in anxiety level (Figures S2F–S2H). GLUT3cKO mice had similar overall activity levels compared with controls, as shown by the number of open and closed arm entries and total distance in the plus maze (Figures S2G and S2H), along with a similar total number of ambulatory movements and rearing on open field activity testing (Figure S2I). There was no difference in withdrawal latency on hot plate testing (Figure S2J), further indicating intact sensorimotor function in GLUT3cKO mice. Therefore, GLUT3 loss in CA1 neurons markedly worsens spatial learning and memory, providing strong in vivo evidence that neurons must import glucose directly to function normally.
To begin to determine how decreased glucose uptake compromises neuronal function and health, we examined the impact of GLUT3cKO on neuronal survival. Loss of GLUT3 did not alter the density of NeuN-positive cells within CA1 in 15-month-old-mice (Figure S3A). However, analysis of brain volume using T2-weighted MRI in live 10- to 14-month-old mice (Figure 3A) showed that female GLUT3cKO mice had decreased hippocampal volume versus controls, while GLUT3cKO had no impact in males. The difference in hippocampal volume was independent of mouse age (Figure S3B). To further assess this change, we performed high-resolution T2-weighted MRI of postmortem brains in 19-month-old mice (Figure 3B), imaged in the intact skull to preserve overall brain structure and minimize potential experimental damage to the brain during dissection. Once again, GLUT3cKO female mice had smaller hippocampal and CA1 volumes than controls. The small decrease in CA1 volume in female GLUT3cKO mice may reflect synaptodendritic rarefaction in addition to some neuronal death.30–32 Nonetheless, these findings indicate that postnatal loss of GLUT3 does not have large effects on the survival of CA1 neurons, suggesting that GLUT3cKO primarily disrupts neuronal function rather than survival. Total brain volume and thalamus volume were also smaller in female GLUT3cKO mice compared with wild-type (WT) controls, while ventricle size was similar (Figure 3C).
Figure 3. Female GLUT3cKO mice have smaller CA1 and total brain volumes, and decreased HP [1-13C]lactate-to-pyruvate ratios.
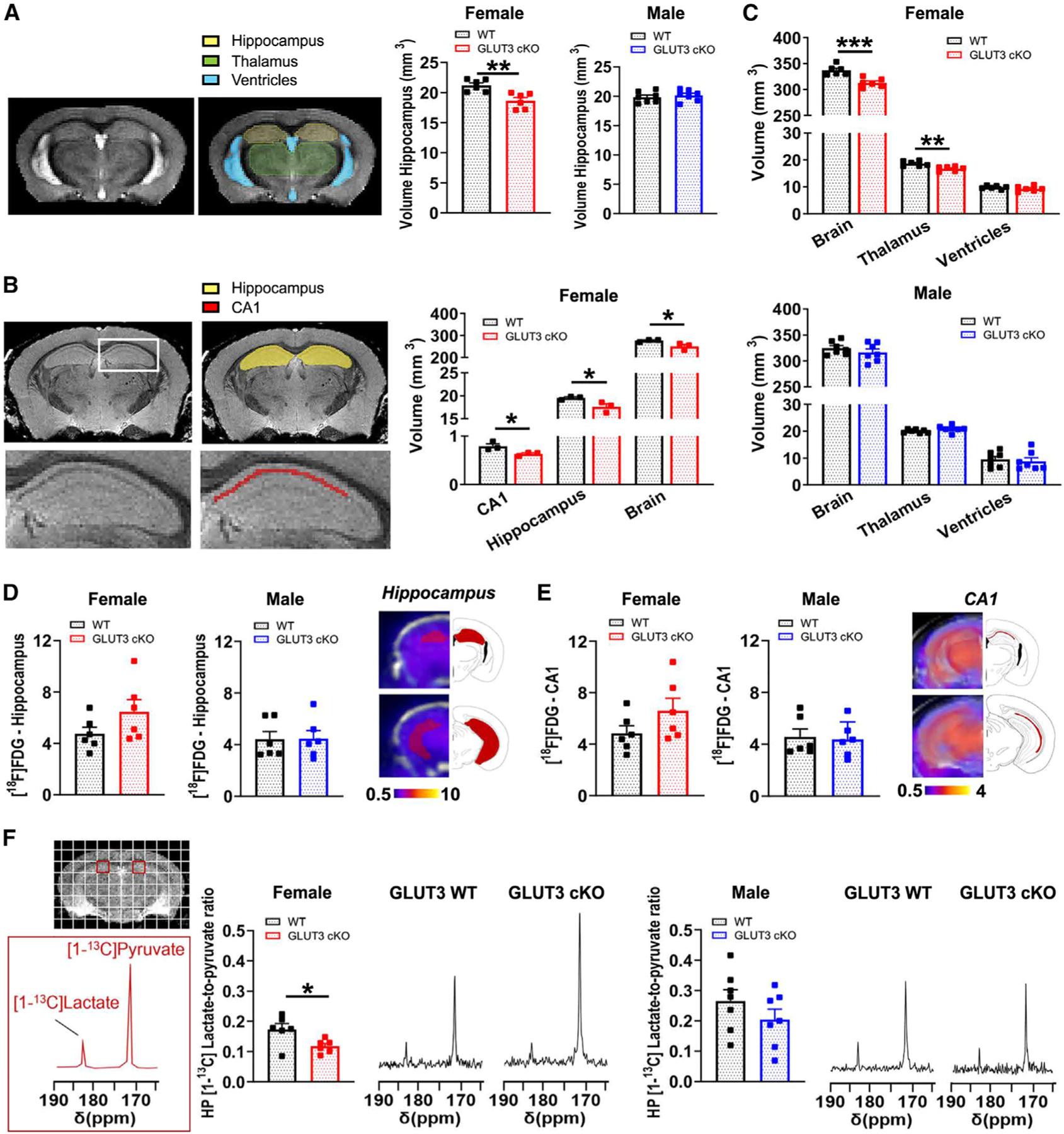
(A) Representative in vivo T2-weighted images of 10- to 14-month-old mice used for volumetric analyses. The hippocampal volume was significantly smaller in female GLUT3cKO mice. Data are means ± SEM; n = 6–7 mice/group.
(B) Total brain and thalamus volumes calculated from in vivo T2-weighted images of 10- to 14-month-old mice were smaller in female GLUT3cKO mice, while ventricle volume was unchanged. n = 6–7 mice/group.
(C) Representative ex vivo T2-weighted images of 19-month-old mice used for volumetric analyses. CA1, hippocampal, and entire brain volumes were smaller in female GLUT3cKO mice. n = 3 mice/group.
(D and E) [18F]FDG-PET signal from the hippocampus (D) and specifically from the CA1 area (E) was similar between 12- and 14-month-old females and males. n = 6 mice/group.
(F) Representative 13C spectra of 8- to 14-month-old mice showing HP [1-13C]pyruvate and HP [1-13C]lactate levels from a region containing CA1 (red square) for female and male mice. HP [1-13C]lactate-to-pyruvate ratios were significantly lower in female GLUT3cKO mice. n = 6–7 mice/group.
*p % 0.05,**p % 0.01,***p % 0.001 by unpaired t tests.
Although loss of Cre-expressing cortical neurons might explain the decrease in total brain volume, GLUT3KO mice also displayed decreased thalamus volume, a region where GLUT3 was not knocked out in neuronal cell bodies.33 This suggests either volume loss due to shrinkage of neurites from Cre-expressing neurons that project to the thalamus34 or a non-cell-autonomous effect. Moreover, since the CamKCre mice express Cre from day P19,25,26 at roughly the same time that the mouse brain reaches adult size (≈P20),35 this presumably represents volume loss rather than a developmental change. Finally, although changes in the activity of CA1 hippocampal neurons can influence peripheral glucose levels,36 we found no significant changes in blood glucose levels between GLUT3cKO and WT mice (Figure S3C).
In vivo metabolic and anatomical imaging shows sex-specific requirement for neuronal GLUT3
To determine the impact of GLUT3cKO on glucose metabolism in vivo, we first assessed total brain glucose uptake using [18F] FDG-PET. GLUT3cKO did not affect the [18F]FDG-PET signal in either the hippocampus or CA1 of 11- to 15-month-old male or female mice (Figures 3D and 3E), consistent with prior findings that postnatal day 15 pups lacking GLUT3 in the brain also have normal FDG-PET.7 This suggests that glucose uptake by glia may constitute the majority of the [18F]FDG-PET signal, precluding detection of neuronal glucose uptake in vivo. We therefore used HP 13C MRSI to measure pyruvate conversion into lactate. Although the contributions of neurons and glia to the HP 13C signal also cannot be directly distinguished by 13C MRSI, combining this method with neuron-specific transgenic models enabled us to assess how changing glucose metabolism within neurons affects metabolism in different brain regions. In the hippocampus, HP [1-13C] lactate-to-pyruvate ratios were significantly lower in female GLUT3cKO versus WT mice (Figures 3F and S3D). Males showed a similar trend. No significant differences were found in the thalamus (Figure S3E), where GLUT3 is not deleted in neuronal cell bodies.
To investigate the molecular basis for decreased pyruvate-to-lactate conversion in CA1, we examined expression of proximal genes that regulate pyruvate metabolism. Pyruvate flux toward lactate can be modulated by lactate dehydrogenase (LDH)37–39 and pyruvate dehydrogenase (PDH),40,41 as well as monocarboxylate transporters (MCTs),42 which are responsible for pyruvate uptake and lactate efflux. However, we did not observe any changes in the expression of LDH, PDH, or MCT isoforms (Figures S3F–S3H). The conversion of pyruvate into lactate by LDH may also depend on the coenzyme NAD(H),43–45 with an increased NADH/NAD+ ratio associated with increased HP [1-13C]pyruvate-to-lactate flux. However, no change in the expression of GAPDH, which plays a prominent role in NAD metabolism,46 was detected (Figure S3I).
Neuronal requirement for glycolysis in vivo
The finding that GLUT3cKO mice develop severe memory loss provides strong in vivo evidence that neurons must import glucose directly to function normally. However, this does not prove they require glycolysis, as neurons have been hypothesized to metabolize glucose primarily through the PPP under physiologic conditions.8,9 To gain insight into the neuronal requirement for glycolysis, we analyzed the effect of postnatal deletion of PKM1,47 the predominant neuronal isoform of PK48,49 that catalyzes the final step in glycolysis.
We bred PKM1lox/lox mice with CamKIIα (CamKCre) mice (Figure 4A). PKM1cKO (PKM1lox/lox; CamKCre) mice were the progeny of PKM1lox/lox and PKM1wt/lox; CamKCre. PKM1WT included control mice (PKM1wt/lox and PKM1lox/lox) lacking the Cre transgene. PKM1cKO mice were born in roughly normal Mendelian proportions (control 42.4%, PKM1 heterozygotes 29.3%, PKM1cKO 28.3%, n = 191 mice total), and no differences in survival were noted (Figure S4A). PKM1cKO mice of both sexes had similar body weights compared with controls through 12 months of age (Figure 4B). Loss of PKM1 expression in essentially all CA1 neurons was confirmed by immunofluorescence (Figure 4C).
Figure 4. Female mice have an age-dependent neuronal requirement for glycolysis.
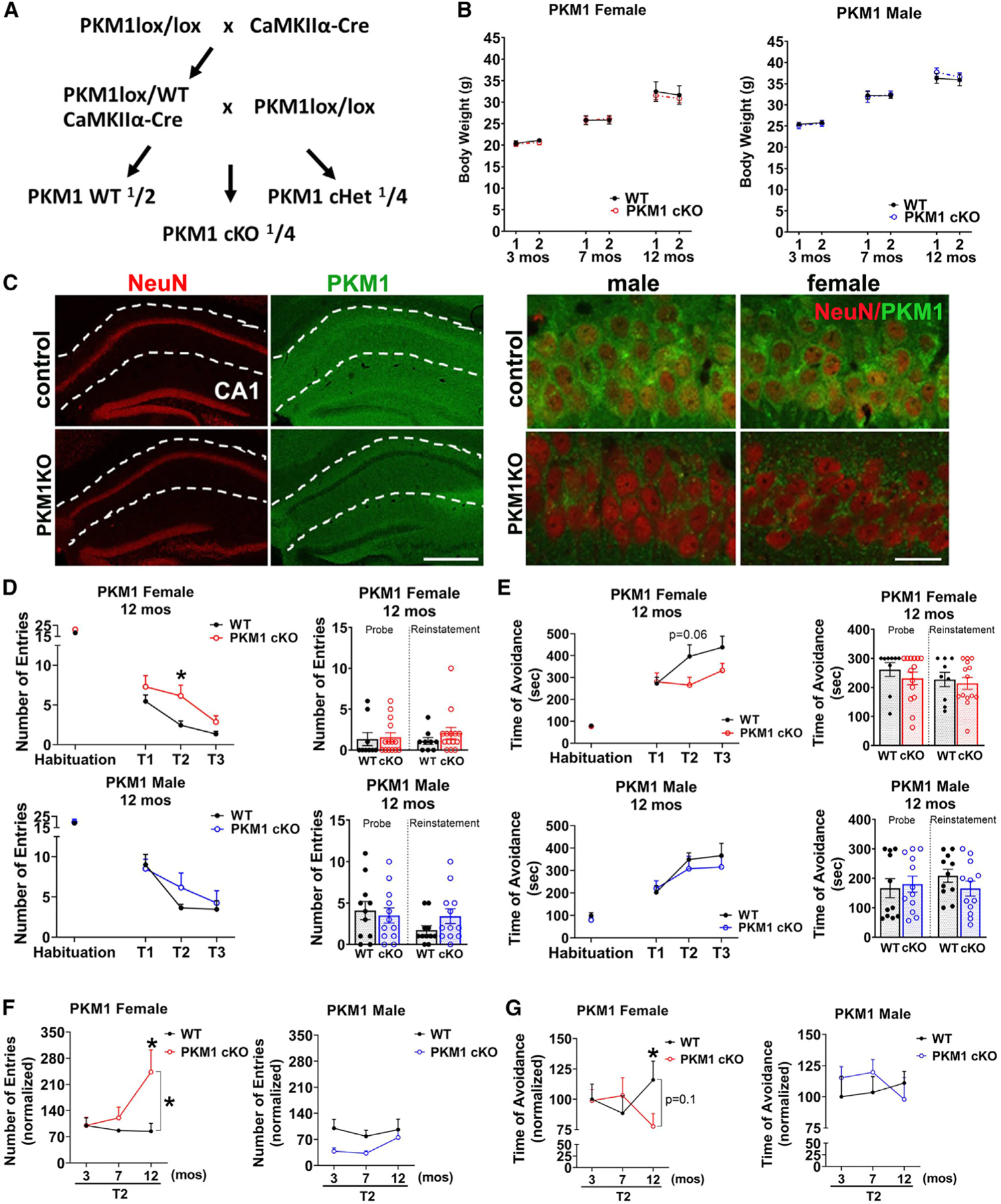
(A) Breeding scheme to generate mice with conditional postnatal deletion of PKM1 in CA1 and other forebrain neurons.
(B) PKM1cKO mice have similar weights to controls. Repeated weight measurements at 3, 7, and 12 months of age. Data are means ± SEM; n = 10–11 PKM1 WT and 14 KO females, and 11 or 12 WT and 13 KO males at each time point.
(C) PKM1 immunofluorescence shows loss of PKM in CA1 neurons in PKM1cKO mice. Sections from 12-month-old mice are stained with NeuN (red) and PKM1 (green). Scale bar, 400 mm (left), 40 mm (right).
(D–G) Female PKM1cKO mice develop age-dependent spatial learning and memory deficits as shown by active place avoidance.
(D and E) Female PKM1cKO mice have increased entrances into the aversive zone (D), and a trend of decreased maximal time of avoidance of this zone (E) at 12 months of age (p = 0.06). No deficits were observed in males.
(F and G) Longitudinal analysis shows change in second time point (T2) of active place avoidance testing, with each mouse normalized to the mean control value at 3 months. PKM1cKO females are equivalent to controls at 3 and 7 months of age, but 12-month-old PKM1cKO mice enter the aversive zone more frequently than controls (F), and avoid it for less time (G), whereas no deficits were observed in males (see Figure S4 for full data from 3 to 7 months). n = 10–11 WT, 14 KO females, and 11 or 12 WT and 13 KO males, each compiled from three cohorts.
*p % 0.05 by Welch ANOVA with Dunnett’s T3 multiple comparisons test (D and E) and Welch’s t tests (F and G). Brackets in graphs (F and G) show significance of linear mixed modeling for the interaction of genotype and age (F and G).
We next examined how PKM1cKO affects spatial learning and memory assessed by active place avoidance. Both male and female PKM1cKO mice had normal memory at 3 and 7 months (Figures S4B–S4E). However, female (but not male) PKM1cKO mice developed learning and memory loss by 12 months, as indicated by increased entrances to the aversive zone (Figures 4D and 4F) and decreased maximal time of avoidance of the aversive zone (Figures 4E and 4G).
Female (but not male) PKM1cKO mice also showed deficits on elevated plus maze testing. PKM1cKO female mice showed a trend of increased time in the open arm (Figure S4F), and total distance moved was increased at 7 months (Figure S4G). Female PKM1cKO had more open and closed arm entries than controls (Figures S4G and S4H), suggesting the difference in open arm time could be driven by hyperactivity. Indeed, female PKM1cKO moved a greater distance in open field testing and had increased rearing (Figure S4I). In contrast, male and female PKM1cKO mice had normal withdrawal latency on hot plate testing (Figure S4J), indicating preserved sensory function. Therefore, complete PKM1 loss worsens memory and produces hyperactivity in female mice, providing in vivo evidence that postnatal neurons must metabolize glucose by glycolysis and suggesting a sex-specific difference in the requirement and/or mechanism of glycolysis.
To determine how decreased glycolysis can compromise the function of CA1 neurons, we first examined the impact of PKM1cKO on neuronal survival. However, there was no change in the density of NeuN-positive cells within CA1 at 12 months in either female or male PKM1cKO mice (Figure S5A). In addition, total brain volume and hippocampus, thalamus, and ventricle size assessed by in vivo T2-weighted MRI in live 11- to 15-month-old mice did not differ between female or male PKM1cKO and PKM1WT mice (Figures 5A and S5B). Therefore, postnatal loss of PKM1 disrupts neuronal function rather than survival in female mice.
Figure 5. Female PKM1cKO mice have increased metabolic conversion of HP pyruvate to lactate.
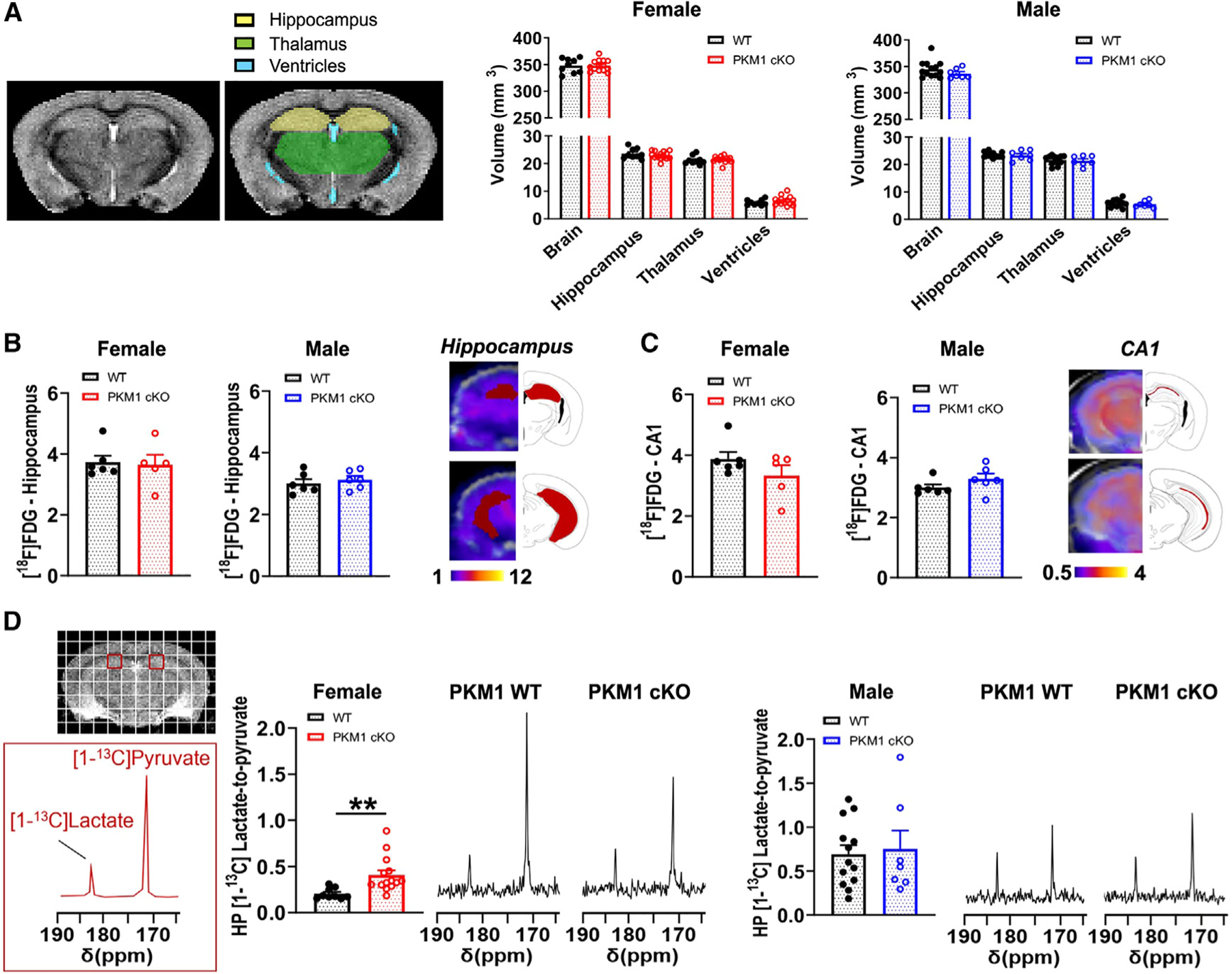
(A) Representative in vivo T2-weighted images of 11- to 15-month-old mice used for volumetric analyses showed no differences between female or male PKM1cKO and PKM1WT mice for the entire brain, hippocampus, thalamus, or ventricles. Data are means ± SEM. n = 9 PKM1WT, 14 KO females, and n = 7 PKM1WT, 13 KO males.
(B and C) There were no differences in [18F]FDG-PET signal between 11- and 14-month-old mice PKM1WT and PKM1cKO mice in the hippocampus (B) or CA1 (C). n = 5–6 mice/group.
(D) Representative 13C spectra of 11- to 15-month-old mice showing HP [1-13C]pyruvate and HP [1-13C]lactate levels from a region containing CA1 (red square). HP [1-13C]lactate-to-pyruvate ratios were markedly higher in female PKM1cKO versus PKM1WT mice, but were similar in males. n=9 PKM1WT, 14 KO females and seven PKM1WT, 13 KO males. **p%0.01 by unpaired t tests.
PKM1-mediated glycolysis is critical in neurons from female mice
[18F]FDG-PET imaging did not detect any significant differences in glucose uptake in the total hippocampus, or specifically in CA1, in either male or female PKM1cKO mice (Figures 5B and 5C). We used HP 13C MRSI to measure pyruvate conversion into lactate. In female PKM1cKO mice, a significant increase in the rate of pyruvate-to-lactate conversion (i.e., increased HP [1-13C]lactate-to-pyruvate ratio) was observed in the CA1 area (Figure 5D), while no changes were detected in male PKM1cKO mice. We also observed an increased rate of pyruvate-to-lactate conversion in the thalamus of female PKM1cKO mice (Figure S5D), even though thalamic neurons had grossly normal levels of PKM1 expression (Figure S5E). This likely reflects contributions of neurites from Cre-expressing neurons projecting to the thalamus,34 although non-cell-autonomous effects could also contribute.
Notably, PKM1cKO had no impact on cerebral blood volume and flow as measured by MRI methods, suggesting that these parameters had no impact on the HP metabolite levels (Figure S5F). Decreased pyruvate production from glucose in PKM1cKO mice may lead to more import of HP [1-13C] pyruvate into cells or an upregulation of LDH activity, either of which would increase HP [1-13C] pyruvate-to-lactate conversion. In addition, compensation by the PKM2 isoform could also lead to a relative increase in HP [1-13C] pyruvate-to-lactate conversion.50 However, the relative contributions of these factors to increasing HP [1-13C] pyruvate-to-lactate conversion remain to be delineated.
To gain insight into how male and female neurons may differentially compensate for the absence of PKM1, we assessed PKM2 levels. Although PKM2 is normally expressed in glia rather than neurons,51 it can be upregulated in vivo to compensate for loss of PKM1.47 While PKM1 levels were decreased in both male and female PKM1cKO mice, no significant changes in PKM2 levels were detected in either sex (Figure S5G), although this does not exclude an increase in PKM2 activity or a compensatory increase in PKM2 in astrocytes.
GLUT3cKO and PKM1cKO do not alter post-synaptic electrophysiology under standard high-glucose conditions
We next investigated the consequences of PKM1 or GLUT3 deficiency on the function of CA1 pyramidal cells. First, we used hippocampal slices from 20- to 23-month-old mice to interrogate the CA3-CA1 pathway by stimulating Schaffer collaterals and recording the response in both the dendritic layer (str. Radiatum, field postsynaptic potentials [fPSPs]) and the pyramidal cell layer (str. Pyramidale, local field potentials [LFPs]) (Figure S6A). We found that loss of either GLUT3 or PKM1 did not affect the presynaptic dynamics, seen as paired-pulse ratio at varying interpulse intervals (Figures S6B and S6H), the post-synaptic response to increasing stimulation intensities (Figures S6C and S6I), or cellular excitability (population spike integral vs. fPSP slope relationship; Figures S6D and S6J). Network function in response to repetitive synaptic stimulation (30 s, 10-Hz train) was also unaltered, with knockout (KO) slices exhibiting a robust response comparable to that in WT both in dendritic (Figures S6E and S6K) and cellular (Figures S6F, S6G, S6L, and S6M) layers. These findings suggest that PKM1 or GLUT3 deficiency in CA1 pyramidal cells does not significantly alter their post-synaptic electrophysiological function under standard electrophysiologic conditions, including high glucose concentrations (10 mM), although we did not assess function under physiologic brain glucose levels (≈1–1.5 mM). In addition, these recordings assess the post-synaptic response in CA1 dendritic and somatic layers and therefore would not detect any potential changes downstream at the CA1 pyramidal cells’ axonal boutons. The latter requires interrogation of the output of CA1, which is technically challenging in ex vivo brain slice preparations.
GLUT3cKO neurons have decreased glucose uptake and levels that are unresponsive to changes in external glucose
Our behavioral studies indicate a requirement for glucose uptake and glycolysis for CA1 neuronal function. Our finding that GLUT3cKO decreased brain size and body weight in female mice, while PKM1cKO did not, also suggests that neurons require glucose uptake for functions besides glycolysis, although differences in the ability to compensate for GLUT3 versus PKM1 loss may also contribute. To gain insight into how GLUT3 influences glucose metabolism in neurons, we examined the impact of GLUT3cKO on glycolytic gene expression in CA1 neurons, but this was unchanged (Figure 6A).
Figure 6. Neurons require glucose uptake and glycolysis to maintain ATP at the synapse.
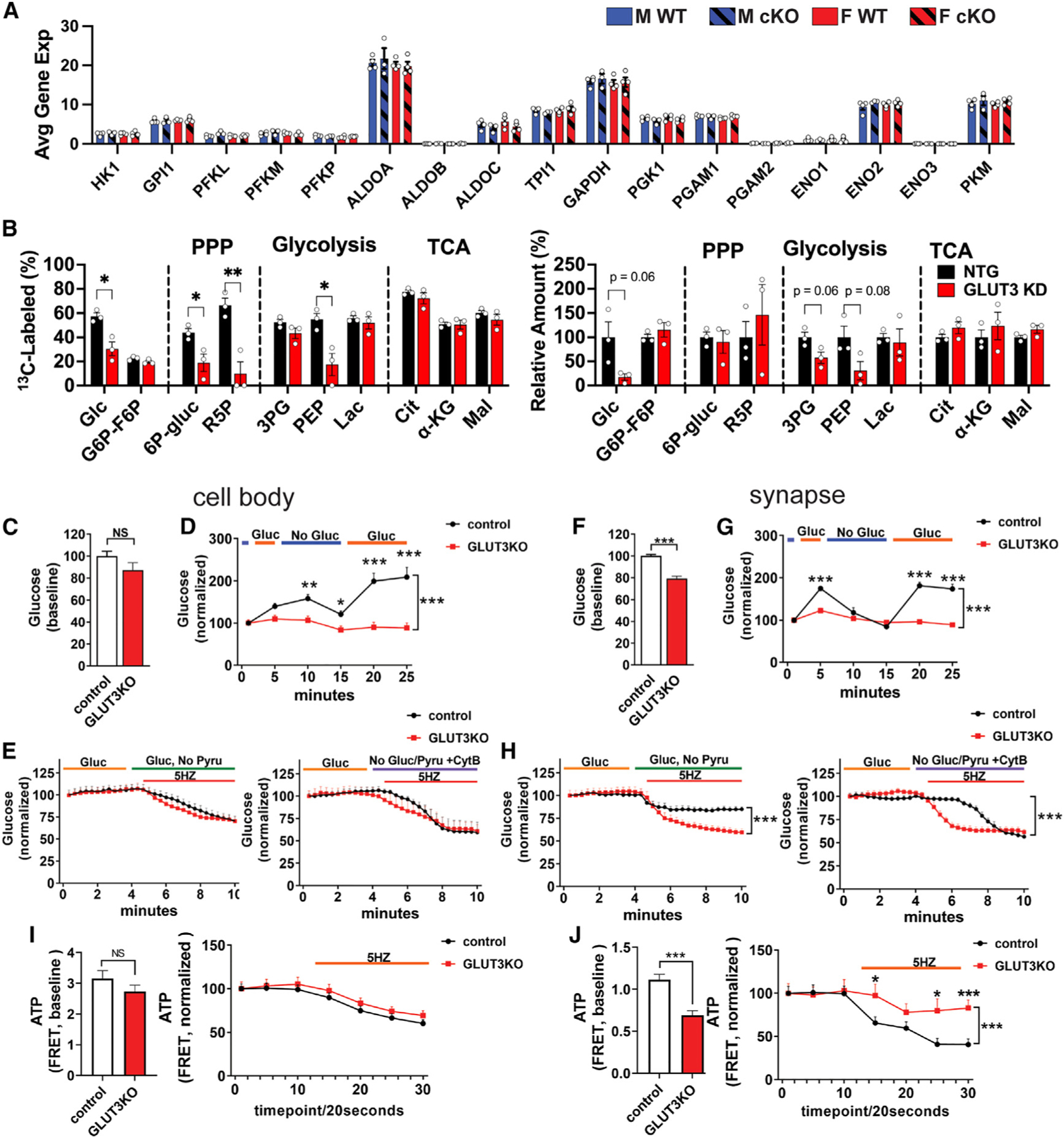
(A) Expression of glycolytic genes in GLUT3cKO CA1 neurons. Data are means ± SEM. n = 3–4 mice/group, compiled from 20 to 36 capture areas/mouse in CA1.
(B) Effect of GLUT3 KD in iPSC-derived neurons incubated for 24 h with 1.5 mM [U-13C]glucose on percentage of glucose-derived metabolites and total metabolites. The corresponding isotopologue data are shown in Figure S7C. Identical NTG controls (1.5 mM [U-13C]glucose) are shown in Figures S7B and S7C. n = 3 samples/group.
(C–H) GLUT3 KO disrupts glucose homeostasis in individual neurons. GLUT3lox/lox neurons were co-transfected with a fluorescent glucose sensor (iGlucoSnFR-mRuby) and either Cre (to delete GLUT3, GLUT3KO) or empty vector (control), as well as BFP-synaptophysin to identify synaptic boutons.
(C and D) GLUT3KO neurons had similar basal glucose levels to controls at the cell body (C), but their glucose levels were less responsive to changes in the extracellular glucose (D).
(E) Glucose levels decreased similarly in GLUT3KO and control neurons with electrical stimulation (5 Hz, 5.5 min) to increase neural activity (left), and the speed and extent of decrease was somewhat greater when glucose uptake was blocked with cytochalasin B (right) n = 8–10 coverslips/group (two or three cells/coverslip) from three independent experiments.
(D and E) Control and GLUT3cKO glucose values are normalized to the starting point.
(F and G) The synapses of GLUT3KO neurons had lower basal glucose levels (F), and their glucose levels were less responsive to changes in the extracellular glucose than controls (G).
(H) Glucose levels in GLUT3KO synapses decreased to a greater extent in GLUT3cKO versus control synapses, in response to electrical stimulation (left). Blocking all glucose uptake with cytochalasin B caused glucose levels in controls to drop to GLUT3KO levels (right). n = 8–10 coverslip/group, three to five synapses/coverslip from three independent experiments.
(I) GLUT3KO neurons have similar basal ATP levels at cell bodies (left), and in response to electrical stimulation (5 Hz, 5.5 min) to increase the ATP demand (right). n = 11–13 coverslips/group, two or three cells/coverslip from five independent experiments.
(J) GLUT3KO synaptic boutons have decreased ATP levels (left), and their ATP levels decrease less in response to stimulation (5 Hz, 5.5 min). n = 6–7 coverslips/group, three to five synapses/coverslip from three independent experiments.
ns, not significant; *p < 0.05; **p < 0.01; ***p < 0.001, by unpaired t tests (C, F, I, J) or two-way ANOVA with Tukey’s multiple comparison (A and B). Brackets in graphs show significance of linear mixed modeling for the interaction of genotype and time (D, E, G, H, I right, J right) with Sidak’s multiple comparison.
We hypothesized that decreased glucose uptake across physiologically relevant concentrations would decrease the levels of cytosolic glucose and downstream glucose metabolites. We therefore used targeted and untargeted metabolomics to examine the impact of lowering GLUT3 levels with CRISPRi on glucose metabolites. iPSC lines co-expressing an inducible dCas9-KRAB and either GLUT3 (GLUT3 KD) or an NTG (control) sgRNA were differentiated into neurons, and then cultured for 11 days, at which time GLUT3 KD neurons showed near-complete KD of GLUT3 expression (Figure S7A).
Neurons were then incubated with [U-13C]glucose for 24 h prior to harvesting for metabolomics.23 In control cells, the incorporation of 13C-labeled metabolites into downstream glycolytic, TCA, and PPP metabolites decreased with decreasing extracellular [U-13C]glucose, spanning concentrations typically found in cell culture and in vivo in physiologic and pathologic conditions (0.15–20.5 mM) (Figure S7B).
Interestingly, intracellular glucose was decreased in GLUT3KD neurons incubated with [U-13C]glucose for 24 h at the physiological 1.5 mM glucose concentration but not at 20.5 mM glucose (Figure S7B). Specifically, the percentage of glucose labeled by [U-13C] decreased from ≈60% in the NTG neurons to 30% in the GLUT3 KD neurons, while total levels dropped to <20% of control values (Figures 6B and S7C), consistent with decreased glucose uptake due to GLUT3 KD. Moreover, GLUT3 KD neurons had markedly decreased labeled fractions of the glycolytic metabolite PEP, as well as the PPP metabolites 6P-gluconate, and ribose 5-phosphate (R5P). There were no changes in either the labeled or total fractions of TCA metabolites. Indeed, despite taking in less [U-13C]glucose, neurons with GLUT3 KD still derived an amount of citrate and lactate from [U-13C]glucose equivalent to control neurons (>70% and 50% respectively). This suggests that, when glucose is limiting, neurons prioritize metabolism of glucose metabolites through the TCA cycle and aerobic glycolysis, rather than through the PPP. In contrast, in higher glucose levels typically used for cell culture, GLUT3 KD had little or no impact on either labeled or total levels of glucose or downstream glycolytic, PPP, or TCA metabolites, suggesting that sufficient glucose can enter neurons through other GLUTs under these conditions (Figure S7B).
We next examined relative glucose levels in individual neurons in culture. GLUT3lox/lox hippocampal neurons were co-transfected with a glucose sensor (iGlucoSnFR-mRuby) and either Cre (to delete GLUT3, GLUT3KO) or empty vector (control). We found that GLUT3KO neurons had similar basal glucose levels to controls at the cell body (Figure 6C). However, in contrast to controls, their glucose levels were unresponsive to changes in the extracellular glucose level, consistent with a profound deficit in glucose uptake in the absence of GLUT3 (Figure 6D). We next examined the impact of electrical field stimulation (5 Hz, 5.5 min) to increase the energy requirements of neurons by increasing neural activity.30,52,53 At the cell body, when extracellular glucose was the sole energy substrate, glucose levels decreased to a similar extent in control and GLUT3KO neurons (Figure 6E). As expected, the magnitude and speed of drop was somewhat greater when all glucose uptake was blocked (no glucose and cytochalasin B). These results suggest that, at the cell body, GLUT3KO neurons decrease their rate of glucose consumption to match decreased uptake, such that glucose levels decrease at a similar rate to controls.
We next examined the impact of GLUT3KO on glucose levels at the synapse, where GLUT3 may be enriched.54 At baseline, GLUT3KO neurons had lower glucose level at the synapse than controls (Figure 6F), and their glucose levels were unresponsive to changes in the extracellular glucose level (Figure 6G). Moreover, in the presence of extracellular glucose, intracellular glucose levels declined far more rapidly and to a greater extent in GLUT3KO neurons than controls in response to electrical field stimulation (Figure 6H, left). This suggests that, at the nerve terminal, GLUT3KO neurons consume glucose rapidly in response to electrical stimulation and are unable to replenish their supply, resulting in a marked decline in glucose levels. Blocking all glucose uptake eventually caused glucose levels in control neurons to drop to GLUT3KO levels (Figure 6H, right).
Neurons require glycolysis to maintain synaptic ATP levels
Our electrophysiology data indicate that CA1 neuronal cell bodies in GLUT3cKO slices have sufficient ATP levels to support increased neural activity. However, these studies were unable to provide insight into ATP levels at the synapse, which may be particularly vulnerable to energy failure30,52 because the proportional increase in energy expenditure with neural activity may be greater in synapses than the cell body. To determine if GLUT3KO decreases glucose-derived ATP production in individual neurons, including at the synapse, GLUT3lox/lox and WT neurons were co-transfected with either Cre or empty vector, and an ATP-fluorescence resonance energy transfer (FRET) sensor made of the ε subunit of the bacterial FoF1-ATP synthase (an ATP-binding protein) flanked by mApple and mClover, which is sensitive to physiologic changes in ATP levels.55,56 GLUT3KO had no impact on ATP levels at the cell body in basal conditions (30 mM glucose, 10 mM pyruvate), in which cells could derive ATP from both glycolysis and aerobic respiration (Figure 6I). Moreover, GLUT3KO ATP levels dropped similarly to controls with sustained electrical field stimulation (5 Hz, 5.5 min). However, basal ATP levels were lower in GLUT3KO nerve terminals than in controls, and ATP levels in GLUT3KO nerve terminals failed to decline further during sustained electrical stimulation (Figure 6J), suggesting that GLUT3KO neurons either fail to fire in response to the electrical stimulation despite consuming glucose or that they rely on alternative fuel sources to maintain ATP levels. These findings show that neurons rely on glycolysis to maintain ATP levels at the synapse, consistent with the hypothesis that glycolysis is important for rapid response to increased ATP demand to support neural activity.13
Compensatory pathways in GLUT3KO neurons
Although GLUT3KO disrupts energy metabolism and neuronal function, neurons can nonetheless survive and metabolize glucose without GLUT3. To delineate potential compensatory mechanisms, we used spatial genomics to examine gene expression in CA1 neurons in 7-month-old GLUT3cKO and WT mice. Targeted examination revealed no compensatory changes in other GLUTs (Figure 7A). There were also no increases in the expression of TCA genes in GLUT3cKO neurons (Figure 7B). To search for other compensatory mechanisms, we performed an unbiased analysis of differentially expressed genes in CA1 neurons. The resulting hits were enriched with genes related to either carbohydrate or mitochondrial energy metabolism (Figures 7C–7E). The mitochondrial genes included Iars2, which encodes mitochondrial isoleucine-tRNA synthetase and whose mutation produces a range of clinical disorders with impaired respiratory chain function, including Leigh syndrome57,58; Echdc2 (enoyl-CoA hydratase domain-containing protein 2), a mitochondrial protein involved in beta oxidation of fatty acids and valine catabolism,59,60 and whose deficiency can also cause Leigh syndrome61; and melanocyte inducing transcription factor (Mitf), which regulates both PDH62 and PGC1a,63 while increasing oxidative phosphorylation,64 boosting ATP levels under respiratory conditions23 and protecting against glucose restriction.65 Indeed, Mitf expression was increased by immunofluorescence in CA1 neurons from GLUT3cKO mice (Figure S7D).
Figure 7. Sex-dependent and independent neuronal responses to decreased glucose uptake.
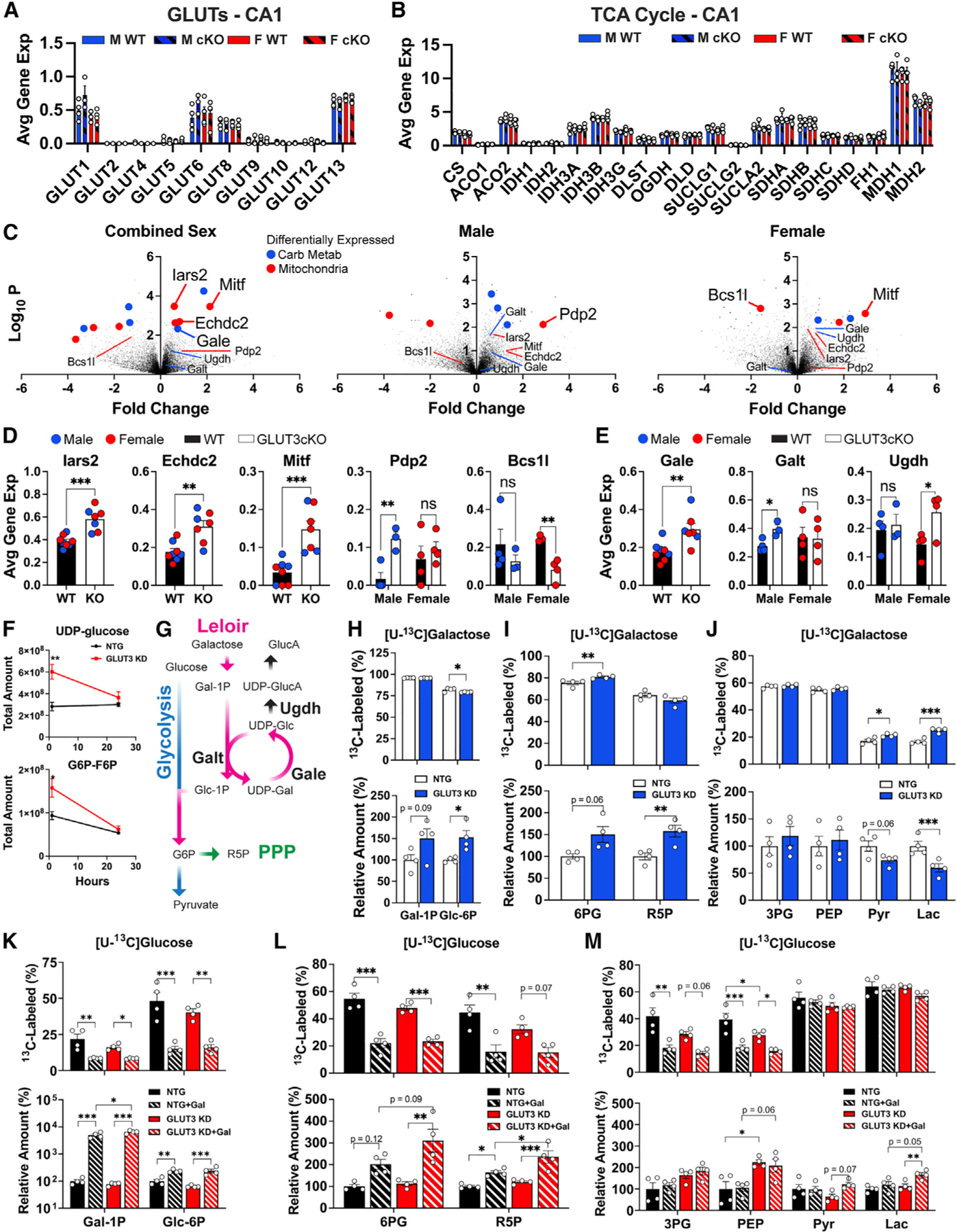
(A) Impact of GLUT3cKO on expression of other GLUTs in CA1 neurons. Data are means ± SEM, n = 3–4 mice/group, each compiled from 20 to 36 capture areas in CA1 per mouse.
(B) Expression of TCA genes in CA1 neurons. n = 3–4 mice/group.
(C) Volcano plots of differentially expressed genes between GLUT3cKO and WT mice. n = 7–8 (combined sexes), three or four (males only) and four (females only) mice/group. Blue and red points indicate genes related to carbohydrate metabolism and mitochondria, respectively.
(D) Gene expression of top hits Iars2, Echdc2, Mitf, Pdp2, and Bcs1l for CA1 neurons. n = 3–4 mice/group. Data points are shown for male (blue) and female (red) mice.
(E) Expression of galactose metabolism hits Gale, Galt, and Ugdh for CA1 neurons. n = 3–4 mice/group. Data points for male (blue) and female (red) mice.
(F) Impact of GLUT3 KD on targeted and untargeted metabolite levels of UDP-glucose and G6P-F6P following incubation with [U-13C]glucose for 1 and 24 h n = 3 samples/group/time point.
(G) Schema of potential compensatory pathway for decreased glucose uptake, by which galactose is metabolized into glucose-6-phosphate.
(H–J) Relative amounts and [U-13C]galactose fractional labeling of Leloir, PPP, and glycolytic metabolites in GLUT3 KD and NTG neurons after 24 h of culture. n = 4 samples/group.
(K–M) Relative total amounts and [U-13C]glucose fractional labeling of Leloir, PPP, and glycolytic pathway metabolites in GLUT3 KD neurons relative to NTG neurons when treated with either 1.5 mM [U-13C]glucose or 1.5:1.5 mM [U-13C]glucose:galactose for 24 h. n = 4 samples/group.
*p < 0.05, **p < 0.01, ***p < 0.001 by unpaired t test and two-way ANOVA with either Tukey’s multiple comparisons test (D, E, H–M) or Sidak’s multiple comparisons test (F).
Although most hits had similar effects on male and female GLUT3KO neurons, we also identified several genes with sex-specific effects (Figures 7D and 7E). In particular, Pdp2 (pyruvate dehydrogenase phosphatase catalytic subunit 2), which diverts metabolism to oxidative phosphorylation, was lower at baseline in male versus female CA1 neurons, while GLUT3cKO increased Pdp2 expression to similar levels as those in female WT and GLUT3cKO neurons. Another mitochondrial gene, Bcs1l (mitochondrial chaperone BCS1), needed for the assembly of mitochondrial respiratory chain complex III, was decreased in female GLUT3cKO neurons. Bcs1l immunofluorescence was also decreased in CA1 neurons from female GLUT3cKO mice (Figure S7D). Bcs1l expression may be downregulated in response to an overall decrease in mitochondrial respiration in GLUT3cKO mice, but further investigation is required to delineate the impacts of these gene expression changes on ATP production when glucose is limiting.
Pathway analysis of CA1 gene expression in GLUT3cKO mice also identified an enrichment of galactose metabolism as a potential compensatory mechanism (Table S2). In particular, Gale (UDP-galactose-epimerase), which plays a key role in the catabolism of galactose into glucose-6-phosphate such that galactose can enter the glycolytic pathway via the Leloir pathway,66 was upregulated in GLUT3cKO mice (Figure 7E). Galactose is also imported across the cell membrane via several glucose transporters, including GLUT3.67 Two genes involved in the metabolism of UDP-glucose demonstrated sex-specific differences with GLUT3cKO: Galt (galactose-1-phosphate uridyltransferase) and Ugdh (UDP-glucose 6-dehydrogenase). Expression of Galt, which uses UDP-glucose for galactose catabolism, was higher only in male mice, whereas Ugdh, which uses UDP-glucose for glucuronate biosynthesis, was higher only in female mice (Figure 7E).
Targeted metabolomics of GLUT3 KD neurons at 1 and 24 h post incubation with [U-13C]glucose revealed increased amounts of UDP-glucose and glucose/fructose-6P (G6P-F6P) after 1 h (Figure 7F). As the metabolites in the media were consumed over the 24-h period, the total amount of UDP-glucose and G6P-F6P in GLUT3 KD neurons declined. The change in total UDP-glucose amount between the 1- and 24-h time points indicates a higher rate of UDP-glucose consumption relative to its biosynthesis; the increased expression of Gale could reflect a response to meet the catabolic demand for UDP-glucose through its increased production. Given that the B27 supplement used in the neuronal differentiation media contains galactose (0.04 mM in media; Figure S7E), we investigated whether human neurons can metabolize galactose via glycolysis and the TCA cycle. Incubation with [U-13C]galactose resulted in robust 13C-incorporation by both NTG and GLUT3 KD neurons into galactose-1P, glucose-6P, as well as into glycolytic, PPP, and TCA metabolites (Figures 7G–7J and S7F). Indicative of upregulated galactose metabolism in response to GLUT3 KD, total levels of galactose-1P and glucose-6P in GLUT3 KD neurons (≈80% KD by rtPCR) cultured with [U-13C]galactose were higher than in NTG neurons (Figure 7H). GLUT3 KD neurons also had slightly higher fractions of 13C-labeling in alpha-ketoglutarate (a-KG) and malate, relative to controls (Figure S7F).
To further assess the interplay between glucose and galactose, we examined the impact of equimolar amounts of galactose and glucose on glucose metabolism. Addition of galactose increased the total levels of galactose-1P and glucose-6P, while markedly decreasing the percentage labeling of galactose-1P and glucose-6P following a 24-h incubation with [U-13C]glucose (Figure 7K), consistent with galactose competing with glucose for conversion into these metabolites. Moreover, the addition of galactose contributed to the increased levels of PPP metabolites 6P-gluconate and ribose-5P (Figure 7L). Interestingly, while the addition of galactose decreased the fractional labeling from [U-13C]glucose in upstream glycolytic metabolites 3PG and PEP, labeling in both pyruvate and lactate remained unchanged (Figure 7M). However, the total amounts of pyruvate and lactate increased ~60% with the addition of galactose only in the GLUT3 KD neurons. The total amounts of TCA metabolites citrate, ⍺-KG, and malate increased in both NTG and GLUT3 KD neurons with the addition of galactose despite the fractional labeling from glucose remaining the same (Figure S7G). The contrast in fractional [U-13C]glucose labeling between the PPP and glycolysis with the addition of galactose illustrates that galactose is preferentially shunted through the PPP, while glucose is directed through glycolysis to the TCA cycle. Taken together, the differentially expressed metabolic and mitochondrial genes suggest a multi-pronged compensatory response to GLUT3cKO involving a shift toward mitochondrial respiration and enhanced galactose metabolism.
DISCUSSION
It has been proposed that neurons derive most or all of their ATP from aerobic respiration,68 and the extent to which neurons directly take up and metabolize glucose through glycolysis, and whether this is required, especially under physiologic conditions, is unknown. Here we show that neurons can indeed take up and metabolize glucose through glycolysis and use this to produce the majority of their TCA metabolites. Moreover, we show they require glucose uptake to maintain ATP levels at the synapse, and normal neuronal function in vivo. Our findings also reveal systems-level sex differences in glucose metabolism and identify increased galactose metabolism as a mechanism to compensate for impaired neuronal glucose uptake.
Neurons require direct glucose uptake and metabolism through glycolysis
Multiple lines of evidence suggest that neurons directly import glucose in culture.6,52,53,69 However, direct confirmation of this has remained elusive because of the presence of glia in primary rodent neuron cultures, which limits the ability to distinguish glial from neuronal uptake. Here, we used metabolomics on near homogeneous human neuron cultures to demonstrate that neurons do indeed directly import glucose. Moreover, we show that GLUT3 is required for normal glucose uptake, but the effect is most robust when glucose levels are at physiologic brain levels. At the higher glucose levels typically used for neuronal culture, glucose could still enter GLUT3KO neurons and reach WT levels. The mechanism may involve other GLUTs, although we found no evidence of upregulation of other GLUTs in GLUT3cKO mice.
What are the consequences of decreased glucose import into neurons? We found that GLUT3KO neurons are less metabolically responsive to changes in extracellular glucose, and that decreased glucose import in GLUT3KO neurons results in a marked decrease in glucose levels during neural activity. Our metabolomics data also suggest that, when glucose uptake is limited, neurons may prioritize maintaining flux of glucose metabolites through the TCA cycle at the expense of both aerobic glycolysis (lactate) and the PPP.
Ultimately, decreased glucose uptake into neurons impairs function, as mice with postnatal deletion of GLUT3 develop spatial learning and memory and deficits. Our findings in GLUT3cKO mice are consistent with and build on findings from Shin et al., who observed memory deficits in male mice in a similar model of GLUT3 deficiency.7 These deficits may result from synaptic dysfunction, as we found that GLUT3 deficiency compromised ATP levels at the nerve terminal but not the cell body. Notably, cell body and dendrite function may also be compromised in vivo, especially as decreased glucose levels in GLUT3KO neurons were only apparent at low glucose levels. However, we did not detect deficits ex vivo under higher (10 mM) glucose conditions.
Another important question is how many of the above changes can be attributed to deficits in glycolysis versus other glucose-supported functions, such as the PPP. Even if glucose is taken up by neurons, neurons have been hypothesized to metabolize glucose primarily through the PPP due to insufficient phosphofructokinase to support glycolysis.8,9 Moreover, neuronal glycolysis is reported to increase in response to certain pathologic stressors, and pharmacologically inhibiting this increase may be neuroprotective.10–12 Here, we show that human neurons do indeed metabolize glucose through glycolysis and can derive most of their TCA metabolites from glucose. Moreover, we show that postnatal deletion of PKM1 in neurons in vivo resulted in memory loss from 12 months of age in female mice, while males were unaffected at this age. This was accompanied by increased conversion of HP [1-13C]pyruvate to [1-13C]lactate, indicating a functionally significant disruption in glycolytic metabolism upon PKM1 deletion, compelling evidence that neurons (or at least neurons from females) require glycolysis for normal function.
Importantly, our findings do not exclude the lactate shuttle as an important contributor to the bioenergetic function of neurons. Indeed, considerable evidence shows both that neurons can support neural activity with either respiration or glycolysis, and that neurons and other cell types can readily switch between fuel sources depending on their energy requirements and local substrate availability.6,23,52,53 However, our data indicate that the lactate shuttle is unable to upregulate and fully support neuronal energy requirements on its own.
Sex-specific requirements for glucose uptake and glycolysis
Interestingly, the PKM1 metabolic deficits detected by HP 13C MR occurred exclusively in females, consistent with the memory deficit specific to female PKM1cKO mice. Interpretation of this sex-specific HP change is complex, but likely reflects not only the primary disruption in neuronal glycolysis but also potential compensatory changes in glial metabolism and neuro-glial coupling (lactate shuttle). The HP pyruvate and lactate signals encompass vascular, neuronal, and glial components, and the MR acquisition parameters (in particular flip angles70) are likely to play a crucial role in the relative contribution of each to the HP lactate-to-pyruvate ratios measured by MR. Although future studies are needed to fully understand the mechanisms driving the HP changes, this represents (to our knowledge) only the second report of HP metabolic imaging detecting sex differences in vivo,63 and the first one in the brain. Our data support the potential of HP metabolic imaging to reveal changes in brain energy metabolism with aging and disease.71 Notably, [18F]FDG-PET imaging did not detect any changes following either GLUT3 or PKM1 deletion, which might be due to high background levels, masking changes occurring in the relatively small CA1 area, and might also indicate that most [18F]FDG-PET signal originates from glial cells.72
We hypothesize that PKM1cKO males may also eventually develop motor deficits, and point out that the human neurons with PKM KD (both PKM1 and M2 isoforms), in which we observed metabolic differences, are male in origin. Nonetheless, neurons in female mice were more susceptible to PKM1 inhibition in our paradigm, perhaps suggesting sex-specific differences in compensatory mechanisms. These differences may also apply to other tissues, as loss of PKM2 promotes hepatocellular cancer at a far greater rate in male than female mice.73
GLUT3cKO mice also developed broad sex-specific changes. First, GLUT3cKO female mice had decreased body weight, consistent with a prior finding that GLUT3 deletion in neurons and glia during early embryogenesis decreased body weight in females but not males.74 Remarkably, our study replicates these results when GLUT3 is deleted in neuronal populations not normally associated with weight regulation. Although we have not excluded the possibility that changes in body weight resulted from memory deficits leading to decreased food consumption, the memory changes occurred in both male and female mice, while the changes in body weight and brain volume were only in females. Moreover, the memory changes involved only the GLUT3cKO mice, while the weight changes also occurred in heterozygotes. An independent study showed that mice with embryonic deletion of GLUT3 throughout the brain had decreased body weight at postnatal day 15 (males and females were analyzed together), despite increased food intake,7 supporting that the changes in body weight in our model are unlikely to result from insufficient food intake. Interestingly, hippocampal sharp wave-ripples were recently found to regulate peripheral glucose concentrations via a pathway projecting to the lateral septum and hypothalamus,36 raising the possibility that GLUT3cKO might influence food consumption through alternate mechanisms. However, the role of this pathway in our observations is unclear, and peripheral glucose levels were unchanged in GLUT3cKO mice.
Metabolic rewiring in response to glucose deficiency
A critical question is how neurons compensate when glucose is limiting in vivo. Indeed, although the capacity for metabolic rewiring has been considered limited in neurons,75 broad compensatory mechanisms to maintain TCA metabolites are observed in the setting of mitochondrial dysfunction.76 Here, we identify two potential mechanisms of neuronal compensation. First, we show that GLUT3cKO neurons upregulate several mitochondrial genes that can promote mitochondrial respiration, including Mitf, Iars2, and Echdc2. Upregulation of these genes may reflect an attempt to boost mitochondrial respiration by increasing the utilization of available glucose and by boosting respiration generated from other fuel sources.
Interestingly, another gene that may promote mitochondrial respiration, PDP2, was increased in male (but not female) GLUT3cKO neurons, perhaps contributing to the increased susceptibility of female mice to GLUT3 loss. Indeed, considerable evidence indicates that males and females (mice and humans) have fundamental differences in glucose and energy metabolism,77 and neurons and skeletal muscle from females rely more on fatty acid b-oxidation as an energy source than those from males.78,79 However, the molecular basis for these differences is poorly understood. Another gene that responded differently between sexes to GLUT3cKO was Bcs1l, a mitochondrial protein responsible for the final assembly of complex III; mutations to Bcs1l have been causally linked to complex III deficiency,80 impaired b-oxidation,81 and hypoglycemia.82 Additional research is required to determine whether the differential expression of such genes may underlie some of the differences in energy metabolism between sexes.
In addition to genes that regulate respiration, we also identified changes in genes regulating galactose metabolism. Galactose catabolism via the Leloir pathway yields glucose 6-phosphate to feed into both glycolysis and the PPP.66 We show that boosting galactose metabolism is an alternate mechanism to bypass glucose deficiency. Notably, GLUT3 not only mediates glucose transport, it also transports other carbohydrates, including galactose.83 As such, upregulation of galactose metabolism genes in the setting of GLUT3 loss of function may, in part, be a compensatory response to maintain intraneuronal galactose levels. However, our finding that galactose import is upregulated in response to GLUT3KD indicates that galactose can enter neurons through other mechanisms, and shows that neurons do indeed upregulate galactose metabolism to meet energy demands when glucose is insufficient. Indeed, GLUT3 has an extremely low affinity for galactose, and galactose is a substrate for other GLUTs (GLUT1, GLUT8) that hippocampal neurons express at lower levels.84,85 As such, our data provide some of the first evidence that neurons normally metabolize galactose, suggesting that galactose may contribute to normal brain energy homeostasis and may be able to compensate when glucose metabolism is disrupted. However, given the changes in memory, brain size, and body weight in GLUT3cKO mice, it is clear that this mechanism was not sufficient to compensate for decreased glucose uptake in CA1 neurons, at least with physiologic levels of energy substrates.
Implications for neurologic and neurodegenerative diseases
Our findings also suggest that impairments in neuronal glucose metabolism may directly contribute to neuronal dysfunction and degeneration in a range of conditions of insufficient brain glucose, such as hypoglycemia, stroke, and Alzheimer’s disease (AD). In AD, changes in glucose metabolism could result from a primary disruption of glucose uptake or glycolysis, or may reflect secondary compensatory changes to other metabolic deficits or to changes in neural activity that affect energy requirements. Interestingly, brains of patients with AD have markedly decreased levels of GLUT3 in the hippocampus and cortex even when normalized for synaptic loss,86 raising the possibility that decreased GLUT3 contributes to the decreased glucose uptake in AD. Moreover, overexpression of human GLUT3 is protective in a model of amyotrophic lateral sclerosis.87 As such, the findings presented here provide important insights into the basic biology of neuronal glucose metabolism and how changes in glucose metabolism may contribute to neurologic disease.
Limitations of the study
The metabolomics experiments prove that human neurons metabolize glucose through glycolysis. However, the pure neuronal population studied lacks glial cells, circuits, and three-dimensional interactions found in vivo that likely influence metabolism. Therefore, the relative flux of glucose metabolites through glycolysis versus the PPP, and through downstream glycolytic pathways, may differ between the human neurons studied and human and mouse neurons in vivo.
Although our paper focuses on changes in neurons, the HP pyruvate and MRI studies in vivo lack cell-type resolution. We have begun to address this by postnatally deleting GLUT3 and PKM1 specifically in neurons. These studies show that neurons need glycolysis to maintain normal brain energy metabolism and volume. However, they do not definitively prove that the changes in either HP pyruvate metabolism or brain volume directly result from changes in the neurons themselves rather than via non-cell-autonomous effects on glia. Moreover, the requirement for neuronal glycolysis does not provide insight into whether neurons also require astrocyte-derived lactate and, if so, the balance of energy derived from neuronal glucose metabolism versus astrocyte-derived lactate.
Last, our work begins to address the key question of how neurons compensate when glucose is limiting. We identify a compensatory upregulation of galactose metabolism that fuels downstream energy metabolites as well as several changes in the expression of metabolism-based genes, but additional studies are required to determine which of these changes are most critical in restoring energy.
STAR☆METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to Ken Nakamura (ken.nakamura@gladstone.ucsf.edu).
Materials availability
Information for resources and reagents are available from the lead contact upon request.
Data and code availability
Visium datasets are available in Tables S3 and S4. Metabolomics data sets are available in Tables S5, S6 and S7.
This paper does not report original code.
Further information and requests for reagents should be directed to and will be fulfilled by the lead contact, Ken Nakamura (ken.nakamura@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal experimental procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals, as adopted by the National Institutes of Health, and with approval of the University of California, San Francisco Institutional Animal Care and Use Committee. Animals used in this project were up to 20 months of age. All mice were housed in a state-of-the-art barrier facility managed by the UCSF Laboratory Animal Resource Center (LARC). Animal care and use in this research are covered under the UCSF ‘‘Assurance of Compliance with PHS Policy on Humane Care and Use of Laboratory Animals by Awardee Institutions’’ number A3400–01. Experiments were performed on age-matched mice of either sex, with the specific number of males and females matched between groups in each experiment (typically divided (50:50 between males and females). All mouse lines were maintained on a C57Bl/6 background (The Jackson Laboratory; RRID:IMSR_JAX:000664).
Generation of iPSC lines and iPSC-derived neuronal cultures
Human WTC-11 induced pluripotent stem cells expressing both doxycycline inducible neurogenin 2 (Ngn2) and inducible CRISPRi (dCas9-KRAB) machinery were received from Martin Kampmann.22 Lentivirus expressing sgRNA targeting PKM sgRNA sequence GCCGCAGCGGGATAACCTT) or GLUT3 (sgRNA sequence GGTAAGACTTTGGATCCTTCCTG), or non-targeting control (GCTGCATGGGGCGCGAATCA) were produced by the UCSF Viracore. iPSCs were incubated with lentivirus for 24h with 8ug/mL polybrene. After two days of culture in StemFlex medium (ThermoFisher A3349401), transfected iPSCs were selected with 1 μg/mL puromycin for 1 day.
iPSCs were cultured in StemFlex media in a 0.1 mg/mL Matrigel (Corning 356234) coated culture flask. Differentiation of the iPSCs was divided into 2 parts: pre-differentiation and maturation. Pre-differentiation consisted of 3 days of culture in pre-differentiation media and maturation consisted of 11 days in neuronal differentiation media. Pre-differentiation media contained Knockout DMEM/F12 (Gibco 12660012), 1X NEAA (Gibco 11140050), 1X N2 supplement (Gibco 17502048), 10 ng/mL NT-3 (Stemcell Tech 78074), 10 ng/mL BDNF (Stemcell Tech 78005), and 1 μg/mL of laminin (Gibco 23017015). On the first day of differentiation, iPSCs were passaged onto a 0.1 mg/mL Matrigel coated culture flask with pre-differentiation media containing doxycycline (2 μg/mL) and ROCK Inhibitor (10 nM). The pre-differentiation media was then refreshed on the third day of pre-differentiation. On the fourth day of differentiation, pre-differentiated neurons were passaged into 5 μg/mL laminin coated 6-well plates at a density of 500,000 cells/well with the neuronal differentiation media; 20 μM of trimethoprim was used to induce the target knockdowns (PKM or GLUT3) 2 days after the transition to the neuronal differentiation media.22 Neuronal differentiation media contained 1:1 of DMEM/F12 (Gibco 11320033) and Neurobasal A media (Gibco 10888022), 1X NEAA (Gibco 11140050), 0.5X N2 supplement (Gibco 17502048), 0.5X B27 supplement (Gibco 17504044), 0.5X GlutaMAX (Gibco 35050061), 10 ng/mL NT-3 (Stemcell Tech 78074), 10 ng/mL BDNF (Stemcell Tech 78005), and 1 μg/mL of laminin (Gibco 23017015). The neuronal differentiation media was refreshed once a week to feed the neurons.
Primary neuronal cultures
Primary hippocampal cultures were dissected from early postnatal (P0 to P1) mouse pups.30,92 Neurons were transfected using electroporation (Lonza, VVSI-1001 with the relevant plasmids and cultured in MEM Eagle’s with Earle’s BSSMedium (UCSF Cell Culture Facility, CCFAC001) supplemented with 5% fetal bovine serum (Hyclone, SH3007103), 1% Glutamax (Gibco 175007), 21mM glucose, 2% B27 supplement (Gibco 17504–044), and 0.1% serum extender (Fisher 355006). Neurons were plated at 600–900 cells/mm2, and uridine (10 μM, Sigma U3003) and 5-fluoro-2-deoxycytidine (10 μM, SigmaF0503) were added on day 4–5 to minimize glial overgrowth. Neurons were cultured at 37°C with 5% CO2 for 9–11 days before live imaging or fixation.
METHOD DETAILS
Metabolomics
Human iPSC-derived neurons were matured for a total of 12 days, including a 24 h incubation with media containing a [U-13C] metabolic probe prior to metabolite collection. The base media for targeted metabolomics contained Neurobasal A media without glucose or pyruvate (Gibco A2477501), 1X NEAA (Gibco 11140050), 0.5X N2 supplement (Gibco 17502048), 0.5X B27 supplement (Gibco 17504044), 0.5X GlutaMAX (Gibco 35050061), 10 ng/mL NT-3 (Stemcell Tech 78074), 10 ng/mL BDNF (Stemcell Tech 78005), and 1 μg/mL of laminin (Gibco 23017015). The media was supplemented with either [U-13C]glucose (Cambridge Isotope Labs CLM-1396–1) or [U-13C]galactose (Sigma-Aldrich 605379) for targeted metabolomics. To extract cellular metabolites, cells were washed in ammonium acetate (150 mM, pH 7.3) at 4C, and then incubated with 80% methanol at −80C for 20 min, prior to centrifugation at 14,000 g for 15 min. To extract cell media metabolites, the 13C-labeled media contained in each well post-incubation was collected and spun at 14,000 g for 10 min. The clarified media was then added to 80% methanol chilled to 4C, followed up with a 1 h incubation at −80C. Media samples were then centrifuged at 14,000 g for 10 min post incubation. Metabolite supernatants were dried in a Labconco CentriVap prior to storage at −80C. Sample metabolites were quantified at the UCLA Metabolomics Core. Metabolomic media conditions are detailed below.
| Condition | Metabolic probe | Description |
|---|---|---|
| 1 | [U-13C]glucose 20.5 mM | Differentiation media with basal level of glucose |
| 2 | [U-13C]glucose 1.5 mM | Physiologically relevant brain glucose level |
| 3 | [U-13C]glucose 1.5 mM | 1:1 glucose:galactose media tracing glucose |
| 4 | [U-13C]galactose 1.5 mM | Galactose replacing all glucose from condition 2 |
Galactose media measurements
A colorimetric galactose assay kit (Sigma Aldrich MAK012) was used to measure the galactose content of neuronal differentiation media supplement B27 (Gibco 17504044) using the protocol included with the kit. Samples were measured using a Molecular Devices Microplate Reader at an absorbance of 570 nm.
Behavioral testing
Elevated plus maze
The elevated plus maze (Kinder Scientific) consists of two open arms (without walls) and two closed arms (with walls) elevated 63 cm above the floor. Mice were acclimated to a dimly-lit testing room for 1 h prior to testing and mice were allowed to freely explore the maze for 10 min. The time, distance, and entries into the open and closed arms are calculated based on infrared photobeam breaks.
Open field
Spontaneous locomotor activity was assessed in the open field test (Flex-Field, San Diego Instruments) using standard procedures.93 Mice were placed in the center of a clear acrylic chamber and allowed to freely explore for 15 min. The photobeams that line the perimeter of the chamber detect horizontal and vertical movements. Ambulatory and fine movements are separated recorded. Fine movements are defined as breaking 2 beams consecutively and ambulatory movements are defined as breaking 3 beams or more.
Active place avoidance
Visual-spatial learning and memory was measured by the active place avoidance task (BioSignal Corp).94 Mice were trained to a new aversive zone for each time point. A habituation trial was conducted on the first day to determine baseline activity, followed by 3 days of shock training to the aversive zone. A probe and reinstatement trial was conducted 24 h after the last day of training. The number of entries, shocks, and the maximum time of avoidance to the aversive zone are calculated based on the video tracking software (Tracker).
Hot plate
Nociception was measured on the hot plate test (IITC). The surface of the aluminum hot plate was heated to 52°C and mice were placed in an enclosure atop the hot plate. The response latency is determined by a hindpaw lick, flick, or jump. The maximum latency was 30 s to prevent injury.
Histology and immunostaining
For in vivo histology, mice were perfused with phosphate-buffered saline (PBS) and then 4% paraformaldehyde (PFA). Brains were postfixed in 4% PFA overnight, and cryoprotected in 30% sucrose. Brain sections were set at 30–40 μm and prepared using a sectioning microtome (Leica SM2000 R). For immunofluorescence, brain sections were washed 3X in PBS, blocked 1 h at room temperature with blocking buffer (5% goat serum (Abcam, Cat #ab7481), 0.2% Triton in PBS), and incubated with primary antibodies at 4°C overnight with blocking buffer. The following antibodies were used: rabbit anti-PKM1 (1:400; Cell signaling Cat #7067, RRID: AB_2715534); rabbit anti-PKM2 (1:400; Novus Cat# NBP1–48308, RRID AB_10011057); chicken anti-MAP2 (1:1000; Abcam Cat# ab5392, RRID:AB_2138153); mouse anti-NeuN (1:1000; Millipore Cat# MAB377, RRID:AB_2298772); rabbit anti-NeuN (1:1000; Abcam Cat# ab177487, RRID: AB_2532109); rabbit anti-BCS1L (1:1000; Novus Cat# NBP2–92916); rabbit anti-Mitf (1:1000; Abcam Cat# ab122982, RRID: AB_10902226). Sections were rinsed 3X in PBS and incubated 2h at RT with the following secondary antibodies: Alexa Fluor 350, 488, 594, or 647 anti-mouse, chicken, or rabbit IgG (1:500; Invitrogen). Sections were imaged on a Zeiss LSM880 confocal microscope with Airyscan detector.
Neuronal density was calculated by dividing the total count number of NeuN and DAPI positive cells within CA1 region by the CA1 region area. Quantification of the density was performed blind to genotype with MetaMorph software (version 7.7.3.0; Universal Imaging, RRID:SciRes_000136).
In vivo MR acquisitions
All in vivo MR experiments were conducted on a 14.1 T vertical MR system (Agilent Technologies, Palo Alto, CA) equipped with 100 G/cm gradients and a dual tune 1H-13C volume coil (ØI = 40 mm, for HP 13C MRSI and T2-weighted MRI) or a single tuned 1H proton coil (ØI = 40 mm, for dynamic susceptibility contrast (DSC) MRI and T2-weighted MRI). For each imaging session, mice were anesthetized using isoflurane (2% in O2) and a 27 G catheter was secured in the tail vein to allow for intravenous (iv) injection of the HP probe or gadolinium contrast agent. Animals were then positioned in a dedicated cradle under constant anesthesia and placed in the MR bore; respiration was continuously monitored during all acquisitions to ensure animal well-being. First, axial T2-weighted images were acquired for adequate positioning of the grid used for HP 13C acquisitions. The following parameters were used: echo time (TE)/repetition time (TR) = 20/1200 m, slice thickness = 1.8 mm, number of averages = 2, matrix = 256 × 256, field of view (FOV) = 30 × 30 mm2. For HP 13C MRSI acquisitions, 24 μL of [1-13C]pyruvate preparation, was hyperpolarized using a Hypersense DNP polarizer (Oxford Instruments) for one approximately hour.40 After polarization, HP [1-13C]pyruvate was rapidly dissolved in isotonic buffer (pH~7) to a final concentration of 80 mM. A final volume of 300 μL of the HP [1-13C]pyruvate solution was then injected iv over 12 sec through the tail vein catheter. 2D 13C MRSI data were acquired 18 s after injection of the HP [1-13C]pyruvate solution, using the following parameters: TE/TR = 0.56/68 m; spectral width = 4006 Hz; 256 points; flip angle (FA) = 10°; FOV = 30 × 30 mm2;5 mm slice thickness. Next, axial T2-weighted Fast Spin Echo images were acquired for the evaluation of brain volume, using the following parameters: TE/TR = 12/2000 m, echo train = 8, slice thickness = 0.5 mm, number of averages = 8, matrix = 256 × 256, FOV = 30 × 30 mm2. For DSC MR images, a high contrast single slice axial T2-weighted Fast Spin Echo image was acquired using the following parameters: TE/TR = 30/3000 m, slice thickness = 2 mm, matrix = 128 × 128, FOV = 20 × 20 mm2. Next, a series of single slice axial T2-weighted Fast Spin Echo images were acquired, using the following parameters: TE/TR = 30/1000 m, slice thickness = 2 mm, matrix = 128 × 128, FOV = 20 × 20 mm2, 150 repetitions. In order to obtain a pre-contrast baseline, the first 25 s of the dynamic MR acquisition were performed before the administration of the contrast agent (Gadavist, Bayer). At 25 s, an iv bolus of 1 mmol/kg of body weight of Gadavist was delivered through the tail vein catheter.
Ex vivo MRI acquisitions
Mice were euthanized and perfused with a phosphate buffered saline (PBS) solution followed by a 4% paraformaldehyde (PFA) solution. Ex vivo MR experiments were conducted on a 14.1 T vertical MR system (Agilent Technologies, Palo-Alto, CA) equipped with a single tuned 1H proton coil (ØI = 20 mm). Ex vivo specimens were submerged in a Fluorinert (Sigma-Aldrich) solution and high-resolution 3D gradient echo images were acquired from the intact brain left in the skull using the following parameters: TE/TR = 15/75 m, number of averages = 6, matrix = 256 × 256 × 256, FOV = 12.8 × 12.8 × 12.8mm2.
MR data analysis
HP 13C MRSI datasets were analyzed using the SIVIC software (http://sourceforge.net/apps/trac/sivic/) and custom-built programs written in MATLAB (MATLAB R2011b, The MathWorks Inc.). The k-space dimensions were zero-filled by a factor of two resulting in a 32 × 32 matrix. The area under the curve (AUC) of HP [1-13C]pyruvate and AUC of HP [1-13C]lactate lorentzian fits were measured and the corresponding HP [1-13C]lactate-to-pyruvate ratio was calculated for each voxel. Regions of interest (ROIs) including the hippocampus and the thalamic area were defined on T2-weighted images and HP [1-13C]lactate-to-pyruvate ratios were reported as mean of the corresponding voxels. For analyses of in vivo MRI data, ROIs were manually delineated on T2-weighted images according to the Franklin and Paxinos anatomical mouse brain atlas using AMIRA software (Mercury Computer systems, San Diego, USA) and the corresponding ROI volume calculated. Specifically, the ROIs consisted of the entire brain, the ventricles and the hippocampus. In a similar manner, for analyses of ex vivo anatomical MRI, ROIs were manually delineated over the entire brain, the hippocampus and the CA1 regions, and the corresponding volume calculated for each ROI. For analyses of DSC images, Dynamic Susceptibility Contrast MR Analysis (DSCoMAN, version 1.0) plugin of ImageJ 1.52p (http://imagej.nih.gov.ij) was used to create cerebral blood volume (CBV) and cerebral blood flow (CBF) maps. ROIs including the hippocampus, thalamic areas and corpus callosum were manually delineated on the high contrast T2-weighted image acquired prior to the DSC acquisitions. These ROIs were then propagated onto the CBV and CBF maps and mean relative CBV and CBF values were obtained. In order to compare animals and groups, CBV and CBF values of the hippocampus and thalamic areas were normalized to the values obtained in the corpus callosum.
In vivo PET acquisitions
All scans were performed on a dedicated small animal PET/computerize tomography (CT) scanner (Inveon, Siemens Healthcare, Malvern, PA, USA). Mice received 71 ± 4.5 μCi of [18F]-fluorodeoxyglucose (FDG) in 0.1 mL volume iv via tail vein. Fifty-five minutes after radiotracer administration, static PET images were collected (10-min acquisition time), followed by a 10-min CT acquisition for attenuation correction of PET reconstruction. Animals were kept awake at room temperature during the 55 min uptake time. The animals were placed under anesthesia (2% isoflurane in O2) during the injections and scans.
PET data analysis
PET images were reconstructed using the ordered subsets expectation maximization (OSEM) algorithm, provided by the manufacturer. The resulting PET images had a 128 × 128 × 159 matrix with a voxel size of 0.776 × 0.776 × 0.796 mm3. CT images were reconstructed using a cone-beam Feldkamp reconstruction algorithm (COBRA, Exxim Computing Corporation, Pleasanton, CA, USA). The reconstructed CT images had a 512 × 512 × 662 matrix size with an isotropic voxel size of 0.191 mm3. The co-registered attenuation map from CT, obtained via a pre-derived rigid transformation matrix, was used for attenuation correction of the PET data. MR anatomical templates were created using the open source registration toolkit ANTS (https://github.com/ANTsX/ANTs) for each genotype and sex using in vivo T2-weighted images, leading to the creation of 8 MR templates: female GLUT3 WT, female GLUT3 KO, male GLUT3 WT, male GLUT3 KO, female PKM1 WT, female PKM1 KO, male PKM1 WT, male PKM1 KO. The PET/CT images of each individual mouse was co-registered to the corresponding MR template using VivoQuant 4.0 software (Invicro). Hippocampus and CA1 ROIs were manually delineated using the MR template and corresponding mean percent-injected dose per grams (%ID/g) values were obtained.
Electrophysiology
Animals were anesthetized with isofluorane and decapitated. The brains were rapidly removed from the skull and placed in the ice-cold (2–5°C) slicing solution. Slicing solution contained (in mM): 110 choline chloride, 2.5 KCl, 26 NaHCO3, 10 MgCl2, 1.25 NaH2PO4, 0.5 CaCl2, 10 glucose, 3 Na Pyruvate, 1 L-Ascorbic acid, pH 7.4.
300 μm-thick sagittal slices were cut from both hemispheres using a Vibratome (VT1200, Leica) and transferred to a 95% O2-CO2 vapor interface holding chamber (BSK5, Scientific Systems Design Inc. Canada) containing artificial cerebrospinal fluid (ACSF) where they were allowed to recover at 34°C for 1 h and held at room temperature (20–22°C) afterward. ACSF contained (in mM): 126 NaCl, 2.5 KCl, 1.5 CaCl2, 1.5 MgCl2, 26 NaHCO3, 1.25 NaH2PO4, 10 glucose, and 1.5 L-Ascorbic acid, pH 7.4. For recordings, slices were transferred to a submerged chamber (RC-27LD, Warner Instruments, USA) where they were continuously superfused on both sides with oxygenated ACSF at 32 °C at a flow rate of 10 mL/min.
Local Field Potentials (LFPs) were elicited by orthodromic stimulation of Schaffer collaterals by concentric bipolar stimulating electrode (FHC, Inc., USA) connected to a constant voltage isolated stimulator (DS2A-MKII, Digitimer North America) and placed in CA2 str. radiatum. Repetitive network activity was induced by a 300-pulse 10Hz stimulus train delivered to Schaffer collaterals; stimulus intensity was chosen to elicit a str. Pyramidale population spike amplitude at 50% of maximum. LFPs were recorded with a glass borosilicate microelectrodes filled with ACSF and placed in CA1 str. Radiatum (dendritic post-synaptic potentials, fPSPs), with the resulting LFP population response simultaneously in str. Pyramidale. Signals were sampled and digitized by MultiClamp 700B amplifier and Digidata 1550 acquisition system with pClamp10 software (Molecular Devices, USA), and analyzed using IgorPro6 software (Wavemetrics Inc., USA) running custom macros. fPSP slopes were analyzed as the linear fit slope values between 10% and 90% of fPSP peak. Input-output relationships were recorded as the fPSP slope values in response to increasing stimulation intensity (5–25mV), as well as the LFP population spike integral vs. fPSP slopes.
Live imaging of glucose or ATP
Live imaging was performed in Tyrode’s buffer (127mM NaCl, 10mM HEPES, 30mM glucose, 10 mM pyruvate, 2.5 mM KCl, 2mM CaCl2 and 2mM MgCl2, pH 7.4) at room temperature with a 40X objective on a Nikon Ti-E inverted microscope with an iXon EMCCD camera (Andor Technology, Belfast, UK) and a perfusion valve system (VC-8, Warner Instruments, Hamden, CT, USA) controlled by MetaMorph Software. Field stimulations (5 Hz, 300s) were performed with an A385 current isolator and an SYS-A310 accupulser signal generator (World Precision Instruments).
For imaging experiments using the cytosolic glucose sensor (iGlucoSnFR-mRuby expressed under the synapsin-1 promoter, kind gift of Jonathan Marvin, HHMI) sequential images were taken from FITC (490/20 ex, 535/50 em) and Texas Red (572/35 ex, 632/60 em) channels (Chroma, 89014). The glucose sensor fluorescence was normalized to mRudy fluorescence to control for expression level. Total glucose uptake was blocked with cytochalasin B (10 μM, Sigma-Aldrich). For measurements with the ATP fluorescence resonance energy sensor (Mendelsohn et al., 2018), sequential images were taken from mClover (490/20 ex, 535/50 em), mApple (572/35 ex, 632/60 em), and FRET channels (490/20 ex, 632/60 em). Synaptic boutons were identified based on co-localization with BFP-synaptophysin. The FRET/donor ratio was calculated for each bouton following the formula: , is the background-corrected fluorescence intensity measured in that given channel. (donor bleed through) and (direct excitation of the acceptor) were calculated by expressing clover and mApple individually and determining the ratios of and independently.95,96
Blood glucose level measurement
Blood samples were collected under non-fasted condition and obtained from the tail tip. The blood glucose level was measured using a glucometer (Cat #37321, Ermaine Laboratories).
Spatial gene expression
Spatial transcriptomics were acquired with Visium spatial gene expression kits (10X Genomics 1000184, 1000193, 1000215). Sample preparation, sample imaging, and library generation were completed in accordance with 10X Spatial Gene Expression protocols included with the kit. Briefly, fresh brain tissue was flash frozen in an isopentane bath cooled to −80C using dry ice. The brain tissue was then embedded in Optimal Cutting Temperature (OCT) compound (Tissue-Tek 62550–12). A cryostat was used to obtain a 10 μm thin section from the hippocampus that was then mounted onto a 10X spatial gene expression slide. Sections were stained with Gfap (Millipore MAB3402), NeuN (Abcam ab177487), and Hoechst 33342 (Invitrogen H1399) before imaging on a Leica Aperio Versa slide scanner. The cDNA libraries were generated at the Gladstone Genomics Core. Libraries were sequenced at the UCSF Center for Advanced Technology on an Illumina NovaSeq 600 on an SP flow cell. Alignment of the sequencing data with spatial data from the Visium slide was completed with the 10X Space Ranger software. The 10X Loupe Browser software was then used to identify four anatomical regions of interest: (1) CA1, (2) CA3, (3) dentate gyrus, and (4) thalamus. RNA capture areas corresponding to each anatomical region were selected for analysis based on their spatial proximity to the anatomical regions and on the expression of known genetic markers. CA1 genetic markers included Fibcd1 and Wfs1; CA3 markers included Nrip3 and Bok; dentate gyrus markers included Calb1 and Prox1; and the thalamus marker was Synpo2. Roughly 15–20 neuronal cell bodies fit into a single capture area for CA1 with roughly 28 capture areas for CA1. Gene expression levels were exported to GraphPad Prism and Microsoft Excel. Gene rankings for hit analysis were established using the fold change score (FCS) and signal-to-noise score (SNS). Equations for these scores are given as:
where is the average gene expression, is the standard deviation, and is the p value derived from an t test. Genes with a p value <0.05 that also appeared in the top 20% for both scoring metrics were highlighted as differentially expressed genes of interest. The expression levels for these genes of interest were probed in both CA1 and the thalamus to identify a subset of genes with CA1-specific changes. Pathway analysis was done on the differentially expressed genes using the Enrichr webtool89–91
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses including the n, what n represents, description of error bars, statistical tests used and level of significance are found in the figure legends. Linear mixed modeling and either Welch’s ANOVA with Dunnett’s T3 multiple comparison test or Welch’s t-tests were used to compare WT versus KO animals. Unpaired t-tests were used to compare metabolomics data within a single time point, whereas two-way ANOVA with Sidak’s multiple comparison was used for comparisons across timepoints. Mann-Whitney U test was used for electrophysiology data. Visium spatial transcriptomics data of targeted gene sets was analyzed by one or two-way ANOVA with either Sidak’s or Tukey’s multiple comparisons test. Determination of top differentially expressed genes used a threshold of the top 20% of differentially expressed genes that were ranked according to the defined SNS and FCS metrics. All analyses were performed using GraphPad Prism version 8.4.3 and Microsoft Excel.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Rabbit monoclonal anti-PKM1 | Cell signaling | Cat #7067; RRID: AB_2715534 |
| Rabbit monoclonal anti-PKM2 | Novus | Cat# NBP1–48308; RRID: AB_10011057 |
| Chicken monoclonal anti-MAP2 | Abcam | Cat# ab5392; RRID: AB_2138153 |
| Mouse monoclonal anti-NeuN | Millipore | Cat# MAB377; RRID: AB_2298772 |
| Rabbit monoclonal anti-NeuN | Abcam | Cat# ab177487; RRID: AB_2532109 |
| Rabbit anti-BCS1L | Novus | Cat# NBP2–92916 |
| Rabbit anti-Mitf | Abcam | Cat# ab122982; RRID: AB_10902226 |
| Anti-rabbit IgG Alexa Fluor 488 | Thermo Fisher Scientific | A11034; RRID: AB_2576217 |
| Anti-mouse IgG Alexa Fluor 594 | Thermo Fisher Scientific | A-11037; RRID: AB_2534095 |
| Anti-chicken IgG Alexa Fluor 350 | Thermo Fisher Scientific | PI62272 |
| Hoechst | Invitrogen | H1399 |
| Bacterial and virus strains | ||
|
| ||
| Lentivirus (2ND gen) PKM sgRNA | UCSF Viracore | This study |
| Lentivirus (2ND gen) GLUT3 sgRNA | UCSF Viracore | This study |
| Lentivirus (2ND gen) NTG sgRNA | UCSF Viracore | This study |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Paraformaldehyde | Electron Microscopy Science | Cat# 30525–89-4 |
| HEPES | Sigma Aldrich | Cat #3375 |
| D-Glucose | Sigma Aldrich | Cat# G7021 |
| Sodium Pyruvate | Sigma Aldrich | Cat# P2262 |
| Cytochalasin B | Sigma Aldrich | Cat# C2743 |
| 2-Deoxy-D-glucose | Sigma Aldrich | Cat# D8375 |
| Rotenone | Sigma Aldrich | r8875–1g |
| Iodoacetic Acid | Thermo Fisher Scientific | Cat# 35603 |
| Sucrose | Sigma Aldrich | Cat# 9378 |
| Phosphate buffered saline | UCSF Media Production | CCFAL001 |
| Isopentane | Sigma Aldrich | Cat# PHR1661 |
| Triton X-100 | Sigma Aldrich | Cat# T8787 |
| Vectashield antifade mounting medium | Vector Laboratories | Cat# H-1000–10 |
| Prolong gold antifade mountant | Thermo Fisher Scientific | Cat# P36941 |
| Prolong diamond antifade mountant | Thermo Fisher Scientific | Cat# P36970 |
| Goat serum | Thermo Fisher Scientific | Cat# 16210064 |
| MEM Eagle’s with Earle’s BSS Medium | UCSF Cell Culture Facility | CCFAC001 |
| Fetal bovine serum | Hyclone | SH3007103 |
| Glutamax | Gibco | Cat# 175007 |
| Serum extender | Thermo Fisher Scientific | 355006 |
| Uridine | Sigma Aldrich | Cat# U3750 |
| 5-fluoro-2-deoxycytidine | Sigma Aldrich | Cat# F0503 |
| [U-13C]glucose | Cambridge Isotope Labs | CLM-1396–1 |
| [U-13C]Galactose | Sigma Aldrich | Cat# 605379 |
| Neurobasal A media | Gibco | A2477501 |
| NEAA | Gibco | 11140050 |
| N2 supplement | Gibco | 17502048 |
| B27 supplement | Gibco | 17504044 |
| GlutaMAX | Gibco | 35050061 |
| NT-3 | Stemcell Tech | 78074 |
| BDNF | Stemcell Tech | 78005 |
| Laminin | Gibco | 23017015 |
| Ammonium acetate | Sigma Aldrich | Cat# 32301 |
| Methanol | Sigma Aldrich | Cat# 34860 |
| StemFlex medium | ThermoFisher | A3349401 |
| Matrigel | Corning | Cat# 356234 |
| DMEM/F12 | Gibco | Cat#12660012 |
| NEAA | Gibco | Cat#11140050 |
| Doxycycline hyclate | Sigma Aldrich | D9891 |
| ROCK Inhibitor Y27632 | Stemcell Tech | 72302 |
| [U-13C] glucose | Cambridge Isotope Labs | CLM-1396–1 |
| Gadavist | Bayer | 768511 |
| [1-13C]pyruvate | Sigma-Aldrich | 677175 |
| [18F]-fluorodeoxyglucose | University of California San Francisco Radiopharmaceutical Facility | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| Glucometer | Ermaine Laboratories | Cat# 37321 |
| Visium spatial gene expression slide & reagent kit | 10X Genomics | 1000184 |
| Dual index Kit TT Set A | 10X Genomics | 1000215 |
| Lonza electroporation Kit | Amaxa | Cat# VVSI-1001 |
| Optimal Cutting Temperature | Tissue-Tek | Cat# 62550–12 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Mouse primary neuronal culture | N/A | This study |
| Human: WTC-11 + NGN2 + dCAS9-KRAB | Martin Kampmann UCSF. | Tian et al.22 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| GLUT3lox/lox | Jeff Rathmell, Vanderbilt | Contat et al.24 |
| PKM1lox/lox | Mathew Vander Heiden, MIT | Davidson et al.47 |
| CamKIIα | Jackson Laboratories | 005359 |
|
| ||
| Oligonucleotides | ||
|
| ||
| PKM sgRNA GCCGCAGCGGGATAACCTT | IDT | Horlbeck et al.88 |
| GLUT3 sgRNA GGTAAGACTTTGGATCCTTCCTG | IDT | Horlbeck et al.88 |
| NTG sgRNA GCTGCATGGGGCGCGAATCA | IDT | Semple et al.35 Horlbeck et al.88 |
|
| ||
| Recombinant DNA | ||
|
| ||
| iGlucoSnFR-mRuby | Jonathan Marvin, HHMI | N/A |
| ATP fluorescence resonance energy sensor | N/A | Mendelsohn et al.56 |
|
| ||
| Software and algorithms | ||
|
| ||
| Prism 8.4.3 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| MetaMorph 7.7.3.0 | Universal Imaging | RRID: SciRes_000136 |
| SIVIC software | N/A | http://sourceforge.net/apps/trac/sivic/ |
| MATLAB R2011b | The MathWorks Inc | N/A |
| AMIRA software | Mercury Computer | N/A |
| ImageJ | NIH | http://imagej.nih.gov.ij |
| VivoQuant 4.0 software | Invicro | N/A |
| Dynamic Susceptibility Contrast MR Analysis | DSCoMAN | version 1.0 |
| ANTS | N/A | https://github.com/ANTsX/ANTs |
| pClamp10 software | Molecular Devices | N/A |
| IgorPro6 software | Wavemetrics Inc | N/A |
| 10X Space Ranger software | 10X Genomics | https://support.10xgenomics.com/spatial-gene-expression/software/downloads/latest |
| 10X Loupe Browser software | 10X Genomics | https://www.10xgenomics.com/products/loupe-browser/downloads |
| Enrichr webtool | Avi Ma’ayan Lab | Chen et al.89 Kuleshov et al.90 Xie et al.91 |
|
| ||
| Other | ||
|
| ||
| LSM 880 Confocal Laser Scanning Microscope with Airyscan | Zeiss | N/A |
| SM2000 R microtome | Leica | N/A |
| Labconco CentriVap Concentrator | Labconco | N/A |
| Video tracking | Tracker | N/A |
| 14.1 T vertical MR system | Agilent Technologies | N/A |
| Hypersense DNP polarizer | Oxford Instruments | N/A |
| PET/computerize tomography (CT) scanner | Siemens Healthcare | Inveon |
| COBRA Feldkamp reconstruction algorithm | Exxim Computing Corporation | N/A |
| VT1200 Vibratome | Leica | N/A |
| BSK5 vapor interface holding chamber | Scientific Systems Design Inc. Canada | N/A |
| RC-27LD submerged chamber | Warner Instruments | N/A |
| fDS2A-MKII voltage isolated stimulator | Digitimer North America | N/A |
| Ti-E inverted microscope | Nikon | N/A |
| iXon EMCCD camera | Andor Technology | N/A |
| VC-8 Perfusion valve system | Warner Instruments | N/A |
| A385 Current isolator | World Precision Instruments | N/A |
| SYS-A310 Accupulser signal generator | World Precision Instruments | N/A |
| Aperio Versa slide scanner | Leica | N/A |
Highlights.
Neurons take up glucose and metabolize it by glycolysis to supply TCA metabolites
HP 13C MRS shows disrupted brain energy when neuronal glucose metabolism is disrupted
Mice require neuronal glucose uptake and glycolysis for learning and memory
Galactose metabolism is upregulated to compensate for disrupted glucose metabolism
ACKNOWLEDGMENTS
We thank Jonathan Marvin (HHMI, Janelia) for providing the glucose sensor, Jessica Speckart and Julia Holtzman and the Gladstone Behavioral core for assistance with behavioral testing and data analysis, and Michael Gil for comments on the manuscript. We also thank Johanna ten Hoeve, Thomas G. Graeber, and the UCLA Metabolomics Center for their assistance with metabolomics studies and data processing. We also thank Blaise Ndjamen and Anna Torrent Moreno of the Gladstone Histology and Light Microscopy core, Eric Chow and the UCSF Center for Advanced Technology, Mylinh Bernardi and the Gladstone Genomics Core, as well as Kathryn Claiborn for helping edit the manuscript and Erica Delin for administrative assistance. This work was supported by National Institutes of Health RF1 AG064170 (to K.N. and M.M.C.), R01 AG065428 (K.N.), AG065428–03S1 (K.N., M.K.), R01 NS102156 (M.M.C.), R21 AI153749 (M.M.C., C.G.), National Institute on Aging R01 AG061150 (M.Z.), R01 AG071697 and P01 AG073082 (Y.H.), R01 CA168653 and R35 CA242379 (M.G.V.H.), R01 DK105550 (J.C.R.), K01AG078485 (N.B.), and National Institutes of Health RR18928 (Gladstone Institutes). It was also supported by the UCSF Bakar Aging Research Institute (BARI) (K.N.), the Alzheimer’s Association (H.L. and L.M.LP), a BrightFocus Foundation Award (L.M.LP), a Berkelhammer Award for Excellence in Neuroscience (Y.J.S.), Hillblom Center and Bakar Aging Research Institute Graduate Fellowship (J.Y.), and a Chan Zuckerberg Initiative Neurodegeneration Challenge Network Ben Barres Early Career Acceleration Award (M.K.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2023.112335.
DECLARATION OF INTERESTS
M.G.V.H. discloses that he is a scientific advisor for Agios Pharmaceuticals, iTeos Therapeutics, Sage Therapeutics, Faeth Therapeutics, DRIOA Ventures, and Auron Therapeutics. Y.H. discloses that he is a co-founder and scientific advisory board member of GABAeron.
INCLUSION AND DIVERSITY
We support inclusive, diverse, and equitable conduct of research.
REFERENCES
- 1.Bélanger M, Allaman I, and Magistretti PJ (2011). Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab 14, 724–738. 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 2.Patel AB, Lai JCK, Chowdhury GMI, Hyder F, Rothman DL, Shulman RG, and Behar KL (2014). Direct evidence for activity-dependent glucose phosphorylation in neurons with implications for the astrocyte-to-neuron lactate shuttle. Proc. Natl. Acad. Sci. USA 111, 5385–5390. 10.1073/pnas.1403576111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lundgaard I, Li B, Xie L, Kang H, Sanggaard S, Haswell JDR, Sun W, Goldman S, Blekot S, Nielsen M, et al. (2015). Direct neuronal glucose uptake heralds activity-dependent increases in cerebral metabolism. Nat. Commun 6, 6807. 10.1038/ncomms7807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ivanov AI, Malkov AE, Waseem T, Mukhtarov M, Buldakova S, Gubkina O, Zilberter M, and Zilberter Y (2014). Glycolysis and oxidative phosphorylation in neurons and astrocytes during network activity in hippocampal slices. J. Cereb. Blood Flow Metab 34, 397–407. 10.1038/jcbfm.2013.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maher F (1995). Immunolocalization of GLUT1 and GLUT3 glucose transporters in primary cultured neurons and glia. J. Neurosci. Res 42, 459–469. 10.1002/jnr.490420404. [DOI] [PubMed] [Google Scholar]
- 6.Ashrafi G, Wu Z, Farrell RJ, and Ryan TA (2017). GLUT4 mobilization supports energetic demands of active synapses. Neuron 93, 606–615.e3. 10.1016/j.neuron.2016.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shin BC, Cepeda C, Estrada-Sánchez AM, Levine MS, Hodaei L, Dai Y, Jung J, Ganguly A, Clark P, and Devaskar SU (2018). Neural deletion of glucose transporter isoform 3 creates distinct postnatal and adult neurobehavioral phenotypes. J. Neurosci 38, 9579–9599. 10.1523/JNEUROSCI.0503-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, and Bolaños JP (2009). The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C–Cdh1. Nat. Cell Biol 11, 747–752. 10.1038/ncb1881. [DOI] [PubMed] [Google Scholar]
- 9.Bolaños JP, Almeida A, and Moncada S (2010). Glycolysis: a bioenergetic or a survival pathway? Trends Biochem. Sci 35, 145–149. 10.1016/j.tibs.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Rodriguez-Rodriguez P, Fernandez E, Almeida A, and Bolaños JP (2012). Excitotoxic stimulus stabilizes PFKFB3 causing pentose-phosphate pathway to glycolysis switch and neurodegeneration. Cell Death Differ 19, 1582–1589. 10.1038/cdd.2012.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burmistrova O, Olias-Arjona A, Lapresa R, Jimenez-Blasco D, Eremeeva T, Shishov D, Romanov S, Zakurdaeva K, Almeida A, Fedichev PO, et al. (2019). Targeting PFKFB3 alleviates cerebral ischemia-reperfusion injury in mice. Sci. Rep 9, 11670. 10.1038/s41598-019-48196-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lopez-Fabuel I, Garcia-Macia M, Buondelmonte C, Burmistrova O, Bonora N, Alonso-Batan P, Morant-Ferrando B, Vicente-Gutierrez C, Jimenez-Blasco D, Quintana-Cabrera R, et al. (2022). Aberrant upregulation of the glycolytic enzyme PFKFB3 in CLN7 neuronal ceroid lipofuscinosis. Nat. Commun 13, 536. 10.1038/s41467-022-28191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Díaz-García CM, Mongeon R, Lahmann C, Koveal D, Zucker H, and Yellen G (2017). Neuronal stimulation triggers neuronal glycolysis and not lactate uptake. Cell Metab 26, 361–374.e4. 10.1016/j.cmet.2017.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ardenkjaer-Larsen JH, Fridlund B, Gram A, Hansson G, Hansson L, Lerche MH, Servin R, Thaning M, and Golman K (2003). Increase in signal-to-noise ratio of > 10,000 times in liquid-state NMR. Proc. Natl. Acad. Sci. USA 100, 10158–10163. 10.1073/pnas.1733835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brindle KM, Bohndiek SE, Gallagher FA, and Kettunen MI (2011). Tumor imaging using hyperpolarized 13C magnetic resonance spectroscopy. Magn. Reson. Med 66, 505–519. 10.1002/mrm.22999. [DOI] [PubMed] [Google Scholar]
- 16.Chaumeil MM, Najac C, and Ronen SM (2015). Studies of metabolism using (13)C MRS of hyperpolarized probes. Methods Enzymol 561, 1–71. 10.1016/bs.mie.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 17.Nelson SJ, Kurhanewicz J, Vigneron DB, Larson PEZ, Harzstark AL, Ferrone M, van Criekinge M, Chang JW, Bok R, Park I, et al. (2013). Metabolic imaging of patients with prostate cancer using hyperpolarized [1-(1)(3)C]pyruvate. Sci. Transl. Med 5, 198ra108. 10.1126/scitranslmed.3006070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallagher FA, Woitek R, McLean MA, Gill AB, Manzano Garcia R, Provenzano E, Riemer F, Kaggie J, Chhabra A, Ursprung S, et al. (2020). Imaging breast cancer using hyperpolarized carbon-13 MRI. Proc. Natl. Acad. Sci. USA 117, 2092–2098. 10.1073/pnas.1913841117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park I, Larson PEZ, Gordon JW, Carvajal L, Chen HY, Bok R, Van Criekinge M, Ferrone M, Slater JB, Xu D, et al. (2018). Development of methods and feasibility of using hyperpolarized carbon-13 imaging data for evaluating brain metabolism in patient studies. Magn. Reson. Med 80, 864–873. 10.1002/mrm.27077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang C, Ward ME, Chen R, Liu K, Tracy TE, Chen X, Xie M, Sohn PD, Ludwig C, Meyer-Franke A, et al. (2017). Scalable production of iPSC-derived human neurons to identify tau-lowering compounds by high-content screening. Stem Cell Rep 9, 1221–1233. 10.1016/j.stemcr.2017.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Pak C, Han Y, Ahlenius H, Zhang Z, Chanda S, Marro S, Patzke C, Acuna C, Covy J, et al. (2013). Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798. 10.1016/j.neuron.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tian R, Gachechiladze MA, Ludwig CH, Laurie MT, Hong JY, Nathaniel D, Prabhu AV, Fernandopulle MS, Patel R, Abshari M, et al. (2019). CRISPR interference-based platform for multimodal genetic screens in human iPSC-derived neurons. Neuron 104, 239–255.e12. 10.1016/j.neuron.2019.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennett NK, Nguyen MK, Darch MA, Nakaoka HJ, Cousineau D, Ten Hoeve J, Graeber TG, Schuelke M, Maltepe E, Kampmann M, et al. (2020). Defining the ATPome reveals cross-optimization of metabolic pathways. Nat. Commun 11, 4319. 10.1038/s41467-020-18084-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Contat C, Ancey PB, Zangger N, Sabatino S, Pascual J, Escrig S, Jensen L, Goepfert C, Lanz B, Lepore M, et al. (2020). Combined deletion of Glut1 and Glut3 impairs lung adenocarcinoma growth. Elife 9, e53618. 10.7554/eLife.53618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horn KE, Glasgow SD, Gobert D, Bull SJ, Luk T, Girgis J, Tremblay ME, McEachern D, Bouchard JF, Haber M, et al. (2013). DCC expression by neurons regulates synaptic plasticity in the adult brain. Cell Rep 3, 173–185. 10.1016/j.celrep.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 26.Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, and Tonegawa S (1996). Subregion- and cell type-restricted gene knockout in mouse brain. Cell 87, 1317–1326. [DOI] [PubMed] [Google Scholar]
- 27.Bergsman JB, and Tsien RW (2000). Syntaxin modulation of calcium channels in cortical synaptosomes as revealed by botulinum toxin C1. J. Neurosci 20, 4368–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miller JA, Guillozet-Bongaarts A, Gibbons LE, Postupna N, Renz A, Beller AE, Sunkin SM, Ng L, Rose SE, Smith KA, et al. (2017). Neuropathological and transcriptomic characteristics of the aged brain. Elife 6, e31126. 10.7554/eLife.31126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lesburguères E, Sparks FT, O’Reilly KC, and Fenton AA (2016). Active place avoidance is no more stressful than unreinforced exploration of a familiar environment. Hippocampus 26, 1481–1485. 10.1002/hipo.22666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shields LY, Kim H, Zhu L, Haddad D, Berthet A, Pathak D, Lam M, Ponnusamy R, Diaz-Ramirez LG, Gill TM, et al. (2015). Dynamin-related protein 1 is required for normal mitochondrial bioenergetic and synaptic function in CA1 hippocampal neurons. Cell Death Dis 6, e1725. 10.1038/cddis.2015.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marty S, and Peschanski M (1995). Effects of target deprivation on the morphology and survival of adult dorsal column nuclei neurons. J. Comp. Neurol 356, 523–536. 10.1002/cne.903560404. [DOI] [PubMed] [Google Scholar]
- 32.Li Z, Okamoto KI, Hayashi Y, and Sheng M (2004). The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 119, 873–887. [DOI] [PubMed] [Google Scholar]
- 33.Tonegawa S, Tsien JZ, McHugh TJ, Huerta P, Blum KI, and Wilson MA (1996). Hippocampal CA1-region-restricted knockout of NMDAR1 gene disrupts synaptic plasticity, place fields, and spatial learning. Cold Spring Harb. Symp. Quant. Biol 61, 225–238. [PubMed] [Google Scholar]
- 34.Wang X, Zhang C, Szábo G, and Sun QQ (2013). Distribution of CaMKIIalpha expression in the brain in vivo, studied by CaMKIIalpha-GFP mice. Brain Res 1518, 9–25. 10.1016/j.brainres.2013.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Semple BD, Blomgren K, Gimlin K, Ferriero DM, and Noble-Haeusslein LJ (2013). Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol 106–107, 1–16. 10.1016/j.pneurobio.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tingley D, McClain K, Kaya E, Carpenter J, and Buzsáki G (2021). A metabolic function of the hippocampal sharp wave-ripple. Nature 597, 82–86. 10.1038/s41586-021-03811-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Macdonald EB, Begovatz P, Barton GP, Erickson-Bhatt S, Inman DR, Cox BL, Eliceiri KW, Strigel RM, Ponik SM, and Fain SB (2021). Hyperpolarized (13)C magnetic resonance spectroscopic imaging of pyruvate metabolism in murine breast cancer models of different metastatic potential. Metabolites 11, 274. 10.3390/metabo11050274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perkons NR, Kiefer RM, Noji MC, Pourfathi M, Ackerman D, Siddiqui S, Tischfield D, Profka E, Johnson O, Pickup S, et al. (2020). Hyperpolarized metabolic imaging detects latent hepatocellular carcinoma domains surviving locoregional therapy. Hepatology 72, 140–154. 10.1002/hep.30970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varma G, Seth P, Coutinho de Souza P, Callahan C, Pinto J, Vaidya M, Sonzogni O, Sukhatme V, Wulf GM, and Grant AK (2021). Visualizing the effects of lactate dehydrogenase (LDH) inhibition and LDH-A genetic ablation in breast and lung cancer with hyperpolarized pyruvate NMR. NMR Biomed 34, e4560. 10.1002/nbm.4560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guglielmetti C, Najac C, Didonna A, Van der Linden A, Ronen SM, and Chaumeil MM (2017). Hyperpolarized (13)C MR metabolic imaging can detect neuroinflammation in vivo in a multiple sclerosis murine model. Proc. Natl. Acad. Sci. USA 114, E6982–E6991. 10.1073/pnas.1613345114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Harris RA, Lone A, Lim H, Martinez F, Frame AK, Scholl TJ, and Cumming RC (2019). Aerobic glycolysis is required for spatial memory acquisition but not memory retrieval in mice. eNeuro 6, ENEURO.0389–18.2019. 10.1523/ENEURO.0389-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rao Y, Gammon S, Zacharias NM, Liu T, Salzillo T, Xi Y, Wang J, Bhattacharya P, and Piwnica-Worms D (2020). Hyperpolarized [1-(13)C] pyruvate-to-[1-(13)C]lactate conversion is rate-limited by monocarboxylate transporter-1 in the plasma membrane. Proc. Natl. Acad. Sci. USA 117, 22378–22389. 10.1073/pnas.2003537117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Day SE, Kettunen MI, Gallagher FA, Hu DE, Lerche M, Wolber J, Golman K, Ardenkjaer-Larsen JH, and Brindle KM (2007). Detecting tumor response to treatment using hyperpolarized 13C magnetic resonance imaging and spectroscopy. Nat. Med 13, 1382–1387. 10.1038/nm1650. [DOI] [PubMed] [Google Scholar]
- 44.Brindle KM (2015). Imaging metabolism with hyperpolarized (13)C-labeled cell substrates. J. Am. Chem. Soc 137, 6418–6427. 10.1021/jacs.5b03300. [DOI] [PubMed] [Google Scholar]
- 45.Witney TH, Kettunen MI, Day SE, Hu DE, Neves AA, Gallagher FA, Fulton SM, and Brindle KM (2009). A comparison between radiolabeled fluorodeoxyglucose uptake and hyperpolarized (13)C-labeled pyruvate utilization as methods for detecting tumor response to treatment. Neoplasia 11, 574–582. 10.1593/neo.09254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie N, Zhang L, Gao W, Huang C, Huber PE, Zhou X, Li C, Shen G, and Zou B (2020). NAD(+) metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther 5, 227. 10.1038/s41392-020-00311-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Davidson SM, Schmidt DR, Heyman JE, O’Brien JP, Liu AC, Israelsen WJ, Dayton TL, Sehgal R, Bronson RT, Freinkman E, et al. (2022). Pyruvate kinase M1 suppresses development and progression of prostate adenocarcinoma. Cancer Res 82, 2403–2416. 10.1158/0008-5472.CAN-21-2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci 34, 11929–11947. 10.1523/JNEUROSCI.1860-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tech K, Tikunov AP, Farooq H, Morrissy AS, Meidinger J, Fish T, Green SC, Liu H, Li Y, Mungall AJ, et al. (2017). Pyruvate kinase inhibits proliferation during postnatal cerebellar neurogenesis and suppresses medulloblastoma formation. Cancer Res 77, 3217–3230. 10.1158/0008-5472.CAN-16-3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park I, Mukherjee J, Ito M, Chaumeil MM, Jalbert LE, Gaensler K, Ronen SM, Nelson SJ, and Pieper RO (2014). Changes in pyruvate metabolism detected by magnetic resonance imaging are linked to DNA damage and serve as a sensor of temozolomide response in glioblastoma cells. Cancer Res 74, 7115–7124. 10.1158/0008-5472.CAN-14-0849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Casson RJ, Wood JPM, Han G, Kittipassorn T, Peet DJ, and Chidlow G (2016). M-Type pyruvate kinase isoforms and lactate dehydrogenase A in the mammalian retina: metabolic implications. Invest. Ophthalmol. Vis. Sci 57, 66–80. 10.1167/iovs.15-17962. [DOI] [PubMed] [Google Scholar]
- 52.Pathak D, Shields LY, Mendelsohn BA, Haddad D, Lin W, Gerencser AA, Kim H, Brand MD, Edwards RH, and Nakamura K (2015). The role of mitochondrially derived ATP in synaptic vesicle recycling. J. Biol. Chem 290, 22325–22336. 10.1074/jbc.M115.656405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rangaraju V, Calloway N, and Ryan TA (2014). Activity-driven local ATP synthesis is required for synaptic function. Cell 156, 825–835. 10.1016/j.cell.2013.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vannucci SJ, Maher F, and Simpson IA (1997). Glucose transporter proteins in brain: delivery of glucose to neurons and glia. Glia 21, 2–21. 10.1002/(sici)1098-1136. [DOI] [PubMed] [Google Scholar]
- 55.Imamura H, Nhat KPH, Togawa H, Saito K, Iino R, Kato-Yamada Y, Nagai T, and Noji H (2009). Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc. Natl. Acad. Sci. USA 106, 15651–15656. 10.1073/pnas.0904764106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mendelsohn BA, Bennett NK, Darch MA, Yu K, Nguyen MK, Pucciarelli D, Nelson M, Horlbeck MA, Gilbert LA, Hyun W, et al. (2018). A high-throughput screen of real-time ATP levels in individual cells reveals mechanisms of energy failure. PLoS Biol 16, e2004624. 10.1371/journal.pbio.2004624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vona B, Maroofian R, Bellacchio E, Najafi M, Thompson K, Alahmad A, He L, Ahangari N, Rad A, Shahrokhzadeh S, et al. (2018). Expanding the clinical phenotype of IARS2-related mitochondrial disease. BMC Med. Genet 19, 196. 10.1186/s12881-018-0709-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Perli E, Giordano C, Tuppen HAL, Montopoli M, Montanari A, Orlandi M, Pisano A, Catanzaro D, Caparrotta L, Musumeci B, et al. (2012). Isoleucyl-tRNA synthetase levels modulate the penetrance of a homoplasmic m.4277T>C mitochondrial tRNA(Ile) mutation causing hypertrophic cardiomyopathy. Hum. Mol. Genet 21, 85–100. 10.1093/hmg/ddr440. [DOI] [PubMed] [Google Scholar]
- 59.Ferdinandusse S, Friederich MW, Burlina A, Ruiter JPN, Coughlin CR 2nd, Dishop MK, Gallagher RC, Bedoyan JK, Vaz FM, Waterham HR, et al. (2015). Clinical and biochemical characterization of four patients with mutations in ECHS1. Orphanet J. Rare Dis 10, 79. 10.1186/s13023-015-0290-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Du J, Li Z, Li QZ, Guan T, Yang Q, Xu H, Pritchard KA, Camara AKS, and Shi Y (2013). Enoyl coenzyme a hydratase domain-containing 2, a potential novel regulator of myocardial ischemia injury. J. Am. Heart Assoc 2, e000233. 10.1161/JAHA.113.000233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharpe AJ, and McKenzie M (2018). Mitochondrial fatty acid oxidation disorders associated with short-chain enoyl-CoA hydratase (ECHS1) deficiency. Cells 7, 46. 10.3390/cells7060046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sharkia I, Hadad Erlich T, Landolina N, Assayag M, Motzik A, Rachmin I, Kay G, Porat Z, Tshori S, Berkman N, et al. (2017). Pyruvate dehydrogenase has a major role in mast cell function, and its activity is regulated by mitochondrial microphthalmia transcription factor. J. Allergy Clin. Immunol 140, 204–214.e8. 10.1016/j.jaci.2016.09.047. [DOI] [PubMed] [Google Scholar]
- 63.Guo W, Ma J, Yang Y, Guo S, Zhang W, Zhao T, Yi X, Wang H, Wang S, Liu Y, et al. (2020). ATP-citrate lyase epigenetically potentiates oxidative phosphorylation to promote melanoma growth and adaptive resistance to MAPK inhibition. Clin. Cancer Res 26, 2725–2739. 10.1158/1078-0432.CCR-19-1359. [DOI] [PubMed] [Google Scholar]
- 64.Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, et al. (2013). Oncogenic BRAF regulates oxidative metabolism via PGC1alpha and MITF. Cancer Cell 23, 302–315. 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ferguson J, Smith M, Zudaire I, Wellbrock C, and Arozarena I (2017). Glucose availability controls ATF4-mediated MITF suppression to drive melanoma cell growth. Oncotarget 8, 32946–32959. 10.18632/oncotarget.16514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Holden HM, Rayment I, and Thoden JB (2003). Structure and function of enzymes of the Leloir pathway for galactose metabolism. J. Biol. Chem 278, 43885–43888. 10.1074/jbc.R300025200. [DOI] [PubMed] [Google Scholar]
- 67.Mueckler M, and Thorens B (2013). The SLC2 (GLUT) family of membrane transporters. Mol. Aspects Med 34, 121–138. 10.1016/j.mam.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hamberger A, and Hyden H (1963). Inverse enzymatic changes in neurons and glia during increased function and hypoxia. J. Cell Biol 16, 521–525. 10.1083/jcb.16.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Díaz-García CM, Meyer DJ, Nathwani N, Rahman M, Martínez-François JR, and Yellen G (2021). The distinct roles of calcium in rapid control of neuronal glycolysis and the tricarboxylic acid cycle. Elife 10, e64821. 10.7554/eLife.64821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Larson PEZ, and Gordon JW (2021). Hyperpolarized metabolic MRI-acquisition, reconstruction, and analysis methods. Metabolites 11, 386. 10.3390/metabo11060386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Le Page LM, Guglielmetti C, Taglang C, and Chaumeil MM (2020). Imaging brain metabolism using hyperpolarized (13)C magnetic resonance spectroscopy. Trends Neurosci 43, 343–354. 10.1016/j.tins.2020.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zimmer ER, Parent MJ, Souza DG, Leuzy A, Lecrux C, Kim HI, Gauthier S, Pellerin L, Hamel E, and Rosa-Neto P (2017). [(18)F] FDG PET signal is driven by astroglial glutamate transport. Nat. Neurosci 20, 393–395. 10.1038/nn.4492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dayton TL, Gocheva V, Miller KM, Israelsen WJ, Bhutkar A, Clish CB, Davidson SM, Luengo A, Bronson RT, Jacks T, and Vander Heiden MG (2016). Germline loss of PKM2 promotes metabolic distress and hepatocellular carcinoma. Genes Dev 30, 1020–1033. 10.1101/gad.278549.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shin BC, Cepeda C, Eghbali M, Byun SY, Levine MS, and Devaskar SU (2021). Adult glut3 homozygous null mice survive to demonstrate neural excitability and altered neurobehavioral responses reminiscent of neurodevelopmental disorders. Exp. Neurol 338, 113603. 10.1016/j.expneurol.2021.113603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Goodpaster BH, and Sparks LM (2017). Metabolic flexibility in health and disease. Cell Metab 25, 1027–1036. 10.1016/j.cmet.2017.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Motori E, Atanassov I, Kochan SMV, Folz-Donahue K, Sakthivelu V, Giavalisco P, Toni N, Puyal J, and Larsson NG (2020). Neuronal metabolic rewiring promotes resilience to neurodegeneration caused by mitochondrial dysfunction. Sci. Adv 6, eaba8271. 10.1126/sciadv.aba8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tramunt B, Smati S, Grandgeorge N, Lenfant F, Arnal JF, Montagner A, and Gourdy P (2020). Sex differences in metabolic regulation and diabetes susceptibility. Diabetologia 63, 453–461. 10.1007/s00125-019-05040-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Du L, Hickey RW, Bayir H, Watkins SC, Tyurin VA, Guo F, Kochanek PM, Jenkins LW, Ren J, Gibson G, et al. (2009). Starving neurons show sex difference in autophagy. J. Biol. Chem 284, 2383–2396. 10.1074/jbc.M804396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Christianto A, Baba T, Takahashi F, Inui K, Inoue M, Suyama M, Ono Y, Ohkawa Y, and Morohashi KI (2021). Sex differences in metabolic pathways are regulated by Pfkfb3 and Pdk4 expression in rodent muscle. Commun. Biol 4, 1264. 10.1038/s42003-021-02790-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Levéen P, Kotarsky H, Mörgelin M, Karikoski R, Elmér E, and Fellman V (2011). The GRACILE mutation introduced into Bcs1l causes postnatal complex III deficiency: a viable mouse model for mitochondrial hepatopathy. Hepatology 53, 437–447. 10.1002/hep.24031. [DOI] [PubMed] [Google Scholar]
- 81.Kotarsky H, Keller M, Davoudi M, Levéen P, Karikoski R, Enot DP, and Fellman V (2012). Metabolite profiles reveal energy failure and impaired beta-oxidation in liver of mice with complex III deficiency due to a BCS1L mutation. PLoS One 7, e41156. 10.1371/journal.pone.0041156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Purhonen J, Grigorjev V, Ekiert R, Aho N, Rajendran J, Pietras R, Truvé K, Wikström M, Sharma V, Osyczka A, et al. (2020). A spontaneous mitonuclear epistasis converging on Rieske Fe-S protein exacerbates complex III deficiency in mice. Nat. Commun 11, 322. 10.1038/s41467-019-14201-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Colville CA, Seatter MJ, Jess TJ, Gould GW, and Thomas HM (1993). Kinetic analysis of the liver-type (GLUT2) and brain-type (GLUT3) glucose transporters in Xenopus oocytes: substrate specificities and effects of transport inhibitors. Biochem. J 290, 701–706. 10.1042/bj2900701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maher F, Davies-Hill TM, and Simpson IA (1996). Substrate specificity and kinetic parameters of GLUT3 in rat cerebellar granule neurons. Biochem. J 315, 827–831. 10.1042/bj3150827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Reagan LP, Rosell DR, Alves SE, Hoskin EK, McCall AL, Charron MJ, and McEwen BS (2002). GLUT8 glucose transporter is localized to excitatory and inhibitory neurons in the rat hippocampus. Brain Res 932, 129–134. 10.1016/s0006-8993(02)02308-9. [DOI] [PubMed] [Google Scholar]
- 86.Simpson IA, Chundu KR, Davies-Hill T, Honer WG, and Davies P (1994). Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann. Neurol 35, 546–551. 10.1002/ana.410350507. [DOI] [PubMed] [Google Scholar]
- 87.Manzo E, Lorenzini I, Barrameda D, O’Conner AG, Barrows JM, Starr A, Kovalik T, Rabichow BE, Lehmkuhl EM, Shreiner DD, et al. (2019). Glycolysis upregulation is neuroprotective as a compensatory mechanism in ALS. Elife 8, e45114. 10.7554/eLife.45114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, and Weissman JS (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife 5, e19760. 10.7554/eLife.19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–W97. 10.1093/nar/gkw377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, et al. (2021). Gene set knowledge discovery with Enrichr. Curr. Protoc 1, e90. 10.1002/cpz1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Berthet A, Margolis EB, Zhang J, Hsieh I, Zhang J, Hnasko TS, Ahmad J, Edwards RH, Sesaki H, Huang EJ, and Nakamura K (2014). Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. J. Neurosci 34, 14304–14317. 10.1523/JNEUROSCI.0930-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Johnson ECB, Ho K, Yu GQ, Das M, Sanchez PE, Djukic B, Lopez I, Yu X, Gill M, Zhang W, et al. (2020). Behavioral and neural network abnormalities in human APP transgenic mice resemble those of App knock-in mice and are modulated by familial Alzheimer’s disease mutations but not by inhibition of BACE1. Mol. Neurodegener 15, 53. 10.1186/s13024-020-00393-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Arguello A, Meisner R, Thomsen ER, Nguyen HN, Ravi R, Simms J, Lo I, Speckart J, Holtzman J, Gill TM, et al. (2021). Iduronate-2-sulfatase transport vehicle rescues behavioral and skeletal phenotypes in a mouse model of Hunter syndrome. JCI Insight 6, e145445. 10.1172/jci.insight.145445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Xia Z, and Liu Y (2001). Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys. J 81, 2395–2402. 10.1016/S0006-3495(01)75886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shields LY, Li H, Nguyen K, Kim H, Doric Z, Garcia JH, Gill TM, Haddad D, Vossel K, Calvert M, and Nakamura K (2021). Mitochondrial fission is a critical modulator of mutant APP-induced neural toxicity. J. Biol. Chem 296, 100469. 10.1016/j.jbc.2021.100469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Visium datasets are available in Tables S3 and S4. Metabolomics data sets are available in Tables S5, S6 and S7.
This paper does not report original code.
Further information and requests for reagents should be directed to and will be fulfilled by the lead contact, Ken Nakamura (ken.nakamura@ucsf.edu).