

Abstract
Gliomas are the most prevalent and devastating primary malignant brain tumors in adults. Despite substantial advances in understanding glioma biology, there have been no regulatory drug approvals in the US since bevacizumab in 2009 and tumor treating fields in 2011. Recent phase III clinical trials have failed to meet their prespecified therapeutic primary endpoints, highlighting the need for novel therapies. The poor prognosis of glioma patients, resistance to chemo-radiotherapy, and the immunosuppressive tumor microenvironment underscore the need for the development of novel therapies. Gene therapy-based immunotherapeutic strategies that couple the ability of the host immune system to specifically kill glioma cells and develop immunological memory have shown remarkable progress. Two adenoviral vectors expressing Ad-HSV1-TK/GCV and Ad-Flt3L have shown promising preclinical data, leading to FDA approval of a non-randomized, phase I open-label, first in human trial to test safety, cytotoxicity, and immune-stimulatory efficiency in high-grade glioma patients (NCT01811992). This review provides a thorough overview of immune-stimulatory gene therapy highlighting recent advancements, potential drawbacks, future directions, and recommendations for future implementation of clinical trials.
Keywords: gene therapy, Ad-hCMV-TK + Ad-hCMV-Flt3L, glioma, immune-suppression, immunogenic cell death, dexamethasone, suicide gene therapy, oncolytic virotherapy, immune checkpoint inhibitors
Graphical abstract
Lowenstein and colleagues demonstrate that their dual-vector gene therapy remodels the TME by causing glioma cell death, and DC recruitment and differentiation, thereby unleashing an immunologically “hot” microenvironment that favors anti-glioma therapeutic responses. Combining this therapy with immune checkpoint inhibitors, DC vaccines, and CAR-T cells, will further enhance glioma treatment.
Introduction
Gliomas are extremely aggressive tumors with a particularly poor prognosis, and glioblastoma, IDH wild-type, grade 4 (GBM) remains an incurable glioma with an overall survival of 16–18 months.1 Regardless of continued promising preclinical results, there has been little progress in improving clinical outcomes for GBM patients over the last two decades.2,3 Glioma cells’ ability to invade brain tissue means that surgical resection alone cannot remove all glioma cells, resulting in tumor relapse and poor long-term therapeutic outcomes.4 Even with constant advances in surgical and imaging techniques, there has been little improvement in survival.3,4 Therefore, there is an urgent need for novel therapeutic options to enhance the prognosis for glioma patients.
Viral vector-based gene therapy is a versatile and promising treatment option for this devastating disease.5,6,7 Gene therapy uses viral vectors, which can be given locally during the initial surgery and may kill glioma cells that are difficult to remove, thus possibly reducing recurrence rates and improving therapeutic responses. Viral vectors are often employed for gene therapy in GBM clinical trials because of their high transfection efficiency of tumor cells. Adenoviral vectors are the most commonly used viral vectors in gene therapy trials (22 trials), followed by HSV1 vectors (8 trials), and retroviral vectors (4 trials), as well as various other viruses such as parvovirus-H1, measles virus, Newcastle disease, and poliovirus, etc., used in a smaller number of trials.7 A succinct and comprehensive overview of the clinical trials is shown in Table 1. Recently, we concluded an open-label, non-randomized phase I clinical trial, where we combined cytotoxic and immune-stimulatory gene therapy with standard of care (SOC) for glioma. This involves maximal surgical resection, followed by chemoradiation and adjuvant chemotherapy (ClinicalTrials.gov identifier: NCT01811992).8,9 Mechanistic details of our dual viral-vector gene therapy strategy are illustrated in Figure 1. No dose-limiting toxicity was encountered.8 Furthermore, most gene therapy strategies mediated by viral vectors have been demonstrated to be safe clinically.5,6,10,11,12 In addition, in some gene therapy trials, histopathological assessments of paired tissue (consisting of primary and secondary tissues from the same patient) have shown an enhanced recruitment and activation of immune cells in the secondary tissue following treatment.5,6,8 Todo and co-workers conducted a phase II trial aimed at treating residual or recurrent glioblastoma using up to six doses of oncolytic teserpaturev (HSV-1 G47Δ; Delytact) directly into the tumor, resulting in encouraging 1-year survival rates.5,6 The approval of this therapeutic approach for patients with recurrent glioblastoma in Japan, the first ever for gene therapy holds great potential for uncovering new clinical and molecular insights.
Table 1.
Comprehensive overview of clinical trials for viral vector-based therapies in brain malignancies (1992–2023)
| ClinicalTrials.Gov identifier | Clinical phase | Viral vector | Interventions | Administration scheme | Participants | Trial status | Age | Pathology |
|---|---|---|---|---|---|---|---|---|
| NCT00634231 | 1 | adenovirus | AdV-TK + valacyclovir | injections into the tumor bed during surgical procedure | 8 | completed (2010–2021) | child, adult | malignant glioma, recurrent ependymoma |
| NCT01811992 | 1 | adenovirus | Ad-hCMV-TK + Ad-hCMV-Flt3L + valacyclovir | injections into the tumor cavity during surgical procedure | 19 | completed (2014–2021) | adult | malignant glioma, glioblastoma multiforme |
| NCT02026271 | 1 | adenovirus | Ad-RTS-hIL-12 + veledimex | injection into the wall of resection cavity | 40 | completed (2014–2021) | adult | glioblastoma multiforme, anaplastic oligoastrocytoma |
| NCT03679754 | 1 | adenovirus | Ad-RTS-hIL-12 + veledimex | veledimex prior to resection and Ad injection during surgery | 36 | completed (2018–2021) | adult | recurrent glioblastoma |
| NCT03636477 | 1 | adenovirus | Ad-RTS-hIL-12 + veledimex + nivolumab | administered nivolumab 1 week preoperatively, followed by veledimex prior to resection, and Ad injection during surgery | 21 | completed (2018–2021) | adult | glioblastoma |
| NCT03576612 | 1 | adenovirus | AdV-TK + valacyclovir + nivolumab | AdV injections into the wall of the resection cavity, followed by valacyclovir 1–3 days post-surgery, and nivolumab injections 2 weeks post-surgery | 36 | active, not recruiting (2018–) | adult | malignant glioma |
| NCT05139056 | 1 | adenovirus | neural stem cells expressing CRAd-S-pk7 | NSC carrier into the tumor cavity | 36 | recruiting (2021–) | adult | recurrent high-grade glioma, astrocytoma, oligodendroglioma |
| NCT03330197 | 1 and 2 | adenovirus | Ad-RTS-hIL-12 + veledimex | veledimex prior to resection and Ad injection during surgery | 6 | completed (2017–2021) | child, adult | pediatric brain tumor, DIPG |
| NCT03596086 | 1 and 2 | adenovirus | AdV-(HSV-TK) + valacyclovir | injections into the tumor cavity during surgical procedure | 62 | recruiting (2018–) | adult | glioblastoma, astrocytoma grade III |
| NCT03603405 | 1 and 2 | adenovirus | AdV-(HSV-TK) + valacyclovir | Injections into the tumor cavity during surgical procedure | 62 | recruiting (2018–) | adult | glioblastoma, anaplastic astrocytoma |
| NCT04006119 | 2 | adenovirus | Ad-RTS-hIL-12 + veledimex + cemiplimab-Rwlc | administered cemiplimab-rwlc 1 week preoperatively, followed by veledimix prior to resection, and Ad injection during surgery | 40 | completed (2019–2023) | adult | glioblastoma |
| NCT00870181 | 2 | adenovirus | ADV-TK | intrarterial cerebral infusion | 47 | completed (2009–2013) | adult | malignant glioma, glioblastoma multiforme |
| NCT00589875 | 2 | adenovirus | AdV-TK + valacyclovir | AdV injections into the tumor bed post-surgical resection | 52 | completed (2007-2017) | adult | malignant glioma, glioblastoma multiforme, anaplastic astrocytoma |
| NCT00805376 | 1 | oncolytic adenovirus | DNX-2401 (Delta-24-RGD-4C) | intratumoral injection | 37 | completed (2009–2015) | adult | brain tumor, CNS diseases |
| NCT02197169 | 1 | oncolytic adenovirus | DNX-2401 + IFN-γ | OV injection into the tumor followed by IFN-γ | 37 | completed (2014–2018) | adult | glioblastoma, gliosarcoma |
| NCT03178032 | 1 | oncolytic adenovirus | DNX-2401 | virus infusion through the cerebellar peduncle | 12 | active, not recruiting (2017–) | child | brainstem glioma, DIPG |
| NCT03714334 | 1 | oncolytic adenovirus | DNX-2440 | administered virus during surgical procedures using a cannula | 24 | recruiting (2018–) | adult | glioblastoma |
| NCT03896568 | 1 | oncolytic adenovirus | Ad5-DNX-2401 | mesenchymal stem cells loaded with a tumor-selective OV, DNX-2401, administered via intra-arterial injection | 36 | recruiting (2019–) | adult | IDH1 WT allele, recurrent glioblastoma, recurrent anaplastic astrocytoma |
| NCT01956734 | 1 | oncolytic adenovirus | DNX-2401 + temozolomide | intratumoral OV injection into the tumor cavity followed by temozolomide | 31 | completed (2013–2017) | adult | glioblastoma, recurrent glioblastoma |
| NCT01582516 | 1 and 2 | oncolytic adenovirus | Delta-24-RGD | virus administered intratumorally using four catheters | 20 | completed (2010–2014) | adult | recurrent glioblastoma |
| NCT04758533 | 1 and 2 | oncolytic adenovirus | AloCELYVIR (mesenchymal allogenic cells + ICOVIR-5) | weekly intravenous infusion of AloCELYVIR for 8 weeks | 12 | recruiting (2021–) | child, adult | DIPG, medulloblastoma, glioma |
| NCT02798406 | 2 | oncolytic adenovirus | DNX-2401 + pembrolizumab | intratumoral OV injection into the tumor cavity followed by intravenous pembrolizumab | 49 | completed (2016–2021) | adult | glioblastoma, gliosarcoma, malignant glioma |
| NCT00157703 | 1 | oncolytic HSV-1 | G207 | intratumoral injection into the tumor cavity | 9 | completed (2005–2008) | adult | malignant glioma |
| NCT02457845 | 1 | oncolytic HSV-1 | G207 | a single dose of G207 infused through catheters into the tumor | 13 | active, not recruiting (2016–) | child, adult | progressive or recurrent supratentorial neoplasms |
| NCT03911388 | 1 | oncolytic HSV-1 | G207 | a single dose of G207 infused through catheters into the tumor | 15 | recruiting (2019–) | child | recurrent or refractory cerebellar brain tumors |
| NCT02062827 | 1 | oncolytic HSV-1 | M032 (NSC 733972) | a single dose of M032 infused through catheters into the tumor | 24 | active, not recruiting (2013–) | adult | recurrent glioblastoma, progressive glioblastoma, anaplastic astrocytoma |
| NCT03152318 | 1 | oncolytic HSV-1 | rQNestin34.5v.2 + cyclophosphamide | cyclophosphamide was administered intravenously two days pre-operatively followed by OV Injections into the tumor cavity during a surgical procedure | 62 | recruiting (2017–) | adult | recurrent malignant glioma, astrocytoma, oligodendroglioma |
| NCT05084430 | 1 and 2 | oncolytic HSV-1 | M032 (NSC 733972) + pembrolizumab | oncolytic HSV that expresses IL12 administered into the tumor bed followed by pembrolizumab intravenous injection of a total of three combined doses | 28 | recruiting (2021–) | adult | glioblastoma, gliosarcoma, anaplastic astrocytoma |
| NCT00028158 | 1 and 2 | oncolytic HSV-1 | G207 | administered virus during surgical resection into the tumor | 65 | completed (2001–2003) | adult | glioma, astrocytoma, glioblastoma |
| NCT04482933 | 2 | oncolytic HSV-1 | G207 | intratumoral infusion through four silastic catheters | 40 | not yet recruiting (2023–) | child, adult | recurrent pediatric high-grade glioma |
| NCT03043391 | 1 | oncolytic poliovirus | polio/rhinovirus recombinant (PVSRIPO) | administered intratumorally via convection-enhanced delivery using an intracerebral catheter | 12 | active, not recruiting (2017–) | child, adult | recurrent malignant glioma, astrocytoma, ATRT, oligodendroglioma |
| NCT00390299 | 1 | oncolytic MV | MV-CEA (carcinoembryonic antigen expressing) | injection into the tumor cavity | 23 | completed (2006–2019) | adult | anaplastic astrocytoma, oligodendroglioma |
| NCT02962167 | 1 | oncolytic MV | MV-NIS | injections into the tumor bed post-surgical resection | 46 | recruiting (2017–) | child, adult | recurrent medulloblastoma, recurrent ATRT |
| NCT01174537 | 1 and 2 | oncolytic NDV | Newcastle disease virus | injection into the tumor cavity | 0 | withdrawn (2011) | adult | glioblastoma, sarcoma, neuroblastoma |
| NCT02986178 | 2 | oncolytic-poliovirus | Polio/rhinovirus recombinant (PVSRIPO) | administered intratumorally via convection-enhanced delivery using an intracerebral catheter | 122 | active, not recruiting (2017–) | adult | malignant glioma |
| NCT01156584 | 1 | retrovirus | Toca 511 vector + Toca FC | single, stereotactic, transcranial, intratumoral or intravenous injection | 54 | completed (2010–2016) | adult | glioblastoma, anaplastic astrocytoma, oligodendroglioma |
| NCT01985256 | 1 | retrovirus | Toca 511 vector + Toca FC | administered intravenously and then intracranially | 17 | completed (2014–2016) | adult | glioblastoma, anaplastic astrocytoma, oligodendroglioma |
| NCT00001328 | 1 | cell line producing retrovirus | G1TKSVNa.53 producer cell line + cytovene | intratumoral injection | 15 | completed (1992–2010) | adult | brain neoplasm |
| NCT02414165 | 2 and 3 | retrovirus | Toca 511 vector + Toca FC + lomustine + bevacizumab | injection into the tumor cavity | 403 | terminated (2015–2020) | adult | glioblastoma, anaplastic astrocytoma |
Figure 1.

Mechanistic details of the dual viral-vector combined cytotoxic and immune-stimulatory gene therapy
After surgical resection, first-generation adenoviral vectors (Ads) encoding HSV1-thymidine kinase (TK) and HSV1-FMS-like tyrosine kinase 3 ligand (Flt3L) are injected into the tumor cavity, followed by administration of the prodrug ganciclovir (GCV) (rodents), or valacyclovir (VCV) (humans). Ad-TK selectively targets dividing tumor cells without damaging non-dividing stromal cells. (1A) Ad-TK induces glioma cell death in the presence of the prodrug (VCV), which is administered systemically. (1B) Flt3L promotes recruitment and differentiation of DCs in the glioma TME. Tumor cells infected with Ad-Flt3L express Flt3L, which is released into the circulation, inducing DC expansion, migration, and accumulation in the TME from the bone marrow. (2) Tumor cells infected with Ad-TK express TK protein that phosphorylates GCV to GCV/VCV-monophosphate (GCVp/VCVp), which is further phosphorylated by cellular kinases to the tri-phosphorylated form (GCVp3/VCVp3; purine analog), which selectively inhibits DNA synthesis, leading to DNA breaks and apoptosis in proliferating glioma cells. Phosphorylated GCV/VCV is passively transported to surrounding TK-non-expressing cells via gap junction intercellular communication inducing cell death. This bystander effect enhances the antitumor effect of Ad-TK gene therapy. (3) The expression of TK in the presence of GCV/VCV also leads to the release of glioma-specific antigens and DAMPs, such as HMBG1, calreticulin, and ATP from dying glioma cells. (4) DCs are recruited into the TME by Flt3L and take up tumor antigens released from the dying glioma cells, with DAMPs further stimulating immune responses. These DAMPs bind their corresponding receptors expressed on DCs, where HMGB1 binds to TLR2 to promote cytokine production and tumor antigen cross-presentation, extracellular ATP binds to the purinergic receptor P2X7R to promote DC recruitment, while calreticulin binds to the CD91 receptor involved in immunosurveillance. (5) DCs loaded with glioma-specific antigens migrate to the cervical draining lymph node (DLN) where they present the antigens to naive T cells on MHC I, resulting in the priming and clonal expansion of glioma antigen-specific effector T cells (Teff) with anti-glioma immunity. (6) Primed CD8+ effector T cells enter the circulation from the DLN to the TME and kill glioma cells through the production of granzyme B, perforin, and effector cytokine IFN-ɣ. (7) Continued exposure of T cells to glioma antigens promotes immunological memory, resulting in the inhibition of tumor recurrence through the presence of memory T cells (CD103 and CD69) that facilitate the ongoing anti-glioma immune response especially as the tumor recurs. The figure was created with BioRender.com.
Notably, Goertsen et al. developed novel adeno-associated virus (AAV) capsids (AAV.CAP-B10 and AAV.CAP-B22) that can pass through the blood-brain barrier (BBB) with neuronal specificity in both rodents and non-human primates. This breakthrough opens up new opportunities for basic research and therapeutic applications that were previously unachievable with naturally occurring serotypes.13 In a study by Yao et al., they demonstrated that an AAV vector (AAV.CPP.16) with an improved capacity to cross the BBB could be used for systemic curative anti-tumor gene therapy against GBM in rodents.14 Taken altogether, the overwhelming safety of viral vector-based gene therapies has implications for GBM therapy in the US and will likely seek approval from the US Food and Drug Administration (FDA) in the coming years.
Both clinical and preclinical research have provided evidence supporting the use of viral vector-based gene therapy, either as a standalone treatment or in combination with other therapeutic approaches. In this context, we discuss the detailed developmental pathway from rodents to glioma patients. We also address the challenges and recommendations regarding anti-glioma immune responses that are emerging as viral vector-based therapeutic strategies develop into immunotherapy medications. In addition, we emphasize how combining gene therapy with other immunotherapies may be able to overcome obstacles to effective glioma treatment. The aim of this review is to shed light on the current status of gene therapies and immunotherapies, and to provide recommendations for developing more effective combination therapeutic strategies for the treatment of brain tumors.
Current state of glioma therapies
The SOC for newly diagnosed GBM is maximum safe tumor resection, followed by a course of radiation (totaling 60 Gy through fractionated focal irradiation over 6 weeks) and concurrent chemotherapy utilizing temozolomide (TMZ). The current SOC has remained unchanged since 2005, with the median overall survival being 14.6 months, as opposed to 12.1 months with radiation alone.15 A better outcome and benefit from TMZ chemotherapy is predicted by O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation.16 The historical survival rate for glioblastoma is generally ∼16 months, although advanced medical centers throughout the US record survival rates between 19 and 22 months.17,18 In the US, tumor-treating fields (TTFs) have been added as an optional therapy for GBM treatment.
The TTF transmits low intensity alternating electric fields to the tumors, which has demonstrated an improvement in both progression-free survival and overall survival rates from 4 to 6.7 months and 16 to 20.9 months, respectively, in TMZ-alone vs. the TTF-TMZ group, with an HR of 0.63 and a 95% CI of 0.53–0.76, and p value of 0.001 in a phase III trial (NCT00916409).19
Despite advances in treating GBM, the vast majority of patients eventually experience a relapse. The management of recurring or progressing tumors typically involves personalized approaches, taking into account factors such as the patient’s neurological condition, time since diagnosis, and previous treatments received. Second-line treatment options may include surgery, radiotherapy, chemotherapy with alkylating agents, and antiangiogenic therapy with bevacizumab.8,10,15,16,19,20,21,22 Numerous clinical trials have been conducted to address this gap in GBM treatment, exploring over 100 different targeted drugs to date. Unfortunately, none of these treatments have demonstrated consistent clinical benefit.10,12,15,19,22,23 Thus, there is much hope that immunovirotherapeutics will provide novel treatments. It is worth noting that patients with recurring GBM who have received previous radiotherapy and chemotherapy have a higher mutational burden, potentially making them more immunogenic than those who have not undergone treatment. This has led to increased confidence and hope in the potential of immunotherapy as a treatment for recurrent GBM. However, the complex interplay between the immune system and the cerebral parenchyma presents significant challenges that must be addressed to develop effective immunotherapies.24
Suicide gene therapy for high-grade gliomas: A multifaceted approach
Suicide gene therapy (SGT) is widely used in both preclinical and clinical settings to treat high-grade gliomas. SGT is a two-step process: first, cancer cells are transduced with a vector carrying a suicide gene which encodes an enzyme such as thymidine kinase or cytosine deaminase (CD), capable of converting a prodrug (ganciclovir/5-fluorocytosine) into a toxic metabolite. In the second step, the corresponding prodrug is administered, triggering cell death upon enzymatic catalysis and cell division.25,26 An ideal prodrug should be an optimal substrate for the enzyme, induce cell death with minimal or no off-target toxicity, efficiently cross the BBB, and activate the bystander effect. The bystander effect plays a crucial role in SGT, as it allows for the elimination of non-transduced (bystander) cells through the intercellular transfer of prodrug metabolites.27,28,29 Therefore, achieving complete eradication of malignant cells would be possible at least (in theory) by transducing only a fraction of the tumor. Emerging evidence suggests that certain SGT approaches, including our dual-vector thymidine kinase (TK) and Flt3L therapy, can induce immunogenic cell death stimulating an anti-tumor immune response and enhanced treatment efficacy (Figure 2).9,25,30,31,32
Figure 2.

The cellular organization of immunologically “cold” and “hot” gliomas
Combined cytotoxic and immune-stimulatory gene therapy remodel the immunosuppressive TME to an inflamed hot glioma microenvironment. The GBM cold immunosuppressive TME is characterized by low infiltration of CD8+ T cells, DCs, and NK cells but high infiltration of immunosuppressive MDSCs, M2-polarized macrophages, and Tregs. The cold TME also has low expression and presentation of tumor antigens, which hinders the success of immunotherapy. However, combined cytotoxic and immune-stimulatory gene therapy remodels the TME to an immunostimulatory hot TME that attracts effector CD8+ T cells, DCs, NK cells, and fewer Tregs and MDSCs. Glioma-specific antigens and DAMPs released from dying glioma cells trigger immunogenicity and adjuvanticity, respectively. Ad-Flt3L injections promote the recruitment, infiltration, and activation of DCs, thereby enhancing antigen presentation, priming, and clonal expansion of glioma-specific CD8+ cytotoxic effector T cells. The figure was created with BioRender.com.
Nishiyama et al. demonstrated the application of this concept in cancer treatment by delivering CD from Escherichia coli, followed by administration of 5-fluorocytosine. This resulted in a significant reduction in tumor burden in a syngeneic EA285 rat glioma model.26 Moolten and Wells introduced SGT using TK from HSV-1 for cancer treatment.33 CD and HSV1-TK are the most commonly used suicide genes for the treatment of high-grade gliomas. Interestingly, compared with other adenovirus-encoding pro-apoptotic cytokines, such as TRAIL, FasL, and TNF-α, our approach using Ad-TK was more effective and less toxic.34 Several other SGTs have been developed as well, although they are beyond the scope of our current discussion.
What limits the efficacy of SGT?
Initially, preclinical studies primarily focused on the development of cells producing the HSV1-βgal vectors, which were used to transduce neurons. These in vitro studies showed promising results as all cells were successfully transduced, suggesting potential positive outcomes in the case of 100% target cell transduction. However, when considering tumor cells, 100% transduction is very difficult to achieve especially in vivo.35 In a study conducted on 13 glioma patients who underwent surgical resection followed by radiation therapy, 7 received an additional treatment involving gene therapy using HSV1-TK. Gene therapy was administered through direct intracerebral injection of retrovirus vector-producing cells during tumor surgery, and was followed by systemic administration of ganciclovir (GCV). The results of the study showed that the group receiving gene therapy exhibited an antitumor immune response, whereas the control group did not.36 However, in Rainov’s phase III clinical trial, patients were divided into two groups: one receiving standard therapy (surgery and radiotherapy) and the other receiving standard therapy combined with adjuvant gene therapy during surgery. Unfortunately, the addition of adjuvant gene therapy did not lead to reduced tumor progression or overall survival.37 Nevertheless, this trial demonstrated the feasibility and favorable biosafety profile of the gene therapy strategy employed. The lack of therapeutic efficacy in this trial has been attributed to the delayed SOC radiotherapy.37
Current state of immunotherapy modalities for glioma
For a long time, the central nervous system (CNS) was considered to be immune privileged. This means that foreign tissue or cells transplanted into the brain could engraft successfully, even though they would be rejected by the host immune system if implanted in other parts of the body.38,39 Studies have shown that the concept of the brain being an immune-privileged organ is not entirely accurate, as immune cells have been found to enter the brain in both healthy and diseased states.39,40 Immune cells access the CNS through various barriers, including the BBB, blood-meningeal barrier, and the blood-cerebrospinal fluid (CSF) barrier.41,42 Unlike most other organs, the brain has two distinct immune compartments: one located within the brain parenchyma and the other within the ventricles and meninges.39,41 Both compartments differ in how they interact with the systemic immune system. The brain parenchyma is protected from the immune system due to the lack of classical lymphatic drainage, while the ventricles and meninges offer a direct pathway for lymphatic drainage. This pathway enables the passage of solutes and immune cells from the brain to the cervical lymph nodes, initiating a systemic immune response. However, during inflammation and glioma, lymphocytes have an increased capacity to infiltrate the brain.41,42,43,44,45 Immunotherapeutic strategies have been successful in treating certain types of malignancy, such as melanoma,46 non-small cell lung carcinoma,47 and kidney48,49 and bladder cancers.49 Due to this success, researchers have been exploring several immune-based approaches in glioma preclinical models and in several clinical trials (Figure 3), including immune checkpoint inhibitors, myeloid targeted therapies, dendritic cell (DC) vaccines, CAR-T cells, and oncolytic and gene therapy viral vectors.5,6,10,11,50
Figure 3.

Potential combination strategies for inducing anti-glioma immune responses
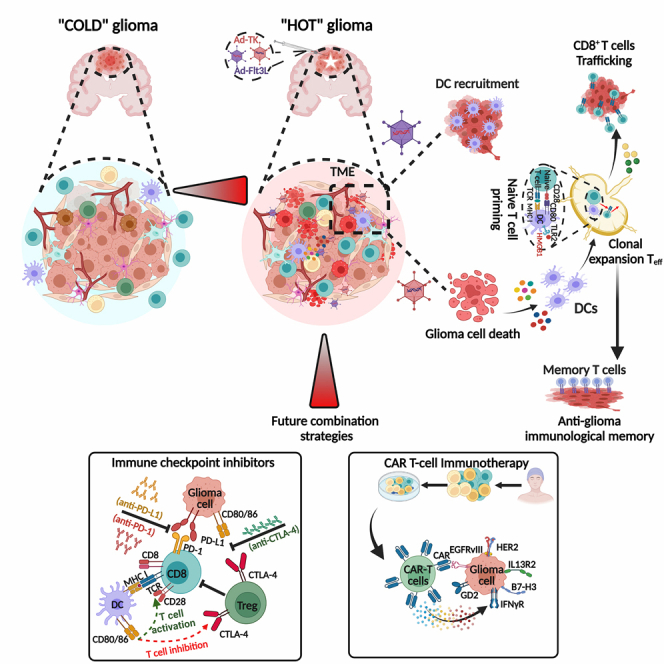
This figure explores the potential of immune checkpoint inhibitors, DC vaccines, immunovirotherapy, and CAR-T cells, and their implications for future clinical trials when combined with our TK + Flt3L gene therapy. The top left panel shows the immune checkpoint inhibitors, such as pembrolizumab (anti-PD-1), avelumab (anti-PD-L1), and ipilimumab (anti-CTLA-4), which aim to combat T cell exhaustion or immunosuppression by blocking immune checkpoints, thereby restoring T cell functionality and enhancing anti-glioma immunity. The top right panel shows the activation and recruitment of tumor neo-antigen-specific T cells into the TME to selectively target and kill glioma cells in response to vaccination. The current state-of-the-art vaccination approaches educate T cells to target glioma neo-antigens, such as injection of ex-vivo-developed matured DCs loaded with either autologous tumor cell lysate (DC vaccines; DCVax-L) or peptides, injection of synthetic peptides plus immune adjuvant (peptide vaccines; CDX-110, NeoVax, and NOA-16) and intratumoral oncolytic virotherapy (localized vaccination). In the bottom left panel, various oncolytic virotherapy strategies, including DNX-2401, PVS-RIPO, and HSV-1-G47Δ, are depicted. These treatments enhance the recruitment and functionality of immune cells, shift primed CD8+ immune cells toward an anti-glioma phenotype, and thereby reduce immune suppression within the TME. The activation of anti-glioma immunity by oncolytic virotherapy/gene therapy often leads to the production of various pro-inflammatory cytokines, which further help to generate an immune-stimulatory hot TME (Figures 1 and 2). The bottom right panel illustrates CAR immunotherapies, which involve genetically engineering a patient’s own T cells or non-patient NK-92 cells to express neo-antigen-specific CARs. These CARs are expanded in culture and then transferred to the patient adoptively. Once infused into the patient, the CAR-T cells recognize tumor-specific antigenic peptides and execute effector functions, such as releasing antitumor cytokines (perforin, granzyme-B, and interferons), leading to the eradication of target cells. Several glioma-specific cell surface antigens are being investigated for their potential as targets for CAR-T cell therapy. These antigens include IL13R2, EGFR/EGFRvIII, HER2, disialoganglioside GD2, and B7-H3. Promising results have been reported in preclinical research and clinical trials. The figure was created with BioRender.com.
Immunotherapy with CAR-T cells has revolutionized the treatment of B cell leukemia or lymphoma.51 Despite its limited efficacy in GBM and other solid tumors, researchers are generating preclinical and clinical data to optimize CAR-T cell therapy for GBM (NCT03423992).52 CARs are MHC-independent synthetic receptors with an extracellular tumor-antigen-specific binding domain and intracellular signaling domains. When CAR-T cells recognize target ligands, they carry out effector functions, such as releasing antitumor cytokines and eradicating target cells. A number of promising glioma-specific cell surface antigens have been tested, including IL13R2, EGFR/EGFRvIII, HER2, disialoganglioside GD2, and B7-H3, in preclinical research and clinical trials (Figure 3).53,54 Impaired T cell trafficking to the glioma, immunosuppressive tumor microenvironment (TME), and antigen heterogeneity are the primary limitations to CAR-T cell efficacy in GBM. As this field advances, addressing these impediments is crucial for the successful implementation in the clinic.
To enhance clinical outcomes in GBM, the introduction of nanoparticle-based treatments that can penetrate the BBB is of high relevance. We have recently shown that targeting CXCR4 signaling using synthetic protein nanoparticles coated with the transcytotic peptide (iRGD) loaded with AMD3100 (CXCR4 antagonist) decreased infiltration of immunosuppressive CXCR4+ M-MDSCs (monocytic myeloid-derived suppressor cells) to the glioma immune microenvironment, sensitizing glioma toward radiotherapy.55 Recent work from our lab has shown that galectin-1 regulates the expression of exosomal miR-1983 from glioma cells. This miR-1983 possesses a 5′-UGUUU-3′ sequence at its 3′ end, which activates Toll-like receptor 7 (TLR7) and triggers downstream signaling via MyD88-IRF5/IRF7-IFN-β to eradicate glioma.56 These microRNAs could be utilized in the development of glioma-specific nanoparticles to induce natural killer (NK)-mediated innate immunity and combine with cytotoxic and immune-stimulatory gene therapy (Ad-TK + Ad-Flt3L) to bolster the innate and adaptive immune systems simultaneously.
Continued investigations into inhibiting specific immunosuppressive factors in primary brain tumor patients have resulted in several approaches, often in combination with other treatments. Unfortunately, efforts to inhibit TGF-β using antisense oligonucleotides or blocking antibodies, as well as TGF-β receptor 1 (TGFβR1) kinase inhibitors, have not proven successful in demonstrating any survival benefits.57 The mammalian target of rapamycin (mTOR) regulates various physiological and pathological cellular functions such as apoptosis, proliferation, and autophagy by controlling several signaling pathways.58,59,60,61 The mTOR pathway is frequently activated in GBM and facilitates tumor growth by regulating immune cells differentiation and function.62 The PTEN/PI3K/AKT/mTOR pathway has been identified as a significant contributor to the development and progression of GBM. Despite previous attempts to target this pathway with PI3K, AKT, or mTORC1 inhibitors, no improvement in the outcome of GBM patients was observed.59 However, available evidence indicates that the dual mTORC1/2 blocking using AZD8055 or AZD2014 augments the radiosensitivity, thereby offering a promising therapeutic approach for treating GBM.60 However, the systemic inhibition of mTOR can cause immune suppression and other dose-limiting adverse effects.63 Alternatively, confining the pharmacological effects of mTOR inhibitors to the CNS could widen their therapeutic window. To achieve this, Zhang et al. recently developed a binary approach that consists of brain-permeable mTOR inhibitor (RapaLink-1) with a brain-impermeant ligand of FK506-binding protein 12 (RapaBlock) to reduce systemic off-target effects.64 This combination allows for brain-specific mTOR inhibition through the intracellular protein FKBP12. Using this drug combination in a glioblastoma xenograft model resulted in tumor regression without causing detectable systemic toxicity.64 Such a strategy may overcome the limitations of systemic mTOR inhibition that have been observed thus far, such as immune suppression, growth inhibition, and metabolic disorders.58,59,64 This suggests that dual targeting in combination with gene therapy could be studied as a potential anti-invasive approach for the treatment of GBM in future clinical trials.
To instigate immune exclusion, gliomas often use the immune-suppressing enzyme indoleamine 2,3-dioxygenase (IDO).65 Various clinical trials are underway to evaluate IDO inhibitors in adult and pediatric brain tumors (NCT02502708, NCT02052648, NCT02327078, NCT05106296; ClinicalTrials.gov). However, the ECHO-301 trial’s phase III results (NCT02752074) have dampened enthusiasm for these agents, as they showed no clinical benefit from combining IDO inhibition with immune checkpoint blockade in metastatic melanoma.66 Although there are ongoing phase I and II trials investigating arginase inhibitors for advanced and metastatic solid tumors (NCT02903914), none of them are specifically targeted toward brain tumors. Inhibiting specific immunosuppressive mediators has exhibited successful results in preclinical animal models, but it has not yet demonstrated promising results in GBM patients. This lack of efficacy may be attributed to various factors, including poor penetration of some therapeutic molecules into the brain stroma, use of anti-inflammatory steroids such as dexamethasone, or the diverse range of immunosuppressive mechanisms displayed by GBM.
Glioma cells that have been subjected to radio- or chemotherapy often exhibit increased cytosolic DNA levels.67 The cyclic GMP-AMP (cGAMP) synthase/stimulator of interferon gene (cGAS/STING) DNA sensing pathway has shown “promise” as an immunotherapy target due to its ability to stimulate the innate immune response locally. Upon cGAMP production by cGAS, it binds to STING, activating STING signaling, and leading to the production of pro-inflammatory cytokines, including type I interferons (IFNs).68 In the TME, the presence of these cytokines and T cell infiltration are characteristic features of a “hot” tumor, which is generally more receptive to immunotherapy (Figure 2). To achieve better immunotherapy responses, researchers are focusing on converting “cold” TMEs, which lack T cells, into hot TMEs (Figure 2).69 Prior studies have shown that appropriate type I IFN production and T cell recruitment require cGAS expression in tumor cells and STING activation in DCs, TAMs, or endothelial cells. However, Low et al.70 utilized scRNA-seq transcriptomic and methylation profiling to demonstrate that the epigenetic silencing of STING expression in GBM cells alone resulted in a cold TME, rendering GBM suppressive to immunotherapy despite the continued expression of STING in stromal, myeloid, and endothelial cells.70 Our recent work has also shown that H3.3-G34R mutations, which are found in pediatric high-grade gliomas, contribute to genomic instability, leading to the activation of cGAS/STING pathway, which triggers the immune system and enhances the efficacy of DNA-damaging therapies.71
T cell exhaustion is a major limitation to effective anti-tumor immune responses, characterized by the loss of effector functions, altered metabolic and transcriptional profiles, and the expression of inhibitory receptors, such as PD-1, CTLA-4, LAG-3, and TIM-3 (transmembrane immunoglobulin mucin-3) (Figure 3).72,73 Inhibitory cytokines within the TME, including IL-10 and TGF-β, also contribute to T cell exhaustion.72,73 Clinical trials targeting hallmarks of exhaustion are underway (NCT02658981, NCT04588987, NCT05345002).74 Dexamethasone suppresses naive T cells and impairs immunotherapy efficacy, and is regularly prescribed to glioma patients to reduce cerebral swelling and alleviate symptoms.75 It would be ideal if it would be possible to optimize dexamethasone administration without compromising immunotherapy efficacy. Optimizing dexamethasone administration and developing alternative therapies for brain inflammation that do not interfere with the immune system are crucial for effective immunotherapy against glioma.76,77,78
Herein, we describe studies conducted on two well-studied immune checkpoint inhibitors, namely CTLA-4 and PD-1, and their contribution to T cell dysfunction in glioma (Figure 3). Although primed T cells can migrate to the site of glioma during tumor progression, they tend to lose their functionality. This leads to their eventual exhaustion, inactivation, or ineffectiveness in rejecting gliomas.72,73,74 This emphasizes the need to revive dysfunctional T lymphocytes to offer clinical benefits to glioma patients. Therefore, there is significant interest in preventing or reversing overall T cell dysfunctionality to combat the immunosuppressive TME, thereby increasing the number of effector T cells in the brain. Furthermore, we provide the theoretical framework underpinning the development of immunostimulatory gene therapy exhibiting a robust immune response against glioma-specific antigens to target the gliomas. We summarize its strengths and weaknesses as tested in the clinical setting until now, and discuss future directions for these promising gene therapies achieved with Ad-hCMV-TK/GCV and Ad-hCMV-Flt3L (Clinicaltrials.gov: NCT01811992).8
Promises and challenges of immune checkpoint inhibitors
Immune-inhibitory mechanisms limit the cytotoxic efficacy of effector T cells. Thus, antibodies blocking immune regulatory checkpoints are now employed to potentiate the anti-tumor cytotoxic activity of T cells (Figure 3).79,80 Multiple forms of cancer, including melanoma, lung cancer, and upper gastrointestinal tumors, have been successfully treated with inhibitory checkpoint immunotherapies,46,47,49,66 where positive results have been obtained in a significant percentage of patients. However, primary malignant gliomas did not respond to currently available immune checkpoint inhibitors (Figure 3).73 Moreover, the therapeutic resistance of malignant gliomas is also seen vis-a-vis other novel therapeutic strategies. The clinical trials that utilized PD-1 blockade in primary and recurrent GBM patients without selecting for MGMT methylation status,81 along with the use of vaccines targeting tumor-associated antigens (Figure 3) (NCT03893903, NCT04842513, NCT01920191, NCT01130077, NCT01903330), or administration of replication-competent retroviruses (Toca 511/Toca FC), failed to demonstrate any significant clinical benefits for glioma patients.81,82
DC vaccination strategies have been shown to induce antitumor immune responses in human patients undergoing clinical trials for GBM.50,83 The very recent phase III clinical trial that was non-randomized and involved DC vaccination loaded with autologous tumor lysate (DCVax-L) for primary and recurrent GBM patients demonstrated significant clinical benefits (Figure 3).50 The efficacy of DC vaccination seems to rely on the techniques used to generate tumor cell lysates and conditioned DCs. To enhance the therapeutic efficacy and antitumor immunity induced by DC vaccination, we postulated that modifying the TME through in situ Ad-TK/GCV + Ad-Flt3L gene therapy could be a promising approach. Our findings demonstrated that the combination of in situ immune-stimulatory gene therapy with DC vaccination led to long-term survival in approximately 90% of the animals. This represented a significant improvement compared with either therapy alone and indicated that gene therapy enhanced the therapeutic efficacy and antitumor immune responses induced by DC vaccination.84 These findings support starting novel phase I clinical trials to assess the safety and efficacy of this combined approach (DC vaccination + gene therapy).
Research on the glioma microenvironment and tumor-infiltrating leukocytes indicates that TME is both diverse and immunosuppressive, which contributes to immunotherapy resistance.72,73,74 Glioma TME is characterized by the production of immunosuppressive cytokines, including IL-10, TGF-β, IDO-1, CSF-1, arginase, and prostaglandin E2. Moreover, there is recruitment of immunosuppressive cells, such as MDSCs and CD163+ M2 macrophages. The exclusion of anti-tumor immune effector cells, such as the CD154+ CD8+ T cell also occurs, along with the expression of cell-surface inhibitory ligands, such as PD-1, PD-L1, and CTLA-4. In the TME of GBM, the CD45+ cells exhibit a reduced presence of CD3+ T cells and an abundance of myeloid cells, including microglia, monocytes, and macrophages.72,73,74,85 Glioma-associated macrophages have elevated levels of immunosuppressive phenotypic markers and are associated with poor survival. Therefore, effective immunotherapeutic approaches must involve modifying macrophage features targeting other inhibitory mechanisms that impede therapeutic interventions.86,87,88
In glioma, certain T cell dysfunctionality that inhibits the immune response can be reversed by reducing immunosuppressive cells or preventing T cell exhaustion.89,90 The aim of therapy targeting immune checkpoint pathways is to reverse the immunosuppressive TME, promote anti-tumor immunity, and ultimately restore the function of cytotoxic CD8+ T cells (Figure 3). However, clinical trials using nivolumab, a monoclonal antibody that targets PD-1, failed to show clinical benefit in patients with recurrent GBM when compared with bevacizumab treatment (which targets angiogenesis).81,91
At present, over 40 “phase I/II” clinical trials are underway to evaluate the efficacy of immune checkpoint inhibition in glioma. These trials involve the use of monoclonal antibodies that specifically target PD-1, such as pembrolizumab, cemiplimab, or nivolumab or PD-L1, such as avelumab, durvalumab, or atezomab in conjunction with SOC (Figure 3).92 Anti-PD-1 immunotherapy has demonstrated clinical efficacy in a subset of patients with recurrent GBM with BRAF- and PTPN11-activating mutations, which promote MAPK/ERK pathway activation. ERK1/2 activity in recurrent GBM is a predictor of response to PD-1 blockage and is correlated with unique myeloid cell phenotype.93
Viral vector-based therapies for glioma
The development of viral therapies for use as an alternative cancer treatment has picked up momentum over the past ten years. In 2015, the FDA approved IMLYGIC (talimogene laherparepvec), a genetically modified oncolytic viral therapy (HSV-1) that treats locally unresectable cutaneous, subcutaneous, and nodal lesions in patients with recurrent melanoma.94 Promising safety and efficacy outcomes of an open-label, single-arm phase III clinical trial involving intravesical nadofaragene firadenovec (Adstiladrin) offer a realistic alternative to chemotherapy for BCG-unresponsive non-muscle-invasive bladder cancer.95 On 16 December, 2022, the FDA approved the first non-replicating adenovirus-based gene therapy for high-risk, non-muscle-invasive bladder cancer.95,96
However, remarkable genetic and signaling heterogeneity of solid neoplasms, including glioma, constitute a serious therapeutic challenge.97,98,99 Keeping this in consideration, the pleiotropic mechanisms of killing dividing tumor cells employed by virotherapies are hypothesized to be beneficial in tackling glioma heterogeneity. Recent studies have further documented an effective role of virotherapies in reverting the TME from cold immunosuppressed tumors to an inflamed hot state, allowing for a more diverse immune response (Figure 2).5,6,10,13,100,101 Virotherapies can trigger an immune-stimulatory response through the activation of pathogen-associated molecular patterns, damage-associated molecular patterns (DAMPs), and pattern recognition receptors. Furthermore, evidence supporting the activation of M1 macrophages through TLRs signaling in response to the release of DAMPs such as HMGB1, calreticulin, and heat shock proteins is shown in Figure 1.102,103,104 Gene therapies have demonstrated safety and enhanced infiltration of immune cells, suggesting improved systemic immunity after treatment, as shown by immunophenotyping of matched primary and secondary surgery biopsies of the patients enrolled in NCT01811992.7,8,89,105,106,107
One of the most exciting recent steps forward is the use of teserpaturev (G47Δ; Delytact), a new anti-GBM oncolytic virus, which was recently licensed for human therapy in Japan.6 In this phase II clinical trial, the oncolytic HSV G47Δ was stereotactically injected into 19 patients with recurrent GBM. The reported 1-year survival rate was 84% vs. 15% in the prespecified control group, and the overall median survival from the first G47Δ injection was reported as 20.2 and 28.8 months from the initial surgery.6 In contrast to previous viral trials, which all used a single stereotactic injection to deliver the treatment, the G47Δ virus used in this study was delivered to the tumor up to six times over the course of 5 months, along with tumor biopsies.6,108 Furthermore, analysis of successive tumor biopsies showed a clear correlation between the dosage schedule and the increase in effector T cell infiltration (CD4+ and CD8+) as well as reduction in FOXP3+ cells (Figure 3).5,6
Prior to the G47Δ viral therapy success, Desjardins and colleagues developed engineered oncolytic poliovirus (PVSRIPO), which obtained FDA breakthrough therapy designation following a trial involving 61 patients with biopsy-proven recurrent glioma (NCT01491893).10 Although this trial failed to find a significant difference in median survival (12.5 months) compared with control groups (11.3 months), the 2-year overall survival was 24%, with five patients reaching the 3-year landmark.10 However, clinical benefits from larger controlled clinical trials are still expected. Table 1 provides a comprehensive overview of clinical trials for viral vector-based therapies in brain malignancies conducted between 1992 and 2023.
The administration of two adenoviral vectors expressing HSV1-TK and Flt3L is considered safe and well tolerated in patients with newly diagnosed high-grade glioma (Figure 1).8 Further efficacy trials, in conjunction with blocking the suppressive TME, are warranted, considering the encouraging evidence of immune infiltration of effector cytotoxic T cells (CD8+) from the successive biopsies using multiplex immunocytochemical analysis.8
It is likely that additional research into these approaches will exploit new administration regimens and therapeutic combinations that will enhance the prognosis and outcome for high-grade glioma. Longitudinal analysis into these virotherapies trials will further provide exciting novel clinical and molecular insights. In this review, we describe numerous preclinical and clinical studies with promising and encouraging results, including our combined cytotoxic and immune-stimulatory gene therapy for glioma (Table 1).8
Speculations on the potential limitations of some gene therapy clinical trials
The ASPECT trial, which was conducted by Westphal et al., investigated the safety and efficacy of locally administered adenovirus-mediated HSV-TK (sitimagene ceradenovec) followed by intravenous ganciclovir in patients with newly diagnosed resectable glioblastoma.109
This and many other gene therapy clinical trials using HSV1-TK provided valuable insights into the potential of this treatment approach for glioblastoma patients. This opened the path toward the recent clinical findings, which demonstrated the feasibility and tolerability of an adenoviral vector expressing an inducible form of IL-12, injected into the resection cavity walls of patients with recurrent high-grade glioma.110 Chiocca et al. conducted a subsequent phase I trial combining this treatment with nivolumab, an immune checkpoint inhibitor.111 The combination approach exhibited promising systemic immune responses (increase in the percentage of CD3+CD8+ T cells in peripheral blood from day 0 to 28), which were controlled by the oral administration of veledimex, which stimulates the expression of IL-12. This trial highlighted the importance of tightly controlling the release of potent proinflammatory cytokines such as IL-12 to balance toxicity and effectiveness.110 Below, we speculate on the limitations of some of these trials.
Anti-viral immunity: one possible factor contributing to the suboptimal results of PVS-RIPO could be the presence of pre-existing immunity against viral components used in these therapies. Patients with prior exposure or immunity to the components of the viral vector or the poliovirus used in PVS-RIPO might have had a reduced response to the treatment. An improved understanding of the brain TME will pave the way for enhanced and individualized OV therapies.112
Immunological challenges: PVS-RIPO and Toca511 involve immunological mechanisms for glioma treatment. PVS-RIPO uses a modified poliovirus to stimulate an immunological response, while Toca511 uses a retroviral vector for gene delivery and immune activation. The immunosuppressive glioma microenvironment may have impeded PVS-RIPO and Toca511 therapy diminishing their effectiveness. Factors such as the presence of immune checkpoints, immunosuppressive cells (Treg cells and MDSCs), and the limited infiltration of immune effector cells could have contributed to the lack of sustained major clinical benefit of PVS-RIPO and Toca511.
Considering these immunological challenges, a potential future direction for enhancing clinical benefits should involve a combinatorial approach. Combinations of our cytotoxic and immune-stimulatory gene therapy approach with checkpoint inhibitors could potentially reverse the immunosuppressive TME and enhance the immune responses. In addition, inhibiting immunosuppressive cells, particularly MDSCs, within the glioma microenvironment could eliminate their inhibitory effects on immune cells.89,105 We have shown in in vivo models that these strategies enhance our dual-vector therapy.89,105,107
Interestingly, tissue obtained from successive surgeries (pre- and post-viral vector injections with SOC) by us (Umemura et al.)9 and others (Lang e al.)113 have demonstrated the presence of immune cells infiltrating the tumors in these phase I trials.9,113 Spatially resolved multiplex immunohistochemistry in eight patients subjected to two surgeries showed an increase in CD8+ T cells expressing activation markers granzyme B and CD107a.9 An increase in pDCs was also observed, which is consistent with our preclinical findings.114,115
In addition, five out of the eight patients subjected to re-section demonstrated reduced proportions of Tregs in the recurrent TME.9 Interestingly, persistent HSV1-TK expression for up to 17 months after dual-vector administration was observed upon recurrence. Five out of eight patients had HSV1-TK immunoreactive cells within the resected recurrence tissue. These results support extended valacyclovir administration in larger phase II clinical trials.9
Molecular profiling of pre- and post-DNX-2401-treated tissue revealed the infiltration of CD4+ and CD8+ T cells into the tumor, accompanied by a notable reduction in T cell exhaustion marker TIM-3 expression.113 This reduction suggests that DNX-2401 holds promise in overcoming specific aspects of T cell exhaustion, thus indicating its potential to induce an immune-mediated anti-glioma response. To further explore the potential of DNX-2401, a phase I/II clinical trial is currently underway to assess its effectiveness in combination with pembrolizumab (NCT02798406).116
The ultimate hurdle to successful glioma immunotherapy: Dexamethasone-mediated immunosuppression
Dexamethasone is a double-edged sword. It is necessary to reverse deadly brain edema, and it also inhibits potentially curative immune responses. Dexamethasone has been shown to limit the clinical benefit of immune checkpoint inhibitor-treated glioblastoma patients. Lorgulescu et al., found that initiating dexamethasone at the start of immunotherapy treatment posed the greatest risk to overall survival. In 181 patients receiving anti-PD-1 or anti-PD-L1 therapy, using dexamethasone at baseline had twice the risk of mortality.76 Recently, Jain’s group found that Losartan reduces edema induced by immune checkpoint blockade and increases survival in mouse models of GBM.117 In addition, the Boswellia serrata-derived herbal extract, 5-Loxin, effectively impedes the expression of vascular endothelial growth factor. This leads to a reduction in perilesional edema in brain tumor patients who are undergoing fractionated radiation therapy.118 However, further testing is required to determine the general efficacy of these new drugs in GBM patients.
Another approach to tackle the immunosuppression associated with dexamethasone would be to engineer T cells with resistance to dexamethasone, followed by bone marrow adoptive transfer to patients, thereby providing a bone marrow resistant to dexamethasone. The potential development of a high-affinity mutated form of glucocorticoid receptor that could bind dexamethasone without nuclear translocation (i.e., a dominant negative receptor) could be explored. Finally, glucocorticoid receptor β (GRβ) has been associated with glucocorticoid resistance through dominant negative regulation of GRα.119 By creating a dominant negative specific receptor, one could potentially sequester dexamethasone in the cytoplasm and thus reduce its deleterious effects on T cell function. Another avenue to consider is the development of small molecules (i.e., J9 [SIH-182]) that can reverse dexamethasone-mediated immunosuppression in T cells.120 These molecules could potentially target dexamethasone-mediated immunosuppression and restore T cell responsiveness to immunotherapy.120,121 Furthermore, the engineering of CAR-T cells with permanently disrupted GRs) has shown promise as a strategy for engineering CAR-T cells resistant to dexamethasone inhibition for the treatment of GBM.122,123 Exploring the manipulation of GRβ expression or activity could provide additional avenues for overcoming dexamethasone-induced immunosuppression and potential clinical success.
Preclinical models for testing translational efficacy: How predictive are they?
The existing preclinical models exhibit limited predictive capacity regarding the effectiveness of translational therapeutics in clinical trial outcomes. The commonly used serum-culture-based patient-derived xenografts do not accurately represent the genetic composition, invasive characteristics, and proliferative index of human GBM.25 They also lack an intact immune system and may lose important genetic lesions over time. Genetically engineered murine models of GBM provide a more accurate model of gliomagenesis, exhibiting histological and molecular similarities to human GBM. However, these models have limitations such as variable reproducibility, extended tumor latency, and incomplete knowledge of tumor localization. Recent advances in imaging techniques offer potential for utilizing genetically engineered models in preclinical evaluation of neuro-oncology therapeutics. The presence of quiescent glioma cells possessing stem cell-like properties presents challenges for SGTs that target actively dividing cells. To overcome these limitations, it is crucial to develop animal models that encompass histopathological resemblance (invasion pattern), biochemical similarity, intact tumor-host interactions (non-immunogenic tumor), intracranial location, accurate tumor mapping, predictable growth patterns, and high reproducibility. Unfortunately, a GBM animal model with all these features does not exist. In addition, comprehensive testing of novel therapies is lacking in patient-derived primary spheroid models and glioma stem cell lines, which are considered the new standards for preclinical studies in gliomas. Therefore, it is recommended to conduct experiments using a variety of tumor models to better predict the clinical outcomes of translational therapies.
Delivery route for gene therapy vectors into the brain and brain tumors
Systemic delivery of viral vectors for gene therapy encounters challenges, including off-target effects in non-CNS tissues and limited bioavailability in the CNS. Intranasal delivery of cell-based vectors has shown promise in preclinical glioma models.124,125 However, the current predominant method for SGT involves multiple intracranial injections directly into the resection cavity after surgery. This approach is suboptimal due to the heterogeneous nature of the surrounding tissue, which contains infiltrating tumor cells and brain tissue. Lack of control over the extent of tumor tissue resected contributes to trial failures and interpatient variability, which complicates data interpretation. Convection-enhanced delivery (CED) is an advanced technique that uses catheters and a micropump to achieve continuous low-pressure flow into solid tissues, including brain tumors. While CED is effective for targeting larger tissue areas, its application in resection cavities is not optimized since primary tumors are typically treated through neurosurgery, the SOC. Therefore, for future clinical trials, CED implementation is currently limited to injecting vectors into recurrent tumors or inoperable primary tumors.
Systemic administration of AAVs is another option, but their efficacy can be compromised by the immune system’s response. Localized gene therapy directly in the tumor bed/resection cavity is preferred, and efforts are being made to extend its availability for longer durations. Recent advances in AAV capsids (AAV.CAP-B10 and AAV.CAP-B22) have enabled the crossing of the BBB with neuronal specificity, opening up new possibilities for research and therapeutic applications.13 Improved AAV vectors (AAV.CPP.16 and AAV.CPP.21) have demonstrated the potential for systemic curative anti-tumor gene therapy against mouse models of glioma and showed enhanced transduction efficiency in non-human primates (cynomolgus monkeys).14
Comprehensive exploration of cytotoxic and immune-stimulatory gene therapy
Ad-Flt3L as a single agent in vivo
The field of cancer immunotherapy has developed exciting advancements for some cancer types over the past decade. Efforts are underway to develop immunotherapeutic interventions that can reverse the immunosuppressive TME and enhance the success of immunotherapies against glioma. The effectiveness of immunotherapies relies on the presence of functional T cells within tumors. However, the immunosuppressive nature of the glioma TME limits T cell infiltration and their cytotoxic efficacy. DCs play a crucial role in shaping immune responses due to their ability to present antigens to naive T cells. Earlier, the brain has been considered an immune-privileged site.38 It comprises antigen-presenting cells such as perivascular macrophages, pericytes, and microglial cells. However, evidence suggests the absence of DCs from the brain parenchyma proper.41 Instead, DCs are present in areas such as the meninges, choroid plexus, and brain ventricles, as well as in inflamed brain tissue.38,41 With this in mind, we proposed that enhancing the infiltration of DCs into the TME could have potential therapeutic benefits.
Our very first study was focused on evaluating the efficacy of recombinant adenoviral vectors encoding human soluble FMS-like tyrosine kinase 3 ligand (RadhsFlt3L) against intracranial syngeneic CNS-1 gliomas in Lewis rats, and in mouse models. Flt3L is a naturally occurring glycoprotein stimulating early hematopoietic progenitors via the Flt3 receptor. The Flt3L/Flt3 pathway plays a crucial role in the development and function of DCs, and its potential to enhance cancer immunotherapy has been comprehensively reviewed in both preclinical and clinical settings.126 The rationale for using Flt3L ligand is its ability to stimulate the expansion of DCs, as well as activate NK cells, B cells and T cells. Through a series of experiments, we observed that the administration of 8 × 107 PFU RadhsFlt3L resulted in a remarkable 70% survival rate. This positive outcome was attributed to the enhanced infiltration of OX62+MHCII+DCs in rats and CD11C+33D1+MHCII+F4/80+DEC205- in mice. In addition, our neuropathological analysis demonstrated tumor rejection in conjunction with animal long-term survival. This study represents the pioneering use of Flt3L to stimulate the anti-glioma responses in experimental rodent model.127
Combination therapies
Numerous clinical trials have explored gene therapy as a potential treatment for glioma, with SGT employing a prodrug activation system, specifically the HSV1-TK gene combined with the prodrug GCV, being the most widely used approach. While this approach was found to be highly effective in most preclinical models, the bystander effect in clinical trials was lower than anticipated. This suggests that there may have been inadequate vector distribution and/or low transduction efficiency.
Ad-HSV1-TK + Ad-Flt3L
Furthermore, we employed a combinatorial approach to enhance the anti-tumor immune response induced by RadhsFlt3L. This involved incorporating a conditionally cytotoxic SGT utilizing adenoviral vectors expressing HSV1-TK, together with systemically administered GCV. We demonstrated that monotherapy using either conditional cytotoxicity or immune-stimulation alone failed to show significant regression in the CNS-1 syngeneic intracranial glioma Lewis rat’s model. However, when various immunostimulatory therapies were combined with conditional cytotoxicity, only the RadFlt3L + HSV1-TK showed prolonged survival in over 80% of experimental animals, while RadCD40L and RadIL-12 did not produce significant effects. To identify the involvement of specific immune cell types, we depleted either macrophages or CD4+ cells prior to administering the combined therapy resulting in reduced therapeutic efficacy. Our data further suggested that the depletion of CD8+ or NK cells did not inhibit the survival in the RAdFlt3L + HSV1-TK treated groups. In conclusion, the combination of immune-stimulation (RadFlt3L) and conditional cytotoxic gene therapy (HSV1-TK) significantly enhanced the long-term survival and tumor regression in gliomas.31 These findings form the basis for the ongoing clinical trial (NCT01811992), representing the first study of its kind (The Lancet Oncology, in press).9
Chemotherapy employing TMZ in combination with radiotherapy (IR) prolongs the survival of glioma patients’.15,16,19 However, the 2-year survival rate was 27.3%, 3-year was 16%, 4-year was 12.1%, and 5-year was just 5% with IR + TMZ, compared with 10.9%, 4.4%, 3%, and 1.9% with IR alone.16 Given these data, we were curious to determine the therapeutic efficacy of TMZ when administered in combination with Ad-HSV1-TK + Ad-Flt3L. Our findings indicated that, despite the well-documented immune-inhibitory impacts of TMZ, its administration did not hamper the anti-glioma immunity achieved by combined Ad-HSV1-TK/GCV + Ad-Flt3L gene therapy. This study further supported the eventual clinical implementation of our gene therapy in glioma patients already receiving TMZ as a first-line chemotherapy treatment.128 Importantly, the anti-glioma immune response was greatly augmented, as was the long-term survival in the glioma mouse model.
Reversion of brain tumor-induced behavioral deficits
To investigate whether combined gene-therapy reverses the behavioral abnormalities produced by the growing intracranial tumor 3 days’ post-treatment with Ad-Flt3L + Ad-HSV1-TK in the presence of GCV, we evaluated the behavior of glioma-bearing rats by analyzing amphetamine-induced rotational behavior. Their brains were harvested to analyze neuropathology. Our results suggested that intracranial glioma induces behavioral deficits that can be restored by combined immune-stimulatory gene therapy. The data from the long-term survivors showed the complete recovery of the brain architecture and established its safety and efficacy for glioma treatment. The data further suggested that rotational behavior can serve as a substitute for both tumor growth and the effectiveness of the treatment.129
Release of HMGB1 from dying tumor cells mediates TLR2 activation
Cell death caused by various treatments can facilitate the immune systems’ response to antigens, as tumor antigens and signals released during cell death can help in the uptake of antigens by DCs and promote their maturation. Radiation-caused cell death in a tumor can trigger cytotoxic T lymphocyte (CTL) responses, leading to prolonged immunity against glioma. However, the release of endogenous TLR ligands from dying cells can also result in either tolerance or autoimmunity. We discovered that high-mobility group box 1 (HMGB1) acts as an endogenous TLR2 agonist secreted from dying glioma cells both in vitro and in vivo in response to radiation, TMZ, and adenoviral vector Ad-TK/GCV. As a result, HMGB1 from tumors activates endogenous TLR2 signaling, initiating a cytotoxic CD8+ T cell-mediated immune response against gliomas (Figure 1). We observed that inhibiting HMGB1 activity using glycyrrhizin or specific anti-HMGB1-neutralizing antibodies in vivo prevented Flt3L/TK-induced brain tumor regression.103 We demonstrated that combined gene delivery in the presence of systemic GCV promotes the release of putative endogenous TLR ligands by killing glioma cells, and infiltrating DCs in the brain elicit powerful CD8+ T cell-dependent systemic anti-glioma immunity and immunological memory.103 Compared with other Ads encoding pro-apoptotic cytokines, such as TRAIL, FasL, and TNF-α, our approach using Ad-TK was more effective and less toxic.34,103
Anti-glioma immunological memory in vivo
Most immunotherapeutic approaches have obtained disappointing results because of unavoidable glioma recurrences. Keeping into consideration the high rate of tumor recurrence in glioma patients, it is of utmost importance that novel therapies suppress recurrent tumors. Therefore, we were interested to assess whether combined gene therapy induces immunological memory in various intracranial glioma rat and mouse models (i.e., intracranial CNS-1, recurrent intracranial CNS-1, intracranial F98, intracranial 9L, and intracranial CNS-1 flank model).
We utilized a combinatorial therapeutic approach (Ad-TK/GCV + Ad-Flt3L) that not only activates the anti-tumor immune response by stimulating DCs and macrophages but also generates CD8+ T cell-mediated immunological memory that combats recurrent glioma in an experimental rat model (Figure 1). Our work demonstrated that long-term survivors who eliminated the re-challenged tumor had higher tumor antigen-specific T cell precursors in the spleen and exhibited a delayed-type hypersensitivity response against the tumor antigen. These results corroborate the role of cellular immunity in mediating anti-glioma immunological memory response. In addition, CD8+ T cell depletion showed that CD8+ memory T cells mediate the elimination of recurrent tumors. The presence of circulating antibodies against tumor antigens also suggests the possible involvement of humoral immunity against the intracranial glioma.130
We conducted further tests to investigate whether Flt3L could enhance the recognition of neo-antigens in the brain stroma and overcome immune privilege. Our findings revealed that the expression of the influenza glycoprotein hemagglutinin (HA) as a surrogate neo-antigen within the brain stroma failed to elicit systemic immunity. However, when Flt3L was co-expressed, it successfully induced an immune response. We also observed that depletion of CD4+CD25+ Tregs, either alone or in combination with Flt3L administration, resulted in a synergistic effect that enhanced anti-HA immune responses. Our results further illustrate that Flt3L expression in the brain parenchyma does not induce autoimmunity. However, the immune response triggered by Treg depletion was also insufficient to induce behavioral deficits. These results further demonstrate that Flt3L can be used in the clinical setting because it overrides brain immune privilege and facilitates immune responses against neo-antigens without showing the sign of autoimmune neuropathology.131 In addition, we also modified the CNS1 cells to express the HA as a substitute tumor neo-antigen. We then observed that animals that survived long-term after treatment with WT-CNS1+ Flt3L/TK and were re-challenged with CNS1-HA cells, showed the presence of T cells specific to HA without the need for additional gene therapy. This finding provides further evidence that Flt3L/TK can generate glioma-specific immunological memory, which can also trigger immunity against surrogate neo-antigens to prevent glioma recurrence.132 Thus, Flt3L is able to break brain immune privilege but does not induce a generalized immune response against brain tissue.131,132
Mechanism of action of combined Ad-TK + Ad-Flt3L immune-stimulatory gene therapy: The role of B cells
Immunovirotherapy is a therapeutic approach aimed at delaying or inhibiting tumor recurrences by inducing the differentiation and proliferation of cytotoxic T cells and accelerating antigen presentation to naive T cells (Figure 1). However, glioma heterogeneity, high mutation rate, and their highly infiltrative nature make vaccination efficacy against a limited set of tumor antigens challenging. To address these challenges, we developed a combined gene therapy approach involving intratumoral administration of Ad-TK and Ad-Flt3L. This approach kills dividing tumor cells and simultaneously primes an immune response against released tumor antigens by recruiting DCs in the brain TME. Our earlier preclinical work showed that this combined immune-stimulatory gene therapy elicits an anti-glioma immune response and immunological memory, with cytotoxic CD8+ T cells playing a crucial role in brain tumor regression. While the role of B cells in anti-glioma immunity is not well understood, we investigated whether B cells are necessary to stimulate antitumor immune response by Ad-TK + Ad-Flt3L treatment.
To determine the role of B cells in the anti-glioma immune response triggered by Ad-TK + Ad-Flt3L gene therapy, we conducted an experiment using transgenic mice deficient in total B cells or marginal zone B cells, which were depleted by antibodies. We found that the combined gene therapy failed to induce glioma suppression when implanted intracranially in these mice. In addition, the absence of antigen-specific T cell clonal expansion in mice deficient in B cells (Igh6−/−) indicated that B cells were necessary to elicit the anti-glioma immune response. Collectively, we postulated that B cells may serve as antigen-presenting cells to augment the clonal expansion of glioma antigen-specific T cells and mediate the suppression of glioma within the CNS.133
It has been well documented that tumor impedes plasmacytoid DCs (pDCs) function by releasing anti-inflammatory cytokines (TGF-β, VEGF, and IL-10) in the TME, which weaken the tumor-infiltrating potential of pDCs to trigger antitumor immunity. We demonstrated that pDCs were recruited within the glioma TME in response to Ad-TK/GCV + Ad-Flt3L treatment and stimulated the production of inflammatory cytokines, which eventually facilitate T cell proliferation (Figure 1). The activated pDCs trigger the IFN-α release to complete the immune circuit during the anti-glioma immunity in response to combined gene therapy. Accordingly, pDC manipulation could be beneficial to further potentiate ongoing glioma immunotherapies.114
Targeting immunosuppressive MDSCs
Animals implanted with glioma tumors exhibited an immunosuppressive phenotype, both in the glioma TME and systemically. MDSC infiltration was documented in the serum as well as in the TME of glioma patients.134 Prior research has shown that MDSCs have the ability to suppress anti-tumor immune responses in humans and GBM models through various means, including the inhibition of T cell activation and expansion, the promotion of immunosuppression, and the hindrance of T cell migration. Addressing these mechanisms is crucial for the enhancement of immunotherapies.89,105 Next, we wished to test whether combining MDSC depletion or checkpoint blockade would enhance the efficacy of our combined Ad-TK/Flt3L + GCV gene therapy. Our data revealed that more than 40% of tumor-infiltrating immune cells were MDSCs and express T cell-suppressive molecules, viz. IL-4Ra, inducible nitric oxide synthase (iNOS), arginase, PD-L1, and CD80. There was significant increase in median survival in the MDSC-depleted group co-administered with combined gene therapy in the mouse glioma model. Moreover, combining checkpoint blockade, such as anti-PD-L1 or anti-CTLA-4, highly enhanced the efficacy of combined Ad-TK/GCV + Ad-Flt3L gene virotherapy.89,135
Our group previously demonstrated that depletion of immunosuppressive MDSCs led to a significant improvement in the efficacy of an immune-mediated gene therapy strategy.89 More recently, we showed that, in comparison with mIDH1 glioma, wtIDH1 glioma had a higher abundance of suppressive CD11b+Ly6G+ granulocytic MDSCs and greater expression of PD-L1, iNOS, and Arg1. In addition, murine mIDH1 glioma cells were found to secrete significantly more G-CSF than their wtIDH1 counterparts, which was determined to be due to H3K4me3 enrichment in the Csf3 gene encoding G-CSF. The expression of G-CSF was epigenetically activated by mIDH1 in glioma stem/progenitor-like cells, promoting the reprogramming of myeloid cells within the mIDH1 glioma TME. This increased secretion of G-CSF led to the expansion of pre-neutrophils and neutrophils while reducing the immunosuppressive phenotype of PMN-MDSCs present in the mIDH1 TME.105 In a wtIDH1 mouse model, the combined use of immune-stimulatory gene therapy Ad-TK/GCV and Ad-Flt3L along with recombinant G-CSF (rG-CSF) resulted in a significant survival benefit compared with TK/Flt3L or rG-CSF alone.105 These findings suggest that patients with mIDH1 glioma may benefit from immunotherapy due to the G-CSF produced by their tumors, while patients without the mutation may benefit from combining treatment with G-CSF and immunotherapy.105
Revitalizing T cell functionality: Reprogram glioma-induced immunosuppression
The effectiveness of immunovirotherapy is hampered by T cell dysfunction, which can be attributed to insufficient infiltration of T cells and the immunosuppressive TME. There are various factors that contribute to T cell dysfunctionality, such as exhaustion, senescence, tolerance, anergy, and ignorance. In addition, metabolic factors such as tumor-induced nutrient competition and hypoxia can impair T cell function.105,136,137,138 Glioma cells can also upregulate or bind immune checkpoint molecules on exhausted T cells, restricting their capacity to lyse tumor cells. To overcome this challenge, antibodies that inhibit checkpoint interactions can prevent the exhausted phenotype and promote reactive T cells to engage in anti-glioma immunity (Figure 3).139,140
The CD8+ T cells play an important role in inhibiting tumor progression, but their function can be inhibited by myeloid cells or activated by combined immunostimulatory gene therapy.141 There is spatial heterogeneity within tumors, where CD8+ T cells are often located in the tumor center and CD4+ T cells are found in perivascular areas.142,143 The relationships between T cell subtypes are complex, and their function can shift from a stimulatory to suppressive phenotype over time. Therefore, using these intricate relationships as therapeutic targets can be challenging. However, improved immunologic analytical tools and single-cell transcriptomics have enhanced the relevance of various T cell subtypes in glioma progression, providing a promising avenue for future therapeutic development. We predict that, in the future, we will be able to combine these therapeutic strategies with immune-stimulatory gene therapy.
Enhancing SOC with immunovirotherapy: A powerful combination
Recent immunovirotherapeutics including AdV-TK + valacyclovir,11,12,108 combined Ad-HSV1-TK + Ad-Flt3L,8,31,105,107 PVSRIPO,144 oncolytic DNX-2401,100 oncolytic HSV-1 G207,101 and HSV-1 G47Δ (Delytact),5,6 showed promising clinical benefits. It is very likely that, in the near future, immunovirotherapeutic will become part of the SOC for adult and pediatric glioma. Due to the remarkable spatial and temporal tumor heterogeneity, and immune-editing, it seems that an exclusive “miracle drug” comprising a “one-size-fits-all” is unlikely to emerge for glioma interventions.113,144
GBMs are characterized by an immunosuppressive TME with abundant myeloid cells and fewer effector T cells, rendering them cold tumors that exhibit a limited immunotherapy response (Figure 2). The infiltration of Treg cells further exacerbates the immunosuppressive TME.24,106 We have shown that endogenous G-CSF in mIDH1 low-grade glioma improved the efficacy of immunostimulatory gene therapy. The combination of gene therapy and G-CSF treatment significantly improved the survival of mice with wtIDH1 glioma.105 However, in mice with the IDH1 mutation, the effect was profound, with 90% of the mice surviving long-term and remaining tumor-free. These findings were supported by the TCGA datasets, which showed that patients with mIDH1 gliomas also had higher expressions of G-CSF.105 Interestingly, the re-programming of bone marrow granulopoiesis via G-CSF represents a promising approach to modulating MDSC function and enhancing the efficacy of immune-stimulatory gene therapy in cancer patients.105
Activation of the cGAS/STING pathway to elicit a potent immune response: Combination approach
Both IDH1 and H3.3-G34R mutations in adult and pediatric gliomas, respectively, have been found to epigenetically increase the DNA damage response, resulting in genetic instability and the emergence of extrachromosomal DNA in the cytoplasm.71,145 This phenomenon activates the cGAS/STING pathway in glioma cells, leading to the activation of immune cells including NK cells and DCs. STING pathway activation significantly remodels the glioma TME, promoting a robust immune response and NK cell-mediated glioma regression with prolonged immune memory.69 We anticipate that, in the future, STING agonists could be combined with immune-stimulatory gene therapy to further enhance their effectiveness.
Recommending long-term valacyclovir treatment
Hossain et al. demonstrated that recurrent tumor cells carrying HSV-TK retain functional HSV-TK, suggesting that a single-dose of GCV application may not have been sufficient to effectively target slowly dividing or quiescent glioma cells.30 Furthermore, they observed significantly enhanced survival with long-term GCV administration in rodents.30 Surprisingly, tumor tissue collected during tumor recurrence revealed persistent expression of HSV1-TK for a duration of up to 17 months after our dual-vector administration approach (NCT01811992).9 Among the patients who underwent surgeries for tumor recurrences, five out of eight individuals exhibited the presence of HSV1-TK immunoreactive cells in the obtained tissue samples.8,9 Thus, in preclinical models and clinical trials of HSV-TK-mediated gene therapy, continuous or long-term treatment with valacyclovir should be considered as an alternative to usual short-term valacyclovir treatment.
Challenges
Although immunovirotherapy has emerged as a rapidly advancing field in the treatment of GBM, achieving consistent and sustained therapeutic benefit remains a challenge. Several obstacles need to be overcome to improve its efficacy, including.
-
(1)
Limited response to immunovirotherapy seen in GBM treatment can be attributed to the use of steroids (such as dexamethasone) to manage cerebral edema.
-
(2)
Insufficient infiltration of T cells (scarcity of immune cells) and antigen-presenting cells (such as DCs) in the glioma microenvironment.
-
(3)
The immunosuppressive TME, including the presence of MDSCs, limits immunotherapy for glioma.
-
(4)
Mechanisms that allow GBM to evade immune surveillance, such as T cell dysfunction, T cell exhaustion, or upregulation of immune checkpoint molecules that impair the anti-glioma immune response.
-
(5)
Spatiotemporal heterogeneity in GBM tumors exhibit extensive spatial, genetic, and phenotypic diversity, leading to variations in their response to immunotherapy.
-
(6)
Unknown longevity of transgene expression in humans.
-
(7)
Existing preclinical models exhibit limited predictive capacity regarding the human effectiveness of translational therapeutics in clinical trial outcomes.
-
(8)
Lack of consensus regarding the most effective route for therapeutic delivery.
Future directions
Addressing these challenges through ongoing research and clinical trials will be pivotal in enhancing the effectiveness of immunotherapy in the treatment of GBM and providing better therapeutic options for patients. Future directions include.
-
(1)
Developing strategies to either replace dexamethasone or inhibit its deleterious effect on T cell function.
-
(2)
Explore combination approaches that enhance infiltration of T cells and DCs to the TME (e.g., Flt3L, IL-12).
-
(3)
Finding ways to reprogram immunosuppressive TME (cells or mediator molecules) to enhance immunotherapy (e.g., MDSCs, G-CSF).
-
(4)
Developing strategies to revert T cell dysfunction by inhibiting checkpoint interactions and enhancing the tumoricidal capacity of cytotoxic T cells.
-
(5)
Improving our understanding of the relevance of different T cell subtypes in glioma progression using advanced single-cell and spatial transcriptomics.
-
(6)
Exploit the longevity of transgene expression to improve the efficacy of gene therapy (i.e., continuous administration of valacyclovir treatment for enhanced targeting of slowly dividing or quiescent glioma cells).
-
(7)
Utilizing multiple tumor models that closely resemble the characteristics of human gliomas to enhance predictive accuracy. In addition, exploring novel therapies in patient-derived primary spheroid models and glioma stem cell lines can provide valuable insights.
-
(8)
Implementing localized gene therapy directly in the tumor bed (CED) or intracranial injection into the resection cavity, as preferred, and leveraging advancements in AAV capsids for the potential systemic delivery and crossing of the BBB and achieving tumor specificity. Develop a consensus on optimal glioma delivery routes.
Conclusions
Immunovirotherapy shows promise as a potential treatment for glioma, but it still needs to overcome challenges to achieve successful outcomes through a multipronged approach. Combination therapies, including gene therapy, immune checkpoint inhibitors, CAR-T cells, and other therapeutics, have shown signals of improved survival benefits in preclinical and clinical settings. However, going forward, it will be essential for the immunotherapeutic approaches to the treatment of gliomas to address the challenges related to dexamethasone’s effects on T cell function. To develop advanced combination therapies and enhance clinical benefits for glioma patients, it is essential to understand the interplay between tumor cells, immune cells, and the TME. By stimulating a robust anti-glioma immune response and neutralizing the immunosuppressive TME, future therapeutic advancements can be achieved.
The dual-vector approach using Ad-hCMV-TK and Ad-hCMV-Flt3L gene therapy has proven to be safe and feasible in treating challenging high-grade gliomas, which are traditionally resistant to conventional treatments. The approach targets dividing residual malignant glioma cells that persist after surgery, potentially triggering targeted anti-glioma immune responses. Tumors are also likely to express the transgene (HSV1-TK) for up to 17 months post viral-vector administration. This strongly suggests that continuous administration of valacyclovir could further enhance the cytotoxic impact of Ad-HSV-TK, leading to improved patient outcomes. Notably, this dual-vector approach can be safely combined with SOC chemoradiation for newly diagnosed high-grade glioma patients. These promising findings have the potential to enhance overall survival and quality of life and warrant further exploration in larger phase IB/II clinical trials.
Acknowledgments
This work was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) (grant nos. R37-NS094804, R01-NS105556, R01-NS122536, R01-NS124167, R01-NS122165, and R21-NS123879-01 to M.G.C.), NIH/NINDS (grant nos. R01-NS076991, R01-NS096756, R01-NS082311, R01-NS122234, and R01-NS127378 to P.R.L.), National Institute of Biomedical Imaging and Bioengineering (R01-EB022563), Rogel Cancer Center at The University of Michigan (G023089 to M.G.C.), National Cancer Institute (grant no. R01-CA243916 to P.R.L.), Ian's Friends Foundation (grant no. G024230), Leah’s Happy Hearts Foundation (grant no. G013908), Pediatric Brain Tumor Foundation (grant no. G023387), Smiles for Sophie Forever Foundation, Phase One Foundation, Los Angeles, CA. to M.G.C. and P.R.L., and ChadTough Foundation (grant no. G023419 to M.G.C. and P.R.L.).
Author contributions
S.M.F. wrote this manuscript, with overall guidance and revisions of M.G.C. and P.R.L. S.M.F. prepared the figures under the guidance of M.G.C. and P.R.L. S.M.F., P.R.L., and M.G.C. reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
Declaration of interests
The authors declare no competing interests.
References
- 1.Wen P.Y., Weller M., Lee E.Q., Alexander B.M., Barnholtz-Sloan J.S., Barthel F.P., Batchelor T.T., Bindra R.S., Chang S.M., Chiocca E.A., et al. Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020;22:1073–1113. doi: 10.1093/neuonc/noaa106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weller M., van den Bent M., Preusser M., Le Rhun E., Tonn J.C., Minniti G., Bendszus M., Balana C., Chinot O., Dirven L., et al. EANO guidelines on the diagnosis and treatment of diffuse gliomas of adulthood. Nat. Rev. Clin. Oncol. 2021;18:170–186. doi: 10.1038/s41571-020-00447-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bagley S.J., Kothari S., Rahman R., Lee E.Q., Dunn G.P., Galanis E., Chang S.M., Nabors L.B., Ahluwalia M.S., Stupp R., et al. Glioblastoma clinical trials: Current landscape and opportunities for improvement. Clin. Cancer Res. 2022;28:594–602. doi: 10.1158/1078-0432.CCR-21-2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aldape K., Brindle K.M., Chesler L., Chopra R., Gajjar A., Gilbert M.R., Gottardo N., Gutmann D.H., Hargrave D., Holland E.C., et al. Challenges to curing primary brain tumours. Nat. Rev. Clin. Oncol. 2019;16:509–520. doi: 10.1038/s41571-019-0177-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Todo T., Ino Y., Ohtsu H., Shibahara J., Tanaka M. A phase I/II study of triple-mutated oncolytic herpes virus G47Δ in patients with progressive glioblastoma. Nat. Commun. 2022;13:4119. doi: 10.1038/s41467-022-31262-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Todo T., Ito H., Ino Y., Ohtsu H., Ota Y., Shibahara J., Tanaka M. Intratumoral oncolytic herpes virus G47Δ for residual or recurrent glioblastoma: A phase 2 trial. Nat. Med. 2022;28:1630–1639. doi: 10.1038/s41591-022-01897-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varela M.L., Comba A., Faisal S.M., Argento A., Franson A., Barissi M.N., Sachdev S., Castro M.G., Lowenstein P.R. Gene therapy for high grade glioma: The clinical experience. Expert Opin. Biol. Ther. 2023;23:145–161. doi: 10.1080/14712598.2022.2157718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Umemura Y., Orringer D., Junck L., Heth J., Sagher O., Leung D., Mammoser A., Hervey-Jumper S., Varela M.L., Comba A., et al. Combined cytotoxic and immune therapy for primary adult high-grade glioma. medRxiv. 2022 doi: 10.1101/2022.11.04.22281950. Preprint at. [DOI] [PubMed] [Google Scholar]
- 9.Umemura Y., Orringer D., Junck L., Varela M.L., West M.E.J., Faisal S.M., Comba A., Heth J., Sagher O., Leung D., et al. Combined cytotoxic and immune-stimulatory gene therapy for primary adult high-grade glioma: a phase 1, first-in-human trial. Lancet Oncol. 2023 doi: 10.1016/S1470-2045(23)00347-9. https://doi.org/10.1016/S1470-2045(23)00347-9 [DOI] [PubMed] [Google Scholar]
- 10.Desjardins A., Gromeier M., Herndon J.E., 2nd, Beaubier N., Bolognesi D.P., Friedman A.H., Friedman H.S., McSherry F., Muscat A.M., Nair S., et al. Recurrent glioblastoma treated with recombinant poliovirus. N. Engl. J. Med. 2018;379:150–161. doi: 10.1056/NEJMoa1716435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiocca E.A., Aguilar L.K., Bell S.D., Kaur B., Hardcastle J., Cavaliere R., McGregor J., Lo S., Ray-Chaudhuri A., Chakravarti A., et al. Phase IB study of gene-mediated cytotoxic immunotherapy adjuvant to up-front surgery and intensive timing radiation for malignant glioma. J. Clin. Oncol. 2011;29:3611–3619. doi: 10.1200/JCO.2011.35.5222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wheeler L.A., Manzanera A.G., Bell S.D., Cavaliere R., McGregor J.M., Grecula J.C., Newton H.B., Lo S.S., Badie B., Portnow J., et al. Phase II multicenter study of gene-mediated cytotoxic immunotherapy as adjuvant to surgical resection for newly diagnosed malignant glioma. Neuro Oncol. 2016;18:1137–1145. doi: 10.1093/neuonc/now002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Goertsen D., Flytzanis N.C., Goeden N., Chuapoco M.R., Cummins A., Chen Y., Fan Y., Zhang Q., Sharma J., Duan Y., et al. AAV capsid variants with brain-wide transgene expression and decreased liver targeting after intravenous delivery in mouse and marmoset. Nat. Neurosci. 2022;25:106–115. doi: 10.1038/s41593-021-00969-4. [DOI] [PubMed] [Google Scholar]
- 14.Yao Y., Wang J., Liu Y., Qu Y., Wang K., Zhang Y., Chang Y., Yang Z., Wan J., Liu J., et al. Variants of the adeno-associated virus serotype 9 with enhanced penetration of the blood-brain barrier in rodents and primates. Nat. Biomed. Eng. 2022;6:1257–1271. doi: 10.1038/s41551-022-00938-7. [DOI] [PubMed] [Google Scholar]
- 15.Stupp R., Mason W.P., van den Bent M.J., Weller M., Fisher B., Taphoorn M.J.B., Belanger K., Brandes A.A., Marosi C., Bogdahn U., et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 16.Stupp R., Hegi M.E., Mason W.P., van den Bent M.J., Taphoorn M.J.B., Janzer R.C., Ludwin S.K., Allgeier A., Fisher B., Belanger K., et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 17.Melnick K.F., Miller P., Carmichael E., McGrath K., Ghiaseddin A., Tran D.D., Rahman M. The trial effect in patients with glioblastoma: Effect of clinical trial enrollment on overall survival. J. Neurooncol. 2022;159:479–484. doi: 10.1007/s11060-022-04083-8. [DOI] [PubMed] [Google Scholar]
- 18.Grossman S.A., Ye X., Piantadosi S., Desideri S., Nabors L.B., Rosenfeld M., Fisher J., NABTT CNS Consortium Survival of patients with newly diagnosed glioblastoma treated with radiation and temozolomide in research studies in the United States. Clin. Cancer Res. 2010;16:2443–2449. doi: 10.1158/1078-0432.CCR-09-3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stupp R., Taillibert S., Kanner A., Read W., Steinberg D., Lhermitte B., Toms S., Idbaih A., Ahluwalia M.S., Fink K., et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA. 2017;318:2306–2316. doi: 10.1001/jama.2017.18718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weller M., van den Bent M., Tonn J.C., Stupp R., Preusser M., Cohen-Jonathan-Moyal E., Henriksson R., Le Rhun E., Balana C., Chinot O., et al. European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017;18:e315–e329. doi: 10.1016/S1470-2045(17)30194-8. [DOI] [PubMed] [Google Scholar]
- 21.Weller M., Cloughesy T., Perry J.R., Wick W. Standards of care for treatment of recurrent glioblastoma--are we there yet? Neuro Oncol. 2013;15:4–27. doi: 10.1093/neuonc/nos273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilbert M.R., Dignam J.J., Armstrong T.S., Wefel J.S., Blumenthal D.T., Vogelbaum M.A., Colman H., Chakravarti A., Pugh S., Won M., et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014;370:699–708. doi: 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.dos Santos M.A., Pignon J.P., Blanchard P., Lefeuvre D., Levy A., Touat M., Louvel G., Dhermain F., Soria J.C., Deutsch E., Le Teuff G. Systematic review and meta-analysis of phase I/II targeted therapy combined with radiotherapy in patients with glioblastoma multiforme: Quality of report, toxicity, and survival. J. Neurooncol. 2015;123:307–314. doi: 10.1007/s11060-015-1802-5. [DOI] [PubMed] [Google Scholar]
- 24.Faisal S.M., Comba A., Varela M.L., Argento A.E., Brumley E., Abel C., 2nd, Castro M.G., Lowenstein P.R. The complex interactions between the cellular and non-cellular components of the brain tumor microenvironmental landscape and their therapeutic implications. Front. Oncol. 2022;12 doi: 10.3389/fonc.2022.1005069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hossain J.A., Marchini A., Fehse B., Bjerkvig R., Miletic H. Suicide gene therapy for the treatment of high-grade glioma: past lessons, present trends, and future prospects. Neurooncol. Adv. 2020;2:vdaa013. doi: 10.1093/noajnl/vdaa013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishiyama T., Kawamura Y., Kawamoto K., Matsumura H., Yamamoto N., Ito T., Ohyama A., Katsuragi T., Sakai T. Antineoplastic effects in rats of 5-fluorocytosine in combination with cytosine deaminase capsules. Cancer Res. 1985;45:1753–1761. [PubMed] [Google Scholar]
- 27.Miletic H., Fischer Y., Litwak S., Giroglou T., Waerzeggers Y., Winkeler A., Li H., Himmelreich U., Lange C., Stenzel W., et al. Bystander killing of malignant glioma by bone marrow-derived tumor-infiltrating progenitor cells expressing a suicide gene. Mol. Ther. 2007;15:1373–1381. doi: 10.1038/sj.mt.6300155. [DOI] [PubMed] [Google Scholar]
- 28.Miletic H., Fischer Y.H., Giroglou T., Rueger M.A., Winkeler A., Li H., Himmelreich U., Stenzel W., Jacobs A.H., von Laer D. Normal brain cells contribute to the bystander effect in suicide gene therapy of malignant glioma. Clin. Cancer Res. 2007;13:6761–6768. doi: 10.1158/1078-0432.CCR-07-1240. [DOI] [PubMed] [Google Scholar]
- 29.Li S., Tokuyama T., Yamamoto J., Koide M., Yokota N., Namba H. Potent bystander effect in suicide gene therapy using neural stem cells transduced with herpes simplex virus thymidine kinase gene. Oncology. 2005;69:503–508. doi: 10.1159/000091032. [DOI] [PubMed] [Google Scholar]
- 30.Hossain J.A., Latif M.A., Ystaas L.A.R., Ninzima S., Riecken K., Muller A., Azuaje F., Joseph J.V., Talasila K.M., Ghimire J., et al. Long-term treatment with valganciclovir improves lentiviral suicide gene therapy of glioblastoma. Neuro Oncol. 2019;21:890–900. doi: 10.1093/neuonc/noz060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ali S., King G.D., Curtin J.F., Candolfi M., Xiong W., Liu C., Puntel M., Cheng Q., Prieto J., Ribas A., et al. Combined immunostimulation and conditional cytotoxic gene therapy provide long-term survival in a large glioma model. Cancer Res. 2005;65:7194–7204. doi: 10.1158/0008-5472.CAN-04-3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hossain J.A., Riecken K., Miletic H., Fehse B. Cancer suicide gene therapy with TK.007. Methods Mol. Biol. 2019;1895:11–26. doi: 10.1007/978-1-4939-8922-5_2. [DOI] [PubMed] [Google Scholar]
- 33.Moolten F.L., Wells J.M. Curability of tumors bearing herpes thymidine kinase genes transferred by retroviral vectors. J. Natl. Cancer Inst. 1990;82:297–300. doi: 10.1093/jnci/82.4.297. [DOI] [PubMed] [Google Scholar]
- 34.Candolfi M., Yagiz K., Foulad D., Alzadeh G.E., Tesarfreund M., Muhammad A.K.M.G., Puntel M., Kroeger K.M., Liu C., Lee S., et al. Release of HMGB1 in response to proapoptotic glioma killing strategies: efficacy and neurotoxicity. Clin. Cancer Res. 2009;15:4401–4414. doi: 10.1158/1078-0432.CCR-09-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Geller A.I., Breakefield X.O. A defective HSV-1 vector expresses Escherichia coli beta-galactosidase in cultured peripheral neurons. Science. 1988;241:1667–1669. doi: 10.1126/science.241.4873.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rainov N.G., Kramm C.M., Banning U., Riemann D., Holzhausen H.J., Heidecke V., Burger K.J., Burkert W., Körholz D. Immune response induced by retrovirus-mediated HSV-tk/GCV pharmacogene therapy in patients with glioblastoma multiforme. Gene Ther. 2000;7:1853–1858. doi: 10.1038/sj.gt.3301311. [DOI] [PubMed] [Google Scholar]
- 37.Rainov N.G. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum. Gene Ther. 2000;11:2389–2401. doi: 10.1089/104303400750038499. [DOI] [PubMed] [Google Scholar]
- 38.Galea I., Bechmann I., Perry V.H. What is immune privilege (not)? Trends Immunol. 2007;28:12–18. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 39.Lowenstein P.R. Immunology of viral-vector-mediated gene transfer into the brain: An evolutionary and developmental perspective. Trends Immunol. 2002;23:23–30. doi: 10.1016/s1471-4906(01)02063-4. [DOI] [PubMed] [Google Scholar]
- 40.Negi N., Das B.K. CNS: Not an immunoprivilaged site anymore but a virtual secondary lymphoid organ. Int. Rev. Immunol. 2018;37:57–68. doi: 10.1080/08830185.2017.1357719. [DOI] [PubMed] [Google Scholar]
- 41.Engelhardt B., Vajkoczy P., Weller R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017;18:123–131. doi: 10.1038/ni.3666. [DOI] [PubMed] [Google Scholar]
- 42.Louveau A., Plog B.A., Antila S., Alitalo K., Nedergaard M., Kipnis J. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J. Clin. Invest. 2017;127:3210–3219. doi: 10.1172/JCI90603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aspelund A., Antila S., Proulx S.T., Karlsen T.V., Karaman S., Detmar M., Wiig H., Alitalo K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015;212:991–999. doi: 10.1084/jem.20142290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Louveau A., Herz J., Alme M.N., Salvador A.F., Dong M.Q., Viar K.E., Herod S.G., Knopp J., Setliff J.C., Lupi A.L., et al. CNS lymphatic drainage and neuroinflammation are regulated by meningeal lymphatic vasculature. Nat. Neurosci. 2018;21:1380–1391. doi: 10.1038/s41593-018-0227-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Louveau A., Smirnov I., Keyes T.J., Eccles J.D., Rouhani S.J., Peske J.D., Derecki N.C., Castle D., Mandell J.W., Lee K.S., et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523:337–341. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hodi F.S., O'Day S.J., McDermott D.F., Weber R.W., Sosman J.A., Haanen J.B., Gonzalez R., Robert C., Schadendorf D., Hassel J.C., et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gross N.D., Miller D.M., Khushalani N.I., Divi V., Ruiz E.S., Lipson E.J., Meier F., Su Y.B., Swiecicki P.L., Atlas J., et al. Neoadjuvant cemiplimab for stage II to IV cutaneous squamous-cell carcinoma. N. Engl. J. Med. 2022;387:1557–1568. doi: 10.1056/NEJMoa2209813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Motzer R.J., Tannir N.M., McDermott D.F., Arén Frontera O., Melichar B., Choueiri T.K., Plimack E.R., Barthélémy P., Porta C., George S., et al. Nivolumab plus ipilimumab versus sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018;378:1277–1290. doi: 10.1056/NEJMoa1712126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parikh M., Powles T. Immune checkpoint inhibition in advanced bladder and kidney cancer: Responses and further management. Am. Soc. Clin. Oncol. Educ. Book. 2021;41:e182–e189. doi: 10.1200/EDBK_323835. [DOI] [PubMed] [Google Scholar]
- 50.Liau L.M., Ashkan K., Brem S., Campian J.L., Trusheim J.E., Iwamoto F.M., Tran D.D., Ansstas G., Cobbs C.S., Heth J.A., et al. Association of autologous tumor lysate-loaded dendritic cell vaccination with extension of survival among patients with newly diagnosed and recurrent glioblastoma: A phase 3 prospective externally controlled cohort trial. JAMA Oncol. 2023;9:112–121. doi: 10.1001/jamaoncol.2022.5370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sterner R.C., Sterner R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021;11:69. doi: 10.1038/s41408-021-00459-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lin Q., Ba T., Ho J., Chen D., Cheng Y., Wang L., Xu G., Xu L., Zhou Y., Wei Y., et al. First-in-human trial of EphA2-redirected CAR T-cells in patients with recurrent glioblastoma: A preliminary report of three cases at the starting dose. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.694941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Majzner R.G., Theruvath J.L., Nellan A., Heitzeneder S., Cui Y., Mount C.W., Rietberg S.P., Linde M.H., Xu P., Rota C., et al. CAR T cells targeting B7-H3, a pan-cancer antigen, demonstrate potent preclinical activity against pediatric solid tumors and brain tumors. Clin. Cancer Res. 2019;25:2560–2574. doi: 10.1158/1078-0432.CCR-18-0432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Prapa M., Chiavelli C., Golinelli G., Grisendi G., Bestagno M., Di Tinco R., Dall'Ora M., Neri G., Candini O., Spano C., et al. GD2 CAR T cells against human glioblastoma. NPJ Precis. Oncol. 2021;5:93. doi: 10.1038/s41698-021-00233-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Alghamri M.S., Banerjee K., Mujeeb A.A., Mauser A., Taher A., Thalla R., McClellan B.L., Varela M.L., Stamatovic S.M., Martinez-Revollar G., et al. Systemic delivery of an adjuvant CXCR4-CXCL12 signaling inhibitor encapsulated in synthetic protein nanoparticles for glioma immunotherapy. ACS Nano. 2022;16:8729–8750. doi: 10.1021/acsnano.1c07492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shah D., Comba A., Faisal S.M., Kadiyala P., Baker G.J., Alghamri M.S., Doherty R., Zamler D., Nuñez G., Castro M.G., Lowenstein P.R. A novel miR1983-TLR7-IFNbeta circuit licenses NK cells to kill glioma cells, and is under the control of galectin-1. Oncoimmunology. 2021;10 doi: 10.1080/2162402X.2021.1939601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brandes A.A., Carpentier A.F., Kesari S., Sepulveda-Sanchez J.M., Wheeler H.R., Chinot O., Cher L., Steinbach J.P., Capper D., Specenier P., et al. A phase II randomized study of galunisertib monotherapy or galunisertib plus lomustine compared with lomustine monotherapy in patients with recurrent glioblastoma. Neuro Oncol. 2016;18:1146–1156. doi: 10.1093/neuonc/now009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mecca C., Giambanco I., Donato R., Arcuri C. Targeting mTOR in glioblastoma: Rationale and preclinical/clinical evidence. Dis. Markers. 2018;2018 doi: 10.1155/2018/9230479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chinnaiyan P., Won M., Wen P.Y., Rojiani A.M., Werner-Wasik M., Shih H.A., Ashby L.S., Michael Yu H.H., Stieber V.W., Malone S.C., et al. A randomized phase II study of everolimus in combination with chemoradiation in newly diagnosed glioblastoma: Results of NRG Oncology RTOG 0913. Neuro Oncol. 2018;20:666–673. doi: 10.1093/neuonc/nox209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luchman H.A., Stechishin O.D.M., Nguyen S.A., Lun X.Q., Cairncross J.G., Weiss S. Dual mTORC1/2 blockade inhibits glioblastoma brain tumor initiating cells in vitro and in vivo and synergizes with temozolomide to increase orthotopic xenograft survival. Clin. Cancer Res. 2014;20:5756–5767. doi: 10.1158/1078-0432.CCR-13-3389. [DOI] [PubMed] [Google Scholar]
- 61.Saxton R.A., Sabatini D.M. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dumas A.A., Pomella N., Rosser G., Guglielmi L., Vinel C., Millner T.O., Rees J., Aley N., Sheer D., Wei J., et al. Microglia promote glioblastoma via mTOR-mediated immunosuppression of the tumour microenvironment. EMBO J. 2020;39 doi: 10.15252/embj.2019103790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pallet N., Legendre C. Adverse events associated with mTOR inhibitors. Expert Opin. Drug Saf. 2013;12:177–186. doi: 10.1517/14740338.2013.752814. [DOI] [PubMed] [Google Scholar]
- 64.Zhang Z., Fan Q., Luo X., Lou K., Weiss W.A., Shokat K.M. Brain-restricted mTOR inhibition with binary pharmacology. Nature. 2022;609:822–828. doi: 10.1038/s41586-022-05213-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhai L., Bell A., Ladomersky E., Lauing K.L., Bollu L., Nguyen B., Genet M., Kim M., Chen P., Mi X., et al. Tumor cell IDO enhances immune suppression and decreases survival independent of tryptophan metabolism in glioblastoma. Clin. Cancer Res. 2021;27:6514–6528. doi: 10.1158/1078-0432.CCR-21-1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Long G.V., Dummer R., Hamid O., Gajewski T.F., Caglevic C., Dalle S., Arance A., Carlino M.S., Grob J.J., Kim T.M., et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019;20:1083–1097. doi: 10.1016/S1470-2045(19)30274-8. [DOI] [PubMed] [Google Scholar]
- 67.Duan Q., Zhang H., Zheng J., Zhang L. Turning cold into hot: Firing up the tumor microenvironment. Trends Cancer. 2020;6:605–618. doi: 10.1016/j.trecan.2020.02.022. [DOI] [PubMed] [Google Scholar]
- 68.Zhang X., Bai X.C., Chen Z.J. Structures and mechanisms in the cGAS-STING innate immunity pathway. Immunity. 2020;53:43–53. doi: 10.1016/j.immuni.2020.05.013. [DOI] [PubMed] [Google Scholar]
- 69.Berger G., Knelson E.H., Jimenez-Macias J.L., Nowicki M.O., Han S., Panagioti E., Lizotte P.H., Adu-Berchie K., Stafford A., Dimitrakakis N., et al. STING activation promotes robust immune response and NK cell-mediated tumor regression in glioblastoma models. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2111003119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Low J.T., Chandramohan V., Bowie M.L., Brown M.C., Waitkus M.S., Briley A., Stevenson K., Fuller R., Reitman Z.J., Muscat A.M., et al. Epigenetic STING silencing is developmentally conserved in gliomas and can be rescued by methyltransferase inhibition. Cancer Cell. 2022;40:439–440. doi: 10.1016/j.ccell.2022.04.009. [DOI] [PubMed] [Google Scholar]
- 71.Haase S., Banerjee K., Mujeeb A.A., Hartlage C.S., Núñez F.M., Núñez F.J., Alghamri M.S., Kadiyala P., Carney S., Barissi M.N., et al. H3.3-G34 mutations impair DNA repair and promote cGAS/STING-mediated immune responses in pediatric high-grade glioma models. J. Clin. Invest. 2022;132 doi: 10.1172/JCI154229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ravi V.M., Neidert N., Will P., Joseph K., Maier J.P., Kückelhaus J., Vollmer L., Goeldner J.M., Behringer S.P., Scherer F., et al. T-cell dysfunction in the glioblastoma microenvironment is mediated by myeloid cells releasing interleukin-10. Nat. Commun. 2022;13:925. doi: 10.1038/s41467-022-28523-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee A.H., Sun L., Mochizuki A.Y., Reynoso J.G., Orpilla J., Chow F., Kienzler J.C., Everson R.G., Nathanson D.A., Bensinger S.J., et al. Neoadjuvant PD-1 blockade induces T cell and cDC1 activation but fails to overcome the immunosuppressive tumor associated macrophages in recurrent glioblastoma. Nat. Commun. 2021;12:6938. doi: 10.1038/s41467-021-26940-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guo Q., Shen S., Guan G., Zhu C., Zou C., Cao J., Cheng W., Xu X., Yu J., Lin Z., et al. Cancer cell intrinsic TIM-3 induces glioblastoma progression. iScience. 2022;25 doi: 10.1016/j.isci.2022.105329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Giles A.J., Hutchinson M.K.N.D., Sonnemann H.M., Jung J., Fecci P.E., Ratnam N.M., Zhang W., Song H., Bailey R., Davis D., et al. Dexamethasone-induced immunosuppression: mechanisms and implications for immunotherapy. J. Immunother. Cancer. 2018;6:51. doi: 10.1186/s40425-018-0371-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Iorgulescu J.B., Gokhale P.C., Speranza M.C., Eschle B.K., Poitras M.J., Wilkens M.K., Soroko K.M., Chhoeu C., Knott A., Gao Y., et al. Concurrent dexamethasone limits the clinical benefit of immune checkpoint blockade in glioblastoma. Clin. Cancer Res. 2021;27:276–287. doi: 10.1158/1078-0432.CCR-20-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Swildens K.X., Sillevis Smitt P.A.E., van den Bent M.J., French P.J., Geurts M. The effect of dexamethasone on the microenvironment and efficacy of checkpoint inhibitors in glioblastoma: a systematic review. Neurooncol. Adv. 2022;4:vdac087. doi: 10.1093/noajnl/vdac087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koch M.S., Zdioruk M., Nowicki M.O., Griffith A.M., Aguilar E., Aguilar L.K., Guzik B.W., Barone F., Tak P.P., Tabatabai G., et al. Systemic high-dose dexamethasone treatment may modulate the efficacy of intratumoral viral oncolytic immunotherapy in glioblastoma models. J. Immunother. Cancer. 2022;10 doi: 10.1136/jitc-2021-003368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bouffet E., Larouche V., Campbell B.B., Merico D., de Borja R., Aronson M., Durno C., Krueger J., Cabric V., Ramaswamy V., et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J. Clin. Oncol. 2016;34:2206–2211. doi: 10.1200/JCO.2016.66.6552. [DOI] [PubMed] [Google Scholar]
- 80.Arrieta V.A., Dmello C., McGrail D.J., Brat D.J., Lee-Chang C., Heimberger A.B., Chand D., Stupp R., Sonabend A.M. Immune checkpoint blockade in glioblastoma: From tumor heterogeneity to personalized treatment. J. Clin. Invest. 2023;133 doi: 10.1172/JCI163447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Omuro A., Brandes A.A., Carpentier A.F., Idbaih A., Reardon D.A., Cloughesy T., Sumrall A., Baehring J., van den Bent M., Bähr O., et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro Oncol. 2023;25:123–134. doi: 10.1093/neuonc/noac099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cloughesy T.F., Petrecca K., Walbert T., Butowski N., Salacz M., Perry J., Damek D., Bota D., Bettegowda C., Zhu J.J., et al. Effect of vocimagene amiretrorepvec in combination with flucytosine vs standard of care on survival following tumor resection in patients with recurrent high-grade glioma: A randomized clinical trial. JAMA Oncol. 2020;6:1939–1946. doi: 10.1001/jamaoncol.2020.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ogino H., Taylor J.W., Nejo T., Gibson D., Watchmaker P.B., Okada K., Saijo A., Tedesco M.R., Shai A., Wong C.M., et al. Randomized trial of neoadjuvant vaccination with tumor-cell lysate induces T cell response in low-grade gliomas. J. Clin. Invest. 2022;132 doi: 10.1172/JCI151239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mineharu Y., King G.D., Muhammad A.K.M.G., Bannykh S., Kroeger K.M., Liu C., Lowenstein P.R., Castro M.G. Engineering the brain tumor microenvironment enhances the efficacy of dendritic cell vaccination: Implications for clinical trial design. Clin. Cancer Res. 2011;17:4705–4718. doi: 10.1158/1078-0432.CCR-11-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Reardon D.A., Brandes A.A., Omuro A., Mulholland P., Lim M., Wick A., Baehring J., Ahluwalia M.S., Roth P., Bähr O., et al. Effect of nivolumab vs bevacizumab in patients with recurrent glioblastoma: The CheckMate 143 phase 3 randomized clinical trial. JAMA Oncol. 2020;6:1003–1010. doi: 10.1001/jamaoncol.2020.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wei J., Chen P., Gupta P., Ott M., Zamler D., Kassab C., Bhat K.P., Curran M.A., de Groot J.F., Heimberger A.B. Immune biology of glioma-associated macrophages and microglia: functional and therapeutic implications. Neuro Oncol. 2020;22:180–194. doi: 10.1093/neuonc/noz212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.de Groot J., Penas-Prado M., Alfaro-Munoz K., Hunter K., Pei B.L., O'Brien B., Weathers S.P., Loghin M., Kamiya Matsouka C., Yung W.K.A., et al. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro Oncol. 2020;22:539–549. doi: 10.1093/neuonc/noz185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Goswami S., Walle T., Cornish A.E., Basu S., Anandhan S., Fernandez I., Vence L., Blando J., Zhao H., Yadav S.S., et al. Immune profiling of human tumors identifies CD73 as a combinatorial target in glioblastoma. Nat. Med. 2020;26:39–46. doi: 10.1038/s41591-019-0694-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kamran N., Kadiyala P., Saxena M., Candolfi M., Li Y., Moreno-Ayala M.A., Raja N., Shah D., Lowenstein P.R., Castro M.G. Immunosuppressive myeloid cells' blockade in the glioma microenvironment enhances the efficacy of immune-stimulatory gene therapy. Mol. Ther. 2017;25:232–248. doi: 10.1016/j.ymthe.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zeng J., See A.P., Phallen J., Jackson C.M., Belcaid Z., Ruzevick J., Durham N., Meyer C., Harris T.J., Albesiano E., et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013;86:343–349. doi: 10.1016/j.ijrobp.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Omuro A., Vlahovic G., Lim M., Sahebjam S., Baehring J., Cloughesy T., Voloschin A., Ramkissoon S.H., Ligon K.L., Latek R., et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018;20:674–686. doi: 10.1093/neuonc/nox208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cordell E.C., Alghamri M.S., Castro M.G., Gutmann D.H. T lymphocytes as dynamic regulators of glioma pathobiology. Neuro Oncol. 2022;24:1647–1657. doi: 10.1093/neuonc/noac055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Arrieta V.A., Chen A.X., Kane J.R., Kang S.J., Kassab C., Dmello C., Zhao J., Burdett K.B., Upadhyayula P.S., Lee-Chang C., et al. ERK1/2 phosphorylation predicts survival following anti-PD-1 immunotherapy in recurrent glioblastoma. Nat. Cancer. 2021;2:1372–1386. doi: 10.1038/s43018-021-00260-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Andtbacka R.H.I., Kaufman H.L., Collichio F., Amatruda T., Senzer N., Chesney J., Delman K.A., Spitler L.E., Puzanov I., Agarwala S.S., et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015;33:2780–2788. doi: 10.1200/JCO.2014.58.3377. [DOI] [PubMed] [Google Scholar]
- 95.Boorjian S.A., Alemozaffar M., Konety B.R., Shore N.D., Gomella L.G., Kamat A.M., Bivalacqua T.J., Montgomery J.S., Lerner S.P., Busby J.E., et al. Intravesical nadofaragene firadenovec gene therapy for BCG-unresponsive non-muscle-invasive bladder cancer: A single-arm, open-label, repeat-dose clinical trial. Lancet Oncol. 2021;22:107–117. doi: 10.1016/S1470-2045(20)30540-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cripe T.P. Cancer gene therapy bears fruit. Mol. Ther. 2023;31:303. doi: 10.1016/j.ymthe.2023.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Favero F., McGranahan N., Salm M., Birkbak N.J., Sanborn J.Z., Benz S.C., Becq J., Peden J.F., Kingsbury Z., Grocok R.J., et al. Glioblastoma adaptation traced through decline of an IDH1 clonal driver and macro-evolution of a double-minute chromosome. Ann. Oncol. 2015;26:880–887. doi: 10.1093/annonc/mdv127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McGranahan N., Favero F., de Bruin E.C., Birkbak N.J., Szallasi Z., Swanton C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015;7:283ra54. doi: 10.1126/scitranslmed.aaa1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.McGranahan N., Swanton C. Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell. 2015;27:15–26. doi: 10.1016/j.ccell.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 100.Gállego Pérez-Larraya J., Garcia-Moure M., Labiano S., Patiño-García A., Dobbs J., Gonzalez-Huarriz M., Zalacain M., Marrodan L., Martinez-Velez N., Puigdelloses M., et al. Oncolytic DNX-2401 virus for pediatric diffuse intrinsic pontine glioma. N. Engl. J. Med. 2022;386:2471–2481. doi: 10.1056/NEJMoa2202028. [DOI] [PubMed] [Google Scholar]
- 101.Friedman G.K., Johnston J.M., Bag A.K., Bernstock J.D., Li R., Aban I., Kachurak K., Nan L., Kang K.D., Totsch S., et al. Oncolytic HSV-1 G207 immunovirotherapy for pediatric high-grade gliomas. N. Engl. J. Med. 2021;384:1613–1622. doi: 10.1056/NEJMoa2024947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tian L., Xu B., Chen Y., Li Z., Wang J., Zhang J., Ma R., Cao S., Hu W., Chiocca E.A., et al. Specific targeting of glioblastoma with an oncolytic virus expressing a cetuximab-CCL5 fusion protein via innate and adaptive immunity. Nat. Cancer. 2022;3:1318–1335. doi: 10.1038/s43018-022-00448-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Curtin J.F., Liu N., Candolfi M., Xiong W., Assi H., Yagiz K., Edwards M.R., Michelsen K.S., Kroeger K.M., Liu C., et al. HMGB1 mediates endogenous TLR2 activation and brain tumor regression. PLoS Med. 2009;6:e10. doi: 10.1371/journal.pmed.1000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ma R., Li Z., Chiocca E.A., Caligiuri M.A., Yu J. The emerging field of oncolytic virus-based cancer immunotherapy. Trends Cancer. 2023;9:122–139. doi: 10.1016/j.trecan.2022.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Alghamri M.S., McClellan B.L., Avvari R.P., Thalla R., Carney S., Hartlage C.S., Haase S., Ventosa M., Taher A., Kamran N., et al. G-CSF secreted by mutant IDH1 glioma stem cells abolishes myeloid cell immunosuppression and enhances the efficacy of immunotherapy. Sci. Adv. 2021;7:eabh3243. doi: 10.1126/sciadv.abh3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Banerjee K., Núñez F.J., Haase S., McClellan B.L., Faisal S.M., Carney S.V., Yu J., Alghamri M.S., Asad A.S., Candia A.J.N., et al. Current approaches for glioma gene therapy and virotherapy. Front. Mol. Neurosci. 2021;14 doi: 10.3389/fnmol.2021.621831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Faisal S.M., Mendez F.M., Nunez F., Castro M.G., Lowenstein P.R. Immune-stimulatory (TK/Flt3L) gene therapy opens the door to a promising new treatment strategy against brainstem gliomas. Oncotarget. 2020;11:4607–4612. doi: 10.18632/oncotarget.27834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chiocca E.A., Nassiri F., Wang J., Peruzzi P., Zadeh G. Viral and other therapies for recurrent glioblastoma: Is a 24-month durable response unusual? Neuro Oncol. 2019;21:14–25. doi: 10.1093/neuonc/noy170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Westphal M., Ylä-Herttuala S., Martin J., Warnke P., Menei P., Eckland D., Kinley J., Kay R., Ram Z., ASPECT Study Group Adenovirus-mediated gene therapy with sitimagene ceradenovec followed by intravenous ganciclovir for patients with operable high-grade glioma (ASPECT): A randomised, open-label, phase 3 trial. Lancet Oncol. 2013;14:823–833. doi: 10.1016/S1470-2045(13)70274-2. [DOI] [PubMed] [Google Scholar]
- 110.Chiocca E.A., Yu J.S., Lukas R.V., Solomon I.H., Ligon K.L., Nakashima H., Triggs D.A., Reardon D.A., Wen P., Stopa B.M., et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aaw5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chiocca E.A., Gelb A.B., Chen C.C., Rao G., Reardon D.A., Wen P.Y., Bi W.L., Peruzzi P., Amidei C., Triggs D., et al. Combined immunotherapy with controlled interleukin-12 gene therapy and immune checkpoint blockade in recurrent glioblastoma: An open-label, multi-institutional phase I trial. Neuro Oncol. 2022;24:951–963. doi: 10.1093/neuonc/noab271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Groeneveldt C., van den Ende J., van Montfoort N. Preexisting immunity: Barrier or bridge to effective oncolytic virus therapy? Cytokine Growth Factor Rev. 2023;70:1–12. doi: 10.1016/j.cytogfr.2023.01.002. [DOI] [PubMed] [Google Scholar]
- 113.Lang F.F., Conrad C., Gomez-Manzano C., Yung W.K.A., Sawaya R., Weinberg J.S., Prabhu S.S., Rao G., Fuller G.N., Aldape K.D., et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: Replication and immunotherapeutic effects in recurrent malignant glioma. J. Clin. Oncol. 2018;36:1419–1427. doi: 10.1200/JCO.2017.75.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Candolfi M., King G.D., Yagiz K., Curtin J.F., Mineharu Y., Muhammad A.K.M.G., Foulad D., Kroeger K.M., Barnett N., Josien R., et al. Plasmacytoid dendritic cells in the tumor microenvironment: Immune targets for glioma therapeutics. Neoplasia. 2012;14:757–770. doi: 10.1593/neo.12794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Curtin J.F., King G.D., Barcia C., Liu C., Hubert F.X., Guillonneau C., Josien R., Anegon I., Lowenstein P.R., Castro M.G. Fms-like tyrosine kinase 3 ligand recruits plasmacytoid dendritic cells to the brain. J. Immunol. 2006;176:3566–3577. doi: 10.4049/jimmunol.176.6.3566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Nassiri F., Patil V., Yefet L.S., Singh O., Liu J., Dang R.M.A., Yamaguchi T.N., Daras M., Cloughesy T.F., Colman H., et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: A phase 1/2 trial. Nat. Med. 2023;29:1370–1378. doi: 10.1038/s41591-023-02347-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Datta M., Chatterjee S., Perez E.M., Gritsch S., Roberge S., Duquette M., Chen I.X., Naxerova K., Kumar A.S., Ghosh M., et al. Losartan controls immune checkpoint blocker-induced edema and improves survival in glioblastoma mouse models. Proc. Natl. Acad. Sci. USA. 2023;120 doi: 10.1073/pnas.2219199120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Warnick R.E. Treatment of adverse radiation effects with Boswellia serrata after failure of pentoxifylline and vitamin E: Illustrative cases. J. Neurosurg. Case Lessons. 2023;5 doi: 10.3171/CASE22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Min J., Perera L., Krahn J.M., Jewell C.M., Moon A.F., Cidlowski J.A., Pedersen L.C. Probing dominant negative behavior of glucocorticoid receptor beta through a hybrid structural and biochemical approach. Mol. Cell. Biol. 2018;38 doi: 10.1128/MCB.00453-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cantley A.M., Welsch M., Ambesi-Impiombato A., Sanchez-Martin M., Kim M.Y., Bauer A., Ferrando A., Stockwell B.R. Small molecule that reverses dexamethasone resistance in T-cell acute lymphoblastic leukemia (T-ALL) ACS Med. Chem. Lett. 2014;5:754–759. doi: 10.1021/ml500044g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Olivas-Aguirre M., Torres-López L., Pottosin I., Dobrovinskaya O. Overcoming glucocorticoid resistance in acute lymphoblastic leukemia: Repurposed drugs can improve the protocol. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.617937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Brown C.E., Rodriguez A., Palmer J., Ostberg J.R., Naranjo A., Wagner J.R., Aguilar B., Starr R., Weng L., Synold T.W., et al. Off-the-shelf, steroid-resistant, IL13Ralpha2-specific CAR T cells for treatment of glioblastoma. Neuro Oncol. 2022;24:1318–1330. doi: 10.1093/neuonc/noac024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rafiq S., Hackett C.S., Brentjens R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020;17:147–167. doi: 10.1038/s41571-019-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Carvalho L.A., Teng J., Fleming R.L., Tabet E.I., Zinter M., de Melo Reis R.A., Tannous B.A. Olfactory ensheathing cells: A Trojan Horse for glioma gene therapy. J. Natl. Cancer Inst. 2019;111:283–291. doi: 10.1093/jnci/djy138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Li G., Bonamici N., Dey M., Lesniak M.S., Balyasnikova I.V. Intranasal delivery of stem cell-based therapies for the treatment of brain malignancies. Expert Opin. Drug Deliv. 2018;15:163–172. doi: 10.1080/17425247.2018.1378642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cueto F.J., Sancho D. The Flt3L/Flt3 axis in dendritic cell biology and cancer immunotherapy. Cancers (Basel) 2021;13 doi: 10.3390/cancers13071525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Ali S., Curtin J.F., Zirger J.M., Xiong W., King G.D., Barcia C., Liu C., Puntel M., Goverdhana S., Lowenstein P.R., Castro M.G. Inflammatory and anti-glioma effects of an adenovirus expressing human soluble Fms-like tyrosine kinase 3 ligand (hsFlt3L): Treatment with hsFlt3L inhibits intracranial glioma progression. Mol. Ther. 2004;10:1071–1084. doi: 10.1016/j.ymthe.2004.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Candolfi M., Yagiz K., Wibowo M., Ahlzadeh G.E., Puntel M., Ghiasi H., Kamran N., Paran C., Lowenstein P.R., Castro M.G. Temozolomide does not impair gene therapy-mediated antitumor immunity in syngeneic brain tumor models. Clin. Cancer Res. 2014;20:1555–1565. doi: 10.1158/1078-0432.CCR-13-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.King G.D., Kroeger K.M., Bresee C.J., Candolfi M., Liu C., Manalo C.M., Muhammad A.G., Pechnick R.N., Lowenstein P.R., Castro M.G. Flt3L in combination with HSV1-TK-mediated gene therapy reverses brain tumor-induced behavioral deficits. Mol. Ther. 2008;16:682–690. doi: 10.1038/mt.2008.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ghulam Muhammad A.K.M., Candolfi M., King G.D., Yagiz K., Foulad D., Mineharu Y., Kroeger K.M., Treuer K.A., Nichols W.S., Sanderson N.S., et al. Antiglioma immunological memory in response to conditional cytotoxic/immune-stimulatory gene therapy: humoral and cellular immunity lead to tumor regression. Clin. Cancer Res. 2009;15:6113–6127. doi: 10.1158/1078-0432.CCR-09-1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Larocque D., Sanderson N.S.R., Bergeron J., Curtin J.F., Girton J., Wibowo M., Bondale N., Kroeger K.M., Yang J., Lacayo L.M., et al. Exogenous fms-like tyrosine kinase 3 ligand overrides brain immune privilege and facilitates recognition of a neo-antigen without causing autoimmune neuropathology. Proc. Natl. Acad. Sci. USA. 2010;107:14443–14448. doi: 10.1073/pnas.0913496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.King G.D., Muhammad A.K.M.G., Larocque D., Kelson K.R., Xiong W., Liu C., Sanderson N.S.R., Kroeger K.M., Castro M.G., Lowenstein P.R. Combined Flt3L/TK gene therapy induces immunological surveillance which mediates an immune response against a surrogate brain tumor neoantigen. Mol. Ther. 2011;19:1793–1801. doi: 10.1038/mt.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Candolfi M., Curtin J.F., Yagiz K., Assi H., Wibowo M.K., Alzadeh G.E., Foulad D., Muhammad A.K.M.G., Salehi S., Keech N., et al. B cells are critical to T-cell-mediated antitumor immunity induced by a combined immune-stimulatory/conditionally cytotoxic therapy for glioblastoma. Neoplasia. 2011;13:947–960. doi: 10.1593/neo.11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gielen P.R., Schulte B.M., Kers-Rebel E.D., Verrijp K., Petersen-Baltussen H.M.J.M., ter Laan M., Wesseling P., Adema G.J. Increase in both CD14-positive and CD15-positive myeloid-derived suppressor cell subpopulations in the blood of patients with glioma but predominance of CD15-positive myeloid-derived suppressor cells in glioma tissue. J. Neuropathol. Exp. Neurol. 2015;74:390–400. doi: 10.1097/NEN.0000000000000183. [DOI] [PubMed] [Google Scholar]
- 135.Hardcastle J., Mills L., Malo C.S., Jin F., Kurokawa C., Geekiyanage H., Schroeder M., Sarkaria J., Johnson A.J., Galanis E. Immunovirotherapy with measles virus strains in combination with anti-PD-1 antibody blockade enhances antitumor activity in glioblastoma treatment. Neuro Oncol. 2017;19:493–502. doi: 10.1093/neuonc/now179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chuntova P., Chow F., Watchmaker P.B., Galvez M., Heimberger A.B., Newell E.W., Diaz A., DePinho R.A., Li M.O., Wherry E.J., et al. Unique challenges for glioblastoma immunotherapy-discussions across neuro-oncology and non-neuro-oncology experts in cancer immunology. Meeting Report from the 2019 SNO Immuno-Oncology Think Tank. Neuro Oncol. 2021;23:356–375. doi: 10.1093/neuonc/noaa277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nduom E.K., Weller M., Heimberger A.B. Immunosuppressive mechanisms in glioblastoma. Neuro Oncol. 2015;17:vii9–vii14. doi: 10.1093/neuonc/nov151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Woroniecka K.I., Rhodin K.E., Chongsathidkiet P., Keith K.A., Fecci P.E. T-cell dysfunction in glioblastoma: Applying a new framework. Clin. Cancer Res. 2018;24:3792–3802. doi: 10.1158/1078-0432.CCR-18-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Collier J.L., Weiss S.A., Pauken K.E., Sen D.R., Sharpe A.H. Not-so-opposite ends of the spectrum: CD8(+) T cell dysfunction across chronic infection, cancer and autoimmunity. Nat. Immunol. 2021;22:809–819. doi: 10.1038/s41590-021-00949-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Mirzaei R., Sarkar S., Yong V.W. T cell exhaustion in glioblastoma: Intricacies of immune checkpoints. Trends Immunol. 2017;38:104–115. doi: 10.1016/j.it.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 141.Farhood B., Najafi M., Mortezaee K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019;234:8509–8521. doi: 10.1002/jcp.27782. [DOI] [PubMed] [Google Scholar]
- 142.Shekarian T., Zinner C.P., Bartoszek E.M., Duchemin W., Wachnowicz A.T., Hogan S., Etter M.M., Flammer J., Paganetti C., Martins T.A., et al. Immunotherapy of glioblastoma explants induces interferon-gamma responses and spatial immune cell rearrangements in tumor center, but not periphery. Sci. Adv. 2022;8 doi: 10.1126/sciadv.abn9440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Comba A., Faisal S.M., Dunn P.J., Argento A.E., Hollon T.C., Al-Holou W.N., Varela M.L., Zamler D.B., Quass G.L., Apostolides P.F., et al. Spatiotemporal analysis of glioma heterogeneity reveals COL1A1 as an actionable target to disrupt tumor progression. Nat. Commun. 2022;13:3606. doi: 10.1038/s41467-022-31340-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mosaheb M.M., Dobrikova E.Y., Brown M.C., Yang Y., Cable J., Okada H., Nair S.K., Bigner D.D., Ashley D.M., Gromeier M. Genetically stable poliovirus vectors activate dendritic cells and prime antitumor CD8 T cell immunity. Nat. Commun. 2020;11:524. doi: 10.1038/s41467-019-13939-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Núñez F.J., Mendez F.M., Kadiyala P., Alghamri M.S., Savelieff M.G., Garcia-Fabiani M.B., Haase S., Koschmann C., Calinescu A.A., Kamran N., et al. IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aaq1427. [DOI] [PMC free article] [PubMed] [Google Scholar]