

Abstract
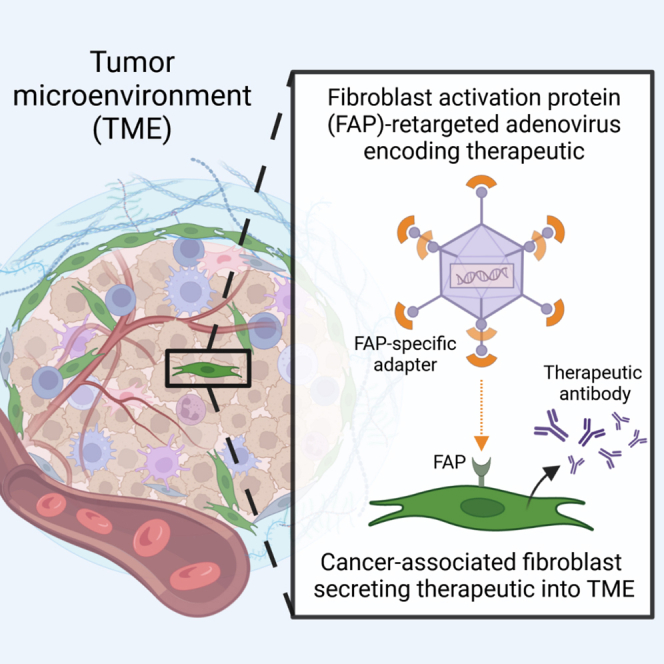
Fibroblast activation protein (FAP) is a cell surface serine protease that is highly expressed on reactive stromal fibroblasts, such as cancer-associated fibroblasts (CAFs), and generally absent in healthy adult tissues. FAP expression in the tumor stroma has been detected in more than 90% of all carcinomas, rendering CAFs excellent target cells for a tumor site-specific adenoviral delivery of cancer therapeutics. Here, we present a tropism-modified human adenovirus 5 (Ad5) vector that targets FAP through trivalent, designed ankyrin repeat protein-based retargeting adapters. We describe the development and validation of these adapters via cell-based screening assays and demonstrate adapter-mediated Ad5 retargeting to FAP+ fibroblasts in vitro and in vivo. We further show efficient in vivo delivery and in situ production of a therapeutic payload by CAFs in the tumor microenvironment (TME), resulting in attenuated tumor growth. We thus propose using our FAP-Ad5 vector to convert CAFs into a “biofactory,” secreting encoded cancer therapeutics into the TME to enable a safe and effective cancer treatment.
Keywords: adenovirus vectors, cell delivery, tumor microenvironment, cancer—gene therapy, cancer—targeted therapy
Graphical abstract
Plückthun and colleagues present a tropism-modified adenoviral vector with specificity for FAP on cancer-associated fibroblasts, enabling a targeted delivery of anti-cancer therapeutics, which can be produced in situ in the tumor microenvironment. An in situ drug production may help reduce off-site effects, thereby preventing systemic toxicities and increasing local therapeutic efficacy.
Introduction
Over the last few decades, cancer therapy has benefited substantially from immunotherapeutic approaches involving monoclonal antibodies (mAbs) and recombinant cytokines with anti-tumor activity. However, systemic application of such agents is often still restricted due to severe dose-limiting toxicities as well as off-target effects, both hampering therapeutic efficacy and safety.1,2 One promising strategy to overcome these limitations is to deliver the genetic information for the in situ production of the effector proteins utilizing suitable delivery systems such as viral vectors. One of the most commonly used viral vector is derived from human adenovirus serotype 5 (Ad5), a non-enveloped double-stranded DNA virus consisting of three major capsid proteins: the hexon, penton, and fiber.3,4 In comparison with other viral vectors, mainly derived from lentivirus or adeno-associated virus (AAV), Ad5-based vectors bring the following superior characteristics for clinical use: (1) they do not integrate into the host cell genome and are therefore safe, (2) they efficiently transduce dividing and non-dividing cells, and (3) they have a large packaging capacity of up to 37 kilobases (kb).5 The large packaging capacity allows for a delivery of multiple payload genes and renders adenoviral vectors very attractive for combination cancer immunotherapy.
Despite all favorable features, tissue-specific delivery of Ad5-based vectors is still compromised in vivo. One hurdle is the strong liver tropism of Ad5, especially upon systemic administration, mainly resulting from interactions of the hexon’s hypervariable regions (HVRs) with host molecules, causing vector sequestration by liver-resident cells.6,7,8 Another limitation of Ad5 vectors is represented by the virion’s natural cell specificity and difficulties in efficiently redirecting the vector to the desired tissue. To overcome these challenges, binding to cellular receptors that are naturally involved in viral cell entry needs to be prevented, while introducing specificity for cell surface molecules located on the cell type of choice. Cell entry of Ad5 occurs in sequential steps: first, the knob domain of the fiber protein binds to its high affinity, primary attachment receptor termed coxsackievirus and adenovirus receptor (CAR).9,10 Then, an arginine-glycine-aspartate motif in the penton base binds to integrins on the cell surface, which initiates the cellular virus uptake by clathrin-mediated endocytosis.11,12
Previous retargeting efforts have aimed at modifying Ad5 interactions through various manipulations of the viral capsid, including genetic mutations, chemical modifications, and genetic fusions of targeting peptides mainly to the fiber protein.13,14,15,16,17 However, these attempts have been accompanied with reduced transduction efficiency and insufficient vector targeting. To address these drawbacks, we have previously developed a generic, protein-based de- and retargeting platform for Ad5 vectors consisting of two parts: (1) a hexon-binding, single-chain variable fragment (scFv)-based trimeric shield that protects the vector from undesired host factor interactions accounting for its in vivo liver tropism.18 In addition, this shield protects the vector from immune clearance, thereby helping to overcome yet another hurdle, namely the widespread pre-existing humoral immunity against Ad5 in the human population.19,20 (2) A bispecific, designed ankyrin repeat protein (DARPin)-based modular adapter binding trivalently to the fiber knob, thereby preventing CAR interactions and thus reducing CAR-mediated cell transduction, while allowing to introduce a defined cell specificity by incorporating appropriate targeting moieties.21,22,23 Importantly, this bispecific adapter does not require genetic fusion to the fiber protein and can hence be applied to any Ad5-derived vector, including high-capacity Ad5 vectors.
By applying DARPin adapters specific for tumor cell markers, such as human epidermal growth factor receptor 2 (HER2), to a shielded HVR7-mutated Ad5 vector, we recently demonstrated efficient vector detargeting from the liver and other organs, as well as successful vector retargeting to tumor cells in vivo.18 Following the validation of our shielded and retargeted adenoviral (SHREAD) delivery platform, we aim to expand our retargeting system to stromal cells in the tumor microenvironment (TME) to enable alternative vector targeting and tumor treatment strategies. One of the most abundant stromal cell types in the TME are cancer-associated fibroblasts (CAFs), which evolve alongside cancer cells during tumor development and progression.24,25 Importantly, CAFs express the cell surface glycoprotein fibroblast activation protein α (FAP) that is selectively present in reactive stromal fibroblasts and generally absent in healthy adult tissues.26,27 More importantly, FAP on CAFs was found to be overexpressed in more than 90% of all carcinomas, including lung, colorectal, prostate, and breast cancer,28 and was therefore established as a promising target for tumor therapy, imaging, and diagnosis.29,30,31,32,33,34,35,36,37,38,39,40,41,42 These characteristics render CAFs an attractive cell type for adenoviral vector retargeting via FAP, to then utilize CAFs as a “biofactory” for the local production and secretion of cancer therapeutics in the TME.
Here, we present a cell-based screening approach following ribosome display selections to generate FAP-specific DARPin adapters that enable retargeting of Ad5-derived vectors to fibroblasts via FAP. We describe the characterization of the retargeting adapters, and then provide in vitro and in vivo data demonstrating adapter-mediated Ad5 retargeting to FAP+ fibroblasts. Furthermore, we show efficient in vivo delivery of a therapeutic payload to FAP+ fibroblasts in the TME resulting in reduced tumor growth. These data prove feasibility of our concept: to deliver genes encoding biomolecules with anti-tumor activity through a FAP-retargeted Ad5 to CAFs in the tumor stroma.
Results
Ribosome display selection of DARPins against hFAP
FAP is a 170 kDa type II transmembrane serine protease with a large C-terminal extracellular domain containing the enzyme’s active site.43 To redirect our Ad5 vector to CAFs via FAP, we built on our previously developed modular adapter consisting of (1) a retargeting DARPin mediating cell specificity, (2) the fiber knob-binding DARPin 1D3, and (3) the trimerizing protein SHP derived from lambdoid phage 21 (Figure 1A). To generate the retargeting DARPin with specificity for FAP, recombinant human FAP (hFAP) was used and ribosome display selections were essentially performed as described before,44 but using a high-throughput set-up (see materials and methods). A total of four selection rounds yielded 380 DARPin candidates, which were subsequently analyzed for hFAP binding. Notably, these candidates contained mainly two (N2C) or three (N3C) internal repeats with randomized surface flanked by capping repeats.
Figure 1.

Generation and validation of DARPin-based hFAP-specific adenoviral retargeting adapters
(A) Schematic representation of a bispecific trimeric DARPin adapter for adenoviral retargeting. The retargeting DARPin (orange) with specificity for a selected cell surface molecule (e.g., FAP) is fused via a long flexible linker to the knob-binding DARPin 1D3 (green) that in turn is fused via a short linker to the trimerizing protein SHP from lambdoid phage (yellow). The bispecific trimeric DARPin adapter forms a highly stable clamp around the fiber knob (red) to block natural cellular interactions and redirect adenoviral tropism to selected cells (e.g., FAP+ cells). (B) Flow cytometry analysis of hFAP expression of the parental HT1080 and HT1080hFAP cell line upon hFAP antibody staining. (C) Cell-based adapter binding assay on target and non-target cells. The purified “Top 9” adapters constructed with the selected hFAP-specific DARPins were titrated on hFAP+ HT1080hFAP and hFAP– HT1080 cells in the concentration range of 0.1–100 nM. Binding was detected via flow cytometry by staining of the His-tagged adapter, and specific binding signals were determined as ΔMFI = MFI (HT1080hFAP cells) − MFI (HT1080 cells). The non-binding control adapter E3_5 was applied as a negative binding control. Bars represent specific binding signals of single point measurements. Representative data of two independent experiments are shown. MFI, mean fluorescent intensity. (D) Transduction of target and non-target cells by hFAP adapter-retargeted Ad5. Recombinant Ad5 encoding iRFP670 was pre-incubated with the “Top 9” hFAP adapters (colored filled bars) or the E3_5 blocking adapter (black empty bar) and tested for transduction of hFAP+ HT1080hFAP and hFAP– HT1080 cells in comparison with the untargeted Ad5 (black filled bar) at a multiplicity of infection (MOI) of 20 plaque-forming units (PFU)/cell. Transduction was assessed from cellular expression of iRFP670 detected by flow cytometry. Dashed lines indicate cut-off levels above which functional and hFAP-specific adapters were identified. Bars represent mean transduction level of two biological replicates ± standard deviation (SD). Statistics: unpaired t test; ∗p < 0.05, ∗∗p < 0.005; p values are indicated for each sample with respect to the untargeted Ad5 for HT1080hFAP cells or with respect to the E3_5-Ad5 for HT1080 cells. Representative data of three independent experiments are shown. (E) Flow cytometry analysis for CAR expression of the HT1080 and HT1080hFAP cell line in comparison with the positive control HeLa cell line upon CAR antibody staining.
To allow a high-throughput binding analysis, we established a flow cytometry-based cell-binding assay using suitable target and non-target cell lines. The parental HT1080 fibrosarcoma cell line (presenting no endogenous hFAP) and a stably transfected clone with high expression of hFAP, termed “HT1080hFAP cell line,”39 were used (Figure 1B). We screened all 380 DARPins for hFAP binding on cells, using crude Escherichia coli extracts, and identified 91 putative binders, testing first the target cell line only (Figure S1A). Then, this subpopulation was assessed for binding on both the target and non-target cell lines, revealing 25 specific binders as determined by the ratio of specific and unspecific binding signal, the latter set by the unselected, non-binding DARPin E3_545 (Figure S1B). Sequencing of the 25 specific binders identified 20 unique binders with high diversity, of which 11 were N3C and 9 were N2C proteins.
We expressed the selected 20 binders in small-scale E. coli cultures and purified them using immobilized metal ion affinity chromatography (IMAC). We confirmed the correct molecular mass via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (Figure S1C), and applied analytical size-exclusion chromatography (SEC) to select monodisperse proteins (Figure S1D). In addition, we re-tested the purified proteins for hFAP binding on target and non-target cells, leading to the selection of 10 well-behaved, hFAP-specific DARPins (Figure S1E). Among these 10 DARPins, particularly candidate no. 6 yielded very high binding signals even at subnanomolar concentrations, thereby outperforming all other selected DARPins. To further investigate binding to hFAP, we measured the binding kinetics by surface plasmon resonance for this DARPin and determined a KD value of 163 pM (Figure S2).
Generation and validation of DARPin-based hFAP adapters
To generate retargeting adapters for an hFAP-mediated cell transduction by Ad5, we used the selected hFAP-specific DARPins and incorporated them as retargeting module into our previously developed Ad5 adapter.22 Protein analysis via SDS-PAGE and analytical SEC of the obtained adapters upon small-scale expression in E. coli and IMAC purification resulted in the selection of nine adapters. We subsequently tested these adapters for hFAP binding on (hFAP+) HT1080hFAP and (hFAP−) HT1080 cells. Our cell-based binding analysis showed specificity for all nine adapters, and found different adapter binding characteristics upon titration in a concentration range of 0.1–100 nM (Figure 1C), confirming our previous data obtained with the single DARPins.
We hypothesized that not only the binding affinity but also the binding epitope, and/or other factors, play a role in the internalization process of the adenovirus into the cell. Therefore, we decided to test all of the selected adapters for their capability to promote an hFAP-dependent cell transduction.
To this end, we made use of both the HT1080hFAP and HT1080 cell lines and performed cell transduction experiments with hFAP adapter-retargeted Ad5 (further on termed “hFAP-Ad5”) in comparison with the untargeted Ad5. In addition, we included the non-targeting control adapter E3_5 with the Ad5 (further on termed “E3_5-Ad5”), which should block the fiber knob from interactions with cell surface molecules22 as it carries the non-binding DARPin E3_5. Cell transduction was determined via expression of the near-infrared fluorescent protein (iRFP) 670 encoded on the Ad5 vector and was detected via flow cytometry.
Six out of nine hFAP adapters (nos. 3, 4, 5, 6, 8, and 20) demonstrated a clear increase in the transduction of HT1080hFAP cells (60%–90% transduced cells) when compared with the untargeted Ad5 (43% transduced cells; marked by the dashed line) (Figure 1D). Out of these six retargeting adapters, two (nos. 5 and 8) also showed a major increase in transduction of hFAP– HT1080 cells (20%–45% transduced cells) when compared with the E3_5-Ad5 (9% transduced cells; marked by the dashed line), suggesting an unspecific, hFAP-independent transduction. Notably, these two adapters were both constituted of retargeting DARPins with two internal repeats (N2C), whereas the remaining adapters were constituted of retargeting DARPins with three internal repeats (N3C). As a result, we excluded these two adapters due to low specificity and selected four well-performing retargeting adapters (nos. 3, 4, 6, and 20) that enable an hFAP-mediated adenoviral cell transduction.
Interestingly, transduction levels observed with adapter no. 3 (constructed of a DARPin with comparably lower affinity) were equally high as those observed with adapters nos. 4, 6, and 20 (all constructed of DARPins with comparably higher affinity), thus supporting our assumption that binding affinity alone does not determine a suitable Ad5 retargeting adapter.
Of further note, transduction levels of hFAP– HT1080 cells were overall reduced in comparison with the transduction levels of hFAP+ HT1080hFAP cells. A possible explanation could be the cellular expression levels of the respective adenoviral attachment/entry molecule (CAR for untargeted Ad5 vs. hFAP for hFAP-Ad5) depicted in Figures 1B and 1E, thereby affirming our fiber knob adapter-based retargeting strategy.
hFAP adapters enable Ad5 transduction of human fibroblasts
Having proven target specificity and functionality on the engineered fibrosarcoma cell line, we next aimed to validate the adapters for the retargeting of Ad5 via FAP in a more relevant setting using normal human fibroblasts. We chose the Detroit 551 (D551) human embryonic skin fibroblast cell line that endogenously expresses medium to high levels of hFAP (Figure 2A) and low levels of the primary Ad5 attachment receptor CAR (Figure 2B). Transduction of D551 cells by hFAP-Ad5, control E3_5-Ad5, or untargeted Ad5 revealed a massive increase (10- to 60-fold) in transduction when using the hFAP retargeting adapter for two different multiplicities of infection (MOIs) (referring to plaque-forming units [PFU]/cell) (Figure 2C). At an MOI of 20, all four hFAP adapter-retargeted Ad5 reached cell transduction levels of 42%–58%, whereas the untargeted Ad5 showed only 5% cell transduction. The increased transduction efficiency mediated by the hFAP adapter was even more prominent at an MOI of 2 with transduction levels of 4%–12% for all hFAP-Ad5 and only 0.5% for the untargeted Ad5. For the control E3_5-Ad5 we consistently observed a reduced transduction (10% at MOI = 20; 0.7% at MOI = 2) in comparison with all hFAP-Ad5, demonstrating that the increased transduction efficiency seen with the hFAP adapter was indeed mediated via the hFAP-specific retargeting DARPin.
Figure 2.

Selected hFAP adapters mediate adenoviral transduction of hFAP-expressing human fibroblasts
Flow cytometry analysis for (A) hFAP expression and (B) CAR expression of the human fibroblast D551 cell line upon antibody staining. The HeLa cell line served as a positive control in the CAR expression analysis. (C) Transduction of human fibroblasts by hFAP adapter-retargeted Ad5. Recombinant Ad5 encoding iRFP670 was pre-incubated with the selected “Top 4” hFAP adapters or the E3_5 blocking adapter and analyzed at two different MOIs (PFU/cell) for transduction of hFAP+ D551 cells in comparison with the untargeted Ad5. Transduction levels were determined via cellular expression of iRFP670 detected by flow cytometry. Bars represent mean transduction level of two biological replicates ± SD. Statistics: unpaired t test; ∗p < 0.05, ∗∗p < 0.005; p values are indicated for each sample with respect to the untargeted Ad5. Representative data of two independent experiments are shown. (D) Incubation time-dependent transduction of human fibroblasts by hFAP adapter-retargeted and untargeted Ad5. Recombinant Ad5 encoding TdTomato was pre-incubated with the hFAP adapter no. 6 and analyzed for transduction of hFAP+ D551 cells in comparison with the untargeted Ad5 after 4, 24, and 48 h incubation time of the (retargeted) adenoviral vector with the cells at an MOI of 10 (PFU/cell). Transduction was measured via cellular expression of TdTomato detected by flow cytometry. Representative data of two biological replicates of one experiment are shown.
Observing that the untargeted Ad5 only marginally transduced D551 cells, as opposed to the hFAP-Ad5, when incubated for 6 h with the cells, we wondered whether the transduction levels would increase upon a longer incubation time of the vector with the cells. Therefore, we analyzed the transduction of D551 cells by hFAP adapter-retargeted and untargeted Ad5 upon 24 and 48 h vector incubation time, as well as after only 4 h vector incubation (Figure 2D). For this experiment, we selected the so-far overall best-performing adapter no. 6 for Ad5 retargeting and chose an MOI of 10 (PFU/cell) based on our prior experiments. Furthermore, we used a TdTomato-encoding Ad5 since we found the signal intensity of this fluorescent protein to be stronger than the one detected with iRFP670 (data not shown). After 4 h incubation time, the hFAP-Ad5 yielded 9.4% transduced cells, whereas the untargeted Ad5 yielded 0.3% transduced cells. For hFAP-Ad5, the transduction signal kept increasing over the time course, reaching 52.3% and 81.8% transduced cells after 24 and 48 h incubation time, respectively. In contrast, the untargeted Ad5 yielded only 0.6% and 2.4% transduced cells after 24 and 48 h incubation time, respectively, showing that even after a long duration the fibroblasts were rarely transduced by untargeted Ad5. Conversely, with the help of the hFAP adapter a considerable fraction of fibroblasts could be transduced by Ad5 after only a short duration. In line with the cellular hFAP and CAR expression levels (Figures 2A and 2B), these data prove that the Ad5 transduction was truly hFAP dependent and mediated via the hFAP retargeting adapter. Given that the human fibroblast D551 cell line is seemingly not susceptible to a natural Ad5 infection, our findings demonstrate the advantage of the hFAP adapter that enables Ad5 transduction of otherwise untransducible cells.
Identification of a mouse FAP-specific adapter
Following our hFAP adapter screen and validation, we sought to generate an Ad5 adapter with specificity for murine FAP, enabling us to study vector retargeting to murine fibroblasts in vivo in mice. An initial sequence similarity search with the Basic Local Alignment Search Tool (BLAST) (www.uniprot.org) revealed that the extracellular domains of human and murine FAP harbor 90.1% amino acid sequence identity, suggesting that our previously selected, hFAP-specific DARPins might be cross-reactive. To test this, we analyzed our well-behaved single DARPins (see Figure S1E) for binding to recombinant mouse FAP (mFAP) in an enzyme-linked immunosorbent assay (ELISA). Seven out of 18 hFAP DARPins indeed showed cross-reactivity to mFAP, as defined by a binding signal that was greater than the signal of our negative binding control (Figure 3A). To confirm cross-reactivity and ensure mFAP specificity on the cell surface, we analyzed binding using suitable target and non-target cells, as done before. Here, we used the mFAP-expressing, murine NIH3T3mFAP fibroblast cell line that had been created by lentiviral transduction of the FAP– NIH3T3 parental cell line with murine FAP40 (Figure 3B). Our cell-based binding analysis revealed that only candidate no. 6 of the seven presumably cross-reactive DARPins bound specifically to mFAP on cells, as shown by a high binding signal on target cells and no binding signal on non-target cells, while the remaining six DARPins consistently showed a higher binding signal on non-target than on target cells (Figure 3C). This observation was confirmed with the corresponding DARPin-based adapters, which were as well tested for binding on NIH3T3mFAP and NIH3T3 cells (Figure S3). Therefore, we selected candidate no. 6 as both mFAP-specific and human/mouse cross-reactive adapter and proceeded to the in vitro Ad5 retargeting analysis.
Figure 3.

Retargeting of Ad5 to murine fibroblasts using a human/mouse cross-reactive FAP DARPin
(A) Analysis of hFAP DARPins for binding to mFAP via ELISA. Purified DARPins, previously selected to be hFAP specific, were analyzed via ELISA for cross-reactivity to mFAP. The unselected, non-binding DARPin E3_5 was applied as a negative binding control. The maltose-binding protein (MBP)-specific DARPin off7 and recombinant MBP were applied as a technical positive binding control. The dashed line indicates a cut-off signal set on the negative binding control to select mFAP-binding DARPins. Bars represent mean transduction level of two biological replicates ± SD. Statistics: unpaired t test; ∗p < 0.05, ∗∗p < 0.005; p values are indicated for each sample with respect to the E3_5 control DARPin. (B) Flow cytometry analysis of mFAP expression of the NIH3T3 and NIH3T3mFAP cell line with mFAP antibody staining. (C) Cell-based DARPin binding assay on target and non-target cells. Purified DARPins selected as mFAP binders by ELISA were analyzed for binding on mFAP+ NIH3T3mFAP and mFAP− NIH3T3 cells. Binding was detected by flow cytometry upon FLAG tag antibody staining of the FLAG-tagged DARPin. The unselected, non-binding DARPin E3_5 was applied as a negative binding control. Bars represent specific binding signal of single point measurements. Representative data at 1 μM DARPin concentration of a titration experiment are shown. MFI, mean fluorescent intensity. (D) Transduction of target and non-target cells by mFAP adapter-retargeted Ad5. Recombinant Ad5 encoding iRFP670 was pre-incubated with the mFAP adapter no. 6 or the E3_5 blocking adapter and tested for transduction of mFAP+ NIH3T3mFAP and mFAP– NIH3T3 cells in comparison with the untargeted Ad5 at an MOI of 2 (PFU/cell). Transduction levels were determined via cellular expression of iRFP670 detected by flow cytometry. Bars represent mean transduction level of two biological replicates ± SD. Representative data of three independent experiments are shown. (E) Flow cytometry analysis for CAR expression of the NIH3T3 and NIH3T3mFAP cell line in comparison with the positive control A549 cell line upon CAR antibody staining.
To test the selected mFAP-specific adapter for Ad5 retargeting to mFAP+ cells in vitro, we used an iRFP670-encoding Ad5 and analyzed transduction levels of NIH3T3mFAP and NIH3T3 cells upon incubation with mFAP-Ad5, untargeted Ad5, or control E3_5-Ad5 by flow cytometry. For NIH3T3mFAP cells, we detected a transduction rate of 52% for the mFAP-Ad5, 43% for the untargeted Ad5, and 22% for the control E3_5-Ad5 (Figure 3D), demonstrating an increase in transduction when using the mFAP-specific adapter and a decrease in transduction when using the E3_5 blocking adapter. For NIH3T3 cells, we detected similar transduction levels for the mFAP-Ad5 and the untargeted Ad5 (25% and 28%, respectively) and a decreased transduction for the control E3_5-Ad5 (14%). Although the data verify the functionality of the mFAP adapter, a prominent retargeting effect as seen with the human fibroblast cell line (Figure 2D) was not found with the murine cell line tested here. A possible explanation for this observation can be provided through the high CAR expression levels depicted in Figure 3E, showing that both NIH3T3mFAP and NIH3T3 cells are highly positive for the primary Ad5 attachment receptor CAR. Consequently, high transduction levels can be achieved via both cell surface molecules, FAP and CAR, hampering a clear assessment of the contribution of the mFAP adapter. Nonetheless, since on mFAP+ cells we observed a decrease of transduction with the E3_5 control adapter (blocking CAR interaction) and an increase with the mFAP adapter compared with the untargeted Ad5, we conclude that the mFAP adapter is functional and promotes the retargeting of Ad5 to murine fibroblasts via FAP. Importantly, mFAP adapter-retargeted Ad5 did moreover not affect cell viability upon transduction, as analyzed in our in vitro viability/toxicity assay (Figure S4).
Establishment of a FAP+ fibroblast-enriched tumor mouse model
Having selected and validated the mFAP-specific adapter in cell-based assays, we aimed to investigate its ability to mediate Ad5 retargeting to FAP+ cells in vivo. We chose the subcutaneous NCI-N87 human gastric cancer xenograft model utilizing immunodeficient SCID/beige mice. These mice are characterized by a lack of T and B lymphocytes, as well as by deficient natural killer cells, which facilitates tumor engraftment.46,47 NCI-N87 tumor cells overexpress HER2 but do not express FAP (Figure S5A). Therefore, vector retargeting to FAP+ stromal cells (and vector detargeting from tumor and other non-target cells) could be well monitored in this tumor mouse model.
However, when we initially performed preliminary in vivo experiments to establish the tumor xenograft model, we found that the overall FAP expression in the whole tumor was below 5% (Figure S5B). Reasonable modifications of the experimental protocol (e.g., number of tumor cells injected; time of tumor growth; age of the animal) did not significantly augment the FAP abundance. We observed the same limitation also for other subcutaneous tumor mouse models, including syngeneic models using immunocompetent C57BL/6 and BALB/c mouse strains. This is a major difference to human carcinomas, which are commonly rich in CAFs.48 Since a rather low abundance of FAP+ cells might impede a reliable Ad5 retargeting analysis, we thus established a FAP+ fibroblast-enriched subcutaneous tumor xenograft model, in which NCI-N87 tumor cells were co-injected with NIH3T3mFAP cells. These murine fibroblasts had been engineered to co-express mFAP+ and the green fluorescent protein (GFP), enabling their detection even without cell surface marker staining (Figure S5C). By varying the ratio of tumor cells to fibroblasts, this model then allowed an in vivo analysis of the cell specificity as well as the cell selectivity of the retargeted vector.
Successful retargeting of Ad5 to FAP+ fibroblasts in vivo
To analyze mFAP adapter-mediated Ad5 retargeting in vivo, we co-injected NCI-N87 and NIH3T3mFAP cells subcutaneously into the flank of SCID/beige mice, using 100-fold more tumor cells than fibroblasts to not saturate the tumor with stromal cells and enable a fair retargeting analysis. After tumor establishment, and before administering the adenoviral vector, we examined the cellular composition of the tumor via flow cytometry (Figure S5D). On the day of vector injection, the tumor was comprised of 50% NIH3T3mFAP cells (GFP+), 30% tumor cells (HER2+), 5% immune cells (CD45+), and 15% other cells (unstained) (Figure S5E). Subsequently, we injected mFAP adapter-retargeted or untargeted TdTomato-encoding Ad5 intratumorally, and analyzed cell transduction of harvested tumors via flow cytometry 3 days post injection.
We observed a significant increase in transduction of mFAP+ fibroblasts (46% FAP-Ad5 vs. 32% untargeted Ad5) and a considerable decrease in transduction of tumor cells (16% FAP-Ad5 vs. 24% untargeted Ad5) when applying the mFAP adapter to Ad5 (Figures 4A and 4B), demonstrating a clear preference of the adapter-retargeted vector for targeted fibroblasts over off-target tumor cells. Regarding the transduction of immune cells, similar levels (20%) were detected for both FAP-Ad5 and untargeted Ad5.
Figure 4.

Successful retargeting of Ad5 to FAP-expressing fibroblasts in vivo
(A) HER2-overexpressing NCI-N87 tumor cells and GFP-labeled, mFAP-expressing NIH3T3mFAP fibroblast cells were co-injected subcutaneously into the flank of SCID/beige mice. After tumor establishment (200 mm3 tumor volume), mice were treated intratumorally with 3 × 109 PFU FAP-retargeted or untargeted Ad5 encoding TdTomato. Three days post injection, tumors were harvested and analyzed by flow cytometry. Transduced cells were detected via TdTomato expression and further characterized by cell surface marker staining or GFP expression. Each data point represents a single mouse. Bars represent mean ± SD of five mice per group. Statistics: unpaired t test; ∗p < 0.05. Representative data of two independent experiments are shown. (B) Quantification of (A), indicating mean values of transduced cells. (C) Immunohistochemical analysis of (A) to investigate the cell specificity of FAP-retargeted Ad5. Representative immunofluorescence images of tumor tissues stained for HER2 (cyan) and counter-stained with DAPI (blue) for nuclei staining. FAP+ cells were detected via GFP expression (green), and cells transduced with Ad5 were detected via TdTomato expression (magenta). Scale bars 1 mm.
To further investigate cell specificity of the vector, we performed an immunofluorescence staining of the tumor tissues (Figure 4C). Here, we again observed a strong preference of the FAP-retargeted Ad5 for mFAP+ fibroblasts, confirming our previous findings. As shown in the fluorescence microscopy images, the TdTomato signal of FAP-Ad5 predominantly co-localized with the GFP signal of mFAP+ fibroblasts. We therefore conclude that the adenoviral tropism could successfully be redirected to FAP+ stromal cells in vivo through the mFAP-specific DARPin adapter.
Efficient in vivo delivery of active anti-cancer therapeutics using FAP-Ad5
We next explored whether the FAP-retargeted Ad5 would be functional for the in vivo delivery of cancer-attacking biomolecules to the TME. We used the FAP+ fibroblast-enriched subcutaneous NCI-N87 xenograft model in immunodeficient SCID/beige mice established here. This HER2-dependent human xenograft is sensitive to anti-HER2 therapy, allowing us to study a FAP-Ad5-mediated gene delivery of the mAb trastuzumab (TZB) (brand name Herceptin), for which an anti-tumor efficacy has been shown previously in this model.49
Accordingly, tumor-bearing mice received a single intratumoral injection of FAP-retargeted TZB-encoding Ad5 (termed FAP-Ad5-TZB) and were then monitored for tumor growth for 19 days (Figure 5A). For comparison, one group of tumor-bearing mice received a single intratumoral injection of clinical-grade recombinant TZB (termed TZB (Herceptin) 1×), analogous to FAP-Ad5-TZB treatment, and another group received weekly repeating intratumoral injections of clinical-grade recombinant TZB for a total of three doses (termed TZB (Herceptin) 3×). Tumor-bearing mice treated with PBS served as a control group.
Figure 5.

Efficient in vivo delivery of anti-cancer therapeutics using FAP-Ad5
(A) Growth analysis of HER2+ tumor xenografts upon Ad5-mediated treatment with trastuzumab (TZB) or with TZB (Herceptin) as a protein. HER2-overexpressing NCI-N87 tumor cells and NIH3T3mFAP cells were co-injected subcutaneously into the flank of SCID/beige mice for tumor establishment. At a tumor volume of 50 mm3, mice were treated intratumorally with 9 × 108 PFU FAP-retargeted Ad5 encoding TZB (FAP-Ad5-TZB; n = 5), or one single dose of 200 μg of TZB (Herceptin) as a protein (n = 3), or three doses of 200 μg of TZB (Herceptin) as a protein (n = 3), or PBS (n = 5). Arrows indicate time points of injection for the corresponding treatment. Data points represent mean ± SD. Statistics: unpaired t test; ∗p < 0.05, ∗∗p < 0.005, ∗∗∗p < 0.0005; p values are indicated in black color for FAP-Ad5-TZB with respect to PBS and in blue color for FAP-Ad5-TZB with respect to TZB (Herceptin) 1×; results for TZB (Herceptin) 1× and TZB (Herceptin) 3× with respect to PBS were statistically non-significant (p > 0.05). (B) Tumor weights of harvested tumors from (A) 19 days post injection. Each data point represents a single mouse. Statistics: unpaired t test; ∗p < 0.05, ∗∗p < 0.005. (C) Detection of TZB within harvested tumors from (A) 19 days post injection. Representative immunofluorescence images of tumor tissues stained for TZB (cyan) and counter-stained with DAPI (blue) for nuclei staining. Scale bars 1 mm.
We observed an immediate reduction in tumor growth throughout the first 3 days post injection for all TZB (Herceptin)-treated mice, whereas for FAP-Ad5-TZB-treated mice we observed the effect with a short delay throughout days 4–7 (Figure 5A). This short delay is presumably due to the initially required cellular expression of TZB after adenoviral transduction and the therefore delayed accumulation of the drug. Thereafter, tumors of all TZB-treated mice restarted growing, but interestingly much faster for TZB (Herceptin)-treated mice than for FAP-Ad5-TZB-treated mice, for which a significant delay in outgrowth could be detected. Tumor growth of TZB (Herceptin)-treated mice could moreover be slowed down through further injections of the drug (see TZB (Herceptin) 3×), leading us to the hypothesis that the profound delay in outgrowth seen for FAP-Ad5-TZB-treated mice is most likely attributed to a continuous production and secretion of the anti-cancer therapeutic throughout the study.
In line with these findings, tumors of FAP-Ad5-TZB-treated mice examined after tumor harvest were significantly smaller than those of PBS- or TZB (Herceptin)-treated mice (Figures 5B and S6).
To confirm the presence of TZB within the tumor, we analyzed tumor tissues via immunofluorescence staining to detect the therapeutic mAb. TZB could be detected in tumors of TZB (Herceptin)-treated as well as FAP-Ad5-TZB-treated mice but not in tumors of PBS-treated mice (Figure 5C), correlating the presence of the drug with the observed biological effect. Remarkably, TZB was found to be present in various areas throughout the whole tumor for FAP-Ad5-TZB-treated mice, whereas for TZB (Herceptin)-treated mice TZB was found to be rather restricted to one spot within the tumor.
Taken together, our data demonstrate efficient in vivo delivery of a therapeutic payload to FAP+ fibroblasts in the TME by FAP-Ad5. The locally secreted biomolecule delayed tumor growth, thereby proving functionality of the FAP adapter to retarget Ad5 vectors to CAFs in the tumor stroma for cancer therapeutic applications.
Discussion
Adenoviral vectors derived from Ad5 are one of the most widely used vectors for the delivery of gene therapeutics, and have shown great clinical promise in ex vivo and in vivo gene therapy.3 However, in vivo gene delivery is still restricted due to the natural adenoviral tropism and insufficient vector targeting. In the present study, we developed a DARPin-based adapter with specificity for FAP to enable Ad5 retargeting to CAFs and thereby advance Ad5-mediated cancer gene therapy.
Using ribosome display together with a cell-based screening approach, we selected 10 FAP-specific DARPins, partly with subnanomolar binding affinity, which served as targeting moiety in our modular adapter. Further selection and characterization of the eventually generated adapters yielded four candidates with specificity for hFAP. These four adapters enabled FAP-mediated and FAP-specific Ad5 transduction of FAP+ human fibroblasts in vitro, mimicking CAFs in the tumor stroma. Importantly, transduction of these fibroblasts by untargeted Ad5 was negligible (<5% cells transduced, Figure 2C), even after 48 h of vector incubation. These FAP+ human fibroblasts expressed only low levels of the primary Ad5 cell attachment/entry receptor CAR, as confirmed via flow cytometry (Figure 2B). This experimental finding thus demonstrates the great advantage of our FAP retargeting adapter that enables gene delivery to FAP+ fibroblasts, and potentially other FAP+ cells, that are otherwise not transducible by Ad5 via CAR.
Among the final selected best-performing adapters, three showed exclusive binding to hFAP, whereas one showed binding to both hFAP and mFAP. This human/mouse cross-reactive adapter allowed us to perform preclinical experiments in mice, which can later be translated to patient-derived tumors. Our in vivo analysis of the cross-reactive FAP adapter demonstrated successful Ad5 retargeting to FAP+ fibroblasts in the TME in tumor-bearing mice upon intratumoral vector administration. We observed a clear preference of the FAP-retargeted Ad5 for FAP+ fibroblasts and detected a decrease in FAP– tumor cell transduction. Quantification of the cell populations present in the TME showed that at the time point of vector administration 50% of all cells consisted of target cells (fibroblasts), whereas the other 50% were composed of tumor cells, immune cells, and other non-target cells. The observed cell transduction within this heterogeneous TME required a satisfactory cell-selectivity of the vector, which was indeed achieved through our retargeting module.
Nonetheless, we did observe some residual off-targeting for FAP-Ad5 to the immune cell population, matching the transduction levels of the untargeted Ad5. We hypothesize that this off-targeting to immune cells mainly results from interactions of the hexon with macrophages, which are still abundant in the immunodeficient mouse model used here.50,51 Particularly, HVR1 of the hexon is implicated in vector sequestration by macrophages through charge-dependent interactions with scavenger receptors located on these cells.50,51 In this study, we did not apply the vector shield that has previously shown to mask HVR1 and other HVRs of the hexon.18 The similar off-targeting to immune cells observed with both vectors could therefore indicate a hexon-dependent interaction promoting immune cell transduction, rather than a fiber knob-dependent interaction modifiable through our adapter molecule.
Future studies using an HVR1-mutated vector, and/or applying our capsid shield, could be used to investigate our hypothesis further. In this context, it would furthermore be of interest to investigate the amount of transduced target cells upon systemic administration of the shielded and FAP-retargeted Ad5 vector, which has not been studied here. Alemany et al.52 showed that unshielded and unmodified Ad5 is rapidly cleared from blood by liver-resident macrophages (Kupffer cells) in mice. The originally developed PEGylation strategy reduced the clearance rate but also reduced infectivity.52 This problem has been overcome by developing a tight-binding but reversible protein shield based on a trimerized anti-hexon scFv, which enabled a predominant transduction of tumor tissue upon systemic vector administration.18 While these experiments were carried out with adapters for tumor cell-specific surface proteins, it is reasonable to assume that similar results will be obtained with stromal cell-specific retargeting adapters such as developed here. However, further investigations will have to confirm this.
In contrast to our present study that comprises an in vivo retargeting analysis, Kuklik et al.53 recently presented an in vitro study on the retargeting of an AAV2-based vector to CAFs via FAP. AAV2-based vectors equally rank among the most commonly used vectors for clinical gene therapy. However, AAV2 vectors harbor a relatively small packaging capacity (4.7 kb for AAV2 vs. 37 kb for Ad5), and are therefore restricted in payload size. Thus, high-capacity Ad5 vectors harbor a much larger packaging capacity and therefore allow for a delivery of large therapeutic gene cargos, with the opportunity of encoding regulatory elements and multiple genes. This important characteristic distinguishes our vector platform from other gene delivery approaches.
By using a HER2-overexpressing mouse tumor xenograft model, we could furthermore test in vivo gene delivery of the clinically relevant mAb TZB to FAP+ stromal fibroblasts by our FAP-retargeted Ad5. FAP-Ad5-delivered TZB showed superior efficacy over clinical-grade recombinant TZB (Herceptin) applied as a protein via the same route of administration. We attribute this superiority of FAP-Ad5-delivered TZB to a presumably continuous production and secretion of the drug by transduced fibroblasts throughout the study, leading to a more even biodistribution within the TME. We had previously studied the production of TZB by the tumor cells themselves upon delivery via a HER2-retargeted Ad5 to HER2+ tumor cells, and observed a long duration of production.54 Indeed, TZB had been found in the TME 61 days post intratumoral vector injection, indicating a continuous secretion of the therapeutic mAb, which shows a half-life of 28 days in humans but of only 8 days in mice.55,56 The sustained biomolecule expression constitutes another benefit of our adenoviral delivery platform in comparison with a direct drug administration, particularly for molecules with short half-life. This characteristic and the concept of a local drug production to improve bioavailability and reduce off-target toxicities reaffirmed our aim of installing a biofactory in the TME.
To this end, the stromal cell-targeted (paracrine) delivery approach presented here might exhibit certain advantages over our previous tumor cell-targeted (autocrine) delivery: (1) expression of the anti-cancer agent in the TME might be of longer duration in the paracrine approach since the biofactory (transduced CAFs) would not be therapeutically affected by the encoded agent. In contrast, in an autocrine approach, expression of the anti-cancer agent might be limited by the lifetime of the transduced (tumor) cells, as they are producer cells but at the same time affected by the action of the therapeutic. (2) Targeting the vector to CAFs might also be beneficial regarding the spatial distribution and/or abundance of stromal cells, particularly in stroma-rich tumors, which are common for human cancers.48 (3) Given that FAP is expressed in more than 90% of all carcinomas,28 a stromal cell-targeted delivery via FAP on CAFs would moreover allow to treat a variety of human cancers, independent of the existence of specific/targetable tumor cell markers or suitable retargeting adapters. This might additionally be a promising strategy to avoid antigen escape of even those tumors cells that carry defined markers.
In conclusion, the FAP-specific adapter presented herein expands our Ad5 retargeting system, and enables a stromal cell-targeted delivery of anti-cancer biomolecules to the TME with potential benefits over existing, tumor cell-targeted delivery strategies. The FAP adapter can be combined with our scFv-based capsid shield to overcome pre-existing immunity against the vector.18,20 More importantly, it can be applied to any Ad5-derived vector including high-capacity Ad5 vectors, which allows the simultaneous delivery of multiple payloads for combination cancer therapy as shown by Brücher et al.57 Thus, our FAP-retargeted Ad5 delivery platform may help advance current cancer treatment options involving biologics with required local or prolonged production. Beyond cancer therapy, our FAP-Ad5 vector could potentially be applied to other medical disorders with FAP involvement, such as chronic inflammatory diseases and fibrosis.58,59,60,61,62
Materials and methods
Chemicals
All chemicals were purchased from Sigma-Aldrich unless stated otherwise.
Cell lines
The cell lines HT1080 and HT1080hFAP were generated as described previously39 and kindly provided by Prof. Christian Münz (University of Zürich). HT1080 and HT1080hFAP cells were cultured in complete RPMI medium (RPMI 1640 GlutaMAX [Thermo Fisher Scientific] supplemented with 10%, v/v, fetal bovine serum [FBS] [BioConcept]; 100 U/mL penicillin, and 100 μg/mL streptomycin [Sigma-Aldrich]) containing 200 μg/mL G418 (Carl Roth) and 150 μg/mL Hygromycin B (Invitrogen). NIH3T3 and NIH3T3mFAP cell lines were generated as described previously40 and kindly provided by Prof. Ellen Puré (University of Pennsylvania). NIH3T3 and NIH3T3mFAP cells were cultured in complete RPMI medium. HEK293, D551, HeLa, A549, and NCI-N87 cell lines were purchased from the American Type Culture Collection. HEK293, D551, HeLa, and A549 cells were cultured in complete DMEM medium (DMEM-high glucose [Sigma-Aldrich] supplemented with 10%, v/v, FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin), and NCI-N87 cells were cultured in complete RPMI medium. All cell lines were maintained at 37°C and 5% CO2 in a humidified atmosphere, and routinely tested and confirmed negative for mycoplasma contamination.
Ribosome display selection of DARPins against hFAP
Ribosome display selections were essentially performed as described previously,44 but using a semi-automatic KingFisher Flex MTP 96-well platform. Biotinylated recombinant hFAP (Sino Biological) was used for the selection, in alternating selection rounds, on either MyOne T1 streptavidin-coated beads (Pierce) or Sera-Mag neutravidin-coated beads (GE Healthcare), and a total of four selection rounds were performed. After E. coli transformation, binders were directly analyzed for hFAP binding by performing flow cytometry-based cell-binding assays described below.
Cloning, expression, and purification of DARPins and adapters
DARPins were cloned into the pQIq backbone containing an N-terminal His6-tag and a C-terminal FLAG tag. Adapters were cloned into the pQIq backbone containing an N-terminal His10-tag with a 3C protease cleavage site. For DNA propagation, the E. coli XL1-Blue strain was transformed with the corresponding plasmids.
For high-throughput screening, DARPins and adapters were expressed in a small-scale format (1 mL culture volume, 96-deep-well plate [Thermo Fisher Scientific]) in the E. coli strain XL1-Blue. Upon cell lysis, proteins were purified via immobilized-metal ion affinity chromatography (IMAC) using a HisPur Cobalt Spin Plate (Thermo Fisher Scientific) equilibrated with equilibration buffer (50 mM Na2HPO4/NaH2PO4, 300 mM NaCl [pH 7.4]). After washing with wash buffer (50 mM Na2HPO4/NaH2PO4, 300 mM NaCl, 15 mM imidazole [pH 7.4]), proteins were eluted in elution buffer (50 mM Na2HPO4/NaH2PO4, 300 mM NaCl, 250 mM imidazole [pH 7.4]) and further buffer exchanged to PBS (pH 7.4) using a Pall AcroPrep filter plate (Pall). Purity and molecular weights were analyzed via SDS-PAGE and analytical SEC using a Superdex 75 5/150 GL column (GE Healthcare).
For in vivo experiments, selected adapters were expressed in large-scale format (500 mL culture volume) in the E. coli strain BL21. Upon cell lysis, protein purification was carried out via IMAC using Ni-NTA agarose (QIAGEN) equilibrated with TBSEq (50 mM Tris-HCl, 400 mM NaCl [pH 7.4]). After washing with TBSW (50 mM Tris-HCl, 400 mM NaCl, 20 mM imidazole, 10%, v/v, glycerol [pH 7.4]), adapters were eluted in TBSE (50 mM Tris-HCl, 400 mM NaCl, 250 mM imidazole [pH 7.4]). Purified adapters were then incubated with 3C protease (kindly provided by Dr. Fabian Brandl, University of Zurich) for removal of the His10-tag and dialyzed against PBS (pH 7.4) for buffer exchange. Adapters were then further purified via preparative SEC using a Superdex 200 10/300 GL column (GE Healthcare). Purity and molecular weights were analyzed as described above. Prior to in vivo injection into mice, purified adapters were assessed for endotoxin content using the EndoSafe Portable Test System (Charles River Laboratories) and test cartridges with 0.5–0.005 EU/mL sensitivity, to confirm that endotoxin levels would not exceed the limit for endotoxin content for injectable products recommended by the Food and Drug Administration.
Flow cytometry-based cell-binding assay for binder screening
For a high-throughput target-binding analysis of DARPins and adapters, we performed flow cytometry-based cell-binding assays using suitable target and non-target cells as follows. Cells were harvested by trypsinization and washed with ice-cold PBS-B (DPBS, Sigma-Aldrich) containing 1%, w/v, bovine serum albumin (BSA). A total of 1 × 105 cells resuspended in PBS-B was added per well of a 96-well plate (V-bottom, non-treated surface; Thermo Fisher Scientific) and kept on ice. Purified DARPins or adapters diluted in DPBS (or crude E. coli extracts in a final dilution of 1:100) were added to the cells and incubated for 60 min on ice. Thereafter, cells were washed with ice-cold PBS-B and subsequently stained for 30 min on ice with appropriate antibodies diluted in PBS-B to detect the bound DARPin or adapter. Mouse anti-FLAG M2-FITC antibody (1:200, Sigma-Aldrich, F4049) was used for DARPin detection. Mouse anti-His (1:200, QIAGEN, 34670) and goat anti-mouse IgG-AF488 (1:1,000, Invitrogen, A11001) were used for adapter detection. Upon antibody staining, cells were washed with ice-cold PBS-B and then fixed with fixation buffer (DBPS containing 2%, w/v, paraformaldehyde for 10 min at room temperature [RT]). After a final wash step, cells were resuspended in PBS-B. Samples were measured on a BD FACSCanto II (BD Biosciences) using the high-throughput sampler, and flow cytometry analysis was performed using FlowJo software (BD Biosciences).
ELISA
mFAP-binding DARPins were screened via ELISA using recombinant mFAP (R&D Systems), and recombinant maltose-binding protein (MBP) (kindly provided by Joana Marinho, University of Zurich) for an MBP binder as a positive control. Target protein was coated overnight (O/N) at 4°C on MaxiSorp 96-well plates (Nunc). ELISA-blocking buffer (PBS containing 0.5%, w/v, BSA) was used for blocking performed for 2 h at RT while shaking on a plate shaker. Plates were washed with ELISA-PBS-T (PBS containing 0.05%, v/v, Tween 20). Samples were diluted (100 nM DARPin concentration) in ELISA-PBS-TB (ELISA-PBS-T containing 0.5%, w/v, BSA) and incubated for 1 h at 4°C while shaking. DARPins were detected with rabbit anti-FLAG antibody (1:5,000, GenScript, A01868) and goat anti-rabbit-AP antibody (1:10,000, Sigma-Aldrich, A3687), both diluted in ELISA-PBS-TB and incubated for 1 h at 4°C while shaking. Substrate solution consisting of 3 mM p-nitrophenyl phosphate (pNPP) diluted in pNPP buffer (50 mM NaHCO3, 50 mM MgCl2) was used for final detection. Absorbance at 405 nm was measured on a Tecan Infinite M1000 plate reader (Tecan).
Affinity measurements using surface plasmon resonance
Binding kinetics of selected DARPins were determined as described previously63 using recombinant biotinylated hFAP (Sino Biological). In brief, recombinant biotinylated hFAP was diluted in PBS (pH 7.4) and immobilized on a SPP NAHC200M Sensor Chip (XanTec Bioanalytics) to a level of 1,000 resonance units. DARPins were diluted in running buffer (PBS [pH 7.4] containing 0.005%, v/v, Tween 20) covering a concentration range of 0.1–10 nM. A 1:1 Langmuir binding model was used to fit the data measured on a ProteOn XPR36 instrument (Bio-Rad) using ProteOn Manager Software (version 3.1.0.6, Bio-Rad).
Viral vector generation and purification
All Ad5 vectors used in this study were E1/E3 deleted and thus replication deficient. Furthermore, all Ad5 vectors harbored four genetic mutations in hexon HRV7 (I421G, T423N, E424S, and L426Y) to reduce coagulation factor X-mediated liver tropism, as described previously by Schmid et al.18 Vectors were generated as described before18,54 using the AdEasy Adenoviral Vector System (Agilent). Vector purification was performed via ultracentrifugation using two sequential cesium chloride gradients. In some instances, generated vectors were reamplified and purified by Vector Biolabs (Malvern, PA).
Flow cytometry analysis
Cell marker expression was determined via flow cytometry upon antibody staining as follows. For cell lines, cells were harvested by trypsinization, washed with ice-cold FACS-buffer (DPBS containing 1%, w/v, BSA and 0.1%, w/v, NaN3), and resuspended in ice-cold FACS buffer to proceed with the antibody staining. Antibody staining was performed by incubating 1 × 105 to 1 × 106 cells with the appropriate antibody (see list below) for 20 min on ice. Cells were then washed with ice-cold FACS buffer and fixed with fixation buffer for 10 min at RT. After a final wash step, cells were resuspended in FACS buffer and stored at 4°C until being analyzed at the flow cytometer.
For harvested solid tumors, tumors were minced with a scalpel, incubated in digestion medium (RPMI 1640 GlutaMAX supplemented with 2%, v/v, FBS, 1 mg/mL collagenase IV [Gibco], 0.5 mg/mL hyaluronidase [ITW Reagents], and 0.5 mg/mL DNase I [Merck]) for 30 min at 37°C, and passed through a 70-μm mesh to yield a single-cell suspension. Cells were collected by centrifugation (500 × g for 5 min at 4°C) and washed with ice-cold DPBS. Thereafter, red blood cell lysis was performed using ACK Lysing Buffer (Thermo Fisher Scientific) following the manufacturer’s instructions. ACK Lysing Buffer was diluted by adding a 10-fold excess of DPBS containing 5 mM EDTA, followed by an incubation for 10 min on ice. Subsequently, cells were washed with ice-cold DPBS and subjected to a live/dead staining using the LIVE/DEAD Fixable Violet Dead Cell Stain Kit (Invitrogen). Then, IgG Fc receptors were blocked using TruStainFcX PLUS (BioLegend) followed by the antibody-staining procedure described above.
For multicolor flow cytometry panels, single-stained controls or UltraComp eBeads Plus (Thermo Fisher Scientific) were used for compensation. Fluorescence minus one and further proper controls were included in each experiment.
Flow cytometers used in this study included BD FACSCanto II, BD LSRFortessa, BD FACSymphony 5L (all BD Biosciences), and Cytek Aurora 5L (Cytek Biosciences). Flow cytometry analysis was performed using FlowJo software (BD Biosciences).
Antibodies used for flow cytometry
Antibodies used for flow cytometry included: mouse anti-human FAP-APC (R&D Systems, FAB3715A), sheep anti-human FAP (R&D Systems, AF3715), mouse anti-mouse FAP (Merck, MABC1145), mouse anti-CAR (Millipore, 05–644), mouse anti-HER2-AF488 (BioLegend, 324410), mouse anti-HER2-AF647 (BioLegend, 324412), rat anti-mouse CD45-AF700 (BioLegend, 103127), donkey anti-sheep IgG-CF488A (Sigma-Aldrich, SAB4600038), goat anti-mouse IgG-AF647 (Invitrogen, A21235), and goat anti-mouse IgG-AF488 (Invitrogen, A11001). Optimal antibody dilutions were determined via titration or adopted from the manufacturer’s recommendation.
In vitro Ad5 transduction assay
Totals of 1 × 104 or 5 × 104 cells were seeded per well of a 96- or 24-well cell culture (Corning), respectively. The Ad5 vector was pre-incubated with the corresponding adapter (10-fold molar excess of adapter over knob) diluted in DPBS for 1 h on ice. Untargeted vector was treated accordingly by substituting the adapter by DPBS only. For transduction, cell medium was changed to fresh complete medium and (retargeted) Ad5 vector was added to the cells at the indicated MOI (referring to PFU/cell). Cells were incubated for 6 h (unless stated otherwise) at 37°C and 5% CO2 before performing another medium change to remove the viral vector. Cells were further incubated for 20–26 h at 37°C and 5% CO2 and then harvested by trypsinization. If needed, antibody staining to detect cell surface markers was performed as described above. After washing, cells were fixed with fixation buffer for 10 min at RT and thereafter stored in FACS buffer at 4°C until being analyzed at the flow cytometer (instruments listed above). Transduction was measured via the cellular expression of a fluorescent reporter protein (iRFP670 or TdTomato) for which the corresponding DNA had been delivered by the Ad5 vector.
Animal experiments
Eight- to 9-week-old female Fox Chase SCID/beige mice (CB17.Cg-PrkdcscidLystbg-J/Crl; Charles River) were used for the in vivo studies. All animal experiments were performed in accordance with the Swiss animal protection law and with approval of the Cantonal Veterinary Office (Zurich, Switzerland).
In vivo Ad5 retargeting study
Mice were subcutaneously injected into the left flank with 1 × 106 NCI-N87 and 1 × 104 NIH3T3mFAP cells in 100 μL DPBS containing 50% Matrigel (Corning). Tumors were measured with a caliper and tumor volumes calculated from V = 0.5 × length × width × width. Nineteen days after tumor engraftment (reaching approximately 200 mm3 tumor volume), mice were intratumorally injected with 50 μL of indicated Ad5 vector (3 × 109 PFU in DPBS) encoding TdTomato. For FAP-retargeted Ad5, vector had been pre-incubated with the corresponding adapter (10-fold molar excess of adapter over knob) diluted in DPBS for 1 h on ice. Three days post injection, mice were sacrificed, and tumors were harvested and processed for subsequent analysis via flow cytometry (see above) or immunohistochemistry (IHC) (see below).
In vivo Ad5 therapeutic delivery study
Mice were subcutaneously injected into the left flank with 1 × 106 NCI-N87 and 1 × 105 NIH3T3mFAP cells in 100 μL DPBS containing 50% Matrigel. Tumor volumes were determined as stated above. Nine days after tumor engraftment (reaching approximately 50 mm3 tumor volume), mice were intratumorally injected with 50 μL of (1) FAP-Ad5-TZB (FAP-retargeted Ad5 encoding TZB; for Ad5 retargeting procedure see above; 9 × 108 PFU in DPBS), (2) Herceptin (Roche; 200 μg in DPBS), or (3) PBS (DPBS). One group of mice received repeated intratumoral injections (one injection per week) of Herceptin (200 μg in DPBS) adding up to a total of three injections. Nineteen days post (first) injection, mice were sacrificed and tumors were harvested, weighed, and processed for subsequent analysis via IHC (see below).
IHC
IHC was performed using cryosections (10-μm thick) of frozen tumor tissues embedded in O.C.T. compound (Thermo Fisher Scientific). Cryosections were fixed with acetone for 10 min at −20°C, washed with IHC-PBS-T (PBS [pH 7.4] containing 0.1%, v/v, Tween 20), and blocked with IHC blocking buffer (IHC-PBS-T with 10% normal goat serum [Cell Signaling Technology]) for 1 h at RT. Sections were incubated with primary antibody diluted in IHC blocking buffer O/N at 4°C. After washing, sections were incubated with appropriate secondary antibodies diluted in IHC blocking buffer for 1 h at 4°C, washed, and counterstained with DAPI (Thermo Fisher Scientific; 300 nM final concentration) for 5 min at RT. After final washing, sections were mounted with ProLong Gold antifade mountant (Thermo Fisher Scientific) and analyzed using the THUNDER Imager Microscope (Leica). Antibodies used for IHC analysis included rat anti-HER2 (1:500, Thermo Fisher Scientific, MA1-82367), goat anti-rat IgG-AF647 (1:1,000, Amersham Biosciences, 4418), and goat anti-human IgG-AF647 (1:1,000, Thermo Fisher Scientific, A21445).
Data and code availability
The data supporting the findings of this study are available from the corresponding author, A.P., upon reasonable request.
Acknowledgments
We are grateful to Joana Marinho, Thomas Reinberg, Anna-Lena Schinke, and Sven Furler for DARPin selection and hit validation, and to Dr. Jonas Schäfer and Dr. Birgit Dreier for supervising the activity. We thank Prof. Ellen Puré and Prof. Christian Münz for providing cell lines, Dr. Lukas Becker for helping with protein purification and for scientific discussions, Nina Schumacher and Faye Menard for helping with tumor processing during in vivo studies, Dr. Fabian Brandl for assisting with surface plasmon resonance analysis and for providing reagents, Dr. Markus Schmid for providing materials and reagents, Jonas Kolibius for scientific discussions and critical reading of the manuscript, and Dr. Sheena N. Smith for helpful discussions during the initiation of the project and for providing materials and reagents. We are grateful to the Zurich Integrative Rodent Physiology (ZIRP) team, especially to Dr. Svende Pfundstein for training and technical assistance during in vivo experiments, as well as to Dr. Petra Seebeck for assistance with the animal license application. We acknowledge the Flow Cytometry Facility of the University of Zurich for training and maintenance of the instruments. We further acknowledge the Laboratory Animal Services Center of the University of Zurich for training, animal caretaking, and experimental equipment. This project was funded by the Schweizerische Nationalfonds Sinergia grant CRSII5_170929 and by grant 310030_192689 (both to A.P.). The graphical abstract was created with BioRender.com.
Author contributions
Conceptualization, K.P.H. and A.P.; methodology, K.P.H. and F.K.; investigation, K.P.H., M.v.G., and G.N.-D.: validation, K.P.H.; formal analysis, K.P.H.; project administration, K.P.H.; visualization, K.P.H. and P.C.F.; resources, M.v.G., P.C.F., and L.B.; writing – original draft, K.P.H.; writing – review & editing, M.v.G., P.C.F., F.K., G.N.-D., L.B., and A.P.; supervision, A.P.; funding acquisition, A.P.
Declaration of interests
A.P. is a cofounder and shareholder of Vector BioPharma, which is commercializing the retargeted, shielded adenovirus delivery technology.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.08.018.
Supplemental information
References
- 1.Sharma P., Allison J.P. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 2.Berraondo P., Sanmamed M.F., Ochoa M.C., Etxeberria I., Aznar M.A., Pérez-Gracia J.L., Rodríguez-Ruiz M.E., Ponz-Sarvise M., Castañón E., Melero I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer. 2019;120:6–15. doi: 10.1038/s41416-018-0328-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang F., Qin Z., Lu H., He S., Luo J., Jin C., Song X. Clinical translation of gene medicine. J. Gene Med. 2019;21 doi: 10.1002/jgm.3108. [DOI] [PubMed] [Google Scholar]
- 4.Russell W.C. Adenoviruses: update on structure and function. J. Gen. Virol. 2009;90:1–20. doi: 10.1099/vir.0.003087-0. [DOI] [PubMed] [Google Scholar]
- 5.Bulcha J.T., Wang Y., Ma H., Tai P.W.L., Gao G. Viral vector platforms within the gene therapy landscape. Signal Transduct. Target. Ther. 2021;6:53. doi: 10.1038/s41392-021-00487-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kalyuzhniy O., Di Paolo N.C., Silvestry M., Hofherr S.E., Barry M.A., Stewart P.L., Shayakhmetov D.M. Adenovirus serotype 5 hexon is critical for virus infection of hepatocytes in vivo. Proc. Natl. Acad. Sci. USA. 2008;105:5483–5488. doi: 10.1073/pnas.0711757105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waddington S.N., Mcvey J.H., Bhella D., Parker A.L., Barker K., Atoda H., Pink R., Buckley S.M.K., Greig J.A., Denby L., et al. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell. 2008;132:397–409. doi: 10.1016/j.cell.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 8.Alba R., Bradshaw A.C., Mestre-Francés N., Verdier J.M., Henaff D., Baker A.H. Coagulation factor X mediates adenovirus type 5 liver gene transfer in non-human primates (Microcebus murinus) Gene Ther. 2012;19:109–113. doi: 10.1038/gt.2011.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tomko R.P., Xu R., Philipson L. HCAR and MCAR: the human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. USA. 1997;94:3352–3356. doi: 10.1073/pnas.94.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bergelson J.M., Cunningham J.A., Droguett G., Kurt-Jones E.A., Krithivas A., Hong J.S., Horwitz M.S., Crowell R.L., Finberg R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 11.Wickham T.J., Mathias P., Cheresh D.A., Nemerow G.R. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell. 1993;73:309–319. doi: 10.1016/0092-8674(93)90231-e. [DOI] [PubMed] [Google Scholar]
- 12.Meier O., Greber U.F. Adenovirus endocytosis. J. Gene Med. 2004;6:S152–S163. doi: 10.1002/jgm.553. [DOI] [PubMed] [Google Scholar]
- 13.Krasnykh V.N., Mikheeva G.V., Douglas J.T., Curiel D.T. Generation of recombinant adenovirus vectors with modified fibers for altering viral tropism. J. Virol. 1996;70:6839–6846. doi: 10.1128/jvi.70.10.6839-6846.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Douglas J.T., Rogers B.E., Rosenfeld M.E., Michael S.I., Feng M., Curiel D.T. Targeted gene delivery by tropism-modified adenoviral vectors. Nat. Biotechnol. 1996;14:1574–1578. doi: 10.1038/nbt1196-1574. [DOI] [PubMed] [Google Scholar]
- 15.Michael S.I., Hong J.S., Curiel D.T., Engler J.A. Addition of a short peptide ligand to the adenovirus fiber protein. Gene Ther. 1995;2:660–668. [PubMed] [Google Scholar]
- 16.Hofherr S.E., Shashkova E.V., Weaver E.A., Khare R., Barry M.A. Modification of adenoviral vectors with polyethylene glycol modulates in vivo tissue tropism and gene expression. Mol. Ther. 2008;16:1276–1282. doi: 10.1038/mt.2008.86. [DOI] [PubMed] [Google Scholar]
- 17.Barry M.A., Rubin J.D., Lu S.C. Retargeting adenoviruses for therapeutic applications and vaccines. FEBS Lett. 2020;594:1918–1946. doi: 10.1002/1873-3468.13731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmid M., Ernst P., Honegger A., Suomalainen M., Zimmermann M., Braun L., Stauffer S., Thom C., Dreier B., Eibauer M., et al. Adenoviral vector with shield and adapter increases tumor specificity and escapes liver and immune control. Nat. Commun. 2018;9:450. doi: 10.1038/s41467-017-02707-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sumida S.M., Truitt D.M., Lemckert A.A.C., Vogels R., Custers J.H.H.V., Addo M.M., Lockman S., Peter T., Peyerl F.W., Kishko M.G., et al. Neutralizing antibodies to adenovirus serotype 5 vaccine vectors are directed primarily against the adenovirus hexon protein. J. Immunol. 2005;174:7179–7185. doi: 10.4049/jimmunol.174.11.7179. [DOI] [PubMed] [Google Scholar]
- 20.Barouch D.H., Kik S.V., Weverling G.J., Dilan R., King S.L., Maxfield L.F., Clark S., Ng'ang'a D., Brandariz K.L., Abbink P., et al. International seroepidemiology of adenovirus serotypes 5, 26, 35, and 48 in pediatric and adult populations. Vaccine. 2011;29:5203–5209. doi: 10.1016/j.vaccine.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dreier B., Mikheeva G., Belousova N., Parizek P., Boczek E., Jelesarov I., Forrer P., Plückthun A., Krasnykh V. Her2-specific multivalent adapters confer designed tropism to adenovirus for gene targeting. J. Mol. Biol. 2011;405:410–426. doi: 10.1016/j.jmb.2010.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dreier B., Honegger A., Hess C., Nagy-Davidescu G., Mittl P.R.E., Grütter M.G., Belousova N., Mikheeva G., Krasnykh V., Plückthun A. Development of a generic adenovirus delivery system based on structure-guided design of bispecific trimeric DARPin adapters. Proc. Natl. Acad. Sci. USA. 2013;110:E869–E877. doi: 10.1073/pnas.1213653110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freitag P.C., Brandl F., Brücher D., Weiss F., Dreier B., Plückthun A. Modular adapters utilizing binders of different molecular types expand cell-targeting options for adenovirus gene delivery. Bioconjug. Chem. 2022;33:1595–1601. doi: 10.1021/acs.bioconjchem.2c00346. [DOI] [PubMed] [Google Scholar]
- 24.Kalluri R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer. 2016;16:582–598. doi: 10.1038/nrc.2016.73. [DOI] [PubMed] [Google Scholar]
- 25.D'Arcangelo E., Wu N.C., Cadavid J.L., Mcguigan A.P. The life cycle of cancer-associated fibroblasts within the tumour stroma and its importance in disease outcome. Br. J. Cancer. 2020;122:931–942. doi: 10.1038/s41416-019-0705-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rettig W.J., Chesa P.G., Beresford H.R., Feickert H.J., Jennings M.T., Cohen J., Oettgen H.F., Old L.J. Differential expression of cell surface antigens and glial fibrillary acidic protein in human astrocytoma subsets. Cancer Res. 1986;46:6406–6412. [PubMed] [Google Scholar]
- 27.Rettig W.J., Garin-Chesa P., Beresford H.R., Oettgen H.F., Melamed M.R., Old L.J. Cell-surface glycoproteins of human sarcomas: differential expression in normal and malignant tissues and cultured cells. Proc. Natl. Acad. Sci. USA. 1988;85:3110–3114. doi: 10.1073/pnas.85.9.3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garin-Chesa P., Old L.J., Rettig W.J. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc. Natl. Acad. Sci. USA. 1990;87:7235–7239. doi: 10.1073/pnas.87.18.7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Welt S., Divgi C.R., Scott A.M., Garin-Chesa P., Finn R.D., Graham M., Carswell E.A., Cohen A., Larson S.M., Old L.J., et al. Antibody targeting in metastatic colon cancer: a phase I study of monoclonal antibody F19 against a cell-surface protein of reactive tumor stromal fibroblasts. J. Clin. Oncol. 1994;12:1193–1203. doi: 10.1200/JCO.1994.12.6.1193. [DOI] [PubMed] [Google Scholar]
- 30.Scott A.M., Wiseman G., Welt S., Adjei A., Lee F.T., Hopkins W., Divgi C.R., Hanson L.H., Mitchell P., Gansen D.N., et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin. Cancer Res. 2003;9:1639–1647. [PubMed] [Google Scholar]
- 31.Jansen K., Heirbaut L., Cheng J.D., Joossens J., Ryabtsova O., Cos P., Maes L., Lambeir A.M., De Meester I., Augustyns K., Van der Veken P. Selective inhibitors of Fibroblast Activation Protein (FAP) with a (4-quinolinoyl)-glycyl-2-cyanopyrrolidine scaffold. ACS Med. Chem. Lett. 2013;4:491–496. doi: 10.1021/ml300410d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kratochwil C., Flechsig P., Lindner T., Abderrahim L., Altmann A., Mier W., Adeberg S., Rathke H., Röhrich M., Winter H., et al. (68)Ga-FAPI PET/CT: Tracer uptake in 28 different kinds of cancer. J. Nucl. Med. 2019;60:801–805. doi: 10.2967/jnumed.119.227967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Millul J., Bassi G., Mock J., Elsayed A., Pellegrino C., Zana A., Dakhel Plaza S., Nadal L., Gloger A., Schmidt E., et al. An ultra-high-affinity small organic ligand of fibroblast activation protein for tumor-targeting applications. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2101852118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Santos A.M., Jung J., Aziz N., Kissil J.L., Puré E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J. Clin. Invest. 2009;119:3613–3625. doi: 10.1172/JCI38988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams S., Miller G.T., Jesson M.I., Watanabe T., Jones B., Wallner B.P. PT-100, a small molecule dipeptidyl peptidase inhibitor, has potent antitumor effects and augments antibody-mediated cytotoxicity via a novel immune mechanism. Cancer Res. 2004;64:5471–5480. doi: 10.1158/0008-5472.CAN-04-0447. [DOI] [PubMed] [Google Scholar]
- 36.Brennen W.N., Rosen D.M., Wang H., Isaacs J.T., Denmeade S.R. Targeting carcinoma-associated fibroblasts within the tumor stroma with a fibroblast activation protein-activated prodrug. J. Natl. Cancer Inst. 2012;104:1320–1334. doi: 10.1093/jnci/djs336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deng L.J., Wang L.H., Peng C.K., Li Y.B., Huang M.H., Chen M.F., Lei X.P., Qi M., Cen Y., Ye W.C., et al. Fibroblast Activation Protein alpha activated tripeptide bufadienolide antitumor prodrug with reduced cardiotoxicity. J. Med. Chem. 2017;60:5320–5333. doi: 10.1021/acs.jmedchem.6b01755. [DOI] [PubMed] [Google Scholar]
- 38.Kakarla S., Chow K.K.H., Mata M., Shaffer D.R., Song X.T., Wu M.F., Liu H., Wang L.L., Rowley D.R., Pfizenmaier K., Gottschalk S. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol. Ther. 2013;21:1611–1620. doi: 10.1038/mt.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schuberth P.C., Hagedorn C., Jensen S.M., Gulati P., Van Den Broek M., Mischo A., Soltermann A., Jüngel A., Marroquin Belaunzaran O., Stahel R., et al. Treatment of malignant pleural mesothelioma by fibroblast activation protein-specific re-directed T cells. J. Transl. Med. 2013;11:187. doi: 10.1186/1479-5876-11-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang L.C.S., Lo A., Scholler J., Sun J., Majumdar R.S., Kapoor V., Antzis M., Cotner C.E., Johnson L.A., Durham A.C., et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014;2:154–166. doi: 10.1158/2326-6066.CIR-13-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Claus C., Ferrara C., Xu W., Sam J., Lang S., Uhlenbrock F., Albrecht R., Herter S., Schlenker R., Hüsser T., et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci. Transl. Med. 2019;11 doi: 10.1126/scitranslmed.aav5989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xin L., Gao J., Zheng Z., Chen Y., Lv S., Zhao Z., Yu C., Yang X., Zhang R. Fibroblast Activation Protein-alpha as a target in the bench-to-bedside diagnosis and treatment of tumors: A narrative review. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.648187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aertgeerts K., Levin I., Shi L., Snell G.P., Jennings A., Prasad G.S., Zhang Y., Kraus M.L., Salakian S., Sridhar V., et al. Structural and kinetic analysis of the substrate specificity of human fibroblast activation protein alpha. J. Biol. Chem. 2005;280:19441–19444. doi: 10.1074/jbc.C500092200. [DOI] [PubMed] [Google Scholar]
- 44.Hanes J., Plückthun A. In vitro selection and evolution of functional proteins by using ribosome display. Proc. Natl. Acad. Sci. USA. 1997;94:4937–4942. doi: 10.1073/pnas.94.10.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Binz H.K., Stumpp M.T., Forrer P., Amstutz P., Plückthun A. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 2003;332:489–503. doi: 10.1016/s0022-2836(03)00896-9. [DOI] [PubMed] [Google Scholar]
- 46.Bosma G.C., Custer R.P., Bosma M.J. A severe combined immunodeficiency mutation in the mouse. Nature. 1983;301:527–530. doi: 10.1038/301527a0. [DOI] [PubMed] [Google Scholar]
- 47.Roder J., Duwe A. The beige mutation in the mouse selectively impairs natural killer cell function. Nature. 1979;278:451–453. doi: 10.1038/278451a0. [DOI] [PubMed] [Google Scholar]
- 48.Micke P., Strell C., Mattsson J., Martín-Bernabé A., Brunnström H., Huvila J., Sund M., Wärnberg F., Ponten F., Glimelius B., et al. The prognostic impact of the tumour stroma fraction: A machine learning-based analysis in 16 human solid tumour types. EBioMedicine. 2021;65 doi: 10.1016/j.ebiom.2021.103269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Harada S., Yanagisawa M., Kaneko S., Yorozu K., Yamamoto K., Moriya Y., Harada N. Superior antitumor activity of trastuzumab combined with capecitabine plus oxaliplatin in a human epidermal growth factor receptor 2-positive human gastric cancer xenograft model. Mol. Clin. Oncol. 2015;3:987–994. doi: 10.3892/mco.2015.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu Z., Tian J., Smith J.S., Byrnes A.P. Clearance of adenovirus by Kupffer cells is mediated by scavenger receptors, natural antibodies, and complement. J. Virol. 2008;82:11705–11713. doi: 10.1128/JVI.01320-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Khare R., Reddy V.S., Nemerow G.R., Barry M.A. Identification of adenovirus serotype 5 hexon regions that interact with scavenger receptors. J. Virol. 2012;86:2293–2301. doi: 10.1128/JVI.05760-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alemany R., Suzuki K., Curiel D.T. Blood clearance rates of adenovirus type 5 in mice. J. Gen. Virol. 2000;81:2605–2609. doi: 10.1099/0022-1317-81-11-2605. [DOI] [PubMed] [Google Scholar]
- 53.Kuklik J., Michelfelder S., Schiele F., Kreuz S., Lamla T., Müller P., Park J.E. Development of a bispecific antibody-based platform for retargeting of capsid modified AAV vectors. Int. J. Mol. Sci. 2021;22:8355. doi: 10.3390/ijms22158355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith S.N., Schubert R., Simic B., Brücher D., Schmid M., Kirk N., Freitag P.C., Gradinaru V., Plückthun A. The SHREAD gene therapy platform for paracrine delivery improves tumor localization and intratumoral effects of a clinical antibody. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2017925118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Levêque D., Gigou L., Bergerat J.P. Clinical pharmacology of trastuzumab. Curr. Clin. Pharmacol. 2008;3:51–55. doi: 10.2174/157488408783329931. [DOI] [PubMed] [Google Scholar]
- 56.Li Z., Li Y., Chang H.P., Chang H.Y., Guo L., Shah D.K. Effect of size on solid tumor disposition of protein therapeutics. Drug Metab. Dispos. 2019;47:1136–1145. doi: 10.1124/dmd.119.087809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brücher D., Kirchhammer N., Smith S.N., Schumacher J., Schumacher N., Kolibius J., Freitag P.C., Schmid M., Weiss F., Keller C., et al. iMATCH: an integrated modular assembly system for therapeutic combination high-capacity adenovirus gene therapy. Mol. Ther. Methods Clin. Dev. 2021;20:572–586. doi: 10.1016/j.omtm.2021.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rovedatti L., Di Sabatino A., Knowles C.H., Sengupta N., Biancheri P., Corazza G.R., Macdonald T.T. Fibroblast activation protein expression in Crohn's disease strictures. Inflamm. Bowel Dis. 2011;17:1251–1253. doi: 10.1002/ibd.21446. [DOI] [PubMed] [Google Scholar]
- 59.Bauer S., Jendro M.C., Wadle A., Kleber S., Stenner F., Dinser R., Reich A., Faccin E., Gödde S., Dinges H., et al. Fibroblast activation protein is expressed by rheumatoid myofibroblast-like synoviocytes. Arthritis Res. Ther. 2006;8:R171. doi: 10.1186/ar2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Croft A.P., Campos J., Jansen K., Turner J.D., Marshall J., Attar M., Savary L., Wehmeyer C., Naylor A.J., Kemble S., et al. Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature. 2019;570:246–251. doi: 10.1038/s41586-019-1263-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Acharya P.S., Zukas A., Chandan V., Katzenstein A.L.A., Puré E. Fibroblast activation protein: a serine protease expressed at the remodeling interface in idiopathic pulmonary fibrosis. Hum. Pathol. 2006;37:352–360. doi: 10.1016/j.humpath.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 62.Levy M.T., Mccaughan G.W., Abbott C.A., Park J.E., Cunningham A.M., Müller E., Rettig W.J., Gorrell M.D. Fibroblast activation protein: a cell surface dipeptidyl peptidase and gelatinase expressed by stellate cells at the tissue remodelling interface in human cirrhosis. Hepatology. 1999;29:1768–1778. doi: 10.1002/hep.510290631. [DOI] [PubMed] [Google Scholar]
- 63.Binz H.K., Amstutz P., Kohl A., Stumpp M.T., Briand C., Forrer P., Grütter M.G., Plückthun A. High-affinity binders selected from designed ankyrin repeat protein libraries. Nat. Biotechnol. 2004;22:575–582. doi: 10.1038/nbt962. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are available from the corresponding author, A.P., upon reasonable request.