

Significance
Lung fibrosis is a progressive, lethal disease with limited treatment options available that do not provide cure. Hence, a better understanding of the inflammatory pathophysiology and molecular mechanisms of tissue remodeling is mandatory. In this study, we uncover that externalized histones from neutrophil extracellular traps act profibrotic by shifting the balance of critical cytokines. Externalized histones activate platelets to release TGFβ1, which subsequently antagonizes antifibrotic Interleukin-27 (IL-27) production from macrophages via multiple intracellular signaling pathways. The importance of these mechanisms is highlighted by the observation that blocking monoclonal antibodies against externalized histones protect from excessive collagen matrix deposition in injured lungs.
Keywords: cytokines, innate immunity, macrophages, platelets, pulmonology
Abstract
Externalized histones erupt from the nucleus as extracellular traps, are associated with several acute and chronic lung disorders, but their implications in the molecular pathogenesis of interstitial lung disease are incompletely defined. To investigate the role and molecular mechanisms of externalized histones within the immunologic networks of pulmonary fibrosis, we studied externalized histones in human and animal bronchoalveolar lavage (BAL) samples of lung fibrosis. Neutralizing anti-histone antibodies were administered in bleomycin-induced fibrosis of C57BL/6 J mice, and subsequent studies used conditional/constitutive knockout mouse strains for TGFβ and IL-27 signaling along with isolated platelets and cultured macrophages. We found that externalized histones (citH3) were significantly (P < 0.01) increased in cell-free BAL fluids of patients with idiopathic pulmonary fibrosis (IPF; n = 29) as compared to healthy controls (n = 10). The pulmonary sources of externalized histones were Ly6G+CD11b+ neutrophils and nonhematopoietic cells after bleomycin in mice. Neutralizing monoclonal anti-histone H2A/H4 antibodies reduced the pulmonary collagen accumulation and hydroxyproline concentration. Histones activated platelets to release TGFβ1, which signaled through the TGFbRI/TGFbRII receptor complex on LysM+ cells to antagonize macrophage-derived IL-27 production. TGFβ1 evoked multiple downstream mechanisms in macrophages, including p38 MAPK, tristetraprolin, IL-10, and binding of SMAD3 to the IL-27 promotor regions. IL-27RA-deficient mice displayed more severe collagen depositions suggesting that intact IL-27 signaling limits fibrosis. In conclusion, externalized histones inactivate a safety switch of antifibrotic, macrophage-derived IL-27 by boosting platelet-derived TGFβ1. Externalized histones are accessible to neutralizing antibodies for improving the severity of experimental pulmonary fibrosis.
Pulmonary fibrosis is a devastating disease with frequent progression to respiratory failure and death (1). Insights into its molecular pathogenesis are imperative to devise better therapies in the future. The excess accumulation of extracellular matrix components (ECM) by myofibroblasts is shaped by a chronic inflammation encompassing complex interactions of multiple cell types including neutrophils, platelets, alveolar macrophages (AM), and T cells (2, 3). In general, these hematopoietic cells possess powerful effector functions and can communicate via cytokines such as TGFβ and IL-27.
Transforming growth factor β (TGFβ) plays a central role in aggravating lung fibrosis (2, 4). Activated TGFβ1-3 isoforms bind to the TGFβ type II receptor (TGFbRII) for initiating TGFβ type I receptor (TGFbRI) phosphorylation (5). Subsequently, the TGFbRI/TGFbRII complex phosphorylates DNA-binding SMAD2/SMAD3 transcription factors and also activates non-SMAD pathways (e.g., p38 MAPK, JNK, PI3K-Akt) to promote ECM formation (5).
Interleukin-27 (IL-27), a member of the IL-12 family, is a heterodimeric cytokine formed by noncovalent association of the subunits p28 (IL-27p28) and EBI3 (6). EBI3 can also assemble with p35 to form IL-35, designating IL-27p28 as the signature subunit of IL-27 (7). Macrophages and dendritic cells are major sources of IL-27 (6). IL-27 binds to its heterodimeric receptor consisting of a ligand binding-chain, IL-27RA (WSX-1), and a signal-transducing chain, gp130, both expressed in lymphocytes (7, 8).
Externalized histones are released either as major components of neutrophil extracellular traps (NETs) or by dying nonimmune cells (9). Hypercitrullination of nuclear histones precedes NET formation and is catalyzed by protein arginine deiminase 4 (PAD4) (10). Citrullinated histone H3 in plasma, biofluids, and tissues is a specific and reliable biomarker for the presence of NETs (11–13). Externalized histones perpetuate tissue injury by mediating cytotoxicity in endothelial cells and lung epithelial cells (14, 15). The cytotoxic activity of histone H4 is the highest of all histone proteins (14, 15). In addition, externalized histones act as damage-associated molecular patterns by ligation with pattern recognition receptors (PRRs), e.g., Toll-like receptors 2 and 4 (9). These events initiate activation of platelets (16), immune cell chemotaxis (14, 15), and the release of chemokines/cytokines (15). NETs and externalized histones are involved in the pathophysiology of acute lung injury (15, 17, 18), sepsis (14), infection (19, 20), autoimmune disease (21), thrombosis (16, 22), and pulmonary fibrosis (23, 24).
The central objective of the presented study was to characterize the role and involvement of externalized histones in the cellular and molecular networks of pulmonary fibrosis. Our data demonstrate that fibrotic tissue remodeling is intensified by externalized histones via a cytokine-dependent mechanism, that involves release of platelet-derived TGFβ1 to suppress anti-fibrotic IL-27 production of macrophages.
Methods
Ethics.
The deidentified bronchoalveolar lavage fluids (BALF) from idiopathic pulmonary fibrosis (IPF) patients (21 males, 8 females, age: 64.3 ± 10.7 y; SI Appendix, Table S1) and healthy volunteers (5 males, 5 females, age: 40.3 ± 18.8 y) were obtained from the UGMLC/DZL Gießen Biobank. The samples had been collected prior to the current study with written informed consent and approval from the ethics committee of the Justus-Liebig University, Germany. All studies with mice were approved by the State Investigation Office of Rhineland-Palatinate and the Institutional Animal Care and Use Committee of Boston University.
Pulmonary Fibrosis.
Lung injury and subsequent pulmonary fibrosis were induced by intratracheal bleomycin of naïve or bone marrow chimeric mouse strains with collection of BALF and lung tissue at indicated time points. Additional details on the genetically engineered mouse strains and extended methods are provided as SI Appendix.
Isolation and Cultures of Cells.
Macrophages and MLE-12 cells were obtained and cultured as described before (6, 15, 25). Lentiviral transductions of shRNAs were performed using reagents from Sigma-Aldrich. Platelets were isolated from freshly drawn anticoagulated mouse blood and their reactivity studied by light transmission aggregometry. Further details are available in SI Appendix.
Flow Cytometry and Imaging Flow Cytometry.
Flow cytometry protocols of immune cells were used as reported before (6, 26). Details on antibodies for surface markers, intracellular IL-27p28 staining and imaging of NETs are presented in SI Appendix.
Protein Detection.
IL-27p28, IL-10, TGFβ1 and citrullinated histone H3 were detected by ELISA (20). Other mediators and phospho-p38 MAPKThr180/Tyr182 were quantified by bead-based assays (Luminex-200) (25). Collagen I–V in lungs was measured by the SIRCOL assay (Biocolor). The hydroxyproline assay kit was from Sigma-Aldrich. GFP reporter fluorescence in BALF was measured by Fluoroskan Ascent FL (Thermo Scientific), Ascent Software 2.6. Further details are available in SI Appendix.
Reverse Transcription PCR.
RNA isolation, reverse transcription, and PCR were performed as described before (27). Additional details including primer sequences are available in SI Appendix.
Chromatin Immunoprecipitation (ChIP).
The Magna ChIP kit (Millipore) was used on sonicated DNA of formaldehyde fixed macrophages. Additional details and antibodies are listed in SI Appendix.
Histology.
Paraffin-embedded lung sections for light microscopy were stained according to the Masson–Goldner’s Trichrome protocol. Further details are available in SI Appendix.
Reagents.
Information on reagents, antibodies, and recombinant proteins used in this study are listed as tables in SI Appendix.
Statistical Analysis.
Data are shown as mean, and error bars represent SEM GraphPad Prism Version 9 was used for statistical analysis. Student’s t test was used to compare two groups, and one-way ANOVA was used for more than two groups. The human samples were analyzed by nonparametric Mann–Whitney test because normality tests indicated a nonnormal distribution. The numbers of patient samples or mice per group in vivo are indicated by circles in the figures. The numbers of independent biological replicate experiments in vitro were typically 2–4 and are specified in the figure legends. Values of P < 0.05 were considered significant. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Results
Release of Externalized Histones during Pulmonary Fibrosis.
The hypothesis that externalized histones contribute to the pathogenesis of lung fibrosis relies on the prerequisite that these factors are present under such conditions. Therefore, we initially analyzed cryopreserved human BALF samples for the presence of citrullinated histone H3 as a surrogate marker for NETs. In fact, significantly higher amounts of citH3 (P < 0.01) were detectable in BALF from human patients with IPF (n = 29) as compared to healthy controls (n = 10) (Fig. 1A). The mean of optical densities values for citH3 by nonquantitative ELISA was approximately fivefold higher in the IPF group than in the healthy subjects. The clinical characteristics of IPF patients are shown in SI Appendix, Table S1.
Fig. 1.

Cellular sources of externalized histones during pulmonary fibrosis. (A) Presence of externalized citrullinated histone H3 in BALF samples of healthy human controls (n = 10) and patients with IPF (n = 29), ELISA with optical density (OD) measurements at 405 nm. (B and C) Time course of citrullinated histone H3 (B) and Histone H4 (C) in cell-free BALF of C57BL/6 J mice (n ≥ 4/group for each time point) after treatment with bleomycin i.t. or NaCl 0.9% i.t. (sham; negative control), ELISA. (D) NET formation detected as externalized citrullinated histone H3 on nonpermeabilized Ly6G+CD11b+ neutrophils in BALF of C57BL/6 J wild-type (WT) mice 2 d after bleomycin i.t. (n = 6 mice/group), flow cytometry with pregating on CD11b. Absolute cell numbers in the quadrants of interest are shown in blue font. (E) Visualization of NETs from BALF cells, which were obtained 2 d after bleomycin i.t. and stained for citrullinated externalized histone H3 (green) together with Ly6G (red) for neutrophils, ImageStreamX Mark II imaging flow cytometry. (F) PAD4−/− mice and WT mice were subjected to bleomycin-induced lung injury and citrullinated H3 was detected in cell-free BALF after 2 d. (G) Detection of green fluorescence in cell-free BALF of histone H2B-eGFP fusion protein reporter mice (H2B-eGFP+/−) 2 d after bleomycin administration as compared to WT control mice. Fluorescence of a FITC-labeled control antibody was used for normalization (=100% value). (H) Chimeric mice were generated by transplantation of H2B-eGFP+/− donor bone marrow into irradiated WT recipients. In the control groups, syngeneic bone marrow was transplanted. After 5 wk, all mice received bleomycin i.t. and H2B-eGFP fluorescence was detected in cell-free BALF another 2 d later. Numbers of mice in frames F–H are indicated by circles; A: Mann–Whitney test, B and C: one-way ANOVA, F–H: Student’s t test, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To model the appearance of externalized histones in a defined experimental setting of lung fibrosis, C57BL/6 J (wild type; WT) mice received bleomycin i.t. or sterile NaCl 0.9% i.t. as control/sham. Externalized citrullinated histone H3 (citH3) was increased in cell-free BALF after bleomycin-induced lung injury with maximal amounts detectable at day 2 followed by a subsequent decline continuing up to day 10 (Fig. 1B). Histone H4 peaked at day 3 and subsequently declined until day 10 after injury (Fig. 1C). The citH3 was present on Ly6G+ neutrophils indicating NET formation (Fig. 1D). These NETs were further visualized by imaging flow cytometry of nonpermeabilized Ly6G+ neutrophils (red), and the eruptive citH3 (green), which showed a variety of cone-like and spear-like shapes (Fig. 1E; see SI Appendix, Fig. S1 for gating strategy). The release of citH3 was reduced in PAD4−/− mice, which is consistent with the concept that PAD4 is essential for generation of citrullinated extracellular traps (Fig. 1F).
Altogether, these data indicated that recruited lung neutrophils released NETs as a source of externalized histones early in the course of experimental pulmonary fibrosis. However, stromal lung cells may also contribute to externalized histones because of bleomycin-mediated and/or perpetuating histone-mediated cytotoxicity (SI Appendix, Fig. S2A).
To test whether nonhematopoietic cells generate externalized histones, we employed transgenic reporter mice with human histone H2B fused to eGFP. The cell-free BALF of H2B-eGFP+/− mice showed increased green fluorescence after intratracheal bleomycin as compared to baseline signals in WT mice (Fig. 1G). Next, chimeric mice were generated by transplantation of donor H2B-eGFP+/− bone marrow into lethally irradiated WT mice (H2B-eGFP+/−WT; SI Appendix, Fig. S2B), with WT and H2B-eGFP+/−H2B-eGFP+/− transplantations as controls. The engraftment efficacy was confirmed in Ly6G+CD11b+ neutrophils from bone marrow (SI Appendix, Fig. S2C). Bleomycin-induced lung injury resulted in green fluorescence of cell-free BALF from chimeric WT mice (Fig. 1H). However, these signals from H2B-eGFP+/−WT mice were substantially lower as compared to H2B-eGFP+/−H2B-eGFP+/− positive controls, suggesting that externalized histones can also be released by nonhematopoietic cells. The presence of H2B-eGFP fusion proteins did not significantly change the severity of lung injury as assessed by alveolar albumin leakage (SI Appendix, Fig. S2D).
In summary, the finding that externalized histones were released from different cellular sources suggested that their direct blockade (using neutralizing anti-histone antibodies) could be more promising than specific inhibition of NET formation (using PAD4 inhibitors).
Neutralization of Externalized Histones Improves Pulmonary Fibrosis by Limiting TGFβ1 Release.
To study the functional relevance of externalized histones in pulmonary fibrosis, we sought to neutralize the most cytotoxic histone H4 by monoclonal blocking antibodies (clone BWA3; specificity for H2A and H4) (14, 15). Treatment with anti-H2A/H4 antibodies versus isotype IgG1k antibodies markedly reduced the severity of ECM deposition in WT mouse lungs during fibrosis as documented by representative histology (Fig. 2A), and their quantitative whole slide image analysis (Fig. 2B). The mRNA levels for collagen I were substantially lower in lung tissue of anti-H2A/H4 antibody-treated mice (Fig. 2C). The amounts of collagen I–V protein decreased significantly on the anti-histone regimen (Fig. 2D). Hydroxyproline concentrations in the BALF and lung tissues were also lowered after H2A/H4 neutralization compared to IgG1k controls at 4 wk after bleomycin-induced injury (Fig. 2 E and F), indicating reduced lung fibrosis. The hydroxyproline assays confirmed histology findings (Fig. 2 A and B) and other collagen detection methods using RT-PCR and SIRCOL assay (Fig. 2 C and D).
Fig. 2.

Externalized histones promote pulmonary fibrosis. (A) Histology of lungs from WT mice which received either monoclonal anti-H2A/H4 antibodies (clone: BWA3, 250 µg/mouse i.v.) or mouse IgG1κ isotype control antibodies simultaneously with i.t. bleomycin (n = 3–6/group). Masson–Goldner’s trichrome staining of lungs was performed after 4 wk (Scale bar, 50 μm). (B) Quantification of lung collagen deposition from scanned whole slides from mice in frame A. (C) ECM expression as evaluated by RT-PCR for collagen I (col1a2) mRNA in lung homogenates during bleomycin-induced fibrosis. (D) SIRCOL assay for collagen I–V in lung tissue of anti-histone antibody-treated mice or control IgG1k-treated mice after bleomycin i.t. (E and F) Hydroxyproline concentrations in BALF and lung tissue of anti-histone antibody-treated mice as compared to IgG1k controls during fibrosis, 4 wk. (G and H) TGFβ1 and IL-27p28 concentrations in BALF of anti-histone antibody-treated mice as compared to IgG1k controls at 2 wk or 4 wk after bleomycin, ELISA. Horizontal dashed lines indicate groups of samples which were analyzed on separate days and ELISA plates. Experiments in all frames were done with C57BL/6 J (WT) mice and analyzed 4 wk (frame A–F) or 2 wk (stated in frame G and H) after bleomycin i.t. (1 U/kg body weight). Numbers of mice in frames B–H are indicated by circles; Student’s t test or one-way ANOVA; *P < 0.05, **P < 0.01, ns: not significant.
In addition, anti-H2A/H4 antibodies reduced the expression of fibroblast-specific protein-1 (fsp-1), α-smooth muscle actin (α-sma), vimentin, slug, snail, and zeb2 as markers of epithelial-to-mesenchymal transition (SI Appendix, Fig. S3 A and B).
To further investigate the mechanisms of how externalized histones promote fibrosis, we broadly screened the mouse BAL fluids for differences in the production of cytokines. Notably, neutralization of externalized histones resulted in approximately twofold lower concentrations of TGFβ1 in BALF at 2 wk and 4 wk after bleomycin (Fig. 2G), suggesting that externalized histones increased TGFβ1 during lung fibrosis. Secondly, anti-H2A/H4 antibodies resulted in a reciprocal upregulation of IL-27p28 in BALF after 2 wk, while IL-27p28 was not detectable after 4 wk (Fig. 2H). The appearance of IL-27p28 and TGFβ1 preceded the ECM accumulation in lungs, further suggesting that these mediators play a causative role in pathogenesis of fibrosis (SI Appendix, Fig. S4 A–C). Lung fibrosis was observed after bleomycin injury through Masson–Goldner’s trichrome and Hematoxylin and Eosin staining (SI Appendix, Fig. S4 D and E).
Externalized Histones Activate Platelets to Release TGFβ1 for Aggravating Fibrosis.
Platelets are a prominent source of TGFβ and platelets interact with NETs (17, 28). We found CD41+CD49b+ platelets to moderately increase in lungs after bleomycin administration as studied by flow cytometry (SI Appendix, Fig. S5A). In addition, accumulation of CD42b+ platelets in lungs after bleomycin injury was corroborated by histology and quantitative image analysis after brightfield immunohistochemistry (SI Appendix, Fig. S5 B and C). Externalized/purified histones mediated the aggregation of isolated platelets through interaction with TLR2 and TLR4 receptors (SI Appendix, Fig. S5D) (29). Incubation of washed platelets with endotoxin-free, purified histones for 10 min at 37 °C resulted in a fourfold increase of TGFβ1 release in supernatants as compared to unstimulated controls, although histones were found to be less potent than thrombin (Fig. 3A). There was a significant increase in TGFβ1 release from platelets after incubation with recombinant human histones (H2A, H2B, H3, H4), citrullinated histone H3, and human thrombin (positive control) compared to unstimulated negative controls (Fig. 3B). Next, we generated mice with platelet-specific genetic deletion of TGFβ1. In contrast to constitutive TGFβ1−/− mice, which develop an early lethal inflammatory syndrome, the ΔPltTGFβ1 mice were healthy and fertile. As expected, the isolated platelets from these mice showed defective release of TGFβ1 as compared to littermate controls after thrombin activation (Fig. 3C). In addition, untreated platelet-specific TGFβ1-deficient mice displayed ~80% reduction of TGFβ1 in plasma (Fig. 3D), while platelet aggregation was not altered (SI Appendix, Fig. S5E). When purified histones were administered intratracheally to platelet-specific TGFβ1-deficient mice, lower concentrations of TGFβ1 were noted in BALF after 8Â h compared to TGFβ1fl/fl controls, while levels were significantly higher compared to sham (Fig. 3E). BALF from platelet-specific TGFβ1-deficient mice showed no detectable TGFβ1 levels before and after bleomycin injury while elevated amounts were observed in TGFβ1fl/fl and WT mice compared to their respective controls (Fig. 3F). These experiments suggested that platelets are a major source of TGFβ1 in response to bleomycin injury and externalized histones in vivo, although high doses of purified histones may trigger a release also from other cell types.
Fig. 3.

TGFβ1 is released by platelets in response to externalized histones. (A) Platelets were isolated from C57BL/6 J mice and activated with purified histones (50 µg/mL) or thrombin (100 ng/mL) for 10 min before quantification of TGFβ1 in supernatants. (B) TGFβ1 release from platelets after incubation with recombinant human histones (H2A, H2B, H2, H4), citrullinated histone H3, human thrombin, and untreated (Ctrl), 10 min. (C) Isolated platelets from mice with platelet-specific gene deletion of TGFβ1 (ΔPltTGFβ1) mice and littermate controls (TGFβ1fl/fl) were activated with thrombin (100 ng/mL) for 10 min before quantification of TGFβ1 in supernatants. (D) Plasma concentrations of TGFβ1 in mice with platelet-specific deletion compared to littermate controls, 4 wk after bleomycin. (E) ΔPltTGFβ1 mice and TGFβ1fl/fl control mice received purified histones i.t. (100 µg/mouse) or PBS i.t. (sham) and TGFβ1 was detected in BALF after 8 h. (F) TGFβ1 in BALF of mice with platelet-specific deletion compared to TGFβ1fl/fl littermate controls and C57BL/6 J (WT) mice, 24 h after bleomycin. TGFβ1 was measured by ELISA in all frames. Frames A–C are representative of three independent experiments and frames D–F were done with numbers of mice as indicated by circles; Student’s t test or one-way ANOVA; **P < 0.01, ***P < 0.001, ****P < 0.0001, ns: not significant.
In bleomycin-induced fibrosis, platelet-specific TGFβ1-deficient mice showed markedly reduced interstitial collagen depositions in their lungs together with a lower degree of alveolar simplifications in Masson-Goldner’s trichrome histology (Fig. 4A). These changes were corroborated in quantitative image analysis compared to TGFβ1fl/fl control group mice (Fig. 4B). The accumulations of collagen I mRNA and collagen I–V protein were diminished in lung tissue of mice with deletion of TGFβ1 in platelets (Fig. 4 C and D). On the other hand, TGFβ1 deficiency resulted in higher concentrations of IL-27p28 in BALF (Fig. 4E).
Fig. 4.

Platelet-derived TGFβ1 drives pulmonary fibrosis. (A) Histology of lung sections 4 wk after bleomycin-induced fibrosis. ΔPltTGFβ1 lungs were compared to TGFβ1fl/fl control lungs, Masson–Goldner’s trichrome staining (Scale bar, 50 µm), n = 6/group. (B) Quantification of lung collagen deposition from scanned whole slides from mice in frame A. (C) RT-PCR for collagen I gene expression in lung homogenates of ΔPltTGFβ1 mice and TGFβ1fl/fl control mice during bleomycin-induced fibrosis. (D) Collagen I–V protein content in lungs of ΔPltTGFβ1 mice and TGFβ1fl/fl mice, SIRCOL assay. (E) IL-27p28 in BALF of ΔPltTGFβ1 and TGFβ1fl/fl mice during bleomycin-induced lung fibrosis, ELISA. (F) Supernatants of activated washed platelets from ΔPltTGFβ1 mice or TGFβ1fl/fl control mice were transferred to cell cultures of C57BL/6 J bone marrow-derived macrophages (BMDM) before stimulation with LPS for 24 h and quantification of IL-27p28 release by ELISA. Frames B–E (4 wk of bleomycin i.t.) were done with numbers of mice as indicated by circles and frame F is representative of three independent experiments; Student’s t test or one-way ANOVA; *P < 0.05, **P < 0.01, ***P < 0.001.
To further study the interference of TGFβ1 with IL-27p28 release, we transferred supernatants from platelets with TGFβ1 deletion and controls to cultures of TLR4/LPS-activated macrophages (Fig. 4F). Platelet supernatants from TGFβ1 competent control mice strongly antagonized IL-27p28 release, while this effect was mitigated with platelet supernatants from platelet-specific TGFβ1-deficient mice (Fig. 4F).
TGFβ1 Attenuates Macrophage-Derived IL-27p28 Release via TGFβ Receptor Type I/II Signaling.
To better characterize the relationship between TGFβ1 and IL-27p28, we used cultured macrophages from C57BL/6 J mice. Macrophages of several origins such as AM, peritoneal macrophages (PEM), and BMDM displayed strong inhibition of TLR4-induced release of IL-27p28 by recombinant TGFβ1 (Fig. 5A). Similar effects were observed in the AM cell line, MH-S, and in RAW264.7 macrophages (Fig. 5A). TGFβ1 antagonized IL-27p28 production in both M1 and M2 polarized macrophages (SI Appendix, Fig. S6A). Collectively, this suggested that suppression of IL-27p28 by TGFβ1 is a fundamental phenomenon regardless of macrophage origin or polarization.
Fig. 5.
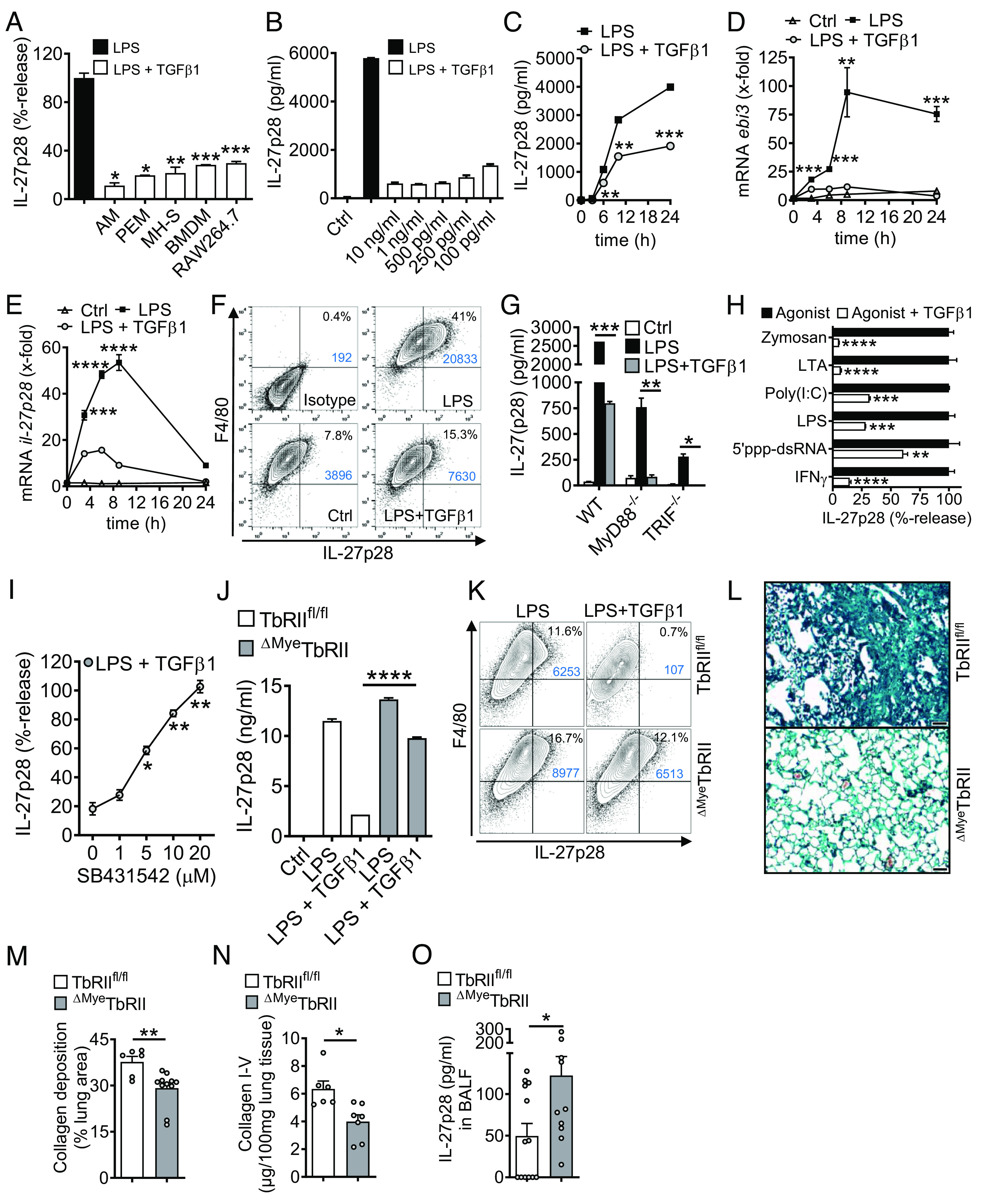
TGFβ receptor type I/II signaling mediates suppression of IL-27p28 in macrophages. (A) Mononuclear phagocytic cells were incubated with LPS (100 ng/mL) ± TGFβ1 (10 ng/mL) followed by detection of IL-27p28 in supernatants; C57BL/6 J-derived AM and PEM, mouse AM cell line (MH-S), C57BL/6 J BMDM and RAW264.7 macrophage cell line, 24 h. LPS was set as 100% for each cell type. (B) TLR4/LPS-activated BMDM were incubated with different concentrations of TGFβ1 before quantification of IL-27p28, 24 h. (C) Time course of IL-27p28 release by LPS-activated BMDM ± TGFβ1. (D) RT-PCR for ebi3 mRNA after LPS ± TGFβ1 in BMDM. (E) RT-PCR for il-27p28 mRNA after LPS ± TGFβ1 in BMDM. (F) Flow cytometry for intracellular IL-27p28 in BMDM, 12 h, pregated on CD11b+ cells. (G) Inhibition of IL-27p28 release by TGFβ1 in BMDM from C57BL/6 J (WT), MyD88−/− or TRIF−/− mice, 24 h. (H) Suppression of IL-27p28 release by TGFβ1 in BMDM activated with several agonists; Zymosan (TLR2), LTA (TLR2), Poly(I:C) (TLR3), LPS (TLR4), 5′ppp-dsRNA (RIG-I) and IFNγ, 24 h. (I) Reversal of TGFβ1-mediated IL-27p28 suppression by increasing concentrations of the TGFβ receptor I signaling inhibitor, SB431542, in TLR4/LPS-activated BMDM, 24 h. LPS alone was used for normalization (=100% value). (J) IL-27p28 from BMDM of TGFβ receptor II floxed (TbRIIfl/fl) mice and ΔMyeTbRII mice after LPS ± TGFβ1, 24 h. (K) Flow cytometry of intracellular IL-27p28 in BMDM from TbRIIfl/fl or ΔMyeTbRII mice, 12 h. (L) Lung histology of bleomycin-induced fibrosis in TbRIIfl/fl or ΔMyeTbRII mice, 4 wk, Masson–Goldner’s trichrome staining (Scale bar, 50 µm). (M) Quantification of lung collagen deposition from scanned whole slides from mice in frame L. (N) Collagen I–V in lungs of TbRIIfl/fl mice and ΔMyeTbRII mice after bleomycin i.t, 4 wk, SIRCOL assay. (O) IL-27p28 in BALF of bleomycin-treated TbRIIfl/fl mice and ΔMyeTbRII mice, 4 wk. Frames A-CG-JO: ELISA. Representatives of three independent experiments (frames A–K) or n ≥ 6 mice/group (frames L–O); Student’s t test or one-way ANOVA; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
TGFβ1 (in dosages from 10 ng/mL to 100 pg/mL) was a potent and consistent suppressor of IL-27p28 (Fig. 5 B and C). The expression of mRNA for both IL-27 subunits, il-27p28 and ebi3, was antagonized by TGFβ1 (Fig. 5 D and E). TGFβ1 decreased intracellular IL-27p28 protein in F4/80+CD11b+ macrophages (41% vs. 15.3%; Fig. 5F). TGFβ1 interfered with both the MyD88/TRIF pathways to suppress TLR4-induced IL-27p28 (Fig. 5G). IL-27p28 release was activated by agonists for TLR2, TLR3, TLR4, RIG-I, and by IFNγ, all of which were antagonized by TGFβ1 (Fig. 5H).
TGFβ1 ligates with the heteromeric TGFβRI/TGFβRII receptor complex. SB431542, a small molecule inhibitor for TGFβRI signaling via ALK5 (and ALK4/ALK7), nullified the inhibition by TGFβ1 in a dose-dependent manner (Fig. 5I). F4/80+CD11b+ macrophages from mice with myeloid cell-specific TGFβRII-deficiency were resistant to TGFβ1-mediated suppression of IL-27p28 (Fig. 5 J and K). In pulmonary fibrosis, the genetic ablation of TGFβRII resulted in protection from histologic ECM deposition and in lower amounts of collagen in lungs (Fig. 5 L–N). The concentrations of IL-27p28 in BALF were higher in myeloid cell–specific TGFβRII-deficient mice, which provided additional evidence that TGFβ1 suppressed the release of IL-27p28 by macrophages during pulmonary fibrosis (Fig. 5O).
TGFβ1 Antagonizes Macrophage-Derived IL-27p28 Production by Orchestration of Multiple Mechanisms.
To further investigate the intracellular mechanisms how TGFβ1 inhibits IL-27p28, we turned our attention to SMAD3. We confirmed that TGFβ1 mediated rapid phosphorylation of SMAD3/2 at Ser465/Ser467 and Ser423/Ser425 in F4/80+CD11b+ macrophages (SI Appendix, Fig. S6 B and C). Next, we performed lentiviral stable transductions of shRNAs for silencing of SMAD3 in RAW264.7 macrophages. Two independent shRNA sequences decreased smad3 mRNA expression to 39% and 34% of basal levels, along with reduced frequencies of F4/80+CD11b+SMAD3+ RAW264.7 macrophages as compared to nontarget shRNA or nontransduced controls (SI Appendix, Fig. S6 D and E). The SMAD3 silencing significantly diminished the efficacy of TGFβ1 to suppress IL-27p28 release by TLR4-activated macrophages (Fig. 6A). Similar findings for il-27p28 mRNA suggested the involvement of SMAD3 as a regulator of gene expression (Fig. 6B). We identified putative SMAD3 binding sites (i.e., CAGA boxes) in the promoter regions of il-27p28 and ebi3 by computational analysis (SI Appendix, Fig. S6 F and G). Chromatin immunoprecipitation (ChIP) assays demonstrated that TGFβ1 reversed the TLR4/LPS-induced loss of SMAD3 binding to the corresponding promoter regions of il-27p28 and ebi3 (Fig. 6 C and D). In summary, these findings suggested a requirement of SMAD3 for suppression of IL-27 by TGFβ1.
Fig. 6.

TGFβ1-mediated antagonism of IL-27p28 release via SMAD3, p38, TTP, and IL-10. (A) IL-27p28 release with reduced activity of TGFβ1 in lentiviral transduced RAW264.7 macrophages with shRNA knockdown of SMAD3 (two independent shRNAs) as compared to nontarget shRNA (Ctrl), 24 h, ELISA. (B) TGFβ1 partially fails to suppress il-27p28 mRNA expression in transduced RAW264.7 macrophages after treatments with LPS ± TGFβ1, 6 h, RT-PCR. (C) TGFβ1 restores SMAD3 binding to the il-27p28 promoter region, BMDM, 3 h, ChIP assay. (D) TGFβ1 restores SMAD3 binding to the ebi3 promoter region, BMDM, 3 h, ChIP assay. (E) Effects of several p38 MAPK inhibitors (SB 203580 [10 µM], PH-797804 [10 nM], BIRB796 [50 nM]) on IL-27p28 release in BMDM after LPS ± TGFβ1, 24 h, ELISA. (F) Reduced capacity of TGFβ1 to suppress IL-27p28 release in BMDM from tristetraprolin-deficient (TTP−/−) mice as compared to WT mice, ELISA, 24 h. (G) Flow cytometry of F4/80+CD11b+IL-27p28+ BMDM from TTP−/− mice and WT mice after LPS ± TGFβ1, 12 h, pregated on CD11b. (H) IL-27p28 release from BMDM incubated with LPS ± TGFβ1 in the copresence of blocking IL-10 receptor antibodies (α-IL10R; 10 µg/mL) or rat IgG1 isotype control antibodies (10 µg/mL), ELISA. (I) Defective suppression of IL-27p28 by TGFβ1 in BMDM from IL-10−/− mice as compared to WT mice. BMDM were from C57BL/6 J (WT) mice in all frames unless specified otherwise; LPS (100 ng/mL), TGFβ1 (10 ng/mL). Data in each frame are representative of three independent experiments; Student’s t test or one-way ANOVA; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001; ns: not significant.
A noncanonical signaling pathway (SMAD-independent) of TGFβ1 is phosphorylation of p38 MAPK, which we confirmed to occur at Thr180/Tyr182 in macrophages (SI Appendix, Fig. S6H). Selective small molecule inhibitors (SB 203580, PH-797804, BIRB796) for p38 MAPK increased IL-27p28 release and reversed its TGFβ1-mediated inhibition (Fig. 6E). Hence, these data suggested that TGFβ1 recruited p38 MAPK as an inhibitory signaling pathway for limiting IL-27p28 production.
Tristetraprolin (TTP; zfp36) is a zinc finger protein homolog and downstream target of p38 MAPK, which can mediate some actions of TGFβ (30). To test whether TTP plays a role in production of IL-27p28, we obtained macrophages from TTP-deficient mice. In fact, TGFβ1 was less potent to antagonize IL-27p28 release in TTP-deficient macrophages (Fig. 6F). The data are presented as relative values for better comparison, because the TTP-deficient macrophages produced approximately twofold higher amounts of IL-27p28 upon activation. In flow cytometry, activated F4/80+CD11b+ macrophages deficient of TTP showed more intracellular IL-27p28 protein, but lower responsiveness to TGFβ1 as compared to WT macrophages (Fig. 6G).
We have reported that IL-10 antagonizes IL-27 production (6). In fact, TGFβ1 promoted the release of IL-10 in low concentrations from TLR4/LPS-activated macrophages at later time points (>6 h) (SI Appendix, Fig. S6I). Blockade of IL-10R signaling by monoclonal antibodies attenuated the TGFβ1-mediated inhibition of IL-27p28 (Fig. 6H). In addition, TGFβ1 was less potent to suppress IL-27p28 in IL-10-deficient macrophages (Fig. 6I). This suggested that an autocrine/paracrine IL-10 loop contributed to a sustained downmodulation of IL-27p28 by TGFβ1.
Susceptibility of IL-27RA-Deficient Mice During Pulmonary Fibrosis.
To further shed light into the role of IL-27, we studied IL-27RA−/− mice in bleomycin-induced pulmonary fibrosis. Histological analysis and quantification using whole slide scanning of trichrome stained lung sections of IL-27RA−/− mice showed more severe accumulation of ECM with alveolar simplification as compared to WT mice and a significant increase in collagen deposition (Fig. 7 A and B). The expression of collagen I mRNA was substantially increased in IL-27RA−/− mice (Fig. 7C). Accordingly, collagen I–V proteins were also elevated in IL-27RA−/− mice as compared to WT mice during pulmonary fibrosis (Fig. 7D). A reduction in the numbers of protective CD4+IFNγ+ Th1 cells was contrasted by increased profibrotic CD4+IL-17A+ Th17 cells in lungs of IL-27RA−/− mice (Fig. 7E). Finally, we used mice with dual deletion of IL-17 isoforms A and F. These IL-17A−/−/IL-17F−/− mice displayed lower accumulation of collagen I–V during pulmonary fibrosis (Fig. 7F). Hence, this finding supported the concept of profibrotic effects of T cell derived IL-17 family members and their suppression by IL-27 (7, 31).
Fig. 7.

Protective role of IL-27RA during pulmonary fibrosis. (A) Lung sections of IL-27RA−/− mice and WT mice during bleomycin-induced pulmonary fibrosis, Masson–Goldner’s trichrome staining, n ≥ 6/group (Scale bar, 50 µm). (B) Quantification of collagen deposition using scanned whole slides from mice in frame A. (C) Expression of collagen I mRNA in lung tissue of IL-27RA−/− and WT mice after bleomycin, RT-PCR. (D) SIRCOL protein assay for collagen I–V in lung tissue of the experiment described under C. (E) Flow cytometry of CD4+ T cells in BALF of bleomycin i.t.-treated IL-27RA−/− mice as compared to WT mice. (F) Collagen accumulation after bleomycin-induced pulmonary fibrosis in IL-17A−/−IL-17F−/− double-knockout mice as compared to WT mice, SIRCOL assay. All frames were analyzed at 4 wk after bleomycin and frames B–F were done with numbers of mice as indicated by circles; Student’s t test or one-way ANOVA; *P < 0.05, **P < 0.01.
Discussion
In the present report, we advance the concept of externalized histones as drivers of tissue remodeling and fibrosis in lungs. Externalized histones hijack a cellular circuit linking platelets with macrophages for steering toward an imbalance between profibrotic TGFβ1 and antifibrotic IL-27 activities (SI Appendix, Fig. S7) (32). Externalized histones were elevated in IPF patients (Fig. 1A), and their neutralization was protective in experimental pulmonary fibrosis (Fig. 2).
Extracellular traps contain histones as part of three-dimensional web-like scaffolds formed by neutrophils (NETs), macrophages (METs), eosinophils, mast cells, and nonhematopoietic resident cells (9, 33). Our data with chimeric H2B-eGFP mice suggest that resident lung cells may contribute to the total histone burden in the early phase of injury leading to pulmonary fibrosis, although we cannot exclude incomplete chimerism in this model.
We used a blocking antibody specific for an epitope corresponding to a region of high sequence similarity between H2A and H4 (34) to mitigate the severity of pulmonary fibrosis. Histone H4 displays a ~10-fold higher cytotoxic activity for endothelial and lung epithelial cells than other histones H1, H2A, H2B, and H3 (14, 15). Therapeutic strategies that would universally block all core histones might be even more effective to confine TGFβ (Fig. 3B). Negatively charged nonanticoagulant heparin was recently used to neutralize the positively charged externalized histones. This treatment reduced collagen I expression and CCl4-mediated liver fibrosis in mice (35).
The finding that externalized histones liberate platelet TGFβ1 in the lung is in accordance with the concept of NETs as inducers of platelet activation, plasmatic coagulation, and thrombosis (16, 22). The interactions of NETs and platelets are bidirectional. Platelets activate NET formation through TLR4 on neutrophils during bacterial sepsis and lung injury (17, 28). Platelet trapping in bleomycin-treated lungs correlates with ECM depositions (36). Platelet depletion or platelet inhibition reduces bleomycin-induced lung fibrosis (37–39). Platelets and mononuclear phagocytes are important sources of TGFβ (40, 41). Platelet contaminations may mislead to overestimation of monocyte/macrophage-derived TGFβ (42). In fact, our studies underscore the relevance of platelet-derived TGFβ1 in the development of pulmonary fibrosis (43).
TGFβ1 released by platelets controlled the production of IL-27 by macrophages. Disruption of TGFβ signaling in macrophages resulted in higher IL-27 and less pulmonary fibrosis (Fig. 5 L–O), suggesting that TGFβ curbs a protective IL-27-dependent macrophage phenotype. In lung fibrosis, macrophages can powerfully antagonize fibrogenesis by mediators, induction of myofibroblast apoptosis, and degradation of ECM components (2, 44).
TGFβ1 reversed SMAD3 promotor binding to p28/ebi3 genes (Fig. 6 C and D). The observed effect of TLR4 agonist may be explained by a TLR4-IRAK1-ERK1/2 pathway activating c-Jun as a corepressor of SMAD3 and phosphorylation of alternative sites (Thr179/Ser208) in the SMAD3 linker region (45). The inhibitory pathway of p38 MAPK is recruited by TLR4 and further augmented by TGFβ1 to counterbalance the activating pathways (NFκB, IRF-1/-3/-9) for IL-27 production (7, 25). The MAPK inhibitor, SB203580, can also attenuate TGFβ1-induced expression and activation of SMAD3 (46), providing additional rationale for its IL-27 enhancing efficacy (Fig. 6E). TGFβ1 enforced IL-10 to suppress IL-27 (Fig. 6 H and I). The IL-10 promoter contains SMAD3-binding elements (47), and p38 MAPK can increase IL-10 production (48). TTP is a regulator of mRNA stability and TGFβ signaling (30). TTP is phosphorylated by p38 MAPK (49), and blocks the nuclear translocation of NFκB (50). Altogether, these observations suggest that TGFβ1 (liberated from platelets in response to externalized histones) orchestrates multiple intracellular signaling mechanisms to limit an antifibrotic IL-27-dependent macrophage response.
Injections of recombinant mouse IL-27 resulted in alleviated pulmonary fibrosis after bleomycin (51, 52), which is consistent with our findings that IL-27RA−/− mice showed increased ECM deposition with deranged T cell subsets suggesting that IL-27 acts antifibrotic (Fig. 7). The increased Th17 cells in IL-27RA−/− mice with fibrosis were in accordance with IL-27 suppressing Th17 cells, as shown in neuroinflammation (7). In fact, IL-27 inhibits Th17 cell responses in mice and humans (7, 53, 54). Bleomycin-induced lung fibrosis is improved in IL-17A−/− mice or using neutralizing anti-IL-17A antibodies (55, 56). This is in accordance with our observation that IL-17AF−/− double-knockout mice showed lower collagen depositions in lungs after bleomycin. In contradiction to the above concept of antifibrotic IL-27, one study described that antibody-mediated neutralization of IL-27 increased ECM deposition during bleomycin-induced lung fibroses (57).
NETs are released in lung injury, COPD, GvHD after lung transplantation, ANCA-associated vasculitis, and cystic fibrosis (58, 59). An involvement of PAD4 was reported in age-related organ fibrosis (60), although the interpretation is complicated because of NET-independent functions of PAD4 in the regulation of pluripotency and proliferation (61, 62). In line with our findings in Fig. 1F, another group reported reduced NET formation and lung fibrosis in PAD4−/− mice (23). NETs directly promote differentiation, proliferation, migration, and collagen production in cultured human lung fibroblasts (63). NETs can induce epithelial-to-mesenchymal transition, although the contribution of EMT for replenishing myofibroblast populations is debatable (64–67).
The prevalence of IPF (~0.5 to 2 per 10,000 persons) corresponds to ~30,000 patients in the United States with a life expectancy of 2–6 y after diagnosis (68). To alleviate this large-scale clinical problem, the targeting of externalized histones may help to restore a protective cytokine balance and promises therapeutic potential for the future.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
This work was supported by the Federal Ministry of Education and Research (01EO1503 to W.R., M.B., and K.S.), the Deutsche Forschungsgemeinschaft (BO3482/3-3, BO3482/4-1 to M.B., CRC829 to S.A.E., CRC156 TP KS01 to M.P.R., CRC1066 TP B18 to M.P.R., CRC1292/2 (Project No. 318346496) TP01 to T.B., TP02 to W.R., TP15 to A.W., and TP21N to M.P.R., TRR355/1 (Project No. 490846870) A09 and A10 to T.B., A08 and B05 to A.W., GU769-/4-2 (SPP 1468 “Immunobone”) to M.G., and TRR332 TP C6 to M.G.), the NIH (1R01HL141513, 1R01HL139641, and 1R01HL166588 to M.B.) a Marie Curie Career Integration Grant of the European Union (Project 334486 to M.B.). The authors are responsible for the contents of this publication. We thank Dr. Jürgen Roes for generously providing the TbRIIfl/fl mouse strain. We thank Markus Dudek, Anja Hasenberg, Sophie Henneberg, Fabien Meta, Frédéric-Kurt Norheimer, Johannes Platten, Laura Roßmann, Robert Rümmler, Norman F. Russkamp and Foruzandeh Samangan for help and assistance with experiments. We thank Anna Tseng, Aoife Kateri O’Connell, and Dr. Nicholas Crossland for help and assistance with histology and imaging. M.B. thanks Dr. Jay Mizgerd for mentorship and reading the manuscript.
Author contributions
D.R.R., A.S., J.R., T.B., M.P.R., M.G., A.W., B.G., P.A.W., W.R, K.S., and M.B. designed research; D.R.R., A.S., J.R., K.S., F.M., S.S., and M.B. performed research; S.A.E., T.B., H.K., M.P.R., G.C., C.R., B.G., I.P., A.G., D.S., M.G., A.W., W.R., K.S., and M.B. contributed new reagents/analytic tools; D.R.R., A.S., J.R., F.M., G.C., S.S., K.S., and M.B. analyzed data; and D.R.R., A.S., and M.B. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
This article is a PNAS Direct Submission. R.D. is a guest editor invited by the Editorial Board.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Sgalla G., Kulkarni T., Antin-Ozerkis D., Thannickal V. J., Richeldi L., Update in pulmonary fibrosis 2018. Am. J. Respir. Crit. Care Med. 200, 292–300 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Wynn T. A., Integrating mechanisms of pulmonary fibrosis. J. Exp. Med. 208, 1339–1350 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smigiel K. S., Parks W. C., Macrophages, wound healing, and fibrosis: Recent insights. Curr. Rheumatol. Rep. 20, 17 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Leask A., Abraham D. J., TGF-beta signaling and the fibrotic response. FASEB J. 18, 816–827 (2004). [DOI] [PubMed] [Google Scholar]
- 5.Massague J., TGFbeta signalling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bosmann M., et al. , Interruption of macrophage-derived IL-27(p28) production by IL-10 during sepsis requires STAT3 but not SOCS3. J. Immunol. 193, 5668–5677 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bosmann M., Ward P. A., Modulation of inflammation by interleukin-27. J. Leukocyte Biol. 94, 1159–1165 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villarino A., et al. , The IL-27R (WSX-1) is required to suppress T cell hyperactivity during infection. Immunity 19, 645–655 (2003). [DOI] [PubMed] [Google Scholar]
- 9.Allam R., et al. , Histones from dying renal cells aggravate kidney injury via TLR2 and TLR4. J. Am. Soc. Nephrol. (JASN) 23, 1375–1388 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li P., et al. , PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 207, 1853–1862 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wong S. L., Wagner D. D., Peptidylarginine deiminase 4: A nuclear button triggering neutrophil extracellular traps in inflammatory diseases and aging. FASEB J. 32, fj201800691R (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Papayannopoulos V., Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 18, 134–147 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Thalin C., et al. , Validation of an enzyme-linked immunosorbent assay for the quantification of citrullinated histone H3 as a marker for neutrophil extracellular traps in human plasma. Immunologic Res. 65, 706–712 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu J., et al. , Extracellular histones are major mediators of death in sepsis. Nat. Med. 15, 1318–1321 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bosmann M., et al. , Extracellular histones are essential effectors of C5aR- and C5L2-mediated tissue damage and inflammation in acute lung injury. FASEB J. 27, 5010–5021 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fuchs T. A., et al. , Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. U.S.A. 107, 15880–15885 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Caudrillier A., et al. , Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Invest. 122, 2661–2671 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abrams S. T., et al. , Circulating histones are mediators of trauma-associated lung injury. Am. J. Respir. Crit. Care Med. 187, 160–169 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brinkmann V., et al. , Neutrophil extracellular traps kill bacteria. Science 303, 1532–1535 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Sanders N. L., et al. , Neutrophil extracellular traps as an exacerbating factor in bacterial pneumonia. Infect. Immun. 90, e0049121 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kessenbrock K., et al. , Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 15, 623–625 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Semeraro F., et al. , Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 118, 1952–1961 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki M., et al. , PAD4 deficiency improves bleomycin-induced neutrophil extracellular traps and fibrosis in mouse lung. Am. J. Respir. Cell Mol. Biol. 63, 806–818 (2020). [DOI] [PubMed] [Google Scholar]
- 24.Yan S., Li M., Liu B., Ma Z., Yang Q., Neutrophil extracellular traps and pulmonary fibrosis: An update. J. Inflamm. (Lond) 20, 2 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bosmann M., et al. , Complement activation product C5a is a selective suppressor of TLR4-induced, but not TLR3-induced, production of IL-27(p28) from macrophages. J. Immunol. 188, 5086–5093 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma A., et al. , IL-27 enhances gammadelta T cell-mediated innate resistance to primary hookworm infection in the lungs. J. Immunol. 208, 2008–2018 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roewe J., et al. , Neuroendocrine modulation of IL-27 in macrophages. J. Immunol. 199, 2503–2514 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Clark S. R., et al. , Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 13, 463–469 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Xu J., Zhang X., Monestier M., Esmon N. L., Esmon C. T., Extracellular histones are mediators of death through TLR2 and TLR4 in mouse fatal liver injury. J. Immunol. 187, 2626–2631 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogawa K., Chen F., Kim Y. J., Chen Y., Transcriptional regulation of tristetraprolin by transforming growth factor-beta in human T cells. J. Biol. Chem. 278, 30373–30381 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Wilson M. S., et al. , Bleomycin and IL-1beta-mediated pulmonary fibrosis is IL-17A dependent. J. Exp. Med. 207, 535–552 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.She Y. X., Yu Q. Y., Tang X. X., Role of interleukins in the pathogenesis of pulmonary fibrosis. Cell Death Discov. 7, 52 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boe D. M., Curtis B. J., Chen M. M., Ippolito J. A., Kovacs E. J., Extracellular traps and macrophages: New roles for the versatile phagocyte. J. Leukocyte Biol. 97, 1023–1035 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Monestier M., Fasy T. M., Losman M. J., Novick K. E., Muller S., Structure and binding properties of monoclonal antibodies to core histones from autoimmune mice. Mol. Immunol. 30, 1069–1075 (1993). [DOI] [PubMed] [Google Scholar]
- 35.Wang Z., et al. , Extracellular histones stimulate collagen expression in vitro and promote liver fibrogenesis in a mouse model via the TLR4-MyD88 signaling pathway. World J. Gastroenterol. 26, 7513–7527 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Piguet P. F., Vesin C., Pulmonary platelet trapping induced by bleomycin: Correlation with fibrosis and involvement of the beta 2 integrins. Int. J. Exp. Pathol. 75, 321–328 (1994). [PMC free article] [PubMed] [Google Scholar]
- 37.Carrington R., Jordan S., Wong Y. J., Pitchford S. C., Page C. P., A novel murine model of pulmonary fibrosis: The role of platelets in chronic changes induced by bleomycin. J. Pharmacol. Toxicol. Methods 109, 107057 (2021). [DOI] [PubMed] [Google Scholar]
- 38.Zhan T., et al. , Cangrelor alleviates bleomycin-induced pulmonary fibrosis by inhibiting platelet activation in mice. Mol. Immunol. 120, 83–92 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Kishi M., et al. , Blockade of platelet-derived growth factor receptor-beta, not receptor-alpha ameliorates bleomycin-induced pulmonary fibrosis in mice. PLoS ONE 13, e0209786 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meyer A., et al. , Platelet TGF-beta1 contributions to plasma TGF-beta1, cardiac fibrosis, and systolic dysfunction in a mouse model of pressure overload. Blood 119, 1064–1074 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khalil N., Corne S., Whitman C., Yacyshyn H., Plasmin regulates the activation of cell-associated latent TGF-beta 1 secreted by rat alveolar macrophages after in vivo bleomycin injury. Am. J. Respir. Cell Mol. Biol. 15, 252–259 (1996). [DOI] [PubMed] [Google Scholar]
- 42.Merino J., Casado J. A., Cid J., Sanchez-Ibarrola A., Subira M. L., The measurement of transforming growth factor type beta (TGF beta) levels produced by peripheral blood mononuclear cells requires the efficient elimination of contaminating platelets. J. Immunol. Methods 153, 151–159 (1992). [DOI] [PubMed] [Google Scholar]
- 43.Bochenek M. L., et al. , Activated endothelial TGFbeta1 Signaling promotes venous thrombus nonresolution in mice via Endothelin-1: Potential role for chronic thromboembolic pulmonary hypertension. Circ. Res. 126, 162–181 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duffield J. S., Lupher M., Thannickal V. J., Wynn T. A., Host responses in tissue repair and fibrosis. Annu. Rev. Pathol. 8, 241–276 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim E. Y., Kim B. C., Lipopolysaccharide inhibits transforming growth factor-beta1-stimulated Smad6 expression by inducing phosphorylation of the linker region of Smad3 through a TLR4-IRAK1-ERK1/2 pathway. FEBS Lett. 585, 779–785 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Xu Q., Tan Y., Zhang K., Li Y., Crosstalk between p38 and Smad3 through TGF-beta1 in JEG-3 choriocarcinoma cells. Int. J. Oncol. 43, 1187–1193 (2013). [DOI] [PubMed] [Google Scholar]
- 47.Kitani A., et al. , Transforming growth factor (TGF)-beta1-producing regulatory T cells induce Smad-mediated interleukin 10 secretion that facilitates coordinated immunoregulatory activity and amelioration of TGF-beta1-mediated fibrosis. J. Exp. Med. 198, 1179–1188 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ma W., et al. , The p38 mitogen-activated kinase pathway regulates the human interleukin-10 promoter via the activation of Sp1 transcription factor in lipopolysaccharide-stimulated human macrophages. J. Biol. Chem. 276, 13664–13674 (2001). [DOI] [PubMed] [Google Scholar]
- 49.Mahtani K. R., et al. , Mitogen-activated protein kinase p38 controls the expression and posttranslational modification of tristetraprolin, a regulator of tumor necrosis factor alpha mRNA stability. Mol. Cell Biol. 21, 6461–6469 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gu L., et al. , Suppression of IL-12 production by tristetraprolin through blocking NF-kcyB nuclear translocation. J. Immunol. 191, 3922–3930 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dong Z., et al. , IL-27 alleviates the bleomycin-induced pulmonary fibrosis by regulating the Th17 cell differentiation. BMC Pulm. Med. 15, 13 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dong Z., et al. , IL-27 attenuates the TGF-beta1-induced proliferation, differentiation and collagen synthesis in lung fibroblasts. Life Sci. 146, 24–33 (2016), 10.1016/j.lfs.2016.01.004. [DOI] [PubMed] [Google Scholar]
- 53.Batten M., et al. , Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat. Immunol. 7, 929–936 (2006). [DOI] [PubMed] [Google Scholar]
- 54.Murugaiyan G., et al. , IL-27 is a key regulator of IL-10 and IL-17 production by human CD4+ T cells. J. Immunol. 183, 2435–2443 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cipolla E., et al. , IL-17A deficiency mitigates bleomycin-induced complement activation during lung fibrosis. FASEB J. 31, 5543–5556 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qu Z., et al. , IL-22 inhibits bleomycin-induced pulmonary fibrosis in association with inhibition of IL-17A in mice. Arthritis Res. Ther. 24, 280 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kim H. S., Go H., Akira S., Chung D. H., TLR2-mediated production of IL-27 and chemokines by respiratory epithelial cells promotes bleomycin-induced pulmonary fibrosis in mice. J. Immunol. 187, 4007–4017 (2011). [DOI] [PubMed] [Google Scholar]
- 58.Porto B. N., Stein R. T., Neutrophil extracellular traps in pulmonary diseases: Too much of a good thing? Front. Immunol. 7, 311 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Negreros M., Flores-Suarez L. F., A proposed role of neutrophil extracellular traps and their interplay with fibroblasts in ANCA-associated vasculitis lung fibrosis. Autoimmun. Rev. 20, 102781 (2021). [DOI] [PubMed] [Google Scholar]
- 60.Martinod K., et al. , Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J. Exp. Med. 214, 439–458 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Christophorou M. A., et al. , Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature 507, 104–108 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nakashima K., et al. , PAD4 regulates proliferation of multipotent haematopoietic cells by controlling c-myc expression. Nat. Commun. 4, 1836 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chrysanthopoulou A., et al. , Neutrophil extracellular traps promote differentiation and function of fibroblasts. J. Pathol. 233, 294–307 (2014). [DOI] [PubMed] [Google Scholar]
- 64.Pandolfi L., et al. , Neutrophil extracellular traps induce the epithelial-mesenchymal transition: Implications in post-COVID-19 fibrosis. Front. Immunol. 12, 663303 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ueha S., Shand F. H., Matsushima K., Cellular and molecular mechanisms of chronic inflammation-associated organ fibrosis. Front. Immunol. 3, 71 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hoyles R. K., et al. , An essential role for resident fibroblasts in experimental lung fibrosis is defined by lineage-specific deletion of high-affinity type II transforming growth factor beta receptor. Am. J. Respir. Crit. Care Med. 183, 249–261 (2011). [DOI] [PubMed] [Google Scholar]
- 67.Bartis D., Mise N., Mahida R. Y., Eickelberg O., Thickett D. R., Epithelial-mesenchymal transition in lung development and disease: Does it exist and is it important? Thorax 69, 760–765 (2014). [DOI] [PubMed] [Google Scholar]
- 68.Ley B., Collard H. R., Epidemiology of idiopathic pulmonary fibrosis. Clin. Epidemiol. 5, 483–492 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.