

Abstract
Objective
Protein kinase C (PKC) influences myocardial contractility and susceptibility to long-term cardiac dysfunction after ischemia–reperfusion injury. In diabetes, PKC inhibition has a protective effect in terms of microvascular dysfunction. SK-channel dysfunction also influences endothelial dysfunction in cardioplegic hypoxia–reoxygenation (CP-H/R). Here, we examine whether acute inhibition of PKC beta protects against CP-H/R–induced coronary endothelial and SK channel dysfunction.
Methods
Isolated mouse coronary arterioles, half pretreated with selective PKC inhibitor ruboxistaurin (RBX), were subjected to hyperkalemic, cardioplegic hypoxia (1 hour), and reoxygenation (1 hour) with Krebs buffer. Sham control vessels were continuously perfused with oxygenated Krebs buffer without CP-H/R. After 1 hour of reoxygenation, responses to the endothelium-dependent vasodilator adenosine-diphosphate (ADP) and the SK-channel activator NS309 were examined. Endothelial SK-specific potassium currents from mouse heart endothelial cells were examined using whole-cell path clamp configurations in response to NS309 and SK channel blockers apamin and TRAM34.
Results
CP-H/R significantly decreased coronary relaxation responses to ADP (P = .006) and NS309 (P = .0001) compared with the sham control group. Treatment with selective PKC beta inhibitor RBX significantly increased recovery of coronary relaxation responses to ADP (P = .031) and NS309 (P = .004) after CP-H/R. Treatment with RBX significantly increased NS309-mediated potassium currents following CP-H/R (P = .0415). Apamin and TRAM34 sensitive currents were significantly greater in CP-H/R + RBX versus CP-H/R mouse heart endothelial cells (P = .0027).
Conclusions
Acute inhibition of PKC beta significantly protected mouse coronary endothelial function after CP-H/R injury. This suggests that acute PKC beta inhibition may be a novel approach for preventing microvascular dysfunction during CP-H/R.
Key Words: protein kinase C, cardioplegia, hypoxia–reoxygenation, ruboxistaurin, vascular reactivity
Graphical abstract
Depicts overview of premise, study, and key findings.
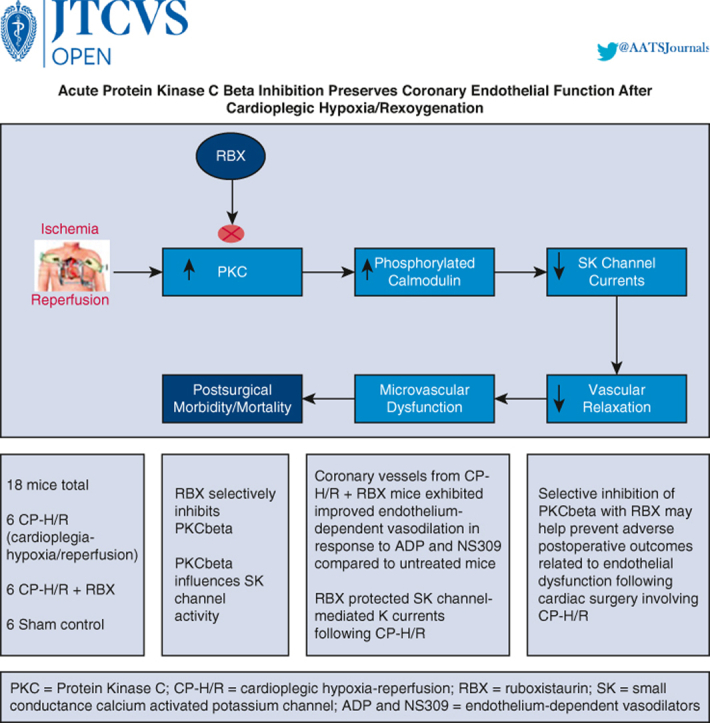
Increased PKC beta activity contributes to postcardiac surgery endothelial dysfunction.
Central Message.
Treatment of tissue with a PKC beta inhibitor protects endothelial function and microvascular responsiveness to endothelium-derived vasodilators following cardioplegic hypoxia–reoxygenation.
Perspective.
Pretreatment with PKC beta inhibitors may be a useful therapeutic tool for preventing microvascular dysfunction following ischemia–reperfusion injury during cardiac surgery involving cardioplegia/cardiopulmonary bypass.
Coronary artery bypass grafting is the most common cardiac surgical procedure performed in the United States.1 Most coronary artery bypass grafting protocols are “on-pump,” using cardioplegic (CP) arrest and cardiopulmonary bypass (CPB) to stop the heart while preserving the body's circulation using an external heart–lung machine.2 Despite decades of extensive innovation and refinement of novel cardioplegic solutions and optimizing techniques for cardioprotection during CPB, the risk of postoperative CP hypoxia/reoxygenation (H/R) injury remains high following CP/CPB.3
Following CP-H/R, the coronary microcirculation exhibits increased propensity for vasospasm and myocardial malperfusion, with altered responsiveness to vasoactive substances such as serotonin, endothelin-1, thromboxaneA2, and neuropeptide Y.3,4 Generalized endothelial dysfunction disrupts the balance between vasoconstriction and vasodilation through markedly impaired microvascular responses to endothelium-derived vasodilators such as nitric oxide, prostacyclin, and endothelium-derived hyperpolarizing factors (EDHFs).3,5 EHDF promotes vasodilation through the opening of small-conductance calcium-activated potassium channels (SK channels), which facilitate endothelial cell hyperpolarization and vascular smooth muscle relaxation.6 We and others have previously found that CP-H/R injury inactivates/inhibits endothelial SK channels, observed through decreased responsiveness to various SK-channel activators such as NS309.7
Protein kinase C (PKC) is a potential mediator of cardiovascular pathology in settings of ischemia and hypoxia-related injury, such as heart failure and myocardial infarction. Members of the PKC family can be broadly subdivided into 3 subgroups: classical PKCs (alpha, beta, and gamma); novel PKCs (delta, epsilon, eta, theta), and atypical PKCs (zeta, iota, and lambda).8 The classical PKCs are activated by calcium and phospholipids in contrast to novel and atypical isozymes, which are calcium-independent and activated only by specific lipids.9
Transgenic mice engineered to overexpress PKC alpha exhibit decreased cardiac contractility, whereas PKC alpha knockout mice exhibit increased cardiac contractility and were less susceptible to heart failure after long-term pressure overload stimulation or induction of ischemia–reperfusion injury.10,11 Phosphorylation of PKC alpha at site threonine 638 positively correlated with increased left ventricular volume and reduced ejection fraction in pig models of heart failure following ischemia–reperfusion.12 In contrast, PKC beta/gamma knockout mice exhibited more severe heart failure following long-term pressure overload.11 Increased activity of the PKC delta and epsilon isoforms have been observed in the myocardium of patients following CPB and cardioplegic arrest.13
Pharmacologic inhibition of PKC alpha/beta/gamma in mice using Ro-32 to 0432 and Ro-31 to 8220 increases cardiac contractility and confers partial protection against long-term decompensation and dilated cardiomyopathy after myocardial infarction.14 Likewise, application of Ro-32 to 0432, Ro-31 to 8220, and ruboxistaurin (RBX; LY 333531, 2007), another PKC inhibitor that specifically targets PKC beta, protects against heart failure, and reduced left ventricular ejection fraction in pig models of myocardial infarction.12,15
Comparatively little work exists concerning the effects of PKC modulation in the microcirculation. In addition, although RBX has shown promise in the treatment of diabetic complications in which PKC plays a prominent role, including diabetic retinopathy, diabetic neuropathy, and diabetic nephropathy, few studies have investigated whether treatment with PKC inhibitors such as RBX might improve coronary microvascular function in the context of CP-H/R. In this study, we use a rodent model of CP-H/R to test whether treatment with RBX protects against microvascular endothelial dysfunction in mouse small coronary arteries.
Finally, PKC is involved in numerous downstream cell-signaling pathways, including many involving cell membrane ion channels. For example, our group has implicated PKC beta in NADH-mediated SK channel dysfunction in human coronary artery endothelial cells, and other studies have shown that PKC-mediated signaling pathways influence cardiac sodium channel activity.16 Hence, we also examined the effect of RBX on SK-channel recording activity before and after CP-H/R.
Methods
Animal Models and Heart Tissue Collection
We used eighteen 16-week-old C57BL/6J mice in this study, with equal parity between male and female mice. The Institutional Animal Care and Use Committee of Rhode Island Hospital approved all the experiments outlined in this section (original approval date: May 18, 2018; newest approval date: April 26, 2021; internal reference number: 502118 [original], 501721 [new]). Each mouse underwent a thoracotomy to access the heart following anesthesia administration with inhaled isoflurane. Hearts were removed from the thorax by gently lifting at the apex and severing the great vessels. Following removal, hearts were immediately placed either in cold (4 °C) Krebs buffer (1119 NaCl, 25 NaHCO3, 4.6 KCl, 1.2 KH2PO4, 1.2 MgSO4, 1.8 CaCl2, 11 glucose, in mM, pH 7.4) for microvessel experiments or cell culture medium to prepare for endothelial cell isolation.
Experimental groups
Six mouse hearts (3 from male mice, 3 from female mice) were assigned to each of 3 groups: sham controls, CP-H/R, and CP-H/R + RBX.
Microvessel dissection and CP-H/R simulation
Coronary arterial microvessels (70-100 μm in diameter) from the left anterior descending artery–dependent subepicardial region of the left ventricle were dissected manually from isolated mouse hearts according to previously described protocols17 (Figure 1, A). Following dissection, microvessels were placed in a microvessel organ chamber, where they were cannulated with 2 glass micropipettes (measuring 40-80 μm in diameters), secured with 10-0 nylon monofilament sutures, and pressurized in a no-flow state. The microvessel chamber initially contained aerated (95% O2, 5% CO2) Krebs buffer solution maintained at 37 °C. Vessels were allowed to remain under these conditions for 60 minutes for stabilization before initiation of experiments. Two consecutive 1-hour long exposures to oxygenated Krebs solution were the only conditions to which the sham control vessel group was exposed. An inverted microscope (Olympus CK2; Olympus Optical) was used to project microvessel images onto a monitor. Internal luminal diameters were measured using an electronic dimension analyzer.
Figure 1.

Experimental design for microvascular reactivity and endothelial experiment protocols. A, Mouse coronary arterial microvessels (70-100 μm in internal diameter) from the left anterior descending artery dependent subepicardial region of the left ventricle were harvested and assigned to 3 groups: normoxia (sham controls), cardioplegic hypoxia–reoxygenation (1 hour), and cardioplegic hypoxia–reoxygenation (1 hour) with ruboxistaurin treatment. Following normoxia, cardioplegic hypoxia–reoxygenation, and/or ruboxistaurin treatment, microvascular responses to ADP, NS309, and SNP were measured. B, Mouse heart endothelial cells (MHECs) were harvested, and divided into the same 3 groups as were used for the microvascular reactivity study, with the key difference being 3 hours of cardioplegic hypoxia–reoxygenation. Following treatments, patch clamp methods were used to record whole cell potassium currents. ADP, Adenosine diphosphate; SNP, sodium nitroprusside.
In order to simulate the effects of ischemic cardioplegia during cardiac surgeries, an in vitro CP-H/R model of microvessel injury was used. A modified St Thomas hyperkalemic cardioplegic solution, consisting of 110 NaCl, 16 MgCl2, 10 NaHCO3, 20 KCl, 1.5 CaCl2 (in mM, pH 7.4), bathed microvessels in the experimental groups for 5 minutes before onset of hypoxia. Following this, coronary arterioles were cooled in ice and then set again in St Thomas solution for 60 minutes under hypoxic conditions at 4 °C (bubbling a 5% CO2, 95% N2 gas mixture through the chamber). After 60 minutes of hypoxia, vessels were reoxygenated for 60 minutes at 37 °C (using 95% O2, 5% CO2 gas mixture). Importantly, the partial pressure of oxygen in the hypoxic cardioplegia reached a stable level below 30 mm Hg within 2 minutes of changing the bubbling gas. This helped to prevent true anoxic conditions because a small amount of oxygen continuously diffused into the circulated cardioplegic solution from the atmosphere. In the CP-H/R + RBX group, the microvessels were bathed with 50 nM of RBX 5 minutes before and during hypoxia and reoxygenation.
Microvascular reactivity assessment
Following exposures/treatments in the microvessel chambers, microvessels were preconstricted with the endothelial vasoconstrictor endothelin-1 to between 30% and 50% of baseline vessel diameter, following a protocol that has been published previously.17, 18, 19 Importantly, in our microvessel study, vessels were predilated at constant pressure of 40 mm Hg. Because of this, we needed to preconstrict vessels by using endothelin-1 or U46619 before effectively studying vasodilatory responses. Once stable vasoconstriction was achieved, vasodilators were applied to test microvascular relaxation, including the SK channel activator NS309 (10−9-10−5 M), endothelium-dependent vasodilator adenosine diphosphate (ADP; 10−9-10−4 M), and the endothelium-independent vasodilator sodium nitroprusside (SNP; 10−9-10−4 M).20 Drugs were applied in random order, and each vessel was exposed to 1 or 2 interventions.
CP-H/R model of endothelial cells
Mouse heart endothelial cells (MHECs) were isolated from harvested mouse hearts (n = 4) and cultured in EGM-2 MV medium (Lonza Biosciences) (Figure 1, B). Endothelial cell biomarkers, such as CD31, were used to confirm that cultured cells were indeed endothelial cells. The CP-H/R model of MHECs is as follows: following isolation, MHECS were placed in a sealed chamber perfused with a high-nitrogen gas mixture (95% N2, 5% CO2) for 3 hours. After this, MHECs were transferred to a normoxic culture incubator. For the RBX treatment group, 50 nM RBX was added before hypoxia and before reoxygenation.
Endothelial cell potassium currents: patch clamp recordings
Whole-cell patch clamp configurations were used to assess endothelial potassium currents from MHECs. Before recordings, MHECs were washed twice with calcium-free Dulbecco's Modified Eagle Medium. Recording of potassium currents was performed in voltage-clamp mode using an Axon Axopatch-200B amplifier, Axon Digidata 1550B A/D converter, and pClamp 11 software (all provided by Molecular Devices). A bath solution consisted of 5 KCl, 140 NaCl, 2 MgCl2, 1 CaCl2, 10 HEPES, 30 glucose (all in mM, at pH 7.4, 22 °C). Patch pipettes were filled with pipette solution that consisted of 20 KCl, 1 MgCl2, 10 HEPES, 110 K-Aspartate, 8.5 CaCl2, 0.01 niflumic acid, and 10 BAPTA (all in mM, at pH 7.2, calculated free calcium at 400 nmol/L). Stepping in 20-mV increments from a –50 mV holding potential, using 150-millisecond test pulses in the range of –100 to 100 mV, were used to obtain current-voltage potassium current recordings. The sampling rate was 10 kHz, and low-pass filter frequency was 2 kHz. For current-time potassium current recordings, MHEC membrane potentials were held at 100 mV. The effects of the SK-channel activator NS309 on whole cell potassium currents were examined. To test specificity of SK-channel activation, the SK2/SK3 channel blocker apamin (10–7 M) and the SK4 blocker TRAM34 (10–6 M) were applied.
Chemicals
ADP, SNP, NS309, apamin, and TRAM34 were purchased from Sigma-Aldrich.
Data Analysis and Statistics
Data are presented as mean ± standard deviation of the mean. Percent relaxation of preconstricted vessel diameters reflect degrees of microvascular responsiveness. The Shapiro–Wilk test was used to assess normality of the data. One- or two-way analyses of variance with post-hoc tests were used to analyze microvascular reactivity, patch-clamp, and PKC activity data using GraphPad Prism 7 (GraphPad Software).
Results
Acute PKC Inhibition With RBX Improved Coronary Arteriolar Endothelium-Dependent Relaxation Responses Following CP-H/R
Across all 3 groups, there were no significant differences in baseline microvessel diameter for harvested mouse coronary arterioles. Before vasodilatory response testing, microvessels were preconstructed using endothelin 1. NS309, ADP, and SNP all promoted dose-dependent vasodilatory effects (Figure 2, Figure 3, Figure 4, respectively). Coronary arteriolar responses to the endothelium-dependent vasodilators NS309 (10−5 M, P = .0001) (Figure 2) and ADP (10−4 M, P = .006) (Figure 3) were significantly decreased in the CP-H/R group versus the sham control (Krebs buffer only) group. Treatment with the selective PKC beta inhibitor RBX significantly protected the coronary vasorelaxation responses of the CP-H/R + RBX group to NS309 (P = .004) (Figure 2) and ADP (P = .031) (Figure 3) compared with the untreated CP-H/R group. Curiously, there were no statistically significant differences in vessel relaxation responses to the endothelium-independent vasodilator SNP among all groups (Figure 4).
Figure 2.

Coronary endothelial responses of mouse microvessels to endothelium-dependent vasodilator NS309. A, Dose-dependent vasodilation of sham control mouse microvessels and vessels undergoing CP-H/R with or without RBX treatment in response to the SK channel activator NS309 (10–9-10–5 M). B, Bar graph shows vasodilation response to NS309 10–5 M. CP-H/R, Microvessels undergoing hypoxia/reoxygenation; RBX + CP-H/R, microvessels pretreated with RBX before hypoxia and reoxygenation; RBX, ruboxistaurin.
Figure 3.

Coronary endothelial responses of mouse microvessels to endothelium-dependent vasodilator ADP. A, Sham control (no CP-H/R) and CP-H/R vessels with or without RBX treatment in response to the endothelium-dependent vasodilator ADP (10–9-10–4 M). B, Bar graph shows vasodilatory response to ADP 10–5 M. ADP, Adenosine diphosphate; CP-H/R, microvessels undergoing hypoxia/re-oxygenation; RBX + CP-H/R, microvessels pretreated with RBX before hypoxia and reoxygenation; RBX, ruboxistaurin.
Figure 4.

Coronary endothelial responses of mouse microvessels to endothelium-independent vasodilator SNP. A, Sham control (no CP-H/R) and CP-H/R vessels with or without RBX treatment in response to the endothelium-independent vasodilator SNP (10–9-10–4 M). B, Bar graph shows vasodilatory response to SNP 10–5 M. SNP, Sodium nitroprusside; CP-H/R, Microvessels undergoing hypoxia/reoxygenation; RBX + CP-H/R, microvessels pretreated with RBX before hypoxia and reoxygenation; RBX, ruboxistaurin.
RBX Increased Endothelial SK Currents After CP-H/R
Whole-cell potassium currents for sham control, CP-H/R, and CP-H/R + RBX MHECs can be seen in Figure 5, A (CP-H/R = MHECs undergoing hypoxia/reoxygenation, RBX + CP-H/R = MHECs pretreated with RBX before hypoxia and reoxygenation, RBX = ruboxistaurin, PKC beta inhibitor). Application of the SK-channel activator NS309 increased potassium currents in sham control (untreated with CP-H/R or RBX), CP-H/R and CP-H/R + RBX MHECs. These effects were abolished with coapplication of the SK-channel inhibitors TRAM34 and apamin, demonstrating that the NS309-induced potassium current is mediated by SK channel activity (Figure 5, A). NS309-sensitive potassium currents were significantly lower in CP-H/R versus sham control MHECs (P = .049) and CP-H/R versus CP-H/R + RBX MHECS (P = .0415) (Figure 5, B and C). Likewise, the apamin + TRAM34-sensitive potassium currents, which we have demonstrated are SK-mediated potassium currents, were significantly greater in CP-H/R + RBX MHECs versus CP-H/R MHECs (P = .0027) and in sham control versus CP-H/R MHECs (P = .0032) (Figure 5, D and E).
Figure 5.

RBX significantly increases SK-channel currents of mice heart endothelial cells (MHECs) in CP-H/R model. A, Representative traces of the whole cell currents of MHECs at holding potential of –50 mV and test potentials from –100 to +100 mV in 20-mV increments. B, Whole-cell I-V relationships sensitive to NS309 (1 μM) in MHECs of sham control and CP-H/R with or without RBX (50 nM) treatment. C, Box plots show NS309-sensitive component of potassium current at +100 mV in sham control MHECs and CP-H/R MHECs treated with or without RBX. ∗P = .0490, sham control (n = 6) versus CP-H/R (n = 7); P = .0415, CP-H/R (n = 7) versus CP-H/R + RBX (n = 6). D, whole-cell I-V relationships sensitive to TRAM34 (10 μM) + apamin (100 nM) in sham control MHECs and CP-H/R MHECs with or without RBX treatment. E, Box plots shows TRAM34 + apamin-sensitive component of potassium current at +100 mV in control MHECs and CP-H/R MHECs treated with or without RBX (n = 5/group). ∗∗P = .0032, control (n = 6) versus CP-H/R (n = 7); P = .0027, CP-H/R (n = 7) versus CP-H/R + RBX (n = 6). CP-H/R, MHECs undergoing hypoxia/reoxygenation; CP-H/R + RBX, MHECs pretreated with RBX before hypoxia and reoxygenation; RBX, ruboxistaurin.
Discussion
The family of SK channels contains 4 broad subtypes: SK1 (KCa2.1), SK2 (KCa2.2), SK3 (KCa2.3), and SK4 (IK1) (KCa3.1). SK 1 channels predominantly localize to neurons, although some reports suggest that they may also be found in atrial tissue.21,22 SK2 may be found in cardiomyocytes, neurons, and endothelial cells.21,23,24 SK3 and SK4 are strongly found on endothelial cells, where they contribute to EDHF-mediated vasodilation.25,26 Although the specific identity of EDHF remains a matter of debate, most prevailing theories suggest that EDHF ultimately elevates intracellular calcium levels in endothelial cells.
Figure 6 provides an overview of SK channel-mediated endothelial hyperpolarization. Calcium binds to calmodulin, which itself can bind constitutively to the C termini of SK channels. This leads to a conformational change in the calmodulin–SK-channel calmodulin binding domain that opens the SK-channel pore, permitting efflux of potassium and endothelial hyperpolarization.27 Endothelial hyperpolarization may then be transferred to vascular smooth muscle in several ways, including direct communication with vascular smooth muscle cells via myoendothelial gap junctions or activation of inward rectifier potassium channels and sodium–potassium ATPases on vascular smooth muscle cells.28
Figure 6.

Schematic of SK-channel–mediated vasodilation. A, CP-H/R increases PKC activity, which phosphorylates SK channels. This results in decreased SK-channel calcium sensitivity and reduces SK-channel opening, leading to reduced potassium currents and reduced endothelial hyperpolarization. This diminishes microvascular relaxation. B, RBX inhibits PKC, preventing PKC-mediated inhibition of SK-channel activity, thereby promoting endothelial hyperpolarization and coronary microvascular smooth muscle relaxation. SK channels are cyan on EC membranes. EC, Endothelial cell; CP-H/R, cardioplegic hypoxia–reoxygenation; PKC, protein kinase C; RBX, ruboxistaurin.
Our group has previously provided evidence of altered SK-channel activity following CPB. Post-CP/CPB relaxation responses to endothelium-dependent vasodilators substance P and ADP, both of which have been implicated in EDHF/SK channel–mediated endothelial hyperpolarization, and the SK-channel activator NS309 were significantly reduced versus pre-CP/CPB in human atrial microvessels.29 Similarly, mouse small coronary artery endothelial cells undergoing CP-H/R exhibited reduced SK-channel currents (patch clamp recordings) and considerable intracellular calcium overload, which strongly correlated with the diminished SK currents.19
Pretreatment of MHECs and human coronary artery endothelial cells with NS309 before CP-H/R has a protective effect on SK channel activity and preserves SK-channel currents.7 In line with these findings, the current study shows that RBX pretreatment can also protect SK-channel currents in MHECs following CP-H/R, which is associated with preserved endothelium-dependent vasodilation. This provides further support for the notion that SK-channel dysfunction is a key mediator of general endothelial dysfunction following CP-H/R. Therefore, correcting SK-channel dysfunction may subsequently provide significant therapeutic benefit.
PKC is an important regulator of SK-channel activity through phosphorylation of SK-channel–bound calmodulin.30 PKC phosphorylation of SK-channel–bound calmodulin significantly reduces the sensitivity of the calmodulin–SK-channel complex to calcium, thereby reducing the channel's open probability.30 The specific direction of change in PKC expression following CP-H/R may vary depending on the specific isoform involved.
Earlier, we discussed studies suggesting that PKC delta and epsilon isoforms were increased in the myocardium following CP-H/R.13 However, other studies report decreased PKC alpha activity in human coronary and skeletal microvessels following CP/CPB.31 In an earlier study, inhibition of PKC alpha with safingol reduced phenylephrine induced vasoconstriction in human coronary and skeletal muscle arterioles following CP/CPB.31 In contrast, activation of PKC alpha with tamoxifen increased phenylephrine induced vasoconstriction.
In the current study, we show that inhibition of PKC beta with RBX improves microvascular relaxation in an endothelium-dependent manner, evidenced by the strong protection of ADP and NS309 induced relaxation responses following CP-H/R injury (cf. Figure 7). We can further link inhibition of PKC beta with protection of SK channel activity based on patch clamp results of preserved SK channel-dependent potassium currents in CP-H/R + RBX MHECs. Thus, inhibiting PKC beta with RBX may provide a novel approach for targeting SK-channel hypoactivity following CP-H/R, mitigating against the development of postoperative endothelial dysfunction.
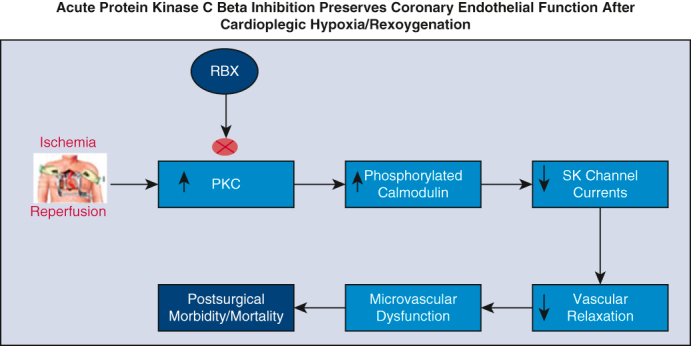
Figure 7.

Depicts overview of premise, study, and key findings. RBX, Ruboxistaurin; PKC, protein kinase C; CP-H/R, cardioplegic hypoxia–reoxygenation; ADP, adenosine diphosphate.
Importantly, our study showed that CP-H/R treatment did not affect microvascular relaxation responses to the endothelium-independent vasodilator SNP, and that RBX had no impact on SNP-mediated vasodilation after CP-H/R. This suggests that CP-H/R injury may spare endothelium-independent vasomodulatory systems, with damage predominantly dealt to endothelium-dependent systems through mechanisms such as altered PKC activity. Future studies will be needed to better characterize and elucidate the mechanisms of SNP and endothelium-independent vasodilation in the setting of CP-H/R.
Because vessels in our microvessel study were predilated at a constant pressure of 40 mm Hg, we needed to preconstrict vessels using endothelin-1 before studying vasodilatory responses. Endothelin-1 alone has been shown to activate PKC and reduce cardiomyocyte contractility during periods of ischemia and reperfusion.32 Importantly, previous research has shown significant increases in endothelin-1 release in the myocardium and peripheral blood following CP/CPB.33,34 We have demonstrated previously that CP/CPB leads to activation of PKC alpha and beta in human coronary microvessels without the presence of endothelin-1.13 Our group has also shown that elevated endothelin-1 levels in conjunction with CP/CPB synergistically increase PKC activity.35 Our patch clamp studies also demonstrate here that selective PKC inhibition with RBX improves endothelial SK-channel function in the absence of endothelin-1, which provides direct evidence for a non-endothelin-1–related role of PKC activity in CP-H/R.
A few limitations of the current study are worth mentioning. First, as mentioned previously, because our protocol, which has been used in many previous investigations referred to in this study, involves predilating vessels at 40 mm Hg, we needed to use endothelin-1 to preconstrict vessels before studying vasodilatory responses. We did not investigate which specific SK-channel subtype accounts for the preserved SK-dependent potassium currents following RBX treatment; more detailed studies of SK-channel activity will be required. Next, the current study was done using MHECs from mouse models of CP-H/R injury; additional studies using human coronary microvessels will be needed to verify that the observations of this study translate appropriately to humans.
The absence of continuous sanguineous perfusion is another limitation of the mouse model, as it is very difficult to collect enough mouse blood to continuously perfuse vessels. A clinically relevant larger animal model, such as pig, should be used to test our in vitro study in the future. This would also allow for examining the effect of PKC beta inhibition on postcardiac arrest function in a large animal intact heart model. Future studies may also investigate potential roles for downstream effector molecules of PKC, such as ERK, in CP-H/R–related endothelial dysfunction. Finally, it will be useful to see how well RBX protects endothelial function following CP-H/R in animal models or humans with pre-existing microvascular complications, such as diabetes or hypertension, who make up a significant portion of the cardiac surgery patient base.
In conclusion, acute protein kinase C beta inhibition with RBX improves endothelium-dependent vasodilation and endothelial SK channel function following CP-H/R injury (Figure 7). This study provides important initial evidence for the clinical potential of RBX in patients prior to undergoing cardiac surgery involving CP/CPB for protection against postoperative endothelial dysfunction.
Conflict of Interest Statement
The authors reported no conflicts of interest.
The Journal policy requires editors and reviewers to disclose conflicts of interest and to decline handling or reviewing manuscripts for which they may have a conflict of interest. The editors and reviewers of this article have no conflicts of interest.
Acknowledgments
The authors thank Julie Braza of the CPVB COBRE Service Cores at Ocean State Research Institute, Providence VA Medical Center, for assistance with endothelial cell isolation and culture.
Footnotes
This research project was mainly supported by the National Institutes of Health (NIH) 1R01HL127072-01A1 and 1R01 HL136347-01 and National Institute of General Medical Science (NIGMS) of 5P20-GM103652 (Pilot Project) to J.F. and 5P20-GM103652 to E.O.H.. This work was supported in part by R01HL136347 to 04S1 to J.F. and R01HL46716 and R01HL128831 to F.W.S.
References
- 1.Melly L., Torregrossa G., Lee T., Jansens J.L., Puskas J.D. Fifty years of coronary artery bypass grafting. J Thorac Dis. 2018;10:1960–1967. doi: 10.21037/jtd.2018.02.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carvajal C., Goyal A., Tadi P. StatPearls. StatPearls publishing; 2022. Cardioplegia. Accessed December 12, 2022. https://www.ncbi.nlm.nih.gov/books/NBK554463/ [PubMed] [Google Scholar]
- 3.Feng J., Kant S., Sellke F.W. Microvascular dysfunction following cardioplegic arrest and cardiopulmonary bypass. Vessel Plus. 2021;5:30. doi: 10.20517/2574-1209.2021.57. [DOI] [Google Scholar]
- 4.Ruel M., Khan T.A., Voisine P., Bianchi C., Sellke F.W. Vasomotor dysfunction after cardiac surgery. Eur J Cardiothorac Surg. 2004;26:1002–1014. doi: 10.1016/j.ejcts.2004.07.040. [DOI] [PubMed] [Google Scholar]
- 5.Vanhoutte P.M., Shimokawa H., Feletou M., Tang E.H. Endothelial dysfunction and vascular disease—a 30th anniversary update. Acta Physiol. 2017;219:22–96. doi: 10.1111/apha.12646. [DOI] [PubMed] [Google Scholar]
- 6.Liu Y., Kabakov A.Y., Xie A., Shi G., Singh A.K., Sodha N.R., et al. Metabolic regulation of endothelial SK channels and human coronary microvascular function. Int J Cardiol. 2020;312:1–9. doi: 10.1016/j.ijcard.2020.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Z., Shi G., Liu Y., Xing H., Kabakov A.Y., Zhao A.S., et al. Coronary endothelial dysfunction prevented by small-conductance calcium-activated potassium channel activator in mice and patients with diabetes. J Thorac Cardiovasc Surg. 2020;160:e263–e280. doi: 10.1016/j.jtcvs.2020.01.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breitkreutz D., Braiman-Wiksman L., Daum N., Denning M.F., Tennenbaum T. Protein kinase C family: on the crossroads of cell signaling in skin and tumor epithelium. J Cancer Res Clin Oncol. 2007;133:793–808. doi: 10.1007/s00432-007-0280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Steinberg S.F. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88:1341–1378. doi: 10.1152/physrev.00034.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ravichandran V.S., Patel H.J., Pagani F.D., Westfall M.V. Cardiac contractile dysfunction and protein kinase C–mediated myofilament phosphorylation in disease and aging. J Gen Physiol. 2019;151:1070–1080. doi: 10.1085/jgp.201912353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Q., Chen X., Macdonnell S.M., Kranias E.G., Lorenz J.N., Leitges M., et al. Protein kinase C{alpha}, but not PKC{beta} or PKC{gamma}, regulates contractility and heart failure susceptibility: implications for ruboxistaurin as a novel therapeutic approach. Circ Res. 2009;105:194–200. doi: 10.1161/CIRCRESAHA.109.195313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sharp T.E., III, Kubo H., Berretta R.M., Starosta T., Wallner M., Schena G.J., et al. Protein kinase C inhibition with ruboxistaurin increases contractility and reduces heart size in a swine model of heart failure with reduced ejection fraction. JACC Basic Transl Sci. 2017;2:669–683. doi: 10.1016/j.jacbts.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sodha N.R., Clements R.T., Bianchi C., Sellke F.W. Cardiopulmonary bypass with cardioplegic arrest activates protein kinase C in the human myocardium. J Am Coll Surg. 2008;206:33–41. doi: 10.1016/j.jamcollsurg.2007.06.308. [DOI] [PubMed] [Google Scholar]
- 14.Hambleton M., York A., Sargent M.A., Kaiser R.A., Lorenz J.N., Robbins J., et al. Inducible and myocyte-specific inhibition of PKCalpha enhances cardiac contractility and protects against infarction-induced heart failure. Am J Physiol Heart Circ Physiol. 2007;293:H3768–H3771. doi: 10.1152/ajpheart.00486.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ladage D., Tilemann L., Ishikawa K., Correll R.N., Kawase Y., Houser S.R., et al. Inhibition of PKCα/β with ruboxistaurin antagonizes heart failure in pigs after myocardial infarction injury. Circ Res. 2011;109:1396–1400. doi: 10.1161/CIRCRESAHA.111.255687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu M., Shi G., Yang K.C., Gu L., Kanthasamy A.G., Anantharam V., et al. Role of protein kinase C in metabolic regulation of the cardiac Na+ channel. Heart Rhythm. 2017;14:440–447. doi: 10.1016/j.hrthm.2016.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sabe S.A., Kononov M.A., Bellam K.G., Sodha N., Ehsan A., Jackson W.F., et al. Poorly controlled hypertension is associated with increased coronary myogenic tone in patients undergoing cardiac surgery with cardiopulmonary bypass. J Thorac Cardiovasc Surg. 2022;165:e256–e267. doi: 10.1016/j.jtcvs.2022.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J., Partovian C., Li J., Hampton T.G., Metais C., Tkachenko E., et al. Modulation of microvascular signaling by heparan sulfate matrix: studies in syndecan-4 transgenic mice. Microvasc Res. 2002;64:38–46. doi: 10.1006/mvre.2002.2399. [DOI] [PubMed] [Google Scholar]
- 19.Song Y., Xing H., He Y., Zhang Z., Shi G., Wu S., et al. Inhibition of mitochondrial reactive oxygen species improves coronary endothelial function after cardioplegic hypoxia/reoxygenation. J Thorac Cardiovasc Surg. 2021;164:e207–e226. doi: 10.1016/j.jtcvs.2021.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xing H., Zhang Z., Shi G., He Y., Song Y., Liu Y., et al. Chronic inhibition of mROS protects against coronary endothelial dysfunction in mice with diabetes. Front Cell Dev Biol. 2021;9 doi: 10.3389/fcell.2021.643810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tuteja D., Xu D., Timofeyev V., Lu L., Sharma D., Zhang Z., et al. Differential expression of small-conductance Ca2+-activated K+ channels SK1, SK2, and SK3 in mouse atrial and ventricular myocytes. Am J Physiol Heart Circ Physiol. 2005;289:H2714–H2723. doi: 10.1152/ajpheart.00534.2005. [DOI] [PubMed] [Google Scholar]
- 22.Sailer C.A., Kaufmann W.A., Marksteiner J., Knaus H.G. Comparative immunohistochemical distribution of three small-conductance Ca2+-activated potassium channel subunits, SK1, SK2, and SK3 in mouse brain. Mol Cell Neurosci. 2004;26:458–469. doi: 10.1016/j.mcn.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 23.Deardorff A.S., Romer S.H., Deng Z., Bullinger K.L., Nardelli P., Cope T.C., et al. Expression of postsynaptic Ca2+-activated K+ (SK) channels at C-bouton synapses in mammalian lumbar motoneurons. J Physiol. 2013;591:875–897. doi: 10.1113/jphysiol.2012.240879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y., Cole V., Lawandy I., Ehsan A., Sellke F.W., Feng J. Decreased coronary arteriolar response to KCa channel opener after cardioplegic arrest in diabetic patients. Mol Cell Biochem. 2018;445:187–194. doi: 10.1007/s11010-017-3264-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stankevicius E., Dalsgaard T., Kroigaard C., Beck L., Boedtkjer E., Misfeldt M.W., et al. Opening of small and intermediate calcium-activated potassium channels induces relaxation mainly mediated by nitric-oxide release in large arteries and endothelium-derived hyperpolarizing factor in small arteries from rat. J Pharmacol Exp Therapeut. 2011;339:842–850. doi: 10.1124/jpet.111.179242. [DOI] [PubMed] [Google Scholar]
- 26.Bellien J., Thuillez C., Joannides R. Contribution of endothelium-derived hyperpolarizing factors to the regulation of vascular tone in humans. Fundam Clin Pharmacol. 2008;22:363–377. doi: 10.1111/j.1472-8206.2008.00610.x. [DOI] [PubMed] [Google Scholar]
- 27.Kant S., Sellke F., Feng J. Metabolic regulation and dysregulation of endothelial small conductance calcium activated potassium channels. Eur J Cell Biol. 2022;101 doi: 10.1016/j.ejcb.2022.151208. [DOI] [PubMed] [Google Scholar]
- 28.Griffith T.M. Endothelium-dependent smooth muscle hyperpolarization: do gap junctions provide a unifying hypothesis? Br J Pharmacol. 2004;141:881–903. doi: 10.1038/sj.bjp.0705698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Feng J., Liu Y., Clements R.T., Sodha N.R., Khabbaz K.R., Senthilnathan V., et al. Calcium-activated potassium channels contribute to human coronary microvascular dysfunction after cardioplegic arrest. Circulation. 2008;118(14 Suppl):S46–S51. doi: 10.1161/circulationaha.107.755827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Allen D., Fakler B., Maylie J., Adelman J.P. Organization and regulation of small conductance Ca2+-activated K+ channel multiprotein complexes. J Neurosci. 2007;27:2369–2376. doi: 10.1523/JNEUROSCI.3565-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sodha N.R., Feng J., Clements R.T., Bianchi C., Boodhwani M., Ramlawi B., et al. Protein kinase C alpha modulates microvascular reactivity in the human coronary and skeletal microcirculation. Surgery. 2007;142:243–252. doi: 10.1016/j.surg.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 32.Mukherjee R., Apple K.A., Squires C.E., Kaplan B.S., McLean J.E., Saunders S.M., et al. Protein kinase C isoform activation and endothelin-1 mediated defects in myocyte contractility after cardioplegic arrest and reperfusion. Circulation. 2006;114:I308–I313. doi: 10.1161/CIRCULATIONAHA.105.001388. [DOI] [PubMed] [Google Scholar]
- 33.Verma S., Maitland A., Weisel R.D., Fedak P.W., Li S.H., Mickle D.A., et al. Increased endothelin-1 production in diabetic patients after cardioplegic arrest and reperfusion impairs coronary vascular reactivity: reversal by means of endothelin antagonism. J Thorac Cardiovasc Surg. 2006;123:1114–1119. doi: 10.1067/mtc.2002.121972. [DOI] [PubMed] [Google Scholar]
- 34.Sharma A.C., Fogelson B.G., Nawas S.I., Vigneswaran W.T., Sam A.D., II, Alden K.J., et al. Elevated coronary endothelin-1 but not nitric oxide in diabetics during CABG. Ann Thorac Surg. 1999;67:1659–1663. doi: 10.1016/s0003-4975(99)00287-8. [DOI] [PubMed] [Google Scholar]
- 35.Feng J., Liu Y., Khabbaz K.R., Hagberg R., Sodha N.R., Osipov R.M., et al. Endothelin-1-induced contractile responses of human coronary arterioles via endothelin-A receptors and PKC-alpha signaling pathways. Surgery. 2010;147:798–804. doi: 10.1016/j.surg.2009.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]