

Abstract
Obesity, defined as a body mass index (BMI) ≥ 30, is an established risk factor for breast cancer among women in the general population after menopause. Whether elevated BMI is a risk factor for women with a germline mutation in BRCA1 or BRCA2 is less clear because of inconsistent findings from epidemiological studies and a lack of mechanistic studies in this population. Here, we show that DNA damage in normal breast epithelia of women carrying a BRCA mutation is positively correlated with BMI and with biomarkers of metabolic dysfunction. In addition, RNA sequencing showed obesity-associated alterations to the breast adipose microenvironment of BRCA mutation carriers, including activation of estrogen biosynthesis, which affected neighboring breast epithelial cells. In breast tissue explants cultured from women carrying a BRCA mutation, we found that blockade of estrogen biosynthesis or estrogen receptor activity decreased DNA damage. Additional obesity-associated factors, including leptin and insulin, increased DNA damage in human BRCA heterozygous epithelial cells, and inhibiting the signaling of these factors with a leptin-neutralizing antibody or PI3K inhibitor, respectively, decreased DNA damage. Furthermore, we show that increased adiposity was associated with mammary gland DNA damage and increased penetrance of mammary tumors in Brca1+/− mice. Overall, our results provide mechanistic evidence in support of a link between elevated BMI and breast cancer development in BRCA mutation carriers. This suggests that maintaining a lower body weight or pharmacologically targeting estrogen or metabolic dysfunction may reduce the risk of breast cancer in this population.
INTRODUCTION
Inheriting a pathogenic mutation in the DNA repair gene breast cancer gene 1 (BRCA1) or breast cancer gene 2 (BRCA2) is causally linked to the development of breast and ovarian cancer in women (1, 2). Although there is strong evidence linking obesity to the development of hormone receptor–positive breast cancer after menopause in the general population (3), there are conflicting results in BRCA mutation carriers. Some studies have found that maintaining a lower body weight or weight loss in young adulthood is associated with delayed onset of breast cancer (4, 5). Other studies have reported that adiposity or elevated body weight in adulthood is associated with increased cancer risk (6–9). Conversely, some reports indicate that increased body mass index (BMI) in young adulthood may have protective effects and that risk is modified by menopausal status (9–11). The lack of clarity on the role of BMI and risk of breast cancer development in BRCA mutation carriers limits the ability of clinicians to provide evidence-based guidance on prevention strategies beyond prophylactic surgical intervention.
Weight gain and obesity, defined as having a BMI ≥ 30, are often coupled with metabolic syndrome, insulin resistance, and changes to adipose tissue, including that of the breast microenvironment (12–15). Obesity-induced changes to breast adipose tissue include dysregulation of hormone and adipokine balance and increased production of inflammatory mediators (16). For example, estrogen biosynthesis is increased in obese breast adipose tissue due to overexpression of aromatase, which catalyzes the conversion of androgens to estrogens, in adipose stromal cells (17–19). In addition, excessive expansion of adipocytes leads to hypoxia, lipolysis, and altered adipokine production, including a higher leptin-to-adiponectin ratio (15, 16, 20). These changes to the breast microenvironment may have important implications for breast carcinogenesis given that breast epithelial cells are embedded in this milieu and engage in epithelium-adipose cross-talk (21).
BRCA1 and BRCA2 are critical for their role in homologous recombination–mediated repair of DNA double-strand breaks (22). Mutations in either BRCA1 or BRCA2 cause a defect in DNA repair, which can lead to an accumulation of DNA damage and, consequently, tumorigenesis (23, 24). Studies have linked obesity or metabolic syndrome to DNA damage, including in leukocytes (25), skeletal muscle (26), peripheral blood mononuclear cells (27), and pancreatic β cells (28), but no studies have examined the relationship between obesity and DNA damage in normal breast epithelial cells.
Here, we show that BMI and markers of metabolic dysfunction are positively correlated with DNA damage in normal breast epithelia of women carrying a BRCA mutation, a finding that is extended to the fallopian tubes of BRCA mutation carriers. To identify obesity-associated drivers of DNA damage, we used RNA sequencing (RNA-seq) of whole breast tissue and of isolated breast epithelial organoids from BRCA mutation carriers, along with ex vivo and in vitro studies with BRCA1 and BRCA2 mutant primary tissues and cell lines. In vivo studies in Brca1 heterozygous knockout mice were also conducted to determine whether high-fat diet (HFD)–induced obesity is associated with epithelial cell DNA damage and greater mammary tumor penetrance relative to mice fed a low-fat diet (LFD). These data presented provide mechanistic evidence supporting an increased risk of breast cancer development in BRCA mutation carriers with elevated BMI and metabolic dysfunction and, importantly, provide clinically relevant strategies for risk reduction.
RESULTS
Elevated BMI positively correlates with breast epithelial cell DNA damage in women carrying a mutation in BRCA1 or BRCA2
To assess the amount of DNA damage in normal breast epithelia associated with BMI in women carrying a BRCA1 or BRCA2 mutation, we constructed tissue microarrays from noncancerous breast tissue obtained from 69 women undergoing mastectomy. The study population included BRCA1 (n = 40) and BRCA2 (n = 29) mutation carriers who had documented BMI (kg/m2) between 19.38 and 44.9 (median 23.9) at the time of surgery, as shown in Table 1. When grouping the population by BMI categories of lower weight (BMI < 25 kg/m2, n = 43) or overweight/obese (BMI ≥ 25.0 kg/m2, n = 26), median age was significantly higher in the group with overweight/obesity compared with the group with lower weight (P = 0.03). Additional clinical features elevated in the group with overweight/obesity compared with the group with lower weight included percentage of participants diagnosed with dyslipidemia (P = 0.01) and with hypertension (P = 0.046). The group with lower weight also had a greater representation of premenopausal versus postmenopausal participants compared with the group with overweight/obesity (P = 0.022). Diagnosis of diabetes, race, presence of invasive tumor, tumor subtype, and BRCA1 versus BRCA2 mutation were not significantly different between the two BMI groups (Table 1).
Table 1. Baseline characteristics of study population based on BMI category.
BMI, body mass index; HR, hormone receptor; HER2, human epidermal growth factor receptor 2; TNBC, triple-negative breast cancer.
| Variables | All (n = 69) | BMI < 25 (n = 43) | BMI ≥ 25 (n = 26) | P |
|---|---|---|---|---|
| BMI, median (range) | 23.9 (19.38–44.9) | 22.2 (19.38–24.7) | 28.8 (25.3–44.9) | <0.001 |
| BRCA mutation, n (%) | 0.6 | |||
| BRCA1 | 40 (58.0%) | 26 (60.5%) | 14 (53.8%) | |
| BRCA2 | 29 (42.0%) | 17 (39.5%) | 12 (46.2%) | |
| Age, median (range) | 40 (25–69) | 39.0 (25–60) | 44.5 (28–69) | 0.03 |
| Diabetes, n (%) | 0.4 | |||
| No | 68 (98.6%) | 43 (100.0%) | 25 (96.2%) | |
| Yes | 1 (1.4%) | 0 (0%) | 1 (3.8%) | |
| Dyslipidemia, n (%) | 0.01 | |||
| No | 62 (89.9%) | 42 (97.7%) | 20 (76.9%) | |
| Yes | 7 (10.1%) | 1 (2.3%) | 6 (23.1%) | |
| Hypertension, n (%) | 0.046 | |||
| No | 61 (88.4%) | 41 (95.3%) | 20 (76.9%) | |
| Yes | 8 (11.6%) | 2 (4.7%) | 6 (23.1%) | |
| Menopausal status, n (%) | 0.022 | |||
| Pre- | 46 (66.7%) | 33 (76.7%) | 13 (50.0%) | |
| Post- | 23 (33.3%) | 10 (23.3%) | 13 (50.0%) | |
| Race, n (%) | 0.1 | |||
| Asian | 1 (1.4%) | 1 (2.3%) | 0 (0.0%) | |
| Black | 2 (2.9%) | 2 (4.7%) | 0 (0.0%) | |
| Other | 2 (2.9%) | 0 (0.0%) | 2 (7.7%) | |
| White | 56 (81.2%) | 33 (76.7%) | 23 (88.5%) | |
| Missing | 8 (11.6%) | 7 (16.3%) | 1 (3.8%) | |
| Invasive tumor present, n (%) | 0.7 | |||
| No | 39 (56.5%) | 25 (58.1%) | 14 (53.8%) | |
| Yes | 30 (43.5%) | 18 (41.9%) | 12 (46.2%) | |
| Tumor subtype, n (%) | >0.9 | |||
| HR+ | 22 (31.9%) | 14 (32.6%) | 8 (30.8%) | |
| HER2+ | 1 (1.4%) | 1 (2.3%) | 0 (0.0%) | |
| TNBC | 9 (13.0%) | 5 (11.6%) | 4 (15.4%) | |
| N/A | 37 (53.6%) | 23 (53.5%) | 14 (53.8%) |
Immunofluorescence (IF) staining for the DNA double-strand break marker γH2AX was performed with nuclear counterstain Hoechst to visualize the number of foci of DNA damage per epithelial cell (Fig. 1A). Among BRCA1 and BRCA2 mutation carriers, but not among women with no identified mutation in BRCA, BMI was positively associated with breast epithelial cell DNA damage as quantified by the number of γH2AX foci/100 cells (Fig. 1, B and C). Age was also found to be positively correlated with DNA damage (Fig. 1D). Although this correlation diminished when adjusting for BMI (P = 0.335; Table 2), BMI remained positively associated with DNA damage when adjusting for age (P = 0.003; Table 2). DNA damage amounts in postmenopausal women trended higher compared with those in premenopausal women (Fig. 1E); however, this difference was not significant. In addition, circulating concentrations of sex hormone–binding globulin (SHBG), which binds estrogens to decrease their bioavailability, were negatively correlated with breast epithelial cell DNA damage (Fig. 1F). This negative association remains significant when adjusting for age but not BMI (P = 0.020 and P = 0.081, respectively; Table 2). Elevated BMI is often coupled to insulin resistance, a hallmark of metabolic dysfunction. Accordingly, fasting serum concentrations of insulin and Homeostasis Model Assessment 2 of Insulin Resistance (HOMA2 IR) values were positively correlated with amounts of breast epithelial cell DNA damage, whereas fasting serum glucose concentrations were not (Fig. 1, G to I). Insulin and HOMA2 IR retained significance after adjustments for either BMI (P = 0.009 and P = 0.010, respectively) or age (P < 0.001 for both; Table 2). No correlation with DNA damage was observed for circulating biomarkers of inflammation, including high-sensitivity C-reactive protein (hsCRP) and interleukin-6 (IL-6) or with crown-like structures (CLS), a histological marker of local breast adipose inflammation (Fig. 1, J to L) (29). These data indicate that among BRCA mutation carriers, elevated BMI is a risk factor for breast epithelial cell DNA damage. Furthermore, specific obesity-associated factors, including insulin resistance and estrogen balance, may be important drivers of this risk.
Fig. 1. BMI and additional clinical characteristics are positively correlated with DNA damage in breast epithelia of women carrying a BRCA mutation.
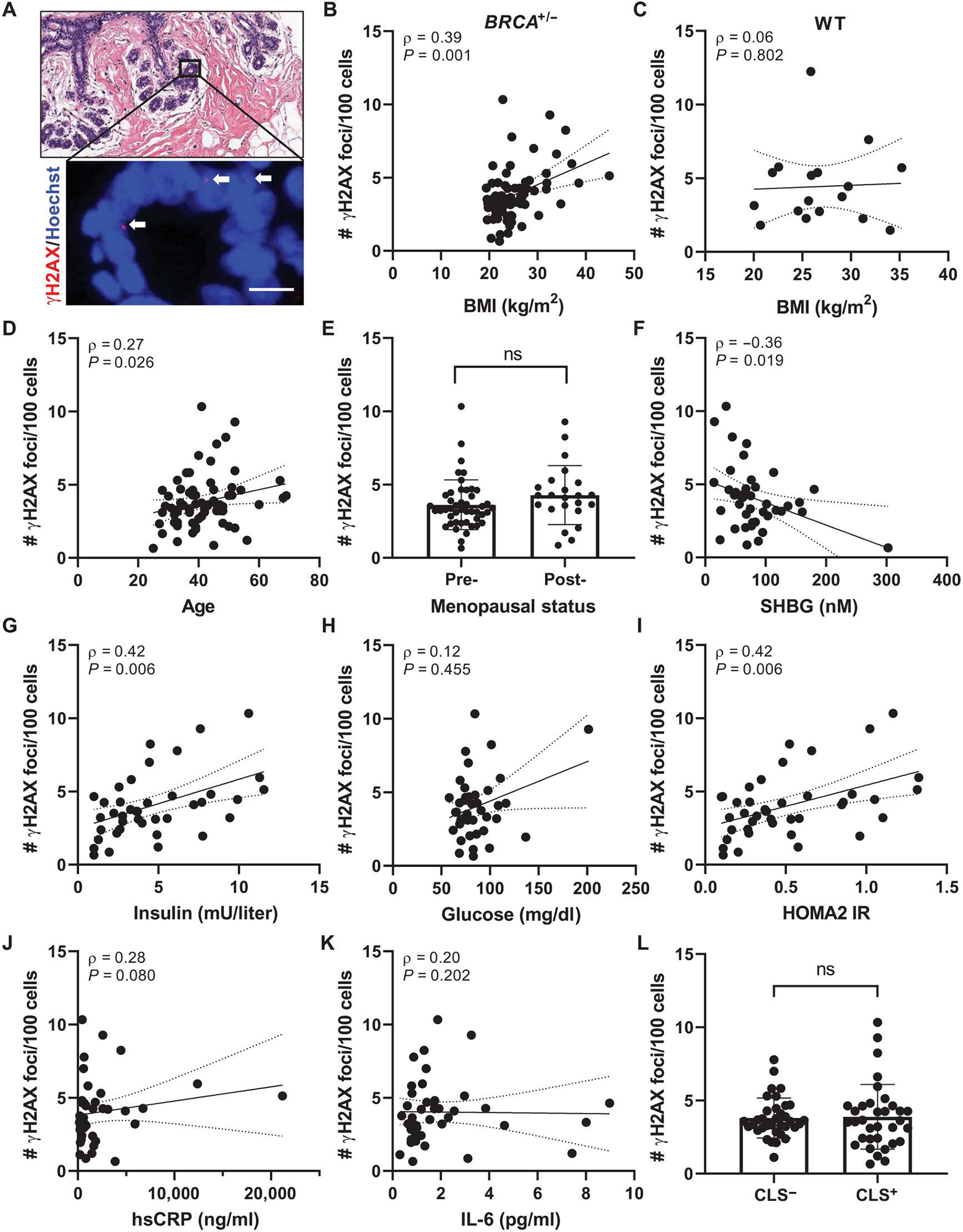
(A) Representative image of tissue microarray section of normal breast epithelium shown by hematoxylin and eosin (H&E) stain (top) and by IF staining (bottom) for γH2AX (red, arrows) colocalizing with Hoechst (blue). Scale bar, 10 μm. (B) Correlation between epithelial cell DNA damage as measured by number of γH2AX foci/100 cells with BMI in BRCA mutation carriers and in (C) age-matched women WT for BRCA (n = 17). # = number. (D) Correlation between epithelial cell DNA damage and age. (E) Average DNA damage in the study population grouped by menopausal status: premenopausal, n = 46, and postmenopausal, n = 23. Epithelial cell DNA damage correlated with circulating serum biomarkers including (F) sex hormone–binding globulin (SHBG), (G) insulin, (H) glucose, (I) Homeostasis Model Assessment 2 of Insulin Resistance (HOMA2 IR), (J) high-sensitivity C-reactive protein (hsCRP), and (K) interleukin-6 (IL-6) in a subset of the study population with available fasting serum at the time of surgery (n = 41). (L) Average DNA damage in the study population when grouped by those exhibiting histological breast adipose tissue inflammation defined as presence of crown-like structures (CLS) versus those with no CLS present (CLS− versus CLS+). Two-tailed Mann-Whitney test was used to determine significant differences in grouped comparisons, and data are presented as means ± SD. Correlation between variables was assessed by Spearman’s rank correlation coefficient (ρ). Associated P value and ρ are shown for continuous variables with 95% confidence intervals. ns, not significant; n = 69 unless otherwise stated.
Table 2.
Association of clinical features and blood biomarkers with DNA damage, adjusting for age or BMI.
| Variables | Correction | P | Correction | P |
|---|---|---|---|---|
| BMI | Age | 0.003 | ||
| Age | BMI | 0.335 | ||
| SHBG (nM) | BMI | 0.081 | Age | 0.020 |
| Insulin (mU/liter) | BMI | 0.009 | Age | <0.001 |
| HOMA2 IR | BMI | 0.010 | Age | <0.001 |
Elevated BMI is associated with differences in gene expression in breast adipose tissue and in breast epithelial cells of BRCA mutation carriers
To identify changes associated with obesity in breast epithelial cells and in the breast adipose microenvironment that may be linked to DNA damage, we conducted RNA-seq studies on both isolated primary breast epithelial cells and noncancerous whole breast tissue obtained from BRCA1 and BRCA2 (BRCA) mutation carriers. RNA-seq was conducted on breast tissue pieces obtained from BRCA mutation carriers with BMI < 25 kg/m2 (n = 64) and with BMI ≥ 25 kg/m2 (n = 67). An unsupervised heatmap was constructed, which showed general clustering of cases with BMI < 25 and clustering of cases with BMI ≥ 25 by gene expression (Fig. 2A). A total of 2329 genes were significantly up-regulated with obesity, and 1866 were significantly down-regulated (table S1; P < 0.05). Ingenuity Pathway Analysis (IPA) identified several pathways that were significantly altered in the cases with BMI ≥ 25, which include pathways associated with obesity and metabolic dysfunction, such as “Phagosome Formation,” “Liver X receptor-retinoid X receptor (LXR/RXR) Activation,” “Tumor Microenvironment Pathway Activation,” and “Estrogen Biosynthesis” (Fig. 2B). A heatmap of genes involved in estrogen regulation showed a significant (P < 0.05) increase in many genes involved in the bioactivity, biosynthesis, and activation of estrogens, including steroid sulfatase, 3β-hydroxysteroid dehydrogenase type 1 (3βHSD1), aldo-keto reductase family 1 member C3 (AKR1C3), aldo-keto reductase family 1 member B15 (AKR1B15), 17β-hydroxysteroid dehydrogenase type 1 (17βHSD1), and aromatase [cytochrome P450 family 19 subfamily A member 1 (CYP19A1)] (Fig. 2C). Conversely, gene expression of 17β-hydroxysteroid dehydrogenase type 8 (17βHSD8), involved in estrogen inactivation, was significantly lower in cases with BMI ≥ 25 relative to cases with BMI < 25 (P < 0.05). Moreover, there were mixed effects of obesity on the expression of genes involved in estrogen catabolism to hydroxylated metabolites and neutralization by catechol-O-methyltransferase (COMT).
Fig. 2. Elevated BMI is associated with significant changes in gene expression in breast adipose tissue and in breast epithelial cells of BRCA mutation carriers.
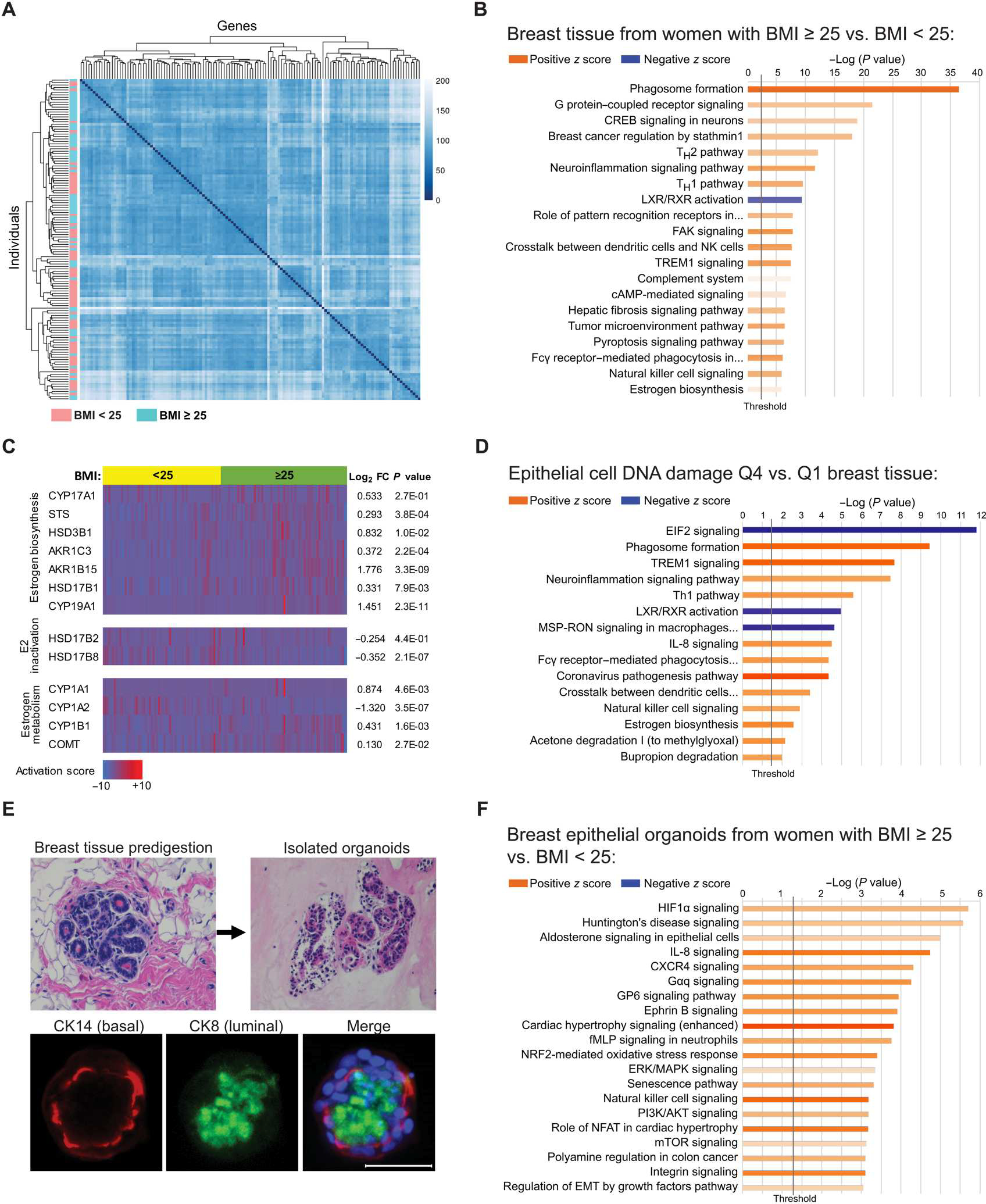
(A) Unsupervised heatmap of whole breast tissue gene expression by RNA-seq in BRCA mutation carriers identified by BMI category of <25 (n = 64, blue) or ≥25 (n = 67, pink). Rows represent individual patients, and columns represent genes. (B) IPA analysis of RNA-seq data showing activation (z score) of the top 20 canonical pathways regulated in breast tissue from BRCA mutation carriers with BMI ≥ 25 compared with carriers with BMI < 25 with an absolute value z score of >0.5. (C) Heatmap of RNA-seq gene expression data generated from breast tissue of BRCA mutation carriers grouped by BMI category of <25 (yellow) or ≥25 (green) showing selected genes associated with estrogen biosynthesis, estradiol (E2) inactivation, and estrogen metabolism. Corresponding gene expression (log2FC) and P values are shown in tissue from women with BMI ≥ 25 relative to BMI < 25. Columns represent individual patients. (D) DNA damage in breast epithelial cells was quantified in tissue sections from n = 61 patients from whom corresponding whole breast tissue RNA-seq data were also available. The cases were stratified by quartile of DNA damage, and the breast tissue gene expression from cases with the highest amount of DNA damage [quartile 4 (Q4)] was compared with that from cases with the lowest amount [quartile 1 (Q1)] of DNA damage. Top 15 canonical pathways regulated in Q4 versus Q1 with an absolute value z score of >2.0 are shown. (E) Representative H&E-stained images of a breast tissue section before digestion and epithelial organoids after isolation. Organoids stained positively for luminal marker cytokeratin 8 (CK8; green) and basal marker cytokeratin 14 (CK14; red) as shown by IF staining merged with Hoechst (blue). Scale bar, 50 μm. (F) IPA analysis of RNA-seq gene expression data showing activation of the top 20 canonical pathways regulated in primary breast epithelial organoids from BRCA mutation carriers with BMI ≥ 25 (n = 9) relative to carriers with BMI < 25 (n = 10) with an absolute value z score of >1.0. The lengths of the bars on all canonical pathway graphs are determined by the Fisher’s exact test. P value with entities that have a −log (P value) > 1.3 is shown.
To explore which changes in the breast microenvironment are associated with DNA damage in breast epithelial cells, we analyzed breast tissue pathway changes in relation to amounts of epithelial cell DNA damage quantified by γH2AX IF staining (n = 61; Fig. 2D). The amount of epithelial cell DNA damage in each case was stratified by quartiles, and breast tissue gene expression was compared in the highest quartile (Q4) relative to the lowest quartile (Q1), independent of BMI (table S2). The top 15 canonical pathways activated in Q4 versus Q1 breast tissue are shown (Fig. 2D), with several pathways being common to both DNA damage and BMI analyses (Fig. 2D versus Fig. 2B). Although the estrogen biosynthesis pathway was found to be activated in tissue from cases with BMI ≥ 25 compared with cases with BMI < 25 (z score = 0.775, P = 1.14 × 10−6; Fig. 2B), a stronger activation score was found when comparing DNA damage Q4 versus Q1 (z score = 2.646, P = 2.7 × 10−3; Fig. 2D), suggesting that tissue estrogen biosynthesis is highly correlated with the amount of breast epithelial cell DNA damage, irrespective of BMI.
Breast epithelial organoids were isolated from BRCA mutation carriers with BMI < 25 (n = 10) or with BMI ≥ 25 (n = 9) at the time of surgery. To validate and characterize the isolated epithelial organoids, we conducted IF staining for cytokeratin 8 (CK8) and cytokeratin 14 (CK14), characteristic markers of luminal and basal epithelial cells, respectively, that are known to comprise the breast epithelium (Fig. 2E). A total of 1144 genes were significantly up-regulated, and 537 genes were significantly down-regulated in organoids from women with BMI ≥ 25 relative to organoids from women with BMI < 25 (P < 0.05; table S3). The top 20 canonical pathways identified by IPA as regulated in the organoids from women with BMI ≥ 25 are shown (Fig. 2F) and include activation of pathways known to be associated with obesity, including “Hypoxia-inducible factor 1 (HIF1α) signaling,” “Interleukin 8 (IL-8) signaling,” “Extracellular signal-regulated kinase/mitogen-activated protein kinase (ERK/MAPK) signaling,” and “Phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) signaling,” among others.
To investigate the mechanisms that would account for differences in the impact of BMI on DNA damage in BRCA mutation carriers versus noncarriers (Fig. 1, B and C), we also conducted RNA-seq on organoids isolated from age-matched women wild type (WT) for BRCA with BMI < 25 (n = 11) or with BMI ≥ 25 (n = 8). A total of 750 genes were significantly up-regulated, and 659 genes were significantly down-regulated in organoids from noncarriers with BMI ≥ 25 relative to organoids from women with BMI < 25 (P < 0.05; table S4). The top 20 canonical pathways regulated in organoids from noncarriers with BMI ≥ 25 showed little overlap with pathways regulated in organoids from BRCA mutation carriers (fig. S1A versus Fig. 2F), with a direct comparison of z scores and pathway activation or deactivation shown in fig. S1B.
Collectively, these RNA-seq studies show that BRCA1 and BRCA2 mutation carriers who have overweight/obesity as defined by BMI ≥ 25 have altered breast epithelial cell and breast adipose microenvironment gene expression compared with carriers with BMI < 25. In addition, these studies provide a rationale for further exploring whether estrogens are a driver of DNA damage in breast epithelial cells from BRCA mutation carriers. Although elevated BMI was also associated with gene expression changes to breast epithelial cells from noncarriers, the changes observed in relation to BMI were distinct from those found in BRCA mutation carriers. This may help to explain differences in correlation data between BMI and DNA damage among these two populations.
Cross-talk between epithelial cells and the breast adipose microenvironment
Given the gene expression changes identified in BRCA heterozygous breast adipose tissue and in breast epithelial cells in association with elevated BMI, we next investigated whether the breast adipose microenvironment drives gene expression in breast epithelial cells. The IPA Upstream Regulator tool was used to identify regulators of gene expression differences in organoids isolated from BRCA mutation carriers with overweight/obesity (BMI ≥ 25) relative to organoids isolated from carriers with lower weight (BMI < 25). To highlight endogenous factors that may be responsible for driving gene expression changes, we filtered results to show the top 20 secreted factors. Among these factors, β-estradiol (E2; an estrogen) was the top predicted upstream regulator (Table 3). A number of additional predicated upstream organoid regulators were significantly up-regulated in breast tissue from BRCA mutation carriers with overweight/obesity (P < 0.05), including several interleukins (IL-2, IL-15, and IL-5), transforming growth factor–β1 (TGF-β1), colony-stimulating factor 1 (CSF1), angiopoietin-2 (ANGPT2), and wingless-type MMTV integration site family member 5A (WNT5A). Some factors, such as insulin, are known to be elevated in obesity but are not produced locally in breast tissue and therefore do not have an observed tissue gene expression in our study. These data suggest that some endogenously produced factors in the breast microenvironment of women with overweight/obesity may interact with neighboring breast epithelial cells to induce gene expression changes and DNA damage.
Table 3.
Predicted upstream regulators of gene expression differences in breast epithelial organoids isolated from BRCA mutation carriers with BMI≥ 25 relative to carriers with BMI < 25 and associated gene expression in whole breast tissue.
| Organoid upstream regulator | Predicted activation state | Activation z score | P value of overlap | Breast tissue RNA-seq (log2 fold change) | P value |
|---|---|---|---|---|---|
| β-Estradiol | Activated | 4.728 | 2.2 × 10−10 | See Fig. 2C | |
| IL-2 | Activated | 3.402 | 3.1 × 10−2 | 0.563 | 2.3 × 10−1 |
| GDF2 | Activated | 3.217 | 4.9 × 10−3 | −0.081 | 9.8 × 10−1 |
| IL-15 | Activated | 3.152 | 1.5 × 10−3 | 0.299 | 4.1 × 10−5 |
| TNF-SF11 | Activated | 3.125 | 3.2 × 10−2 | −0.757 | 9.7 × 10−2 |
| Insulin | Activated | 3.113 | 6.1 × 10−3 | ||
| IL-4 | Activated | 3.016 | 1.9 × 10−3 | −0.25 | 7.4 × 10−1 |
| TGF-B1 | Activated | 2.942 | 6.0 × 10−9 | 0.455 | 2.2 × 10−8 |
| Hydrogen peroxide | Activated | 2.839 | 3.1 × 10−3 | ||
| IL-3 | Activated | 2.674 | 7.6 × 10−4 | −0.122 | 9.7 × 10−1 |
| CSF1 | Activated | 2.602 | 8.9 × 10−3 | 0.35 | 1.4 × 10−6 |
| Lh | Activated | 2.598 | 1.7 × 10−3 | ||
| Dinoprost (PGF2α) | Activated | 2.569 | 2.9 × 10−2 | ||
| IL-5 | Activated | 2.496 | 5.5 × 10−3 | 0.173 | 6.9 × 10−1 |
| ATP | Activated | 2.443 | 8.7 × 10−3 | ||
| MDK | Activated | 2.433 | 2.9 × 10−2 | −0.34 | 4.4 × 10−3 |
| AGT | Activated | 2.345 | 4.2 × 10−3 | −0.56 | 9.6 × 10−4 |
| ANGPT2 | Activated | 2.329 | 1.1 × 10−3 | 0.38 | 9.2 × 10−5 |
| WNT5A | Activated | 2.292 | 1.7 × 10−3 | 0.184 | 1.3 × 10−1 |
| Pyruvic acid | Activated | 2.156 | 1.5 × 10−3 |
Targeting estrogen in breast tissue from BRCA mutation carriers reduces epithelial cell DNA damage
Next, we conducted mechanistic studies to determine whether targeting estrogen signaling or biosynthesis in breast tissue would lead to decreased amounts of breast epithelial cell DNA damage. We first conducted immunohistochemistry (IHC) staining to verify that normal epithelia from BRCA1 and BRCA2 mutation carriers express estrogen receptor α (ERα). Epithelial cells staining positively for ERα were found throughout the epithelium among carriers of BRCA1 or BRCA2 mutations (representative images shown in Fig. 3A, top row). IF staining was then conducted to visualize whether γH2AX foci colocalized with ERα-positive cells. Representative images are shown, which highlight ERα-positive cells frequently staining positively for γH2AX foci (Fig. 3A, bottom row). Next, we tested whether disrupting estrogen signaling through use of the drug fulvestrant, which degrades the ER, would affect amounts of DNA damage in the breast. Breast tissue was obtained from BRCA mutation carriers undergoing surgery (n = 7) and were plated as explants in the presence of fulvestrant (100 μM) or vehicle for 24 hours (Fig. 3B). Explants were formalin-fixed and sectioned for assessment of breast epithelial cell DNA damage by IF staining. A 32.5% reduction in DNA damage was observed overall after treatment with fulvestrant (Fig. 3C).
Fig. 3. Targeting estrogen signaling or production in breast tissue decreases epithelial cell DNA damage in women carrying a mutation in BRCA1 or BRCA2.
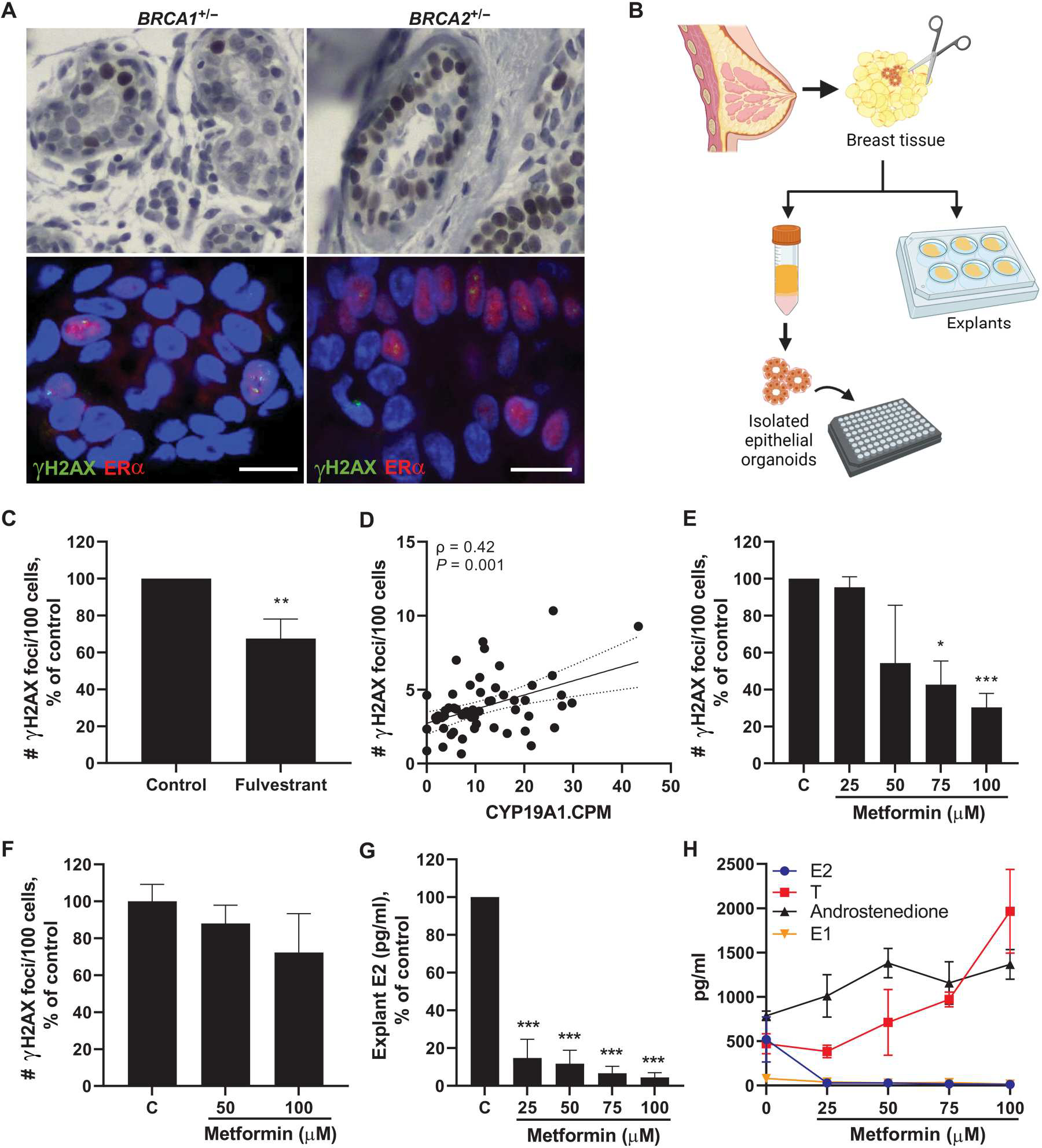
(A) Representative IHC staining of ERα expression in breast epithelia from carriers of a BRCA1 or BRCA2 mutation (top). Representative IF staining showing colocalization of number of γH2AX foci (green) with ERα-positive cells (red) (bottom). Scale bar, 10 μm. (B) Experimental schematic showing collection of breast tissue and plating of explants or isolation of primary breast epithelial organoids for treatment studies. (C) Breast epithelial cell DNA damage assessed by IF (number of γH2AX foci/100 cells) in ex vivo breast adipose tissue explants from BRCA mutation carriers treated with fulvestrant (100 nM) for 24 hours (pooled average of n = 7 patients). (D) Aromatase (CYP19A1) expression in breast tissue from BRCA mutation carriers [RNA-seq counts per million (CPM)] correlated with amount of breast epithelial cell DNA damage in corresponding tissue sections (n = 58). Spearman’s rank correlation coefficient (ρ) and associated P value are shown with 95% confidence intervals. (E) Breast epithelial cell DNA damage in ex vivo breast adipose tissue explants from BRCA mutation carriers treated with metformin (0 to 100 μM) for 24 hours (pooled average of n = 3 patients). (F) DNA damage in isolated primary breast epithelial cells from BRCA mutation carriers treated with metformin (0 to 100 μM) for 24 hours (representative of n = 2 experiments). (G) Average E2 concentrations and (H) overlay of E2, testosterone (T), androstenedione, and estrone (E1) concentrations in ex vivo breast adipose explants after 24-hour treatment with metformin (pooled average of n = 3 patients). Student’s t test was used to determine significant differences from control unless otherwise stated. Data are presented as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Next, we hypothesized that targeting estrogen biosynthesis in the breast by down-regulating aromatase expression would lead to less estrogen exposure to the epithelial cells and, consequently, decreased DNA damage. In support of this hypothesis, RNA-seq data from BRCA1 and BRCA2 mutation carriers showed a positive correlation between breast adipose aromatase expression and the amount of breast epithelial cell DNA damage (Fig. 3D). Because aromatase expression is known to be up-regulated in association with obesity, we conducted additional statistical analyses to adjust for BMI and found that aromatase remained independently positively associated with DNA damage (P = 0.030). To target estrogen biosynthesis, we used metformin, a widely used antidiabetic drug, which has also been shown to decrease aromatase production in the breast by stimulation of adenosine monophosphate (AMP)–activated protein kinase (AMPK) in adipose stromal cells (30, 31). Breast tissue obtained from BRCA mutation carriers (n = 3) was plated as explants and treated with metformin (0 to 100 μM) for 24 hours followed by IF assessment of breast epithelial cell DNA damage. A dose-dependent decrease in DNA damage was observed, with significant differences after 75 and 100 μM metformin treatment (P < 0.05 and P < 0.001, respectively; Fig. 3E). Because metformin is known to decrease aromatase expression in adipose stromal cells surrounding breast epithelial cells, we digested breast tissue to isolate the epithelial cells from their microenvironment (Fig. 3B) and treated them with metformin for 24 hours to determine whether the presence of the breast microenvironment is required for the effect of metformin on DNA damage. Although there was a modest trend for reduction in DNA damage with increasing doses of metformin, these results were not significant (Fig. 3F). Consistently, tissue concentrations of E2 were markedly reduced in breast explants after 24-hour metformin treatment in a dose-dependent manner (Fig. 3G). In addition, testosterone and androstenedione, which are converted to E2 and estrone (E1) by aromatase, respectively, were increased in explants after treatment with metformin, whereas both E1 and E2 decreased (Fig. 3H). These data show that metformin treatment leads to decreased estrogen biosynthesis in breast tissue in association with reduction in epithelial cell DNA damage.
Local and systemic factors contribute to DNA damage in BRCA1 and BRCA2 heterozygous breast epithelial cells
Our data support a paracrine interaction between adipose tissue and breast epithelial cells. Having found a direct role for E2 in mediating DNA damage in primary human tissues, we next explored the role of additional obesity-associated factors. This included those present in breast adipose tissue conditioned medium (CM), as well as recombinant leptin and insulin. To first investigate whether factors derived from breast adipose tissue were able to directly induce DNA damage in BRCA mutant breast epithelial cells, we treated BRCA1 heterozygous knockout MCF-10A cells with CM from reduction mammoplasty or nontumor quadrants of mastectomy tissue (n = 36, BMI: 20.6 to 40.1 kg/m2; Fig. 4A). Breast adipose CM treatment was positively correlated with DNA damage as a function of the patient’s BMI, as measured by IF of γH2AX foci (Fig. 4B). Representative confocal images are shown in fig. S2. To determine whether effects of CM on DNA damage were generalizable to BRCA2 mutation carriers, we tested a subset of CM cases (n = 13) in MCF-10A cells carrying a heterozygous BRCA2 mutation, generated using CRISPR-Cas9 gene editing. A positive correlation between BMI and DNA damage was also observed in these cells (Fig. 4C). Representative confocal images are shown in fig. S2. These studies demonstrate that factors secreted by breast adipose tissue directly stimulate DNA damage in breast epithelial cells. Furthermore, given the lack of ER expression in MCF-10A cells (32), these studies also highlight the existence of additional factors beyond estrogen that may be contributing to DNA damage induction in the setting of obesity in BRCA1 and BRCA2 mutant breast epithelial cells.
Fig. 4. Obesity-induced changes to the local breast adipose microenvironment promote DNA damage in BRCA1 and BRCA2 heterozygous breast epithelial cells.
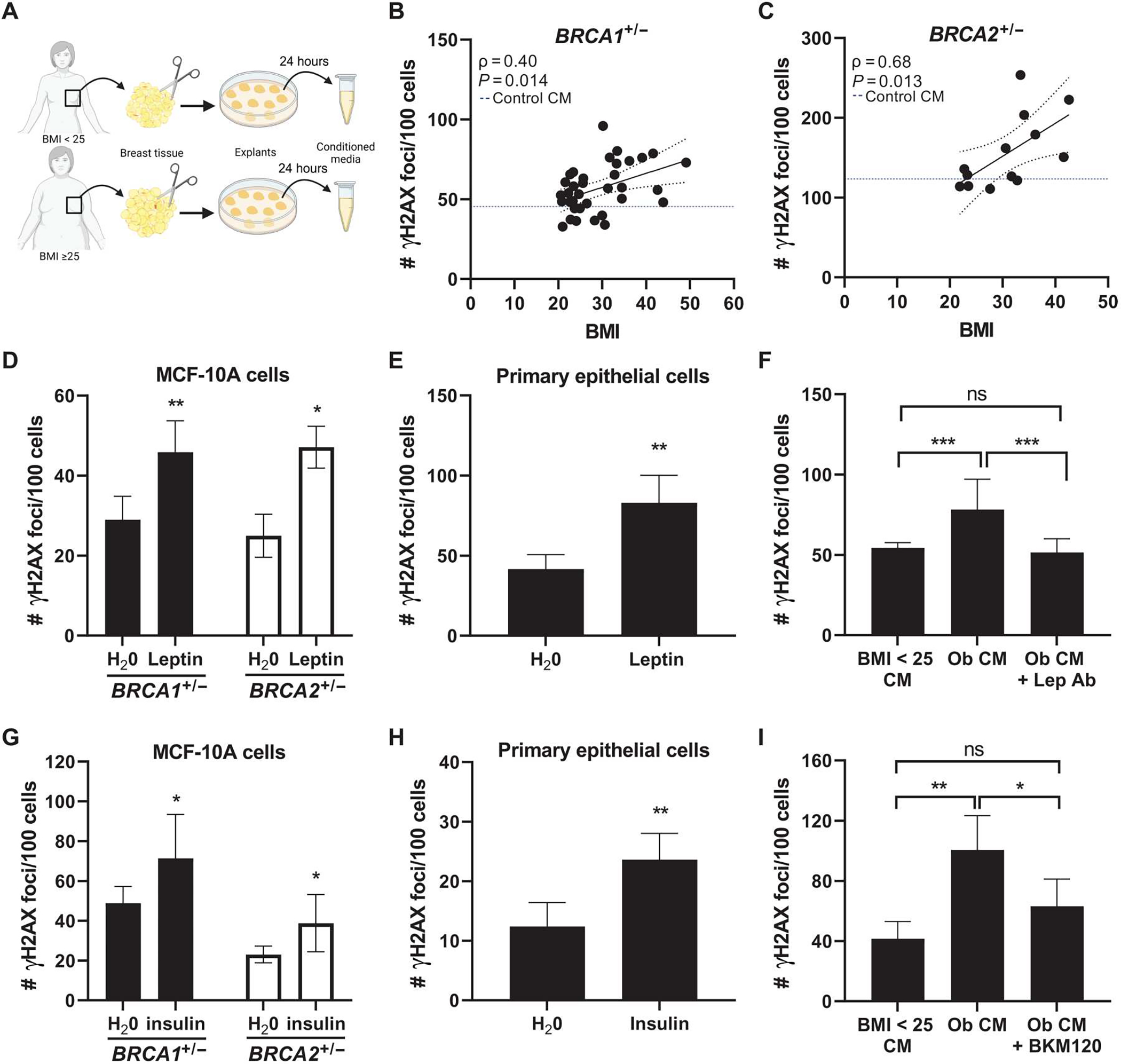
(A) Experimental schematic showing the collection of breast adipose tissue conditioned medium (CM) from women with BMI < 25 and with BMI ≥ 25. (B) MCF-10A cells were treated with CM for 24 hours. DNA damage assessed by IF (number of γH2AX foci/100 cells) is shown correlated with BMI in BRCA1+/− (n = 36 CM cases) and (C) BRCA2+/− (n = 13 CM cases) MCF-10A cells. Blue dotted line represents amount of DNA damage induced by control CM (medium not conditioned by adipose explants). Spearman’s rank correlation coefficient (ρ) and associated P value are shown along with 95% confidence intervals. (D) DNA damage in BRCA1+/− and BRCA2+/− MCF-10A cells and in (E) primary BRCA1+/− breast epithelial cells treated with leptin (400 ng/μl) for 24 hours. (F) DNA damage in BRCA1+/− MCF-10A cells after 24-hour treatment with CM derived from a woman with BMI < 25 (“BMI < 25 CM”), with obesity (“Ob CM”), or with Ob CM in the presence of a leptin-neutralizing antibody (“Lep Ab”). (G) DNA damage in BRCA1+/− and BRCA2+/− MCF-10A cells and in (H) primary BRCA2+/− breast epithelial cells treated with insulin (100 nM) for 24 hours. (I) DNA damage in BRCA1+/− MCF-10A cells after 24-hour treatment with BMI < 25 CM, Ob CM, or Ob CM in the presence of PI3K inhibitor BKM120 (1 μM). Student’s t test was used to determine significant differences in (D) to (I). All experiments in MCF-10A cells were conducted a minimum of two times, with representative results from one experiment shown. Data in primary cells were generated from cells treated in triplicate. Data are presented as means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001.
The expression of leptin, known to be directly correlated with adiposity, was significantly higher in the breast tissue of BRCA mutation carriers with a BMI ≥ 25 compared with those with a BMI < 25 (log2FC = 0.61, P = 3.48 × 10−6; table S1). Studies have found leptin to have promitogenic and antiapoptotic effects in breast cancer cells (33–36). However, its effects on normal breast epithelial cells are less well characterized. Here, we treated both BRCA1 and BRCA2 heterozygous MCF-10A cells with leptin (400 ng/ml) for 24 hours and found a significant induction of DNA damage in both cell lines (P < 0.01 and P < 0.05, respectively; Fig. 4D) and in primary breast epithelial cells (P < 0.01; Fig. 4E). In addition, the ability of CM derived from women with obesity to induce DNA damage in BRCA1 heterozygous breast epithelial cells was blocked when incubating in the presence of a leptin-neutralizing antibody (Fig. 4F).
Next, having identified insulin as positively correlated with DNA damage in tissue microarrays from BRCA mutation carriers, independent of BMI (Fig. 1G and Table 2), and as a top upstream regulator of gene expression in primary breast epithelial organoids isolated from women with BMI ≥ 25 (table 3), we conducted additional mechanistic studies to determine whether insulin can directly induce DNA damage. Treatment of BRCA1 and BRCA2 heterozygous knockout MCF-10A cells with insulin (100 nM) for 24 hours resulted in a significant increase in DNA damage in both cell lines (P < 0.05; Fig. 4G) and in primary breast epithelial cells (P < 0.01; Fig. 4H). Both leptin and insulin have been shown to act through PI3K (37, 38). Treatment of BRCA1 heterozygous breast epithelial cells with a PI3K inhibitor, BKM120 (1 μM), was effective at reducing the ability of CM derived from women with obesity to induce DNA damage (Fig. 4I). These data show that factors produced locally by breast adipose tissue from women with obesity or factors elevated with metabolic dysfunction contribute to the induction of DNA damage in BRCA heterozygous knockout breast epithelial cells.
The effects of breast adipose CM on DNA damage and repair in breast epithelial cells are dependent on a heterozygous BRCA mutation
RNA-seq was performed on BRCA1+/− MCF-10A cells treated with CM derived from women with a BMI < 25 or BMI ≥ 30 (n = 3 per group). Results demonstrated that consistent with DNA damage measurements (fig. S3A), IPA analysis of differentially regulated genes in the cells treated with CM from breast adipose tissue of women with a BMI ≥ 30 relative to BMI < 25 (table S5) showed increased activation of functions associated with DNA damage and genomic instability, including “Formation of micronuclei,” “Chromosomal instability,” and “Breakage of chromosomes” (Table 4). Alternatively, activation of functions associated with DNA repair were decreased, including “Repair of DNA” and “Checkpoint control” (Table 4). Among the 47 genes involved in Repair of DNA that were significantly (P < 0.05) regulated by CM derived from adipose tissue of women with obesity (BMI ≥30 CM), several genes involved in homologous recombination–mediated repair were down-regulated, including meiotic recombination 11 homolog A (MRE11A), BRCA1, BRCA1-interacting helicase 1 (BRIP1), x-ray repair cross complementing 2 (XRCC2), BRCA2, and RAD54L (table S6).
Table 4.
Activation of diseases or functions associated with DNA damage or DNA repair in BRCA1+/− epithelial cells treated with breast adipose tissue CM derived from women with obesity (BMI ≥ 30) relative to women with BMI < 25.
| Categories | Diseases or functions annotation | P value | Predicted activation state | Activation z score | Number of molecules |
|---|---|---|---|---|---|
| Cellular assembly and organization | Formation of micronuclei | 2.53 × 10−6 | Increased | 2.756 | 9 |
| DNA replication, recombination, and repair | Chromosomal aberration | 5.37 × 10−6 | Increased | 2.853 | 31 |
| Chromosomal instability | 2.43 × 10−8 | Increased | 2.603 | 19 | |
| Breakage of chromosomes | 2.88 × 10−5 | Increased | 2.488 | 11 | |
| Cell cycle; DNA replication, recombination, and repair | Checkpoint control | 1.99 × 10−6 | Decreased | −2.756 | 15 |
| Spindle checkpoint | 9.33 × 10−7 | Decreased | −2.035 | 12 | |
| DNA replication, recombination, and repair | Repair of DNA | 4.36 × 10−9 | Decreased | −3.334 | 47 |
| Double-stranded DNA break repair of tumor cell lines | 9.94 × 10−6 | Decreased | −2.241 | 14 | |
| Metabolism of DNA | 2.10 × 10−10 | Decreased | −2.09 | 54 |
To determine whether the effects of breast adipose CM derived from women with obesity are specific to BRCA1 heterozygous epithelial cells, we treated MCF-10A cells WT for BRCA1 with the same CM derived from women with BMI < 25 or BMI ≥ 30 (n = 3 per group), and changes in gene expression were evaluated by RNA-seq (table S7). Unlike in the BRCA1+/− MCF-10A cells, CM from women with a BMI ≥ 30 did not significantly induce DNA damage in WT MCF-10A cells compared with BMI < 25 CM (fig. S3). IPA analysis of differentially regulated genes in cells treated with CM from women with a BMI ≥ 30 relative to BMI < 25 showed a number of overlapping changes in the top 50 regulated “Diseases and Functions” in both WT and BRCA1+/− MCF-10A cells (Fig. 5A). However, when filtering the Diseases and Functions to highlight pathways associated with DNA damage or DNA repair, most functions associated with these pathways were not significantly regulated in the WT MCF-10A, unlike what was observed in BRCA1+/− MCF-10A cells (Fig. 5B).
Fig. 5. Breast adipose CM from women with obesity regulates gene expression and pathways associated with DNA damage and repair more robustly in BRCA1+/− MCF-10A cells compared with WT MCF-10A cells.
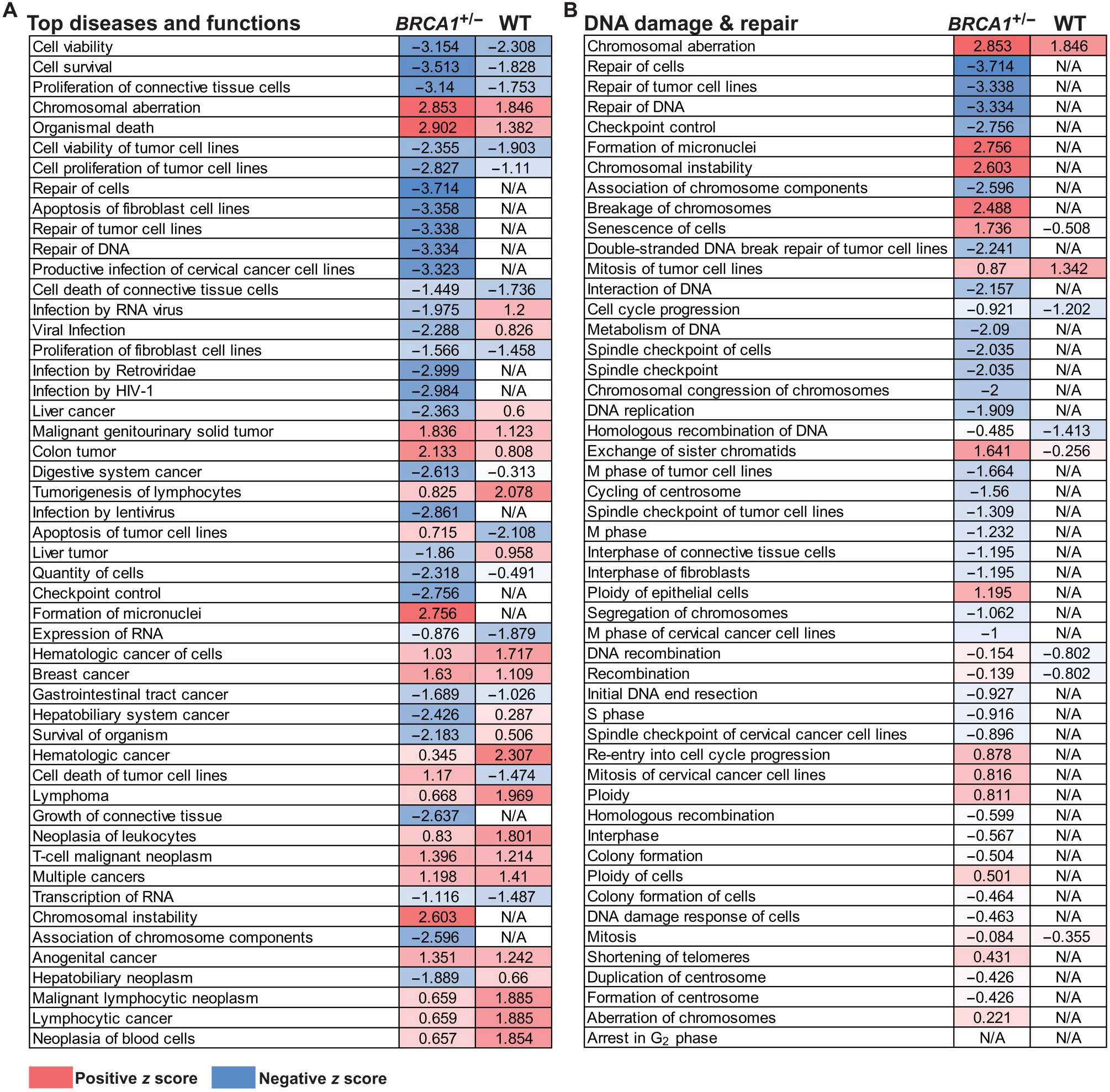
(A) MCF-10A cells carrying a heterozygous BRCA1 mutation (BRCA1+/−) or WT for BRCA were treated with breast adipose CM from women with BMI ≥ 30 (n = 3) or BMI < 25 (n = 3) for 24 hours. RNA-seq was conducted followed by IPA analysis of differentially expressed genes in BMI ≥ 30 relative to BMI < 25 CM-treated cells. Top 50 regulated “Diseases and Functions” are shown with corresponding activation z score in BRCA1+/− versus WT cells. (B) Diseases and functions were filtered to show pathways involved in DNA damage and DNA repair. Activation z scores are color-coded as heatmaps, with gradations of red representing a positive z score and gradations of blue representing a negative z score. Significantly regulated pathways as defined by −log (P value) > 1.3 are shown. Pathways with cells showing no color and a not applicable (“N/A”) z score were not significantly regulated.
HFD feeding is associated with elevated mammary gland DNA damage and early tumor penetrance in female Brca1 heterozygous knockout mice
DNA damage is a known driver of chromosomal defects that can lead to cancer. However, whether the obesity-associated elevation in breast epithelial cell DNA damage is linked to breast cancer penetrance in the setting of a heterozygous BRCA mutation has not been established. To investigate this question, we conducted preclinical studies using genetically modified mice with a whole-body heterozygous loss in Brca1 (Brca1+/−) on a C57BL/6 background. Four-week-old female Brca1+/− mice were randomized to receive LFD or HFD for 22 weeks to produce lean and obese mice, respectively (Fig. 6A). Mice fed HFD gained significantly more weight than LFD fed mice and weighed on average 34.1 versus 23.3 g, respectively, at the time of euthanasia (P < 0.001; Fig. 6B). Overall adiposity was also increased in association with HFD feeding as determined by greater accumulation of subcutaneous and visceral fat compared with the LFD group (fig. S4). To confirm that the HFD-fed mice exhibited altered metabolic homeostasis in our Brca1+/− model of diet-induced obesity, we conducted glucose tolerance tests after 21 weeks on experimental diets. This demonstrated delayed clearance of glucose from blood over 90 min after intraperitoneal injection of glucose in the HFD group compared with LFD-fed mice (Fig. 6, C and D). To determine whether changes observed in the mammary fat pads of Brca1+/− mice in response to feeding were analogous to those seen in the breast tissue of women in relation to obesity, we conducted RNA-seq on inguinal mammary fat pads from LFD and HFD mice harvested at euthanasia (table S8). IPAwas used to identify activation of the top differentially regulated canonical pathways in HFD mammary fat pads relative to LFD, results of which were juxtaposed with regulation of these same pathways in human breast tissue from BRCA mutation carriers with BMI ≥ 25 versus BMI < 25. The top 20 canonical pathways regulated by obesity in the mouse mammary fat pad showed similar regulation patterns compared with human breast tissue from women with overweight/obesity (BMI ≥ 25) (Fig. 6E), suggesting that diet-induced obesity in our Brca1+/− mice can serve as a model system for obesity in women carrying a BRCA mutation with respect to studies of the breast.
Fig. 6. HFD feeding leads to elevated mammary gland DNA damage in association with increased mammary tumor penetrance and decreased tumor latency in Brca1+/− mice.
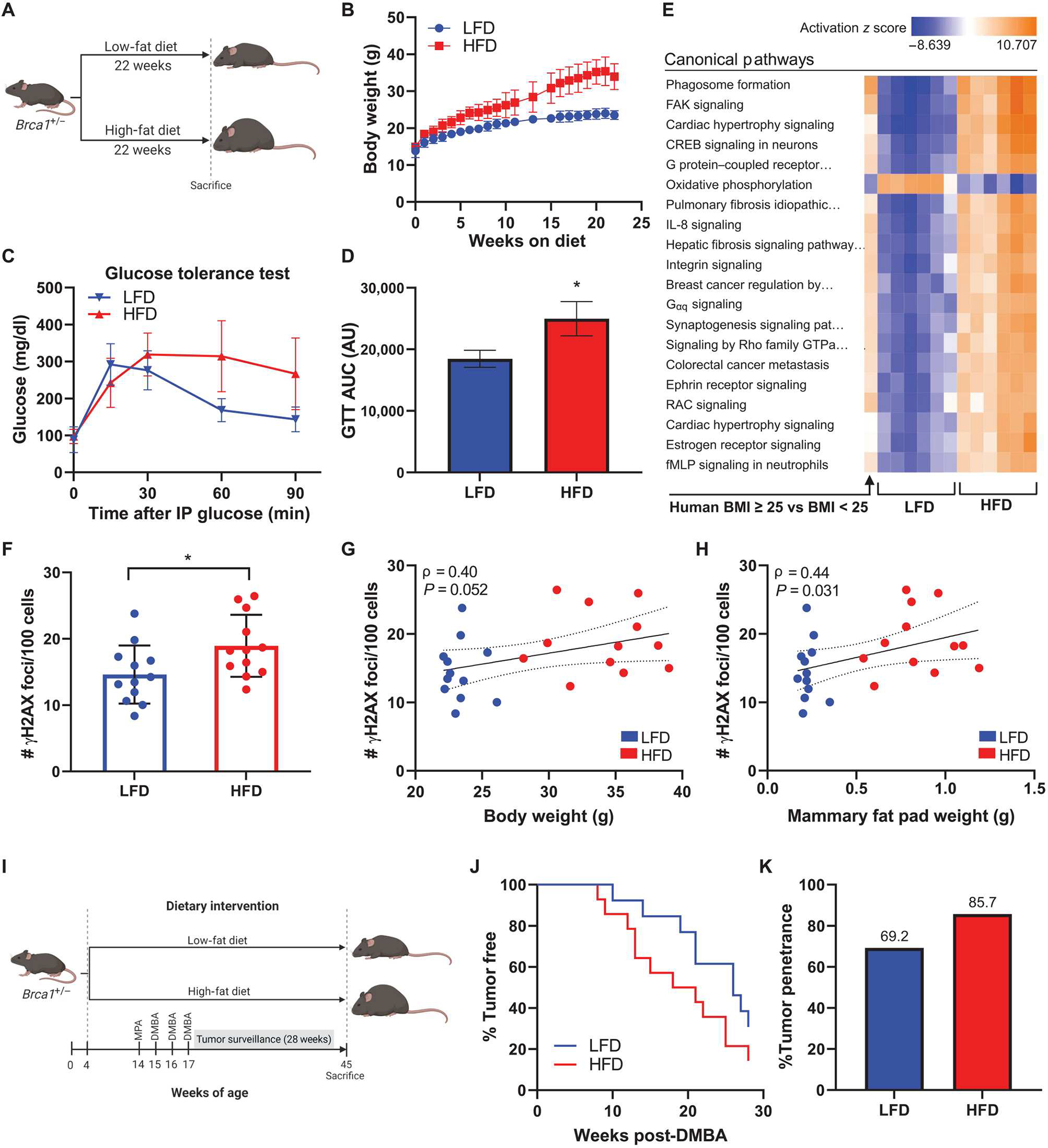
(A) Experimental schematic of diet-induced obesity in C57BL6/J female Brca1+/− mice (n = 12 per group). (B) Average body weight of mice fed LFD or HFD over 22 weeks. (C) Glucose tolerance test conducted 1 week before euthanasia and (D) area under curve (AUC) calculation for each group (means ± SEM). AU, arbitrary units. (E) RNA-seq was conducted on whole mammary fat pad tissue from HFD and LFD mice (n = 6 per group). Activation of top 20 canonical pathways regulated in mammary fat pads from HFD mice compared with LFD mice is shown adjacent to corresponding pathway regulation in breast tissue from BRCA mutation carriers with BMI ≥ 25 versus carriers with BMI < 25 (n = 64 to 67 per group). (F) DNA damage assessed by IF (number of γH2AX foci/100 cells) in mammary glands at the time of euthanasia. (G) Correlation between mammary gland DNA damage and mouse body weight and (H) mammary fat pad weight among all mice. Spearman’s rank correlation coefficient (ρ) and associated P values are shown along with 95% confidence intervals. (I) Experimental schematic of medroxyprogesterone acetate/7,12-dimethylbenz[a]anthracene (MPA/DMBA)–induced tumorigenesis model in female Brca1+/− mice randomized to LFD or HFD groups (n = 13 or 14 per group). (J) Mammary tumor development in LFD and HFD mice shown as percentage of mice tumor-free over the 28-week surveillance period. (K) Overall mammary tumor penetrance at the end of the surveillance period shown as percentage of mice in each group that developed a mammary tumor. Student’s t test was used to determine significance unless otherwise stated. Data are presented as means ± SD unless otherwise stated. *P < 0.05.
IF staining for γH2AX of Brca1+/− mouse mammary glands at euthanasia showed that HFD-fed mice had elevated amounts of mammary gland DNA damage compared with LFD-fed mice (Fig. 6F). These findings are analogous to the increased amounts of DNA damage observed in association with BMI in breast tissues from women carrying a BRCA mutation (Fig. 1B). Furthermore, there was a trend for a positive correlation between DNA damage and body weight, irrespective of diet (Fig. 6G), and a significant positive correlation between DNA damage and mammary fat pad weight (P = 0.031; Fig. 6H). This suggested that the amount of adiposity may be a stronger predictor of DNA damage in mammary epithelium as compared with whole body weight.
Next, we examined whether elevation in mammary gland DNA damage was associated with tumorigenesis. Female Brca1+/− mice were first made obese by HFD feeding for 10 weeks and then were implanted with a subcutaneous medroxyprogesterone acetate (MPA) pellet to sensitize them to mammary tumor development upon exposure to three doses of the carcinogen 7,12-dimethylbenz[a]anthracene (DMBA) (Fig. 6I). Mammary tumors developed earlier in the HFD group compared with the control LFD group (Fig. 6J). In addition, 85.7% of mice in the HFD group developed mammary tumors by the end of the 28-week surveillance period compared with 69.2% of mice in the LFD group (Fig. 6K).
Elevated BMI is associated with DNA damage in the fallopian tube, but not ovary, of BRCA mutation carriers
In addition to elevated breast cancer risk, women carrying a BRCA1 or BRCA2 mutation have high lifetime risk for developing ovarian cancer (1, 2). Because weight gain is associated with increased risk of ovarian cancer in BRCA mutation carriers (39), we extended our studies in the breast to investigate the impact of elevated BMI on DNA damage in the ovarian epithelium and in epithelial cells of the fallopian tube. IF staining for γH2AX was performed with nuclear counterstain Hoechst to quantify the number of foci of DNA damage per epithelial cell in nontumorous ovarian tissue and fallopian tube fimbriae from women carrying a BRCA1 or BRCA2 mutation undergoing prophylactic salpingo-oophorectomy. In the ovarian epithelium, there was no significant increase in DNA damage in the cases with BMI ≥ 25 (n = 9) compared with the cases with BMI < 25 (n = 17) (P = 0.59; Fig. 7A). However, there was a significant increase in DNA damage observed in the epithelial cells of the fallopian tube from women with BMI ≥ 25 (n = 12) compared with women with BMI < 25 (n = 21) (P = 0.03; Fig. 7B).
Fig. 7. BMI is associated with DNA damage in the fallopian tube but not the ovary in women carrying a BRCA mutation.
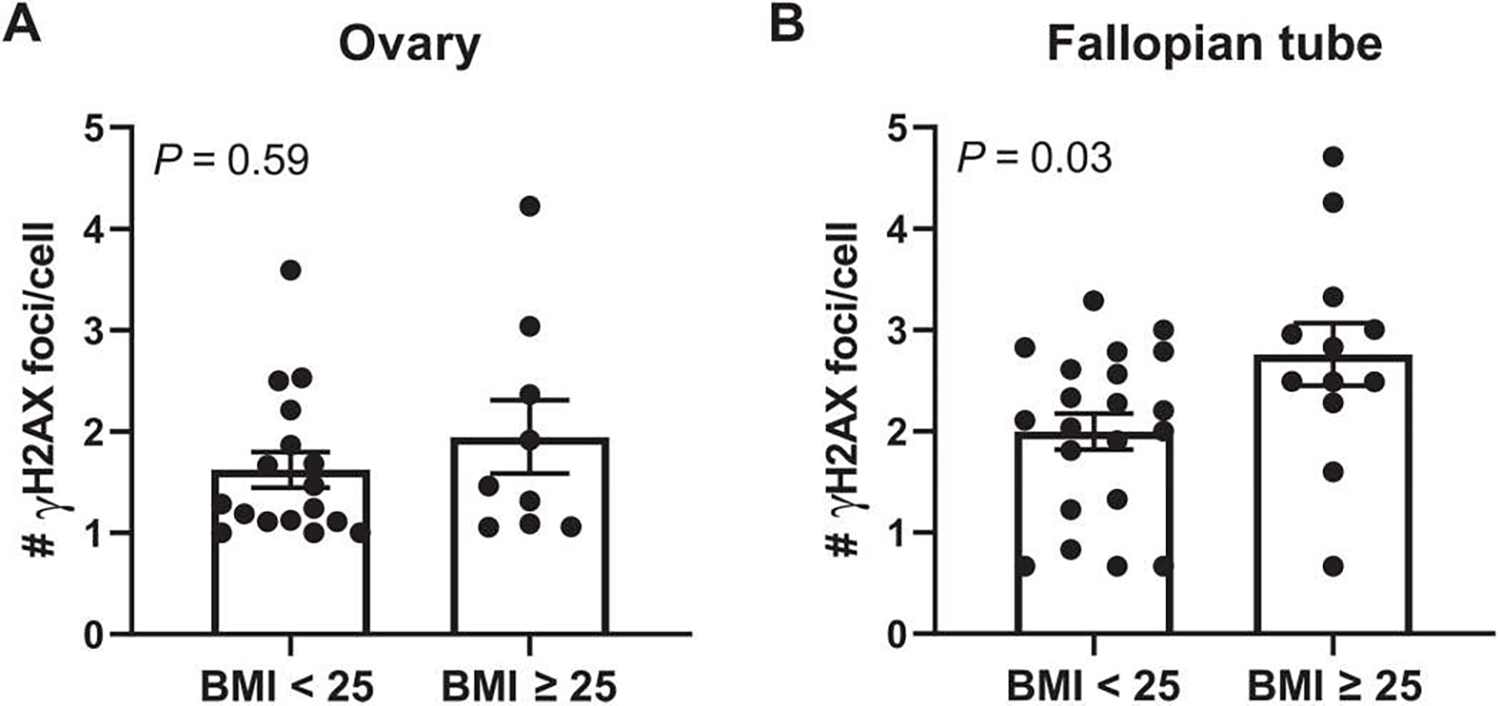
(A) DNA damage assessed by IF (number of γH2AX foci/cell) in epithelial cells of the ovary and in (B) epithelial cells of fallopian tube fimbriae in BRCA mutation carriers grouped by BMI < 25 (n = 17 to 21 per group) or BMI ≥ 25 (n = 9 to 12). Two-tailed Mann-Whitney test was used to determine significant differences (P value) between groups. Data are presented as means ± SEM.
DISCUSSION
The data presented here demonstrate that BMI is positively associated with the accumulation of DNA double-strand breaks in normal breast epithelial cells in carriers of a mutation in BRCA1 or BRCA2. Beyond BMI, insulin and insulin resistance, as measured by HOMA2 IR, were independently associated with DNA damage, irrespective of BMI or age. Accordingly, it is possible that BRCA mutation carriers who are defined as lower weight by BMI, but are hyperinsulinemic or “metabolically obese,” may also be at risk for elevated amounts of DNA damage and, consequently, breast cancer development. Although previous studies have shown that inflammation can lead to DNA damage in both normal and cancerous cells in other tissues (40–43), our data do not support a link between local or systemic inflammation and breast epithelial cell DNA damage.
To our knowledge, this is the first study to conduct transcriptional profiling of noncancerous breast tissue and isolated breast epithelial cells from BRCA mutation carriers with overweight/obesity versus those with lower weight. Although several factors and pathways associated with metabolic dysfunction were shown to be up-regulated in breast tissue and in epithelial cells, the identification of pathways related to estrogen biosynthesis (tissue) and signaling (epithelial cells) was of particular interest given the availability of clinically approved drugs that target estrogen. In addition, previous in vitro studies showed that treatment with estrogen and estrogen metabolites induced DNA damage in BRCA1 heterozygous breast epithelial cells (44), providing further rationale for exploring the role of estrogen as a mediator of obesity-induced DNA damage. Here, we show that fulvestrant, an ER degrader, is effective at reducing epithelial cell DNA damage in breast tissue explants from BRCA mutation carriers. However, this drug is not currently approved for use in the prevention setting, and the side effects may limit its future use for this purpose. Alternatively, metformin is widely prescribed in patients with type II diabetes and has an excellent safety profile that makes this drug a promising option for preventative use in BRCA mutation carriers with excess body weight. We show that metformin was effective at reducing breast epithelial cell DNA damage at clinically relevant concentrations primarily due to effects on the breast adipose microenvironment. Previous studies have shown that metformin decreases adipose stromal cell expression of aromatase through activation of AMPK (30, 31). Our study extends these findings by demonstrating the downstream consequence of down-regulation in aromatase through mass spectrometry studies, which showed marked reduction in E2 in breast tissue after metformin treatment. In addition to reducing estrogen exposure, previous work has shown that metformin treatment reduces endogenous reactive oxygen species (ROS) and associated DNA damage in a mammary epithelial cell line (45), providing an additional possible mechanism for the effects of metformin in our studies.
Epidemiological studies have reported decreased risk of breast cancer in BRCA mutation carriers in association with reduced estrogen exposure achieved by salpingo-oophorectomy surgery, which diminishes ovarian estrogen production, or through treatment with tamoxifen, an ER antagonist in the breast (46–48). Our studies propose estrogen-mediated induction of DNA damage as a possible explanation for the protective effects observed by decreasing estrogen exposure in this population. Estrogen can induce DNA damage through various actions, as reviewed by our group and others (49, 50), including through ligand binding to ERα, which stimulates proliferation and potentially replication stress with ROS production as a by-product of increased cellular respiration. In addition, the metabolism of estrogen yields genotoxic metabolites, a process that produces ROS through redox cycling. These metabolites can directly interact with DNA to form adducts in an ER-independent manner. Given the multiple avenues through which estrogen can induce DNA damage in cells, additional studies are warranted to characterize the mechanisms of estrogen-induced DNA damage in breast epithelial cells from BRCA mutation carriers in the setting of obesity.
We also offer insights into potential mechanisms leading to DNA damage accumulation. RNA-seq analysis of BRCA1 heterozygous MCF-10A cells treated with breast adipose CM from women with obesity relative to CM from women with lower weight showed not only increased activation of pathways associated with DNA damage but also down-regulation of pathways associated with DNA repair. This raises the possibility that obesity may affect DNA repair capacity, which would be detrimental in cells already exhibiting defective DNA repair due to a mutation in BRCA1 or BRCA2. In support of this possibility, MCF-10A cells WT for BRCA did not exhibit increased DNA damage when treated with CM from women with obesity, nor was there a significant impact on pathways associated with DNA damage or repair. This is consistent with our tissue microarray findings showing no association between BMI and breast epithelial cell DNA damage in age-matched nonmutation carriers. It is possible that DNA damage resolution occurs more quickly in cells that are WT for BRCA and that we are not capturing the full extent of potential detrimental effects of obesity in this population. Nevertheless, our data suggest that the impact of overweight/obesity is distinct in BRCA mutation carriers compared with noncarriers with respect to DNA damage and repair. Therefore, although obesity is associated with increased breast cancer risk in postmenopausal women in the general population (3), the mechanisms that drive this risk are likely to be different than the possible mechanisms highlighted in our studies of BRCA mutation carriers, which will have implications on selection of effective risk reduction strategies in these two populations.
Our in vitro studies demonstrate the ability of several obesity-associated factors, including leptin and insulin, to cause the accumulation of DNA damage, suggesting a collective milieu of factors that may contribute to the elevation in DNA damage observed in BRCA mutation carriers in association with BMI. The ability of CM derived from women with obesity to induce damage in BRCA1 heterozygous cells was diminished when treating in the presence of an antibody or drug that inhibits leptin or insulin signaling, respectively. Because insulin signals through PI3K, we used BKM120, a PI3K inhibitor, to disrupt insulin actions in the presence of CM. It is possible that inhibiting PI3K signaling is disrupting not only insulin signaling but also signaling of other factors associated with obesity that act through PI3K, including growth factors or leptin, which collectively contributed to the observed decrease in DNA damage. In addition, growing evidence points to a role for the PI3K pathway in the DNA damage response; however, these studies have been limited to cancer cells (51–54).
Our studies also show a link between obesity-induced DNA damage and tumor development using a Brca1+/− mouse model of diet-induced obesity. HFD-fed mice exhibited elevated mammary gland DNA damage in association with decreased latency and increased overall penetrance of mammary tumors when exposed to the carcinogen DMBA. These data suggest that the elevation in DNA damage that we observed in association with BMI in women carrying a BRCA mutation may also be associated with increased breast cancer penetrance.
Last, our data show that obesity-associated DNA damage may not only be limited to the breast epithelia of BRCA mutation carriers. Although no increase in DNA damage was found in epithelial cells of the ovary in women with overweight/obesity undergoing prophylactic salpingo-oophorectomy, we did observe higher amounts of DNA damage in the epithelial cells of the fallopian tube in association with overweight/obesity. Our results are consistent with reports from recent years that point to the fallopian tube as the likely site of origin of ovarian cancer (55, 56), to be confirmed by ongoing clinical trials of risk-reducing salpingectomy with delayed oophorectomy. In addition, these data highlight a potential mechanism for the link between weight gain and ovarian cancer in this population.
A limitation of our study includes a cohort size of n = 69 in our correlation study of DNA damage and BMI, which prevented us from analyzing effects of BMI separately in BRCA1 and BRCA2 mutation carriers. Although both BRCA1 and BRCA2 are essential for DNA repair, their roles in the DNA damage response are not identical, and each mutation is associated with different subtypes of tumor development. Larger studies assessing the relative effect of BMI on DNA damage in BRCA1 and BRCA2 mutation carriers separately could provide additional information to help personalize risk estimates. Moreover, concentrations of estrogens vary considerably during the menstrual cycle and affect proliferation of breast epithelial cells. Our studies did not account for phase of menstrual cyclewhen assessing DNA damage, which may have led to increased variability in our data, particularly considering our identification of estrogen as a mediator of obesity-induced epithelial cell DNA damage. Last, the extent to which data from our mouse model can be extrapolated to humans is somewhat limited given that we used a carcinogen-induced tumor model, whereas in BRCA mutation carriers, tumors will arise after years of exposure to both endogenous and environmental factors, some of which will act as carcinogens.
Many methodological challenges exist, which explains the lack of agreement in epidemiological studies attempting to ascertain modifiers of breast cancer risk in BRCA mutation carriers, as reviewed by Milne and Antoniou (57). Given the inconsistencies in reported data, the consensus to date is that there is insufficient evidence to determine the effect of body weight on breast cancer risk in BRCA mutation carriers (57–59). Therefore, a strength of our study is the presentation of mechanistic experimental evidence, which helps to elucidate the relationship between body weight and breast cancer risk in this population.
In addition, our findings provide rationale for conducting clinical trials in BRCA mutation carriers with overweight/obesity to test the efficacy of pharmacological interventions that target metabolic health, weight, and estrogens. Identifying which obesity-related factors need to be targeted for risk reduction, if not all, will have a meaningful impact on developing effective risk reduction strategies. Although recently reported results of the phase 3 randomized MA.32 trial (NCT01101438) found that addition of metformin to standard of care in nondiabetic patients with high-risk breast cancer did not improve invasive disease-free survival versus placebo (60), it remains to be determined whether metformin in the preventative setting would be effective at reducing risk of breast cancer, particularly among BRCA mutation carriers and those with metabolic dysfunction. Our studies point toward the potential of metformin in this setting, because it has been shown to reduce weight and cause decreases in circulating concentrations of insulin, leptin, and estrogens (61–63). These studies would help clarify whether accumulation of DNA damage over time is reversible or whether targeted interventions prevent accumulation of further damage. Positive results would offer clinicians actionable evidence-based prevention strategies for patients in this high-risk population who opt to delay or forgo risk-reducing surgery.
MATERIALS AND METHODS
Study design
The objective of this study was to gain insight into the role of obesity and metabolic dysfunction on breast cancer penetrance among carriers of germline mutations in BRCA1 and BRCA2 and to identify clinically relevant prevention strategies. Clinical samples including both archival tissues and prospectively collected tissues from BRCA mutation carriers and nonmutation carriers, as well as cell lines engineered to carry a BRCA1 or BRCA2 heterozygous knockout mutation, and Brca1+/− mouse models were used in support of this objective. All studies using human tissues were conducted in accordance with protocols approved by the Institutional Review Boards of Memorial Sloan Kettering Cancer Center (MSKCC) under protocol no. 10–040 and Weill Cornell Medicine under protocol nos. 1510016712, 1004010984–01, 1612017836, and 20–01021391. Informed consent from each participant was obtained by study investigators before tissue collection. Animal experiments were conducted in accordance with an approved Institutional Animal Care and Use Committee protocol (no. 2018–0058) at Weill Cornell Medicine.
Studies using archival tissues were coded, and DNA damage was analyzed in a blinded fashion. Studies using prospectively collected tissues and in vitro treatment studies were not blinded; however, DNA damage was analyzed by IF staining using methodology to limit bias as described in the “Confocal microscopy and quantification of γH2AX foci” section. Sample size power calculations were performed for human breast tissue microarray construction (BMI versus DNA damage study). In animal studies, sample size power calculations were conducted, and mice were randomized to dietary treatment groups. Any sample exclusion criteria are described in the sections below or in the figure legends. All in vitro studies were replicated a minimum of two times as described in the figure legends.
Human breast tissue microarray construction and study population
Archival paraffin blocks of embedded nontumorous breast tissue were obtained from 69 women carrying a BRCA1 (n = 40) or BRCA2 (n = 29) mutation and from an age-matched subset of women WT for a BRCA mutation (n = 17) who had previously undergone prophylactic or therapeutic mastectomy at MSKCC from 2011 to 2016. Table 1 describes the clinical characteristics of the BRCA mutation carrier study population that were extracted from electronic medical records. BMI was calculated using height and weight recorded before surgery (kg/m2), and menopausal status was determined per criteria established by the National Comprehensive Cancer Network (64). A pathologist reviewed hematoxylin and eosin–stained sections from each block to identify areas enriched in breast epithelium. Cores measuring 1.5 mm in diameter from identified epithelial areas of each case were incorporated into paraffin blocks for the construction of tissue microarrays. Each tissue microarray was constructed with cases representing an equal distribution of clinical characteristics, including BRCA1 or BRCA2 mutation status and BMI. Unstained sections were cut from each tissue microarray and used for quantification of breast epithelial cell DNA damage by IF staining, as described in the section below.
Assessment of DNA damage by IF staining
To quantify epithelial cell DNA damage, we conducted IF staining of the DNA double-strand break marker γH2AX on human tissue sections, mouse mammary gland tissue sections, or plated cells. Antibodies/reagents that were used include primary γH2AX (p Ser139) antibody (Novus Biologicals, #NB100–74435, RRID:AB_1048941) (unless otherwise stated) at 1:300 dilution, goat anti-mouse Alexa Fluor 546 secondary antibody (Life Technologies, #A11030, RRID:AB_2534089) at 1:1000 dilution, Hoechst 33342 nuclear stain (Santa Cruz Biotechnology, #SC-495790) at 1:1000 dilution, CAS block (Life Technologies, #008120), M.O.M. (Mouse-on-Mouse) immunodetection kit (Vector Laboratories, #BMK-2202), and ProLong Gold Antifade Mountant (Invitrogen, #P36934). Full staining procedures for tissue sections, plated cells, and colocalization studies can be found in the Supplementary Materials.
Confocal microscopy and quantification of γH2AX foci
Tissue slides or plated epithelial cells stained with γH2AX and Hoechst were imaged using a Zeiss LSM 880 confocal microscope. Confocal settings were not changed across samples within each experiment. Areas to image were first selected on the basis of identification of regions rich in breast epithelial cells as determined by Hoechst staining before viewing the γH2AX channel to limit any potential bias in image selection. Images were exported to the image analysis software Imaris (Oxford Instruments) for semiauto-mated quantification of γH2AX foci per 100 cells. Imaris analysis settings were programmed to identify and quantify total cell number in each image and to identify number of γH2AX foci colocalizing with nuclei. All Imaris-analyzed images were visually inspected by investigators to ensure appropriate identification of γH2AX foci and exclusion of background staining. A minimum of 100 cells per case or condition were analyzed, and DNA damage was reported as the number of γH2AX foci per 100 cells unless stated otherwise. Any samples with fewer than 100 cells detected were excluded.
Quantification of blood biomarkers
Fasting blood was collected from patients before surgery. Serum was separated by centrifugation, aliquoted, and stored at −80°C. Enzyme-linked immunosorbent assay was used to measure serum concentrations of insulin (Mercodia), hsCRP, glucose, SHBG, and IL-6 (R&D Systems) following the manufacturer’s protocols. Briefly, for each assay, serum samples and standards were aliquoted in duplicates into the provided 96-well microplate precoated with an antibody specific to the biomarker of interest. All wells were then incubated with a 1× solution of enzyme-linked antibody specific to the biomarker of interest, followed by a wash step and incubation with a substrate solution of 3,3′,5,5′-tetramethylbenzidine (TMB) alone or mix of TMB and hydrogen peroxide to detect the bound conjugate. The reaction was stopped with acid-based stop solution, and optical density was read on a spectrophotometer at the indicated wavelength. A standard curve was used to quantify the concentration of each biomarker.
RNA-seq studies and computational analysis
RNA-seq was conducted on samples in four studies including breast tissue from BRCA mutation carriers, isolated breast epithelial organoids from BRCA mutation carriers and noncarriers, breast adipose tissue CM-treated BRCA1 heterozygous or WT MCF-10A cells, and Brca1+/− mouse mammary fat pads. Details on RNA extraction, sequencing methodology, and computational analyses can be found in the Supplementary Materials.
Isolation of primary breast epithelial cells and breast explant studies
For ex vivo tissue explant studies and isolation of breast epithelial cells, breast tissue was obtained from women undergoing breast mammoplasty or mastectomy surgeries at Weill Cornell Medicine and MSKCC from 2017 to 2021. Surgical specimens were transferred from the operating room to a pathologist who evaluated the breast tissue to confirm that the tissue distributed for experimentation was normal and uninvolved with any quadrant where a tumor may have been present. The tissue was then brought to the laboratory and used in the experiments as described below.
Isolation of breast epithelial cells
About 25 ml of breast tissue were used in each organoid preparation, with care taken to dissect out overly fibrous areas or visible blood vessels. The tissue was finely minced and mixed with complete Ham’s F12 medium [Corning, #10–080-CV, supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin] containing a digestion mix of collagenase type 1 (10 mg/ml; Sigma-Aldrich, #C0130) and hyaluronidase (10 μg/ml; Sigma-Aldrich, #H3506) in a total volume of 50 ml. The tissue was digested overnight on a rotator at 37°C and then centrifuged. The supernatant containing free lipid and adipocytes was discarded, and the pellet was washed and reconstituted in medium, followed by incubation at 4°C for 1 hour to ensure inhibition of enzyme activities. After centrifugation, the pellet was treated with red cell lysis buffer (Sigma-Aldrich, #11814389001), repelleted, reconstituted in medium, and then ran through a 100-μm filter followed by a 40-μm filter. Breast epithelial organoids were collected from the top of the 40-μm filter in mammary epithelial cell growth medium with added supplements (PromoCell, #C-21010). Isolated mammary epithelial organoids were snap-frozen in liquid nitrogen for RNA extraction and RNA-seq or plated for in vitro studies.
Ex vivo metformin and fulvestrant explant studies
To examine the role of breast adipose tissue estrogen in mediation of DNA damage in BRCA mutant epithelial cells, we treated breast explants with drugs targeting estrogen signaling (fulvestrant) or production (metformin). One-centimeter breast tissue explants were cut from breast tissue transferred after surgery and were plated in replicate in a 12-well dish. For metformin studies, breast explants from n = 3 participants were cultured in complete Ham’s F12 medium (10% FBS and 1% penicillin-streptomycin) supplemented with either vehicle (methanol) or metformin hydrochloride (25 to 100 μM; Sigma-Aldrich, #PHR1084). For fulvestrant studies, breast explants from n = 7 participants were cultured in basal mammary epithelial cell growth medium + 0.1% bovine serum albumin (BSA) containing either vehicle (ethanol) or 100 μM fulvestrant (Sigma-Aldrich, #I4409).
After 24 hours of treatment at 37°C in a 5% CO2 incubator, explants were snap-frozen in liquid nitrogen or formalin-fixed and paraffin-embedded. Tissue sections were cut from each paraffin block for assessment of breast epithelial cell DNA damage by IF staining.
Collection of breast adipose tissue CM
CM was generated from breast tissue obtained from n = 36 women with BMIs that range from lower weight to obese (20.6 to 49.1 kg/m2). Ten 1-cm explant pieces of breast adipose tissue were cut from each case with a focus on fatty areas containing no visible blood vessels. The pieces were weighed and placed on a 10-cm dish with 10 ml of basal (phenol red–free, serum-free, and supplement mix–free) mammary epithelial cell growth medium (PromoCell, #C-21215) containing 0.1% BSA. The explants were incubated at 37°C for 24 hours. After incubation, the breast adipose tissue CM was collected and centrifuged at 300g. The supernatant was aliquoted and stored at −80°C for use in in vitro treatment studies. Control CM consisted of the same collection medium with the absence of conditioning by adipose explants.
In vitro studies in MCF-10A cells
Noncancerous breast epithelial cell line MCF-10A carrying a BRCA1 heterozygous mutation (185delAG/+) or WT for a BRCA mutation was purchased from Horizon Discovery and has been previously described (65). MCF-10A cells carrying a BRCA2 heterozygous mutation (6174insT/+) were generated in-house using CRISPR-Cas9 gene editing (additional details provided in the Supplementary Materials). Cells were cultured in Dulbecco’s modified Eagle’s medium/F12 (Invitrogen, #11330–032) supplemented with 5% FBS, 1% penicillin-streptomycin, and the following growth factors: epidermal growth factor (20 ng/ml), hydrocortisone (0.5 mg/ml), cholera toxin (100 ng/ml), and insulin (10 μg/ml) (all purchased from Sigma-Aldrich). Cells were serum-starved for 16 hours before treatments.
In CM studies, CM was thawed on ice from each case and diluted to a final concentration of 25% CM. In leptin studies, cells were treated with human recombinant leptin (400 ng/ml; Sigma-Aldrich, #L4146). In leptin neutralization studies, obese CM was preincubated with a leptin-neutralizing antibody (Lep Ab; 13.3 μg/ml; R&D Systems, #AF398, RRID:AB_355347) for 1 hour at 4°C, and then cells were treated with lower weight or obese CM alone or obese CM + Lep Ab. In insulin studies, cells were treated with 100 nM insulin (Sigma-Aldrich, #I1882). To block insulin signaling, cells were pretreated with the PI3K inhibitor BKM120 (1 μM; MedChemExpress, #HY-70063) for 1 hour and then treated with obese CM + BKM120. All treatments were conducted in replicates or triplicates for 24 hours unless otherwise stated. After treatment, all wells were fixed with ice-cold methanol followed by γH2AX IF staining.
Brca1+/− mouse studies
Diet-induced obesity and mammary gland DNA damage
At 4 weeks of age, 24 female Brca1+/− mice on a C57BL/6 background were randomized to one of two groups (n = 12 per group). One group was fed 10 kcal% LFD (12450Bi, Research Diets), and the second group was fed 60 kcal% HFD (D12492i, Research Diets) ad libitum for 22 weeks until euthanasia. One week before euthanasia, all mice were fasted overnight for 12 hours and underwent glucose tolerance tests to confirm obesity-induced metabolic dysfunction. In brief, baseline glucose measurements were taken from tail vein blood drop collection using a handheld glucose meter (Bayer Contour). Mice then received an intraperitoneal injection of glucose (1 g/kg), and tail vein blood glucose concentrations were recorded at 15- to 30-min intervals over 90 min. After the final measurement, respective experimental diets were restarted ad libitum for an additional week before euthanasia. Mice were euthanized using CO2 inhalation, and mammary gland tissue was collected and snap-frozen (inguinal fat pads) for RNA-seq or fixed (thoracic fat pads) in 10% neutral-buffered formalin overnight before paraffin embedding and sectioning for histological assessment of DNA damage.
MPA/DMBA tumor model
To investigate how obesity affects mammary gland tumor development in Brca1+/− mice, we used the same diet-induced obesity model as described above. At 4 weeks of age, 27 female Brca1+/− mice were randomized to one of two groups (n = 13 or 14 per group). One group was fed LFD, and the second group was fed HFD for the duration of the study. At 14 weeks of age (after 10 weeks on experimental diets), all mice were surgically implanted with a 40-mg MPA pellet (90-day continuous release, Innovative Research of America, #NP-161) placed subcutaneously. At 15, 16, and 17 weeks of age, all mice were dosed with the carcinogen DMBA (1 mg/22 g body weight) delivered by oral gavage in corn oil once per week for 3 consecutive weeks. Mammary tumor development and growth were monitored weekly by palpating all five mammary gland pairs and recording tumor presence and size with caliper measurements for 28 weeks after the last dose of DMBA. Mice were euthanized at the end of the 28-week surveillance period or earlier on the basis of ethical end points, including tumor burden reaching 1.5 cm. Mice that did not recover from pellet implantation surgery or displayed morbidity unrelated to mammary tumors were excluded from the study.
Statistical analysis
To assess significant differences in baseline clinical characteristics and categorical variables, the Fisher exact test was used. To test the strength of correlation between DNA damage and continuous variables, nonparametric Spearman’s rank correlation coefficient was used with two-tailed P value to determine significance of correlations. A multivariable linear model was used to test the association between the amount of DNA damage and clinical characteristics adjusting for BMI or age. Two-tailed Mann-Whitney test was performed on clinical data testing significant differences between two groups. Two-tailed Student’s t test was used on in vitro treatment studies and in mouse studies comparing two groups. All results were performed using R (version 4.0.5) or GraphPad Prism 9. Results with a P value of <0.05 were considered statistically significant.
Supplementary Material
Acknowledgments:
We thank D. Otterburn and L. Cohen, Department of Surgery, and P. Ginter, Department of Pathology, at Weill Cornell Medicine for facilitating accessto reduction mammoplasty tissue. Studies were conducted with the support and facilities provided by the Microscopy and Image Analysis Core Facility, Genomics Resources Core Facility, Center for Translational Pathology, and the Research Animal Resource Center at Weill Cornell Medicine. All schematics were created in BioRender.
Funding:
This work was supported by funding from the National Cancer Institute of the NIH under grants 1R01CA215797 to K.A.B., F31CA236306 to P.B., and R35-CA197588 to L.C.C.; by the Anne Moore Breast Cancer Research Fund to K.A.B.; by the National Breast Cancer Foundation under grant ECF-16–004 to K.A.B.; by the Emilie Lippmann and Janice Jacobs McCarthy Research Scholar Award in Breast Cancer to K.A.B.; by the National Center for Advancing Translational Sciences of the NIH under grant KL2-TR-002385 to M.K.F.; by the Breast Cancer Research Foundation to N.M.I., L.C.C., and K.A.B.; and by the National Institute of General Medical Sciences of the NIH under grant T32GM007739 to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD Program and awarded to M.A. The content issolely the responsibility of the authors and does not necessarily represent the official views of the NIH or other funding agencies.
Footnotes
Competing interests: N.M.I. receives consulting fees from Novartis, Pfizer, and Seattle Genetics; honoraria from Curio Science, Cardinal Health, OncLive, and IntrinsiQ Health; and research funding (to institution) from Novartis, SynDevRx, National Cancer Institute, American Cancer Society, Breast Cancer Research Foundation, and the Conquer Cancer Foundation. L.E.D. is a scientific advisor and holds equity in Mirimus Inc. and has received consulting fees and/or honoraria from Volastra Therapeutics, Revolution Medicines, Repare Therapeutics, Fog Pharma, and Frazier Healthcare Partners. B.D.H. is a founder and consultant for Faeth Therapeutics. L.C.C. is a founder of Agios Pharmaceuticals and Petra Pharmaceuticals and an ad hoc advisor to these companies; is a founder of Volastra, Faeth, and Larkspur; is on the scientific advisory boards for Cell Signaling Technology, Volastra, Faeth, and Larkspur; and is an inventor on patents (pending) for combination therapy for PI3K-associated disease or disorder and the identification of therapeutic interventions to improve response to PI3K inhibitors for cancer treatment.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials. A full list of differentially expressed genes used in computational analyses from all RNA-seq studies presented is included in data file S1, and raw RNA-seq data are included in data files S3 to S8. All reasonable requests for transfer of materials, including BRCA2 heterozygous MCF-10A cell line and Brca1+/− C57BL/6 mice generated in this study, will be granted under a Uniform Biological Material Transfer Agreement by contacting the corresponding author. Requests for transfer of materials, including BRCA2 heterozygous MCF-10A cell line and Brca1+/− C57BL/6 mice generated in this study, will be granted under an MTA (Uniform Biological Material Transfer Agreement) by contacting K.A.B. (kab2060@med.cornell.edu).
REFERENCES AND NOTES
- 1.Kuchenbaecker KB, Hopper JL, Barnes DR, Phillips K-A, Mooij TM, Roos-Blom M-J, Jervis S, Van Leeuwen FE, Milne RL, Andrieu N, Goldgar DE, Terry MB, Rookus MA, Easton DF, Antoniou AC, Mcguffog L, Evans DG, Barrowdale D, Frost D, Adlard J, Ong KR, Izatt L, Tischkowitz M, Eeles R, Davidson R, Hodgson S, Ellis S, Nogues C, Lasset C, Stoppa-Lyonnet D, Fricker J-P, Faivre L, Berthet P, Hooning MJ, Van Der Kolk LE, Kets CM, Adank MA, John EM, Chung WK, Andrulis IL, Southey M, Daly MB, Buys SS, Osorio A, Engel C, Kast K, Schmutzler RK, Caldes T, Jakubowska A, Simard J, Friedlander ML, Mclachlan S-A, Machackova E, Foretova L, Tan YY, Singer CF, Olah E, Gerdes A-M, Arver B, Olsson H, Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 317, 2402–2416 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Chen S, Parmigiani G, Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 25, 1329–1333 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K, Body fatness and cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 375, 794–798 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.King MC, Marks JH, Mandell JB, New G, Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 302, 643–646 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Kotsopoulos J, Olopade OI, Ghadirian P, Lubinski J, Lynch HT, Isaacs C, Weber B, Kim-Sing C, Ainsworth P, Foulkes WD, Eisen A, Sun P, Narod SA, Changes in body weight and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res. 7, R833–R843 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bruno E, Oliverio A, Paradiso A, Daniele A, Tommasi S, Terribile DA, Filippone A, Digennaro M, Pilato B, Danza K, Guarino D, Rossi C, Rossi MM, Venturelli E, Giussani M, Peissel B, Pasanisi P, Lifestyle characteristics in women carriers of BRCA mutations: Results from an italian trial cohort. Clin. Breast Cancer 21, e168–e176 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Nkondjock A, Robidoux A, Paredes Y, Narod SA, Ghadirian P, Diet, lifestyle and BRCA-related breast cancer risk among French-Canadians. Breast Cancer Res. Treat. 98, 285–294 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Abbas S, Siddique A, Shahid N, Khan RT, Fatima W, Breast cancer risk associated with BRCA1/2 variants in the Pakistani population. Breast Cancer 26, 365–372 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Manders P, Pijpe A, Hooning MJ, Kluijt I, Vasen HF, Hoogerbrugge N, van Asperen CJ, Meijers-Heijboer H, Ausems MG, van Os TA, Gomez-Garcia EB, Brohet RM; HEBON FE van Leeuwen, M. A. Rookus, Body weight and risk of breast cancer in BRCA1/2 mutation carriers. Breast Cancer Res. Treat. 126, 193–202 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Kim SJ, Huzarski T, Gronwald J, Singer CF, Møller P, Lynch HT, Armel S, Karlan BY, Foulkes WD, Neuhausen SL, Senter L, Eisen A, Eng C, Panchal S, Pal T, Olopade O, Zakalik D, Lubinski J, Narod SA, Kotsopoulos J, Ainsworth P, Bordeleau L, Tung N, Friedman E, Meschino W, Snyder C, Metcalfe K, Warner E, Rosen B, Demsky R, Weitzel JN, Panabaker K, Couch F, Manoukian S, Pasini B, Daly MB, Steele L, Saal H, Fallen T, Wood M, Mckinnon W, Lemire E, Chudley AE, Serfas K, Elser C, Vadaparampil ST, Ginsburg O, Cullinane CA, Blum JL, Ross T, Mauer C, Kwong A, Cybulski C, Mccuaig J, Rayson D, Isaacs C, Prospective evaluation of body size and breast cancer risk among BRCA1 and BRCA2 mutation carriers. Int. J. Epidemiol. 47, 987–997 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian F, Wang S, Mitchell J, Mcguffog L, Barrowdale D, Leslie G, Oosterwijk JC, Chung WK, Evans DG, Engel C, Kast K, Aalfs CM, Adank MA, Adlard J, Agnarsson BA, Aittomäki K, Alducci E, Andrulis IL, Arun BK, Ausems MGEM, Azzollini J, Barouk- Simonet E, Barwell J, Belotti M, Benitez J, Berger A, Borg A, Bradbury AR, Brunet J, Buys SS, Caldes T, Caligo MA, Campbell I, Caputo SM, Chiquette J, Claes KBM, Collée JM, Couch FJ, Coupier I, Daly MB, Davidson R, Diez O, Domchek SM, Donaldson A, Dorfling CM, Eeles R, Feliubadaló L, Foretova L, Fowler J, Friedman E, Frost D, Ganz PA, Garber J, Garcia-Barberan V, Glendon G, Godwin AK, Garcia EBG, Gronwald J, Hahnen E, Hamann U, Henderson A, Hendricks CB, Hopper JL, Hulick PJ, Imyanitov EN, Isaacs C, Izatt L, Izquierdo Á, Jakubowska A, Kaczmarek K, Kang E, Karlan BY, Kets CM, Kim S-W, Kim Z, Kwong A, Laitman Y, Lasset C, Lee MH, Lee JW, Lee J, Lester J, Lesueur F, Loud JT, Lubinski J, Mebirouk N, Meijers-Heijboer HEJ, Meindl A, Miller A, Montagna M, Mooij TM, Morrison PJ, Mouret-Fourme E, Nathanson KL, Neuhausen SL, Nevanlinna H, Niederacher D, Nielsen FC, Nussbaum RL, Offit K, Olah E, Ong K-R, Ottini L, Park SK, Peterlongo P, Pfeiler G, Phelan CM, Poppe B, Pradhan N, Radice P, Ramus SJ, Rantala J, Robson M, Rodriguez GC, Schmutzler RK, Selkirk CGH, Shah PD, Simard J, Singer CF, Sokolowska J, Stoppa-Lyonnet D, Sutter C, Tan YY, Teixeira RM, Teo SH, Terry MB, Thomassen M, Tischkowitz M, Toland AE, Tucker KM, Tung N, Van Asperen CJ, Van Engelen K, Van Rensburg EJ, Wang-Gohrke S, Wappenschmidt B, Weitzel JN, Yannoukakos D, Greene MH, Rookus MA, Easton DF, Chenevix-Trench G, Antoniou AC, Goldgar DE, Olopade OI, Rebbeck TR, Huo D, Height and body mass index as modifiers of breast cancer risk in BRCA1/2 mutation carriers: A Mendelian randomization study. J. Natl. Cancer Inst. 111, 350–364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grundy SM, Obesity, metabolic syndrome, and cardiovascular disease. J. Clin. Endocrinol. Metab. 89, 2595–2600 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Sun K, Kusminski CM, Scherer PE, Adipose tissue remodeling and obesity. J. Clin. Invest. 121, 2094–2101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sell H, Habich C, Eckel J, Adaptive immunity in obesity and insulin resistance. Nat. Rev. Endocrinol. 8, 709–716 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Bhardwaj P, Brown KA, Obese adipose tissue as a driver of breast cancer growth and development: Update and emerging evidence. Front. Oncol. 11, 638918 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brown KA, Metabolic pathways in obesity-related breast cancer. Nat. Rev. Endocrinol. 17, 350–363 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simpson ER, Davis SR, Minireview: Aromatase and the regulation of estrogen biosynthesis—Some new perspectives. Endocrinology 142, 4589–4594 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Simpson ER, Brown KA, Aromatase overexpression in dysfunctional adipose tissue links obesity to postmenopausal breast cancer. J. Steroid Biochem. Mol. Biol. 153, 35–44 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Zahid H, Subbaramaiah K, Iyengar NM, Zhou XK, Chen IC, Bhardwaj P, Gucalp A, Morrow M, Hudis CA, Dannenberg AJ, Brown KA, Leptin regulation of the p53-HIF1α/PKM2-aromatase axis in breast adipose stromal cells: A novel mechanism for the obesity-breast cancer link. Int. J. Obes. 42, 711–720 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu F, He J, Wang H, Zhu D, Bi Y, Adipose morphology: A critical factor in regulation of human metabolic diseases and adipose tissue dysfunction. Obes. Surg. 30, 5086–5100 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marino N, German R, Rao X, Simpson E, Liu S, Wan J, Liu Y, Sandusky G, Jacobsen M, Stoval M, Cao S, Storniolo AMV, Upregulation of lipid metabolism genes in the breast prior to cancer diagnosis. npj Breast Cancer 6, 50 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yoshida K, Miki Y, Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage. Cancer Sci. 95, 866–871 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Venkitaraman AR, Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 108, 171–182 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Kowalska E, Narod SA, Huzarski T, Zajaczek S, Huzarska J, Gorski B, Lubinski J, Increased rates of chromosome breakage in BRCA1 carriers are normalized by oral selenium supplementation. Cancer Epidemiol. Biomarkers Prev. 14, 1302–1306 (2005). [DOI] [PubMed] [Google Scholar]
- 25.Zaki M, Basha W, El-Bassyouni HT, El-Toukhy S, Hussein T, Evaluation of DNA damage profile in obese women and its association to risk of metabolic syndrome, polycystic ovary syndrome and recurrent preeclampsia. Genes Dis. 5, 367–373 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dungan CM, Peck BD, Walton RG, Huang Z, Bamman MM, Kern PA, Peterson CA, In vivo analysis of γH2AX+ cells in skeletal muscle from aged and obese humans. FASEB J. 34, 7018–7035 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fieres J, Fischer M, Sauter C, Moreno-Villanueva M, Burkle A, Wirtz PH, The burden of overweight: Higher body mass index, but not vital exhaustion, is associated with higher DNA damage and lower DNA repair capacity. DNA Repair 114, 103323 (2022). [DOI] [PubMed] [Google Scholar]
- 28.Tay VSY, Devaraj S, Koh T, Ke G, Crasta KC, Ali Y, Increased double strand breaks in diabetic β-cells with a p21 response that limits apoptosis. Sci. Rep. 9, 19341 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris PG, Hudis CA, Giri D, Morrow M, Falcone DJ, Zhou XK, Du B, Brogi E, Crawford CB, Kopelovich L, Subbaramaiah K, Dannenberg AJ, Inflammation and increased aromatase expression occur in the breast tissue of obese women with breast cancer. Cancer Prev. Res. 4, 1021–1029 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown KA, Hunger NI, Docanto M, Simpson ER, Metformin inhibits aromatase expression in human breast adipose stromal cells via stimulation of AMP-activated protein kinase. Breast Cancer Res. Treat. 123, 591–596 (2010). [DOI] [PubMed] [Google Scholar]
- 31.Samarajeewa NU, Ham S, Yang F, Simpson ER, Brown KA, Promoter-specific effects of metformin on aromatase transcript expression. Steroids 76, 768–771 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Soule HD, Maloney TM, Wolman SR, Peterson WD Jr., Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, S. C. Brooks, Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 50, 6075–6086 (1990). [PubMed] [Google Scholar]
- 33.Perera CN, Chin HG, Duru N, Camarillo IG, Leptin-regulated gene expression in MCF-7 breast cancer cells: Mechanistic insights into leptin-regulated mammary tumor growth and progression. J. Endocrinol. 199, 221–233 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Saxena NK, Vertino PM, Anania FA, Sharma D, Leptin-induced growth stimulation of breast cancer cells involves recruitment of histone acetyltransferases and mediator complex to CYCLIN D1 Promoter via Activation of Stat3. J. Biol. Chem. 282, 13316–13325 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strong AL, Ohlstein JF, Biagas BA, Rhodes LV, Pei DT, Tucker HA, Llamas C, Bowles AC, Dutreil MF, Zhang S, Gimble JM, Burow ME, Bunnell BA, Leptin produced by obese adipose stromal/stem cells enhances proliferation and metastasis of estrogen receptor positive breast cancers. Breast Cancer Res. 17, 112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yin N, Wang D, Zhang H, Yi X, Sun X, Shi B, Wu H, Wu G, Wang X, Shang Y, Molecular mechanisms involved in the growth stimulation of breast cancer cells by leptin. Cancer Res. 64, 5870–5875 (2004). [DOI] [PubMed] [Google Scholar]
- 37.Park H-K, Ahima RS, Leptin signaling. F1000Prime Rep. 6, 73 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hopkins BD, Goncalves MD, Cantley LC, Insulin–PI3K signalling: An evolutionarily insulated metabolic driver of cancer. Nat. Rev. Endocrinol. 16, 276–283 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim SJ, Lubinski J, Huzarski T, Moller P, Armel S, Karlan BY, Senter L, Eisen A, Foulkes WD, Singer CF, Tung N, Bordeleau L, Neuhausen SL, Olopade OI, Eng C, Weitzel JN, Fruscio R, Narod SA, Kotsopoulos J, Hereditary G, Weight gain and the risk of ovarian cancer in BRCA1 and BRCA2 mutation carriers. Cancer Epidemiol. Biomarkers Prev. 30, 2038–2043 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A, Cancer-related inflammation, the seventh hallmark of cancer: Links to genetic instability. Carcinogenesis 30, 1073–1081 (2009). [DOI] [PubMed] [Google Scholar]
- 41.Coussens LM, Werb Z, Inflammation and cancer. Nature 420, 860–867 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meira LB, Bugni JM, Green SL, Lee C-W, Pang B, Borenshtein D, Rickman BH, Rogers AB, Moroski-Erkul CA, Mcfaline JL, Schauer DB, Dedon PC, Fox JG, Samson LD, DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Investig. 118, 2516–2525 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kiraly O, Gong G, Olipitz W, Muthupalani S, Engelward BP, Inflammation-induced cell proliferation potentiates DNA damage-induced mutations in vivo. PLOS Genet. 11, e1004901 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Savage KI, Matchett KB, Barros EM, Cooper KM, Irwin GW, Gorski JJ, Orr KS, Vohhodina J, Kavanagh JN, Madden AF, Powell A, Manti L, Mcdade SS, Park BH, Prise KM, Mcintosh SA, Salto-Tellez M, Richard DJ, Elliott CT, Harkin DP, BRCA1 deficiency exacerbates estrogen-induced dna damage and genomic instability. Cancer Res. 74, 2773–2784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Algire C, Moiseeva O, Deschênes-Simard X, Amrein L, Petruccelli L, Birman E, Viollet B, Ferbeyre G, Pollak MN, metformin reduces endogenous reactive oxygen species and associated DNA damage. Cancer Prev. Res. 5, 536–543 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Kauff ND, Domchek SM, Friebel TM, Robson ME, Lee J, Garber JE, Isaacs C, Evans DG, Lynch H, Eeles RA, Neuhausen SL, Daly MB, Matloff E, Blum JL, Sabbatini P, Barakat RR, Hudis C, Norton L, Offit K, Rebbeck TR, Risk-reducing salpingo-oophorectomy for the prevention of BRCA1- and BRCA2-associated breast and gynecologic cancer: A multicenter, prospective study. J. Clin. Oncol. 26, 1331–1337 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Narod SA, Brunet JS, Ghadirian P, Robson M, Heimdal K, Neuhausen SL, Stoppa-Lyonnet D, Lerman C, Pasini B, de los Rios P, Weber B, Lynch H; Hereditary Breast Cancer Clinical Study, Hereditary breast cancer clinical Study Group Tamoxifen and risk of contralateral breast cancer in BRCA1 and BRCA2 mutation carriers: A case-control study. Hereditary Breast Cancer Clinical Study Group. Lancet 356, 1876–1881 (2000). [DOI] [PubMed] [Google Scholar]
- 48.Rebbeck TR, Lynch HT, Neuhausen SL, Narod SA, Van’T Veer L, Garber JE, Evans G, Isaacs C, Daly MB, Matloff E, Olopade OI, Weber BL, Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N. Engl. J. Med. 346, 1616–1622 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Caldon CE, Estrogen signaling and the dna damage response in hormone dependent breast cancers. Front. Oncol. 4, 106 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bhardwaj P, Au CC, Benito-Martin A, Ladumor H, Oshchepkova S, Moges R, Brown KA, Estrogens and breast cancer: Mechanisms involved in obesity-related development, growth and progression. J. Steroid Biochem. Mol. Biol. 189, 161–170 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang TT, Lampert EJ, Coots C, Lee JM, Targeting the PI3K pathway and DNA damage response as a therapeutic strategy in ovarian cancer. Cancer Treat. Rev. 86, 102021 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Juvekar A, Hu H, Yadegarynia S, Lyssiotis CA, Ullas S, Lien EC, Bellinger G, Son J, Hok RC, Seth P, Daly MB, Kim B, Scully R, Asara JM, Cantley LC, Wulf GM, Phosphoinositide 3-kinase inhibitors induce DNA damage through nucleoside depletion. Proc. Natl. Acad. Sci. U.S.A. 113, E4338–E4347 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cardnell RJ, Feng Y, Diao L, Fan YH, Masrorpour F, Wang J, Shen Y, Mills GB, Minna JD, Heymach JV, Byers LA, Proteomic markers of DNA repair and PI3K pathway activation predict response to the PARP inhibitor BMN 673 in small cell lung cancer. Clin. Cancer Res. 19, 6322–6328 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmana J, Rajendran A, Papa A, Spencer K, Lyssiotis CA, Nardella C, Pandolfi PP, Baselga J, Scully R, Asara JM, Cantley LC, Wulf GM, Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov. 2, 1048–1063 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ducie J, Dao F, Considine M, Olvera N, Shaw PA, Kurman RJ, Shih I-M, Soslow RA, Cope L, Levine DA, Molecular analysis of high-grade serous ovarian carcinoma with and without associated serous tubal intra-epithelial carcinoma. Nat. Commun. 8, 990 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Labidi-Galy SI, Papp E, Hallberg D, Niknafs N, Adleff V, Noe M, Bhattacharya R, Novak M, Jones S, Phallen J, Hruban CA, Hirsch MS, Lin DI, Schwartz L, Maire CL, Tille J-C, Bowden M, Ayhan A, Wood LD, Scharpf RB, Kurman R, Wang T-L, Shih I-M, Karchin R, Drapkin R, Velculescu VE, High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 8, 1093 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Milne RL, Antoniou AC, Modifiers of breast and ovarian cancer risks for BRCA1 and BRCA2 mutation carriers. Endocr. Relat. Cancer 23, T69–T84 (2016). [DOI] [PubMed] [Google Scholar]
- 58.Friebel TM, Domchek SM, Rebbeck TR, Modifiers of cancer risk in BRCA1 and BRCA2 mutation carriers: A systematic review and meta-analysis. J. Natl. Cancer Inst. 106, dju055 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Coletta AM, Peterson SK, Gatus LA, Krause KJ, Schembre SM, Gilchrist SC, Arun B, You YN, Rodriguez-Bigas MA, Strong LL, Lu KH, Basen-Engquist K, Diet, weight management, physical activity and ovarian & breast cancer risk in women with BRCA1/2 pathogenic Germline gene variants: Systematic review. Hered. Cancer Clin. Pract. 18, 5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goodwin PJ, Chen BE, Gelmon KA, Whelan TJ, Ennis M, Lemieux J, Ligibel JA, Hershman DL, Mayer IA, Hobday TJ, Bliss JM, Rastogi P, Rabaglio-Poretti M, Mukherjee SD, Mackey JR, Abramson VG, Oja C, Wesolowski R, Thompson AM, Rea DW, Stos PM, Shepherd LE, Stambolic V, Parulekar WR, Effect of metformin vs placebo on invasive disease–Free survival in patients with breast cancer. JAMA 327, 1963–1973 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goodwin PJ, Dowling RJO, Ennis M, Chen BE, Parulekar WR, Shepherd LE, Burnell MJ, Vander Meer R, Molckovsky A, Gurjal A, Gelmon KA, Ligibel JA, Hershman DL, Mayer IA, Whelan TJ, Hobday TJ, Rastogi P, Rabaglio-Poretti M, Lemieux J, Thompson AM, Rea DW, Stambolic V, Effect of metformin versus placebo on metabolic factors in the MA.32 randomized breast cancer trial. NPJ Breast Cancer 7, 74 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goodwin PJ, Parulekar WR, Gelmon KA, Shepherd LE, Ligibel JA, Hershman DL, Rastogi P, Mayer IA, Hobday TJ, Lemieux J, Thompson AM, Pritchard KI, Whelan TJ, Mukherjee SD, Chalchal HI, Oja CD, Tonkin KS, Bernstein V, Chen BE, Stambolic V, Effect of metformin vs placebo on and metabolic factors in NCIC CTG MA.32. J. Natl. Cancer Inst. 107, djv006 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pimentel I, Chen BE, Lohmann AE, Ennis M, Ligibel J, Shepherd L, Hershman DL, Whelan T, Stambolic V, Mayer I, Hobday T, Lemieux J, Thompson A, Rastogi P, Gelmon K, Rea D, Rabaglio M, Ellard S, Mates M, Bedard P, Pitre L, Vandenberg T, Dowling RJO, Parulekar W, Goodwin PJ, The effect of metformin vs placebo on sex hormones in canadian cancer trials Group MA.32. J. Natl. Cancer Inst. 113, 192–198 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gradishar WJ, Anderson BO, Blair SL, Burstein HJ, Cyr A, Elias AD, Farrar WB, Forero A, Giordano SH, Goldstein LJ, Hayes DF, Hudis CA, Isakoff SJ, Ljung BM, Marcom PK, Mayer IA, McCormick B, Miller RS, Pegram M, Pierce LJ, Reed EC, Salerno KE, Schwartzberg LS, Smith ML, Soliman H, Somlo G, Ward JH, Wolff AC, Zellars R, Shead DA, Kumar R; National Comprehensive Cancer Network Breast Cancer Panel, Breast cancer version 3.2014. J. Natl. Compr. Canc. Netw. 12, 542–590 (2014). [DOI] [PubMed] [Google Scholar]
- 65.Konishi H, Mohseni M, Tamaki A, Garay JP, Croessmann S, Karnan S, Ota A, Wong HY, Konishi Y, Karakas B, Tahir K, Abukhdeir AM, Gustin JP, Cidado J, Wang GM, Cosgrove D, Cochran R, Jelovac D, Higgins MJ, Arena S, Hawkins L, Lauring J, Gross AL, Heaphy CM, Hosokawa Y, Gabrielson E, Meeker AK, Visvanathan K, Argani P, Bachman KE, Park BH, Mutation of a single allele of the cancer susceptibility gene BRCA1 leads to genomic instability in human breast epithelial cells. Proc. Natl. Acad. Sci. U.S.A. 108, 17773–17778 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bray NL, Pimentel H, Melsted P, Pachter L, Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; 1000 Genome Project Data Processing Subgroup, The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L, Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, Lagarde J, Veeravalli L, Ruan X, Ruan Y, Lassmann T, Carninci P, Brown JB, Lipovich L, Gonzalez JM, Thomas M, Davis CA, Shiekhattar R, Gingeras TR, Hubbard TJ, Notredame C, Harrow J, Guigo R, The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu X, Holstege H, van der Gulden H, Treur-Mulder M, Zevenhoven J, Velds A, Kerkhoven RM, van Vliet MH, Wessels LF, Peterse JL, Berns A, Jonkers J, Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc. Natl. Acad. Sci. U.S.A. 104, 12111–12116 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu H, Murphy CJ, Karreth FA, Emdal KB, White FM, Elemento O, Toker A, Wulf GM, Cantley LC, Identifying and targeting sporadic oncogenic genetic aberrations in mouse models of triple-negative breast cancer. Cancer Discov. 8, 354–369 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schwenk F, Baron U, Rajewsky K, A cre-transgenic mouse strain for the ubiquitous deletion of loxP-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 23, 5080–5081 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Katti A, Foronda M, Zimmerman J, Diaz B, Zafra MP, Goswami S, Dow LE, GO: A functional reporter system to identify and enrich base editing activity. Nucleic Acids Res. 48, 2841–2852 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zafra MP, Schatoff EM, Katti A, Foronda M, Breinig M, Schweitzer AY, Simon A, Han T, Goswami S, Montgomery E, Thibado J, Kastenhuber ER, Sanchez-Rivera FJ, Shi J, Vakoc CR, Lowe SW, Tschaharganeh DF, Dow LE, Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol. 36, 888–893 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Han S, Baba T, Yanai S, Byun DJ, Morohashi KI, Kim JH, Choi MH, GC-MS-based metabolic signatures reveal comparative steroidogenic pathways between fetal and adult mouse testes. Andrology 9, 400–406 (2021). [DOI] [PubMed] [Google Scholar]
- 78.Moon JY, McNamara KM, Lee JJ, Chung BC, Sasano H, Choi MH, Improved detectability of sex steroids from frozen sections of breast cancer tissue using GC-triple quadrupole-MS. J. Steroid Biochem. Mol. Biol. 178, 185–192 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.