

Abstract
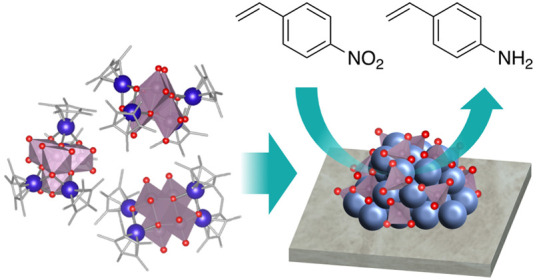
To form high-density metal/oxide interfacial active sites, we developed a catalyst preparation method based on hybrid clustering. An iridium–molybdenum (Ir–Mo) hybrid clustering catalyst was prepared by using the hybrid cluster [(IrCp*)4Mo4O16] (Cp* = η5-C5Me5) as the precursor. The Ir–Mo hybrid clustering catalyst selectively reduced the nitro group in the hydrogenation of 4-nitrostyrene, whereas the coimpregnated Ir–Mo catalyst reduced both the nitro and vinyl groups nonselectively. The hybrid clustering catalyst also exhibited high selectivity, even at a high Ir loading (5 wt %), in contrast to Ir/MoO3, which exhibited high selectivity only at low Ir loadings (<0.3 wt %). In situ X-ray absorption spectroscopy analysis revealed that oxygen vacancies were formed at the Ir/MoOx interface in the presence of H2. We concluded that a high-density Ir/MoOx interface contributes to the preferential adsorption of nitro groups on vacant sites, promoting the selective hydrogenation of nitro groups.
Keywords: nitrostyrene hydrogenation, organometallic polyoxometalate, nanoparticle, iridium, molybdenum, interface
1. Introduction
Supported metal nanoparticles composed of platinum group metals (PGMs) such as Ru, Rh, Pd, and Pt are widely used as catalysts for hydrogenation because of their activity for dissociative adsorption of H2 and the subsequent addition of the dissociated H atoms.1 One of the most challenging problems for PGM catalysts is the control of selectivity in chemoselective hydrogenation, where more than one reducible functional group is present. For example, chemoselective hydrogenation of nitroarenes, an important process in the production of functionalized anilines, requires the development of catalysts that can selectively reduce the nitro group in the presence of other reducible groups such as C=C and C=O.2−4 To enhance selectivity, considerable efforts have been made, including the formation of highly or atomically dispersed species,5−8 confinement of nanoparticles within microporous supports,9,10 introduction of a less active component,11,12 surface modification through strong metal–support interaction (SMSI),13,14 and surface decoration with an additional metal oxide component.15−19 These approaches modulate the preferential adsorption of the nitro group through electronic or steric effects, resulting in selective hydrogenation of the nitro group. Therefore, the formation of a high density of metal/oxide interfacial sites is key to enhancing selectivity.
A straightforward strategy for the efficient formation of metal/oxide interfacial sites is to prepare the catalyst from a PGM precursor with such interfaces. We focused on the possibility of preparing catalysts by using hybrid clustering. Hybrid clusters or organometallic polyoxometalates, formulated as [(M1L)xM2yOn], are composed of metal ions (M1), metal oxide clusters (M2yOn), and organic ligands (L).20 Because the surface oxygen atoms of the metal oxide clusters are coordinated to the metal ions, the hybrid clusters can be viewed as minimal building blocks with M1–O–M2 interfaces. Therefore, we expected the catalysts prepared from the hybrid clusters to have a high density of metal/oxide interfaces, contributing to chemoselective hydrogenation. We previously reported that PGM-based hybrid clustering catalysts exhibited improved activity compared to the corresponding coimpregnated catalysts.21,22 Despite the potential of hybrid clustering catalysts in various applications, there have been few reports on their use in heterogeneous catalysts.23,24
In this study, Ir-M-based hybrid clustering catalysts (M = Mo, V) were prepared by using hybrid clusters [(IrCp*)4Mo4O16] and [(IrCp*)4V6O19] (Cp* = η5-C5Me5) as the precursors (Figure 1). The effect of the preparation method on the catalytic performance was tested using the hydrogenation of 4-nitrostyrene as a model reaction. The formation mechanism of the Ir/MOx interface was studied by using in situ X-ray absorption spectroscopy (XAS). The effect of the Ir/MOx interface on the selectivity is evaluated through in situ XAS and kinetic studies.
Figure 1.

[(IrCp*)4Mo4O16] as a catalyst precursor with Ir–O–Mo interfaces. Hydrogen atoms have been omitted for clarity.
2. Results and Discussion
2.1. Preparation of Hybrid Clustering Catalyst
The hybrid clustering catalysts, Ir4Mo4/Al2O3 and Ir4V6/Al2O3, were prepared by using [(IrCp*)4Mo4O16] and [(IrCp*)4V6O19] as the catalyst precursors, respectively. These precursors were synthesized according to the procedures reported in previous studies25,26 and characterized using positive-ion electrospray ionization mass spectrometry (ESI-MS) and Fourier transform infrared (FT-IR) spectroscopy (Figures S1 and S2). The most intense peaks in the positive-ion ESI mass spectra were assigned to the monovalent proton adducts [H(IrCp*)4Mo4O16]+ and [H(IrCp*)4V6O19]+. The FT-IR spectra exhibited characteristic absorption patterns corresponding to the desired clusters. After the precursor clusters were adsorbed on γ-Al2O3 by impregnation with methanol, the catalysts were prepared through calcination in air at 573 K, followed by reduction under a H2 flow at 573 K. The coimpregnated Ir–Mo and Ir–V catalysts, Ir–Mo/Al2O3 and Ir–V/Al2O3 ([Mo]/[Ir] = 1, [V]/[Ir] = 1.5), and the pristine Ir catalyst, Ir/Al2O3, were prepared using H2IrCl6, (NH4)6[Mo7O24]·4H2O, and NH4VO3 under identical calcination conditions.
According to high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) analysis, the average diameters of Ir4Mo4/Al2O3 and Ir–Mo/Al2O3 were estimated as 1.2 ± 0.2 and 1.3 ± 0.3 nm, respectively, indicating that small nanoparticles were formed regardless of the preparation method (Figure 2).
Figure 2.
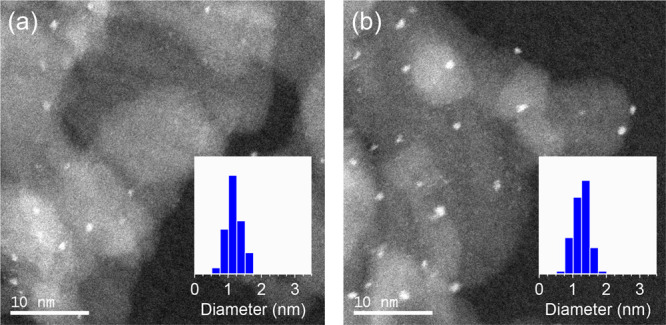
HAADF-STEM images of (a) Ir4Mo4/Al2O3 and (b) Ir–Mo/Al2O3.
The electronic and local structures of Ir4Mo4/Al2O3 and Ir–Mo/Al2O3 were studied through Ir L3- and Mo K-edge XAS. The white-line intensities of the Ir L3-edge X-ray absorption near-edge structure (XANES) spectra were used to determine the oxidation state of Ir. The intensity of Ir4Mo4/Al2O3 was lower than that of Ir–Mo/Al2O3, indicating that the Ir species in Ir4Mo4/Al2O3 were relatively reduced (Figure 3a). The Fourier transform of extended X-ray absorption fine structure (FT-EXAFS) was analyzed by curve-fitting the inverse Fourier transform of the EXAFS spectra. The peak derived from the second coordination sphere of [(IrCp*)4Mo4O16] is assigned to Mo (Figures 3b and S3, Table S1). This is consistent with the crystal structure in which the surface oxygen atoms in the Mo4O16 unit are coordinated to the Ir atoms.25 In contrast, the peak derived from the second coordination sphere of Ir4Mo4/Al2O3 is assigned to Ir. The bond length (2.69 Å) was almost equal to that of the Ir powder (2.71 Å), indicating the presence of metallic Ir species. Although Ir is intrinsically prone to reoxidation upon exposure to air after reduction, metallic species are preserved in Ir4Mo4/Al2O3. Therefore, we concluded that a core–shell structure was formed on Ir4Mo4/Al2O3, where the metallic Ir core was covered with an Ir–Mo mixed oxide shell. In the case of coimpregnated Ir–Mo/Al2O3, no peaks derived from the second coordination sphere were observed. Because the white-line intensity of the XANES spectrum and the oscillation pattern of the EXAFS spectrum matched well with those of Ir2O3·nH2O, we conclude that Ir2O3 nanoparticles were formed on Ir–Mo/Al2O3.
Figure 3.

Ir L3-edge (a) XANES, (b) FT-EXAFS, and (c) Mo K-edge XANES spectra of (i) Ir4Mo4/Al2O3, (ii) Ir–Mo/Al2O3, and (iii) [(IrCp*)4Mo4O16].
The pre-edge peaks in the Mo K-edge XANES spectra were assigned to weak quadrupole-allowed (1s → 4d) and strong dipole-allowed (1s → 5p) transitions, and the local structural disorder of the Mo oxide species reflected the intensity.27,28 The Mo K-edge XANES spectrum of Ir4Mo4/Al2O3 matched well with that of Ir–Mo/Al2O3, indicating that the local structure of Mo was independent of the preparation method (Figure 3c).
2.2. Hydrogenation of 4-Nitrostyrene
The catalytic activities were tested through the chemoselective hydrogenation of 4-nitrostyrene (1) to 4-aminostyrene (2). The substrate had two reducible groups, and the desired product (2) was obtained via the selective hydrogenation of the nitro group. The reaction also afforded two byproducts: 4-nitroethylbenzene (3), formed by the selective hydrogenation of the vinyl group, and 4-aminoethylbenzene (4), formed after complete hydrogenation (Scheme 1). In the initial stage of the reaction, Ir4Mo4/Al2O3 selectively reduced the nitro group to produce 2 (select. > 96%, conv. < 48%) (Figure 4a). In contrast, Ir–Mo/Al2O3 nonselectively reduced both the nitro and C=C groups, and the selectivity toward 2 reached 63% (Figure 4b). The optimized hybrid clustering catalyst with a TiO2 support, Ir4Mo4/TiO2, prepared through calcination at 573 K, followed by H2 reduction at 773 K, exhibited higher activity and selectivity in comparison to the catalyst with an Al2O3 support (Figure 4c). Although the Al2O3-supported catalysts had little influence on the reduction temperature, the TiO2-supported catalysts at a reduction temperature of 773 K exhibited selectivity higher than that at 573 K (Table S2). This is probably due to the reducible nature of TiO2, which results in the formation of an Ir/TiOx interface via SMSI.13,14 The formation of Ir/TiOx contributes to the selectivity by the preferential adsorption of the nitro group and the encapsulation of nonselective Ir–Ir sites.29,30 Ir4Mo4/TiO2 exhibited a high initial selectivity (>99%, conv. < 61%) and maintained its selectivity during the later stages of the reaction (select. 94%, conv. 97%). Although the corresponding coimpregnated catalyst, Ir–Mo/TiO2, also exhibited improved activity and selectivity compared to the Al2O3-supported catalyst, the selectivity significantly decreased as the conversion exceeded ∼70% (Figure 4d). These results suggest that Ir/MoOx is more selective than Ir/TiOx and that the selectivity of the hybrid clustering catalyst can be improved by optimization of the support.
Scheme 1. Hydrogenation of 4-Nitrostyrene (1).

Figure 4.

Time courses of the hydrogenation of 1 catalyzed by (a) Ir4Mo4/Al2O3, (b) Ir–Mo/Al2O3, (c) Ir4Mo4/TiO2, and (d) Ir–Mo/TiO2. Reaction conditions: 1 (0.1 mmol), H2 (0.3 MPa), toluene (1 mL), catalyst (10 mg, Ir: 0.52 mol %), and 303 K.
Enhanced selectivity through hybrid clustering has also been observed for other oxide species such as V. The hybrid clustering Ir–V catalyst, Ir4V6/Al2O3, also exhibited higher selectivity than the coimpregnated Ir–V catalyst, Ir–V/Al2O3; however, the introduction of Mo was more effective than that of V, regardless of the preparation method (Figure 5a, Table S3). A general and straightforward formation method of an Ir/MoOx interface is to deposit the Ir species onto a Mo-oxide support with a high dispersion. Single-atom catalysts (SACs) have demonstrated unique activities in a variety of reactions, including nitrostyrene hydrogenation.5,31 Low metal loadings (e.g., 0.1 wt %) are required to achieve SAC through inhibiting aggregation. To examine the effect of loading on selectivity, the activity of Ir4Mo4/Al2O3 was compared with that of an Ir catalyst supported on MoO3, Ir/MoO3. Ir/MoO3 exhibited high selectivity with low (<0.3 wt %) Ir loadings (Figure 5b). However, the selectivity decreased significantly when the loading increased, probably because of the formation of aggregated Ir species. In contrast, Ir4Mo4/Al2O3 exhibited high selectivity even at a high Ir loading (5 wt %) (Figure 5c). These results indicate that the formation of the Ir/MoOx interface is the key to chemoselective hydrogenation, regardless of the catalyst preparation method, and that hybrid clustering is an efficient method to form such interfacial sites exclusively.
Figure 5.

Initial reaction rate of the hydrogenation of 1 over (a) Ir–Mo- and Ir–V-based catalysts, (b) Ir/MoO3, and (c) Ir4Mo4/Al2O3 with different Ir loadings. Reaction conditions were identical to those cited in the caption of Figure 4 except for the catalyst amount (2–100 mg, Ir: 0.52 mol %). The original data are summarized in Table S3.
2.3. Formation Mechanism of Ir/MoOx Interface
The high selectivity of Ir4Mo4/Al2O3 was observed only when the catalyst was prepared via calcination in air and subsequent reduction under a H2 flow. The optimal temperature for both aerobic calcination and H2 reduction was 573 K for the Al2O3-supported catalyst. The catalysts prepared through calcination at varying temperatures followed by H2 reduction at 573 K exhibited increased selectivity with increasing calcination temperature, reaching a maximum at 573 K (Figure 6a). Subsequently, the selectivity decreased with a further increase in the calcination temperature. In contrast, the effect of the reduction temperature on activity was relatively low (Figure 6b).
Figure 6.

Effect of the preparation conditions on the activity of Ir4Mo4/Al2O3 prepared through (a) calcination at varying temperatures followed by H2 reduction at 573 K and (b) calcination at 573 K followed by H2 reduction at varying temperatures. Reaction conditions were identical with those cited in the caption of Figure 4 except for the reaction time (20 min).
To elucidate the formation mechanism of active sites on Ir4Mo4/Al2O3 through thermal treatment, in situ XANES analysis was conducted under two conditions: (i) calcination of the adsorbed cluster Ir4Mo4/Al2O3_ads under 20% O2/He flow, and (ii) reduction of the calcined catalyst Ir4Mo4/Al2O3_air(573) under H2 flow (Figure S4). The measurements were conducted at 303 K and thereafter at temperatures ranging from 373 to 773 K in increments of 100 K. The white-line intensity of the Ir L3-edge spectra was determined from the maximum normalized absorbance and was used as a measure of the oxidation state of Ir (Figure 7a). Similarly, the edge energy (E0) of the Mo K-edge spectra was determined from the second peak of the first derivative of the normalized absorbance and used as a measure of the oxidation state of Mo (Figure 7b). These intensities and energies increased with increasing oxidation states, as observed for the reference samples of Ir (Ir powder, Ir2O3·nH2O, and IrO2 for Ir0, Ir3+, and Ir4+, respectively) and Mo (MoO2 and MoO3 for Mo4+ and Mo6+, respectively).
Figure 7.

Oxidation states of Ir and Mo during calcination and reduction. (a) White-line intensity of the Ir L3-edge XANES spectra. (b) Edge energy (E0) of the Mo K-edge XANES spectra. The original XANES spectra are shown in Figure S4.
During calcination under a 20% O2/He flow up to 573 K, no significant changes in the oxidation states of Ir and Mo were observed. The oxidation of Ir began at 573 K and a slight change in the oxidation state of Mo was observed. Further increase in the calcination temperature resulted in the formation of Ir4+. Thermogravimetry-differential thermal analysis (TG-DTA) of [(IrCp*)4Mo4O16] under an air flow indicated a weight loss (29.5%) at temperatures ranging from 543 to 599 K (Figure S5). This is attributed to the removal of the Cp* ligand and the reduction of Ir3+ to Ir+ (theoretical value of 29.4%). Thereafter, a gradual weight increase (4.3%) was observed, which was attributed to the oxidation of Ir+ to Ir4+ (theoretical value of 4.9%). Therefore, we concluded that the increase in the oxidation state of Ir and Mo is derived from the decomposition of the precursor cluster and the calcination temperature of 573 K is the minimum temperature required to remove organic ligands without further oxidation. As described previously, the selectivity of Ir4Mo4/Al2O3 was significantly affected by the calcination temperature, and high selectivity was observed only at 573 K (Figure 6a). The organic ligand remained at a lower calcination temperature, and further oxidation proceeded at a higher calcination temperature, resulting in a decrease in the selectivity of the catalyst.
During reduction under a H2 flow, reductions in both Ir and Mo were observed. Selectivity was not significantly affected by the reduction temperature; however, the reduction process was found to be essential for activity (Figure 6b). Ex situ analysis of the optimized catalyst, Ir4Mo4/Al2O3_air(573) H2 (573), revealed that the catalyst was reoxidized after exposure to air (Figures 3 and 7). These results indicate that both the reduction and reoxidation processes contribute to the formation of surface Ir/MoOx interfacial sites.
2.4. Role of Ir/MoOx Interface on the Catalytic Performance
To determine the effect of the Ir/MoOx interface on selectivity, in situ XAS analysis of the catalysts under a H2 flow was conducted (Figure S6). According to the Ir L3-edge XANES spectra, a reduction of Ir was observed for both Ir4Mo4/Al2O3 and Ir–Mo/Al2O3 upon exposure to H2 (Figure 8a). In contrast, according to the Mo K-edge XANES spectra, the reduction of Mo in Ir4Mo4/Al2O3 was observed even at 303 K, whereas the oxidation state of Mo in Ir–Mo/Al2O3 was retained (Figure 8b). The reduction of both Ir and Mo indicates that oxygen vacancies were formed at the Ir/MoOx interface of Ir4Mo4/Al2O3 in the presence of H2. As reported for MoO3-supported or MoOx-modified metal catalysts for deoxygenation32,33 and chemoselective hydrogenation,16,19,34,35 the presence of Mo results in the preferential adsorption of oxygen-containing functional groups. This is because of the reducible nature of Mo, which affects its adsorption behavior through the formation of oxygen vacancies or electronic interactions with the metal species.
Figure 8.

Oxidation states of Ir and Mo under a H2 atmosphere. (a) White-line intensity of Ir L3-edge XANES spectra. (b) Edge energy (E0) of the Mo K-edge XANES spectra. The original XANES spectra are shown in Figure S6.
To determine how the formation of oxygen vacancies contributes to improved selectivity, kinetic studies on the hydrogenation of nitrobenzene (NB) and styrene (ST) were conducted (Scheme 2). The hydrogenation of NB and ST afforded aniline and ethylbenzene as the only products, respectively. The reaction order with respect to the concentration of the substrate ([NB] or [ST]) and the hydrogen pressure (PH2) was estimated from the initial reaction rate under different reaction conditions (Figure S7). For the hydrogenation of NB, the reaction orders with respect to [NB] and PH2 were estimated as 0.04 and 0.97, respectively (Scheme 2a, Table S4). The zero-order kinetics with respect to [NB] indicates that the adsorption of NB is not involved in the rate-determining step (RDS) and that the RDS is a H2-involved step, such as the formation of an oxygen vacancy or the hydrogenation of adsorbed NB. The efficient adsorption of NB can be explained by the formation of oxygen vacancies, where oxygen-containing functional groups are preferentially adsorbed. In contrast, the reaction orders with respect to [ST] and PH2 were estimated as 0.29 and 0.04, respectively, for the hydrogenation of ST (Scheme 2b). This indicates that hydrogen adsorption is not related to the RDS and that the RDS is an ST-involved step, such as the adsorption of ST or the hydrogenation of adsorbed ST. The kinetics of the hydrogenation on coimpregnated Ir–Mo/Al2O3 was completely different from that on Ir4Mo4/Al2O3. Regardless of the substrate (NB or ST), the reaction orders with respect to the substrate were between 0 and 1 and those to PH2 were almost 1 (Figure S8 and Table S4). This implies that the hydrogenation of NB and ST on Ir–Mo/Al2O3 proceeded by a similar mechanism and that the RDS is the reduction of the adsorbed substrate by the hydrogen. The hydrogenation of ST was conducted in the presence of NB to understand the competitive behavior of chemoselective hydrogenation (Figure 9). For Ir4Mo4/Al2O3, ST hydrogenation was completely suppressed in the presence of NB. This is in contrast to Ir–Mo/Al2O3, where ST hydrogenation proceeded in the presence of NB. Moreover, the reaction rates of the separate ST and NB hydrogenations were comparable over Ir–Mo/Al2O3.
Scheme 2. Hydrogenation of (a) Nitrobenzene and (b) Styrene.

The reaction orders were estimated based on the kinetic studies (Figure S7).
Figure 9.

Initial reaction rate of the hydrogenation of the mixture (NB+ST), NB, and ST over (a) Ir4Mo4/Al2O3 and (b) Ir–Mo/Al2O3. Reaction conditions were identical to those cited inthe caption of Figure 4 except for the substrates.
Regarding the effect of the Ir/MoOx interface on the catalytic performance, we anticipate that selectivity is influenced by the preferential adsorption of nitro groups on the oxygen vacancies formed in the presence of H2 (Figure 10). The vacant sites capture and reduce nitro groups rather than vinyl groups and promote the subsequent hydrogenation process to produce anilines. This mechanism is consistent with that of reported PGM catalysts having interfacial active sites with reducible metal oxides.18,36
Figure 10.

Plausible reaction mechanism of hydrogenation of nitroarene on Ir/MoOx interfacial sites.
3. Conclusions
In this study, an Ir–Mo-based catalyst, Ir4Mo4/Al2O3, was prepared by using an Ir–Mo hybrid cluster [(RhCp*)4Mo4O16] (Cp* = η5-C5Me5) as a precursor and applied as a catalyst for the hydrogenation of 4-nitrostyrene. Ir4Mo4/Al2O3 selectively reduced the nitro groups to produce 4-aminostyrene, whereas the coimpregnated catalyst, Ir–Mo/Al2O3, reduced both the nitro and vinyl groups nonselectively. Ir4Mo4/Al2O3 exhibited high selectivity even at a high Ir loading (5 wt %). This is in contrast to Ir/MoO3, which exhibited high selectivity only when the loading was low (<0.3 wt %). The thermal treatment conditions used to prepare Ir4Mo4/Al2O3 strongly affected selectivity. The catalyst should be prepared through calcination in air at a minimal temperature to remove the organic ligands (Cp*) while preventing further oxidation of Ir. Subsequent reduction under H2 is necessary for the catalyst to work effectively, and reoxidation by exposure to air is expected to play an important role in the formation of the Ir/MoOx interface. In situ XAS analysis under H2 revealed the reduction of Mo for Ir4Mo4/Al2O3 even at 303 K, which is explained by the formation of oxygen vacancies at the Ir/MoOx interface. Oxygen vacancies preferentially adsorbed nitro groups over vinyl groups, thereby promoting selective hydrogenation of the nitro groups.
4. Experimental Section
4.1. Chemicals
Pentamethylcyclopentadienyl iridium(III) dichloride dimer, 4-nitrostyrene, and styrene were purchased from TCI. Hydrogen hexachloroiridate(IV) n-hydrate, ammonium molybdate(VI) tetrahydrate, disodium molybdate(VI) dihydrate, ammonium metavanadate, molybdenum(VI) oxide, and nitrobenzene were purchased from Wako Chemicals. Sodium metavanadate was obtained from Sigma-Aldrich. γ-Al2O3 (JRC-ALO-8) and TiO2 (JRC-TIO-17) were supplied by the Catalysis Society of Japan. Before use, γ-Al2O3 and TiO2 were calcined under air at 773 K for 3 h. Deionized water (Milli-Q, >18 MΩ cm) was used in all the experiments.
4.2. Synthesis of Hybrid Clusters
[(IrCp*)4Mo4O16] was synthesized according to a procedure reported in a previous study.25 Na2MoO4·2H2O (1.21 g, 5.00 mmol) was dissolved in water (3 mL), and [IrCp*Cl2]2 (398 mg, 0.500 mmol) was added. Thereafter, the mixture was stirred for 3 h at 353 K. After the reaction, the mixture was concentrated by using a rotary evaporator, and the product was extracted by using dichloromethane (DCM). The DCM layer was washed with water, and the crude product was obtained by evaporating the solvent. The purified product was obtained via recrystallization from DCM/toluene (91% yield). IR (KBr pellets, cm–1): 932, 910, 696, 642, 577, and 536.
[(IrCp*)4V6O19] was synthesized according to a procedure reported in a previous study.26 NaVO3 (610 mg, 5.00 mmol) was added to water (20 mL) and the mixture was stirred for 1 h for complete dissolution. Thereafter, [IrCp*Cl2]2 (398 mg, 0.500 mmol) was added, and the mixture was stirred for 3 h at room temperature. After the reaction, the mixture was concentrated using a rotary evaporator, and the product was extracted using DCM. The DCM layer was washed with water, and the crude product was obtained by evaporating the solvent. The purified product was obtained via recrystallization from DCM/toluene (96% yield). IR (KBr pellet, cm–1): 941, 674, 559, and 499.
4.3. Catalyst Preparation
The hybrid clustering catalyst Ir4Mo4/Al2O3 was prepared as follows: [(IrCp*)4Mo4O16] (12.7 mg, 6.51 μmol) was dissolved in methanol (20 mL), and the solution was added dropwise to a methanolic dispersion of the γ-Al2O3 support (493 mg in 40 mL). The mixture was stirred for 2 h and subsequently dried slowly at 373 K. The resulting solid was dried overnight at 333 K. The catalyst was calcined in static air at 573 K for 1 h, followed by reduction under a H2 flow (10 mL/min) at 573 K for 1 h. The catalyst was stored in the air. Ir4V6/Al2O3 was prepared by using [(IrCp*)4V6O19] as the precursor. Ir–Mo/Al2O3, Ir–V/Al2O3, and Ir/Al2O3 were prepared by using H2IrCl6, (NH4)6[Mo7O24]·4H2O, and NH4VO3. Water was used as a solvent instead of methanol. Ir/MoO3 was prepared by using H2IrCl6 and MoO3. The calculated loading amounts of Ir, Mo, and V were 1.0, 0.50, and 0.40 wt %, respectively ([Mo]/[Ir] = 1, [V]/[Ir] = 1.5).
4.4. Characterization
Positive-ion ESI-MS was conducted with a Shimadzu LCMS-9050. The samples were dissolved in dichloromethane/methanol and electrosprayed at a bias voltage of +4 kV. FT-IR analysis was conducted using a JASCO FT/IR-6100 in transmission mode. The sample was mixed well with KBr and pressed to form a pellet, which was subsequently used for analysis. HAADF-STEM analysis was performed using a JEOL JEM-ARM200F instrument. The samples were prepared by dropping an ethanolic dispersion of the catalyst onto a copper grid with a support membrane and evaporating the solvent. Ir L3-edge and Mo K-edge XAS measurements were conducted using the BL01B1 beamline at SPring-8 of the Japan Synchrotron Radiation Research Institute. The incident X-ray beam was monochromated using a Si(111) or Si(311) double-crystal monochromator for the Ir L3-edge and Mo K-edge measurements, respectively. The measurements were conducted in fluorescence mode using a 19-element Ge solid-state detector at room temperature. Reference samples were ground with boron nitride and pressed into pellets. In situ XAS measurements were conducted in transmission mode using ionization chambers. The pellet sample was placed in a temperature-controlled gas cell equipped with Kapton windows under a gas flow (20% of the mixture of O2/He (50 mL/min) or H2 (50 mL/min)). The temperature was increased from 303 to 373 K and thereafter to 773 K in steps of 100 K. The temperature was maintained for 10 min before each measurement. The XAS data were analyzed using the Rigaku REX2000 program. After background subtraction, the k3-weighted χ spectrum within a k range of 3–17.5 Å–1 was Fourier-transformed into an r space. Curve-fitting analysis was performed on the first (Ir–O) and second (Ir–Mo and Ir–Ir) coordination spheres. The phase shifts and backscattering amplitude functions of the Ir–O, Ir–Mo, and Ir–Ir bonds were extracted from [(IrCp*)4Mo4O16]25 using the FEFF8 program37 by setting σ2 at 0.0036. TG-DTA was conducted using a HITACHI STA7300 TG analyzer. The sample was heated at 353 K for 30 min, and the temperature was increased to 873 K at a ramping rate of 10 K/min under an air flow (200 mL/min). Gas chromatography (GC) analysis was conducted using a Shimadzu GC-2025 gas chromatograph comprising a flame ionization detector equipped with an InertCap 5 capillary column (internal diameter of 0.25 mm and length of 30 m).
4.5. Catalytic Test
The typical procedure for the hydrogenation of 4-nitrostyrene is as follows. The catalyst (10 mg, 0.52 mol % Ir) was placed in a glass tube, and 4-nitrostyrene (0.1 mmol) and toluene (1 mL) were subsequently added. Naphthalene (0.02 mmol) was also added as an internal standard. The glass tube was placed in an autoclave and H2 was introduced. The reaction mixture was stirred at 303 K for a specified reaction time. After the reaction, the catalyst was filtered off and the filtrate was analyzed by GC. The conversion and yield were estimated based on the initial amounts of 4-nitrostyrene. The selectivity and the material balance were calculated according to the formulas below. The material balance was almost equal to 1 (0.95–1.05) in all experiments. The initial reaction rates were estimated from the yield of 4-aminostyrene divided by the product of the total amount of Ir and the reaction time. A representative GC chart of the reaction mixture is shown in Figure S9.
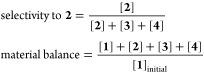 |
Acknowledgments
This research was financially supported by a Grant-in-Aid for Scientific Research (21K14463) from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT). The HAADF-STEM analysis was conducted at the Advanced Characterization Nanotechnology Platform of the University of Tokyo supported by the Nanotechnology Platform of MEXT. The synchrotron radiation experiments were performed with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (2022B1655).
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsorginorgau.3c00017.
Results of characterization and catalytic test (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Vilé G.; Albani D.; Almora-Barrios N.; López N.; Pérez-Ramírez J. Advances in the Design of Nanostructured Catalysts for Selective Hydrogenation. ChemCatChem. 2016, 8, 21–33. 10.1002/cctc.201501269. [DOI] [Google Scholar]
- Blaser H. U.; Steiner H.; Studer M. Selective Catalytic Hydrogenation of Functionalized Nitroarenes: An Update. ChemCatChem. 2009, 1, 210–221. 10.1002/cctc.200900129. [DOI] [Google Scholar]
- Song J.; Huang Z. F.; Pan L.; Li K.; Zhang X.; Wang L.; Zou J. J. Review on Selective Hydrogenation of Nitroarene by Catalytic, Photocatalytic and Electrocatalytic Reactions. Appl. Catal. B Environ. 2018, 227, 386–408. 10.1016/j.apcatb.2018.01.052. [DOI] [Google Scholar]
- Coq B.; Tijani A.; Dutartre R.; Figuéras F. Influence of Support and Metallic Precursor on the Hydrogenation of P-Chloronitrobenzene over Supported Platinum Catalysts. J. Mol. Catal. 1993, 79, 253–264. 10.1016/0304-5102(93)85106-4. [DOI] [Google Scholar]
- Wei H.; Liu X.; Wang A.; Zhang L.; Qiao B.; Yang X.; Huang Y.; Miao S.; Liu J.; Zhang T. FeOx-Supported Platinum Single-Atom and Pseudo-Single-Atom Catalysts for Chemoselective Hydrogenation of Functionalized Nitroarenes. Nat. Commun. 2014, 5, 5634. 10.1038/ncomms6634. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Chang C. R.; Huang Z. Q.; Li J.; Wu Z.; Ma Y.; Zhang Z.; Wang Y.; Qu Y. High Catalytic Activity and Chemoselectivity of Sub-Nanometric Pd Clusters on Porous Nanorods of CeO2 for Hydrogenation of Nitroarenes. J. Am. Chem. Soc. 2016, 138, 2629–2637. 10.1021/jacs.5b11413. [DOI] [PubMed] [Google Scholar]
- Peng Y.; Geng Z.; Zhao S.; Wang L.; Li H.; Wang X.; Zheng X.; Zhu J.; Li Z.; Si R.; Zeng J. Pt Single Atoms Embedded in the Surface of Ni Nanocrystals as Highly Active Catalysts for Selective Hydrogenation of Nitro Compounds. Nano Lett. 2018, 18, 3785–3791. 10.1021/acs.nanolett.8b01059. [DOI] [PubMed] [Google Scholar]
- Liu W.; Feng H.; Yang Y.; Niu Y.; Wang L.; Yin P.; Hong S.; Zhang B.; Zhang X.; Wei M. Highly-Efficient RuNi Single-Atom Alloy Catalysts toward Chemoselective Hydrogenation of Nitroarenes. Nat. Commun. 2022, 13, 3188. 10.1038/s41467-022-30536-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Wang L.; Shao Y.; Wang Y.; Gates B. C.; Xiao F. S. A Pd@Zeolite Catalyst for Nitroarene Hydrogenation with High Product Selectivity by Sterically Controlled Adsorption in the Zeolite Micropores. Angew. Chem., Int. Ed. 2017, 56, 9747–9751. 10.1002/anie.201703938. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Wang N.; Zhang T.; Bai R.; Mayoral A.; Zhang P.; Zhang Q.; Terasaki O.; Yu J. Zeolite-Encaged Single-Atom Rhodium Catalysts: Highly-Efficient Hydrogen Generation and Shape-Selective Tandem Hydrogenation of Nitroarenes. Angew. Chem., Int. Ed. 2019, 58, 18570–18576. 10.1002/anie.201912367. [DOI] [PubMed] [Google Scholar]
- Berguerand C.; Yarulin A.; Cárdenas-Lizana F.; Wärnå J.; Sulman E.; Murzin D. Y.; Kiwi-Minsker L. Chemoselective Liquid Phase Hydrogenation of 3-Nitrostyrene over Pt Nanoparticles: Synergy with ZnO Support. Ind. Eng. Chem. Res. 2015, 54, 8659–8669. 10.1021/acs.iecr.5b02113. [DOI] [Google Scholar]
- Furukawa S.; Takahashi K.; Komatsu T. Well-Structured Bimetallic Surface Capable of Molecular Recognition for Chemoselective Nitroarene Hydrogenation. Chem. Sci. 2016, 7, 4476–4484. 10.1039/C6SC00817H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serna P.; Corma A. Transforming Nano Metal Nonselective Particulates into Chemoselective Catalysts for Hydrogenation of Substituted Nitrobenzenes. ACS Catal. 2015, 5, 7114–7121. 10.1021/acscatal.5b01846. [DOI] [Google Scholar]
- Macino M.; Barnes A. J.; Althahban S. M.; Qu R.; Gibson E. K.; Morgan D. J.; Freakley S. J.; Dimitratos N.; Kiely C. J.; Gao X.; Beale A. M.; Bethell D.; He Q.; Sankar M.; Hutchings G. J. Tuning of Catalytic Sites in Pt/TiO2 Catalysts for the Chemoselective Hydrogenation of 3-Nitrostyrene. Nat. Catal. 2019, 2, 873–881. 10.1038/s41929-019-0334-3. [DOI] [Google Scholar]
- Mitsudome T.; Mikami Y.; Matoba M.; Mizugaki T.; Jitsukawa K.; Kaneda K. Design of a Silver-Cerium Dioxide Core-Shell Nanocomposite Catalyst for Chemoselective Reduction Reactions. Angew. Chem., Int. Ed. 2012, 51, 136–139. 10.1002/anie.201106244. [DOI] [PubMed] [Google Scholar]
- Tamura M.; Yuasa N.; Nakagawa Y.; Tomishige K. Selective Hydrogenation of Nitroarenes to Aminoarenes Using a MoOX-Modified Ru/SiO2 Catalyst under Mild Conditions. Chem. Commun. 2017, 53, 3377–3380. 10.1039/C7CC00653E. [DOI] [PubMed] [Google Scholar]
- Wei H.; Ren Y.; Wang A.; Liu X.; Liu X.; Zhang L.; Miao S.; Li L.; Liu J.; Wang J.; Wang G.; Su D.; Zhang T. Remarkable Effect of Alkalis on the Chemoselective Hydrogenation of Functionalized Nitroarenes over High-Loading Pt/FeOX Catalysts. Chem. Sci. 2017, 8, 5126–5131. 10.1039/C7SC00568G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Qin R.; Wang Y.; Ren J.; Zhou W.; Li L.; Ming J.; Zhang W.; Fu G.; Zheng N. Chemoselective Hydrogenation of Nitroaromatics at the Nanoscale Iron(III)-OH-Platinum Interface. Angew. Chem., Int. Ed. 2020, 59, 12736–12740. 10.1002/anie.202003651. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Fang L.; Fu J.; Wang J.; Jiang Q.; Li T.; Huang J. Highly Selective IrMo/TiO2 Catalyst for Hydrogenation of Nitroarenes. Mol. Catal. 2022, 531, 112705. 10.1016/j.mcat.2022.112705. [DOI] [Google Scholar]
- Putaj P.; Lefebvre F. Polyoxometalates Containing Late Transition and Noble Metal Atoms. Coord. Chem. Rev. 2011, 255, 1642–1685. 10.1016/j.ccr.2011.01.030. [DOI] [Google Scholar]
- Hayashi S.; Shishido T. High-Density Formation of Metal/Oxide Interfacial Catalytic Active Sites through Hybrid Clustering. ACS Appl. Mater. Interfaces 2021, 13, 22332–22340. 10.1021/acsami.1c02240. [DOI] [PubMed] [Google Scholar]
- Hayashi S.; Endo S.; Miura H.; Shishido T. Highly Active and Durable Rh-Mo-Based Catalyst for the NO-CO-C3H6-O2 Reaction Prepared by Using Hybrid Clustering. ACS Mater. Au 2023, 10.1021/acsmaterialsau.3c00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K.; Yamaguchi M.; Shido T.; Ohtani H.; Isobe K.; Ichikawa M. Molecular Modelling of Supported Metal Catalysts: SiO2-Grafted [{(η3-C4H7)2Rh}2V4O12] and [{Rh(C2Me5)}4V6O19] Are Catalytically Active in the Selective Oxidation of Propene to Acetone. J. Chem. Soc. Chem. Commun. 1995, 12, 1301–1303. 10.1039/C39950001301. [DOI] [Google Scholar]
- Ichikawa M.; Pan W.; Imada Y.; Yamaguchi M.; Isobe K.; Shido T. Surface-Grafted Metal Oxide Clusters and Metal Carbonyl Clusters in Zeolite Micropores; XAFS/FTIR/TPD Characterization and Catalytic Behavior. J. Mol. Catal. A Chem. 1996, 107, 23–38. 10.1016/1381-1169(95)00226-X. [DOI] [Google Scholar]
- Hayashi Y.; Toriumi K.; Isobe K. Novel Triple Cubane-Type Organometallic Oxide Clusters: [MCp*MoO4]4•nH2O (M = Rh and Ir; Cp* = C5Me5; n = 2 for Rh and 0 for Ir). J. Am. Chem. Soc. 1988, 110, 3666–3668. 10.1021/ja00219a056. [DOI] [Google Scholar]
- Hayashi Y.; Ozawa Y.; Isobe K. Site-Selective Oxygen Exchange and Substitution of Organometallic Groups in an Amphiphilic Quadruple-Cubane-Type Cluster. Synthesis and Molecular Structure of [(MCp*)4V6O19] (M = Rh, Ir). Inorg. Chem. 1991, 30, 1025–1033. 10.1021/ic00005a028. [DOI] [Google Scholar]
- Yamamoto T. Assignment of Pre-Edge Peaks in K-Edge x-Ray Absorption Spectra of 3d Transition Metal Compounds: Electric Dipole or Quadrupole?. X-Ray Spectrom. 2008, 37, 572–584. 10.1002/xrs.1103. [DOI] [Google Scholar]
- Shishido T.; Asakura H.; Yamazoe S.; Teramura K.; Tanaka T. Structural Analysis of Group V, VI, VII Metal Compounds by XAFS and DFT Calculation. J. Phys. Conf. Ser. 2009, 190, 012073. 10.1088/1742-6596/190/1/012073. [DOI] [Google Scholar]
- Han B.; Guo Y.; Huang Y.; Xi W.; Xu J.; Luo J.; Qi H.; Ren Y.; Liu X.; Qiao B.; Zhang T. Strong Metal-Support Interactions between Pt Single Atoms and TiO2. Angew. Chem., Int. Ed. 2020, 59, 11824–11829. 10.1002/anie.202003208. [DOI] [PubMed] [Google Scholar]
- Xiong M.; Wang G.; Zhao S.; Lv Z.; Xing S.; Zhang J.; Zhang B.; Qin Y.; Gao Z. Engineering of Platinum-Oxygen Vacancy Interfacial Sites in Confined Catalysts for Enhanced Hydrogenation Selectivity. Catal. Sci. Technol. 2022, 12, 2411–2415. 10.1039/D2CY00131D. [DOI] [Google Scholar]
- Yang X.-F.; Wang A.; Qiao B.; Li J.; Liu J.; Zhang T. Single-Atom Catalysts: A New Frontier in Heterogeneous Catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. 10.1021/ar300361m. [DOI] [PubMed] [Google Scholar]
- Prasomsri T.; Nimmanwudipong T.; Román-Leshkov Y. Effective Hydrodeoxygenation of Biomass-Derived Oxygenates into Unsaturated Hydrocarbons by MoO3 Using Low H2 Pressures. Energy Environ. Sci. 2013, 6, 1732–1738. 10.1039/c3ee24360e. [DOI] [Google Scholar]
- Kuwahara Y.; Yoshimura Y.; Haematsu K.; Yamashita H. Mild Deoxygenation of Sulfoxides over Plasmonic Molybdenum Oxide Hybrid with Dramatic Activity Enhancement under Visible Light. J. Am. Chem. Soc. 2018, 140, 9203–9210. 10.1021/jacs.8b04711. [DOI] [PubMed] [Google Scholar]
- Tamura M.; Tokonami K.; Nakagawa Y.; Tomishige K. Selective Hydrogenation of Crotonaldehyde to Crotyl Alcohol over Metal Oxide Modified Ir Catalysts and Mechanistic Insight. ACS Catal. 2016, 6, 3600–3609. 10.1021/acscatal.6b00400. [DOI] [Google Scholar]
- He S.; Xie L.; Che M.; Chan H. C.; Yang L.; Shi Z.; Tang Y.; Gao Q. Chemoselective Hydrogenation of α,β-Unsaturated Aldehydes on Hydrogenated MoOx Nanorods Supported Iridium Nanoparticles. J. Mol. Catal. A Chem. 2016, 425, 248–254. 10.1016/j.molcata.2016.10.016. [DOI] [Google Scholar]
- Wang L.; Guan E.; Zhang J.; Yang J.; Zhu Y.; Han Y.; Yang M.; Cen C.; Fu G.; Gates B. C.; Xiao F.-S. Single-Site Catalyst Promoters Accelerate Metal-Catalyzed Nitroarene Hydrogenation. Nat. Commun. 2018, 9, 1362. 10.1038/s41467-018-03810-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankudinov A. L.; Ravel B.; Rehr J. J.; Conradson S. D. Real-space multiple-scattering calculation and interpretation of x-ray-absorption near-edge structure. Phys. Rev. B 1998, 58, 7565–7576. 10.1103/PhysRevB.58.7565. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this study are available in the published article and its Supporting Information.