

Abstract

Bacteria are orders of magnitude smaller than mammalian cells, and while single cell proteomics (SCP) currently detects and quantifies several thousands of proteins per mammalian cell, it is not clear whether conventional SCP methods will be suitable for bacteria. Here we report on the first successful attempt to detect proteins from individual Escherichia coli bacteria, with validation of our findings by comparison with two bacteria samples and bulk proteomics data. Data are available via ProteomeXchange with the identifier PXD043473.
Keywords: Escherichia coli, bacterial proteome, single cell proteomics
Introduction
Single Cell ProtEomics by Mass Spectrometry (SCoPE-MS) by Budnik and Slavov1 revolutionized in 2017–2018 the nascent field of single cell proteomics (SCP), opening a venue for innovative approaches in ultrasensitive sample preparation and mass spectrometry analysis. The key innovation in SCoPE MS was the use of the carrier proteome (CP) composed of 100–200 cells and multiplexed with single cell samples using isobaric tandem mass tag (TMT). With SCoPE-MS, it is habitually possible to analyze human cells to the depth of 1500–2000 proteins.2,3 There are dozens of recent reports with similar or greater proteome analysis depths owing to continuous advances in sample preparation,4−8 data acquisition expanding the choice of analytical approaches to label-free technique and with data independent acquisition modes,4,9−12 as well as providing optimal data analysis.13,14
The next challenge in single cell proteomics would be to detect proteins from single bacteria. Bacteria come in a wide range of sizes, from the smallest (Mycoplasma gallicepticum with a size of 0.2–0.3 μm) to the largest (a Gram-negative proteobacterium Thiomargarita namibiensis, up to 750 μm).15 Gram-negative Escherichia coli bacteria that are most commonly used in research are much smaller than human cells, but they are easy to grow and easier to lyse than Gram-positive bacteria. The typical diameter of A549 human cell is 11–15 μm,16 with a volume of ≈1000 μm3, while E. coli cells represent a cylinder of 1.0–2.0 μm long with a radius of about 0.5 μm and a volume of ≈1 μm3.17 Other sources give different values for E. coli size ranges, e.g., 2–6 μm long and 1.1–1.5 μm wide.18 This discrepancy is due to the fact that the size and volume of the bacterial cell depend upon the growth rate. In general, faster dividing cells are larger than slower growing bacteria. The dry protein mass of E. coli cells varies from an average value of 148 fg per cell for bacteria dividing every 100 min to 865 fg per cell for those with a 24 min division time, a >5-fold difference.19
The proteome of E. coli contains about 4200 proteins.20 Some of the most abundant proteins in the E. coli proteome are ribosomal proteins. The number of ribosomes is steeply dependent on the growth rate: as one would expect, the faster the growth rate, the more ribosomes that are present.21 It is believed that, under optimal growing conditions, a single E. coli bacterium may have up to 60,000 ribosomes. Assuming such a value, it should be possible to detect and quantify at least most abundant ribosomal proteins (the L7/12 complex is present in four copies per ribosome) by SCoPE-MS, although soluble enzymes (>1000) are also potential target of such analyses. In order to validate protein detection, we compared single bacterium proteomics (SBP) results with the bulk proteome analysis of bacteria, demonstrating that single cell analysis identified proteins that are very abundant in bulk proteomics data.
Materials and Methods
Culturing Bacterial Cells
Escherichia coli BL21(DE3) strain was grown on plates containing LB agar (Sigma), and a single colony was transferred to Terrific Broth (TB) medium. Bacterial growth was monitored by measuring light diffraction at 600 nm on a BioScreen C automated microbiology growth curve analysis system (Bioscreen, Finland) at 37 °C. After 4 h, the bacteria were washed with PBS twice and incubated in PBS containing 5 mM SYTO 9 green fluorescent nucleic acid stain (Thermo Fisher Scientific, S34854) at room temperature (RT) for 15 min. The remaining SYTO 9 dye was removed by centrifugation, and the bacteria were washed with PBS.
Sample Preparation
For bulk proteome analysis, the harvested and washed cells were lysed with two different methods following supplementation with either 100 μL of 25 mM triethylammonuim bicarbonate (TEAB) for probe sonication method or 50 μL of water for the freeze-and-thaw method and sonicated in water bath for 10 min. The cells either were sonicated twice using an ultrasound probe (Vibracell) for 1 min operated with a pulse of 2–2 s (on–off) and amplitude to 20% after or underwent four freeze-and-thaw cycles, being frozen in liquid N2 and then heated on a block heater at 70 °C for 2 min in each cycle. Supernatants were used for determination of protein concentration by the BCA method. Thereafter 25 μg of cell extract was supplemented with 50 mM TEAB to reach a total volume of 100 μL, and proteins were digested without reduction and alkylation by adding 10 μL of 0.1 μg/μL sequencing grade trypsin (Promega) and incubating at 37 °C for ca. 16 h. The resulting peptides were labeled with 500 μg of TMT-10plex (Thermo Fisher Scientific) in 150 μL of dry acetonitrile (ACN) by incubating at RT for 2 h with gentle shaking. The reaction was quenched by adding 15 μL of 5% hydroxylamine (Sigma). The peptides labeled with different TMT labels were pooled together and dried in a vacuum concentrator (Eppendorf).
For single cell proteomics, bacteria were stained with SYTO 9 to facilitate sorting to a 96-well plate in an BD FACSAria III flow cytometer system (BD Biosciences) either one or two cells into wells holding predispensed 5 μL of 100 mM TEAB. In addition, 250 cells to be used as CP were sorted into some wells. As described previously,3 proteins were extracted by four freeze-and-thaw cycles using liquid nitrogen and incubation at 70 °C for 2 min in each cycle with a 2 min interval at RT between each cycle. Proteins were denatured by heating the plates to 90 °C for 10 min. Then 1 or 2 μL of sequencing grade trypsin (Promega) in 50 mM TEAB was added to single/double cells and CP, respectively, and incubated at 37 °C for 16 h. Labeling with TMT-10plex was performed with dispensing in each well 1 μL of a 10 ng/μL reagent solution using a MANTIS liquid handling robot (Formulatrix) and incubating at RT for 1 h with gentle shaking and subsequent quenching for 20 min at RT with 1 μL of 5% hydrocylamine. Labeled peptides from three single and three double cells were pooled together with two CP digests labeled by two different TMT labels and dried in vacuum prior to analysis. Two TMT channels remained unutilized to determine the background noise level.
RPLC-LC-MS/MS Analysis
LC-MS/MS analysis of the bulk proteome sample was performed on an Ultimate 3000 UPLC coupled to a Q Exactive HF hybrid mass spectrometer (Thermo Fisher Scientific). Peptides were loaded onto a trap column (Acclaim PepMap 100 precolumn, 75 μm diameter, 2 cm long, 3 μm C18 beads, 100 Å pores, Thermo Scientific) and separated on an EASY-Spray analytical column (50 cm long, 75 μm i.d., PepMap RSLC C18 2 μm beads, 100 Å pores, Thermo Scientific) using a 120 min long linear gradient from 4% to 26% solvent B (0.1% FA in 98% ACN and 2% water) at a flow rate of 300 nL/min and a column temperature of 55 °C. Mass spectrometry (MS) acquisition method was set as follows: full MS spectra at m/z 375–1700 with a resolution of 120,000 (at 200 m/z), with an automated gain control (AGC) target value of 1 × 106 and 80 ms injection time (IT), with data dependent acquisition (DDA) selection for fragmentation of 18 most abundant ions. MS/MS was performed with an isolation window of 1.4 Th and fragmentation by higher-energy collision dissociation (HCD) at a normalized collision energy (NCE) of 34%, maximum IT of 54 ms, and AGC target of 2 × 105. Fragment ion detection was in the Orbitrap HD analyzer at a resolution of 60,000 with fixed first mass at 110 m/z and with a dynamic exclusion of 45 s.
LC-MS/MS analysis of single cells was performed with a nanoflow UltiMate 3000 UPLC coupled to an Orbitrap Fusion Eclipse Tribrid mass spectrometer equipped with field asymmetric ion mobility device FAIMS Pro (Thermo Fisher Scientific). The peptides were separated on a 25 cm Easy-Spray PepMap C-18 column (Thermo Fisher Scientific) using a gradient from 1%B to 20%B in 75 min and from 1%B to 36%B in 20 min at a 300 nL/min flow rate. FAIMS operated at a compensation voltage cycling every 1.5 s between −50 and −70 V. The mass spectrometer was used in the DDA mode. The precursor ions were recorded at 120,000 resolution in the m/z 250–1500 range, targeting 6 × 105 ions in maximum 100 ms, while MS/MS precursors were isolated with a AGC target of 1 × 105 in maximum 50 ms with a narrow 0.7 Th window and fragmented by HCD at 35% NCE. The fragment ions were detected at 50,000 resolution with dynamic exclusion of 60 s.
Peptides and proteins were identified by searching MS/MS data against the E. coli strain K12 protein database (SwissProt, 4465 entries) by MS Amanda22 using Proteome Discoverer 2.5 software. The precursor and fragment ion mass tolerances were 10 ppm and 0.05 Da, respectively. Variable modifications were set for deamidation of asparagine and glutamine, oxidation of methionine, and “TMT6plex” (+229.163 Da; same as TMT10plex) of lysine and peptide N-termini. The protein list was filtered with an estimated false discovery rate (FDR) of 5% and validation based on q-values using either the Percolator or Target Decoy PSM validator node in Proteome Discoverer. The protein group abundances were calculated as a sum of the peptide abundances determined in Proteome Discoverer (Reporter Ion Quantifier node) based on the detected levels of TMT reporter ions.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE23 partner repository with the data set identifier PXD043473.
Results and Discussion
Bulk Proteomics
In total, 1132 and 1270 bacterial proteins were detected and quantified in bulk analysis across three replicates prepared with freeze-and-thaw and probe sonication, respectively. As we were interested in the most abundant proteins, there was no need to reach a deeper proteome. Among the detected proteins, there were 34 molecules localized in the small subunit of the ribosome and 43 proteins from the large ribosome subunit; eight additional ribosome related proteins were detected. Overall, ribosomal proteins comprise 12.1% of the total protein abundance.
The comparison of the two preparation methods indicated significant differences in extraction efficiency for some protein groups (Figure 1). Probe sonication processed membrane related proteins more effectively, as about 58% of proteins significantly enriched in this approach fell in this category. However, the freeze-and-thaw method provided a higher yield of more than half of the detected proteins, and importantly, one significantly enriched protein was ribosomal (P0A7P5, 50S ribosomal protein L34). Therefore, we decided that the freeze-and-thaw approach is suitable for single bacterium proteomics (SBP) analysis.
Figure 1.

Comparison of sample preparation methods in bulk proteomics. The freeze-and-thaw method extracted 53% more proteins than the probe sonication method did, including a ribosomal protein (L34). The less effectively extracted proteins included mostly membrane proteins (58%) as depicted in the pie chart.
Single Bacterium Proteomics
The overall SBP workflow is presented in Figure 2. Each TMT-10 multiplexed set contained two TMT-channels with 250 bacterial cells each as CP, as well as three single cell and three double cell channels in alternating order. In total, 16 such TMT sets were analyzed. Altogether, 388,641 MS/MS spectra were collected resulting in over 20,000 peptide-spectrum matches (PSMs), among which many mapped on common contaminant proteins (e.g., keratins) as well as trypsin. The TMT labeling efficiency was determined to be 75%, which was somewhat lower than the average labeling efficiency of 85% in our previous work with single mammalian cells,3 but still sufficient for the purpose of this work.
Figure 2.
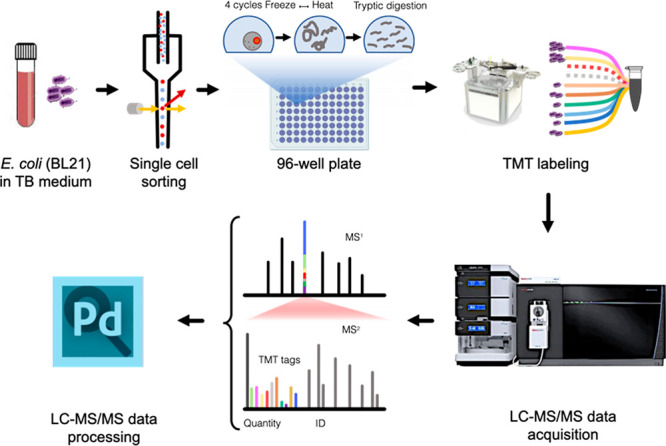
Single bacterium proteomics workflow. The cultured E. coli cells were isolated as single or double cells on 96-well plates by FACS; lysed by freeze-and-thaw, and digested before TMT-10plex labeling. Data acquisition was achieved on an Orbitrap Fusion Eclipse mass spectrometer equipped with FAIMS Pro, collecting MS2 spectra for reporter ion based quantification.
Using Percolator for control of FDR ≤ 0.05, 94 PSMs were annotated in the E. coli database, identifying 29 peptides of 19 bacterial proteins across the 96 single/double cells (Tables 1 and 2). The following three proteins were identified with ≥2 peptides each: elongation factor Tu 2 (P0CE48), glyceraldehyde-3 phosphate dehydrogenase 3 A (P0A9B2), and 2-iminobutanoate/2-iminopropanoate deaminase (P0AF93).
Table 1. Summary of Detected Bacterial Proteins and Peptides Determined by the Percolator Method, Including Channel Occupancy Values Based on the Observation Frequencies in Single, Double, and Carrier Proteome Samplesa.
| protein description | gene name | accession number [position] | annotated sequence | theoretical [MH]+ (Da) | observed m/z (Da) | charge state (z) | retention time (min) | # PSM | protein score (by MS Amanda) | channel occupancy (%) | abundance rank in bulk proteome |
|---|---|---|---|---|---|---|---|---|---|---|---|
| tRNA (guanine-N(1)-)-methyltransferase | trmD | P0A873 [69–75] | [R].DAIHAAK.[A] | 1183.72 | 395.25 | +3 | 31.52 | 3 | 214.27 | 18.75% | |
| glyceraldehyde-3-phosphate dehydrogenase A | gapA | P0A9B2 [218–225] | [K].VLPELNGK.[L] | 1328.82 | 664.92 | +2 | 69.00 | 8 | 221.57 | 50.00% | 1 |
| 2-iminobutanoate/2-iminopropanoate deaminase | ridA | P0AF93 [52–58] | [R].QSLDNVK.[A] | 1033.57 | 517.29 | +2 | 36.19 | 3 | 342.48 | 18.75% | 242 |
| major outer membrane lipoprotein Lpp | lpp | P69776 [53–59] | [R].SDVQAAK.[D] | 1176.70 | 588.85 | +2 | 33.77 | 4 | 190.35 | 25.00% | 466 |
| N-acetylneuraminate lyase | nanA | P0A6L4 [242–248] | [K].VIDLLIK.[T] | 1271.87 | 636.44 | +2 | 86.04 | 4 | 205.75 | 25.00% | |
| 50S ribosomal protein L7/L12 | rplL | P0A7K2 [110–121] | [K].ALEEAGAEVEVK.[-] | 1702.96 | 568.33 | +3 | 63.17 | 16 | 252.42 | 93.75% | 21 |
| elongation factor Tu 2 | tufB | P0CE48 [239–249] | [K].VGEEVEIVGIK.[E] | 1629.98 | 544.00 | +3 | 75.80 | 15 | 203.77 | 75.00% | 11 |
| biotin carboxylase | accC | P24182 [315–324] | [R].IAAGQPLSIK.[Q] | 1456.91 | 728.96 | +2 | 71.29 | 2 | 147.8 | 12.50% | 120 |
| probable cyclic di-GMP phosphodiesterase PdeC | pdeC | P32701 [36–43] | [K].SEVNNQLR.[T] | 1189.64 | 595.33 | +2 | 41.77 | 4 | 177.29 | 25.00% | |
| succinylornithine transaminase | astC | P77581 [353–360] | [K].QISQEAAK.[A] | 1333.77 | 667.39 | +2 | 45.17 | 1 | 189.61 | 6.25% | |
| elongation factor Tu 2 | tufB | P0CE48 [178–188] | [K].ALEGDAEWEAK.[I] | 1676.89 | 559.64 | +3 | 63.80 | 10 | 127.64 | 50.00% | 11 |
| 2-iminobutanoate/2-iminopropanoate deaminase | ridA | P0AF93 [52–58] | [R].QSLDNVK.[A] | 1262.74 | 631.87 | +2 | 44.89 | 1 | 186.3 | 6.25% | 242 |
| ATP-dependent RNA helicase HrpA | hrpA | P43329 [1049–1063] | [R].DSVAIKLFDNPLEQK.[Q] | 2176.23 | 726.08 | +3 | 91.39 | 7 | 104.31 | 25.00% | |
| putative permease PerM | perM | P0AFI9 [179–188] | [K].DKEQMLNAVR.[R] | 1450.74 | 725.88 | +2 | 53.56 | 4 | 172.05 | 18.75% | |
| 4-hydroxybenzoate octaprenyltransferase | ubiA | P0AGK1 [61–79] | [R].AAGCVVNDYADRKFDGHVK.[R] | 2294.15 | 765.39 | +3 | 66.52 | 5 | 194.66 | 12.50% | |
| uncharacterized protein YdaV | ydaV | P77546[180–197] | [K].NEQVVLHQIVDRRTASMR.[S] | 2171.09 | 724.37 | +3 | 81.32 | 11 | 160.15 | 0.00% | |
| outer membrane usher protein HtrE | htrE | P33129[148–159] | [R].LDIDVPQAWVMK.[N] | 1643.90 | 548.64 | +3 | 52.73 | 1 | 136.87 | 0.00% | |
| elongation factor Tu 2 | tufB | P0CE48 [189–205] | [K].ILELAGFLDSYIPEPER.[A] | 2191.18 | 731.07 | +3 | 97.60 | 1 | 120.2 | 0.00% | |
| elongation factor Tu 2 | tufB | P0CE48[125–137] | [R].QVGVPYIIVFLNK.[C] | 1948.20 | 650.07 | +3 | 92.11 | 5 | 118.38 | 0.00% | |
| methylated-DNA-protein-cysteine methyltransferase | ogt | P0AFH0[54–67] | [R].ISATNPGGLSDKLR.[E] | 1428.78 | 714.90 | +2 | 65.42 | 1 | 138.33 | 0.00% | 1081 |
| ATP synthase subunit alpha | atpA | P0ABB0[165–175] | [R].ELIIGDRQTGK.[T] | 1229.68 | 615.35 | +2 | 30.30 | 1 | 228.28 | 0.00% | 33 |
| glyceraldehyde-3-phosphate dehydrogenase A | gapA | P0A9B2[214–225] | [K].AVGKVLPELNGK.[L] | 1224.73 | 408.92 | +3 | 39.82 | 1 | 191.31 | 0.00% | 1 |
| glyceraldehyde-3-phosphate dehydrogenase A | gapA | P0A9B2[226–232] | [K].LTGMAFR.[V] | 795.42 | 398.22 | +2 | 43.10 | 3 | 179.87 | 0.00% | 1 |
| glyceraldehyde-3-phosphate dehydrogenase A | gapA | P0A9B2[5–12] | [K].VGINGFGR.[I] | 819.45 | 410.23 | +2 | 41.50 | 1 | 173.49 | 0.00% | 1 |
| glyceraldehyde-3-phosphate dehydrogenase A | gapA | P0A9B2[218–232] | [K].VLPELNGKLTGMAFR.[V] | 1645.91 | 549.31 | +3 | 68.35 | 2 | 191.49 | 0.00% | 1 |
| glyceraldehyde-3-phosphate dehydrogenase A | gapA | P0A9B2[218–232] | [K].VLPELNGKLTGMAFR.[V] | 1661.90 | 554.64 | +3 | 60.08 | 1 | 395.37 | 0.00% | 1 |
| glyceraldehyde-3-phosphate dehydrogenase A | gapA | P0A9B2[218–232] | [K].VLPELNGKLTGMAFR.[V] | 1662.89 | 554.97 | +3 | 63.85 | 1 | 237.95 | 0.00% | 1 |
| transaldolase A | talA | P0A867[92–99] | [R].VSTEVDAR.[L] | 876.44 | 438.73 | +2 | 19.09 | 2 | 246.62 | 0.00% | 976 |
| chaperone protein DnaK | dnaK | P0A6Y8[167–183] | [K].RIINEPTAAALAYGLDK.[G] | 1816.00 | 606.01 | +3 | 64.08 | 1 | 285.59 | 0.00% | 15 |
Italic = observation only without quantification.
Table 2. Summary of Detected Bacterial Proteins and Peptides Determined by the Targeted Decoy Method, Including Channel Occupancy Values Based on the Observation Frequencies in Single, Double, and Carrier Proteome Samplesa.
| protein description | gene name | accession number [position] | annotated sequence | theoretical [MH]+ (Da) | observed m/z (Da) | charge state (z) | retention time (min) | # PSM | protein score (by MS Amanda) | channel occupancy (%) | abundance rank in bulk proteome |
|---|---|---|---|---|---|---|---|---|---|---|---|
| protein CbrA | cbrA | P31456 [22–33] | [K].LAGKMQVIALDK.[K] | 1761.07 | 587.69 | +3 | 89.87 | 4 | 228.5 | 25.00% | |
| HTH-type transcriptional regulator GadX | gadX | P37639 [243–250] | [R].SAQRLSNR.[D] | 1161.65 | 581.33 | +2 | 30.74 | 2 | 191.41 | 12.50% | |
| ribomal silencing factor RsfS | rsfS | P0AAT6 [1–13] | [-].MQGKALQDFVIDK.[I] | 1952.09 | 651.36 | +3 | 45.44 | 1 | 185.02 | 6.25% | 573 |
| uncharacterized protein YbcK | ybcK | P77698 [194–202] | [R].VKTIELIFK.[L] | 1319.85 | 660.42 | +2 | 75.20 | 13 | 212.92 | 81.25% | |
| prophage integrase IntZ | intZ | P76542 [174–180] | [R].RIGEIFK.[F] | 1320.84 | 660.92 | +2 | 76.86 | 6 | 185.15 | 37.50% | |
| lipoprotein NlpI | nlpI | P0AFB1 [7–26] | [R].WCFVATALTLAGCSNTSWRK.[S] | 2673.40 | 891.81 | +3 | 78.40 | 1 | 180.24 | 6.25% | |
| HTH-type transcriptional regulator AscG | ascG | P24242 [306–324] | [R].LIFMLDGGDFSPPKTFSGK.[L] | 2744.53 | 915.51 | +3 | 85.46 | 1 | 219.47 | 6.25% | |
| 50S ribomal protein L31 type B | ykgM | P0A7N1 [11–30] | [R].TVVFHDTSVDEYFKIGSTIK.[T] | 2744.49 | 686.88 | +4 | 85.49 | 1 | 194.53 | 6.25% | |
| tRNA (guanine-N(1)-)-methyltransferase | trmD | P0A873 [69–75] | [R].DAIHAAK.[A] | 1183.72 | 395.25 | +3 | 31.52 | 2 | 214.27 | 12.50% | |
| ATP-dependent RNA helicase HrpA | hrpA | P43329 [988–993] | [R].SLQDLK.[D] | 1162.71 | 581.86 | +2 | 59.70 | 2 | 221.69 | 12.50% | |
| Transcriptional repressor PifC | pifC | P10030 [2–8] | [M].LSQLNLR.[F] | 1072.67 | 536.84 | +2 | 54.13 | 1 | 195.86 | 6.25% | |
| histidinol-phphate aminotransferase | hisC | P06986 [170–178] | [R].TLLELTRGK.[A] | 1488.95 | 744.97 | +2 | 82.06 | 1 | 181.64 | 31.25% | 945 |
| Probable fructelysine utilization operon transcriptional repressor | frlR | P45544 [179–197] | [R].VVSDKKTIDIFAATRPQAK.[W] | 2318.33 | 773.44 | +3 | 86.67 | 5 | 218.51 | 6.25% | |
| DNA polymerase III subunit delta | holA | P28630 [293–299] | [R].LSQTQLR.[Q] | 1075.63 | 538.32 | +2 | 46.97 | 1 | 186.37 | 6.25% | |
| glutamine-binding periplasmic protein | gln H | P0AEQ3 [222–227] | [K].VNGALK.[T] | 831.51 | 416.26 | +2 | 28.16 | 3 | 206.69 | 18.75% | 135 |
| Kojibie phphorylase | ycjT | P77154 [555–573] | [K].QTILLDYSRAEVNEMQILK.[Q] | 2739.50 | 685.63 | +4 | 82.89 | 1 | 215.42 | 6.25% | |
| glyceraldehyde-3-phphate dehydrogenase A | gapA | P0A9B2 [218–225] | [K].VLPELNGK.[L] | 1328.82 | 664.92 | +2 | 69.48 | 3 | 252.73 | 18.75% | 1 |
| uncharacterized protein YgbA | ygbA | P25728 [45–58] | [R].LDKCVFGEEKPACK.[Q] | 1795.93 | 599.31 | +3 | 66.34 | 2 | 195.97 | 12.50% | |
| 50S ribomal protein L7/L12 | rplL | P0A7K2 [110–121] | [K].ALEEAGAEVEVK.[-] | 1702.96 | 568.33 | +3 | 63.17 | 9 | 252.42 | 56.25% | 21 |
| translation initiation factor IF-2 | infB | P0A705 [1–7] | [-].MTDVTIK.[T] | 1036.59 | 518.80 | +2 | 71.62 | 2 | 201.38 | 12.50% | 48 |
| putative hydrolase YuaR | yuaR | P34211 [159–164] | [K].QLILQK.[I] | 1200.81 | 600.91 | +2 | 66.25 | 1 | 185.42 | 6.25% | |
| nickel import ATP-binding protein NikE | nikE | P33594 [125–136] | [K].SEQLARASEMLK.[A] | 1591.87 | 531.29 | +3 | 71.02 | 1 | 195.59 | 6.25% | |
| antiadapter protein IraM | iraM | P75987 [92–100] | [K].CLHNSCIIK.[I] | 1259.68 | 630.34 | +2 | 84.12 | 1 | 191.4 | 6.25% | |
| major outer membrane lipoprotein Lpp | lpp | P69776 [53–59] | [R].SDVQAAK.[D] | 1176.70 | 588.85 | +2 | 33.77 | 1 | 190.35 | 6.25% | 466 |
| RecBCD enzyme subunit RecC | recC | P07648 [688–706] | [R].QLAPLGFDLMSQKPKRGDR.[S] | 2403.30 | 801.78 | +3 | 85.54 | 1 | 180.46 | 6.25% | |
| Tol-Pal system protein TolA | tolA | P19934 [118–125] | [K].KQAEEAAK.[Q] | 1333.77 | 667.39 | +2 | 45.17 | 1 | 189.61 | 6.25% | 1001 |
| N-acetylneuraminate lyase | nanA | P0A6L4 [242–248] | [K].VIDLLIK.[T] | 1271.87 | 636.44 | +2 | 86.04 | 1 | 205.75 | 6.25% | |
| Tat proofreading chaperone DmsD | dmsD | P69853 [95–113] | [R].ESVLFGDSTLALRQWMREK.[G] | 2725.47 | 909.17 | +3 | 77.51 | 1 | 183.75 | 6.25% | 960 |
| Inner membrane protein YiaV | yiaV | P37683 [163–168] | [R].DIDVAR.[Q] | 917.53 | 459.27 | +2 | 41.97 | 1 | 206.14 | 6.25% | |
| xyle transport system permease protein XylH | xylH | P0AGI4 [268–279] | [R].IYAIGGNLEAAR.[L] | 1477.82 | 739.41 | +2 | 61.15 | 1 | 190.99 | 6.25% | |
| uncharacterized protein YmfJ | ymfJ | P75973 [2–7] | [M].NEQNLK.[H] | 974.55 | 487.77 | +2 | 55.13 | 1 | 184.83 | 6.25% | |
| putative DNA repair helicase RadD | radD | P33919 [253–266] | [R].KGVMIFAATVEHAK.[E] | 1960.15 | 654.05 | +3 | 91.65 | 1 | 193.55 | 6.25% | |
| DNA base-flipping protein | atl | P0AFP2 [67–73] | [R].QVGGVLK.[R] | 930.58 | 465.80 | +2 | 51.98 | 1 | 189.08 | 6.25% | |
| elongation factor Tu 2 | tufB | P0CE48 [239–249] | [K].VGEEVEIVGIK.[E] | 1629.98 | 544.00 | +3 | 74.62 | 2 | 208.23 | 12.50% | 11 |
| HTH-type transcriptional regulator McbR | mcbR | P76114 [81–87] | [R].QLDEINR.[I] | 1117.61 | 559.31 | +2 | 49.29 | 1 | 197.73 | 6.25% | |
| probable bifunctional chitinase/lysozyme | chiA | P13656 [851–862] | [R].NLGMVGIWSIAR.[D] | 1562.86 | 521.62 | +3 | 63.63 | 1 | 181.44 | 6.25% | |
| chaperone protein DnaK | dnaK | P0A6Y8 [415–421] | [K].NTTIPTK.[H] | 1003.60 | 502.30 | +2 | 35.14 | 1 | 198.32 | 6.25% | 15 |
| anaerobic sulfatase-maturating enzyme homologue YdeM | ydeM | P76134 [294–308] | [K].SELKTMNSVQLTAQK.[K] | 1909.01 | 637.00 | +3 | 76.73 | 1 | 190.14 | 6.25% | |
| regulator of sigma-E protease RseP | rseP | P0AEH1 [320–339] | [R].QYGPFNAIVEATDKTWQLMK.[L] | 2586.31 | 862.78 | +3 | 83.12 | 1 | 187.65 | 6.25% | |
| 4-hydroxybenzoate octaprenyltransferase | ubiA | P0AGK1 [61–79] | [R].AAGCVVNDYADRKFDGHVK.[R] | 2294.15 | 765.39 | +3 | 66.52 | 1 | 194.66 | 6.25% | |
| ascorbate-specific PTS system EIIA component | ulaC | P69820[3–11] | [K].LRDSLAENK.[S] | 1045.56 | 523.29 | +2 | 50.41 | 1 | 215.55 | 0.00% | |
| aspartate aminotransferase | aspC | P00509[333–343] | [K].GANRDFSFIIK.[Q] | 1267.68 | 634.34 | +2 | 69.17 | 1 | 222.59 | 0.00% | 29 |
| ATP synthase subunit alpha | atpA | P0ABB0[165–175] | [R].ELIIGDRQTGK.[T] | 1229.68 | 615.35 | +2 | 30.30 | 1 | 228.28 | 0.00% | 33 |
| biotin carboxylase | accC | P24182[315–324] | [R].IAAGQPLSIK.[Q] | 998.59 | 499.80 | +2 | 49.58 | 1 | 181.48 | 0.00% | 120 |
| chaperone protein DnaK | dnaK | P0A6Y8[167–183] | [K].RIINEPTAAALAYGLDK.[G] | 1816.00 | 606.01 | +3 | 64.08 | 1 | 285.59 | 0.00% | 15 |
| chromomal replication initiator protein DnaA | dnaA | P03004[310–316] | [K].ADENDIR.[L] | 1061.54 | 531.27 | +2 | 16.65 | 1 | 194.01 | 0.00% | |
| DNA polymerase III subunit delta | holA | P28630[11–18] | [R].AQLNEGLR.[A] | 901.47 | 451.24 | +2 | 30.19 | 1 | 190.46 | 0.00% | |
| enolase | eno | P0A6P9[406–411] | [K].YNQLIR.[I] | 806.45 | 403.73 | +2 | 30.76 | 1 | 186.21 | 0.00% | 7 |
| excinuclease cho | cho | P76213[101–111] | [K].EQQPLFNKRLR.[R] | 1429.79 | 715.40 | +2 | 58.57 | 1 | 194.27 | 0.00% | |
| glyceraldehyde-3-phphate dehydrogenase A | gapA | P0A9B2[214–225] | [K].AVGKVLPELNGK.[L] | 1224.73 | 408.92 | +3 | 39.82 | 1 | 191.31 | 0.00% | 1 |
| glyceraldehyde-3-phphate dehydrogenase A | gapA | P0A9B2[226–232] | [K].LTGMAFR.[V] | 795.42 | 398.22 | +2 | 43.10 | 1 | 179.87 | 0.00% | 1 |
| glyceraldehyde-3-phphate dehydrogenase A | gapA | P0A9B2[218–232] | [K].VLPELNGKLTGMAFR.[V] | 1645.91 | 549.31 | +3 | 68.35 | 1 | 191.49 | 0.00% | 1 |
| glyceraldehyde-3-phphate dehydrogenase A | gapA | P0A9B2[218–232] | [K].VLPELNGKLTGMAFR.[V] | 1661.90 | 554.64 | +3 | 60.08 | 1 | 395.37 | 0.00% | 1 |
| glyceraldehyde-3-phphate dehydrogenase A | gapA | P0A9B2[218–232] | [K].VLPELNGKLTGMAFR.[V] | 1662.89 | 554.97 | +3 | 63.85 | 1 | 237.95 | 0.00% | 1 |
| high-affinity zinc uptake system protein ZnuA | znuA | P39172[275–286] | [R].MGTLDPLGTNIK.[L] | 1259.67 | 630.34 | +2 | 84.48 | 1 | 208.47 | 0.00% | 591 |
| isoleucine-tRNA ligase | ileS | P00956[432–439] | [R].AQSLKEIK.[G] | 917.53 | 459.27 | +2 | 85.49 | 4 | 191.84 | 0.00% | 72 |
| low-affinity inorganic phphate transporter PitA | pitA | P0AFJ7[332–337] | [K].LSLDQR.[S] | 732.39 | 366.70 | +2 | 29.73 | 1 | 183.61 | 0.00% | |
| nitric oxide reductase FlRd-NAD(+) reductase | norW | P37596[365–373] | [R].MKEAFGLLK.[T] | 1036.59 | 518.80 | +2 | 71.51 | 3 | 215.1 | 0.00% | |
| probable acyltransferase YihG | yihG | P32129[119–128] | [K].HIPMNKYFLK.[Q] | 1291.69 | 646.35 | +2 | 75.18 | 1 | 183.54 | 0.00% | |
| protein YehF | yehF | P33345[31–40] | [K].VGTKGQSQIK.[S] | 1045.60 | 523.30 | +2 | 84.28 | 1 | 203.12 | 0.00% | |
| protein YqfH | yqfH | P0DPP4[2–9] | [M].INQVSVYR.[Q] | 980.50 | 490.76 | +2 | 52.30 | 1 | 187.12 | 0.00% | |
| thiamine kinase | thiK | P75948[5–11] | [R].SNNPITR.[D] | 803.39 | 402.19 | +2 | 19.64 | 1 | 184.47 | 0.00% | |
| Tol-Pal system protein TolA | tolA | P19934[103–108] | [R].LKQLEK.[E] | 758.48 | 379.74 | +2 | 17.05 | 1 | 184.2 | 0.00% | 1001 |
| transaldolase A | talA | P0A867[92–99] | [R].VSTEVDAR.[L] | 876.44 | 438.73 | +2 | 19.09 | 2 | 246.62 | 0.00% | 976 |
| uncharacterized protein YdaV | ydaV | P77546[180–197] | [K].NEQVVLHQIVDRRTASMR.[S] | 2170.10 | 724.04 | +3 | 82.18 | 2 | 190.12 | 0.00% | |
| uncharacterized protein YdaV | ydaV | P77546[180–197] | [K].NEQVVLHQIVDRRTASMR.[S] | 2171.09 | 724.37 | +3 | 82.16 | 2 | 186.46 | 0.00% | |
| uncharacterized protein YdbH | ydbH | P52645[370–384] | [R].SKGRVIDSLDIDEIR.[W] | 1715.93 | 572.65 | +3 | 86.03 | 1 | 186.08 | 0.00% | |
| uncharacterized protein YegR | yegR | P76406[81–87] | [K].MQIVELK.[T] | 860.49 | 430.75 | +2 | 59.24 | 1 | 184.26 | 0.00% | |
| uncharacterized protein YicN | yicN | P0ADL3[27–34] | [K].AIRRLSDR.[L] | 986.59 | 493.80 | +2 | 52.33 | 1 | 275.88 | 0.00% |
Italic = observation only without quantification.
An alternative FDR ≤ 0.05 control was performed with the Target Decoy PSM Validator node in Proteome Discoverer. This approach resulted in 79 PSMs yielding 69 peptides and 60 proteins, with most proteins (16 and 55, respectively) identified by one peptide. Out of these, 12 proteins were the same. Out of the five proteins discovered with ≥2 peptides (Tables 1 and 2), glyceraldehyde-3 phosphate dehydrogenase 3 A with ≥2 peptides was the same in both approaches to FDR control.
An additional FDR control was the peptide abundance rank among all peptides quantified in the bulk analysis. The assumption was made that any peptide detected in single and double cells should be among the top 25% of most abundant peptides in bulk analysis. Yet another FDR control was implemented in the form of the 25% threshold for channel occupancy, which is the percentage of TMT channels among the 96 single and double cells for which nonzero abundance was detected.
With a ≤ 25% rank cutoff for the peptide abundance in bulk analysis and a ≥ 25% channel occupancy, 32 peptides belonging to 12 proteins were detected that passed the FDR ≤ 0.05 threshold in either of the above approaches (Tables 1 and 2). Among these proteins, one ribosomal protein (50S ribosomal protein L7/L12, P0A7K2) was readily detected by both methods as well as translation initiation factor (P0A705), that is a key component of protein synthesis, indirectly related to the ribosomal complex.
The distribution of the relative abundances of the TMT reporter ion for the above peptides in the single- and double cell channels is shown in Figure 3. The mean normalized abundances of eight quantified proteins (glyceraldehyde-3-phosphate dehydrogenase A, 2-iminobutanoate/2-iminopropanoate deaminase, 50S ribosomal protein L7/L12, elongation factor Tu 2, biotin carboxylase detected by the Percolator method and glutamine-binding periplasmic protein, translation initiation factor IF-2, chaperone protein DnaK detected by the Targeted Decoy method) were used together to compare the levels of these proteins in samples with single and double cells. For comparison, the data for the channels corresponding to CP with 250 cells as well as for empty channels are also provided. The average value in the latter was statistically indistinguishable from zero, which indicates the absence of carry-over. The average abundance in the double bacteria channels was (60 ± 10)% higher than that in the single bacterium channels, largely consistent with our expectations. As expected, the average abundance in the CP channels was much higher, even though the difference by a factor of 7 with the single bacterium channels was much below the theoretical value of 250. This discrepancy can be attributed to the compression effect in isobaric labeling,24 as some unlabeled peptides with zero abundances in all TMT reporter channels were also present in the analyzed samples.
Figure 3.
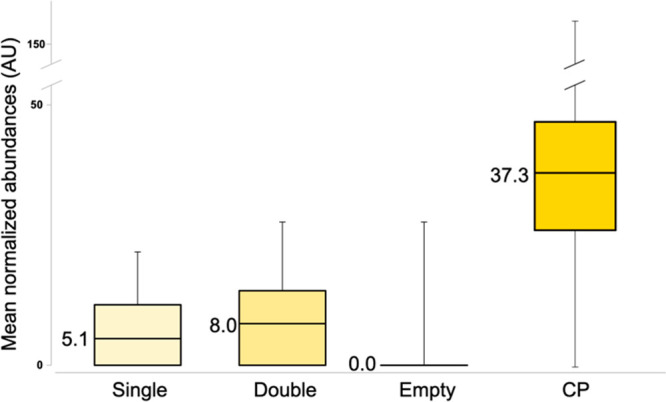
Distributions of the mean abundances calculated with eight bacterial proteins quantified across the data set. The median values are provided for easier comparison.
Conclusions
Summarizing, we have shown that a slightly modified SCoPE MS approach is powerful enough to detect at least a dozen proteins from single bacteria and differentiate quantitatively between one and two bacteria. To the best of our knowledge, this is the first report on MS-based proteomics of single bacterium cells.
Acknowledgments
We would like to thank Marie Ståhlberg for excellent technical assistance.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Special Issue
Published as part of the Journal of the American Society for Mass Spectrometryvirtual special issue “Focus: Asilomar Conference: Single Cell Mass Spectrometry”.
References
- Budnik B.; Levy E.; Harmange G.; Slavov N. SCoPE-MS: Mass Spectrometry of Single Mammalian Cells Quantifies Proteome Heterogeneity during Cell Differentiation. Genome Biol. 2018, 19 (1), 161. 10.1186/s13059-018-1547-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosius V.; Schoof E. M. Recent Advances in the Field of Single-Cell Proteomics. Transl. Oncol. 2023, 27, 101556 10.1016/j.tranon.2022.101556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Végvári Á.; Rodriguez J. E.; Zubarev R. A. Single-Cell Chemical Proteomics (SCCP) Interrogates the Timing and Heterogeneity of Cancer Cell Commitment to Death. Anal. Chem. 2022, 94 (26), 9261–9269. 10.1021/acs.analchem.2c00413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzinger M.; Müller E.; Dürnberger G.; Pichler P.; Mechtler K. Robust and Easy-to-Use One-Pot Workflow for Label-Free Single-Cell Proteomics. Anal. Chem. 2023, 95 (9), 4435–4445. 10.1021/acs.analchem.2c05022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.; Truong T.; Saxton A. J.; Boekweg H.; Payne S. H.; Van Ry P. M.; Kelly R. T. HyperSCP: Combining Isotopic and Isobaric Labeling for Higher Throughput Single-Cell Proteomics. Anal. Chem. 2023, 95 (20), 8020–8027. 10.1021/acs.analchem.3c00906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc A.; Huffman R.; Cantlon J.; Khan S.; Slavov N. Exploring functional protein covariation across single cells using nPOP. Genome Biol. 2022, 23 (1), 261. 10.1186/s13059-022-02817-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshghi A.; Xie X.; Hardie D.; Chen M. X.; Izaguirre F.; Newman R.; Zhu Y.; Kelly R. T.; Goodlett D. R. Sample Preparation Methods for Targeted Single-Cell Proteomics. J. Proteome Res. 2023, 22 (6), 1589–1602. 10.1021/acs.jproteome.2c00429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston S. M.; Webber K. G. I.; Xie X.; Truong T.; Nydegger A.; Lin H.-J. L.; Nwosu A.; Zhu Y.; Kelly R. T. Rapid, One-Step Sample Processing for Label-Free Single-Cell Proteomics. J. Am. Soc. Mass Spectrom. 2023, 34 (8), 1701–1707. 10.1021/jasms.3c00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsburn B. C.; Yuan Y.; Bumpus N. N. Insights into Protein Post-Translational Modification Landscapes of Individual Human Cells by Trapped Ion Mobility Time-of-Flight Mass Spectrometry. Nat. Commun. 2022, 13 (1), 7246. 10.1038/s41467-022-34919-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derks J.; Leduc A.; Wallmann G.; Huffman R. G.; Willetts M.; Khan S.; Specht H.; Ralser M.; Demichev V.; Slavov N. Increasing the Throughput of Sensitive Proteomics by PlexDIA. Nat. Biotechnol. 2023, 41 (1), 50–59. 10.1038/s41587-022-01389-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner A.-D.; Thielert M.; Vasilopoulou C.; Ammar C.; Coscia F.; Mund A.; Hoerning O. B.; Bache N.; Apalategui A.; Lubeck M.; Richter S.; Fischer D. S.; Raether O.; Park M. A.; Meier F.; Theis F. J.; Mann M. Ultra-High Sensitivity Mass Spectrometry Quantifies Single-Cell Proteome Changes upon Perturbation. Mol. Syst. Biol. 2022, 18 (3), e10798 10.15252/msb.202110798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matzinger M.; Mayer R. L.; Mechtler K. Label-Free Single Cell Proteomics Utilizing Ultrafast LC and MS Instrumentation: A Valuable Complementary Technique to Multiplexing. PROTEOMICS 2023, 23 (13–14), 2200162 10.1002/pmic.202200162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderaa C.; Gatto L. The Current State of Single-Cell Proteomics Data Analysis. Curr. Protoc. 2023, 3 (1), e658 10.1002/cpz1.658. [DOI] [PubMed] [Google Scholar]
- Wang B.; Wang Y.; Chen Y.; Gao M.; Ren J.; Guo Y.; Situ C.; Qi Y.; Zhu H.; Li Y.; Guo X. DeepSCP: Utilizing Deep Learning to Boost Single-Cell Proteome Coverage. Brief. Bioinform. 2022, 23 (4), bbac214 10.1093/bib/bbac214. [DOI] [PubMed] [Google Scholar]
- Schulz H. N.; Jorgensen B. B. Big Bacteria. Annu. Rev. Microbiol. 2001, 55, 105–137. 10.1146/annurev.micro.55.1.105. [DOI] [PubMed] [Google Scholar]
- Jiang R.; Shen H.; Piao Y.-J. The Morphometrical Analysis on the Ultrastructure of A549 Cells. Romanian J. Morphol. Embryol. 2010, 51 (4), 663–667. [PubMed] [Google Scholar]
- Phillips R. B.; Kondev J.; Theriot J.. Physical Biology of the Cell; Garland Science, 2009. [Google Scholar]
- Aryal S.Different Size, Shape and Arrangement of Bacterial Cells. Microbiology Info.com. https://microbiologyinfo.com/different-size-shape-and-arrangement-of-bacterial-cells/ (accessed 2023-05-20).
- Lenski R. E.; Travisano M. Dynamics of Adaptation and Diversification: A 10,000-Generation Experiment with Bacterial Populations. Proc. Natl. Acad. Sci. U. S. A. 1994, 91 (15), 6808–6814. 10.1073/pnas.91.15.6808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midha M. K.; Kusebauch U.; Shteynberg D.; Kapil C.; Bader S. L.; Reddy P. J.; Campbell D. S.; Baliga N. S.; Moritz R. L. A Comprehensive Spectral Assay Library to Quantify the Escherichia Coli Proteome by DIA/SWATH-MS. Sci. Data 2020, 7 (1), 389. 10.1038/s41597-020-00724-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley M.Correlates of Smallest Sizes for Microorganisms. In Size Limits of Very Small Microorganisms: Proceedings of a Workshop; National Academies Press, 1999. [PubMed] [Google Scholar]
- Dorfer V.; Pichler P.; Stranzl T.; Stadlmann J.; Taus T.; Winkler S.; Mechtler K. MS Amanda, a Universal Identification Algorithm Optimized for High Accuracy Tandem Mass Spectra. J. Proteome Res. 2014, 13 (8), 3679–3684. 10.1021/pr500202e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Riverol Y.; Bai J.; Bandla C.; García-Seisdedos D.; Hewapathirana S.; Kamatchinathan S.; Kundu D. J.; Prakash A.; Frericks-Zipper A.; Eisenacher M.; Walzer M.; Wang S.; Brazma A.; Vizcaíno J. A. The PRIDE Database Resources in 2022: A Hub for Mass Spectrometry-Based Proteomics Evidences. Nucleic Acids Res. 2022, 50 (D1), D543–D552. 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ow S. Y.; Salim M.; Noirel J.; Evans C.; Rehman I.; Wright P. C. ITRAQ Underestimation in Simple and Complex Mixtures: “The Good, the Bad and the Ugly. J. Proteome Res. 2009, 8 (11), 5347–5355. 10.1021/pr900634c. [DOI] [PubMed] [Google Scholar]