

Abstract
Wolbachia are endosymbiotic and alphaproteobacteria that belong to the order Rickettsiales. They are known to infect half of the insect population and cause host manipulation, and have been categorized into 19 monophyletic lineages called supergroups. Recently, two strains, wCfeJ and wCfeT were isolated from cat fleas (Ctenocephalides felis), but their supergroup relationships were not assigned. In this article, we have attempted to classify these two novel strains and establish their evolutionary lineage (i.e., supergroup designation). For this we performed 16S rRNA similarity analysis and reconstructed 16S rRNA phylogeny of 52 Wolbachia strains (including two novel strains) belong to 19 supergroups. We also performed average nucleotide identity (ANI) and digital DNA-DNA hybridization (dDDH) studies to measure genomic similarity between the two novel genomes. The results revealed that 16S rRNA similarity between the two novel strains is 97.94%, which is below the threshold value of 98.6% and phylogeny shows that they are placed at the two different positions (i.e., showing distinct evolutionary lineages). Further, genomic similarity analysis revealed that the novel genomes have ANI and dDDH values 79% and 22.4% respectively, which were below the threshold value of ANI (95%) and dDDH (70%). These results suggested that the novel strains neither shared a species boundary between them nor with any other previously identified supergroups, which designate them as two new supergroups, namely supergroup V (strain wCfeJ) and supergroup W (strain wCfeT).
Keywords: Wolbachia, Supergroups, 16S rRNA, Phylogeny, Average nucleotide identity (ANI), digital DNA-DNA hybridization (dDDH)
Background:
Wolbachia are alpha-proteobacteria that follows an endosymbiotic life and infect a wide range of arthropods and nematodes [1,2]. These bacteria are gram-negative, obligate and intracellular, and belong to the order Rickettsiales [3]. The genomes of Wolbachia have been analyzed to determine the type and nature of symbiotic relationshipsith their host [4, 5,6,7, 8]. Their nature of relationships in the hosts is reproductive parasite in arthropods, nutritional mutualists in bed bugs, and obligates mutualism in filarial nematodes [9, 10]. Wolbachia mediated all the reproductive manipulation in the host (i.e., mostly arthropods and some nematodes), by means of parthenogenesis, feminization, male-killing, by inducing cytoplasmic incompatibility, and nutritional supplement [11,12,13, 14,15]. It was estimated that Wolbachia infection is up to 40-76% of insects [16,17, 18]. Wolbachia have been classified into distinct monophyletic lineages called supergroups, which first came into the appearance in 1998 [19]. Later, Lo et al popularized this concept [20]. Till now, Wolbachia have been divided into 19 supergroups namely A-F, H-Q, S-U [21,22,, 24]. The 16S rRNA gene is widely used in species determination and identification of new species or strains [25]. Comparison of 16S rRNA gene sequences allows differentiation/delineation of organisms at the genus, species and subspecies level. In the Wolbachia classification, the 16S rRNA gene played an important role in identifying and characterizing the new strains. In this paper, we focused on two recently published and undescribed Wolbachia genomes wCfeT and wCfeJ isolated from cat fleas (Ctenocephalides felis) found in co-infecting mechanism with the same host with different lifestyle such as strain wCfeJ is parasitic and wCfeT is mutualistic [26]. We used 16S rRNA genes to find out the evolutionary lineages of the novel strains in the Wolbachia diversity that currently have 19 supergroups. A total of 50 Wolbachia strains from the existing 19 supergroups and two novel strains were used in this study. Further, we did genomic similarity study on the genomes of the two novel strains using average nucleotide identity (ANI) and digital DNA-DNA hybridization (dDDH) test.
Materials and methods:
Data collection:
For finding the supergroup relationships of the two novel Wolbachia genomes wCfeT and wCfeJ, first, we took 16S rRNA sequences for phylogenetic analysis because it is highly conserved gene and able to show species delineation. 16S rRNA phylogeny is used to check whether two strains wCfeJ and wCfeT cluster with each other or with any other supergroup(s). For this, we used two novel strains plus other 50 strains from 19 supergroups which consist of a total of 52 Wolbachia strains in the study. All the 16S rRNA genes were downloaded from the NCBI database [27]. Details of the 52 Wolbachia strains are given in Table 1.
Table 1. Details of 52 Wolbachia strains used in this study.
| Host Species | Strain | Supergroups | Accession No. |
| Ctenocephalides felis | wCfeT | Novel strain | *NZ_CP051156.1 (HF197_RS05045) |
| Ctenocephalides felis | wCfeJ | Novel strain | *NZ_CP051157.1 (HF196_RS00025) |
| Drosophila melanogaster | wMel | A | *NC_002978.6 (GQX67_RS05935) |
| Nomada panzeri | wNpa | A | *NZ_LYUX01000081.1 (BA050_RS05445) |
| Nomada flava | wNfla | A | *NZ_LYUW01000079.1 (BA052_RS05590) |
| Drosophila simulans | wHa | A | *NC_021089.1 (WHA_RS05510) |
| Drosophila incompta | wInc | A | *CP011148.1 (WG67_RS05300) |
| Carposina sasakii | wCauA | A | *CP041215.1 (wCauA_RS01020) |
| Wolbachia sp. (wRi) | wRi | A | *(NC_012416.1 (WRI_RS06005) |
| Drosophila simulans | wNo | B | *NC_021084.1 (WNO_RS03275) |
| Culex molestus | wPip Mol | B | *NZ_CTEH01000001.1 (WPM_RS00430) |
| Nasonia vitripennis | wVitB | B | *NZ_GL883637.1 (WVB_RS0105690) |
| Aedes albopictus | wAlbB | B | *NZ_RWIK01000001.1 (EJE47_RS00350) |
| Chrysomya megacephala | wMeg | B | *CP021120.1 (CAI20_RS02875) |
| Drosophila mauritiana | wMau | B | *CP034335.1 (EJB00_RS01065) |
| Bemisia tabaci | wBtab | B | *CP016430.1 (BBB02_RS03415) |
| Onchocerca ochengi | wOo | C | AJ010276.1 |
| Onchocerca volvulus | wOvol | C | AF069069.1 |
| Dirofilaria immitis | wDimm | C | KU255236.1 |
| Brugia malayi | wBm | D | *NC_006833.1 (WBM_RS02885) |
| Wuchereria bancrofti | wWb | D | *NZ_NJBR02000071.1 (CCY16_RS03430) |
| Brugia pahangi | wBpah | D | *NZ_CP050521.1 (WBP_RS01130) |
| Litomosoides sigmodontis | wLsig | D | *CP046577 (GOY07_RS01740) |
| Folsomia candida | wFol | E | *NZ_CP015510.2 (ASM33_RS07175) |
| Hypochthonius rufulus | wHruf | E | MN699328.1 |
| Coptotermes acinaciformis | wCaci | F | DQ837197.1 |
| Nasutitermes nigriceps | wNnig | F | DQ837204.1 |
| Cimex lectularius | wCle | F | *NZ_AP013028.1 (WCLE_RS01905) |
| Madathamugadia hiepei | wMhie | F | *NZ_WQMP01000029.1 (GO685_RS01815) |
| Zootermopsi angusticollis | wZangu | H | AY764279.1 |
| Zootermopsi nevadensis | wZnev | H | AY764280.1 |
| Orchopeas leucopus | wOleu | I | AY335924.1 |
| Ctenocephalides felis | wCfe | I | AY157512.1 |
| Myodopsylla gentilis | wMgen | I | AY335918.1 |
| Cruorifilaria tuberocauda | wCtub | J | *CP046579 (GOY13_RS01470) |
| Dipetalonema caudispina | wDcau | J | *CP046580 (GOY14_RS02280) |
| Bryobia sps | wBry | K | EU499316.1 |
| Radopholus similis | wRsim | L | EU833482.1 |
| Pratylenchus penetrans | wPpe | L | *NZ_MJMG01000007.1 (BIY23_RS03360) |
| Cinara cedri | wCed | M | JN384079.1 |
| Macrosiphum euphorbiae | wMeu | M | JN109113.1 |
| Pentalonia nigronervosa | wPni | M | KJ786951 |
| Toxoptera auranti | wTaur | N | JN384094.1 |
| Bemisia tabici | wBt10 | O | KF454771.1 |
| Kaburagia rhushicola | wKM KR | O | MT554837.1 |
| Syringophilopsis turdus | wStur | P | KP114103.1 |
| Torotrogla merulae | wTmer | P | KP114102.1 |
| Torotrogla cardueli | wTcard | Q | KP114101.1 |
| Atemnus politus | wApol | S | *NZ_JAAXCS010000017.1 (HET73_RS02035) |
| Cimex hemipterous | wChem PL13 | T | *NZ_CP061738.1 (ID128_RS02485) |
| Spinturnix mites | wSpin Bat1 | U | KP165041 |
| Spinturnix mites | wSpin Bat2 | U | KP165042 |
| *In cases where accession ID for the genes was not available, accession ID of the genome along with locus tag (in the parenthesis) of the gene has been mentioned |
Sequence similarity measure and Phylogenetic tree reconstruction:
16S rRNA sequence similarity paved a way for species demarcation among all bacterial species. So we firstly performed similarity check on the two novel strains along with other 50 strains using GGDC online server [28]. Further, we did phylogenetic analysis and reconstructed the 16S rRNA phylogeny. We aligned the sequences with CLUSTAL W package [29]. We also performed the model test by using ModelFinder that revealed HKY+F+I+G4 is the best suitable model [30]. Then maximum likelihood (ML) tree was reconstructed by using the IQTREE package with the HKY+F+I+G4 model [31].
ANI measure and dDDH study:
After 16S rRNA similarity and phylogeny, we also measured the genomic similarity of the novel strains. For that, we performed average nucleotide identity (ANI) test and digital DNA-DNA hybridization (dDDH) study. ANI measures nucleotide-level genomic similarity between the coding regions of two genomes and here we attempted to find the divergence of the genomes to check whether two novel genomes are from the same supergroup or belong to different supergroups. We also carried out dDDH analysis to calculate in-silico genome-to-genome comparison using the GGDC tool [28]. The dDDH analysis emerged as an alternative to the tedious wet-lab DNA-DNA hybridization of species delineation. In GGDC tool, we used GBDP (Genome Blast Distance Phylogeny) method to calculate the probability that an inter-genomic distance yielded a dDDH value lower than 70 % considered as a novel species-delimitation threshold.
Results and Discussion:
Sequence similarity and phylogenetic analysis of 16S rRNA gene:
At first, we compared 16S rRNA sequence similarity of the two novel strains and found that similarity is 97.94 %, which is below the previously described threshold for species demarcation 98.6% [32, 33]. This result indicates that the novel strains were not from the same supergroup. Furthermore, we also analysed the sequence similarity of the novel strains with respect to the 50 other Wolbachia strains. Similarity results of the novel strains with other supergroups' starins showed that similarity score of the novel strains is lower than the threshold value of 98.6% (Table 2). Overall, 16S rRNA gene similarity result indicates that the novel strains neither belong to the same supergroup nor belong to any other supergroups.
Table 2. 16S rRNA sequence similarity of the novel strains (wCfeT and wCfeJ) with respect to the 50 Wolbachia strains belong to 19 supergroups.
| Wolbachia strain | Supergroup | 16S rRNA similarity (in %) | |
| wCfeT | wCfeJ | ||
| wRi | A | 98.37 | 98.47 |
| wInc | A | 98.1 | 98.4 |
| wCauA | A | 98.2 | 98.4 |
| wMel | A | 98.34 | 98.34 |
| wNfla | A | 98.47 | 98.27 |
| wNpa | A | 98.47 | 98.27 |
| wHa | A | 98.4 | 98.2 |
| wPip Mol | B | 97.07 | 97.14 |
| wMau | B | 97.07 | 97.01 |
| wNo | B | 97.01 | 96.94 |
| wAlbB | B | 97.27 | 97.2 |
| wMeg | B | 96.94 | 97.01 |
| wBtab | B | 96.87 | 96.94 |
| wVitB | B | 96.74 | 96.67 |
| wOo | C | 96.47 | 96.47 |
| wOvol | C | 96.34 | 96.47 |
| wDimm | C | 96.34 | 97.07 |
| wBm | D | 97.79 | 97.8 |
| wWb | D | 97.6 | 97.94 |
| wBpah | D | 97.34 | 97.8 |
| wLsig | D | 97.27 | 97.54 |
| wHruf | E | 98.38 | 97.94 |
| wFol | E | 98.2 | 98.01 |
| wMhie | F | 97.54 | 97.87 |
| wCle | F | 97.94 | 98.2 |
| wNnig | F | 97.81 | 97.81 |
| wCaci | F | 98.1 | 98.1 |
| wZangu | H | 98.16 | 98.28 |
| wZnev | H | 98.16 | 98.28 |
| wCfe | I | 95.76 | 95.76 |
| wMgen | I | 95.7 | 95.77 |
| wOleu | I | 95.46 | 95.69 |
| wDcau | J | 95.94 | 96.54 |
| wCtub | J | 96.54 | 97.07 |
| wBry | K | 97.47 | 97.26 |
| wRsim | L | 95.97 | 95.38 |
| wPpe | L | 95.93 | 95.41 |
| wMeu | M | 97.78 | 97.59 |
| wPni | M | 97.07 | 96.94 |
| wCced | M | 97.72 | 97.8 |
| wTaur | N | 96.63 | 96.44 |
| wKR KM | O | 96.96 | 96.77 |
| wBtab10 | O | 96.65 | 96.37 |
| wTmer | P | 98.23 | 98.23 |
| wStur | P | 97.88 | 97.88 |
| wTcard | Q | 97.92 | 98 |
| wApol | S | 95.5 | 95.91 |
| wChem PL13 | T | 98.07 | 98.47 |
| wSpin Bat1 | U | 97.69 | 98.27 |
| wSpin Bat2 | U | 97.41 | 97.98 |
Further, 16S rRNA ML phylogenetic tree was reconstructed using the HKY+F+I+G4 model of nucleotide evolution given in Figure 1. In this tree, two novel strains found in the different evolutionary lineages (i.e., not having a common ancestry) indicating that they did not belong to any other previously described supergroups. Here, we found that strain wCfeJ, shows parasitic nature with its hosts, is placed as an outgroup with supergroups C, D, F, J, S, T, and U. The bootstrap value at the node (87.9%) showing the reliability of the node. And strain wCfeT, shows mutualistic nature with its hosts, is placed as an outgroup with supergroups A, B, E, H, I, and N. The bootstrap value at this node is 72.9%, showing reliability of this node. In summary, 16S rRNA gene phylogeny confirms that strains wCfeJ and wCfeT having different lifestyle and has distinct evolutionary lineages.
Figure 1.
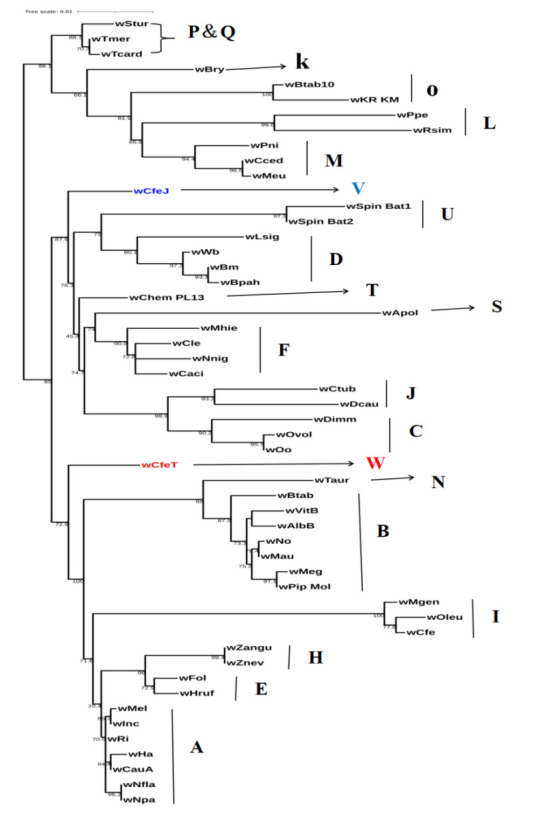
16S rRNA gene phylogeny of 52 Wolbachia strains with 19 supergroups showing position of two novel strains wCfeT (W) and wCfeJ (V). Maximum likelihood tree was reconstructed by using IQTREE with model HKY+F+I+G4. Supergroups are indicated against the strains.
Genome comparison of novel strains:
The supergroups are sub-species level and their genomes are close enough to each other, so genomic divergence analysis is required for supergroup identification. Accordingly, we performed ANI analysis and dDDH study. We found that ANI and dDDH value between strains wCfeT and wCfeJ were found to be 79% and 22.4% respectively. The threshold value for ANI and dDDH are >95% and >70% respectively when the species belong to the same supergroup [32]. This result clearly indicates that the two novel strains are different. Further, the genomic contents of wCfeJ (NZ_CP051157.1) were 1.50Mb genome length; 1228 proteins; 35.2 GC%; 3 rRNA; 34 tRNA; 4 other RNAs; 1,463 genes and 194 pseudogenes. For the wCfeT (NZ_CP051156.1) were 1.20Mb genome length; 1070 proteins; 35.6 GC%; 3 rRNA; 34 tRNA 4 other RNAs; 1,155 genes and 44 pseudogenes. These genomic features show that the novel strains have genomic variations. The genomic analysis results also indicate that the novel strains do not share a species boundary.
Conclusions:
The results of 16S rRNA based similarity analysis and phylogenetic study, and furthermore genomic ANI and dDDH analyses suggested that the novel strains neither shared a species boundary between them nor with any other previously identified supergroups, which designate them as two new supergroups, namely supergroup V (strain wCfeJ) and supergroup W (strain wCfeT). Therefore, our results aid new insights into the Wolbachia diversity and dynamics that will be useful in future comparative studies.
Acknowledgments
The authors thank Aparna Chaturvedi for various useful discussions. AKS thanks the University Grants Commission, India for providing financial support to carry out this research work.
The authors declare that they have no conflict of interest.
Edited by P Kangueane
Citation: Sharma & Som et al. Bioinformation 19(3):336-340(2023)
Declaration on Publication Ethics: The author's state that they adhere with COPE guidelines on publishing ethics as described elsewhere at https://publicationethics.org/. The authors also undertake that they are not associated with any other third party (governmental or non-governmental agencies) linking with any form of unethical issues connecting to this publication. The authors also declare that they are not withholding any information that is misleading to the publisher in regard to this article.
Declaration on official E-mail: The corresponding author declares that official e-mail from their institution is not available for all authors.
License statement: This is an Open Access article which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly credited. This is distributed under the terms of the Creative Commons Attribution License
Comments from readers: Articles published in BIOINFORMATION are open for relevant post publication comments and criticisms, which will be published immediately linking to the original article without open access charges. Comments should be concise, coherent and critical in less than 1000 words.
Bioinformation Impact Factor:Impact Factor (Clarivate Inc 2023 release) for BIOINFORMATION is 1.9 with 2,198 citations from 2020 to 2022 taken for IF calculations.
Disclaimer:The views and opinions expressed are those of the author(s) and do not reflect the views or opinions of Bioinformation and (or) its publisher Biomedical Informatics. Biomedical Informatics remains neutral and allows authors to specify their address and affiliation details including territory where required. Bioinformation provides a platform for scholarly communication of data and information to create knowledge in the Biological/Biomedical domain.
References
- 1.Sironi M, et al. Molecular and Biochemical Parasitology . 1995;74:223. doi: 10.1016/0166-6851(95)02494-8. [DOI] [PubMed] [Google Scholar]
- 2.Werren JH, et al. Proc R Soc Lond B . 1995;261:55. [Google Scholar]
- 3.Harris HL, et al. Symbiosis . 2010;51:37. doi: 10.1007/s13199-010-0065-3. [DOI] [Google Scholar]
- 4.Hoerauf A, et al. Medical Microbiology and Immunology . 2003;192:211. doi: 10.1007/s00430-002-0151-0. [DOI] [PubMed] [Google Scholar]
- 5.Duron O, et al. Heredity . 2007;98:368. doi: 10.1038/sj.hdy.6800948. [DOI] [PubMed] [Google Scholar]
- 6.Hosokawa T, et al. Proc Natl Acad Sci USA . 2010;107:769. [Google Scholar]
- 7.Lindsey ARI, et al. Genes Genomes Genetics . 2016;6:2113. [Google Scholar]
- 8.Badawi M, et al. Genes . 2018;9:290. [Google Scholar]
- 9.Bouchon D, et al. Proc R Soc Lond B . 1998;265:1081. doi: 10.1098/rspb.1998.0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kageyama D, et al. Heredity . 2002;88:444. doi: 10.1038/sj.hdy.6800077. [DOI] [PubMed] [Google Scholar]
- 11.Werren JH, et al. Nat Rev Microbiol . 2008;6:741. doi: 10.1038/nrmicro1969. [DOI] [PubMed] [Google Scholar]
- 12.Cordaux R, et al. Trends in Genetics . 2011;27:332. doi: 10.1016/j.tig.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 13.Miyata M, et al. Biol Lett. 2017;13:20170153. [Google Scholar]
- 14.Beckmann JF, et al. Nat Microbiol . 2017;2:17007. doi: 10.1038/nmicrobiol.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perlmutter JI, et al. PLoS Pathog . 2019;15:e1007936. doi: 10.1371/journal.ppat.1007936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hilgenboecker K, et al. FEMS Microbiology Letters . 2008;281:215. doi: 10.1111/j.1574-6968.2008.01110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zug R, et al. PLoS ONE . 2012;7:e38544. doi: 10.1371/journal.pone.0038544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kajtoch L, et al. Peer J . 2018;6:e4471. doi: 10.7717/peerj.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou W, et al. Proc Biol Sci . 1998;22:265. doi: 10.1098/rspb.1998.0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lo N, et al. Molecular Biology and Evolution . 2002;19:341. doi: 10.1093/oxfordjournals.molbev.a004087. [DOI] [PubMed] [Google Scholar]
- 21.Lefoulon E, et al. BMC Microbiol . 2020;20:188. doi: 10.1186/s12866-020-01863-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laidoudi Y, et al. IJMS . 2020;21:8064. [Google Scholar]
- 23.Konecka E, et al. Infection, Genetics and Evolution . 2021;91:104829. doi: 10.1016/j.meegid.2021.104829. [DOI] [PubMed] [Google Scholar]
- 24.Olanratmanee P, et al. Southeast Asian J Trop Med Public Health . 2021;52:48. [Google Scholar]
- 25.Hassler HB, et al. Microbiome . 2022;10:104. doi: 10.1186/s40168-022-01295-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Driscoll TP, et al. Peer J . 2020;8:e10646. doi: 10.7717/peerj.10646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. https://www.ncbi.nlm.nih.gov/
- 28.Meier-Kolthoff JP, et al. BMC Bioinformatics . 2013;14:60. doi: 10.1186/1471-2105-14-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thompson JD, et al. Nucleic Acids Res . 1994;22:4673. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalyaanamoorthy S, et al. Nat Methods . 2017;14:587. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trifinopoulos J, et al. Nucleic Acids Res . 2016;44:W232. doi: 10.1093/nar/gkw256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim M, et al. International Journal of Systematic and Evolutionary Microbiology . 2014;64:346. [Google Scholar]
- 33.Caudill MT, et al. Microorganisms . 2022;10:605. doi: 10.3390/microorganisms10030605. [DOI] [PMC free article] [PubMed] [Google Scholar]