

Abstract
Background:
Basan syndrome is an autosomal dominant ectodermal dysplasia (ED) with congenital adermatoglyphia, transient neonatal acral bullae, and congenital facial milia. Autosomal dominant adermatoglyphia (ADG) is characterized as adermatoglyphia with hypohidrosis. Recently mutations in the skin-specific isoform of the gene SMARCAD1 have been found in both syndromes.
Objective:
This report proposes to unify these two previously distinct ED, Basan syndrome and ADG, into one syndrome. We offer a new acronym: SMARCAD syndrome (SMARCAD1-associated congenital facial Milia, Adermatoglyphia, Reduced sweating, Contractures, Acral Bullae, and Dystrophy of nails).
Methods:
Sanger sequencing was performed on genomic DNA from a patient with Basan syndrome using primers designed to flank SMARCAD1.
Results:
Sanger sequencing revealed a novel variant, NM_001254949.1:c.−10+2T>G, in the donor splice site of exon 1 of the skin-specific isoform. This variant and the other five previously reported variants in Basan syndrome and ADG are all within the same donor splice site.
Limitations:
As our study was a sporadic case, we were unable to demonstrate segregation of the mutation with the phenotype.
Conclusion:
We conclude that Basan syndrome and ADG are on a phenotypic spectrum of a monogenic syndrome which is better described by the acronym SMARCAD syndrome.
Keywords: adermatoglyphia, Basan syndrome, facial milia, ectodermal dysplasia, SMARCAD1
Introduction
Ectodermal dysplasias (ED) are a group of distinct inherited entities with two or more abnormalities of the major ectodermal structures: hair, nail, teeth, and sweat glands.1 This report proposes to unify two previously distinct ED, Basan syndrome (OMIM 12920) and autosomal dominant adermatoglyphia (ADG) (OMIM 136000), into one condition because they are both caused by mutations in the skin-specific isoform of SMARCAD1. We propose a new acronym: SMARCAD syndrome (SMARCAD1-associated, congenital facial Milia, Adermatoglyphia, Reduced sweating, Contractures, Acral bullae, and Dystrophy of nails).
Basan syndrome is an autosomal dominant ED with variable expression. Less than 10 kindreds have been reported in the literature.1–9 Characteristic features include congenital adermatoglyphia, transient neonatal acral bullae, and congenital facial milia. In addition, many cases also have nail dystrophy, contracture of digits, single transverse palmar crease, acral hyperpigmented macules, palmoplantar keratoderma, and palmoplantar hypohydrosis. ADG is characterized as adermatoglyphia with hypohidrosis; some cases also report palmoplantar hyperkeratosis.10–12
As most syndromes are often initially named based on phenotypic observations, it can be difficult to classify a genetically unified syndrome that has disparate phenotypes without gene isolation. We describe a novel mutation in the skin-specific isoform of SMARCAD1 resulting in Basan syndrome. This mutation is located within the same donor splice site as 5 other mutations that have been reported in patients with ADG and Basan syndrome. We review the literature related to this syndrome to propose a new name that reflects the phenotype more broadly and takes the genotype into account.
Case Report
A 10-day old male, born at 39 weeks via normal spontaneous vaginal delivery, without prenatal complications, presented to the dermatology clinic for evaluation of bilateral heel erosions, which had been bullae at birth. After birth, these bullae ruptured and he developed no further bullae or erosions (Figure 1a). He was also noted to have innumerable congenital milia with the highest density periorally and on the chin (Figure 2a). At his 1-month follow up visit, the bilateral erosions had healed (Figure 1b) and the milia were unchanged (Figure 2b).
Figure 1a:
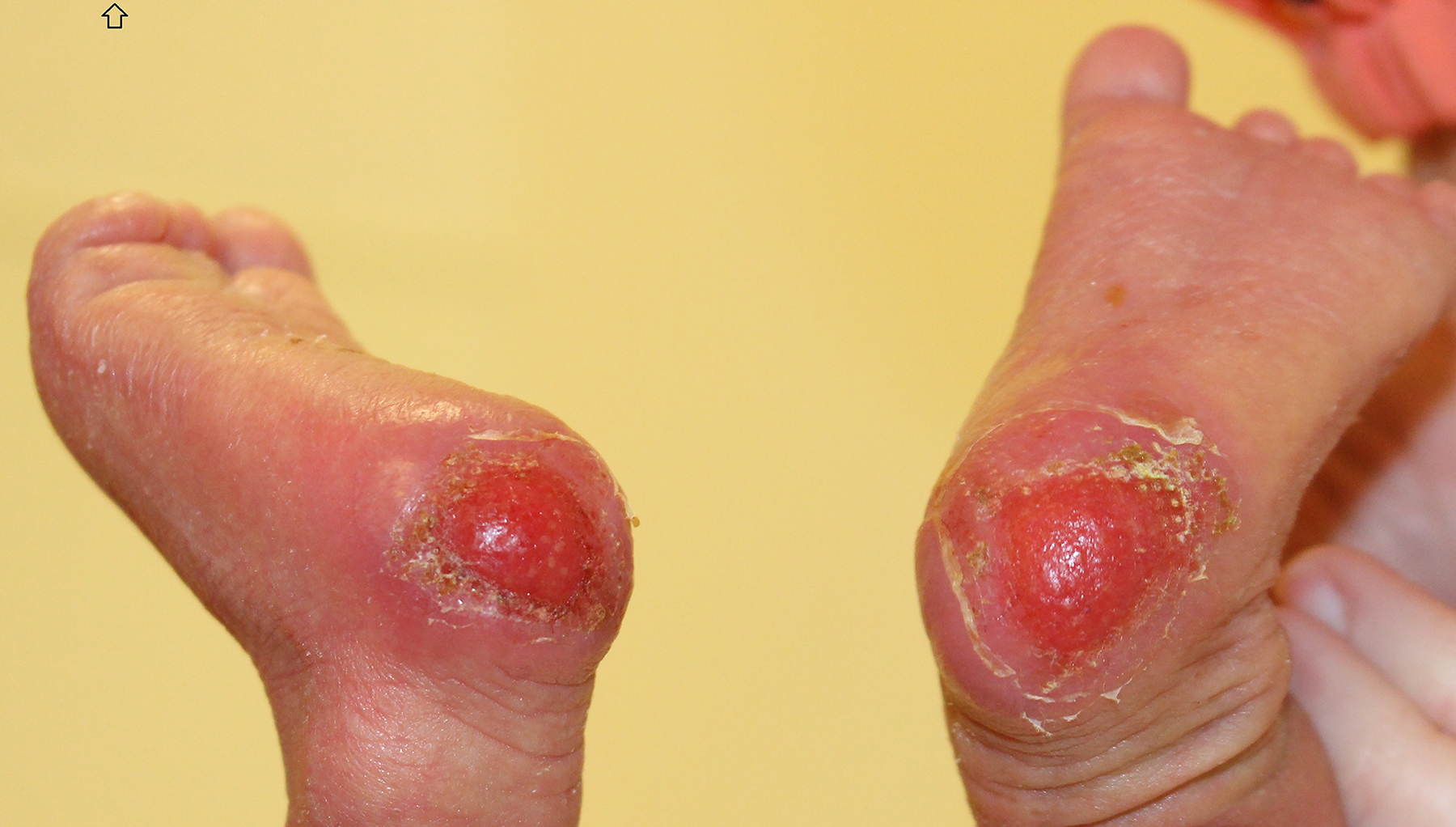
SMARCAD syndrome. Bilateral heel erosions with collarettes of scale from prior bullae at 10 days old.
Figure 2a:
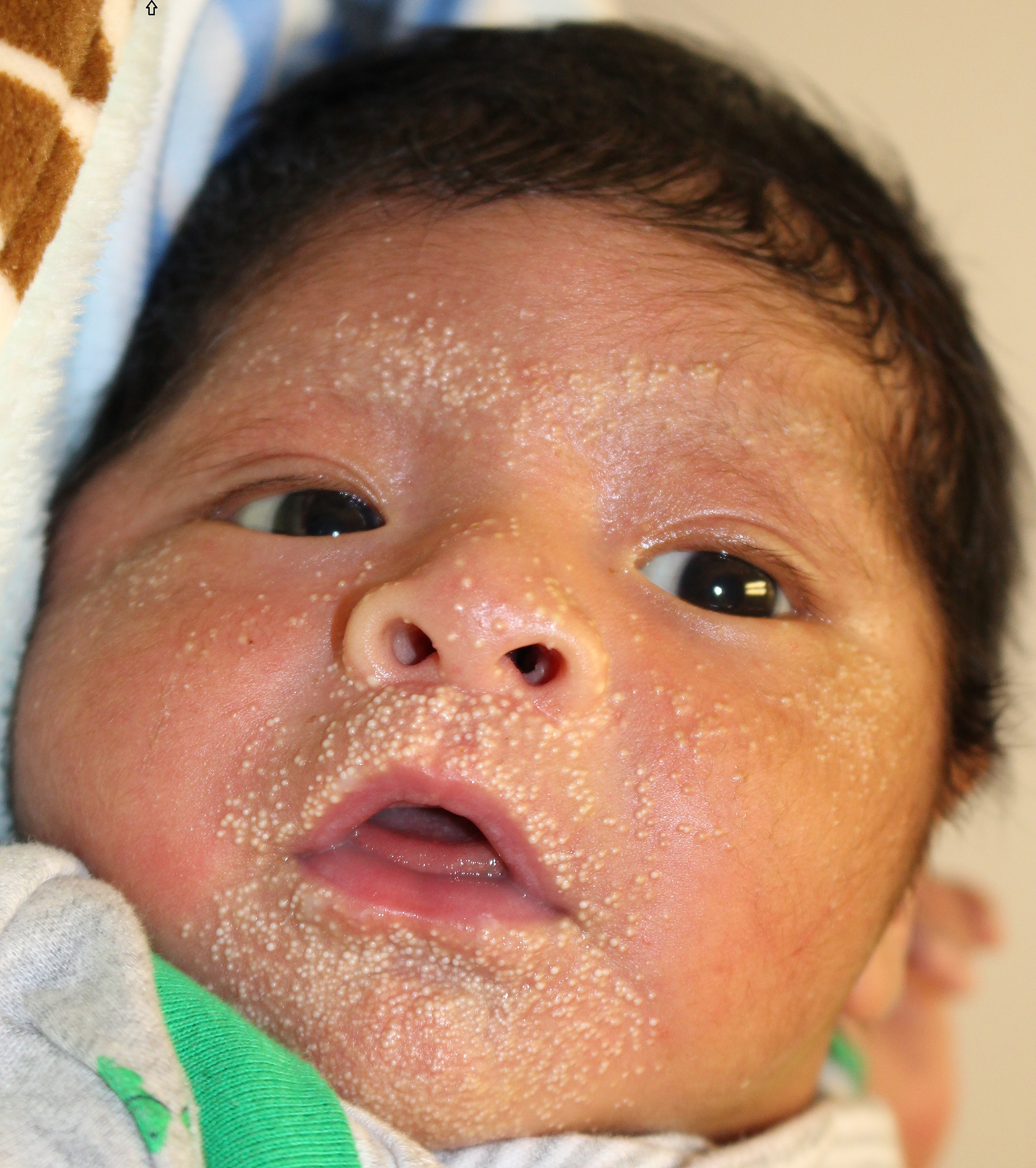
SMARCAD syndrome. Innumerable congenital milia with highest density periorally and on the chin.
Figure 1b:
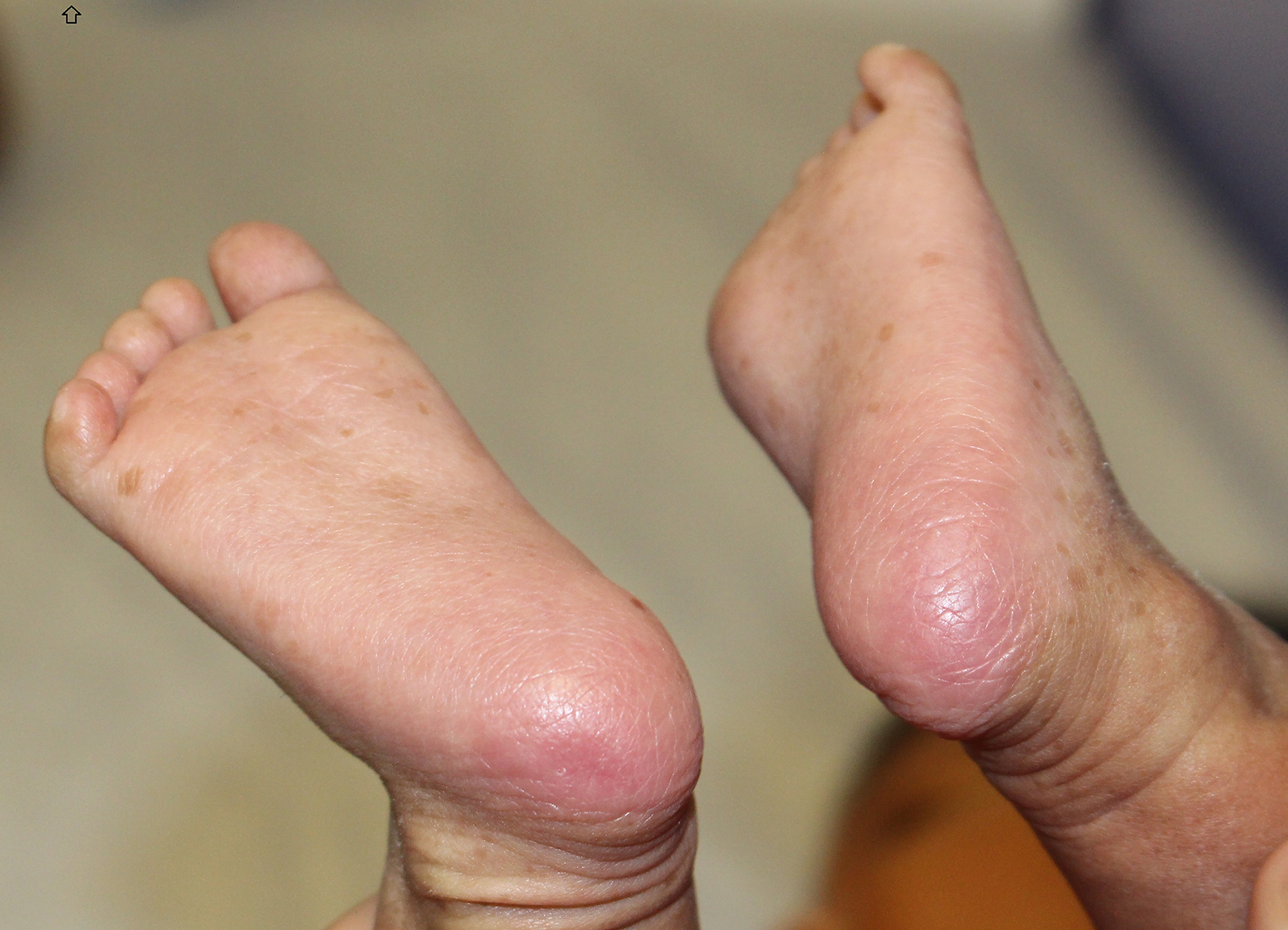
SMARCAD syndrome. Complete resolution of congenital erosions at 1 month old without scarring.
Figure 2b:
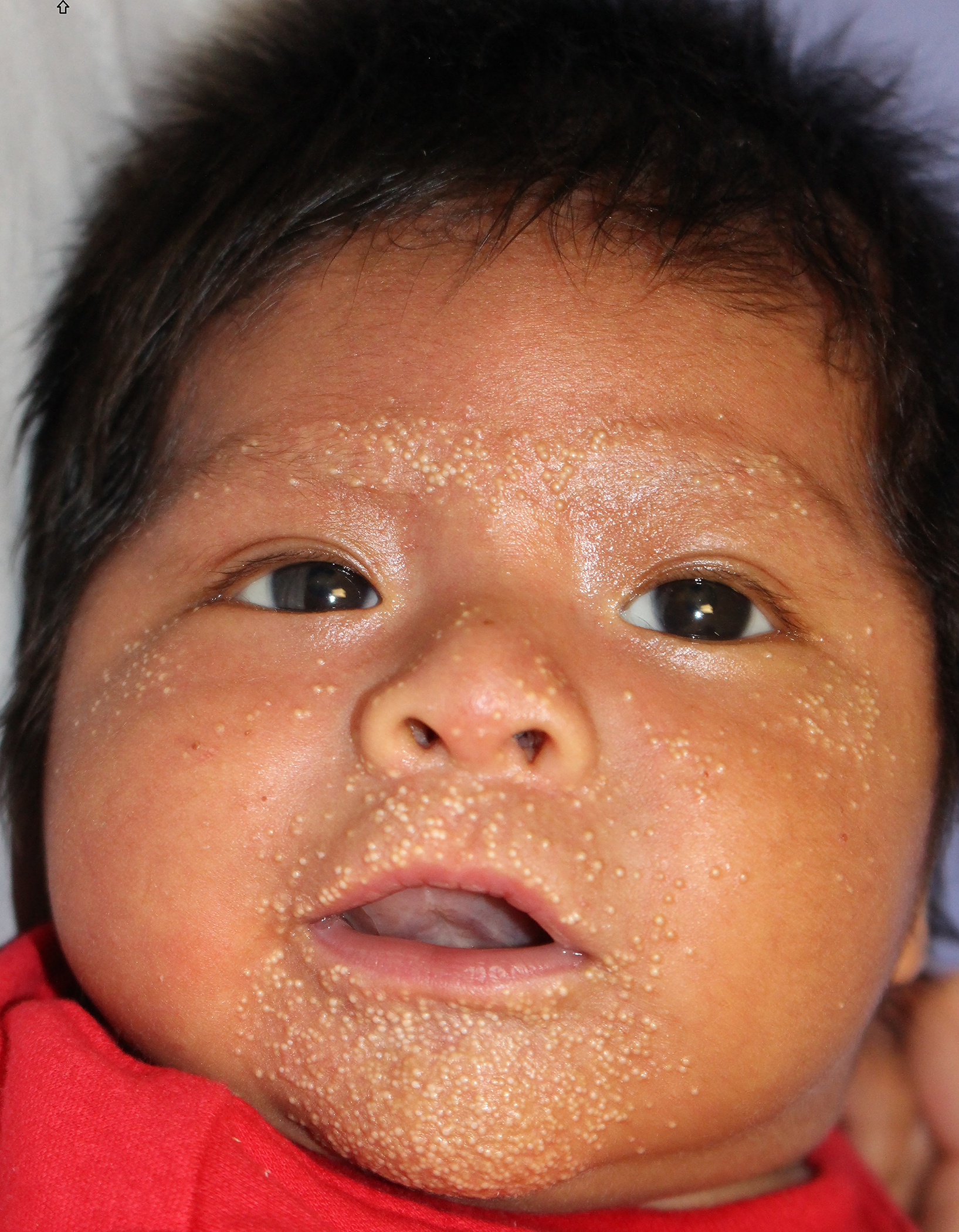
SMARCAD syndrome. Mild decrease in the density of the facial milia after 1 month. The congenital facial milia of SMARCAD syndrome usually regress between 3–6 months of age.
His exam was also significant for adermatoglyphia of his finger and toes, onychorrhexis, hyperpigmented macules on the hands and feet, and a waxy keratoderma with fine wrinkling of the palms and soles (Figure 3). His hair and teeth were unremarkable, and his height and weight were developing normally. Given his clinical features suggestive of Basan syndrome, he was referred to genetics clinic, who confirmed adermatoglyphia (Figure 4) and molecular testing for SMARCAD1 was requested.
Figure 3:
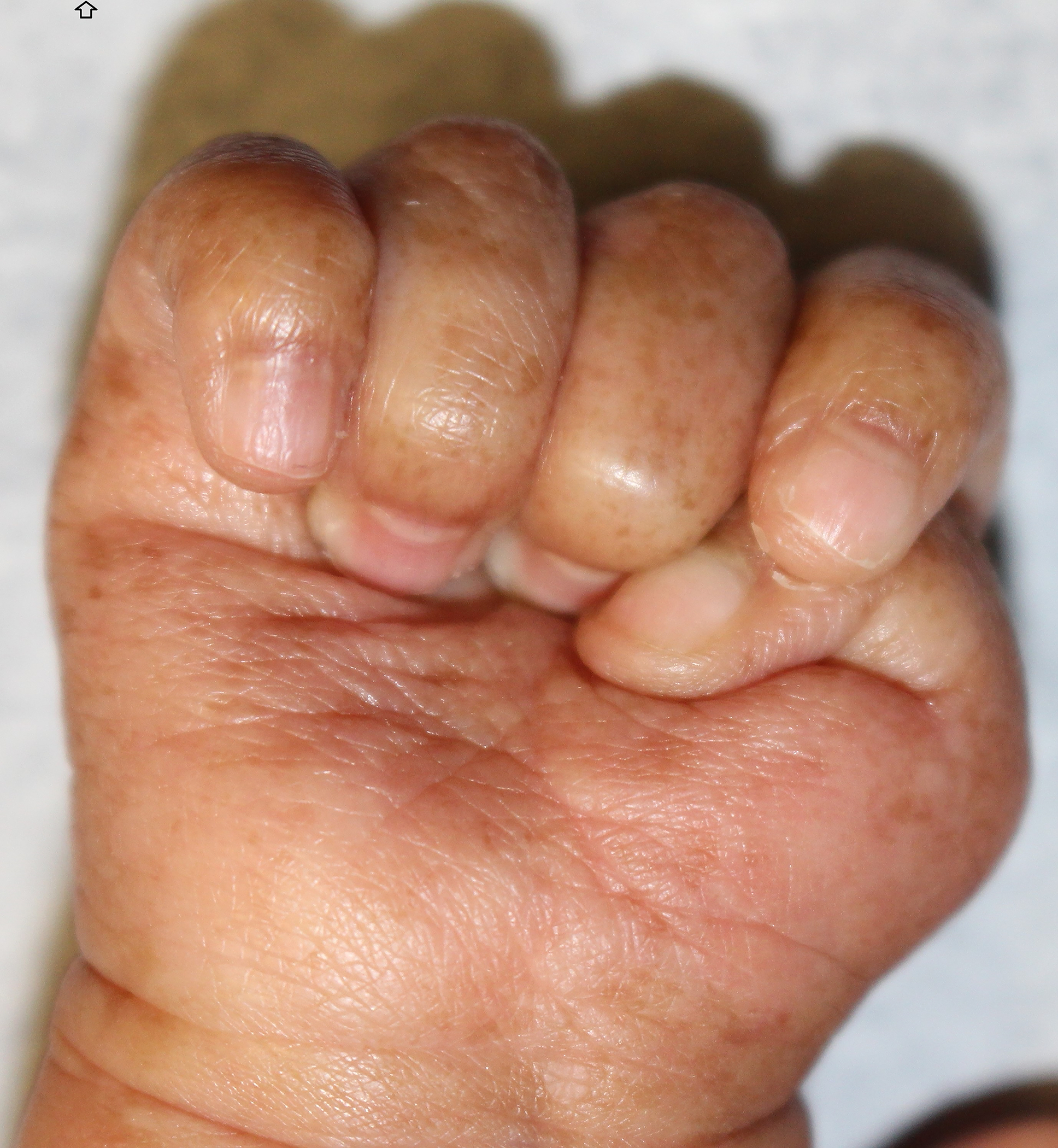
SMARCAD syndrome. Demonstrating onychorrhexis, hyperpigmented macules on both palmar and dorsal hand, and waxy keratoderma with fine wrinkling of the palms.
Figure 4:

SMARCAD syndrome. The patient’s fingerprint (left) shows a complete lack of dermatoglyphs, in contrast to the control’s fingerprint (right).
There was no history of consanguinity. A three-generation family history did not show other affected family members.
Methods
After obtaining informed consent, Sanger sequencing of the skin-specific isoform of SMARCAD1 was performed in a molecular diagnostic laboratory (GeneDx Inc). Using genomic DNA extracted from a peripheral blood sample, a 562bp region including the non-coding exon 1 and its splice donor site of SMARCAD1 was PCR amplified (forward primer: 5’-CACCATGCAGAAAGCAAGAA-3’; reverse primer: 5’-CATCAAGAGTCGCTGCATCT-3’) using standard conditions. Capillary sequencing was performed on an ABI3730 sequencer and bi-directional sequence was assembled, aligned to the reference gene sequence (NM_001254949.1) based on human genome build GRCh37/UCSC hg19 and analyzed for sequence variants. Results were confirmed by capillary sequencing.
Results
Sequencing revealed a novel variant, NM_001254949.1:c.−10+2T>G, in the donor splice site of exon 1 of the skin-specific isoform. When counting from the beginning of the non-coding exon 1, this variant would be named c.378+2T>G, which is similar to the nomenclature given to the five previously reported mutations. The nomenclature we have used (NM_001254949.1:c.−10+2T>G) is preferred as it starts counting from the ATG initiation codon in exon 2, since the first exon is non-coding. Given the ambiguity in mutation nomenclature related to changing conventions, we also provide genomic coordinates, as follows: when using GRCh37/hg19, the variant corresponds to chr4:g.95,174,824T>G, while when using the newer GRCh38/hg38 genome assembly, it corresponds to chr4:g.94,253,673T>G.
Discussion
In 1964, Baird2 was the first to describe a three-generation kindred with 13 affected individuals who all displayed transient congenital facial milia and congenital adermatoglyphia in an autosomal dominant mode of inheritance. He also noted that many of these patients developed hypohidrosis, which was supported by an acral skin biopsy showing lack of skin appendages.2 The following year, Basan7 described what he believed to be a different entity in a kindred of 6 affected individuals with adermatoglyphia, none of whom had congenital facial milia.13 Additional findings included a single transverse palmar crease, nail dystrophy, digital contractures, and positive Minor’s (starch-iodine) test.7 Less than 20 years later, Reed and Schreiner8 described 4 affected individuals in a kindred who demonstrated adermatoglyphia, transient congenital facial milia, nail abnormalities, as well as acral bullae at birth. However, they too concluded that there were enough differences that made the condition in their family distinct from those described by Baird2 and Basan7. In 1993, Limova et al.4 were the first to unify the phenotype after they described 3 individuals in a kindred who demonstrated adermatoglyphia, acral erosions at birth, and mild contractures. They concluded that the differences reflected variable expression of the same entity described by Baird2 and Reed and Schreiner8. Cirillo-Hyland et al.3 re-evaluated an infant from the original kindred reported by Baird2 and reviewed all previously described reports, including the Basan kindred, which they then hypothesized to be one entity. “Basan syndrome” was then coined by Harper et al., in the first edition of their pediatric dermatology textbook.14 Gagey-Caron et al. (2009)6 and Luna et al. (2012)15 were the next to use and solidify “Basan syndrome” as the eponym for OMIM 129200 when reporting their kindreds. Visinoni et al. attempted to classify all ectodermal dysplasias in 2009 and listed OMIM 129200 under the subgroup of abnormalities in nail-sweat glands and named it ‘ED, absent dermatoglyphic pattern, changes in nails, and simian crease.’1
In 2011, Burger et al. published a case report of adermatoglyphia with associated hypohidrosis, which was termed autosomal dominant adermatoglyphia (ADG).12 In addition to adermatoglyphia and hypohidrosis, they described hyperkeratosis of the hands and weight-bearing areas. Nousbeck et al. genotyped this kindred and mapped the disease phenotype to the gene SMARCAD1, a member of the SNF subfamily of the helicase protein superfamily, on 4q22.10 The point mutation, reported as c.378+1G>T (corresponding to c.−10+1G>T using current nomenclature guidelines), was localized to the first donor splice site of the non-coding exon 1, which is unique to the skin-specific isoform of SMARCAD1. Not only did they show that this point mutation segregated with disease phenotype, but that it also resulted in aberrant splicing and thus haploinsufficiency of this isoform. In 2014, Nousbeck genotyped 3 additional kindreds with isolated adermatoglyphia and found 3 novel mutations resulting in disruption of the same conserved donor splice site, 1 for each kindred: c.378+2T>C, c.378+5G>C, and c.378+1G>A.11 Based on Nousbeck et al.’s results, Marks et al. sequenced a kindred with adermatoglyphia, congenital milia, and acral bullae for SMARCAD1 and identified the variant c.378+3A>T, which was one base pair outside the canonical splice donor site for the non-coding exon 1 of the skin-specific isoform. However, while this variant did segregate with the clinical phenotype, unlike Nousbeck et al. they could not demonstrate that it resulted in altered splicing in vitro.9 Li et al. also genotyped a novel kindred with features of adermatoglyphia, congenital milia, and acral bullae. They found the same mutation, c.378+1G>T, that Nousbeck et al. demonstrated in isolated adermatoglyphia, supporting that these phenotypically disparate disorders reflect variability in gene expression of a single disorder.
Our case demonstrates a novel mutation in the skin-specific isoform of SMARCAD1, c.378+2T>G, which is also located in the donor splice site of the first non-coding exon. This variant and the other five previously reported variants in ADG and Basan syndrome are all within four nucleotides of each other. As they are all located in the donor splice site of a non-coding exon, they cannot be identified using whole exome sequencing; rather, Sanger sequencing or whole genome sequencing must be used. Two important points related to the genetic aspect of these variants should be made here. One is that the correct nomenclature for the skin-specific mRNA transcript is NM_001254949. It had previously been erroneously named NM_00112830.10 The latter corresponds to the canonical mRNA transcript, which does not possess the non-coding exon. The second point is that even though we decided to name the variant at the DNA level c.378+2T>G in order to easily compare this novel variant with previously reported ones, the recommendations for the description of DNA sequence variants by the Human Genome Variation Society specify that the coding DNA reference sequence numbering should start with the ATG-translation initiation codon. Thus, the correct nomenclature for the variant itself is c.−10+2T>G, as the translation of the skin-specific isoform does not start until exon 2.
The full length SMARCAD1 isoform is expressed ubiquitously and encodes a protein that is structurally related to the SWI2/SNF superfamily of DNA-dependent ATPases, which are considered to be major regulators of transcriptional activity.10,16 However, little is known about the mechanism of action of the short isoform. The short isoform has a unique 5’-nontranslated exon (exon 1) and is mainly identifiable by RT-PCR in skin fibroblasts (as well as keratinocytes and esophageal tissue to a much lesser degree). Therefore it is referred to as the skin-specific isoform of SMARCAD1.10 In order to elucidate a mechanism of action, Nousbeck et al. downregulated the skin-specific isoform in adult keratinocytes compared to control adult keratinocytes. They found that downregulating the skin-specific isoform of SMARCAD1 affected the expression of eight genes that by pathway analysis were assigned as involved in epidermal growth factor receptor (EGFR) regulation, keratinocyte proliferation and differentiation, and in the pathogenesis of psoriasis.11 It is hypothesized that the point mutations discovered result in aberrant splicing and thus loss-of-function effect on the fibroblasts, which then lead to altered dermatoglyph formation.
Although little is known about the mechanism of dermatoglyph formation, there are three main hypotheses. The first is called the folding hypothesis, which theorizes that the basal layer folds upon itself due to outside mechanical forces, and thus causes the topography of fingerprints; this theory has been rejected by many researchers. The second theory, the nerve hypothesis, theorizes that formation is dictated by the prepattern of nerves or capillaries in the dermis. On the other hand, the fibroblast hypothesis claims that dermatoglyph patterns are first outlined in a prepattern of fibroblasts in the dermis.17 Given that the SMARCAD1 skin-specific isoform is mostly expressed in fibroblasts, the last theory has the most merit. Furthermore, a study in mice found that dermal ridge development was influenced by EGFR signaling, which supports the findings of Nousbeck that haploinsufficiency of the skin-specific isoform of SMARCAD1 alters genes that also influence EGFR signaling.18
Conclusion
Basan Syndrome has been known by many names, including ‘ED, absent dermatoglyphic pattern, changes in nails, and simian crease,’ and ‘adermatoglyphia with congenital facial milia and acral blister, digital contractures, and nail abnormalities.’ We suggest that Basan Syndrome and ADG are not simply allelic disorders, but actually two ends of the phenotypic spectrum of a monogenic syndrome that is better described by the acronym SMARCAD syndrome, for SMARCAD1-associated, congenital facial Milia, Adermatoglyphia, Reduced sweating, Contractures, Acral bullae, and Dystrophy of nails syndrome.
Acknowledgements:
We thank the family of our patient for granting us permission to use their son’s photos for medical education and publication.
Footnotes
Conflicts of Interest: The authors have no conflict of interest to declare.
References:
- 1.Visinoni AF, Lisboa-costa T, Pagnan NAB, Chautard-freire-maia EA, Displasias E. Ectodermal Dysplasias : Clinical and Molecular Review. Am J Med Genet. 2009;Part A(149A):1980–2002. [DOI] [PubMed] [Google Scholar]
- 2.Baird HW. Kindred showing congenital absence of the dermal ridges (fingerprints) and associated anomalies. J Pediatr. 1964;64(May):621–631. [DOI] [PubMed] [Google Scholar]
- 3.Cirillo-Hyland VA, Zackai EH, Honig PJ, Grace KR, Schnur RE. Reevaluation of a kindred with congenital absence of dermal ridges, syndactyly, and facial milia. J Am Acad Dermatol. 1995;32(2 PART 2):315–318. [DOI] [PubMed] [Google Scholar]
- 4.Límová M, Blacker KL, LeBoit PE. Congenital absence of dermatoglyphs. J Am Acad Dermatol. 1993;29(2):355–358. [DOI] [PubMed] [Google Scholar]
- 5.Li M, Wang J, Li Z, et al. Genome-wide linkage analysis and whole-genome sequencing identify a recurrent SMARCAD1 variant in a unique Chinese family with Basan syndrome. Eur J Hum Genet. 2016;25078571(August 2015):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gagey-Caron V SJF. BS. Absence de dermatoglyphes et grains de milium congenitaux” un nouveau cas de syndrome de Basan. Ann Dermatol Venereol. 2009;136(March):419–421. [DOI] [PubMed] [Google Scholar]
- 7.Basan M Ektodermale Dysplasie, fehlendes Papillarmuster. Nagelveraenderungen und Vierfingerfurche. Arch fiir Klin u Exp Dermatologie. 1965;222:546–557. [PubMed] [Google Scholar]
- 8.Reed T, Schreiner RL. Absence of dermal ridge patterns: genetic heterogeneity. Am J Med Genet. 1983;16(1):81–88. [DOI] [PubMed] [Google Scholar]
- 9.Marks KC, Banks WR, Cunningham D, Witman PM, Herman GE. Analysis of two candidate genes for basan syndrome. Am J Med Genet Part A. 2014;164(5):1188–1191. [DOI] [PubMed] [Google Scholar]
- 10.Nousbeck J, Burger B, Fuchs-Telem D, et al. A mutation in a skin-specific isoform of SMARCAD1 causes autosomal-dominant adermatoglyphia. Am J Hum Genet. 2011;89(2):302–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nousbeck J, Sarig O, Magal L, et al. Mutations in SMARCAD1 cause autosomal dominant adermatoglyphia and perturb the expression of epidermal differentiation-associated genes. Br J Dermatol. 2014;171(6):1521–1524. [DOI] [PubMed] [Google Scholar]
- 12.Burger B, Fuchs D, Sprecher E, Itin P. The immigration delay disease: Adermatoglyphia-inherited absence of epidermal ridges. J Am Acad Dermatol. 2011;64(5):974–980. [DOI] [PubMed] [Google Scholar]
- 13.David TJ. Congenital malformations of human dermatoglyphs. Arch Dis Child. 1973;48:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harper J, Oranje A, Prose NE. Textbook of Pediatric Dermatology, 1st Ed. Oxford: Blackwell Science; 2000. [Google Scholar]
- 15.Luna PC, Larralde M. Profuse congenital familial milia with absent dermatoglyphics (Basan’s syndrome): Description of a new family. Pediatr Dermatol. 2012;29(4):527–529. [DOI] [PubMed] [Google Scholar]
- 16.Adra CN, Donato JL, Badovinac R, et al. SMARCAD1, a novel human helicase family-defining member associated with genetic instability: Cloning, expression, and mapping to 4q22-q23, a band rich in breakpoints and deletion mutants involved in several human diseases. Genomics. 2000;69(2):162–173. [DOI] [PubMed] [Google Scholar]
- 17.Kucken M Models for fingerprint pattern formation. Forensic Science International. 2007;171:85–96. [DOI] [PubMed] [Google Scholar]
- 18.Blecher SR, Kapalanga J, Lalonde D. Induction of sweat glands by epidermal growth factor in murine X-linked anhidrotic ectodermal dysplasia. Nature. 1990;345(6275):542–544. [DOI] [PubMed] [Google Scholar]