

Abstract
The neutral amino acid glutamine plays a central role in TGF-β (transforming growth factor-β)–induced myofibroblast activation and differentiation. Cells take up glutamine mainly through a transporter expressed on the cell surface known as solute carrier SLC1A5 (solute carrier transporter 1A5). In the present work, we demonstrated that profibrotic actions of TGF-β are mediated, at least in part, through a metabolic maladaptation of SLC1A5 and that targeting SLC1A5 abrogates multiple facets of fibroblast activation. This approach could thus represent a novel therapeutic strategy to treat patients with fibroproliferative diseases. We found that SLC1A5 was highly expressed in fibrotic lung fibroblasts and fibroblasts isolated from idiopathic pulmonary fibrosis lungs. The expression of profibrotic targets, cell migration, and anchorage-independent growth by TGF-β required the activity of SLC1A5. Loss or inhibition of SLC1A5 function enhanced fibroblast susceptibility to autophagy; suppressed mTOR, HIF (hypoxia-inducible factor), and Myc signaling; and impaired mitochondrial function, ATP production, and glycolysis. Pharmacological inhibition of SLC1A5 by the small-molecule inhibitor V-9302 shifted fibroblast transcriptional profiles from profibrotic to fibrosis resolving and attenuated fibrosis in a bleomycin-treated mouse model of lung fibrosis. This is the first study, to our knowledge, to demonstrate the utility of a pharmacological inhibitor of glutamine transport in fibrosis, providing a framework for new paradigm-shifting therapies targeting cellular metabolism for this devastating disease.
Keywords: pulmonary fibrosis, glutamine metabolism, TGF-β signaling, SLC1A5, bleomycin
Clinical Relevance
We investigated the mechanisms by which TGF-β (transforming growth factor-β)–mediated metabolic regulatory network contributes to lung fibrosis. We provided an entirely new conceptual component for better understanding of the mechanisms driving fibrosis that has the potential to generate new therapeutic approaches for the treatment of idiopathic pulmonary fibrosis.
Fibroproliferative diseases are a leading cause of morbidity and mortality featuring localized and systematic tissue/organ fibrosis (1). Nearly 45% of all deaths in the developed world are caused by chronic inflammatory and fibrogenic disorders (1). Idiopathic pulmonary fibrosis (IPF) is a debilitating, chronic, and irreversible interstitial lung disease with unclear etiology, characterized by aberrant accumulation of extracellular matrix (ECM) proteins in the lungs of individuals between 50 and 85 years old, leading to respiratory failure and death, typically 2–5 years after diagnosis (1, 2). The incidence of IPF has doubled over the past decade (3), and an estimated 50,000 people die each year of IPF in the United States, more deaths than caused by many cancers, including breast cancer (4). Although two U.S. Food and Drug Administration– approved drugs for IPF, nintedanib and pirfenidone (5), have been shown to slow the progression of disease, there is a significant number of side effects that limit therapeutic benefit over time (6). Thus, there is an urgent need to better understand the molecular mechanisms and pathophysiological processes driving IPF to identify novel therapeutic targets and discover effective treatment strategies to improve patient outcomes.
Proliferating cells face a challenge in balancing appropriate concentrations of amino acids required for macromolecular synthesis and growth. As glutamine is the most abundant amino acid in plasma, renewed interest in glutamine metabolism during fibrosis was sparked by the recognition that glutaminolysis controls the activation of myofibroblasts in hepatic fibrosis, nonalcoholic steatohepatitis, cardiac fibrosis, and iatrogenic laryngotracheal stenosis through upregulation of GLS1 (glutaminase 1), YAP1 (Yes-associated protein 1), or nuclear factor erythroid 2–related factor 2 (Nrf2) (7–10). Moreover, recent studies, including our work, have shown that GLS1, which converts glutamine to glutamate, plays a central role in TGF-β (transforming growth factor-β)–induced myofibroblast activation and differentiation and in bleomycin (BLM)–induced experimental pulmonary fibrosis (11–13) Although promising, recent studies also showed that pharmacological inhibition of GLS1 is ineffective in targeting tumors in vivo, even with elevated expression of GLS1, mainly because of metabolic adaptations that render glutaminase dispensable (14). As a hydrophilic amino acid, extracellular glutamine cannot penetrate the plasma membrane without the aid of cell surface glutamine transporters (i.e., solute carrier proteins) (15). ASCT2 (alanine, serine, cysteine transporter 2), encoded by SLC1A5 (solute carrier transporter 1A5) is a Na+-dependent bidirectional amino acid transporter that mainly mediates cellular uptake of large neutral amino acids, specifically glutamine (16). Additional substrates transported by SLC1A5 include alanine, serine, cysteine, threonine, leucine, and asparagine (17). Several studies further demonstrated that SLC1A5 is required for the growth of a variety of cancers and that targeting SLC1A5 by RNA interference, siRNA, or small-molecule inhibitors suppresses cancer cell proliferation in vitro and tumor growth and pulmonary fibrosis in vivo (17–21). The advantages of SLC1A5 as a drug target are as follows: 1) SLC1A5 is expressed on the cell surface and is therefore targetable by both small molecules and therapeutic antibodies; 2) given that SLC1A5 transports metabolites to the rapidly growing cells, inhibiting SLC1A5 for even a short period of time would starve fibrotic fibroblasts while sparing healthy cells; and 3) amino acid transporters have deep binding pockets, making them suitable for the multiple binding of inhibitors. However, little is known currently about the biological impact and mechanisms of regulation of SLC1A5 in pulmonary fibrosis.
In the present study, we found that SLC1A5 was highly expressed in fibrotic lung fibroblasts and that the expression of profibrotic targets, cell migration, and anchorage-independent growth (AIG) by TGF-β required the activity of SLC1A5. Loss or inhibition of SLC1A5 function suppresses mTOR, HIF (hypoxia-inducible factor), and cMyc signaling and impairs mitochondrial function, ATP production, and glycolysis. Pharmacological inhibition of SLC1A5 shifts the profibrotic phenotype to a fibrosis-resolving phenotype in a BLM-treated mouse model of lung fibrosis. Therefore, our results support a novel molecular and metabolic pathway modulating fibrosis resolution and represent a potential new paradigm-shifting approach for therapy targeting cellular metabolism.
Some of the results of these studies have been previously reported in preprint form (https://www.biorxiv.org/content/10.1101/2022.05.23.493168v1).
Methods
A detailed description is provided in the data supplement.
Mice
Wild-type male and female C57BL/6 mice were purchased from Charles River Laboratories and used at 10 weeks of age.
siRNA- and shRNA-mediated Gene Knockdown
Normal human lung fibroblasts (NHLF), IPF fibroblasts, or murine AKR-2B cells were transiently transfected with 40 nM siRNA to SLC1A5, HIF-1α, HIF-2α, cMyc, SMAD2 (SMAD family member 2), SMAD3, mtTFA (mitochondrial transcription factor A) or nontargeting control (Santa Cruz Biotechnology) according to the manufacturer’s protocol. shRNA transfection in NHLFs was performed according to the manufacturer’s instructions (Santa Cruz Biotechnology).
Scratch Assay
For scratch assays, 2 × 105 NHLFs or 1.5 × 105 AKR-2B cells were seeded into 6-well plates containing 10% FBS/Dulbecco’s modified Eagle’s medium (DMEM). After 24 hours of incubation, the monolayer was scraped, and the cultures were then treated with V-9302, L-γ-glutamyl-p-nitroanilide (GPNA) or the indicated siRNA or shRNA and incubated in the presence or absence of TGF-β for 24 hours at 37°C. Images were taken at 24 hours, and the cellular leading edge was quantitated.
Soft Agar Colony Formation Assay
NHLFs (1.25 × 104) were seeded into 6-well plates in the presence or absence of TGF-β (25 ng/ml) with or without V-9302 or GPNA or shRNA targeting SLC1A5. After 7-day growth (peak colony formation) at 37°C, colonies ⩾50 μm in diameter were counted using an optimized closed association rule-mining algorithm in Optronix GelCount (Oxford Optronix).
Seahorse Assay
NHLFs (2 × 105 per well) were seeded into 6-well plates for 24 hours (10% FBS/DMEM). After 24 hours of serum starvation (0.1% FBS/DMEM), cells were pretreated with DMSO (0.1%) or V-9302 (10 μM) at 37 °C for 1 hour, followed by vehicle or TGF-β, and 3,000 cells were seeded into Seahorse assay microplates (Agilent Technologies). Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) was measured on a Seahorse XFp extracellular flux analyzer (Seahorse Bioscience, Agilent Technologies) using the Seahorse XFp Cell Mito Stress Test Kit and XF Glycolysis Stress Test Kit (Agilent Technologies).
BLM Model of Pulmonary Fibrosis
Ten-week-old male and female C57BL/6 mice were administered BLM (3.5 U/kg) or normal saline alone on Day 0. On Day 11, BLM-administered mice were randomly assigned to receive V-9302 (37.5 mg/kg/d) as a soluble intraperitoneal injection (0.2 ml) daily until Day 24. Oxygen saturation as measured by pulse oximetry (SpO2) was determined using the MouseOx monitoring system (Starr Life Sciences) on Days 3, 6, 9, 13, 16, and 21. On Day 25, mice were given pentobarbital (100 mg/kg), and the static and dynamic compliance of the lungs was recorded. After death, lungs were excised, weighed, and collected for hydroxyproline, histopathology, and fibrotic marker expression determination.
Statistics
Statistical significance was calculated using GraphPad Prism 9.3 software. All in vitro data were from a minimum of three biological replicates. In vivo data represented three male and three female mice per group. Results shown are mean ± SEM. Differences between two groups were analyzed using two-tailed unpaired Student’s t tests. When more than two groups were used, statistical analysis was performed using either one-way or two-way ANOVA followed by Tukey’s post hoc test. Statistical significance is denoted in the figures as follows: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. A P value less than 0.05 was considered to indicate statistical significance.
Results
SLC1A5 Expression Is Upregulated in Activated Myofibroblasts
In the present research, we investigated the interrelationship between glutamine transporters and profibrotic TGF-β signaling. Glutamine is transported into cells through plasma membrane glutamine transporters such as SLC1A5, SLC38A1 (solute carrier family 38 member 1), and SLC38A2 (22). In primary human lung fibroblasts (NHLFs), quantitative PCR (qPCR) analysis of transcripts for the important glutamine transporters (solute carrier proteins) showed that SLC1A5 expression was significantly elevated by TGF-β (Figure 1A), and this finding was confirmed and extended by western blot and qPCR analysis, in which elevated expression of SLC1A5 protein and mRNA was observed by TGF-β in NHLFs in a time-dependent manner (Figures 1B and 1C and E1A in the data supplement). This was specific to TGF-β stimulation, as treatment with the TβRI (TGF-β receptor type I) kinase inhibitor SB431542 abolished the response in NHLFs (Figures 1D and E1B). In addition, fibroblasts isolated from lungs of patients with IPF showed significantly enhanced basal SLC1A5 concentrations compared with primary human lung fibroblasts (Figures 1E and 1F). We further investigated whether SLC1A5 knockdown upregulated other amino acid transporters. We stably expressed shRNA targeting SLC1A5 in NHLFs and assessed SLC1A5 protein expression after TGF-β stimulation using three independent pools of shRNA targeting SLC1A5 (see Figure E1C). We did not observe any significant upregulation by TGF-β on other glutamine transporters upon SLC1A5 knockdown, confirming that SLC1A5 suppression is without significant transporter plasticity or redundancy (Figures 1G and 1H). In addition, enhanced SLC1A5 expression was observed in mice that were subjected to fibrosis-inducing BLM administration (Figure 1I).
Figure 1.
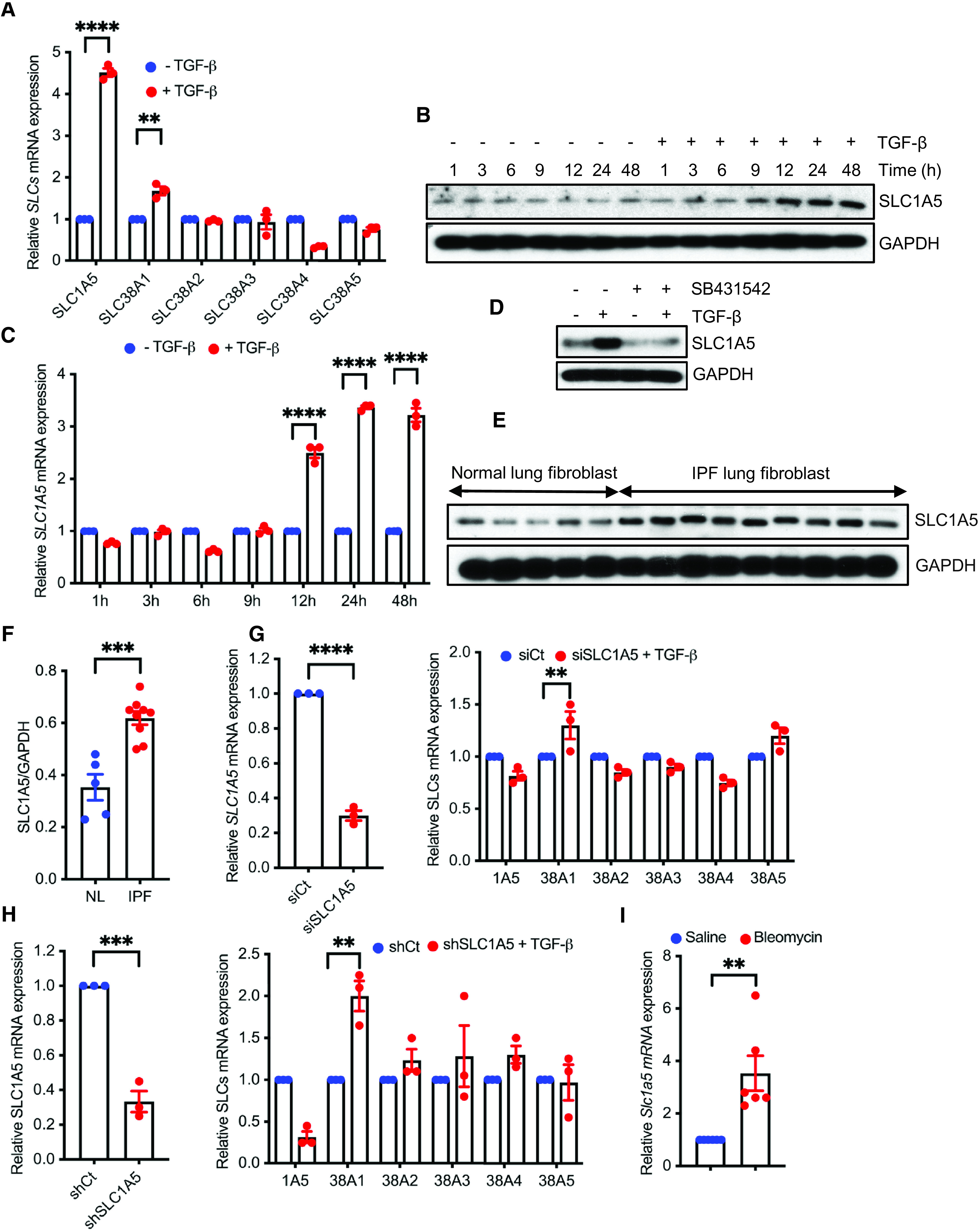
TGF-β (transforming growth factor-β) stimulates SLC1A5 (solute carrier transporter 1A5) and its expression is upregulated in activated fibroblasts. (A) Normal human lung fibroblasts (NHLFs) were treated with 5 ng/ml TGF-β or vehicle (4 mM HCl + 10 mg/ml BSA) for 48 hours, and quantitative PCR (qPCR) analysis of above SLCs was performed. (B) Western blot analysis of SLC1A5 was performed in quiescent NHLFs in the absence (−) or presence (+) of 5 ng/ml TGF-β at the indicated times. (C) NHLFs were treated as in B, and 1 μg RNA was subjected to cDNA synthesis and subsequently qPCR analysis using SLC1A5 primers. (D) NHLFs were treated with DMSO (0.1%) or the TβRI inhibitor SB431542 (10 μM) with or without TGF-β (5 ng/ml, 48-h induction), and protein expression of SLC1A5 was determined using western blotting. (E) Primary lung fibroblasts from patients with idiopathic pulmonary fibrosis (IPF) and healthy control subjects were propagated for three or four passages in cell culture, and western blot analysis for SLC1A5 was performed. (F) Ratios of SLC1A5 to GAPDH in normal and IPF lung fibroblasts. (G) qRT-PCR analysis of important amino acid transporter genes in siRNA mediated knockdown of SLC1A5 in NHLFs.(H) qRT-PCR analysis of important amino acid transporter genes in shRNA mediated knockdown of SLC1A5 in NHLFs. (I) Basal SLC1A5 expression from lungs of saline- and bleomycin-treated mice by qPCR analysis (n = 6). Data generated for qRT-PCR represent mean ± SEM of n = 3 independent experiments. Western blots are representative of three independent experiments. Differences between groups were evaluated using two-way (A, C, and G and H, right) ANOVA with Tukey’s post hoc analysis or unpaired two-tailed Student’s t test (F, I, and G and H, left) using GraphPad Prism 9.3 software. **P < 0.01, ***P < 0.001, and ****P < 0.0001. NL = normal lung; shCt = nontargeting control; shSLC1A5 = Small hairpin RNA (shRNA) against SLC1A5; siCt = nontargeting control; siSLC1A5 = siRNA against SLC1A5; TβRI = TGF-β receptor type I.
Profibrotic TGF-β Signaling Is Dependent on SLC1A5
We next investigated the role of SLC1A5 in profibrotic TGF-β signaling. SLC1A5 expression using siRNA or SLC1A5 activity using V-9302 or GPNA was inhibited, and TGF-β induction of profibrotic molecules was determined using qPCR and western blotting in NHLFs (Figures 2A–2E and E2A–E2C), IPF fibroblasts (Figures 2F and E3), and AKR-2B cells (nontransformed murine fibroblasts) (see Figures E4A–E4D). V-9302 is a newly developed selective inhibitor of SLC1A5 that was effective in suppressing tumor proliferation via inhibition of mTOR signaling (17). GPNA is a potent inhibitor of SLC1A5, as shown previously (23). We performed MTT and XTT assay (cell viability and proliferation assay), which confirmed no significant effect of V-9302 on cellular proliferation (i.e., toxicity) at 10 μM concentrations that significantly affected profibrotic TGF-β signaling (see Figures E5A and E5B). This concentration was subsequently used throughout this study. There were also no significant changes in gene expression predominantly associated with DNA repair (ATM [ATM serine/threonine kinase], ATR [ATR serine/threonine kinase], and BRCA1 [BRCA1 DNA repair associated]), recombination (RAD51 [RAD51 recombinase] and RAD52 [RAD52 homolog, DNA repair protein]), and the cell cycle (CCNB1 [cyclin B1] and CCND1 [cyclin D1]) under V-9302 treatment conditions (see Figure E5C). Although phosphorylation of SMAD2 and SMAD3 occurs independently of SLC1A5 (see Figure E6), expression of profibrotic targets including Col1 (collagen I), FN (fibronectin), CTGF (connective tissue growth factor), and ACTA2 requires the action SLC1A5 (Figures 2A–2F and E2–E4). Next, we tested whether exogenous cell-permeable α-ketoglutarate (α-KG), which is downstream of SLC1A5 in the glutamine metabolism pathway, can restore TGF-β–mediated induction of profibrotic markers upon SLC1A5 silencing. The addition of α-KG overcame the inhibitory effect of SLC1A5 knockdown on the induction of Col1, FN, and ACTA2 by TGF-β (Figures 2G and E7). Collagen facilitates cell–ECM interactions and undergoes cross-linking in the extracellular space. LOX (lysyl oxidase) primarily mediates this process and forms cross-links in ECM proteins. LOX expression is increased in multiple fibrotic diseases and accompanied by increased stiffness and decreased ECM degradation (24). We investigated whether V-9302 treatment altered the expression of key matrix cross-linking and degradation genes. V-9302 treatment downregulated mRNA expression of key matrix cross-linking genes (Figure 2H), whereas expression of key genes associated with matrix degradation and clearance in IPF fibroblasts was upregulated (Figure 2I). Together, these data suggest that myofibroblast differentiation in part depends on the availability of SLC1A5 in TGF-β–mediated fibroblast activation.
Figure 2.
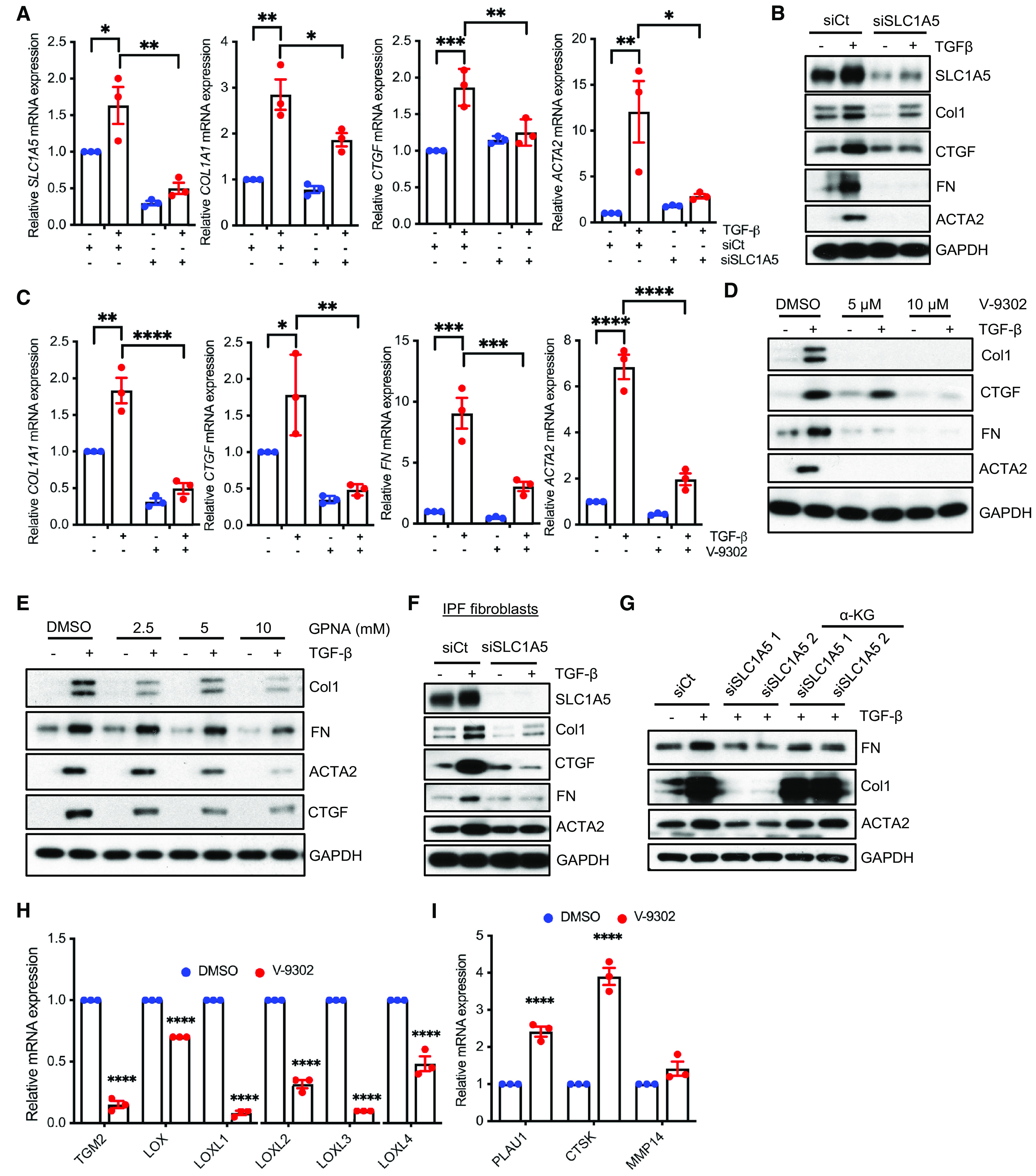
Profibrotic TGF-β signaling is dependent on the action of SLC1A5. (A and B) NHLFs were transfected with nontargeting control (siCt) or siRNA against SLC1A5 (siSLC1A5). Vehicle (−) or TGF-β (+) was directly added to a final concentration of 5 ng/ml, and after 48-hour incubation, qPCR (A) and western blotting (B) of profibrotic molecules were performed. (C) Quiescent NHLFs in 0.1% FBS/Dulbecco’s modified Eagle’s medium (DMEM) were pretreated for 1 hour with vehicle (0.1% DMSO) or 10 μM V-9302 (an SLC1A5 inhibitor) before addition of vehicle (−) (4 mM HCl + 10 mg/ml BSA) or TGF-β (+) (5 ng/ml). After 48-hour incubation, qPCR of the indicated genes was performed. (D) NHLFs in 0.1% FBS/DMEM were pretreated for 1 hour with vehicle (0.1% DMSO) or 5 and 10 μM V-9302 as in C and western blotted for Col1, CTGF, FN, and ACTA2. GAPDH was used as a loading control. (E) Quiescent NHLFs treated with 0.1% DMSO or 2.5, 5, and 10 mM GPNA for 1 hour with the presence or absence of TGF-β (5 ng/ml) for another 48 hours. After 48 hours, cell lysates were prepared and western blotted for indicated proteins. (F) IPF fibroblasts were transfected with either siCt or siSLC1A5 and western blotted as mentioned in A. (G) NHLFs were transfected with control or SLC1A5 siRNA, treated with vehicle or diethyl-2-oxopentanedioate (an esterified form of α-KG; 2 mM) for 24 hours, and then stimulated with vehicle or TGF-β for 24 hours before western blotting for the indicated proteins. (H and I) NHLFs were treated with vehicle (0.1% DMSO) or 10 μM V-9302 for 48 hours. qPCR was performed for ECM cross-linking genes (H), and ECM degradation genes (I). qRT-PCR represents mean ± SEM of n = 3 independent experiments. The western blots are representative of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 (calculated using two-way ANOVA with multiple comparisons by Tukey’s post hoc analysis using GraphPad Prism 9.3 software). α-KG = α-ketoglutarate; Col1 = collagen I; COL1A1 = collagen type I alpha 1 chain; CTGF = connective tissue growth factor; CTSK = cathepsin K; ECM = extracellular matrix; FN = fibronectin; GPNA = L-γ-glutamyl-p-nitroanilide; LOX = lysyl oxidase; LOXL1 = lysyl oxidase like 1; MMP14 = matrix metallopeptidase 14; TGM2 = transglutaminase 2.
Glutamine Uptake, Cell Migration, and AIG Are Dependent on SLC1A5 Activity
As SLC1A5 is a high-affinity glutamine transporter, we sought to investigate the metabolic impact of SLC1A5 inhibition in vitro. To examine the inhibition of glutamine transport by V-9302–mediated or shRNA-mediated knockdown of SLC1A5 in NHLFs, we measured intracellular glutamine concentrations. We found a marked reduction of intracellular glutamine uptake in SLC1A5-inhibited or knocked down cells (Figure 3A). The preceding findings were further extended to examine the effect of SLC1A5 on cell migration and AIG stimulated by TGF-β. SLC1A5 inhibition or knockdown in NHLFs and AKR-2B cells significantly reduced TGF-β–stimulated cell migration (Figures 3B–3D and E8). A similar requirement for SLC1A5 activity was observed for TGF-β–stimulated AIG in soft agar (Figures 3E and 3F).
Figure 3.

Glutamine uptake, cell migration, and anchorage-independent growth (AIG) are dependent on SLC1A5 activity. (A) Glutamine uptake assay in TGF-β–treated (24 h) NHLFs. Results show an increase in glutamine uptake by TGF-β, which is inhibited by the (10 μM) V-9302–mediated (left) or shRNA-mediated knockdown of SLC1A5 (right). (B) (Left) Scratch assays were performed on NHLFs. Red bands indicate the leading edge after 24 hours in the presence (+) or absence (−) of TGF-β (5 ng/ml) alone or containing V-9302 (10 μM) or GPNA (10 mM) and are representative of three separate experiments. (Right) Quantification of wound closure. Data reflect mean ± SEM of n = 3. (C) (Left) NHLFs were transfected with control or SLC1A5 siRNA and then subjected to scratch assay as in B. Red lines indicate the leading edge after 24 hours and are representative of three separate experiments. Data represent mean ± SEM of n = 3. (Right) Quantification of wound closure. (D) (Left) NHLFs stably expressing nontargeting shRNA or shRNA targeting SLC1A5 were treated in the absence (−) or presence (+) of TGF-β for 24 hours and subjected to scratch assay. (Right) Quantification of wound closure. Data reflect mean ± SEM of n = 3. (E and F) Inhibition of SLC1A5 activity by V-9302 and GPNA (E) or shRNA-mediated knockdown of SLC1A5 (F) significantly affects AIG of NHLFs. Quantitative analyses of cell colonies in soft agar are shown. Data reflect mean ± SEM of n = 6. **P < 0.01, ***P < 0.001, and ****P < 0.0001 (calculated using two-way ANOVA with Tukey’s post hoc test using GraphPad Prism 9.3 software).
SLC1A5 Deficiency Suppresses mTOR Signaling and Leads to Transcriptional Reprogramming toward Survival
SLC1A5 was previously shown to be essential for mTOR activation (17). We explored whether SLC1A5 loss caused a reduction in the phosphorylation of mTOR targets. Consistent with this hypothesis, silencing of SLC1A5 or inhibition by V-9302 or GPNA resulted in markedly decreased pS6K, pAkt and p4EBP1 concentrations, suggesting that glutamine deprivation inhibits the mTOR pathway in fibroblasts (Figures 4A and 4B and E9A–E9C). Although TGF-β signals mainly via the SMAD pathway, it also activates other pathways, collectively referred to as “noncanonical” signaling (PI3K/AKT/mTOR), which often complement SMAD action (13). The canonical SMAD signaling pathway is activated within minutes of TGF-β addition, whereas activation of the non-SMAD pathway requires much longer treatment times (25). As SLC1A5 induction occurs relatively late (Figures 1B and 1C), it would not be surprising for TGF-β to stimulate SLC1A5 through both canonical and noncanonical mechanisms. To further identify the pathway(s) required for the induction of SLC1A5 by TGF-β, NHLFs were treated, first, with siRNA that targeted SMAD2, SMAD3, or a nontargeting control (Figure 4C) and, second, with the MAPK (mitogen-activated protein kinase) kinase (MEK), PI3K, AKT, mTORC1, and mTORC1/2 inhibitors U0123, LY294002, MK2206, rapamycin, and torin 1, respectively (Figure 4D), and the induction of SLC1A5 by TGF-β was measured. SMAD3 knockdown as well as inhibition of PI3K and mTORC1/2 abrogated the induction of SLC1A5 by TGF-β (Figures 4C and 4D). As activation of autophagy is known to provide a crucial source of nutrients in response to nutrient depletion, SLC1A5-deficient NHLFs showed a significant induction of autophagy, as demonstrated by elevated expression of LC3BII (microtubule-associated protein 1 light chain 3B II) and beclin-1 (Figures 4E–4G and E10A and E10B). We further confirmed that endoplasmic reticulum stress and unfolded protein response genes were also activated as a compensatory response after inhibition of SLC1A5 (Figure 4H).
Figure 4.
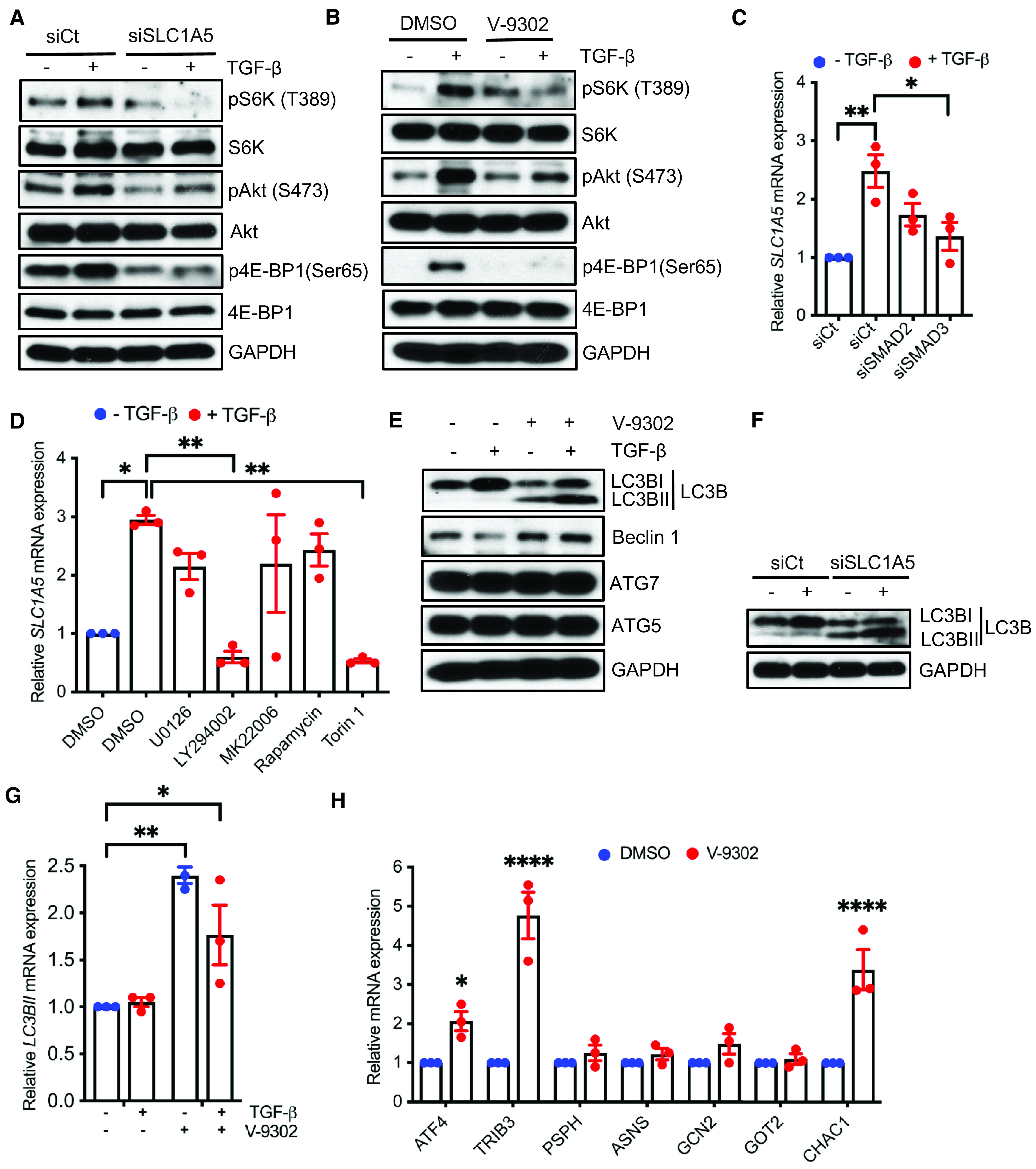
SLC1A5 deficiency suppresses mTOR signaling and leads to transcriptional reprogramming toward survival. (A) NHLFs were transfected with scrambled control or SLC1A5 siRNA and western blotted for the indicative proteins 6 hours before TGF-β or vehicle treatment. (B) NHLFs were treated with V-9302 and stimulated in the absence (−) or presence (+) of TGF-β (5 ng/ml), and indicated proteins were assessed at 6 hours. (C) NHLFs with either siCt or siSMAD2 or siSMAD3 were treated with TGF-β (+) (5 ng/ml) or vehicle (−), and qPCR of SLC1A5 was performed 24 hours after treatment. (D) qPCR for SLC1A5 24 hours after vehicle (−) or TGF-β (+) treatment in the presence of 0.1% DMSO, MEK-ERK1/2 inhibitor U0126 (3 μM), PI3K inhibitor LY294002 (20 μM), Akt inhibitor MK22006 (300 nM), mTORC1 inhibitor rapamycin (100 nM), or mTORC1 + C2 inhibitor torin 1 (200 nM). (E–G) NHLFs were treated as in Figures 2A and 2C, and western blot analysis of autophagic proteins (E and F) or qPCR of LC3BII (G) was performed. (H) qRT-PCR analysis of endoplasmic reticulum stress/unfolded protein response genes in SLC1A5-inhibited NHLFs. All western blots are representative of three separate experiments. Data in C, D, G, and H represent mean ± SEM and n = 3 independent experiments. *P < 0.05, **P < 0.01, and ****P < 0.0001 (calculated by one-way [C and D] or two-way [G and H] ANOVA with Tukey’s post hoc test using GraphPad Prism 9.3 software). ASNS = asparagine synthetase (glutamine-hydrolyzing); ATF4 = activating transcription factor 4; ATG5 = autophagy related 5; CHAC1 = ChaC glutathione-specific gamma-glutamylcyclotransferase 1; GOT2 = glutamic-oxaloacetic transaminase 2; LC3B = microtubule-associated protein 1 light chain 3B; PSPH = phosphoserine phosphatase; siSMAD2 = siRNA targeting SMAD2; siSMAD3 = siRNA targeting SMAD3; TRIB3 = Tribbles pseudokinase 3.
Role of SLC1A5 in HIF and c-Myc Signaling
As HIFs and cMyc control the progression and pathogenesis of fibrosis, and cMyc upregulation has been associated with increased glycolysis and glutaminolysis to support the increased biosynthetic demands (26–28), we next determined whether these transcription factors participate in TGF-β–induced SLC1A5 expression. Silencing HIF1α, HIF2α, and cMyc expression impaired the protein expression of SLC1A5 (Figures 5A and E11A), whereas knockdown or inhibition of SLC1A5 downregulated protein expression of HIF1α, HIF2α, and cMyc (Figures 5B and 5C and E11B and E11C). Furthermore, downregulation of SLC1A5 impaired the gene expression of HIF1α, HIF2α, and cMyc (Figure 5D). Together, the data support a mechanism by which HIF, cMyc, and SLC1A5 constitute a positive feedback loop, sustaining one another’s expression to control fibroblast activation.
Figure 5.
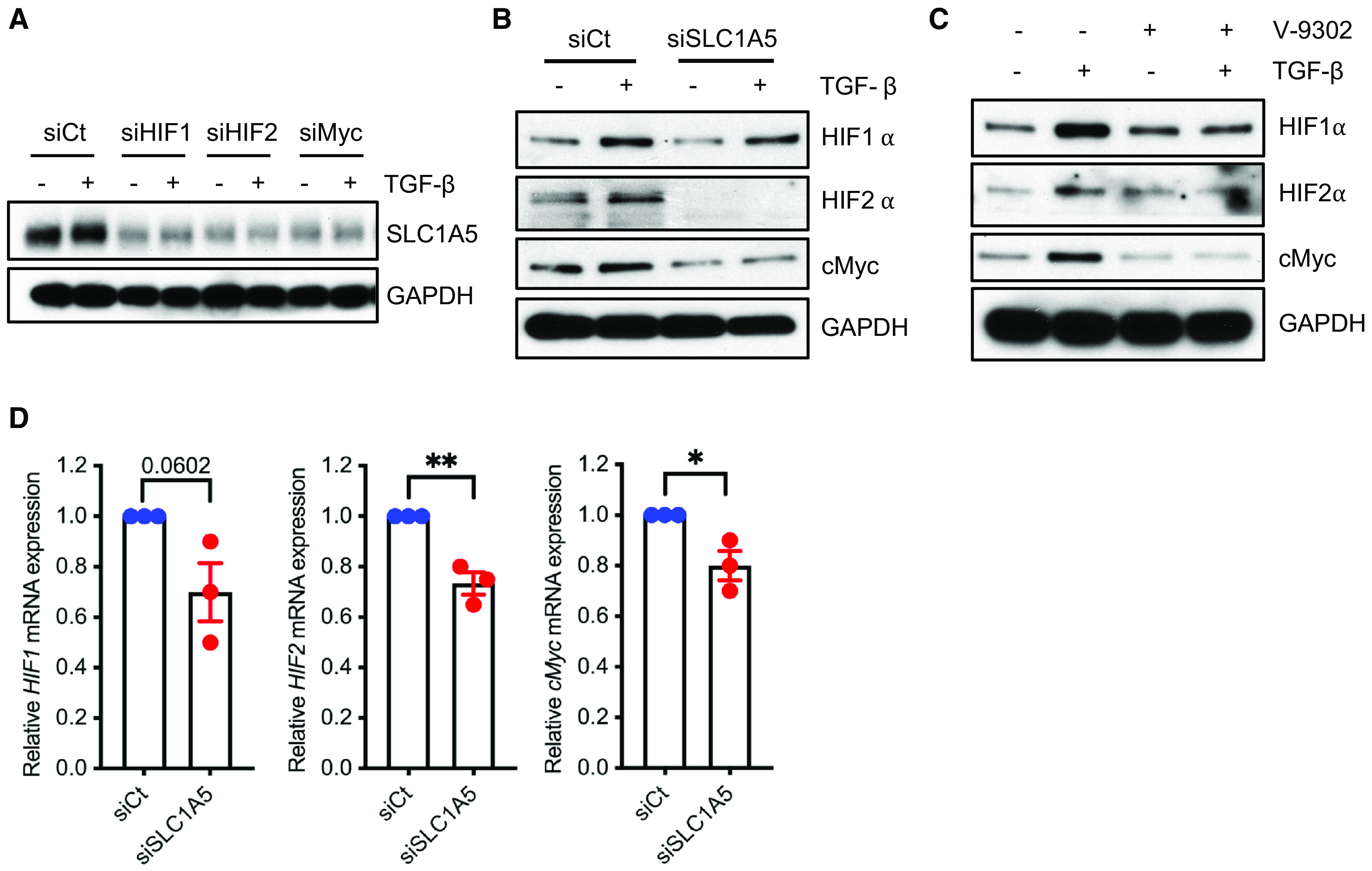
Role of SLC1A5 in HIF (hypoxia-inducible factor) and c-Myc signaling. (A and B) NHLFs were transfected with control or HIF1α, HIF2α, and cMyc siRNA (A) or SLC1A5 siRNA (B). After 36-hour incubation (±5 ng/ml TGF-β), lysates were prepared and western blotted for indicated proteins. (C) NHLFs were treated as in Figure 2C, and western blot analysis was performed. (D) qPCR analysis of HIF1, HIF2, and cMyc genes in SLC1A5-knockdown NHLFs. Western blots are representative of three independent experiments. Differences between two groups were evaluated using unpaired two-tailed Student’s t tests using GraphPad Prism 9.3 software. *P < 0.05 and **P < 0.01.
SLC1A5 Is Critical for Metabolic Reprogramming in Fibroblasts
Myofibroblast differentiation requires metabolic reprogramming characterized by increases in oxidative phosphorylation and glycolysis (29). To determine whether this metabolic reprogramming requires SLC1A5, we analyzed the inhibition of SLC1A5 after TGF-β–induced differentiation on the OCR and the ECAR of myofibroblasts. Inhibition of SLC1A5 by V-9302 impaired the basal and maximal OCRs, implying defective mitochondrial respiration (Figures 6A and 6B). Inhibition of SLC1A5 suppressed the ECAR (Figures 6C and 6D). Furthermore, treatment with rotenone and antimycin (mitochondrial complex I and III inhibitors, respectively) suppressed SLC1A5 and fibrosis marker expression, whereas 3-NP (a mitochondrial complex II inhibitor) did not have a significant effect (Figures 6E and E12). We also examined the effect of SLC1A5 expression on mitochondria-derived ATP production. Downregulation of SLC1A5 significantly decreased ATP production, indicating that ATP production is highly dependent on SLC1A5 (Figure 6F). TFAM (mitochondrial transcription factor A) is involved in mitochondrial DNA transcription and replication, and silencing of TFAM downregulates TGF-β–induced expression of ACTA2 (29). As TFAM is important in maintaining mitochondrial fitness, we examined the roles of mitochondrial fitness and metabolism by silencing TFAM and determined the effect on the expression of SLC1A5. TFAM silencing decreased expression of SLC1A5 (Figure 6G). Silencing of SLC1A5 markedly reduced the expression of TFAM expression in NHLFs (Figure 6H). Taken together, these data suggest that both mitochondrial biogenesis and SLC1A5 expression coordinately support myofibroblast differentiation.
Figure 6.

SLC1A5 is critical for metabolic reprogramming in fibroblasts. (A and B) Quiescent NHLFs were treated for 24 hours with vehicle (DMSO) or V-9302 in the presence or absence of TGF-β, and OCR was assessed using the Seahorse XFp Cell Mito Stress Test Kit on a Seahorse XFp extracellular flux analyzer. Data are presented as individual time points (A) and as averages (B). (C and D) NHLFs were treated as in A, and then glycolysis was measured as ECAR using the Seahorse XF Glycolysis Stress Test Kit. Data represent individual time points (C) and averages (D). (E) NHLFs were pretreated for 1 hour with vehicle (0.1% DMSO) or rotenone, 3-NP, and antimycin (mitochondrial complex I, II, and III inhibitors, respectively) before the addition of vehicle (−) or TGF-β (+) (5 ng/ml). After 48-h incubation, lysates were prepared and western blotted for SLC1A5 and fibrosis marker expression. (F) NHLFs were treated as in Figure 2C, and cellular ATP concentrations were measured. (G and H) NHLFs were transfected with nontargeting or TFAM (mitochondrial transcription factor A) siRNA (G) or SLC1A5 siRNA (H), and expression of SLC1A5 and TFAM was measured. n = 3 independent experiments. All data reflect mean ± SEM. Differences between groups were evaluated using two-way ANOVA with Tukey’s post hoc analysis (B, D, and F) or unpaired Student’s t tests (G and H). *P < 0.05, **P < 0.01, and ****P < 0.0001. ECAR = extracellular acidification rate; FCCP = carbonyl cyanide-p-trifluoromethoxyphenylhydrazone; non-mito = nonmitochondrial; OCR = oxygen consumption rate.
V-9302 Treatment in a Therapeutic Murine Model Ameliorates BLM-induced Fibrosis
As SLC1A5 has been successfully targeted by V-9302 in several tumor models with an optimal dose of 50–75 mg/kg/d (17), we extended our findings to a murine treatment model of lung fibrosis. C57BL/6 male and female mice were intratracheally administered an equal volume of saline (control) or BLM (3.5 U/kg). On Day 11, BLM- and saline-administered mice started receiving treatment with vehicle (PBS supplemented with 2% DMSO) or V-9302 (37.5 mg/kg/d) daily by intraperitoneal injection for 13 days; the animals were killed on Day 25 (Figure 7A). SpO2 is a useful noninvasive measure of overall lung function and morbidity or mortality for experimental pulmonary fibrosis. To accurately measure lung function throughout the study, SpO2 was determined before and every third or fourth day after the initiation of treatment on Day 11. V-9302 stabilized or improved peripheral blood oxygenation during treatment (Figure 7B). Lung compliance is the ability of the lungs to stretch and expand and is the most sensitive parameter for the detection of abnormal pulmonary function. flexiVent (SCIREQ Scientific Respiratory Equipment Inc.) analysis of lung compliance was determined after euthanasia on Day 25. flexiVent analysis indicated a V-9302– dependent improvement of lung compliance (Figure 7C). These physiologic findings further supported beneficial effects of V-9302 with respect to 1) lung weight at study completion (Figure 7D); 2) reduced collagen deposition in the lungs, as assessed by hydroxyproline content (Figure 7E); 3) hematoxylin and eosin and trichrome staining by V-9302 treatment (Figures 7F and 7G and E13); 4) interstitial inflammatory infiltrate and accumulation of foamy macrophages (Figures 7H and 7I) (this is a semiquantitative evaluation considering both the intensity and extent of interstitial inflammatory infiltrate or accumulation of foamy macrophages, which is nonspecific but tends to be associated with obstruction or increased cell damage, i.e., more dead cells producing lipid to be cleaned up by macrophages); 5) significantly reduced inflammatory cytokine gene expression in BLM-induced mice lung treated with V-9302 (see Figure E14); and 6) reduced expression of profibrotic markers after V-9302 treatment (Figure 7J). Importantly, V-9302–treated mice did not show any body weight difference compared with control mice (17). V-9302 also had no demonstrable effect on murine liver and kidney function (see Figures E15 and E16) or blood count parameters (see Figure E17). The intracellular concentration of glutamine in lung tissues was reduced significantly by V-9302 treatment (see Figure E18). We explored whether SLC1A5 inhibition caused a reduction in the phosphorylation of mTOR targets in the lungs of BLM-administered mice. In agreement with the in vitro data, mice treated with BLM and V-9302 exhibited reduced phosphorylation of S6K, Akt, and 4E-BP1 compared with BLM-treated mice, suggesting impaired mTOR signaling (see Figure E19). Furthermore, V-9302 treatment suppressed HIF1α, HIF2α, and cMyc expression in BLM-administered mice (see Figure E20). In addition, V-9302 treatment led to decreased concentrations of ATP, indicating that SLC1A5 is responsible for mitochondrial metabolism (see Figure E21). Taken together, these results demonstrate that pharmacological inhibition of Slc1a5 with V-9302 during the fibrotic phase is sufficient to attenuate BLM-induced pulmonary fibrosis.
Figure 7.
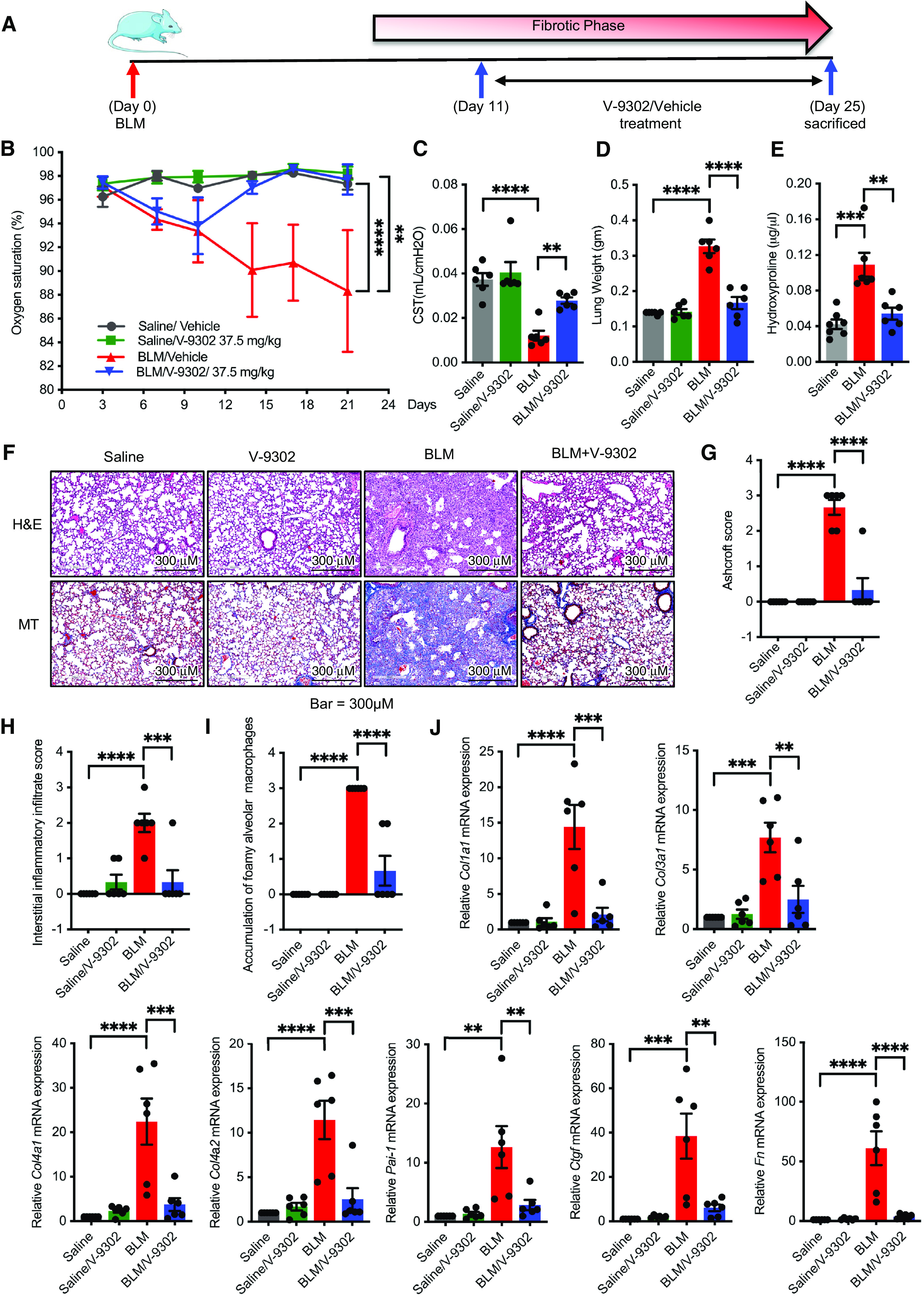
V-9302 treatment in a therapeutic murine model ameliorates bleomycin (BLM)–induced fibrosis. (A) Timelines for in vivo administration of BLM and V-9302. C57BL/6 male or female mice were intratracheally administered with an equal volume of saline (control) or BLM (3.5 U/kg). From Days 11 to 24, mice were treated daily with either vehicle (PBS supplemented with 2% DMSO) or V-9302 (37.5 mg/kg) by intraperitoneal injection. On Day 25, mice were killed. (B) Oxygen saturation was determined on every third day after BLM administration. (C and D) On Day 25, mice were killed, and lungs were subjected to flexiVent determination (C) and measurement of total lung weight (D). (E) Total collagen content determined by hydroxyproline assay. (F–I) H&E staining for histology and MT staining for collagen deposition (blue) were performed (F), and the lung histopathology sections were blindly scored for the degree of fibrosis using the Ashcroft method (G), score for interstitial inflammatory infiltrate (H), and accumulation of foamy macrophages (I). Scoring was performed on the basis of the following criteria: 0 (no fibrosis), 1 (focal/minimal fibrosis), 2 (multifocal/moderate fibrosis), and 3 (confluent/severe fibrosis). Scale bars, 300 μm. (J) qPCR analysis for fibrotic markers Col1a1, Col3a1, Col4a1, Col4a2, Pai-1, Ctgf, and Fn in murine lung tissue harvested on Day 25. Data reflect mean ± SEM of six mice for each group (three male and three female). Differences between groups were determined using two-way ANOVA with Tukey’s post hoc analysis (B) or one-way ANOVA with Tukey’s post hoc analysis (C–E and G–J) using GraphPad Prism 9.3 software. **P < 0.01, ***P < 0.001, and ****P < 0.0001. Col3a1 = collagen, type III, alpha 1; Col4a1 = collagen type IV alpha 1 chain; Col4a1 = collagen type IV alpha 2 chain; CST = lung compliance; H&E = hematoxylin and eosin; MT = Masson’s trichrome.
Discussion
Metabolic dysregulation contributes to the development of chronic lung diseases, including pulmonary fibrosis, and its reliance on glutamine for energy supply is now well acknowledged. Recent studies by our group and others have shown that mitochondrial glutaminase, GLS1, plays a central role in TGF-β–induced myofibroblast activation and differentiation in pulmonary fibrosis (11–13). CB-839 (Calithera Biosciences) is a selective GLS1 inhibitor now being explored in phase 2 clinical trials in multiple liquid and solid tumors (30). Although promising, limitations of this strategy include 1) the presence of more than one isoform of GLS (KGA, GAC, or GLS2); 2) unresolved subcellular localization that can hamper drug availability; and 3) the fact that GLS1 inhibition does not address the extramitochondrial roles of glutamine, including MAPK signaling, the activity of GLS2, and amino acid transporters that require glutamine antiport for their function (e.g., SLC7A5) (17, 31). Therefore, blocking cellular glutamine transport would provide a greater impact on glutamine metabolism than targeting downstream enzymes such as GLS1, which provide extensive biological plasticity and redundancy to proliferating cells to maintain the intracellular glutamine pool (17). SLC1A5 is a neutral bidirectional amino acid transporter belonging to the SLC1 family, is localized in the plasma membrane, has been identified as the most critical plasma membrane glutamine transporter in cancer, and is indispensable for active cell proliferation, mTOR signaling, and redox balance, to name a few (32). Under physiologic conditions, SLC1A5 expression was detected in the lung, skeletal muscle, large intestine, kidney, testis, T cells, brain, and adipose tissue. In polarized intestinal or renal proximal tubule epithelial cells, SLC1A5 was localized to the apical membrane domain. Although a great deal of biological data documenting the medical importance of glutamine transport in cancer are available, there are no reports addressing how metabolic dysregulation by glutamine transporters and cytokine autocrine/paracrine signaling function in an integrated and synergistic manner in the development of fibroproliferative diseases. As fibroblasts are the most important cell type for fibrosis progression and ECM production, in the present study we concentrated only on fibroblasts and revealed critical roles of SLC1A5 in regulating cellular, molecular, and metabolic mechanisms by which SLC1A5 and TGF-β cooperate in regulating myofibroblast activation, tissue repair, and fibrosis resolution by 1) determining the role of SLC1A5 in profibrotic TGF-β signaling; 2) investigating the mechanisms of cell type–specific induction of SLC1A5 by TGF-β and associated cellular, molecular, and metabolic mechanisms; 3) evaluating how the inhibition of SLC1A5’s actions “chemosensitizes” mesenchymal cultures to metabolic reprogramming; and, most important, 4) extending these mechanistic findings to a treatment model of pulmonary fibrosis.
Our data indicate an important relationship between the fibroproliferative actions of TGF-β and the induction of glutamine transporter SLC1A5. We extended these findings to include primary IPF fibroblasts to confirm the generality of the findings. Recently, Schulte and colleagues developed V-9302, a competitive small-molecule antagonist that selectively and potently targets SLC1A5 (17). They further showed a stable V-9302–SLC1A5 interaction by drug affinity responsive target stability (33) and therefore implied that SLC1A5 is a V-9302 target. Pharmacological blockade of SLC1A5 with V-9302 resulted in attenuated cancer cell growth and proliferation, increased cell death, and increased oxidative stress, which collectively contributed to antitumor responses in vitro and in vivo (17). Herein, we showed that V-9302 inhibited SLC1A5-mediated glutamine uptake, and knockdown of SLC1A5 mediated by V-9302 or GPNA or siRNA inhibits fibroblast activation in primary human lung fibroblasts. As depletion of SLC1A5 significantly inhibits the import of essential amino acids (EAAs) into cells, which might induce an EAA shortage and trigger the amino acid stress response signal transduction pathway, we determined that SLC1A5 depletion increased ATF4 (activating transcription factor 4), TRIB3 (Tribbles pseudokinase 3), and CHAC1 (ChaC glutathione-specific gamma-glutamylcyclotransferase 1) expression (all involved in the stress response pathway). An earlier study showed that SLC1A5 deficiency was compensated by increased concentrations of other glutamine transporters (SLC38A1/SLC38A2) in a GCN2 EIF2α kinase (GCN2)–dependent, cell type–specific manner (34). Strikingly, we did not observe any similar increase of SLC38A1/SLC38A2 upon SLC1A5 knockdown, confirming that SLC1A5 is the main transporter of glutamine without significant transporter plasticity or redundancy in fibroblasts. As loss or inhibition of SLC1A5 function depletes glutamine entry into cells and subsequently blocks fibroblast activation, one question remains: whether supplying other amino acids compensates for the loss of SLC1A5 activity or glutamine deficiency. It has been shown that T-cell proliferation is highly sensitive to glutamine concentrations, and this sensitivity is specific, in that glutamine cannot be replaced by supplementing with glutamic acid, proline, or asparagine to restore T-cell proliferation in glutamine-free medium (35). On the other hand, recent studies also suggest that accessibility to other nutrients, particularly nonessential amino acids such as asparagine, aspartate, and arginine, can protect tumor cells from glutamine starvation or from block of glutamine catabolism (36–38). Thus, dissecting differential responses to the loss of SLC1A5 function or limitation of glutamine between fibroblasts and T cells will ensure therapeutic strategies that prevent fibroblast activation while preserving cytotoxic T-cell function. It has been reported that activated fibroblasts self-amplify the fibrotic response by increasing matrix stiffness (39). We asked whether SLC1A5 inhibition influenced the expression of key matrix cross-linking and degradation genes. SLC1A5 antagonism resulted in a shift in transcriptional programs away from a contractile, proliferative, and matrix-depositing state toward a matrix-degrading and softening state potentially linked to fibrosis resolution.
We identified several molecular mechanisms associated with SLC1A5 knockdown or V-9302 treatment. SLC1A5 downregulation or V-9302 exposure resulted in decreased mTOR activity as assessed by pS6K, pAkt, and p4EBP1 concentrations, which is consistent with decreased amino acid metabolism and transport (17, 40). We further determined the roles of MAPK, PI3K, Akt, mTORC1, mTORC2, and SMAD proteins (i.e., pathways all shown to be involved in profibrotic TGF-β signaling) in glutamine transport using a combination of genetic (siRNA) and pharmacological approaches. Our data indicated that TGF-β signaling downstream of PI3K/mTORC2/SMAD2/3 is critical for the induction of SLC1A5, which is consistent with the report that transformed cells with strong PI3K–Akt–mTOR, KRAS, or Myc pathway activation increase their conversion of glutamate to α-KG by glutamate dehydrogenase for metabolism and biosynthesis (41, 42). Several studies have demonstrated a direct link between hypoxia or HIFs in the development of IPF (43, 44), and it is well recognized that the oncogene cMyc enhances glutamine use by directly transactivating the expression of SLC1A5 in cancer (45, 46). Recently, we demonstrated that cMyc is required for TGF-β–induced accumulation of hexokinase 2 in pulmonary fibrosis (47). Here we demonstrated that knockdown of HIF1α, HIF2α, and cMyc prevented the increase in SLC1A5 by TGF-β, whereas siRNA-mediated loss of SLC1A5 downregulated the TGF-β induction of HIF1α, HIF2α, and cMyc, supporting a model of sustained HIF1α, HIF2α, and cMyc induction in TGF-β/SLC1A5–dependent differentiation and activation of myofibroblasts. These findings indicated that HIF1α, HIF2α, and cMyc play distinct roles in glutamine metabolism through SLC1A5 to promote metabolic adaptations. Autophagy is a basic cellular homeostatic process important to cell fate decisions under nutrient-starved conditions. Dysregulation of autophagy affects numerous human diseases, including IPF (48). We observed elevated conversion of LC3-I to LC3-II and beclin-1 after V-9302 exposure in primary human lung fibroblast, which was not unexpected considering the interrelationship among amino acid withdrawal, regulation of mTOR, and autophagy (40). Mitochondria play a central role in energy metabolism, and their dysfunction could induce fibrotic diseases (49). Therefore, understanding the process and mechanism of mitochondrial dysfunction is of great therapeutic value for fibrogenesis. We showed that SLC1A5-inhibited fibroblasts have diminished oxidative phosphorylation and glycolysis, indicating that SLC1A5 is a critical regulator of mitochondrial metabolic reprogramming in fibroblasts.
We next ascertained whether the aforementioned metabolic regulatory network represents a novel approach that can be targeted in a murine treatment model of pulmonary fibrosis. The preclinical efficacy of the SLC1A5 inhibitor V-9302 was assessed in a BLM model of lung fibrosis. V-9302 treatment could robustly block glutamine uptake in a mice tumor model but had no influence in normal tissue and T-cell activation and viability after V-9302 exposure (17). Over the course of V-9302 exposure, liver and kidney histology was similar between C57BL6 male/female mice treated with either V-9302 or vehicle, without any demonstrable effect on murine liver enzymes or circulating blood cell populations. As shown in Figure 7, not only were profibrotic mediators such as Col1a1 (collagen type I alpha 1 chain), Col3a1 (collagen, type III, alpha 1), Col4a1 (collagen type IV alpha 1 chain), Fn, Pai-1 and Ctgf inhibited by V-9302, but SpO2 on room air (an overall indicator of lung physiology that has been shown in various animal models to accurately predict lung function [50]) and lung compliance by flexiVent were similarly improved after SLC1A5 inhibition. Although V-9302 treatment in preclinical studies had no influence on normal tissue or organ homeostasis, without any substantial weight loss, liver or kidney toxicity, or change in T-cell activation and viability (17), further studies are needed to assess the safety and tolerability of different doses of V-9302. The specificity and action of V-9302 in other cell populations (epithelial cells, endothelial cells, and alveolar macrophages) for fibrosis resolution must also be ascertained.
A potential limitation of our study is that about 30 amino acid transporters have been identified in human physiology; however, we show that the fibroproliferative actions of TGF-β reflect an extensive metabolic adaptation through a single transporter, SLC1A5. This finding was surprising given that SLC38A1/2/3 were also reported as glutamine transporters upregulated in cancer with transporter plasticity and redundancy (17). Previous work suggested that SLC1A5 deficiency was compensated by SLC38A1 and SLC38A2 in osteosarcoma cells (34). However, in fibroblasts, we did not observe any significant upregulation of other glutamine transporters upon SLC1A5 knockdown, confirming that there was no significant transporter plasticity or redundancy in these cells. Another potential limitation is that SLC1A5 inhibition by V-9302 in BLM-injured mice is not specific to fibroblasts, and the effects of V-9302 on other lung cell population were not ascertained. As SLC1A5-knockout mice will provide more evidence on the role of SLC1A5 in regulating lung fibrosis, in the future, we will generate conditional knockout of Slc1a5 in fibroblasts. Although we observed that SLC1A5 significantly alters glucose and fatty acid metabolism (data not shown), we believe that further studies will be required to find relevant cross-talk among SLC1A5, glucose, and fatty acid metabolism.
Conclusions
Together, our findings demonstrated that the glutamine transporter SLC1A5 is a gatekeeper for glutamine metabolism and metabolic reprogramming in fibroblasts and that targeting SLC1A5 could be a new strategy for treatment of patients with IPF (see Figure E22). This is the first study, to our knowledge, to demonstrate the utility of a pharmacological inhibitor of glutamine transport in fibrosis, representing a potential new class of therapy targeting cellular metabolism.
Acknowledgments
Acknowledgment
The authors thank Dr. Carol Feghali-Bostwick (Medical University of South Carolina) and Dr. Nathan Sandbo (University of Wisconsin–Madison) for providing primary lung fibroblasts from healthy control subjects and patients with IPF.
Footnotes
Supported by grants from the National Institutes of Health (contract 268201100020C-7-0-1), the Robert N. Brewer Family Foundation, and the Caerus Foundation (A.H.L.); National Heart, Lung, and Blood Institute grant R56HL158549I; the Brewer Family Career Development Award in Support of Idiopathic Pulmonary Fibrosis and Related Interstitial Lung Disease Research; and the Boehringer Ingelheim Discovery Award in Idiopathic Pulmonary Fibrosis/Interstitial Lung Disease (M.C.).
Author Contributions: M.C., D.J.T., and A.H.L. designed the research. M.C., K.J.S., and T.J.K. performed research, with the majority done by M.C. E.S.Y. analyzed mouse lung histology. M.C., D.J.T., and A.H.L. wrote the manuscript. All authors analyzed data and edited the manuscript.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2022-0339OC on July 17, 2023
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1. Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest . 2007;117:524–529. doi: 10.1172/JCI31487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Steele MP, Schwartz DA. Molecular mechanisms in progressive idiopathic pulmonary fibrosis. Annu Rev Med . 2013;64:265–276. doi: 10.1146/annurev-med-042711-142004. [DOI] [PubMed] [Google Scholar]
- 3. Verma S, Slutsky AS. Idiopathic pulmonary fibrosis—new insights. N Engl J Med . 2007;356:1370–1372. doi: 10.1056/NEJMcibr070490. [DOI] [PubMed] [Google Scholar]
- 4. Bors M, Tomic R, Perlman DM, Kim HJ, Whelan TP. Cognitive function in idiopathic pulmonary fibrosis. Chron Respir Dis . 2015;12:365–372. doi: 10.1177/1479972315603552. [DOI] [PubMed] [Google Scholar]
- 5. Raghu G, Selman M. Nintedanib and pirfenidone. New antifibrotic treatments indicated for idiopathic pulmonary fibrosis offer hopes and raises questions. Am J Respir Crit Care Med . 2015;191:252–254. doi: 10.1164/rccm.201411-2044ED. [DOI] [PubMed] [Google Scholar]
- 6. Canestaro WJ, Forrester SH, Raghu G, Ho L, Devine BE. Drug treatment of idiopathic pulmonary fibrosis: systematic review and network meta-analysis. Chest . 2016;149:756–766. doi: 10.1016/j.chest.2015.11.013. [DOI] [PubMed] [Google Scholar]
- 7. Du K, Chitneni SK, Suzuki A, Wang Y, Henao R, Hyun J, et al. Increased glutaminolysis marks active scarring in nonalcoholic steatohepatitis progression. Cell Mol Gastroenterol Hepatol . 2020;10:1–21. doi: 10.1016/j.jcmgh.2019.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Du K, Hyun J, Premont RT, Choi SS, Michelotti GA, Swiderska-Syn M, et al. Hedgehog-YAP signaling pathway regulates glutaminolysis to control activation of hepatic stellate cells. Gastroenterology . 2018;154:1465–1479.e13. doi: 10.1053/j.gastro.2017.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Song J, Meng Y, Wang M, Li L, Liu Z, Zheng K, et al. Mangiferin activates Nrf2 to attenuate cardiac fibrosis via redistributing glutaminolysis-derived glutamate. Pharmacol Res . 2020;157:104845. doi: 10.1016/j.phrs.2020.104845. [DOI] [PubMed] [Google Scholar]
- 10. Tsai HW, Motz KM, Ding D, Lina I, Murphy MK, Benner D, et al. Inhibition of glutaminase to reverse fibrosis in iatrogenic laryngotracheal stenosis. Laryngoscope . 2020;130:E773–E781. doi: 10.1002/lary.28493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bernard K, Logsdon NJ, Benavides GA, Sanders Y, Zhang J, Darley-Usmar VM, et al. Glutaminolysis is required for transforming growth factor-β1-induced myofibroblast differentiation and activation. J Biol Chem . 2018;293:1218–1228. doi: 10.1074/jbc.RA117.000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cui H, Xie N, Jiang D, Banerjee S, Ge J, Sanders YY, et al. Inhibition of glutaminase 1 attenuates experimental pulmonary fibrosis. Am J Respir Cell Mol Biol . 2019;61:492–500. doi: 10.1165/rcmb.2019-0051OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Choudhury M, Yin X, Schaefbauer KJ, Kang JH, Roy B, Kottom TJ, et al. SIRT7-mediated modulation of glutaminase 1 regulates TGF-β-induced pulmonary fibrosis. FASEB J . 2020;34:8920–8940. doi: 10.1096/fj.202000564R. [DOI] [PubMed] [Google Scholar]
- 14. Biancur DE, Paulo JA, Małachowska B, Quiles Del Rey M, Sousa CM, Wang X, et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun . 2017;8:15965. doi: 10.1038/ncomms15965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pochini L, Scalise M, Galluccio M, Indiveri C. Membrane transporters for the special amino acid glutamine: structure/function relationships and relevance to human health. Front Chem . 2014;2:61. doi: 10.3389/fchem.2014.00061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bhutia YD, Babu E, Ramachandran S, Ganapathy V. Amino acid transporters in cancer and their relevance to “glutamine addiction”: novel targets for the design of a new class of anticancer drugs. Cancer Res . 2015;75:1782–1788. doi: 10.1158/0008-5472.CAN-14-3745. [DOI] [PubMed] [Google Scholar]
- 17. Schulte ML, Fu A, Zhao P, Li J, Geng L, Smith ST, et al. Pharmacological blockade of ASCT2-dependent glutamine transport leads to antitumor efficacy in preclinical models. Nat Med . 2018;24:194–202. doi: 10.1038/nm.4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang Z, Liu R, Shuai Y, Huang Y, Jin R, Wang X, et al. ASCT2 (SLC1A5)-dependent glutamine uptake is involved in the progression of head and neck squamous cell carcinoma. Br J Cancer . 2020;122:82–93. doi: 10.1038/s41416-019-0637-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hassanein M, Qian J, Hoeksema MD, Wang J, Jacobovitz M, Ji X, et al. Targeting SLC1a5-mediated glutamine dependence in non-small cell lung cancer. Int J Cancer . 2015;137:1587–1597. doi: 10.1002/ijc.29535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lin J, Yang T, Peng Z, Xiao H, Jiang N, Zhang L, et al. SLC1A5 silencing inhibits esophageal cancer growth via cell cycle arrest and apoptosis. Cell Physiol Biochem . 2018;48:397. doi: 10.1159/000491769. [DOI] [PubMed] [Google Scholar]
- 21. Lian N, Jin H, Zhu W, Zhang C, Qi Y, Jiang M, et al. Inhibition of glutamine transporter ASCT2 mitigates bleomycin-induced pulmonary fibrosis in mice. Acta Histochem . 2022;124:151961. doi: 10.1016/j.acthis.2022.151961. [DOI] [PubMed] [Google Scholar]
- 22. Scalise M, Pochini L, Galluccio M, Console L, Indiveri C. Glutamine transport and mitochondrial metabolism in cancer cell growth. Front Oncol . 2017;7:306. doi: 10.3389/fonc.2017.00306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Geldermalsen M, Wang Q, Nagarajah R, Marshall AD, Thoeng A, Gao D, et al. ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene . 2016;35:3201–3208. doi: 10.1038/onc.2015.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao X, Kwan JYY, Yip K, Liu PP, Liu FF. Targeting metabolic dysregulation for fibrosis therapy. Nat Rev Drug Discov . 2020;19:57–75. doi: 10.1038/s41573-019-0040-5. [DOI] [PubMed] [Google Scholar]
- 25. Rahimi RA, Andrianifahanana M, Wilkes MC, Edens M, Kottom TJ, Blenis J, et al. Distinct roles for mammalian target of rapamycin complexes in the fibroblast response to transforming growth factor-beta. Cancer Res . 2009;69:84–93. doi: 10.1158/0008-5472.CAN-08-2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xiong A, Liu Y. Targeting hypoxia inducible factors-1α as a novel therapy in fibrosis. Front Pharmacol . 2017;8:326. doi: 10.3389/fphar.2017.00326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Qu A, Taylor M, Xue X, Matsubara T, Metzger D, Chambon P, et al. Hypoxia-inducible transcription factor 2α promotes steatohepatitis through augmenting lipid accumulation, inflammation, and fibrosis. Hepatology . 2011;54:472–483. doi: 10.1002/hep.24400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shen Y, Miao N, Wang B, Xu J, Gan X, Xu D, et al. c-Myc promotes renal fibrosis by inducing integrin αv-mediated transforming growth factor-β signaling. Kidney Int . 2017;92:888–899. doi: 10.1016/j.kint.2017.03.006. [DOI] [PubMed] [Google Scholar]
- 29. Bernard K, Logsdon NJ, Ravi S, Xie N, Persons BP, Rangarajan S, et al. Metabolic reprogramming is required for myofibroblast contractility and differentiation. J Biol Chem . 2015;290:25427–25438. doi: 10.1074/jbc.M115.646984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harding JJ, Telli ML, Munster PN, Le MH, Molineaux C, Bennett MK, et al. Safety and tolerability of increasing doses of CB-839, a first-in-class, orally administered small molecule inhibitor of glutaminase, in solid tumors [abstract] J Clin Oncol . 2015;33:2512. [Google Scholar]
- 31. Najumudeen AK, Ceteci F, Fey SK, Hamm G, Steven RT, Hall H, et al. CRUK Rosetta Grand Challenge Consortium The amino acid transporter SLC7A5 is required for efficient growth of KRAS-mutant colorectal cancer. Nat Genet . 2021;53:16–26. doi: 10.1038/s41588-020-00753-3. [DOI] [PubMed] [Google Scholar]
- 32. Liu Y, Zhao T, Li Z, Wang L, Yuan S, Sun L. The role of ASCT2 in cancer: a review. Eur J Pharmacol . 2018;837:81–87. doi: 10.1016/j.ejphar.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 33. Lomenick B, Hao R, Jonai N, Chin RM, Aghajan M, Warburton S, et al. Target identification using drug affinity responsive target stability (DARTS) Proc Natl Acad Sci U S A . 2009;106:21984–21989. doi: 10.1073/pnas.0910040106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bröer A, Gauthier-Coles G, Rahimi F, van Geldermalsen M, Dorsch D, Wegener A, et al. Ablation of the ASCT2 (SLC1A5) gene encoding a neutral amino acid transporter reveals transporter plasticity and redundancy in cancer cells. J Biol Chem . 2019;294:4012–4026. doi: 10.1074/jbc.RA118.006378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol . 2010;185:1037–1044. doi: 10.4049/jimmunol.0903586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pavlova NN, Hui S, Ghergurovich JM, Fan J, Intlekofer AM, White RM, et al. As extracellular glutamine levels decline, asparagine becomes an essential amino acid. Cell Metab . 2018;27:428–438.e5. doi: 10.1016/j.cmet.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Alkan HF, Walter KE, Luengo A, Madreiter-Sokolowski CT, Stryeck S, Lau AN, et al. Cytosolic aspartate availability determines cell survival when glutamine is limiting. Cell Metab . 2018;28:706–720.e6. doi: 10.1016/j.cmet.2018.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lowman XH, Hanse EA, Yang Y, Ishak Gabra MB, Tran TQ, Li H, et al. p53 promotes cancer cell adaptation to glutamine deprivation by upregulating Slc7a3 to increase arginine uptake. Cell Rep . 2019;26:3051–3060.e4. doi: 10.1016/j.celrep.2019.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Haak AJ, Kostallari E, Sicard D, Ligresti G, Choi KM, Caporarello N, et al. Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci Transl Med . 2019;11:eaau6296. doi: 10.1126/scitranslmed.aau6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell . 2009;136:521–534. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jeong SM, Xiao C, Finley LW, Lahusen T, Souza AL, Pierce K, et al. SIRT4 has tumor-suppressive activity and regulates the cellular metabolic response to DNA damage by inhibiting mitochondrial glutamine metabolism. Cancer Cell . 2013;23:450–463. doi: 10.1016/j.ccr.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Csibi A, Fendt SM, Li C, Poulogiannis G, Choo AY, Chapski DJ, et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell . 2013;153:840–854. doi: 10.1016/j.cell.2013.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Tzouvelekis A, Harokopos V, Paparountas T, Oikonomou N, Chatziioannou A, Vilaras G, et al. Comparative expression profiling in pulmonary fibrosis suggests a role of hypoxia-inducible factor-1alpha in disease pathogenesis. Am J Respir Crit Care Med . 2007;176:1108–1119. doi: 10.1164/rccm.200705-683OC. [DOI] [PubMed] [Google Scholar]
- 44. Goodwin J, Choi H, Hsieh MH, Neugent ML, Ahn JM, Hayenga HN, et al. Targeting hypoxia-inducible factor-1α/pyruvate dehydrogenase kinase 1 axis by dichloroacetate suppresses bleomycin-induced pulmonary fibrosis. Am J Respir Cell Mol Biol . 2018;58:216–231. doi: 10.1165/rcmb.2016-0186OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zhao X, Petrashen AP, Sanders JA, Peterson AL, Sedivy JM. SLC1A5 glutamine transporter is a target of MYC and mediates reduced mTORC1 signaling and increased fatty acid oxidation in long-lived Myc hypomorphic mice. Aging Cell . 2019;18:e12947. doi: 10.1111/acel.12947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yue M, Jiang J, Gao P, Liu H, Qing G. Oncogenic MYC activates a feedforward regulatory loop promoting essential amino acid metabolism and tumorigenesis. Cell Rep . 2017;21:3819–3832. doi: 10.1016/j.celrep.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 47. Yin X, Choudhury M, Kang JH, Schaefbauer KJ, Jung MY, Andrianifahanana M, et al. Hexokinase 2 couples glycolysis with the profibrotic actions of TGF-β. Sci Signal . 2019;12:eaax4067. doi: 10.1126/scisignal.aax4067. [DOI] [PubMed] [Google Scholar]
- 48. Patel AS, Lin L, Geyer A, Haspel JA, An CH, Cao J, et al. Autophagy in idiopathic pulmonary fibrosis. PLoS One . 2012;7:e41394. doi: 10.1371/journal.pone.0041394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li X, Zhang W, Cao Q, Wang Z, Zhao M, Xu L, et al. Mitochondrial dysfunction in fibrotic diseases. Cell Death Discov . 2020;6:80. doi: 10.1038/s41420-020-00316-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Verhoeven D, Teijaro JR, Farber DL. Pulse-oximetry accurately predicts lung pathology and the immune response during influenza infection. Virology . 2009;390:151–156. doi: 10.1016/j.virol.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]