

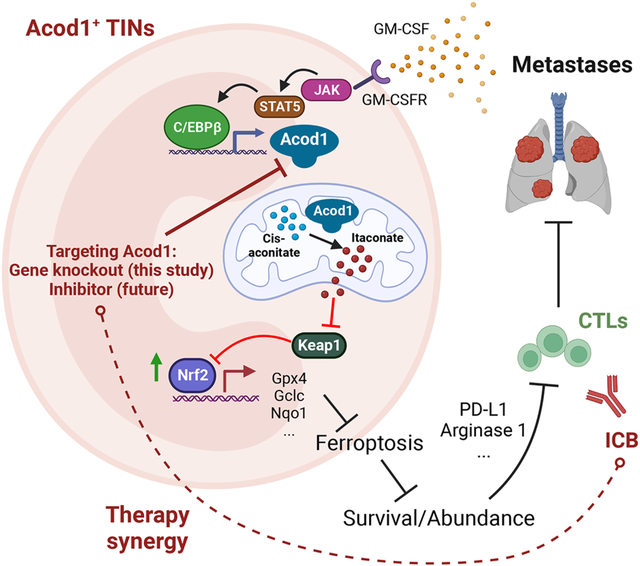
SUMMARY
Metastasis causes breast cancer-related mortality. Tumor-infiltrating neutrophils (TINs) inflict immunosuppression and promote metastasis. Therapeutic debilitation of TINs may enhance immunotherapy, yet it remains a challenge to identify therapeutic targets highly expressed and functionally essential in TINs but under-expressed in extra-tumoral neutrophils. Here, using single-cell RNA-sequencing to compare TINs and circulating neutrophils in murine mammary tumor models, we identified aconitate decarboxylase 1 (Acod1) as the most upregulated metabolic enzyme in mouse TINs and validated high Acod1 expression in human TINs. Activated through the GM-CSF-JAK/STAT5-C/EBPβ pathway, Acod1 produces itaconate which mediates Nrf2-dependent defense against ferroptosis and upholds the persistence of TINs. Acod1 ablation abates TIN infiltration, constrains metastasis (but not primary tumors), bolsters antitumor T cell immunity, and boosts the efficacy of immune checkpoint blockade. Our findings reveal how TINs escape from ferroptosis through the Acod1-dependent immunometabolism switch and establish Acod1 as a target to offset immunosuppression and improve immunotherapy against metastasis.
Keywords: Breast cancer, metastasis, single cell RNA-sequencing, neutrophil, MDSC, Acod1, itaconate, ferroptosis, immune metabolism, immune checkpoint blockade
Graphical Abstract
eTOC blurb
Zhao et al. report the induction of Acod1 in tumor-infiltrating neutrophils (TINs) and its critical role in blunting ferroptosis and sustaining viability for TINs. Acod1 ablation abates TIN density, constrains breast cancer metastasis, bolsters antitumor T cell immunity, and boosts the the anti-metastasis efficacy of immune checkpoint blockade in mice.
INTRODUCTION
Metastasis is the major cause of mortality and morbidity for breast cancer (BC). Immune checkpoint blockade (ICB) drugs, atezolizumab and pembrolizumab, in combination with chemotherapy, are approved to treat patients with PD-L1+ metastatic triple-negative BC (TNBC)1. However, immunotherapy resistance remains a formidable challenge.
Patients with various cancer types, including BC, often exhibit increased neutrophil-to-lymphocyte ratio (NLR) as an independent predictor of mortality2. Immune cell deconvolution identified tumor-infiltrating neutrophils (TINs) as the immune population with the strongest correlation with poor outcomes across 25 cancer types, including BC3.Numerous preclinical studies uphold the pro-tumor and pro-metastasis functions of TINs, whereas the antitumor activities of TINs are often found in early-stage diseases4. Solid tumors upregulate cytokines to induce surged granulopoiesis, and cytokines such as G-CSF and GM-CSF promote the extended survival of TINs5. A predominant pro-tumor/pro-metastasis function of TINs is suppressing effector T cells and natural killer cells6,7, suggesting the equivalent identity between TINs and polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs). For BC, a tumor microenvironment (TME) enriched with TINs is resistant to ICB8. Depletion or therapeutic targeting of immunosuppressive TINs is promising to elicit synergistic efficacy when combined with immunotherapy9. Nonetheless, therapeutic targeting of neutrophils must solve challenges in selectivity and safety10. One possibility is identifying therapeutic targets highly expressed and functionally essential in TINs but absent or under-expressed in extra-tumoral neutrophils.
Aconitate decarboxylase 1 (Acod1) catalyzes the reaction from cis-aconitate (an intermediate of the TCA cycle) to itaconate. Acod1 expression is induced in macrophages by pathogen infection, Toll-like receptor ligands, and specific inflammatory cytokines (e.g. IFNβ, IFNγ, TNF)11,12. Itaconate has an overall anti-inflammatory effect and exerts activities such as inhibition of succinate dehydrogenase and glycolysis, activation of transcription factors Nrf2 and ATF3, and inhibition of the NLRP3 inflammasome11,12. Most studies on Acod1 have been focused on macrophages, leaving the function of Acod1 in neutrophils (especially TINs) elusive.
Here, we applied single-cell RNA sequencing (scRNA-seq) and identified Acod1 to be the most upregulated enzyme-encoding gene in TINs. Acod1 was critical to blunting ferroptosis of TINs and promoting lung metastasis. Acod1 expression ablation in the host improved the anti-metastasis activity of cytotoxic T lymphocytes (CTLs) reinvigorated with ICB therapy. These findings connect neutrophil immunometabolic rewiring to the immunosuppressive barrier in metastasis. Acod1 emerges as a promising biomarker and therapeutic target for TINs. The finding on how TINs avert ferroptosis to stay alive and immunosuppressive supports the ongoing efforts to develop ferroptosis inducers as a potential cancer treatment.
RESULTS
TINs in the mammary TME demonstrate potent immunosuppression.
Consistent with the previous report3, we confirmed that TINs are associated with adverse outcomes in human BC based on immune cell deconvolution (Figure S1A). To study TINs in BC, we used both syngeneic and spontaneous murine mammary tumor models. The C57BL/6-syngeneic line E0771 is often used as a model of TNBC although some consider it luminal B subtype13. MMTV-PyMT transgenic mice, the most commonly used genetically engineered mouse (GEM) model of BC, develop autochthonous mammary adenocarcinoma with pulmonary metastasis14. We developed the cell line Py7160 from an MMTV-PyMT tumor (FVB background). The MMTV-PyMT transgenic mice used in this study are in C57BL/6 background.
First, we characterized neutrophils in these models and confirmed their significant accumulation in the blood, mammary tumors, and lung with metastases (Figure 1A). Metastasized lungs contained 4-fold higher TINs compared with primary tumors. Neutrophils from blood or tumors were purified with αLy6G microbeads (purity >95%) (Figure S1B) and tested for immunosuppression on CD4+ and CD8+ T cells (stimulated with αCD3 and αCD28). TINs from E0771-bearing mice potently suppressed CD4+ and CD8+ T cell proliferation compared with blood neutrophils (BNs) from tumor-free and tumor-bearing mice (Figure 1B). TINs can suppress T cells through various mechanisms including PD-L1 and arginase 1 (Arg1) expression5. We confirmed that PD-L1 and Arg1 were significantly upregulated in TINs isolated from E0771 tumors (Figure S1C and S1D). Inhibition of PD-L1 or Arg1 significantly attenuated the suppression of T cells by TINs (Figure S1E). These TINs are bona fide PMN-MDSCs.
Figure 1. Neutrophils in the mammary TME demonstrate potent immunosuppression and distinct transcriptomic features.
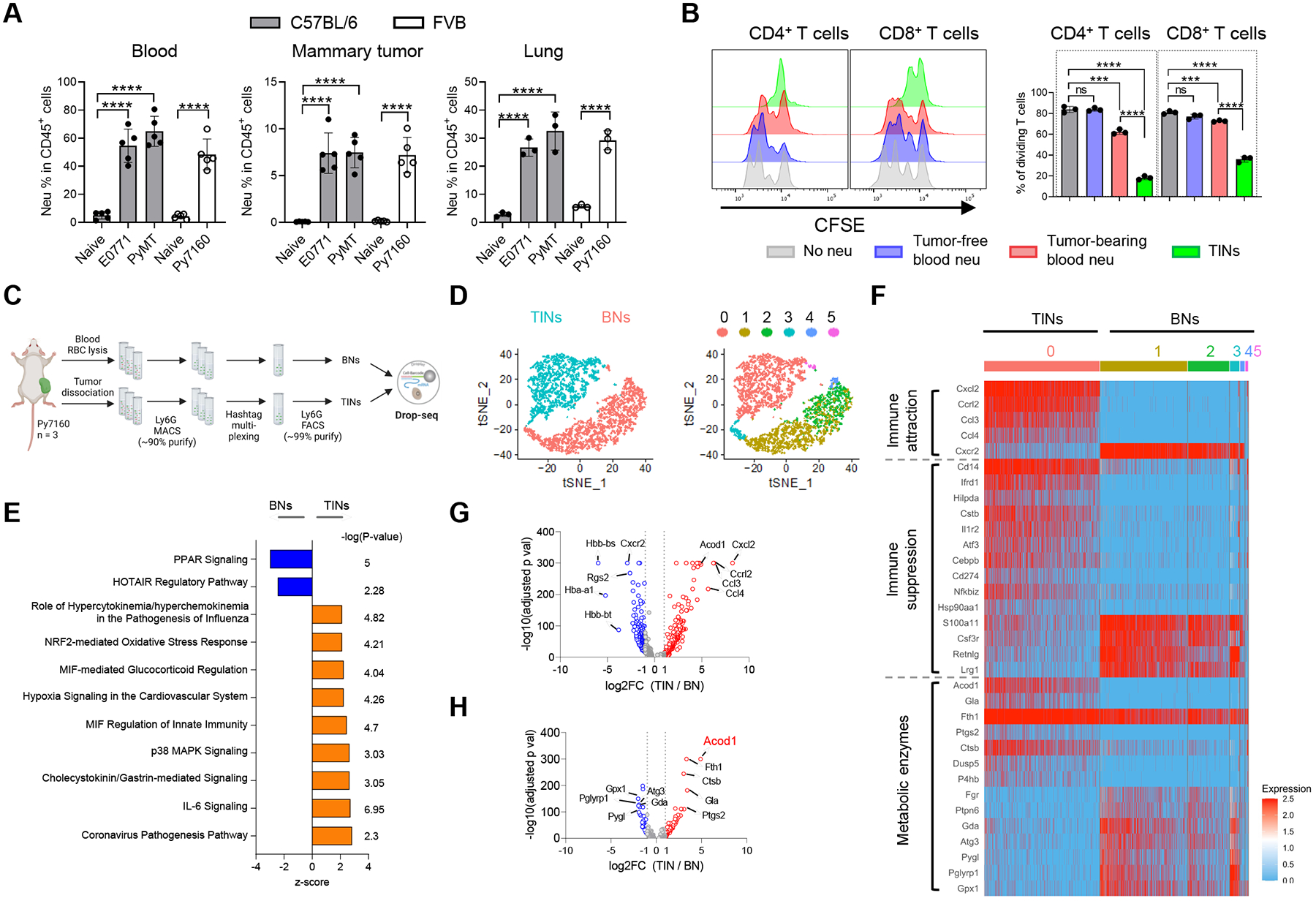
(A) Flow cytometry analysis of neutrophil populations (CD11b+Ly6G+) in blood (n=5), mammary tumors (n=5) and lung metastases (n=3) of mouse BC models. (B) CFSE dilution histograms and proliferating proportions of αCD3/αCD28-stimulated spleen T cell subsets cocultured with neutrophils (1:1 ratio) isolated from tumor-free or E0771 tumor-bearing mice (n=3). (C) Schematic for sample processing, neutrophil sorting and Drop-seq of BNs and TINs from Py7160-bearing mice (n=3). (D) t-distributed stochastic neighbor embedding (t-SNE) plots colored by tissue of origin (left) or cell clusters (right). (E) Top-ranked IPA pathways upregulated in TINs or BNs. (F) Heatmap of most differentially expressed genes by TINs and BNs associated with immune attraction, immune suppression, and metabolic enzymes. (G) Volcano plot of differentially expressed genes (adjusted p values < 0.05). (H) Volcano plot of differentially expressed genes encoding metabolic enzymes (adjusted p values < 0.05). For A and B, data represent mean ± s.e.m.; ns, not significant, ***P<0.001, ****P<0.0001, unpaired two-tailed Student’s t-test. See also Figure S1 and Table S1–S3.
scRNA-seq reveals distinct transcriptomes between TINs and blood neutrophils.
To identify gene expression changes specific to TINs, we sorted Ly6G+ BNs and TINs from Py7160-bearing mice (n=3), multiplexed with cell hashing and profiled with Drop-seq (Figure 1C). In total, 3,645 single cells expressing at least 100 but no more than 2000 genes per cell were further analyzed. BNs and TINs were markedly separated on the tSNE plot, indicating distinct transcriptomic activities (Figure 1D and S1F). Unbiased clustering (resolution = 0.3) identified 6 clusters of the neutrophils, C1-C5 for BNs and C0 for TINs (Figure 1D). Ingenuity pathway analysis (IPA) of differentially expressed genes between BNs and TINs identified enrichment of multiple pathways in TINs, including coronavirus pathogenesis, IL-6 signaling, p38 MAPK signaling, MIF regulation of innate immunity, and NRF2-mediated oxidative stress response (Figure 1E, Table S1). Among the differentially expressed genes, TINs showed upregulation of genes with known functions in immunocyte attraction (Cxcl2, Ccrl2, Ccl3 and Ccl4) and immunosuppression (Ifrd1, Hilpda, Il1r2, Atf3, Cebpb and Cd274) (Figure 1F and 1G, Table S2). To identify metabolism-related gene expression changes, KEGG-catologed metabolic enzyme genes with expression reads in BNs and TINs were analyzed. TINs upregulated a set of enzymes including Ptgs2 which mediates prostaglandin E2 production to induce immunosuppression15 (Figure 1F and 1H, Table S3). Acod1 was the most significantly upregulated enzyme-coding gene (Figure 1H), intriguing us to explore it in TINs.
Acod1 is highly expressed in TINs of primary and metastatic BC in mice and patients.
We examined Acod1 expression in various ways. Acod1 protein was dramatically higher in mammary tumors and metastasized lung compared with blood leukocytes of E0771-bearing mice or tumor-free lung (Figure 2A). We purified neutrophils from different body parts of tumor-bearing mice and utilized qRT-PCR to assess Acod1 expression. Across three models (E0771, Py7160 and MMTV-PyMT), neutrophils from metastasized lung and primary tumors expressed Acod1 at a much higher level than neutrophils from bone marrow, blood, or spleen (Figure 2B). When tumor-free and tumor-bearing mice were compared, neutrophils from blood and bone marrow expressed Acod1 at a similarly negligible level, spleen neutrophils in tumor-bearing mice expressed Acod1 at a moderately higher level, lung neutrophils in metastasis-bearing mice expressed Acod1 at a dramatically higher level (Figure S2A). This result indicates that the elevated Acod1 expression in neutrophils of tumor-bearing mice is due to the influence of the tumor instead of particular tissue environments. To inspect Acod1 expression in different immune populations, we conducted immunofluorescence (IF) co-staining or qRT-PCR of FACS-sorted immune subsets from E0771 tumors. Acod1 expression was restricted to myeloid cells and TINs expressed Acod1 at the highest level (Figure 2C–2E and S2B). Over 80% of Ly6G+ cells expressed Acod1 in E0771-colonized lung (Figure 2F and 2G), whereas Ly6G+ cells in tumor-free lung were undetectable (Figure S2C). Re-analysis of a published transcriptomic dataset of four syngeneic mouse models of mammary tumors8 confirmed that Acod1 was expressed substantially higher in TINs than macrophages or cancer cells (Figure S2D).
Figure 2. Acod1 is highly expressed in TINs of primary and metastatic BC in mice and patients.
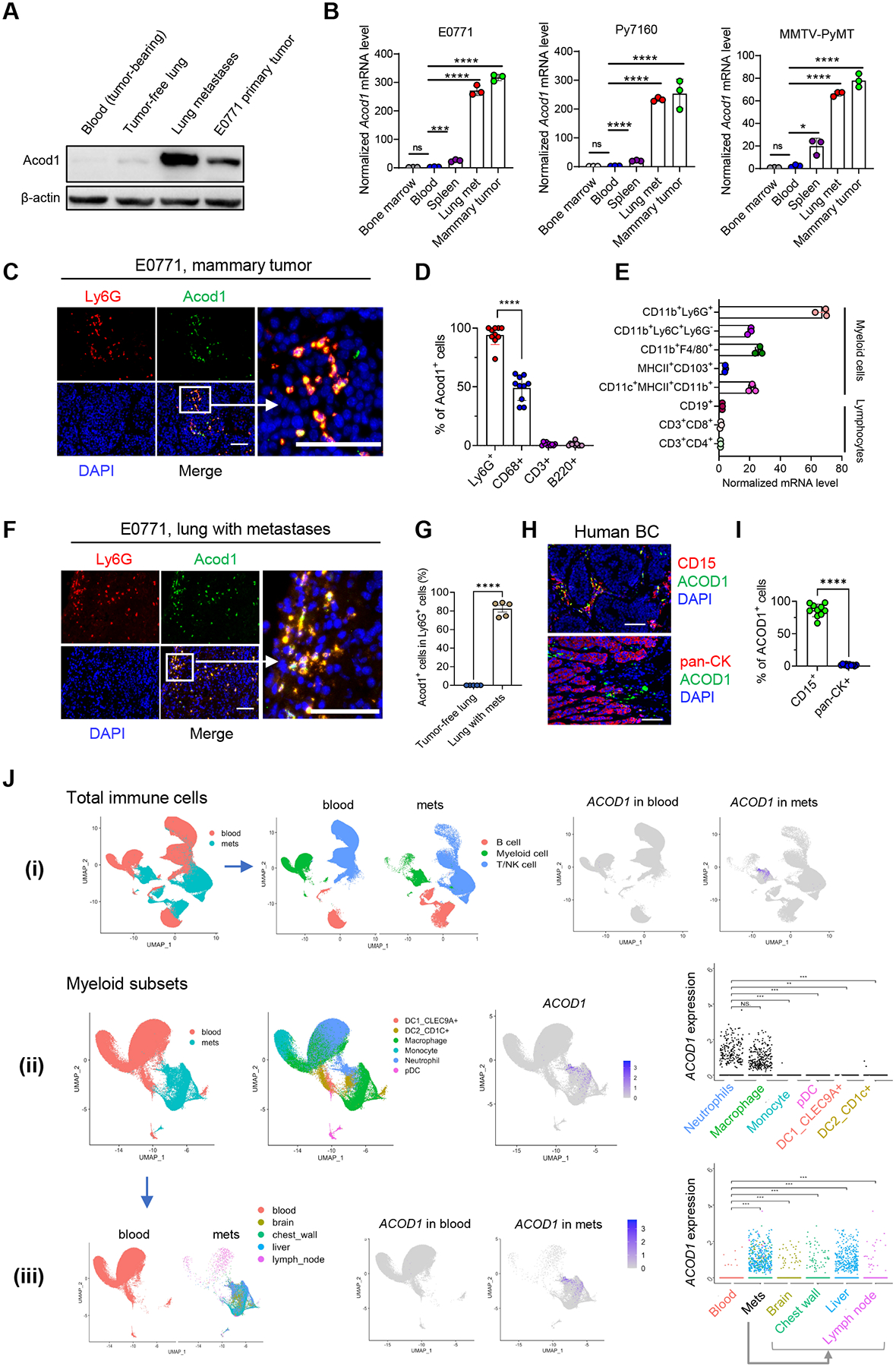
(A) Western blot to assess Acod1 expression in blood or tissues of tumor-free and E0771-bearing mice. (B) qRT-PCR to assess Acod1 expression in neutrophils isolated from tumor-bearing mice of syngeneic and GEM models (n=3). (C) IF co-staining of Acod1 and Ly6G in E0771 primary tumors. (D) Percentage of Acod1+ cells in different immune cells (neutrophils, Ly6G+; macrophages, CD68+; T cells, CD3+; B cells, B220+) based on IF co-staining of E0771 primary tumors (n=10). (E) qRT-PCR quantification of Acod1 in lymphocyte subsets and myeloid subsets sorted from E0771 tumors (n=3). (F) IF co-staining of Acod1 and Ly6G in E0771 lung metastases. (G) Percentage of Acod1+ neutrophils in tumor-free lung or lung with E0771 metastases (n=5). (H-I) Representative image and quantitative result of IF co-staining of ACOD1 and CD15 or pan-Cytokeratin in human BC tissues (n=10). (J) Analysis of scRNA-seq dataset GSE169246 of blood and metastasized tissues of human BC. for ACOD1 expression pattern. (i) UMAP of total CD45+ cells segregated into three main immune populations for blood and metastases (mets) featuring ACOD1 expression (rightmost). (ii) UMAP of myeloid cells segregated into six subsets plotting ACOD1 expression (rightmost). (iii) UMAP of myeloid cells for blood and mets separately featuring ACOD1 expression in different sites (rightmost). In B, D, E, G, I and J, data represent mean ± s.e.m.; ns, not significant, *P<0.05, **P<0.01,***P<0.001, ****P<0.0001, unpaired two-tailed Student’s t-test. In C, F, H, scale bar 100μm. See also Figure S2.
To examine Acod1 expression pattern in human cancers, we first explored the TCGA database and found ACOD1 was highly expressed in multiple cancer types (including BC) compared with normal tissues (Figure S2E). We also observed positive correlations of ACOD1 expression with markers of neutrophils (CD15, CD11b, CD33) and PMN-MDSCs (LOX1) in BC TCGA (Figure S2F). Moreover, GSEA analysis of the BC TCGA data showed that neutrophil gene signatures were enriched in samples with high ACOD1 expression (Figure S2G). Next, we used archived human primary BC specimens and co-stained ACOD1 with CD15 or pan-Cytokeratin, which showed that ACOD1 expression was evident in >85% of CD15+ cells but undetectable in pan-Cytokeratin+ cancer cells (Figure 2H and 2I). To assess the expression of ACOD1 in human BC metastases, we analyzed published scRNA-seq and bulk transcriptomics datasets focused on metastatic BC. For scRNA-seq, we analyzed GSE169246 which profiled blood and various metastases (48 blood, 6 chest wall, 6 liver, 1 brain, 9 lymph node)16. Consistent with results from mouse models, ACOD1 was predominantly expressed by neutrophils and macrophages among immune cells across various metastasis sites but not expressed in the blood (Figure 2J). We also analyzed GSE46141 which profiled 91 BC metastasis samples of six anatomical sites17. Consistent with TCGA primary BC (Figure S2F), metastatic BC showed positive correlations between ACOD1 expression and markers of neutrophils (CD15, CD11b, CD33) and PMN-MDSCs (LOX1) (Figure S2H), and enriched for neutrophil gene signatures in metastases with high ACOD1 expression (Figure S2I). Together, these results demonstrate Acod1 overexpression in TINs of primary and metastatic BC in both mouse models and human patients.
Neutrophil Acod1 promotes lung metastasis in mouse BC models.
To assess the function of Acod1, we designed an experiment using E0771 genetically labeled with mcherry or TR (tk-GFP-luciferase triple reporter) to evaluate primary tumors and lung metastasis in wild-type (WT) and Acod1‒/‒ mice (Figure 3A). Primary tumors grew at similar rates (Figure S3A), yet lung metastasis burden was significantly lower in Acod1‒/‒ than WT (Figure 3B and 3C), corresponding to the extended survival of Acod1‒/‒ mice (Figure 3D). The results remained consistent when metastasis was established with non-labeled E0771 (Figure S3B), ruling out the TR protein as a confounding factor. Conversely, we treated WT mice intravenously (i.v.) injected with E0771-TR with PBS or dimethyl itaconate (DMI), a widely used itaconate derivative (Figure 3E). DMI promoted lung metastasis and shortened survival (Figure 3F–3H), but DMI did not affect primary tumor growth (Figure S3C).
Figure 3. Neutrophil Acod1 promotes lung metastasis in mouse BC models.
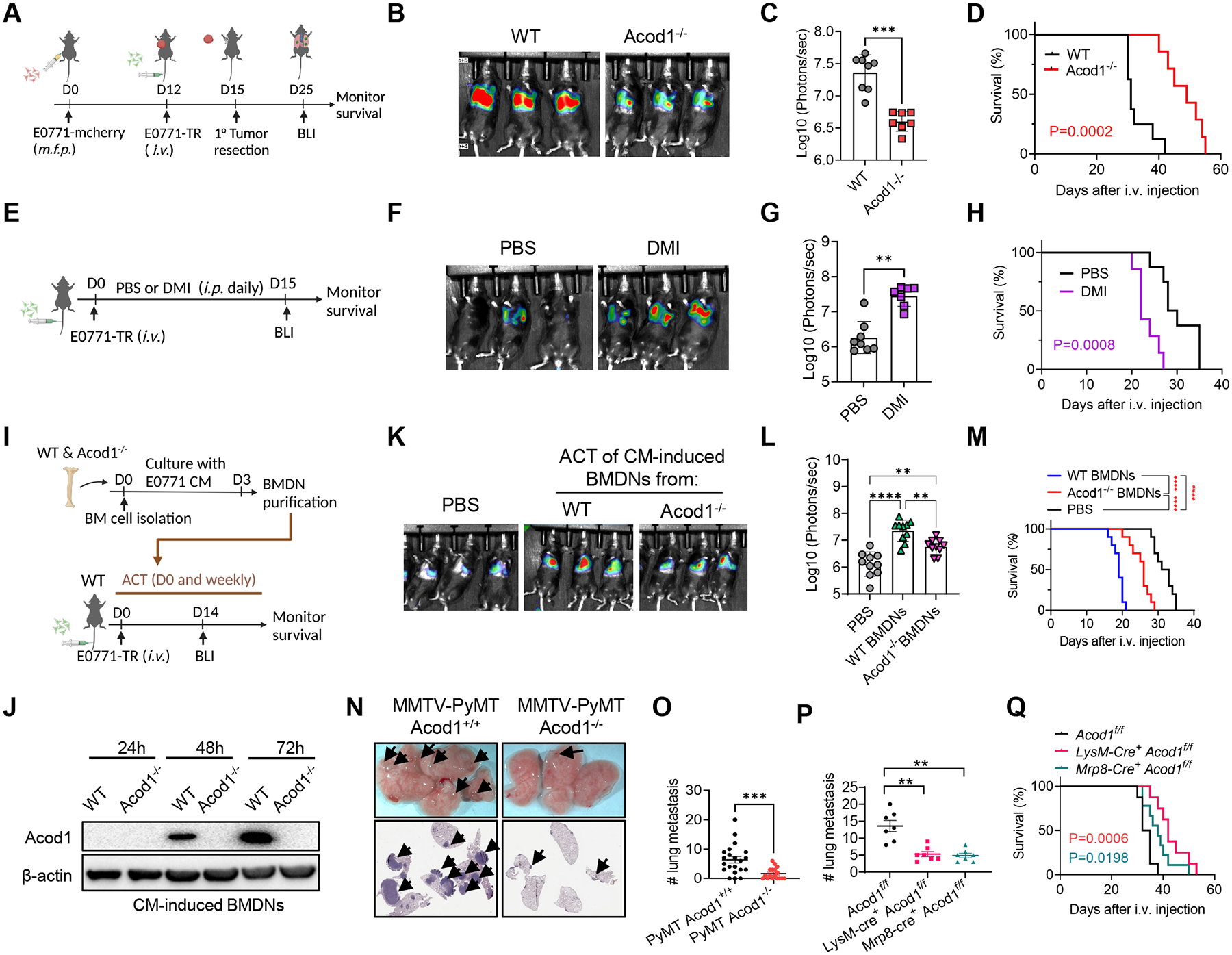
(A) Experimental lung metastasis assay scheme based on E0771. (B-C) Representative BLI images (n=3) and quantification (WT n=8, Acod1‒/‒ n=7) at day 15 after i.v. injection of E0771-TR. (D) Survival curves (WT n=8, Acod1‒/‒ n=7). (E) Assessing the effect of PBS or DMI (100mg kg‒1, daily i.p.) on E0771-TR lung metastasis grown in WT mice. (F-G) Representative BLI images (n=3) and quantification (PBS n= 8, DMI n=7) at day 15 after i.v. injection of E0771-TR. (H) Survival curves (PBS n= 8, DMI n=7). (I) Schematic of ACT of WT or Acod1‒/‒ BMDNs cultured with E0771 CM into WT cohorts i.v. injected of E0771-TR. (J) Western blot of Acod1 for WT or Acod1‒/‒ BMDNs cultured with E0771 CM for 24h, 48h or 72h. (K-L) Representative BLI images and quantification (n=10) at day 14 after i.v. injection of E0771-TR. (M) Survival curves (n=10). (N) Representative photographs and HE staining of lungs in MMTV-PyMT Acod1+/+ and MMTV-PyMT Acod1‒/‒ cohorts when mice died from primary tumors. Arrows denote metastases. (O) Quantification of lung metastasis nodules (MMTV-PyMT Acod1+/+ n=21, MMTV-PyMT Acod1‒/‒ n=17). (P) Quantification of lung metastasis nodules in cohorts Acod1 f/f, LysM-Cre+ Acod1f/f and Mrp8-Cre+ Acod1f/f, 2 weeks after i.v. injection of E0771 (n=7/group). (Q) Survival of Acod1 f/f (n=8), LysM-Cre+ Acod1f/f (n=9) and Mrp8-Cre+ Acod1f/f (n=9) mice after i.v. injection of E0771. For C, G, O and P, data represent mean ± s.e.m.; **P<0.01, ***P<0.001, Mann-Whitney test. For L, data represent mean ± s.e.m.; **P<0.01, ****P<0.0001, one-way ANOVA with Tukey’s multiple comparisons test. For D, H, M and Q, log-rank test with P values labeled or ****P<0.0001. See also Figure S3.
Next, we used neutrophil adoptive cell transfer (ACT). Bone marrow cells from WT and Acod1‒/‒ mice were cultured in the presence of E0771 conditioned medium (CM) for 3 days followed by αLy6G-purification of bone marrow-derived neutrophils (BMDNs) (Figure 3I). The purity and viability of myeloid subsets before and after purification were characterized, and neutrophils with high purity (97%) and viability (91%) were used for ACT (Figure S3D). BMDNs from WT but not Acod1‒/‒ mice showed CM-induced Acod1 expression (Figure 3J). WT mice were i.v. injected with E0771-TR cells, received ACT of the same numbers of viable CM-induced WT and Acod1‒/‒ BMDNs on day 0 and weekly after. A control group was included with PBS injected weekly. Mice receiving CM-induced Acod1‒/‒ BMDNs developed less lung metastasis (Figure 3K and 3L) and survived longer than those receiving CM-induced WT BMDNs (Figure 3M). In contrast, the PBS group developed the least metastasis and survived longer than both BMDN-injected groups. In a second ACT experiment, we used Acod1‒/‒ mice as the recipients and compared the impact of ACT of CM-induced WT and Acod1‒/‒ BMDNs on E0771-TR lung metastasis. The result showed again that mice injected with Acod1‒/‒ BMDNs developed less lung metastasis and survived longer (Figure S3E–S3H). In these two experiments, it was noticeable that the survival of both ACT recipient cohorts was shorter than mice without ACT (e.g., WT in Figure 3D, PBS in Figure 3H and 3M). Conceivably, the exogenous immunosuppression from the weekly neutrophil transfers had accelerated metastasis and shortened survival for both groups. Nonetheless, the separation of the two survival curves between WT and Acod1‒/‒ BMDN transfers upholds the role of Acod1 in neutrophils. Moreover, if the ACT procedure caused an overall induced immune response in the recipient mice, we expect the blood leukocyte counts and cytokine levels to rise in WT and Acod1‒/‒ BMDN transfer groups similarly as in PBS group. However, the change of blood leukocytes only occurred immediately after the neutrophil transfer and a global upregulation of inflammatory cytokines was absent, suggesting an overall immune response is unlikely to explain the overall survival shortening (Figure S3I–S3J).
Syngeneic models are rapid but cannot recapitulate the full spectrum of tumor evolution and multistep metastasis. To address this, we generated MMTV-PyMT Acod1+/+ and MMTV-PyMT Acod1‒/‒ cohorts. Consistent with the result of the E0771 model, Acod1 deficiency did not affect primary tumor onset (Figure S3K), yet significantly reduced the number of lung metastases (Figure 3N and 3O).
To specifically abolish Acod1 expression in neutrophils, we generated three cohorts: LysM-Cre+Acod1f/f, Mrp8-Cre+ Acod1f/f and Ly6G-Cre+ Acod1f/f. While LysM-Cre is active in neutrophils, monocytes/macrophages and some dendritic cells, Mrp8-Cre and Ly6G-Cre are specific to neutrophils18,19. Acod1 was effectively ablated in TINs isolated from E0771 lung metastases in LysM-Cre+Acod1f/f and Mrp8-Cre+ Acod1f/f mice, but only marginally affected in Ly6G-Cre+ Acod1f/f mice (Figure S3L). We confirmed that Ly6G-Cre was restricted to neutrophils by analyzing the peripheral blood of Ly6G-Cre+ tdTomatoLSL mice (Figure S3M). Interestingly, the limited Acod1 knockout by Ly6G-Cre echoes this Cre driver’s known floxed-allele-dependent variation of Cre-recombination4. Concordant with the different Acod1 knockout efficiency, LyzM-Cre+Acod1f/f and Mrp8-Cre+ Acod1f/f mice i.v. injected with E0771 developed less lung metastasis (Figure 3P and Figure S3N) and survived longer compared to Acod1f/f control mice (Figure 3Q), whereas Ly6G-Cre+ Acod1f/f did not show difference (Figure S3O). Primary tumor growth was unaffected in the three cohorts (Figure S3P).
Both whole-body or neutrophil-specific Acod1 loss reduced lung metastasis but had little effect on primary tumors in the BC models, which might reflect the stage-dependent requirement of neutrophils in these models. Indeed, neutrophil depletion with αLy6G antibody delayed animal death caused by E0771 lung metastasis (Figure S3Q) but did not affect primary tumor growth (Figure S3R). This result is consistent with previous reports on the roles of neutrophils and macrophages in MMTV-PyMT or E0771 models6,20 and buttresses the critical function of Acod1 in TIN-dependent lung metastasis.
Tumor-secreted GM-CSF induces Acod1 in neutrophils through the STAT5-C/EBPβ axis.
What drives Acod1 upregulation in TINs? To address this question, we utilized two in vitro models, CM-induced BMDNs (described above) and estrogen-regulated Hoxb8-derived neutrophils (ER-Hoxb8-DNs). ER-Hoxb8-DNs recapitulate the cardinal functions of neutrophils21. We optimized a protocol to generate TIN-like cells by adding tumor cell CM to ER-Hoxb8-DNs (Figure 4A). CM from 4 murine mammary cancer cell lines (E0771, Py7160, 168FARN and 4T1) all induced Acod1 expression in ER-Hoxb8-DNs (Figure 4B) and BMDNs (Figure S4A). We measured cytokine levels in 4T1 and E0771 CM with a mouse cytokine array and detected 8 factors, including GM-CSF, G-CSF and TIMP-1 and chemokines CCL2, CCL5, CXCL1, CXCL2 and CXCL10 from both cell lines (Figure S4B). These cytokines were screened and only GM-CSF significantly upregulated Acod1 in ER-Hoxb8-DNs (Figure 4C).
Figure 4. Tumor-secreted GM-CSF induces Acod1 in neutrophils through the STAT5-C/EBPβ axis.
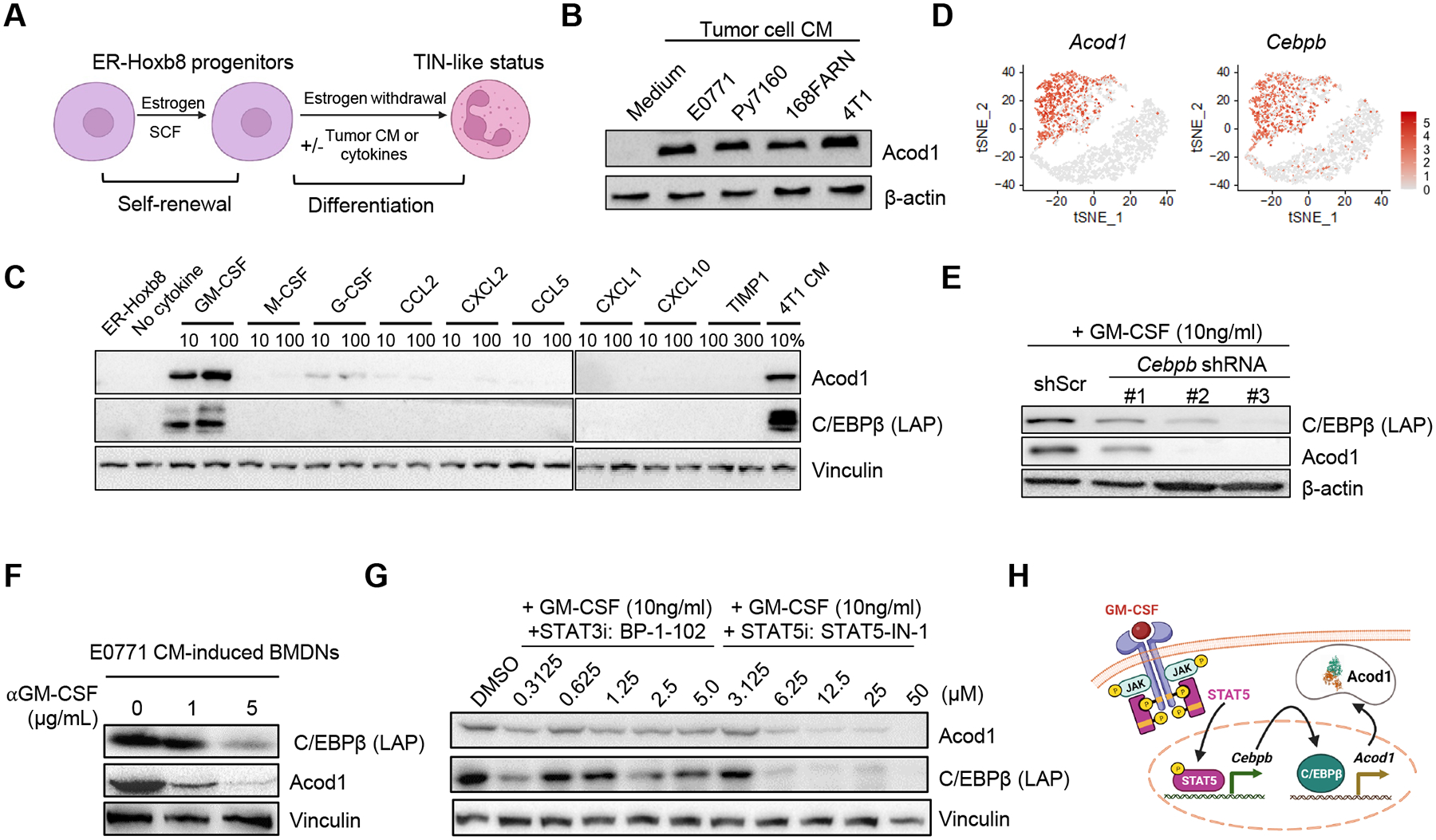
(A) ER-Hoxb8-immortalized mouse myeloid progenitors proliferate in the presence of estrogen (β-estradiol) and stem cell factor (SCF). When estrogen is withdrawn, progenitors differentiate to mature neutrophils (ER-Hoxb8-DNs) and can be induced to a TIN-like status with tumor CM or specific cytokines. (B) Western blot to examine Acod1 expression by ER-Hoxb8-DNs induced with CM from murine mammary cancer cell lines. (C) Western blot of Acod1 and C/EBPβ (LAP) in ER-Hoxb8 progenitor cells (first lane) and ER-Hoxb8-DNs (other lanes) treated with 4T1-expressed cytokines individually (unit ng/ml) or 4T1 CM. (D) t-SNE plot of Acod1 and Cebpb from the Drop-seq data. (E) Western blot of Acod1 and C/EBPβ (LAP) for ER-Hoxb8-DNs with Cebpb knockdown by shRNA (three designs) and induced with GM-CSF. (F) Western blot of Acod1 and C/EBPβ (LAP) for E0771-CM-treated BMDNs with or without αGM-CSF. (G) Western blot of Acod1 and C/EBPβ (LAP) for ER-Hoxb8-DNs induced with GM-CSF in the presence of BP-1–102 (STAT3 inhibitor) or STAT5-IN-1 (STAT5 inhibitor). (H) Schematic of Acod1 upregulation in TINs by tumor-secreted GM-CSF. Western blot results were representative of at least three independent experiments showing consistent patterns. See also Figure S4.
One of GM-CSF downstream transcription factors, CCAAT/enhancer binding protein-β (C/EBPβ), was strongly induced by GM-CSF and tumor CM (Figure 4C). C/EBPβ is required for emergency granulopoiesis and MDSC generation from bone marrow22,23. In our Drop-seq, both Acod1 and Cebpb were exclusively expressed by TINs but not BNs (Figure 4D). Cebpb knockdown with shRNA in ER-Hoxb8-DNs abrogated Acod1 induction by GM-CSF (Figure 4E). Anti-GM-CSF treatment reduced the expression of both C/EBPβ and Acod1 in E0771-CM-induced BMDNs (Figure 4F). Acod1 expression was diminished in BMDNs induced with CM from Csf2-knockout 4T1 and E0771 cells (Figure S4C and S4D), suggesting the crucial role of GM-CSF in inducing Acod1 expression in neutrophils. We should note that, while in our CM-based in vitro experiment, GM-CSF is secreted by tumor cells, in the TME GM-CSF may be produced by various cell types. Additionally, injection of exogenous recombinant GM-CSF in mice dramatically elevated C/EBPβ and Acod1 expression in neutrophils isolated from bone marrow, lung and spleen (Figure S4E). GM-CSF binds to GM-CSF receptor and activates JAK-STAT pathway, particularly JAK2 and STAT3/5 in neutrophils24. We found that GM-CSF treatment induced rapid phosphorylation of STAT3 and STAT5 in ER-Hoxb8-DNs (Figure S4F). STAT5 inhibitor (STAT5-IN-1), but not STAT3 inhibitors (BP-1–102, WP1066 and LLL12), dampened C/EBPβ and Acod1 expression (Figure 4G and Figure S4G). As expected from the GM-CSF/Acod1 axis, without GM-CSF ER-Hoxb8-DNs showed no expression of C/EBPβ and Acod1 regardless of the inhibitors (Figure S4H). These results reveal that tumor-secreted GM-CSF induces Acod1 in TINs through the STAT5-C/EBPβ pathway (Figure 4H).
Acod1 sustains TIN survival by blunting ferroptosis.
Neutrophils in lung metastases exhibit elevated reactive oxygen species (ROS) production compared with normal neutrophils6. Acod1 was reported to downregulate ROS and oxidative stress in macrophages and hepatocytes25–27. To examine whether Acod1 downregulates ROS in TINs, we quantified cellular ROS, lipid ROS and mitochondrial ROS in TINs isolated from E0771 lung metastases developed in WT or Acod1‒/‒ mice. TINs lacking Acod1 showed elevated levels of all three ROS types (Figure 5A–5C). ROS is essential for inducing neutrophil death28. Therefore, increased ROS due to Acod1 loss in TINs may lead to more pronounced death. Indeed, Acod1 loss led to a significant drop in the frequency and viability of TINs in metastasis-bearing mice without affecting the viability of BNs and splenic neutrophils (Figure 5D–5F and S5A–S5B).
Figure 5. Acod1 sustains TIN survival by blunting ferroptosis.
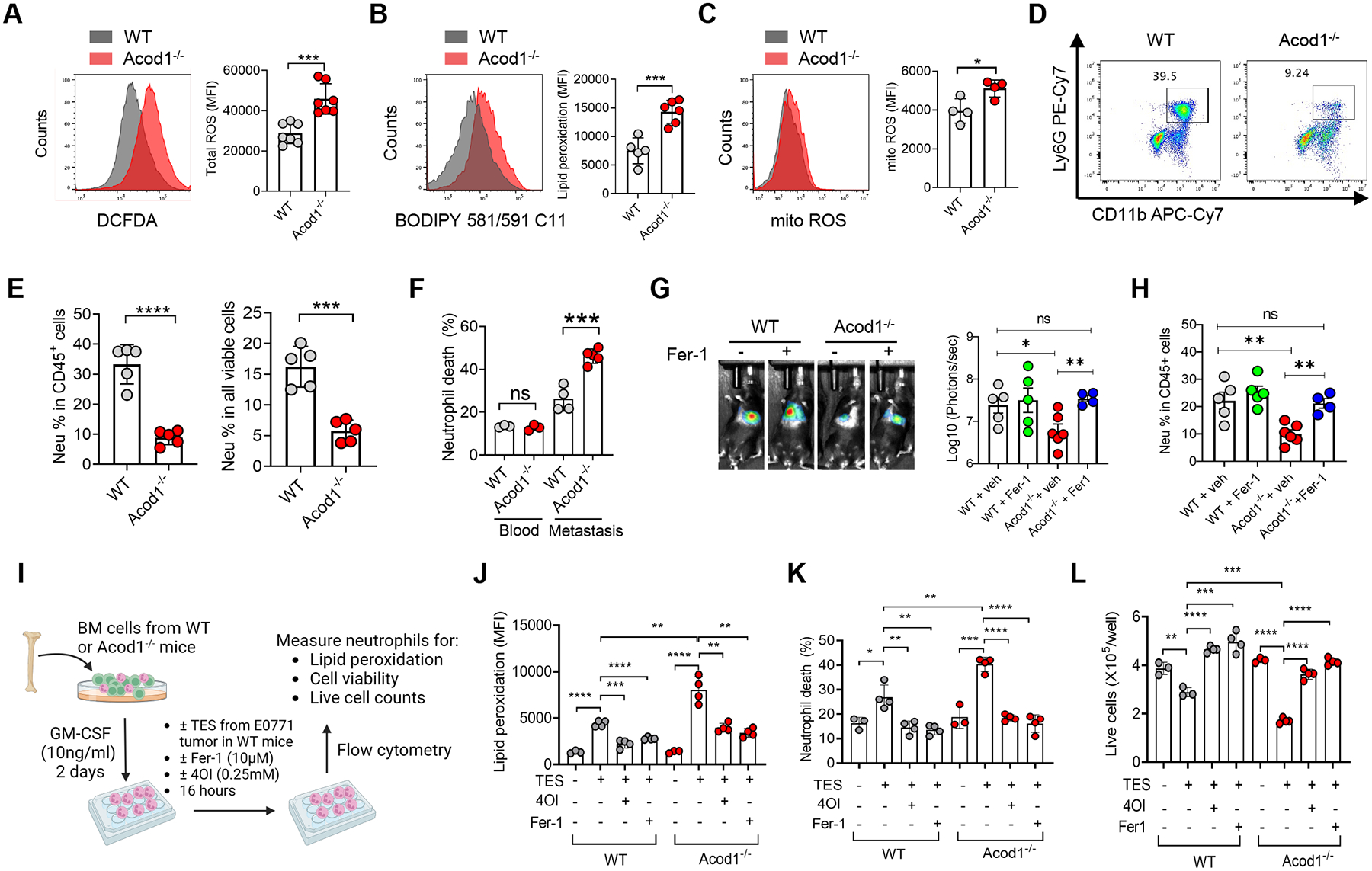
(A-C) Flow cytometry for total cellular ROS (DCFDA) (n=7), lipid ROS (BODIPY 581/591 C11) (n=5–6), and mitochondrial ROS (n=4) in TINs isolated from E0771 lung metastases in WT and Acod1‒/‒ mice. (D) Representative flow cytometry plots to quantify the frequency of CD11b+ Ly6G+ cells in all CD45+DAPI‒ singlets in E0771 lung metastases of WT and Acod1‒/‒ mice. (E) Frequency of CD11b+ Ly6G+ cells in the blood and lungs of E0771 metastasis-bearing WT and Acod1‒/‒ mice (n=5). Cells were gated on CD45+ DAPI‒ singlets (left) or all DAPI‒ singlets (right). (F) Cell death (DAPI+) fraction in total CD45+ CD11b+ Ly6G+ singlets (n=3–5). (G) BLI of WT and Acod1‒/‒ mice treated with vehicle or Fer-1 (2mg kg‒1, i.p., twice weekly) and imaged 14 days after i.v. injection of E0771-TR (n=4–6). (H) TIN percentage in lungs of vehicle or Fer-1 treated cohorts (n=4–6). (I-L) Schematic and results of evaluating the effect of TES (from E0771 mammary tumor grown in WT mice, 20% into culture medium), Fer-1 and 4OI on GM-CSF-induced WT and Acod1‒/‒ BMDNs for measures including lipid peroxidation (BODIPY 581/591 C11), cell death fractions, and viable cell counts (n=4/condition). Data represent mean ± s.e.m.; ns, not significant, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, unpaired two-tailed Student’s t-test (except for G, which used Mann-Whitney test). Experiments were repeated three times with consistent results. See also Figure S5.
Ferroptosis is iron-dependent cell death triggered by excessive ROS burden and lipid peroxidation29. Cancer and immune cells in the TME may undergo ferroptosis in vivo30–33. Based on the higher lipid ROS and lower viability of Acod1-deficient TINs, we hypothesized that Acod1 loss rendered TINs susceptible to ferroptosis, thus ferroptosis inhibition could override the effect of Acod1 loss on TINs. To test the hypothesis, we treated WT and Acod1‒/‒ mice bearing E0771-TR lung metastases with vehicle or ferroptosis inhibitor ferrostatin-1 (Fer-1). Fer-1 markedly recovered metastasis burden (Figure 5G) and TIN abundance (Figure 5H) in Acod1‒/‒ mice to levels comparable with WT mice.
To recapitulate the phenomenon that Acod1 resists TME-induced ferroptosis in neutrophils in vitro, we treated GM-CSF-cultured BM cells from WT or Acod1‒/‒ mice with E0771 mammary tumor explant supernatant (TES) in the presence or absence of Fer-1 and itaconate derivative 4-octyl itaconate (4OI) (Figure 5I). We measured and confirmed that 0.25mM 4OI or DMI added to Acod1‒/‒ BMDNs (no endogenous itaconate) led to dramatically elevated intracellular itaconate (Figure S5C). TES induced more pronounced lipid peroxidation and viability loss of Acod1‒/‒ BMDNs than WT BMDNs (Figure 5J–5L). Critically, this difference was abrogated when cells were treated with either 4OI or Fer-1 (Figure 5J–5L and S5D–S5E).
The comparable histology and low intensity of cleaved Caspase-3 (apoptosis marker) in both normal lung or metastasis-adjacent lung tissue from WT and Acod1‒/‒ mice suggests that the effect of the upregulated ROS by neutrophil Acod1 deletion is limited to neutrophils and unlikely causes damages of normal tissues (Figure S5F–S5G). To assess the impact of Acod1 knockout on normal hematopoiesis, we quantified the stem/progenitor and differentiated hematopoietic cell populations in age-matched tumor-free WT and Acod1‒/‒ mice. The results showed no differences in stem/progenitor populations in the bone marrow, and either no differences (neutrophils, DCs, B and CD8+ T of spleen and blood, splenic monocytes and macrophages, blood CD4+ T) or moderate differences (splenic/blood NK, blood monocytes and macrophages splenic CD4+ T) in differentiated immune cells (Figure S5H), suggesting the potential influence of Acod1 in specific steps of hematopoietic differentiation. Notably, the two populations most relevant in our study (neutrophils and CD8+ T) were unaffected by Acod1 knockout under tumor-free conditions. Taken together, we conclude that Acod1 overexpression protects TINs from ferroptosis, allowing more accumulation of TINs in the metastatic TME.
Acod1 blunts TIN ferroptosis through activating Nrf2-mediated antioxidant response.
In macrophages, itaconate can activate Nrf2 through cysteine alkylation of Keap1 and releasing Keap1-mediated Nrf2 degradation25. Nrf2, as a critical regulator of redox balance and the master transcription factor for antioxidant defense, mitigates lipid peroxidation and resists ferroptosis34. Nrf2 enhances MDSC suppressive activity and tumor infiltration by reducing their oxidative stress and apoptosis rate35. These lines of evidence suggest that Acod1 may rescue TINs from ferroptosis through activating Nrf2-dependent antioxidant response.
To test this idea, we examined the expression of Nrf2 in TINs isolated from E0771 lung metastases of WT and Acod1‒/‒ mice and observed significant depletion of Nrf2 protein by Acod1 loss (Figure 6A), although the mRNA level of Nrf2 was unaltered (Figure 6B). Consistent with the function of Nrf2, Acod1‒/‒ TINs significantly downregulated the expression of antioxidant genes, represented by Gpx4, Gclc and Nqo1, all of which are involved in ferroptosis resistance36,37 (Figure 6C). Treating metastasis-bearing mice with the Nrf2 inhibitor ML385 abolished the differential expression of these genes between WT and Acod1‒/‒ TINs (Figure 6C). Conversely, BMDNs treated with itaconate or its derivatives 4OI and DMI showed higher Nrf2 protein and its transcriptional targets Gpx4, Gclc and Nqo1 (Figure S6A and S6B). Lipid peroxidation in BMDNs primed with E0771 TES was augmented by ML385 (Figure S6C) and diminished by the Nrf2 activator NK252 (Figure S6D) in dose-dependent manners.
Figure 6. Acod1 blunts TIN ferroptosis through activating Nrf2-mediated antioxidant response.
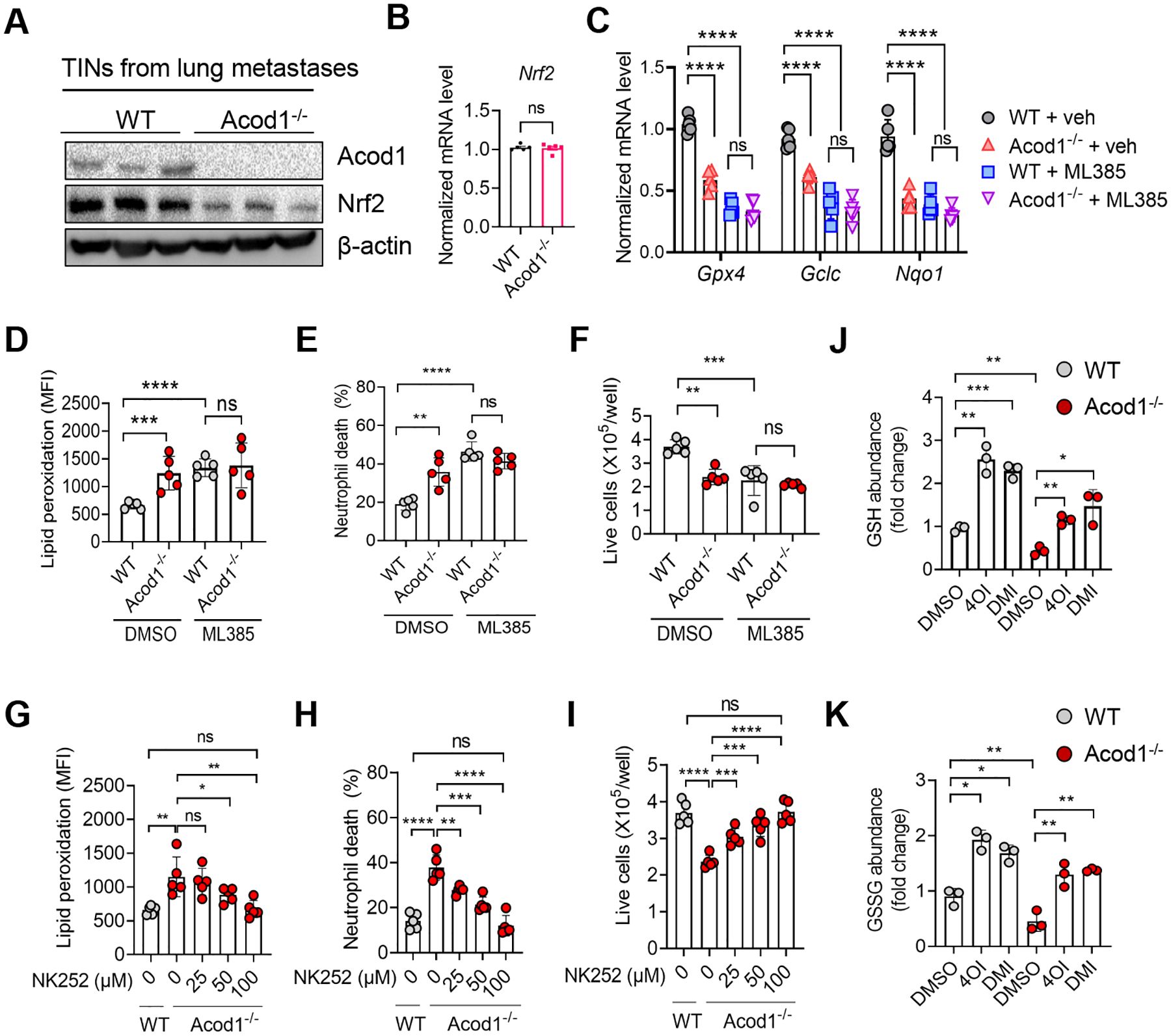
(A) Immunoblot of Acod1 and Nrf2 in TINs isolated from E0771 lung metastases in WT and Acod1‒/‒ mice. (B) qRT-PCR of Nrf2 in TINs isolated from E0771 lung metastases in WT and Acod1‒/‒ mice (n=5). (C) qRT-PCR of Gpx4, Gclc and Nqo1 in TINs isolated from E0771 lung metastases in WT and Acod1‒/‒ mice (n=5) treated with vehicle or ML385 (30mg kg‒1, i.p., daily for 7 days). (D-F) The effect of ML385 (10μM) on E0771 TES-primed WT and Acod1‒/‒ BMDNs for measures including lipid peroxidation (BODIPY 581/591 C11), cell death fractions, and viable cell counts (n=5/condition). (G-I) The effect of NK252 on E0771 TES-primed WT and Acod1‒/‒ BMDNs for measures including lipid peroxidation (BODIPY 581/591 C11), cell death fractions, and viable cell counts (n=5/condition). (J-K) GSH and GSSG abundance in E0771 CM-induced WT and Acod1‒/‒ BMDNs treated with DMSO, 0.25mM 4OI or 0.25mM DMI (n=3). Data represent mean ± s.e.m.; ns, not significant, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, unpaired two-tailed Student’s t-test. Experiments were repeated three times with consistent results. See also Figure S6.
Furthermore, ML385 elevated lipid peroxidation and reduced cell viability of WT TES-stimulated BMDNs to levels comparable to ML385-treated TES-stimulated Acod1‒/‒ BMDNs (Figure 6D–6F). Conversely, NK252 decreased lipid peroxidation and increased cell viability of TES-treated Acod1‒/‒ BMDNs and diminished the difference between WT and Acod1‒/‒ BMDNs at 100uM (Figure 6G–6I). We further assessed the effect of Acod1 loss on the cellular abundance of glutathione, the primary antioxidant metabolite antagonizing ferroptosis. WT BMDNs contained more glutathione than Acod1‒/‒ BMDNs, and exogenous 4OI or DMI dramatically increased glutathione in both BMDNs (Figure 6J–6K). Furthermore, we profiled the metabolomics of WT BMDNs and Acod1‒/‒ BMDNs stimulated with E0771 CM. Both succinate and fumarate increased in abundance to a similar extent, as were the rest of the TCA cycle intermediates (Figure S6E), suggesting that Acod1 deletion in BMDNs “traps” citrate carbon in the TCA cycle that would otherwise have been used to synthesize itaconate. Together, these data elucidate that itaconate produced by Acod1 attenuates TIN ferroptosis by stimulating the Nrf2 antioxidant pathway.
Acod1 extinction boosts adaptive immunity and enhances immunotherapy.
The metastasis-constraining effect of Acod1 ablation depends on adaptive immunity because Rag1‒/‒ Acod1+/+ and Rag1‒/‒ Acod1‒/‒ mice showed similar E0771-TR lung metastasis burden and survival time (Figure 7A–7C). Since Acod1 deficiency debilitates TINs, we tested the effect of Acod1 loss on tumor-infiltrating T cells and the ICB efficacy for lung metastasis. We used the E0771-TR lung metastasis model and observed that while Acod1 extinction or ICB (αPD1 plus αCTLA4) each extended mouse survival, Acod1‒/‒ host treated with ICB showed a significant extension of survival (Figure 7D). Immunophenotyping at day 15 after i.v. injection showed that TIN density in the lung was most reduced by the combination of Acod1 depletion and ICB (Figure 7E). ICB-treated Acod1‒/‒ cohort displayed the most favorable antitumor T cell immunity in the metastatic TME, featured by the highest percentages of total CD8+ T, IFNγ+ CD8+ T, Gzmb+ CD8+ T, and total CD4+ T, but the lowest percentage of Tregs (Figure 7F). Mrp8-cre+Acod1f/f mice were used to limit Acod1 loss to neutrophils in the therapies. ICB-treated Mrp8-cre+Acod1f/f mice survived significantly longer than ICB-treated Acod1f/f littermates (Figure 7G). When ICB-treated Mrp8-cre+Acod1f/f mice were treated with Fer-1 or vehicle, Fer-1 expedited the death of the metastasis-bearing mice (Figure 7H), suggesting that neutrophil-Acod1 loss enhances ICB through upregulating ferroptosis in neutrophils.
Figure 7. Acod1 extinction boosts adaptive immunity and enhances immunotherapy.
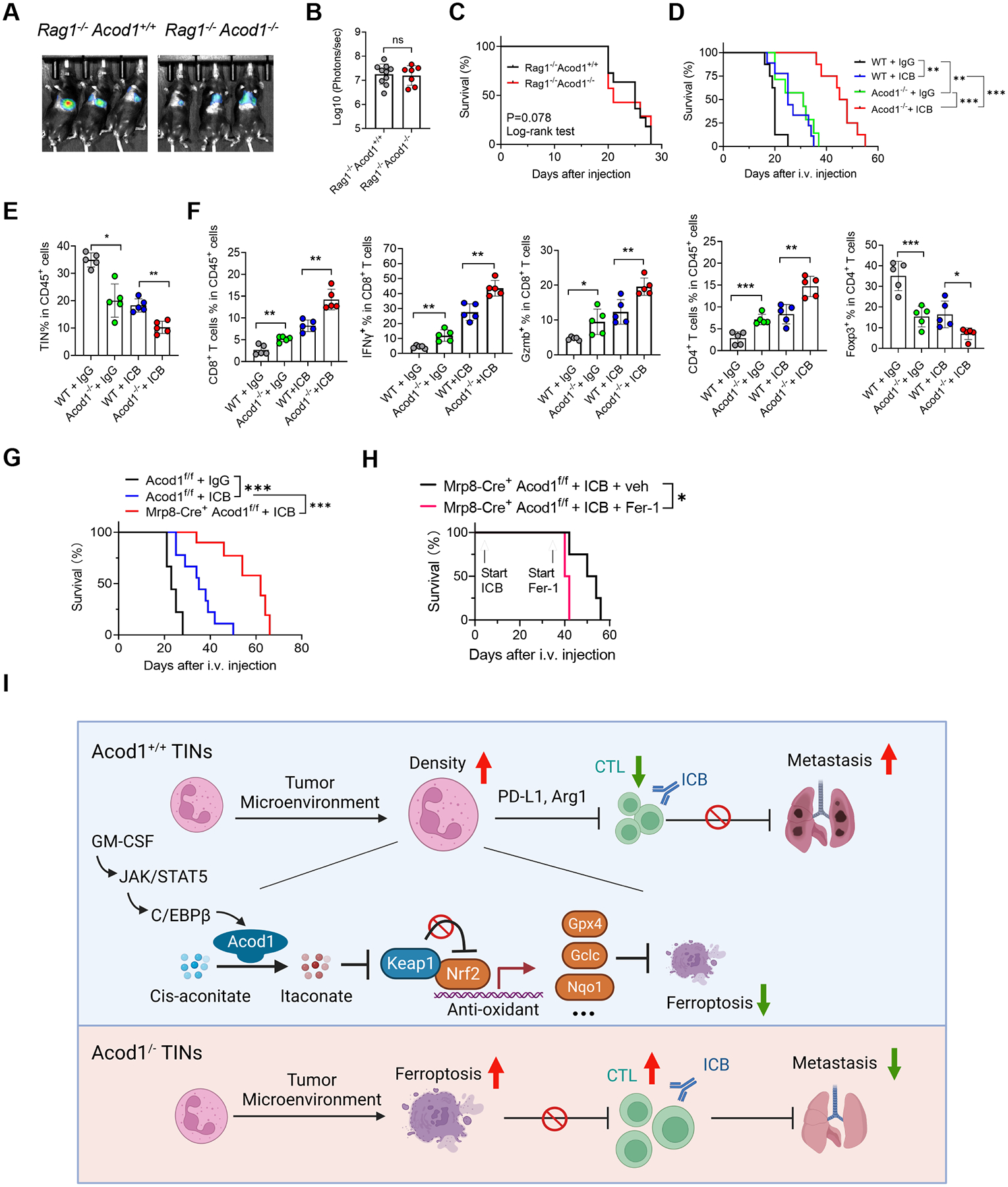
(A-C) Lung metastasis of E0771-TR in Rag1‒/‒Acod+/+ (n=11) and Rag1‒/‒Acod1‒/‒ (n=7) mice, established with the method illustrated in Figure 3A. Shown are representative BLI images (A), BLI quantification (B) and animal survival (C). (D) Survival of WT and Acod1‒/‒ mice (n=8–10) bearing E0771-TR lung metastasis and treated with isotype IgG or ICB (αPDL1 + αCTLA4, 10mg kg‒1 each, i.p., twice/week). (E-F) Flow cytometry to measure frequencies of TINs and T cell subsets from lungs of WT and Acod1‒/‒ interim cohorts 15 days after i.v. injection of E0771-TR (n=5 for each group). (G) Survival of Acod1f/f and Mrp8-cre+Acod1f/f mice bearing E0771-TR lung metastasis and treated with isotype IgG or ICB (αPDL1 + αCTLA4, 10mg kg‒1 each, i.p., twice/week, started three days after i.v. injection). n=10 for each group. (H) Survival of Mrp-8cre+Acod1f/f mice undergoing ICB treatment further treated with or without Fer-1 (2mg kg-1, i.p., once/2 days). Start timepoints of ICB and Fer-1 treatments were indicated, n=4 for each group. (I) Schematic of the mechanism and function of Acod1 in TINs to promote BC lung metastasis (created with BioRender). In B, E and F, data represent mean ± s.e.m.; ns, not significant, *P<0.05, **P<0.01, ***P<0.001, Mann-Whitney test (for B) or unpaired two-tailed Student’s t-test (for E and F). In C, D, G and H, *P<0.05, **P<0.01, ***P<0.001, log-rank test. See also Figure S7.
We were curious whether Acod1 loss directly altered the immunosuppressive capacity of TINs. The expression of PD-L1 and Arg1 was unchanged by Acod1 loss (Figure S7A and S7B). WT and Acod1‒/‒ TINs from lung metastases elicited similar suppression of CD4+ and CD8+ T cells (Figure S7C). Splenic neutrophils from metastasis-bearing WT and Acod1‒/‒ mice showed much weaker suppression on T cells than neutrophils from metastases, and no difference was detected between the two genotypes (Figure S7D). Itaconate was reported to accumulate at ~40μM in the tumor tissue and ~15μM of the tumor interstitial fluid in a murine melanoma model38. We used LC-MS to confirm that the E0771 lung metastasis interstitial fluid had itaconate at 12.6μM (Figure S7E). We treated αCD3/CD28-stimulated CD4+ and CD8+ T cells with itaconate concentration gradients and observed T cell suppression only after itaconate reached 5mM (Figure S7F), far beyond the concentrations reachable in tumor-bearing tissues. Therefore, our result does not support that Acod1 upregulation in TINs directly enhances their immunosuppression or that the secreted itaconate directly inhibits T cells. Instead, our data indicate that Acod1 upregulation upholds the survival and persistence of TINs in metastases to allow sustained execution of immunosuppression through other mechanisms by the TINs.
DISCUSSION
We demonstrate that immunosuppressive TINs manage to survive in the metastatic TME through upregulating Acod1 and generating itaconate, which activates Nrf2-dependent antioxidant response to evade ferroptosis and sustain TIN abundance in metastasis (Figure 7I). GM-CSF activation of the STAT5-C/EBPβ pathway drives Acod1 transcription, and TINs had the highest level of Acod1 compared with other immune populations in the TME. Systemic or neutrophil-specific ablation of Acod1 reduced TIN survival and accumulation, decelerated metastasis growth and extended the mouse survival in immune-competent BC models, whereas itaconate derivative or CM-activated WT (but not Acod1−/−) neutrophils promoted lung metastasis and shortened the animal survival. Host Acod1 loss rectified the antitumor T cell profile and boosted ICB’s effectiveness in treating BC metastasis. Moreover, neutrophil-specific Acod1 loss enhanced ICB therapy, an effect weakened when ferroptosis was inhibited. Finally, ACOD1 is expressed by neutrophils in human BC metastases based on scRNA-seq analysis.
Mounting evidence establishes ferroptosis as a critical tumor suppression mechanism, and tumor cells develop various ways to evade ferroptosis29. Nonetheless, metabolic features of some cancer cells render them fortuitously sensitive to ferroptosis inducers (FINs)29. Emerging evidence reveals that immune cells in the TME are not exempt from ferroptosis and may need to develop ferroptosis-evasion mechanisms to avoid extinction. CD8+ T cells, follicular helper T cells, and Tregs depend on Gpx4 to limit lipid peroxides and defend against ferroptosis39–41. M1-like macrophages are more resistant to FIN-induced ferroptosis than M2-like macrophages thanks to higher nitric oxide production to inhibit lipid peroxidation42. MDSCs (CD11b+ Gr-1+, mixture of PMN-MDSCs and monocytic MDSCs) resist ferroptosis by inhibiting the p53–haeme oxygenase 1 axis43. Recently, Kim et al. reported that PMN-MDSCs employ ferroptosis as an immunosuppression mechanism whereby dying PMN-MDSCs release oxygenated lipids and limit the activity of effector T cells33. Our Drop-seq data also showed upregulation of ferroptosis-related genes in TINs compared with BNs (e.g., Hilpda, Fth1, Ctsb, Ptgs2), suggesting that TINs would have died from ferroptosis if they had failed to activate ferroptosis evasion mechanisms. However, the robust foothold of TINs in the immune cell pool of lung metastasis (~30% of viable CD45+ cells) and the tight regulation of TIN ferroptosis markers and viability by Acod1 status in our results demonstrate that the dramatic Acod1 upregulation is a mechanism used by TINs to defend against ferroptosis and remain a vibrant player in metastasis. Ferroptosis may stochastically transpire in some TINs and may indeed contribute to immunosuppression. However, many ferroptosis-independent immunosuppression mechanisms exist for TINs (e.g. PD-L1 and Arg1). Furthermore, findings from Kim et al. were based on subcutaneous tumors whereas our study focused on metastasis. Overall, we believe that our study and Kim et al. focused on the two sides of the same coin and together reveal the complete picture of the role of ferroptosis in TINs.
Our study identifies the Acod1-itaconate-Keap-Nrf2-antioxidant pathway, initially discovered in macrophages25, in driving the pro-metastasis function of neutrophils. This pathway appears to be a robust immunometabolic switch used by various cell types (macrophages, dendritic cells, hepatocytes, Kupffer cells, and now neutrophils) to protect them from excessive oxidative damages in different pathological conditions, including sepsis, allergy, liver injury, obesity, and cancer26,29,44–46. Very recently, Zhao et al. made the first connection between itaconate upregulation in PMN-MDSCs to immunosuppression of CTLs through a direct itaconate diffusion model where 5mM of exogenous itaconate impeded CD8+ T cells in vitro38. However, both Zhao et al. and our results measured the itaconate level in tumors to be below 40μM, which was less than 0.8% of the exogenous itaconate concentration used in the T cell assays. Therefore, while we should not rule out the possibility of direct inhibition of T cells by itaconate, we believe that Acod1-itaconate-Nrf2-dependent ferroptosis evasion is the predominant mechanism underlying the function of Acod1 in TIN immunosuppression. We should point out a caveat in the in vitro assays where 4OI or DMI were used to support the function of Acod1 in restricting lipid peroxidation and cell death (Figure 5J–5L) and stockpiling glutathione (Figure 6J–6K) in BMDNs, as an additional condition using exogenous itaconate would further strengthen the conclusions.
Our results of the superior antitumor efficacy by combining Acod1 ablation and ICB support the current efforts to develop FINs in combination with ICB therapy. Recent findings of the synergistic antitumor activity by combining cyst(e)inase (a FIN) and ICB30 and the recognition of ferroptosis as a form of immunogenic cell death further support this strategy. Nonetheless, the specific therapeutic target to induce ferroptosis should be deliberated. For example, targeting GPX4 may lead to T cell ferroptosis and sabotage immunotherapy39–41; yet, targeting SLC7A11 may be a viable option because SLC7A11 is dispensable for T cell proliferation in vivo47. Our study highlights Acod1 as a promising immunometabolic target to elicit ferroptosis in TINs and spare extra-tumoral neutrophils (because they are Acod1‒), significantly alleviating the safety concerns. Both Acod1 lentiviral-shRNA48 and the newly identified endogenous Acod1 inhibitor citraconate49 provide exciting opportunities for future development.
Limitations of the study
Although Acod1 level in other immune cells was not as high as in TINs, Acod1 may still regulate other immune cells, especially tumor-associated macrophages, which was not investigated in our study. Future work is needed to co-delete Acod1 and ferroptosis execution genes in neutrophils and other immune cells, as well as using additional ferroptosis inhibitors that target specific cell types, to fully establish the Acod1-ferroptosis relationship in each relevant cell type during metastasis and immunotherapy. The mechanism for itaconate derivatives 4OI and DMI to convert to itaconate inside BMDNs remains unclear. We fully acknowledge that 4-OI was reported to not convert into intracellular itaconate by LPS-activated macrophages50 although conflicting reports also exist51,52, raising the possibility of cell type-specific and context-dependent regulation of itaconate derivatives. We do not exclude that 4OI or DMI can serve as inhibitors of neutrophil activation, a mechanism that would also contribute to the results associated with using them. Our result showing the overall upregulated TCA cycle metabolites upon Acod1 loss in BMDNs does not reflect the inhibition of succinate dehydrogenase by itaconate as seen in LPS-activated macrophages27. A few factors may contribute to this discrepancy, including cell type-specific regulation of the TCA cycle, different kinetics between LPS stimulation and tumor CM stimulation, and difference in absolute itaconate levels, which can be resolved in future studies. The validation of neutrophil ACOD1 expression in human BC metastases should extend beyond in silico analysis and examine clinical metastasis specimens using multiplex staining. Our study was focused on BC; however, ACOD1 may also exert immunomodulatory activities in other carcinomas based on its upregulation in other human cancers.
STAR * METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Xin Lu (xlu@nd.edu).
Materials availability
All the materials generated in this study are available upon reasonable request to the lead contact.
Data and code availability
The Drop-seq data generated in this study were deposited to GEO under accession number GSE216425. The RNA-seq data used to compare Acod1 expression between mammary cancer cells, tumor-infiltrating macrophages and tumor-infiltrating neutrophils in four mouse models were obtained from GSE104765. Gene expression profiles of human breast cancer metastasis tissues were from microarray dataset GSE46141. Single-cell RAN-seq data of human breast cancer metastasis tissue were from GSE169246. The code for scRNA-seq analysis will be provided upon request. Uncropped scans of all western blots and all raw data used to generate graphs are included in Data S1. Further information and requests for resources and reagents should be directed to the lead contact.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All animal work performed in this study was approved by the Institutional Animal Care and Use Committee (IACUC) at University of Notre Dame. All animals were maintained under pathogen-free conditions and cared for in accordance with the International Association for Assessment and Accreditation of Laboratory Animal Care policies and certification. C57BL/6J (RRID: IMSR_JAX:000664), FVB/NJ (RRID:IMSR_JAX:001800), MMTV-PyMT (RRID:IMSR_JAX:022974), Rag1‒/‒ (RRID:IMSR_JAX:002216), Acod1‒/‒ (RRID:IMSR_JAX:029340), LysM-Cre (RRID:IMSR_JAX:004781) and Mrp8-Cre (RRID:IMSR_JAX:021614) mice were purchased from Jackson Laboratory and bred in-house. Acod1f/f allele was a generous gift from Michael Diamond lab at Washington University School of Medicine in St. Louis. Ly6G-Cre allele was a generous gift from Matthaias Gunzer lab at University Hospital Essen. All the mice (except FVB/NJ) are in C57BL/6 background.
Cell lines
Py7160 cell line was generated from a spontaneous tumor of MMTV-PyMT (FVB background). E0771 was received from Siyuan Zhang lab at University of Notre Dame. 4T1 and 168FARN were received from Yibin Kang lab at Princeton University. All these lines were cultured in DMEM (GE Healthcare, SH30243.FS) supplemented with 10% fetal bovine serum (FBS; GE Healthcare, SH30396.03) and 100U/ml penicillin-streptomycin (Cytiva, SV30010).
Human breast cancer specimens
De-identified archived breast cancer formalin-fixed paraffin-embedded (FFPE) blocks were obtained from Harper Cancer Research Institute tissue bank. The clinical information about the samples is provided in Table S4.
METHOD DETAILS
Animal experiments
For orthotopic mammary tumor growth, Py7160, E0771 or E0771-mcherry (106 cells) in 100uL PBS were injected to the fourth mammary fat pads on both flanks of the recipient mice. For experimental metastasis models, E0771-TR (5 ×105 cells) in 100μl PBS were injected into the recipient mice via tail vein at day 12 after mammary fat pad injection of E0771-mcherry. Primary tumors were removed when tumor diameter reached 1cm. Lung metastasis were monitored by bioluminescence imaging (Spectral Ami HT Advanced Molecular Imager) at the indicated time points. For itaconate treatment, mice received PBS or DMI (Sigma-Aldrich, #592498) dosed at 100mg/kg, i.p. daily) 5 days after orthotopic injection or on the same day as i.v. injection. For neutrophil depletion, mice received anti-Ly6G (BioXcell, #BP0075-1) dosed at 5mg/kg, i.p. twice/week 5 days after orthotopic injection or on the same day as intravenous injection. For Acod1 induction in vivo, exogenous recombinant GM-CSF (Biolegend, #576306) was injected i.v. daily (10ug/kg or 50ug/kg) for consecutively 3 days before harvesting neutrophils for western blot validation. For ICB therapy, 3 days after i.v. injection, mice were treated with anti-PD1 (BioLegend, #114116) and anti-CTLA4 (BioLegend, #106207) at 10mg/kg each, i.p., twice a week. All treatments were continued until the specified experimental endpoints. For ferroptosis inhibition, WT and Acod1‒/‒ mice treated with vehicle or Fer-1 at designated timepoints (2mg kg‒1, i.p., twice weekly or once per 2 days)
Tissue collection and single cell preparation
For immune profiling, mammary tumors were harvested when diameter reach ~1cm. Lung metastases were harvested at ~ 2–3 weeks post i.v. injection. Fresh tissue was minced and enzymatically digested in DMEM medium (10ml/g) containing 10% FBS, 1mg/ml collagenase (Sigma-Aldrich, #COLLD-RO) and 0.1mg/ml DNase I (Sigma-Aldrich, #10104159001) for 1–2 hours at 37 °C with gentle agitation. After centrifugation, the pellet was resuspended with 3–5 ml of prewarmed Trypsin/EDTA and incubated for 5 mins at 37 °C. Trypsin activity was stopped with 10 ml of DMEM medium supplemented with 10% FBS and cells were passed through a 70μm cell strainer (BD Biosciences). Cells were centrifuged at 350 × g for 5 min and resuspended in FACS buffer (PBS, 2% FBS and 2 mM EDTA) for subsequent isolation or analysis.
Neutrophil isolation
For TINs, single cell suspension was prepared by enzymatic dissociation method described above. For BNs, whole blood was collected by cardiac puncture into EDTA-containing Eppendorf tubes and treated by RBC lysis buffer to remove red blood cells. MojoSort Mouse Ly6G Selection Kit (BioLegend, #480124) was used to isolate neutrophils according to the manufacturer protocol.
Flow cytometry for immune profiling
Single cell suspension was prepared as described above. Fc receptors were blocked by incubation with anti-mCD16/CD32 (Tonbo Biosciences, #70-0161-U500) for 10 min on ice. Samples were stained for 30 min on ice with one or a combination of the following antibodies: PE-Cy7 anti-CD45 (Tonbo Biosciences, #60-0451-U100), APC anti-CD45 (Tonbo Biosciences, #20-0451-U100), PerCP-Cy5.5 anti-CD11b (Tonbo Biosciences, #65-0112-U100), APC anti-CD11b (Tonbo Biosciences, #20-0112-U100), APC-Cy7 anti-CD11b (Tonbo Biosciences, #25-0112-U100), PE-Cy7 anti-Ly6G (Tonbo Biosciences, #60-1276-U100), PE anti-Ly6G (Tonbo Biosciences, #50-1276-U025), PE anti-CD4 (Tonbo Biosciences, #50-0041-U100), PerCP-Cy5.5 anti-CD8a (Tonbo Biosciences, #65-0081-U100), APC-Cy7 anti-CD3 (Tonbo Biosciences, #25-0032-U100), APC anti-PD-L1 (Tonbo Biosciences, #20-1243-U025). Lineage cocktail biotin antibody (Biolegend, #480050), Percp5.5-Streptavidin (eBioscience, # 45-4317-80), APC anti-c-Kit (Tonbo Bioscience, #201173-U100), PE anti-CD34 (Biolegend, #128609), APC-Cy7 anti-mouse CD16/32 (FcγRII/III) (Biolegend, #101327), Pe-Cy7 anti-Sca-1(Biolegend, #122514).Following staining, cells were washed in FACS buffer (PBS, 2% FBS and 2 mM EDTA). DAPI (Sigma-Aldrich, #D9542) at 0.5ug/ml was used as the viability dye. Samples were run on CytoFLEX S (Beckman Coulter). DAPI+ cells were gated as dead cells, and DAPI‒ cells were gated as viable cells.
To measure T cell cytokine expression, single cell suspensions were prepared from fresh tumor tissues. T cells were enriched by density gradient centrifugation. For cytokine staining, T cells were incubated in culture medium containing PMA (50ng/ml), ionomycin (500ng/ml), Brefeldin A (1: 1000) at 37 °C for 4–6 hours. anti-CD45 (Tonbo Biosciences, # 60-0451-U100), anti-CD3 (Tonbo Biosciences, # 65-0032-U100), and anti-CD4 (Tonbo Biosciences, # 50-0041-U100), anti-CD8 (Tonbo Biosciences, # 50-0081-U100) and Ghost Dye Violet 450 (Tonbo Biosciences, # 13-0863-U100) were added for 30 minutes for surface staining on ice. The cells were washed and resuspended in 1 ml of freshly prepared Foxp3/Transcription Factor Fix/Perm Diluent (Tonbo, #TNB-1022-L160) on ice overnight. After being washed with Foxp3/Perm/Wash buffer (Tonbo Biosciences, #TNB-1213-L150), the cells were stained with anti-Foxp3 (Tonbo Biosciences, #20-5773-U025), anti-IFNg (Tonbo Biosciences, #20-7331-U100), and anti-granzyme B (BioLegend, #515405) for 60 minutes, washed, and resuspended in FACS buffer before running on CytoFLEX S (Beckman Coulter). Flow cytometry data were analyzed using Flowjo v10.8 (FlowJo, RRID: SCR_008520).
Drop-seq and data analysis
Single-cell transcriptomic profiling was performed based on protocol previously described53. Briefly, tumors were harvested from Py7160 bearing mice (n=3) and enzymatically processed into single cell suspension. Blood cells were harvested from the same mice (n=3) with RBC lysed. Neutrophils were enriched to reach >95% purity with MojoSort Mouse Ly6G Selection Kit (BioLegend, #480124). Enriched cells were stained with hashtag antibodies (BioLegend, #155811; 155813; 155815) to enable multiplexing of samples from 3 mice (one hashtag antibody for each mouse). Cells from biological repeats were pooled into two groups (BNs and TINs) and subsequently labeled with flow cytometry antibodies followed by sorting to increase the purity of neutrophils further.
Samples prepared above were loaded on the microfluidic device (fabricated in-house, CAD file from McCarroll Lab website: http://mccarrolllab.org/dropseq/). BNs and TINs were loaded at ~200 cells μl‒1. Single cell suspension and uniquely barcoded microbeads (Chemgenes, MACOSKO201110) suspended in the lysis buffer were co-encapsulated in droplets by the microfluidic device. The droplets serve as compartmentalizing chambers for RNA capture. Once droplet generation was complete, collected droplets were disrupted and RNA-hybridized beads were harvested. Reverse transcription was performed using Maxima H Minus Reverse Transcriptase (Thermo Fisher Scientific, #EP0752) with template switching oligo. cDNA was amplified and PCR products were purified using AMpure Beads (Beckman Coulter). After quantification on a BioAnalazyer High Sensitivity Chip (Agilent), samples were fragmented and amplified for sequencing with the Nextera XT DNA sample prep kit (Illumina). The libraries were purified, quantified, and sequenced on the Illumina NextSeq 500.
Raw Drop-seq data (Fastq files) were aligned and mapped to the mouse mm10 reference genome by STAR aligner. A digital gene expression data matrix was generated with counts of unique molecular identifiers (UMIs) for every detected gene (row) per cell barcode (column). Knee plot, which utilizes the cumulative distribution of reads and identifies an inflection point in the plot, helped us to determine the number of cell barcodes represented in the expression matrix. Based on the data quality, an appropriate number of cell barcodes are selected for downstream analysis. Next, the Seurat R package (satijalab.org/seurat) is used to perform data normalization, dimension reduction (PCA), clustering (resolution=0.3), and differential expression analysis. Cells with either less than 100 genes or more than 2000 genes were excluded. The percentage of reads aligned to mitochondrial genes per cell was calculated and cells with greater than 15% of transcripts derived from mitochondrial genes were filtered out. We obtained 3,645 cells for further analysis.
To analyze genes encoding metabolic enzymes, mouse genes encoding enzymes were accessed from the KEGG database and genes with less than five non-zero expressions in the cells were excluded. We observed a substantial rate of cells that had 0 expressions for most genes and the expressed part of most genes were of one modality, hence a left-truncated Gaussian model was utilized for differential gene expression. The averaged log2 fold change and −log10(adjusted p value) were utilized to generate the volcano plots.
Analysis of published bulk and single-cell transcriptomics dataset
For bulk transcriptomics analysis, FPKM (log2) gene expression data of breast cancer (BRCA) and other cancer types in TCGA were retrieved from UCSC Xena browser. Z-scored data were used to plot the tumor and normal samples, as well as the Pearson correlation of ACOD1 gene expression and selective neutrophil marker gene expression. For gene set enrichment analysis (GSEA), raw counts of ACOD1 gene expression from GDC TCGA BRCA were used in differential expression analysis with edgeR (v3.38.4). BRCA samples were divided into high group (top 373 of total 1098) and low group (bottom 505 of total 1098) based on the median gene expression level of ACOD1. Log2 fold changes of gene expression between ACOD1-high samples and ACOD1-low samples were used to run GSEA (version 4.3.2) on the specified gene sets. The TIN/PMN-MDSC signature was curated based on literature mining (S100A8, S100A9, CD274, CCL4, PTGS2, OLR1, FUT4, CD33, ITGAM, CXCR2, NOS2, HILPDA, STAT5A, CXCL2, STAT3, CSF3R, CD36, ARG1). A similar method was applied to the metastatic BC dataset GSE46141 to analyze gene expression correlations and GSEA enrichment.
Single-cell RNA sequencing analysis of published dataset (GSE169246) was conducted by using the ‘Seurat’ R package on the CD45+ immune cell population of blood and metastasis site samples. Following data quality control and filtering, 439,064 cells were clustered for analysis. Samples from lung metastases were omitted from the analysis due to their significantly lower cell counts compared to other metastatic sites. The total CD45+ immune cells were categorized into NK/T cells, myeloid cells, and B cells using the ‘scType’ R package. From the total population, 71,648 myeloid cells were further annotated into neutrophils, macrophages, monocytes, and dendritic cells using ‘scType’ R packages. ACOD1 jitter plot were plotted by using ‘ggplot2’ R package and the significance among groups were test by using ‘ggsignif’ R package. The markers for cell identity are listed in Table S6.
Western blotting and mouse cytokine array
Cells or fresh tissues were lysed on ice using RIPA buffer supplemented with protease inhibitors (Bimake, #B14012) and phosphatase inhibitors (Roche, #04906845001). Protein concentration was quantified with BCA Protein Assay Kit (VWR, #PI23225). The following primary antibodies were used: β-actin (Santa Cruz, #sc-47778), vinculin (Millipore, #05-38608), mouse Acod1 (Cell Signaling Technology, #17805), Human Acod1(Aviva Systems Biology, #OACA09406), mouse C/EBPβ (BioLegend, #606202), Stat3 (Cell Signaling Technology, #4904), p-Stat3 (Cell Signaling Technology, #9145), Stat5 (Cell Signaling Technology, #94205), p-Stat5 (Cell Signaling Technology, #9314), mouse Nrf2 (Cell Signaling Technology, #12721) and Gpx4 (R&D Systems, #MAB5457). The following secondary antibodies were used: HRP-conjugated goat anti-mouse (Cell Signaling Technology, #7076) and HRP-conjugated goat anti-rabbit (Cell Signaling Technology, #7074). Signals were detected with Clarity Max Western ECL Substrate (Bio-Rad, #1705062). Conditioned medium (CM) was profiled with Proteome Profiler Mouse Cytokine Array Kit, Panel A (R&D Systems, ARY006) to detect secreted cytokines following the manufacturer protocol.
Neutrophil and T cell coculture to measure immunosuppression
Neutrophils were isolated from single cell suspension as described above. T cells were isolated from spleen of C57BL/6J using MojoSort Mouse CD3 Selection Kit (BioLegend, #480100). T cells were labeled with CFSE (Thermo Scientific, #C34570) according to the manufacturer’s instructions. CFSE-labeled T cells were stimulated with plate-bound anti-mouse CD3 antibody (BioLegend, #100302) at 5 μg ml‒1 and anti-CD28 antibody (BioLegend, #102116) at 2 μg ml‒1. Purified neutrophils were cocultured with stimulated T cells at indicated ratios for 48 hours. CFSE signals were analyzed by flow cytometry on gated CD4+ and CD8+ cells. In experiments evaluating PD-L1 or Arg1 blockade, anti-mouse PD-L1 (eBioscience, #14-5983-82) at 10 μg ml‒1 or nor-NOHA (Cayman Chemical, #10006861) at 300 μM was added to the coculture.
Quantitative RT-PCR (qRT-PCR)
RNA was isolated using the RNeasy Kit (BioBasic, BS1361) and reverse transcribed using the All-in-One cDNA Synthesis Kit (Bimake, B24403). qRT-PCR was performed using SYBR Green qPCR Master Mix (Bimake, B21202) on CFX Connect Real-Time PCR Detection System (Bio-Rad). The 2−ΔΔCT method was used to analyze the relative changes in gene expression. Gapdh was used as the house-keeping gene for normalization. Primer sequences are listed in Table S5.
Tumor cell conditioned medium (CM) and tumor explant supernatant (TES)
For CM, tumor cells at ~90% confluent were refreshed with culture medium and incubated for 24 hours. The medium was harvested and passed through 0.22μm filters. For TES, mammary tumors were cut into pieces less than 3mm in diameter and soaked in RPMI1640 with 10% FBS and penicillin-streptomycin. After 16–18 h of incubation at 37 °C, the cell-free supernatant was collected and filtered through 0.22μm filters. CM was typically used as 10% supplement to target culture medium. TES was typically used as 20% supplement to target culture medium. If not used freshly, CM and TES can be stored at ‒80°C.
Bone marrow-derived neutrophil (BMDN) culture and adoptive transfer
Bone marrow cells were flushed with PBS from mouse femur and tibia using 25-gauge needles. The cell suspension was gently disaggregated and passed through 40μm cell strainer to produce single cell suspension. Red blood cells were removed by RBC lysis buffer. Bone marrow cells were then seeded to 6-well plates with 10% tumor cell CM or GM-CSF (BioLegend, #576302) at 10ng/ml for 3 days. For blocking GM-CSF, anti-mouse GM-CSF (Biogend, # 505401) was added. Neutrophils were purified with MojoSort Mouse Ly6G Selection Kit (BioLegend, #480124) as the BMDNs, which were used for subsequent applications. For adoptive transfer, purified BMDNs from WT and Acod1‒/‒ mice were counted with Trypan blue dye and then resuspended with the same density (108 cells/mL) of viable cells. Recipient mice were injected i.v. with BMDNs (107 per mouse) resuspended in 100μl PBS at the designated time points. For leukocyte quantification, blood cells were isolated with EDTA-containing tubes and stained for flow cytometric analysis.
Serum inflammatory cytokine detection
Serum cytokines were quantified using the LEGENDplex mouse inflammation panel (BioLegend, #740446) per the manufacturer’s instructions. All data were collected on CytoFLEX S (Beckman Coulter) and analyzed using LEGENDplex software (BioLegend).
Immunofluorescence (IF) co-staining and fluorescence microscopy
IF co-staining was conducted for either cryosections or FFPE sections. For cryosections, fresh tumor or lung tissues embedded in Tissue–Tek OCT compound (Electron Microscopy Sciences) were sectioned with a cryostat to obtain 5μm sections, which were fixed in 4% paraformaldehyde for 20 min. For FFPE samples, antigen retrieval was performed by heating in a pressure cooker at 95°C for 30 min, followed by 115°C for 1 min in citrate-unmasking buffer (pH 6.0). Permeabilization and blocking was done by covering tissue with a blocking buffer (0.5% saponin + 2% bovine serum albumin + 2% goat serum) for 30 minutes. Next, tissue sections were labeled overnight at 4°C with primary antibodies followed by incubation for 1 hour at room temperature with secondary antibodies and DAPI (Sigma-Aldrich, #D9542) at 1μg ml‒1. Slides were mounted and observed with Leica DMi8. Images were analyzed using Fiji software (RRID:SCR_002285). Primary antibodies for mouse samples: Acod1 (Cell Signaling Technology, #17805), CD11b (BioLegend, #101202), Ly6G (BioXcell, #BP0075-1), CD68 (Bio-rad, #1602), CD3 (BioLegend, #100302), B220 (BioLegend, #103201). Primary antibodies for human samples: rabbit anti-human Acod1 (Aviva Systems Biology, #OACA09406), AF488-conjugated mouse anti-human CD15 (BioLegend, # 301910), mouse anti-human pan-Cytokeratin (BioLegend, #914204). Secondary antibodies were used: AF594-conjugated anti-rabbit (Jackson ImmunoResearch Laboratories, #611-585-215), AF647-conjugated anti-mouse (Jackson ImmunoResearch Laboratories, #115-605-146), AF647-conjugated anti-rabbit (Jackson ImmunoResearch Laboratories, #711-605-152) and AF594-conjugated anti-rat (Jackson ImmunoResearch Laboratories, #712-585-150).
Immunohistochemistry staining
Animal tissues were fixed overnight in 10% formalin and embedded in paraffin. Antigen retrieval was performed by heating in a pressure cooker at 95°C for 30 min, followed by 115°C for 1 min in citrate-unmasking buffer (pH 6.0). IHC staining with cleaved Caspase 3 antibody (Cell Signaling Technology, 9661) was performed as described54. The IHC slides were scanned using an Aperio ScanScope (Leica).
ER-Hoxb8 progenitor cell culture and neutrophil differentiation
ER-Hoxb8 progenitor cell line and the SCF-producing CHO cell line were gifted from David B. Sykes lab at Harvard University. The development of the cell lines was described21. The SCF-CM was generated by collecting supernatant from SCF-producing CHO cells cultured for 4 days after more than 90% confluent. ER-HoxB8 progenitor cell line was cultured in RPMI1640, 10% FBS, 100U/ml penicillin-streptomycin, 1μM β-estradiol (Sigma-Aldrich, #E2758) and 2% SCF-CM. To trigger neutrophil differentiation from ER-HoxB8, β-estradiol was washed away from the ER-HoxB8 culture. The cells were further cultured in RPMI1640, 10% FBS, 100U/ml penicillin-streptomycin, 2% SCF-CM and designated supplements, such as cytokines, chemokines or tumor cell CM, for 5–7 days. Cytokines or chemokines were purchased from BioLegend: GM-CSF (#576302), M-CSF (#576402), G-CSF (#576602), CCL2 (#578402), CXCL2 (#582502), CCL5 (#594202), CXCL1 (#573702), CXCL10 (#573602), TIMP1(#593702). Inhibitors have been used to validate STAT3 or STAT5 signaling involvement. For STAT3 inhibition: BP-1–102 (MCE, #HY-100493), WP1066 (MCE, #HY-15312) and LLL12 (Sigma, #573131). For STAT5 inhibition: STAT5-IN-1 (MCE, #HY-101853).
shRNA knockdown of Cebpb in ER-Hoxb8-DNs
Three MISSION lentiviral shRNA constructions in the pLKO backbone targeting mouse Cebpb (Sigma-Aldrich, #TRCN0000231408, #TRCN0000231410 and #TRCN0000231411) were purchased. A pLKO vector with scramble shRNA was used as control. Lentivirus were generated by transfecting HEK-293T cells with a 4:3:1 ratio of pLKO/psPAX2/pMD2.G with polyethylenimine (Sigma-Aldrich, # 408727). Lentivirus was collected 48 h after transfection. ER-Hoxb8 progenitor cell line (60% confluent) was infected by adding 50% medium volume of the lentivirus mixed with 8 μg/mL polybrene (Sigma-Aldrich, #H9268). Three days later, stable cell line was selected with puromycin (Goldbio, #P-600–500) at 5 μg ml‒1.
CRISPR/cas9-mediated Csf2 knockout cell lines
To generate Csf2 CRPSR/cas9-knockout cells, three different CRISPR/Cas9 sgRNA designs in an all-in-one lentiviral vector (ABM, 16914114) were purchased. Scrambled sgRNA CRISPR/Cas9 All-in-One Lentivector (K010) was used as control. Lentivirus was packaged to infect target cells as described55. After puromycin selection, Csf2 knockout efficiency was validated by quantifying GM-CSF protein levels in tumor CM with ELISA (Biolegend, #432207) per the manufacturer’s instructions.
ROS measurement
For measuring ROS levels in neutrophils isolated from metastases, cells were incubated in PBS containing 2 mM BODIPY 581/591 C11 reagent (Cayman Chemical, #27086) for 30 min at 37°C to measure lipid peroxidation. For cytosolic and mitochondrial ROS measurement, cells were stained with DCFDA (Cayman Chemical, #601520) or mitochondrial ROS detection reagent (Cayman Chemical, #701600) for 20~30 mins according to the manufacturer’s protocol. After incubation with these dyes, cells were washed and stained with antibodies and DAPI before flow cytometry.
For measuring lipid peroxidation in BMDNs, WT and Acod1‒/‒ neutrophils were isolated from bone marrow and seeded into 6-well plate with 10ng ml‒1 GM-CSF (BioLegend, #576302) for 2 days. Next, cell culture was added with DMSO (vehicle), 0.25mM 4OI (Cayman Chemical, #25374), 0.25mM DMI (Sigma, #592498) or 10uM Fer1 (Cayman Chemical Comp, #17729) for additional 16 hours with or without 20% E0771 TES. For some experiments, ML385 (Cayman Chemical, #HY-100523) and NK-252 (Cayman Chemical, #HY-19734) at given concentrations were added to inhibit and activate Nrf2 activity, respectively. Cells were harvested to stain for lipid peroxidation with BODIPY 581/591 C11 reagent (Cayman Chemical, #27086), or stain with DAPI (Sigma-Aldrich, #D9542) at 0.5ug/ml to examine viability with flow cytometry.
Glutathione measurement
WT and Acod1‒/‒ neutrophils were isolated from bone marrow and seeded into 6-well low attachment plate with 10% E0771 CM for 2 days. Next, cell culture was added with DMSO (vehicle), 4OI (0.25mM) or DMI (0.25mM) for additional 16 hours, before the cells were collected for measurement of glutathione (GSH + GSSG) using the Glutathione Assay Kit (Cayman Chemical, #703002) according to the manufacturer’s protocol. The GSH and GSSG concentrations were calculated using a standard curve and normalized to the cell counts in each sample.
Absolute quantification of itaconate in lung interstitial fluid
The lung interstitial fluid extraction method was adapted from previous study56. Briefly, healthy and lung metastasis-bearing mice were euthanized. Lungs were harvested, rinsed with PBS and dried by carefully tapping in a gauze. Subsequently, the organs were placed in a cell strainer with 40μm pores (CELLTREAT, #229481) sitting on top of 50mL centrifuge tube (NEST Scientific, #602052). Edge of cell strainer was cut so the tube could be recapped. The interstitial fluids (around 5 μl per mouse) was collected in the strainer-centrifuge tubes after centrifugation at 400 g, 4 °C for 10 minutes. Interstitial fluid from a few mice was pooled to reach 20μl as one sample. The lung interstitial fluid was stored at ‒80°C.
Frozen lung interstitial fluid was shipped to Metabolomics Core at Rutgers Cancer Institute of New Jersey to measure itaconate concentrations. 13C5 itaconate (4 μM) was spiked into each sample. HILIC separation was performed on a Vanquish Horizon UHPLC system (Thermo Fisher Scientific, Waltham, MA) with an XBridge BEH Amide column (150 mm × 2.1 mm, 2.5 μm particle size, Waters, Milford, MA) using a gradient of solvent A (95%:5% H2O:acetonitrile with 20 mM acetic acid, 40 mM ammonium hydroxide, pH 9.4) and solvent B (20%:80% H2O:acetonitrile with 20 mM acetic acid, 40 mM ammonium hydroxide, pH 9.4). The gradient was 0 min, 100% B; 3 min, 100% B; 3.2 min, 90% B; 6.2 min, 90% B; 6.5 min, 80% B; 10.5 min, 80% B; 10.7 min, 70% B; 13.5 min, 70% B; 13.7 min, 45% B; 16 min, 45% B; 16.5 min, 100% B; and 22 min, 100% B. The flow rate was 300 μl min‒1. The column temperature was set to 25 °C. The autosampler temperature was set to 4 °C, and the injection volume was 5 μl. MS scans were obtained in both negative and positive ion modes with a resolution of 70,000 at m/z 200, in addition to an automatic gain control target of 3 × 106 and m/z scan range of 72 to 1000. Metabolite data was obtained using the MAVEN software package. The ion counts of endogenous itaconate was normalized to the ion counts of 13C5 itaconate to obtain the absolute quantity.
Relative quantification of itaconate in neutrophils
For detecting intracellular itaconate in BMDNs, bone marrow cells from Acod1‒/‒ mice were differentiated with E0771 CM for 3 days and purified with Ly6G magnetic beads as described. Cells were then counted and resuspended with RPMI complete medium and plated into 6-well plate (low attachment) at 2 × 106 cells/mL. BMDNs were treated with itaconate (10mM, 0.25mM), 4OI (0.25mM) or DMI (0.25mM) for 12 hours. To extract metabolites from BMDNs, cells were harvested and washed with PBS. 1 mL ice cold extraction solvent (40:40:20 methanol: acetonitrile : water w/ 0.5% formic acid) was added immediately. After incubating on ice for 5 minutes, 50ul 15% NH4HCO3 was added. Cell lysate mixture was centrifuged at 15,000g for 10 minutes in cold room to pellet cell debris and proteins and transfer 700ul supernatant to 1.5 mL tubes on ice. The samples were stored at −80°C and shipped to Metabolomics Core at Rutgers Cancer Institute of New Jersey to measure relative itaconate concentrations. The LC-MS/MS was performed on a Q Exactive PLUS hybrid quadrupole-orbitrap mass spectrometer coupled to a Vanquish Horizon UHPLC system (Thermo Fisher Scientific, Waltham, MA) with an XBridge BEH Amide column (150 mm × 2.1 mm, 2.5 μm particle size, Waters, Milford, MA). The HILIC separation used a gradient of solvent A (95%:5% H2O:acetonitrile with 20 mM acetic acid, 40 mM ammonium hydroxide, pH 9.4) and solvent B (20%:80% H2O:acetonitrile with 20 mM acetic acid, 40 mM ammonium hydroxide, pH 9.4). The gradient was 0 min, 100% B; 3 min, 100% B; 3.2 min, 90% B; 6.2 min, 90% B; 6.5 min, 80% B; 10.5 min, 80% B; 10.7 min, 70% B; 13.5 min, 70% B; 13.7 min, 45% B; 16 min, 45% B; 16.5 min, 100% B; and 22 min, 100% B. The flow rate was 300 μL/min. The column temperature was set to 25 °C. The autosampler temperature was set to 4 °C, and the injection volume was 5 μL. MS scans were obtained in negative ionization mode with a resolution of 70,000 at m/z 200, in addition to an automatic gain control target of 3 × 106 and m/z scan range of 72 to 1000. The itaconate was monitored by a PRM event (m/z 129.02@HCD53.33). Metabolite data was obtained using the MAVEN software package (mass accuracy window: 5 ppm).
Metabolite profiling
To extract metabolites, 690μL of ice-cold chloroform and methanol (1:1, v/v) was added to 4×106 cells, sonicated in a water bath sonicator for 5 minutes, and incubated on ice for 30 minutes. Then, 310 μL of ice-cold water was added, vortexed, and centrifuged at 15,000xg for 10 minutes at 4°C to induce phase separation. The upper aqueous phase was collected and dried in a vacuum evaporator. Dried extracts were resuspended in pyridine 10mg/mL methoxyamine and derivatized MTBSFA with 1% TMCS57.TBDMS derivatized metabolites were analyzed on an Agilent 5977b GCMS as reported previously57,58. Data were analyzed in MassHunter Quantitative Analysis (v8.0, Agilent Technologies) using chemical standard-verified retention time and m/z of a quantitative and at least one qualitative ion per compound. These were: lactate (RT = 9.3min, quantitative m/z = 261, qualitative m/z = 189), alanine (RT = 9.8min, quantitative m/z = 260, qualitative m/z = 158), citrate (RT = 17.3min, quantitative m/z = 459, qualitative m/z = 431, 357), itaconate (RT = 12.3min, quantitative m/z = 301, qualitative m/z = 343), succinate (RT = 12.1min, quantitative m/z = 289, qualitative m/z = 331, 175), fumarate (RT = 12.3min, quantitative m/z = 287, qualitative m/z = 329), malate (RT = 14.8 min, quantitative m/z = 419, qualitative m/z = 287, 349), and aspartate (RT = 15.2min, quantitative m/z = 418, qualitative m/z = 390, 302).
QUANTIFICATION AND STATISTICAL ANALYSIS
In vitro and in vivo experiments were performed three or more times and conclusions were drawn only when the results were reproducible. One representative result among the replicates was shown in the figures. For non-omics data, statistical analyses were performed using GraphPad Prism v9.3 (RRID: SCR_002798). Unless otherwise mentioned, all data are presented as mean ± s.e.m. (standard error of the mean). We followed this workflow for statistical testing: Shapiro-Wilk test was performed to assess for normality of data distribution: (i) in case of normality, when only two conditions were to test, we performed unpaired t-test; when more than two conditions were to compare, we performed a parametric one-way or two-way ANOVA followed by post hoc test with recommended correction for multiple comparisons to assess the significance among pairs of conditions. (ii) in case of non-normality, when only two conditions were to test, we performed a Mann-Whitney U test; when more than two conditions were to compare, we performed a non-parametric one-way ANOVA followed by recommended test to assess the significance among pairs of conditions. For survival data, log-rank test was used. Sample sizes, error bars, P values, and statistical methods are noted in the figures or figure legends. Statistical significance was defined as P < 0.05.
Supplementary Material
Table S1. Ingenuity Canonical Pathways identified by comparing blood neutrophils (BNs) and tumor-infiltrating neutrophils (TINs). Related to Figure 1.
Table S2. Differentially expressed genes between blood neutrophils (BNs) and tumor-infiltrating neutrophils (TINs). Related to Figure 1.
Table S3. Differentially expressed metabolic enzyme-encoded genes between blood neutrophils (BNs) and tumor-infiltrating neutrophils (TINs). Related to Figure 1.
Table S4. Clinical information for the breast cancer samples used for IF co-staining. Related to STAR Methods.
Table S5. Primer sequences for qRT-PCR and barcode sequence of hashtag antibodies. Related to STAR Methods.
Table S6. Marker genes used for cell annotation in the analysis of scRNA-seq dataset GSE169246. Related to STAR Methods.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Purified anti-β-actin (For WB) | Santa Cruz | Cat# sc-47778 |
| Purified anti-Vinculin (For WB) | Millipore | Cat# 05-386 |
| Purified anti-Mouse Irg1 (For WB and IF) | Cell Signalling Technology | Cat# 17805 |
| Purified anti-Human Acod1 (for IF) | Aviva Systems Biology | Cat# OACA09406 |
| Purified anti-C/EBPβ (For WB) | BioLegend | Cat# 606202 |
| Purified anti-Stat3 (For WB) | Cell Signalling Technology | Cat# 4904P |
| Purified anti-p-Stat3 (For WB) | Cell Signalling Technology | Cat# 9145 |
| Purified anti-Stat5 (D2O6Y) Rabbit mAb (For WB) | Cell Signalling Technology | Cat# 94205T |
| Purified anti-p-Stat5 (For WB) | Cell Signalling Technology | Cat# 9314S |
| Purified anti-Nrf2 (D1Z9C) (For WB) | Cell Signalling Technology | Cat# 12721T |
| Purified anti-Gpx4 Antibody (For WB) | R&D Systems | Cat# MAB5457-SP |
| Purified anti-Ly6G (Clone 1A8; For IF and neutrophil depletion) | BioXcell | Cat# BP0075-1 |
| Purified anti-CD11b (For IF) | BioLegend | Cat# 101202 |
| Purified anti-CD68 (for IF) | Bio-rad | Cat# 1602 |
| Purified anti-CD3 (Clone 145-2C11) (for IF) | BioLegend | Cat# 100302 |
| Purified anti-B220 (Clone RA3-6B2) (for IF) | BioLegend | Cat# 103201 |
| Purified anti-CD3ε [145-2C11] 500 μg (for T cell activation) | BioLegend | Cat# 100302 |
| Purified anti-CD28 (for T cell activation) | BioLegend | Cat# 102102 |
| AF488 anti-hCD15 | BioLegend | Cat# 301910 |
| Purified anti-Pan-Cytokeratin | BioLegend | Cat# 914204 |
| AF647 anti-Rabbit IgG (H+L) | Jackson ImmunoResearch | Cat# 711-605-152 |
| AF647 anti-Mouse IgG (H+L) | Jackson ImmunoResearch | Cat# 115-605-146 |
| AF647 anti-Rat IgG | Jackson ImmunoResearch | Cat# 712-606-153 |
| AF594 anti-Rabbit | Jackson ImmunoResearch | Cat# 611-585-215 |
| AF594 anti-mouse | Jackson ImmunoResearch | Cat# 615-585-214 |
| AF594 anti-Rat IgG | Jackson ImmunoResearch | Cat# 712-585-150 |
| HRP-conjugated anti-mouse IgG | Cell Signalling Technology | Cat# 7076P2 |
| HRP-conjugated anti-rabbit IgG | Cell Signalling Technology | Cat# 7074S |
| PE-Cy7 anti-mLY6G (1A8) | Tonbo Biosciences | Cat# 60-1276-U100 |
| APC anti-h/mCD11b (M1/70) | Tonbo Biosciences | Cat# 20-0112-U100 |
| APC anti-mCD45 (30-F11) | Tonbo Biosciences | Cat# 20-0451-U025 |
| PE-Cy7 anti-mCD45 (30-F11) | Tonbo Biosciences | Cat# 60-0451-U100 |
| PerCP-Cy5.5 anti-mCD45 (30-F11) | Tonbo Biosciences | Cat# 65-0451-U100 |
| PerCP-Cyanine5.5 anti-mCD3 (17A2) | Tonbo Biosciences | Cat# 65-0032-U100 |
| APC-Cy7 anti-mCD3 (17A2) | Tonbo Biosciences | Cat# 25-0032-U100 |
| PE anti-mCD4 (GK1.5) | Tonbo Biosciences | Cat# 50-0041-U100 |
| APC anti-mCD8a (53-6.7) | Tonbo Biosciences | Cat# 20-0081-U100 |
| PerCP-Cy5.5 anti-mCD8a (53-6.7) | Tonbo Biosciences | Cat# 65-0081-U100 |
| APC anti-mIFN gamma (XMG1.2) | Tonbo Biosciences | Cat# 20-7311-U100 |
| APC anti-mFoxp3 (3G3) | Tonbo Biosciences | Cat# 20-5773-U025 |
| Anti-mCD16 + CD32 (2.4G2) | Tonbo Biosciences | Cat# 70-0161-U500 |
| APC Anti-mCD274 (PD-L1) (10F.9G2) | Tonbo Biosciences | Cat# 20-1243-U025 |
| Purified anti-mGM-CSF (for blocking) | Tonbo Biosciences | Cat# 21-8339-U020 |
| Purified anti-Cleaved Caspase-3 (Asp175) Antibody | Cell Signaling Technology | Cat# 9661S |
| Lineage cocktail biotin antibody | Biolegend | Cat# 480050 |
| Percp5.5-Streptavidin | eBioscience | Cat# 45-4317-80 |
| APC anti-c-Kit | Tonbo Bioscience | Cat# 201173-U100 |
| PE anti-CD34 | Biolegend, | Cat# 128609 |
| APC-Cy7 anti-mouse CD16/32 (FcγRII/III) | Biolegend | Cat# 101327 |
| Pe-Cy7 anti-sca-1 | Biolegend | Cat# 122514 |
| Biological samples | ||
| FFPE sample of human breast cancer surgical specimen | Harper Cancer Research Institute at University of Notre Dame | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Collagenase D, from Clostridium histolyticum | Sigma-Aldrich | Cat# COLLD-RO |
| DNase I, Grade II, from bovine pancreas | Sigma-Aldrich | Cat# 10104159001 |
| RIPA Lysis and Extraction Buffer | Thermo Fisher | Cat# 89900 |
| Dapi, for nucleic acid staining | Sigma-Aldrich | Cat# D9542-10MG |
| Recombinant Mouse GM-CSF | BioLegend | Cat# 576302 |
| Recombinant Mouse M-CSF | BioLegend | Cat# 576402 |
| Recombinant Mouse G-CSF | BioLegend | Cat# 574602 |
| Recombinant Mouse CCL2 | BioLegend | Cat# 578402 |
| Recombinant Mouse CXCL2 | BioLegend | Cat# 582502 |
| Recombinant Mouse CCL5 | BioLegend | Cat# 594202 |
| Recombinant Mouse CXCL1 | BioLegend | Cat# 573702 |
| Recombinant Mouse CXCL10 | BioLegend | Cat# 573602 |
| TIMP-1 | BioLegend | Cat# 593702 |
| beta-Estradiol, suitable for cell culture | Sigma-Aldrich | Cat# E2758-250MG |
| STAT5-IN-1 | MedChem Express | Cat# HY-101853 |
| BP-1-102 | MedChem Express | Cat# HY-100493 |
| WP1066 | MedChem Express | Cat# HY-15312 |
| LLL12 | Sigma-Aldrich | Cat# 573131 |
| Dimethyl itaconate,99% | Sigma-Aldrich | Cat# 592498-25G |
| 4-octyl Itaconate | Cayman Chemical | Cat# 25374 |
| Itaconic acid,≥99% | Sigma-Aldrich | Cat# I29204-100G |
| Brefeldin A Solution (1,000X) | VWR | Cat# 420601-BL |
| PMA, for use in molecular biology applications, ≥99% (HPLC) | Sigma-Aldrich | Cat# P1585-25MG |
| Ionomycin, 98% | VWR | Cat# AAJ62448-MCR |
| C11 BODIPY 581/591 | Cayman Chemical | Cat# 27086 |
| APC Annexin V | Tonbo Biosciences | Cat# 20-6409-T100 |
| D-Luciferin, potassium salt | Goldbio | Cat# LUCK-4G |
| Ferrostatin-1 | Cayman Chemical | Cat# 17729 |
| ML385 | MedChem Express | Cat# HY-100523 |
| NK252 | MedChem Express | Cat# HY-19734 |
| Protease inhibitors | Bimake | Cat# B14012 |
| Phosphatase inhibitors | Roche | Cat# 4906845001 |
| Clarity Max™ Western ECL Substrate | Bio-Rad | Cat# 1705062 |
| Critical commercial assays | ||
| CellTrace™ CFSE Cell Proliferation Kit | Invitrogen | Cat# C34554 |
| MojoSort™ Mouse Ly-6G Selection Kit | Biolegend | Cat# 480124 |
| Mitochondrial ROS Detection Assay Kit | Cayman Chemical | Cat# 701600 |
| ROS Detection Cell-Based Assay Kit (DCFDA) | Cayman Chemical | Cat# 601520 |
| MojoSort™ Mouse CD3 T Cell Isolation Kit | Biolegend | Cat# 480024 |
| Foxp3 / Transcription Factor Staining Buffer Kit | Tonbo Biosciences | Cat# TNB-0607-KIT |
| Proteome Profiler Mouse Cytokine Array Kit, Panel A | R&D Systems | Cat# ARY006 |
| GM-CSF ELISA MAX™ Standard ELISA Kit | Biolegend | Cat# 432201 |
| Glutathione Assay Kit | Cayman Chemical | Cat# 703002 |
| BCA Protein Assay Kit | VWR | Cat# PI23225 |
| LEGENDplex™ Mouse Inflammation Panel | Biolegend | Cat# 740446 |
| Deposited data | ||
| Drop-seq raw data comparing mice Py7160 TIN and blood neutrophils | This paper | GEO: GSE216425 |
| Microarray data of human breast cancer metastases | PMID: 24287398 | GEO: GSE46141 |
| Single-cell sequencing of human TNBC metastatic tissue and peripheral blood | PMID: 34653365 | GEO: GSE169246 |
| RNA-seq data for murine breast cancer cells, tumor-infiltrating macrophages and tumor-infiltrating neutrophils | PMID: 31451770 | GEO: GSE104765 |
| PDF file containing uncropped scans of all Western blots | This paper | Data S1 |
| Excel file containing the values used to create all graphs | This paper | Data S1 |
| Experimental models: Cell lines | ||
| Py7160 | This study | N/A |
| E0771 | Siyuan Zhang lab at University of Notre Dame | N/A |
| E0771-TR | This study | N/A |
| 4T1 | Yibin Kang lab at Princeton University | N/A |
| 168FARN | Yibin Kang lab at Princeton University | N/A |
| Nmumg | Yibin Kang lab at Princeton University | N/A |
| ER-Hoxb8 progenitor cell line | David B. Sykes lab at Harvard University | N/A |
| SCF-producing CHO cell line | David B. Sykes lab at Harvard University | N/A |
| GMCSF-producing B16 cell line | David B. Sykes lab at Harvard University | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | RRID:IMSR_JAX:00 0664 |
| Mouse: FVB | The Jackson Laboratory | RRID:IMSR_JAX:00 1800 |
| Mouse: MMTV-PyMT | The Jackson Laboratory | RRID:IMSR_JAX:02 2974 |
| Mouse: Acod1−/− | The Jackson Laboratory | RRID:IMSR_JAX:02 9340 |
| Mouse: MRP8-Cre | The Jackson Laboratory | RRID:IMSR_JAX:02 1614 |
| Mouse: LysM-cre | The Jackson Laboratory | RRID:IMSR_JAX:00 4781 |
| Mouse: Acod1f/f | Michael Diamond lab from Washington University in St. Louis | N/A |
| Mouse: Rag1−/− | The Jackson Laboratory | RRID:IMSR_JAX:00 2216 |
| Mouse: Ly6G-cre | Matthias Gunzer and Anja Hasenberg lab from University Hospital Essen | N/A |
| Oligonucleotides | ||
| qPCR primers | This paper | Table S5 |
| Hashtag antibody barcode sequence | Biolegend | Table S5 |
| Recombinant DNA | ||
| Csf2 sgRNA CRISPR/Cas9 All-in-One Lentivector set (Mouse) | abm | Cat# 16914114 |
| CRISPR Scrambled sgRNA All-in-One Lentiviral Vector (with spCas9) | abm | Cat# K010 |
| Three MISSION lentiviral shRNA constructions in the pLKO backbone targeting mouse Cebpb | Sigma-Aldrich | Cat#TRCN0000231408, TRCN0000231410 and TRCN0000231411) |
| pLKO vector with scramble shRNA | Sigma-Aldrich | Cat# SHC016-1EA |
| psPAX2 | Addgene | Cat# 12260 |
| pMD2.G | Addgene | Cat# 12259 |
| Software and algorithms | ||
| R studio | Posit | https://www.rstudio.com/products/rstudio/ |
| Graphpad Prism 8.0 software | GraphPad Software, Inc. | https://www.graphpad.com |
| FiJi software | https://imagej.net/Fiji/Downloads | RRID:SCR_002285 |
| FlowJo | FlowJo LLC | https://www.flowjo.com |
| Seurat R package v4.0 | Satija Lab | https://satijalab.org/seurat/ |
| Ingenuity Pathway Analysis (IPA) | Qiagen | https://digitalinsights.qiagen.com/products |
| Auro Imaging Software | Spectral Instruments Imaging, LLC | https://spectralinvivo.com/software/ |
Highlights.
Acod1 is an upregulated enzyme in murine and human tumor-infiltrating neutrophils.
Acod1 upregulation is driven by the GM-CSF-JAK/STAT5-C/EBPβ signal pathway.
Itaconate defends against ferroptosis and upholds neutrophils in metastasis.
Acod1 ablation boosts the anti-metastasis efficacy of immune checkpoint blockade.
ACKNOWLEDGMENTS
We would like to thank the Lu lab members for their essential comments and suggestions during this work. We thank Michael Diamond from Washington University School of Medicine for sharing Acod1f/f mice, Matthias Gunzer and Anja Hasenberg from University Hospital Essen for sharing Ly6G-Cre mice, David B. Sykes from Harvard University for sharing ER-Hoxb8 cells, and Kathy D. Miller for consultation of clinical validation experiments. We are grateful for the support from core facilities used in this study, including the Freimann Life Science Center (Teri Highbaugh), Genomics and Bioinformatics Core Facility (Michael Pfrender, Melissa Stephens, Jacqueline Lopez, Brent Harker), Integrated Imaging Facility (Sara Cole, Sarah Chapman), and Harper Cancer Research Institute’s Tissue Biorepository. This work was supported by National Institutes of Health grant R01CA280097 (Xin Lu), Susan G. Komen Foundation grant CCR18548293 (Xin Lu) and METAvivor Early Career Investigator Award (Yun Zhao). Other support included National Institutes of Health grant R01CA248033 (Xin Lu), Department of Defense grants W81XWH2010312 (Xin Lu), W81XWH2010332 (Xin Lu), HT94252310010 (Xin Lu), National Institutes of Health grant P30CA082709 (Jun Wan), and Boler Family Foundation (Xin Lu) at University of Notre Dame.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Figures S1–S7.
REFERENCES
- 1.Latif F, Bint Abdul Jabbar H, Malik H, Sadaf H, Sarfraz A, Sarfraz Z, and Cherrez-Ojeda I (2022). Atezolizumab and pembrolizumab in triple-negative breast cancer: a meta-analysis. Expert review of anticancer therapy 22, 229–235. 10.1080/14737140.2022.2023011. [DOI] [PubMed] [Google Scholar]
- 2.Corbeau I, Jacot W, and Guiu S (2020). Neutrophil to Lymphocyte Ratio as Prognostic and Predictive Factor in Breast Cancer Patients: A Systematic Review. Cancers (Basel) 12. 10.3390/cancers12040958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. (2015). The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 21, 938–945. 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quail DF, Amulic B, Aziz M, Barnes BJ, Eruslanov E, Fridlender ZG, Goodridge HS, Granot Z, Hidalgo A, Huttenlocher A, et al. (2022). Neutrophil phenotypes and functions in cancer: A consensus statement. J Exp Med 219. 10.1084/jem.20220011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao Y, Rahmy S, Liu Z, Zhang C, and Lu X (2020). Rational targeting of immunosuppressive neutrophils in cancer. Pharmacol Ther 212, 107556. 10.1016/j.pharmthera.2020.107556. [DOI] [PubMed] [Google Scholar]
- 6.Li P, Lu M, Shi J, Hua L, Gong Z, Li Q, Shultz LD, and Ren G (2020). Dual roles of neutrophils in metastatic colonization are governed by the host NK cell status. Nat Commun 11, 4387. 10.1038/s41467-020-18125-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veglia F, Sanseviero E, and Gabrilovich DI (2021). Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol 21, 485–498. 10.1038/s41577-020-00490-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim IS, Gao Y, Welte T, Wang H, Liu J, Janghorban M, Sheng K, Niu Y, Goldstein A, Zhao N, et al. (2019). Immuno-subtyping of breast cancer reveals distinct myeloid cell profiles and immunotherapy resistance mechanisms. Nature Cell Biology 21, 1113–1126. 10.1038/s41556-019-0373-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu X, and Lu X (2022). Enhancing immune checkpoint blockade therapy of genitourinary malignancies by co-targeting PMN-MDSCs. Biochim Biophys Acta Rev Cancer 1877, 188702. 10.1016/j.bbcan.2022.188702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Németh T, Sperandio M, and Mócsai A (2020). Neutrophils as emerging therapeutic targets. Nature reviews. Drug discovery, 10.1038/s41573-41019-40054-z. . [DOI] [PubMed] [Google Scholar]
- 11.Peace CG, and O’Neill LA (2022). The role of itaconate in host defense and inflammation. J Clin Invest 132. 10.1172/jci148548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Neill LAJ, and Artyomov MN (2019). Itaconate: the poster child of metabolic reprogramming in macrophage function. Nat Rev Immunol 19, 273–281. 10.1038/s41577-019-0128-5. [DOI] [PubMed] [Google Scholar]
- 13.Le Naour A, Rossary A, and Vasson MP (2020). EO771, is it a well-characterized cell line for mouse mammary cancer model? Limit and uncertainty. Cancer Med 9, 8074–8085. 10.1002/cam4.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Attalla S, Taifour T, Bui T, and Muller W (2021). Insights from transgenic mouse models of PyMT-induced breast cancer: recapitulating human breast cancer progression in vivo. Oncogene 40, 475–491. 10.1038/s41388-020-01560-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prima V, Kaliberova LN, Kaliberov S, Curiel DT, and Kusmartsev S (2017). COX2/mPGES1/PGE2 pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci U S A 114, 1117–1122. 10.1073/pnas.1612920114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Chen H, Mo H, Hu X, Gao R, Zhao Y, Liu B, Niu L, Sun X, Yu X, et al. (2021). Single-cell analyses reveal key immune cell subsets associated with response to PD-L1 blockade in triple-negative breast cancer. Cancer Cell 39, 1578–1593.e1578. 10.1016/j.ccell.2021.09.010. [DOI] [PubMed] [Google Scholar]
- 17.Kimbung S, Kovács A, Bendahl PO, Malmström P, Fernö M, Hatschek T, and Hedenfalk I (2014). Claudin-2 is an independent negative prognostic factor in breast cancer and specifically predicts early liver recurrences. Mol Oncol 8, 119–128. 10.1016/j.molonc.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nair S, Huynh JP, Lampropoulou V, Loginicheva E, Esaulova E, Gounder AP, Boon ACM, Schwarzkopf EA, Bradstreet TR, Edelson BT, et al. (2018). Irg1 expression in myeloid cells prevents immunopathology during M. tuberculosis infection. J Exp Med 215, 1035–1045. 10.1084/jem.20180118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasenberg A, Hasenberg M, Mann L, Neumann F, Borkenstein L, Stecher M, Kraus A, Engel DR, Klingberg A, Seddigh P, et al. (2015). Catchup: a mouse model for imaging-based tracking and modulation of neutrophil granulocytes. Nat Methods 12, 445–452. 10.1038/nmeth.3322. [DOI] [PubMed] [Google Scholar]
- 20.Lin EY, Nguyen AV, Russell RG, and Pollard JW (2001). Colony-stimulating Factor 1 Promotes Progression of Mammary Tumors to Malignancy. J. Exp. Med 193, 727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang GG, Calvo KR, Pasillas MP, Sykes DB, Häcker H, and Kamps MP (2006). Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat Methods 3, 287–293. 10.1038/nmeth865. [DOI] [PubMed] [Google Scholar]
- 22.Hirai H, Zhang P, Dayaram T, Hetherington CJ, Mizuno S, Imanishi J, Akashi K, and Tenen DG (2006). C/EBPbeta is required for ‘emergency’ granulopoiesis. Nat Immunol 7, 732–739. 10.1038/ni1354. [DOI] [PubMed] [Google Scholar]
- 23.Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E, et al. (2010). Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 32, 790–802. 10.1016/j.immuni.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 24.Al-Shami A, Mahanna W, and Naccache PH (1998). Granulocyte-macrophage colony-stimulating factor-activated signaling pathways in human neutrophils. Selective activation of Jak2, Stat3, and Stat5b. J Biol Chem 273, 1058–1063. 10.1074/jbc.273.2.1058. [DOI] [PubMed] [Google Scholar]
- 25.Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E, et al. (2018). Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556, 113–117. 10.1038/nature25986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yi Z, Deng M, Scott MJ, Fu G, Loughran PA, Lei Z, Li S, Sun P, Yang C, Li W, et al. (2020). Immune-Responsive Gene 1/Itaconate Activates Nuclear Factor Erythroid 2-Related Factor 2 in Hepatocytes to Protect Against Liver Ischemia-Reperfusion Injury. Hepatology 72, 1394–1411. 10.1002/hep.31147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, Cervantes-Barragan L, Ma X, Huang SC, Griss T, et al. (2016). Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab 24, 158–166. 10.1016/j.cmet.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geering B, and Simon HU (2011). Peculiarities of cell death mechanisms in neutrophils. Cell Death and Differentiation 18, 1457–1469. 10.1038/cdd.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lei G, Zhuang L, and Gan B (2022). Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer 22, 381–396. 10.1038/s41568-022-00459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al. (2019). CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569, 270–274. 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, Wang Q, Yang M, Qian J, and Yi Q (2021). CD36-mediated ferroptosis dampens intratumoral CD8(+) T cell effector function and impairs their antitumor ability. Cell metabolism 33, 1001–1012 e1005. 10.1016/j.cmet.2021.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu S, Chaudhary O, Rodriguez-Morales P, Sun X, Chen D, Zappasodi R, Xu Z, Pinto AFM, Williams A, Schulze I, et al. (2021). Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity 54, 1561–1577 e1567. 10.1016/j.immuni.2021.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim R, Hashimoto A, Markosyan N, Tyurin VA, Tyurina YY, Kar G, Fu S, Sehgal M, Garcia-Gerique L, Kossenkov A, et al. (2022). Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature. 10.1038/s41586-022-05443-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anandhan A, Dodson M, Schmidlin CJ, Liu P, and Zhang DD (2020). Breakdown of an Ironclad Defense System: The Critical Role of NRF2 in Mediating Ferroptosis. Cell chemical biology 27, 436–447. 10.1016/j.chembiol.2020.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beury DW, Carter KA, Nelson C, Sinha P, Hanson E, Nyandjo M, Fitzgerald PJ, Majeed A, Wali N, and Ostrand-Rosenberg S (2016). Myeloid-Derived Suppressor Cell Survival and Function Are Regulated by the Transcription Factor Nrf2. J Immunol 196, 3470–3478. 10.4049/jimmunol.1501785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. 10.1038/s41586-019-1705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang YP, Mockabee-Macias A, Jiang C, Falzone A, Prieto-Farigua N, Stone E, Harris IS, and DeNicola GM (2021). Non-canonical Glutamate-Cysteine Ligase Activity Protects against Ferroptosis. Cell metabolism 33, 174–189 e177. 10.1016/j.cmet.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao H, Teng D, Yang L, Xu X, Chen J, Jiang T, Feng AY, Zhang Y, Frederick DT, Gu L, et al. (2022). Myeloid-derived itaconate suppresses cytotoxic CD8(+) T cells and promotes tumour growth. Nat Metab. 10.1038/s42255-022-00676-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsushita M, Freigang S, Schneider C, Conrad M, Bornkamm GW, and Kopf M (2015). T cell lipid peroxidation induces ferroptosis and prevents immunity to infection. J Exp Med 212, 555–568. 10.1084/jem.20140857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu C, Sun S, Johnson T, Qi R, Zhang S, Zhang J, and Yang K (2021). The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep 35, 109235. 10.1016/j.celrep.2021.109235. [DOI] [PubMed] [Google Scholar]
- 41.Yao Y, Chen Z, Zhang H, Chen C, Zeng M, Yunis J, Wei Y, Wan Y, Wang N, Zhou M, et al. (2021). Selenium-GPX4 axis protects follicular helper T cells from ferroptosis. Nat Immunol 22, 1127–1139. 10.1038/s41590-021-00996-0. [DOI] [PubMed] [Google Scholar]
- 42.Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, St Croix CM, Mikulska-Ruminska K, Liu B, Shrivastava IH, et al. (2020). Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol 16, 278–290. 10.1038/s41589-019-0462-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu H, Klement JD, Lu C, Redd PS, Yang D, Smith AD, Poschel DB, Zou J, Liu D, Wang PG, et al. (2021). Asah2 Represses the p53-Hmox1 Axis to Protect Myeloid-Derived Suppressor Cells from Ferroptosis. J Immunol 206, 1395–1404. 10.4049/jimmunol.2000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Azzimato V, Chen P, Barreby E, Morgantini C, Levi L, Vankova A, Jager J, Sulen A, Diotallevi M, Shen JX, et al. (2021). Hepatic miR-144 Drives Fumarase Activity Preventing NRF2 Activation During Obesity. Gastroenterology 161, 1982–1997 e1911. 10.1053/j.gastro.2021.08.030. [DOI] [PubMed] [Google Scholar]
- 45.Jaiswal AK, Yadav J, Makhija S, Mazumder S, Mitra AK, Suryawanshi A, Sandey M, and Mishra A (2022). Irg1/itaconate metabolic pathway is a crucial determinant of dendritic cells immune-priming function and contributes to resolute allergen-induced airway inflammation. Mucosal Immunol 15, 301–313. 10.1038/s41385-021-00462-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang H, Chen T, Ren J, Xia Y, Onuma A, Wang Y, He J, Wu J, Wang H, Hamad A, et al. (2021). Pre-operative exercise therapy triggers anti-inflammatory trained immunity of Kupffer cells through metabolic reprogramming. Nature metabolism 3, 843–858. 10.1038/s42255-021-00402-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Arensman MD, Yang XS, Leahy DM, Toral-Barza L, Mileski M, Rosfjord EC, Wang F, Deng S, Myers JS, Abraham RT, and Eng CH (2019). Cystine-glutamate antiporter xCT deficiency suppresses tumor growth while preserving antitumor immunity. Proc Natl Acad Sci U S A 116, 9533–9542. 10.1073/pnas.1814932116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G, et al. (2018). Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med 379, 2108–2121. 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 49.Chen F, Elgaher WAM, Winterhoff M, Büssow K, Waqas FH, Graner E, Pires-Afonso Y, Casares Perez L, de la Vega L, Sahini N, et al. (2022). Citraconate inhibits ACOD1 (IRG1) catalysis, reduces interferon responses and oxidative stress, and modulates inflammation and cell metabolism. Nat Metab 4, 534–546. 10.1038/s42255-022-00577-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Swain A, Bambouskova M, Kim H, Andhey PS, Duncan D, Auclair K, Chubukov V, Simons DM, Roddy TP, Stewart KM, and Artyomov MN (2020). Comparative evaluation of itaconate and its derivatives reveals divergent inflammasome and type I interferon regulation in macrophages. Nat Metab 2, 594–602. 10.1038/s42255-020-0210-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hooftman A, Angiari S, Hester S, Corcoran SE, Runtsch MC, Ling C, Ruzek MC, Slivka PF, McGettrick AF, Banahan K, et al. (2020). The Immunomodulatory Metabolite Itaconate Modifies NLRP3 and Inhibits Inflammasome Activation. Cell Metab 32, 468–478.e467. 10.1016/j.cmet.2020.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liao ST, Han C, Xu DQ, Fu XW, Wang JS, and Kong LY (2019). 4-Octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to exert anti-inflammatory effects. Nat Commun 10, 5091. 10.1038/s41467-019-13078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. 10.1016/j.cell.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang T, Cheng X, Chahoud J, Sarhan A, Tamboli P, Rao P, Guo M, Manyam G, Zhang L, Xiang Y, et al. (2020). Effective combinatorial immunotherapy for penile squamous cell carcinoma. Nat Commun 11, 2124. 10.1038/s41467-020-15980-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhu Y, Zhao Y, Wen J, Liu S, Huang T, Hatial I, Peng X, Al Janabi H, Huang G, Mittlesteadt J, et al. (2023). Targeting the chromatin effector Pygo2 promotes cytotoxic T cell responses and overcomes immunotherapy resistance in prostate cancer. Science immunology 8, eade4656. 10.1126/sciimmunol.ade4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Christen S, Lorendeau D, Schmieder R, Broekaert D, Metzger K, Veys K, Elia I, Buescher JM, Orth MF, Davidson SM, et al. (2016). Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell reports 17, 837–848. 10.1016/j.celrep.2016.09.042. [DOI] [PubMed] [Google Scholar]
- 57.Sheldon RD, Ma EH, DeCamp LM, Williams KS, and Jones RG (2021). Interrogating in vivo T-cell metabolism in mice using stable isotope labeling metabolomics and rapid cell sorting. Nat Protoc 16, 4494–4521. 10.1038/s41596-021-00586-2. [DOI] [PubMed] [Google Scholar]
- 58.Kaymak I, Luda KM, Duimstra LR, Ma EH, Longo J, Dahabieh MS, Faubert B, Oswald BM, Watson MJ, Kitchen-Goosen SM, et al. (2022). Carbon source availability drives nutrient utilization in CD8(+) T cells. Cell Metab 34, 1298–1311.e1296. 10.1016/j.cmet.2022.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Ingenuity Canonical Pathways identified by comparing blood neutrophils (BNs) and tumor-infiltrating neutrophils (TINs). Related to Figure 1.
Table S2. Differentially expressed genes between blood neutrophils (BNs) and tumor-infiltrating neutrophils (TINs). Related to Figure 1.
Table S3. Differentially expressed metabolic enzyme-encoded genes between blood neutrophils (BNs) and tumor-infiltrating neutrophils (TINs). Related to Figure 1.
Table S4. Clinical information for the breast cancer samples used for IF co-staining. Related to STAR Methods.
Table S5. Primer sequences for qRT-PCR and barcode sequence of hashtag antibodies. Related to STAR Methods.
Table S6. Marker genes used for cell annotation in the analysis of scRNA-seq dataset GSE169246. Related to STAR Methods.
Data Availability Statement
The Drop-seq data generated in this study were deposited to GEO under accession number GSE216425. The RNA-seq data used to compare Acod1 expression between mammary cancer cells, tumor-infiltrating macrophages and tumor-infiltrating neutrophils in four mouse models were obtained from GSE104765. Gene expression profiles of human breast cancer metastasis tissues were from microarray dataset GSE46141. Single-cell RAN-seq data of human breast cancer metastasis tissue were from GSE169246. The code for scRNA-seq analysis will be provided upon request. Uncropped scans of all western blots and all raw data used to generate graphs are included in Data S1. Further information and requests for resources and reagents should be directed to the lead contact.