

Abstract
Background and Aims:
Paucity of intrahepatic bile ducts is caused by various etiologies and often leads to cholestatic liver disease. For example, in patients with Alagille syndrome (ALGS), which is a genetic disease primarily caused by mutations in JAG1, bile duct paucity often results in severe cholestasis and liver damage. However, no mechanism-based therapy exists to restore the biliary system in ALGS or other diseases associated with bile duct paucity. Based on previous genetic observations, we investigate whether postnatal knockdown of the glycosyltransferase gene Poglut1 can improve the ALGS liver phenotypes in several mouse models generated by removing one copy of Jag1 in the germline with or without reducing the gene dosage of Sox9 in the liver.
Approach and Results:
Using an antisense oligonucleotide (ASO) established in this study, we show that reducing Poglut1 levels in postnatal livers of ALGS mouse models with moderate to profound biliary abnormalities can significantly improve bile duct development and biliary tree formation. Importantly, ASO injections prevent liver damage in these models without adverse effects. Furthermore, ASO-mediated Poglut1 knockdown improves biliary tree formation in a different mouse model with no Jag1 mutations. Cell-based signaling assays indicate that reducing POGLUT1 levels or mutating POGLUT1 modification sites on JAG1 increase JAG1 protein level and JAG1-mediated signaling, suggesting a likely mechanism for the observed in vivo rescue.
Conclusions:
Our preclinical studies establish ASO-mediated POGLUT1 knockdown as a potential therapeutic strategy for ALGS liver disease and possibly other diseases associated with BD paucity.
Keywords: JAG1, biliary tree, Notch signaling, preclinical study, cholestasis
Graphical Abstract
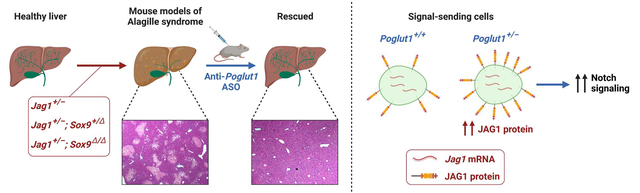
Introduction
Alagille syndrome (ALGS) is a multisystem developmental disorder involving the liver, cardiovascular system, kidney, and skeletal system among other organs.[1,2] ALGS is caused by autosomal dominant mutations in JAG1 (95%) or NOTCH2 (2–3%), which encode key components of the Notch signaling pathway.[3–8] The characteristic feature of ALGS is intrahepatic bile duct (BD) paucity, defined as a significant decrease in the average number of BD per portal vein (PV).[2] BD paucity results in severe cholestasis in many ALGS patients and often leads to end-stage liver disease.
With an estimated frequency of 1:30,000, ALGS is the most common genetic disease resulting in intrahepatic cholestasis.[9] Standard of clinical care for BD paucity is directed towards reducing intrahepatic bile accumulation by altering the flow through surgery or modifying the enterohepatic bile acid transport through inhibitors and binding resins.[10,11] However, similar to other diseases associated with severe BD paucity, the only cure for the liver disease in ALGS is liver transplantation.[11] The shortage of suitable liver donors and the rather long time that patients usually spend on the liver transplant wait list highlight the need for identifying strategies to bypass the requirement for liver transplantation.
A recent study followed 293 ALGS patients who showed features of cholestasis by the age of 5 and had their native liver at the time of enrollment.[12] Prospective analysis of these patients showed that only 24% of them survived to age 18.5 without a liver transplant. This work, which is the largest multi-center natural history study of the ALGS liver disease to date, also reported that ALGS patients exhibit a second wave of liver disease due to fibrosis and portal hypertension in later childhood.[11,12] A more recent multicenter retrospective study analyzed the natural history of the disease in 1,433 patients who were confirmed to have ALGS.[13] In the 1,184 patients who had a history of neonatal cholestasis, a native liver survival rate of 40.3% at 18 years of age was reported.[13] Moreover, 68.9% of patients developed clinically evident portal hypertension by 18.[13] Together, these studies established that the burden of the liver disease and the need for transplantation is very high in ALGS patients.
We previously reported that on a pure C57BL/6 background, mice heterozygous for Jag1 (Jag1+/−) are a genetically representative model for ALGS and are born with significant BD paucity.[14] The BD paucity remains at postnatal day 30 (P30) and is accompanied by ductular reactions, hepatocyte necrosis and fibrosis, resembling chronic hepatobiliary disease.[14,15] We used this model to identify two dosage-sensitive genetic modifiers of the Jag1+/− phenotypes. First, we showed that the Jag1+/− BD paucity can be significantly improved by removing one copy of a glycosyltransferase called “protein O-glucosyltransferase 1” (Poglut1).[14] More recently, we reported that the Jag1+/− liver phenotypes are highly sensitive to the level of Sox9 and established two additional ALGS models (Jag1+/−;Sox9+/flox;Albumin-Cre [hereafter called Jag1+/−;Sox9+/Δ] and Jag1+/−;Sox9flox/flox;Albumin-Cre [hereafter called Jag1+/−;Sox9Δ/Δ]).[15] The BD paucity in these models is more severe than the Jag1+/− model and does not show improvement with age.[15] Therefore, these three models represent the broad range of liver phenotypes observed in ALGS patients.
Here we established an antisense oligonucleotide (ASO) which can efficiently knockdown (KD) Poglut1 in vivo, and found that it improves the liver phenotypes of all three models upon postnatal injection. Cell-based signaling assays suggest that reducing Poglut1 level improves JAG1-mediated signaling by directly affecting JAG1 protein level and signaling. Our data suggest that ASO-mediated KD of POGLUT1 might be a viable therapeutic strategy for ALGS liver disease.
Materials and Methods
ASO-1
The anti-Poglut1 ASO (ASO-1; designed and developed by Ionis Pharmaceuticals) is a 3-10-3 cEt-ASO (GSTSTS mCSASTSASASTSTS mCSTS mCS mCSASA) with full phosphorothioate linkages (PS; “S” subscripts in the sequence) and methylated cytosines (mC). ASO-1 working stocks (5 mg/mL) were diluted in sterile PBS without calcium chloride and without magnesium chloride. Mice were subcutaneously injected with 50 mg/kg of ASO-1 or the same volume of PBS as control.
Mouse strains and breeding
The following strains were used: WT C57BL/6, Jag1dDSL (Jag1+/−)[14,16], Albumin-Cre (Jackson Laboratory #003574)[17], Poglut1+/− (Poglut1+/GT(IST10323G11)TIGM)[18], and Sox9flox/flox (Jackson Laboratory #013106).[19] Animals were kept on a C57BL/6 background for all crosses. All animals were maintained in a temperature-controlled environment at 21°C with 12 hour light-dark cycles in a barrier facility at Baylor College of Medicine per Institutional Animal Care and Use Committee guidelines and under approved animal protocols. Litters were randomly assigned to PBS or ASO-1 injection groups, and all pups from the same litter were injected with either PBS or ASO-1.
Quantification of the BD/PV ratio and fibrosis
The BD/PV ratio and fibrosis were quantified as previously described.[15,20] For details, please see Supporting Materials and Methods.
RNAscope
Liver tissue was fixed overnight to 3 days at 4°C in 4% paraformaldehyde, washed with PBS, equilibrated in 30% sucrose PBS solution for cryoprotection, embedded in O.C.T. (Tissue-Tek®), and stored at −80°C. RNAscope was performed according to ACDbio Multiplex Fluorescent v2 Manual with some modifications. For details, please see Supporting Materials and Methods.
Biliary tree ink injection and quantification of the biliary tree density
Retrograde biliary ink injection was performed as previously described.[21] After clearing, the liver lobes were imaged using a Ziess Axio Zoom.V16 microscope at 20x and reconstructed in Adobe Illustrator. For quantification of the biliary tree density, portions consisting of approximately one third of each left lobe were cropped from the whole image. Using the color threshold tool in ImageJ, each background was filtered out and ink areas were quantified.
Cell culture and Notch signaling assay
Signal-receiving MEFs or COS7 cells were seeded in 24-well plates 8×104 cells/well and cotransfected with the TP-1 luciferase Notch-signaling reporter construct (0.12 μg/well), plasmids expressing WT mouse Notch1 or Notch2 or empty pcDNA3 (0.1 μg/well), and gWIZ β-galactosidase construct (0.06 μg/well). The latter was used for transfection efficiency normalization using HD-FuGENE (Promega, Cat. #E5911) according to the manufacturer’s instructions. After 24 hours, media was removed and signal-sending HEK293T cells (or MEFs) stably expressing JAG1 (1.5×105 cells/well in 1 mL medium) were overlaid on transfected MEFs (or COS7 cells, when MEFs were the signal-sending cells). After 24 hours of co-culture, cells were lysed and luciferase and β-galactosidase assays (Luciferase Assay System; Promega) were performed according to the manufacturer’s instructions. Given the variability in luciferase-based co-culture assays, for each dataset, 3–4 independent experiments were performed, each comprised of 9 samples. For details, please see Supporting Materials and Methods.
Statistical analysis
Analysis of body weights, BD/PV ratios, serum total bilirubin levels, necrosis and fibrosis was carried out with unpaired t-test, t-tests with Welch’s correction, or one-way analysis of variance (ANOVA) with the appropriate multiple comparisons tests (Tukey’s or Dunnett’s) using Prism (GraphPad).
Results
Subcutaneous injection of ASO-1 can efficiently KD Poglut1 in adult mouse livers with no liver toxicity
Our previous work indicated that removing one copy of Poglut1 in the germline or with Albumin-Cre or SM22α-Cre leads to a significant improvement in Jag1+/− liver phenotypes.[14] Since both of these transgenes start CRE-mediated deletion in mouse embryos,[17,22] it was not clear whether decreasing Poglut1 levels after birth would improve their biliary phenotype. To test this notion, which is a prerequisite for therapeutic targeting of POGLUT1 in patients with ALGS, we identified a constrained ethyl (cEt) gapmer anti-Poglut1 ASO (ASO-1) from a cell-based screen. Five weekly subcutaneous injections of ASO-1 at 50 mg/kg/dose into adult (8-week-old) WT mice led to ~95% Poglut1 KD in the liver (Supporting Fig. S1, A and B). Despite efficient Poglut1 KD, the animals did not show growth retardation (Supporting Fig. S1C) or statistically significant abnormalities in liver weight, AST, ALT, or bilirubin (Supporting Fig. S1, D and E). Therefore, we used this anti-Poglut1 ASO for our rescue experiments.
ASO-mediated Poglut1 KD partially rescues the biliary abnormalities in P12 Jag1+/− animals
Based on the strong KD observed with five weekly doses of ASO-1 and because of the robust improvement in the Jag1+/− phenotype upon genetic removal of one copy (50%) of Poglut1,[14] we used two subcutaneous injections of ASO-1 at P1 and P7 (Fig. 1A). This injection regimen led to 46% and 58% reduction in Poglut1 mRNA levels in Jag1+/− and WT livers, respectively, compared to PBS-injected controls (Fig. 1B).
Fig. 1. Early postnatal injection of ASO-1 broadly knocks down Poglut1 in the liver and partially rescues the biliary abnormalities in P12 Jag1+/− animals.
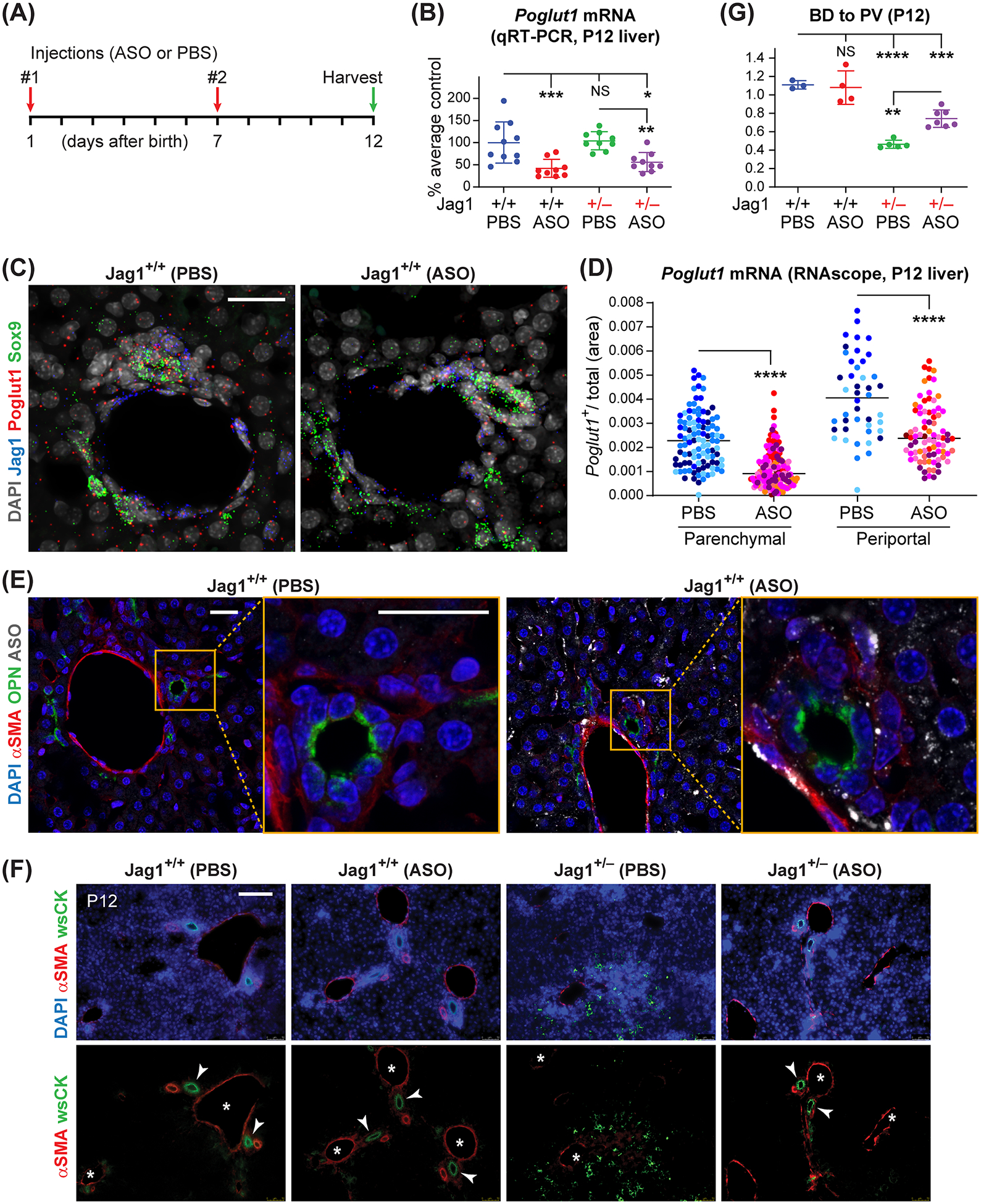
(A) Injection regimen of ASO-1 and PBS in Jag1+/− and WT animals (50 mg/kg/dose). (B) qRT-PCR assays for Poglut1 mRNA levels on WT and Jag1+/− P12 livers injected with ASO or PBS. (C) Liver RNAscope images from ASO- or PBS-injected P12 WT animals, labeled with DAPI and probes for Poglut1, Sox9, and Jag1. (D) Quantification of relative Poglut1+ area of RNAscope images in parenchymal and periportal regions. (E) Liver sections from ASO- or PBS-injected P12 WT animals, stained with antibodies against ASO, osteopontin (OPN) and αSMA. (F) P12 liver sections of each genotype stained with αSMA, wide-spectrum cytokeratin (wsCK) and DAPI. Asterisks mark PVs, arrowheads mark patent BDs. Note the weak αSMA staining, ductular reactions and hypercellularity in the parenchyma of PBS-injected Jag1+/− livers and their rescue upon ASO injections. (G) Average BD to PV ratio in P12 WT and Jag1+/− mice injected with PBS or ASO. In B and G, each circle represents an animal and horizontal lines show mean ± Standard Deviation (SD). In D, circles of the same color represent data from different parenchymal or periportal regions of liver sections from the same animal (n=4 for PBS-injected, n=7 for ASO-injected). NS: not significant, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, using one-way ANOVA with Tukey’s multiple comparisons test (B, G) or unpaired t-test with Welch’s correction (D). Scale bars are 25 μm in C, E, and 100 μm in F.
To examine the expression pattern of Poglut1 in the liver and assess the ability of the ASO to KD Poglut1 in specific liver cell types, we performed RNAscope studies[23] on P12 livers. Poglut1 is expressed in hepatocytes, biliary cells (Sox9+), and periportal mesenchymal cells (Jag1+, Sox9−) (Fig. 1C). Visual inspection of RNAscope images suggests that Poglut1 transcript levels are reduced in parenchymal and periportal regions of ASO-injected livers compared to control livers (Fig. 1C). We next quantified the Poglut1+ pixel area within liver sections and calculated the relative area of the liver covered by Poglut1 transcript signal (after excluding the PV and BD lumens and regions within the image without cells from the total area analyzed). This analysis showed that ASO injections reduce the Poglut1+ area by ~51% in the parenchymal areas, which is an indication of KD in hepatocytes (Fig. 1D). A similar analysis on PV regions to primarily assess KD in the biliary and periportal mesenchymal cells showed a 37% reduction in Poglut1+ area upon ASO injections (Fig. 1D). Antibody staining on PBS-injected and ASO-injected P12 WT livers showed broad ASO uptake by hepatocytes and portal mesenchymal cells (αSMA+), but only limited signal in biliary cells (OPN+) (Fig. 1E). Together, these observations indicate that the cEt ASO used in our studies can efficiently target hepatocytes and periportal mesenchymal cells and potentially the biliary cells to a lesser extent.
Control Jag1+/− animals (PBS-injected) showed a qualitative reduction in the number of patent BDs around PVs, accompanied by weak αSMA staining in the PV mesenchyme and aberrant cytokeratin (CK)-expressing cells and hypercellularity in the liver parenchyma, suggestive of ductular reactions (Fig. 1F).[14,15] Quantification of the patent BDs for each genotype showed that in agreement with our previous reports at P3, P7 and P30,[14,15] the BD to PV ratio is significantly decreased in P12 Jag1+/− livers (PBS-injected) compared to WT controls (Fig. 1G). Importantly, ASO injections improved all of these phenotypes in Jag1+/− mice (Fig 1, F and G), suggesting that postnatal Poglut1 KD can reverse the BD paucity present in late embryonic and early postnatal Jag1+/− animals and reduce the ductular reactions resulting from BD paucity in these animals.
Poglut1 KD in early postnatal period improves biliary development and significantly decreases liver damage in P30 Jag1+/− mice
By P30, Jag1+/− animals exhibit more severe ductular reactions and also some degree of necrosis.[14] To examine whether the ASO-induced improvements observed in P12 Jag1+/− livers persist until a later stage, we analyzed additional cohorts of Jag1+/− mice and their WT siblings injected with the same ASO or PBS regimen as P12 (Fig. 2A). Similar to P12 livers, the ASO significantly reduced Poglut1 mRNA levels in WT (62%) and Jag1+/− (58%) livers (Fig. 2B). Two injections of ASO-1 led to a statistically significant increase in the BD to PV ratio in P30 Jag1+/− mice, accompanied by a strong reduction in ductular reactions and periportal hypercellularity and a statistically significant rescue of liver necrosis and fibrosis in these animals (Fig. 2, C–H). These data provide strong evidence that a sustained improvement in Jag1+/− liver phenotypes can be achieved by a limited number of anti-Poglut1 ASO injections.
Fig. 2. Poglut1 KD in early postnatal period improves biliary development and significantly decreases liver damage in P30 Jag1+/− mice.
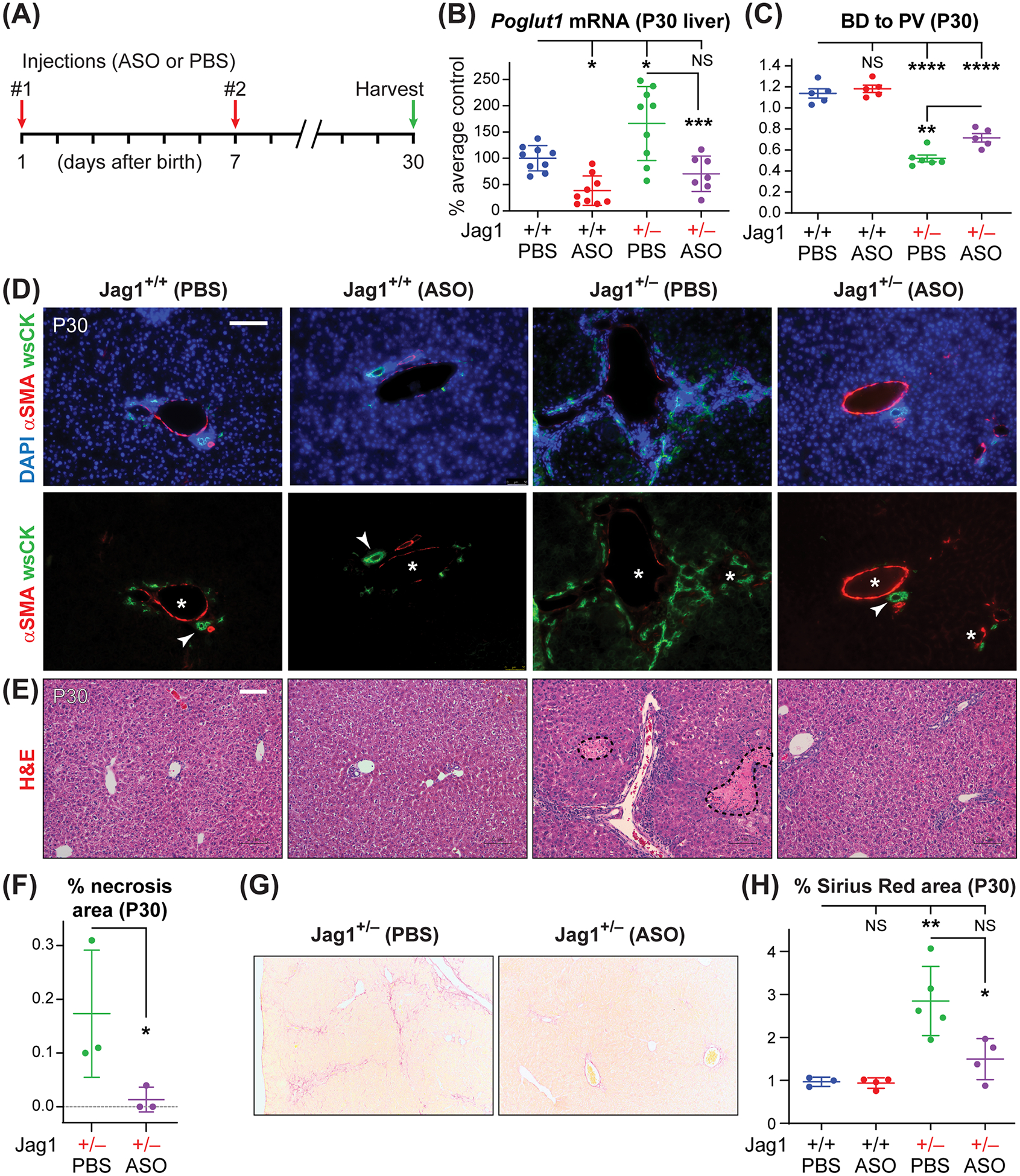
(A) Injection regimen of ASO-1 and PBS in Jag1+/− and WT animals (50 mg/kg/dose). (B) qRT-PCR assays for Poglut1 mRNA levels on WT and Jag1+/− P30 livers injected with ASO or PBS. Note the increase in the Poglut1 mRNA levels in Jag1+/− livers compared to WT livers at this age. (C) Average BD to PV ratio in P30 WT and Jag1+/− mice injected with PBS or ASO. (D) P30 liver sections of each genotype stained with αSMA, wide-spectrum cytokeratin (wsCK) and DAPI. Asterisks mark PVs, arrowheads mark patent BDs. ASO-1 partially rescues the BD to PV ratio in Jag1+/− animals and reduces the ductular reactions and parenchymal hypercellularity observed in control animals. (E) Hematoxylin and eosin (H&E) staining of P30 liver sections from the indicated genotypes. Dashed shapes mark areas of necrosis. (F) Percent area of necrotic regions in H&E staining of liver sections form ASO- and PBS-injected animals. (G) Sirius Red staining of P30 livers from Jag1+/− animals injected with ASO-1 or PBS. (H) The degree of liver fibrosis in the indicated genotypes are measured as the percent area of each liver stained with Sirius Red. In B, C, F and H, each circle represents an animal and horizontal lines show mean ± SD. NS: not significant, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, using one-way ANOVA with Tukey’s multiple comparisons test (B, C, H) or unpaired t-test (F). Scale bars are 100 μm.
Poglut1 KD in early postnatal period improves biliary development and prevents liver damage in ALGS mouse models with severe liver phenotypes
Similar to Jag1+/− animals, two injections of ASO-1 resulted in a sustained significant reduction in liver Poglut1 mRNA levels in models with more severe liver phenotypes (Jag1+/−;Sox9+/Δ and Jag1+/−;Sox9Δ/Δ) (Figs. 3A and 4A; 72% reduction in Jag1+/−;Sox9+/Δ and 66% reduction in Jag1+/−;Sox9Δ/Δ). In both genotypes, ASO injections led to a significant increase in the BD to PV ratio (Figs. 3B and 4B). In agreement with our recent report,[15] the average BD to PV ratio in PBS-injected P30 Jag1+/−;Sox9+/Δ and Jag1+/−;Sox9Δ/Δ animals was lower than that in Jag1+/− mice (Figs. 3B and 4B; compare to Fig. 2C). Nevertheless, upon ASO injections, the average BD to PV ratio rose to the same level as ASO-injected Jag1+/− livers or even higher. These observations suggest that ASO-mediated Poglut1 KD can improve biliary development in ALGS mouse models regardless of the severity of the phenotype.
Fig. 3. ASO-1 injections improve the liver phenotypes in Jag1+/− animals lacking one copy of Sox9 in the liver.
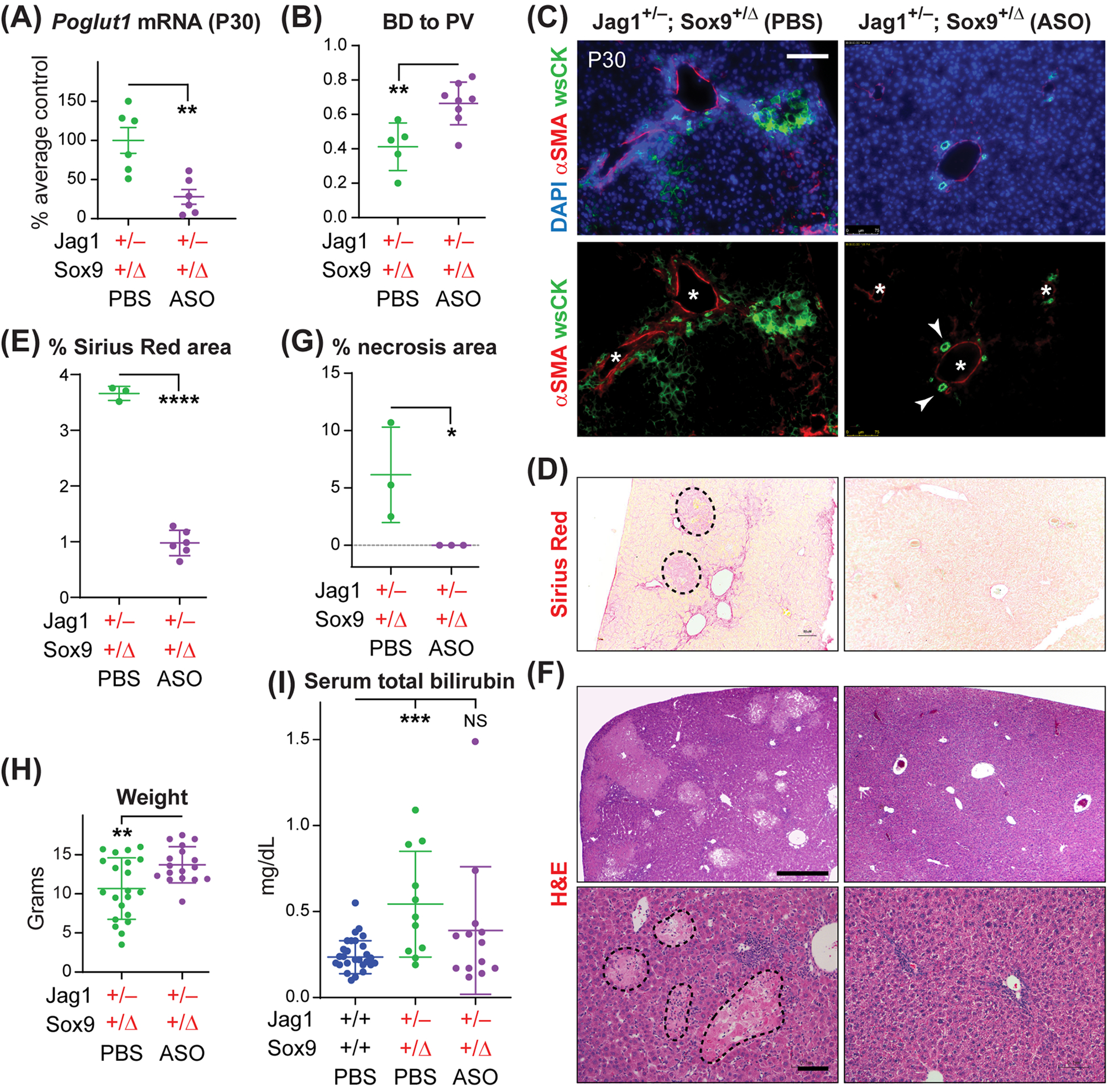
(A) qRT-PCR assays for Poglut1 mRNA levels on Jag1+/−;Sox9+/Δ P30 livers injected with ASO or PBS. (B) Average BD to PV ratio in Jag1+/−;Sox9+/Δ mice injected with PBS or ASO. (C) P30 liver sections of each genotype stained with αSMA, wide-spectrum cytokeratin (wsCK) and DAPI. Asterisks mark PVs, arrowheads mark patent BDs. Scale bar is 100 μm. (D) Sirius Red staining of P30 livers from Jag1+/−;Sox9+/Δ animals injected with ASO-1 or PBS. Necrotic regions (dashed shapes) were excluded from quantification. (E) The degree of liver fibrosis in the indicated genotypes are measured as the percent area of each liver stained with Sirius Red. (F) H&E staining of P30 liver sections from Jag1+/−;Sox9+/Δ animals injected with ASO-1 or PBS. Dashed shapes in the lower-left panel mark areas of necrosis. Scale bars are 500 μm (top panels) and 100 μm (bottom panels). (G) Percent area of necrotic regions in H&E staining of liver sections form the indicated genotypes. (H) Weights for P30 Jag1+/−;Sox9+/Δ animals injected with ASO-1 or PBS. (I), Serum total bilirubin for the indicated genotypes. In all graphs, each circle represents an animal and horizontal lines show mean ± SD. NS: not significant, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, using one-way ANOVA with Tukey’s multiple comparisons test (I) or unpaired t-test (A, B, E, G, H).
Fig. 4. ASO-1 injections improve the liver phenotypes in Jag1+/− animals lacking both copies of Sox9 in the liver.
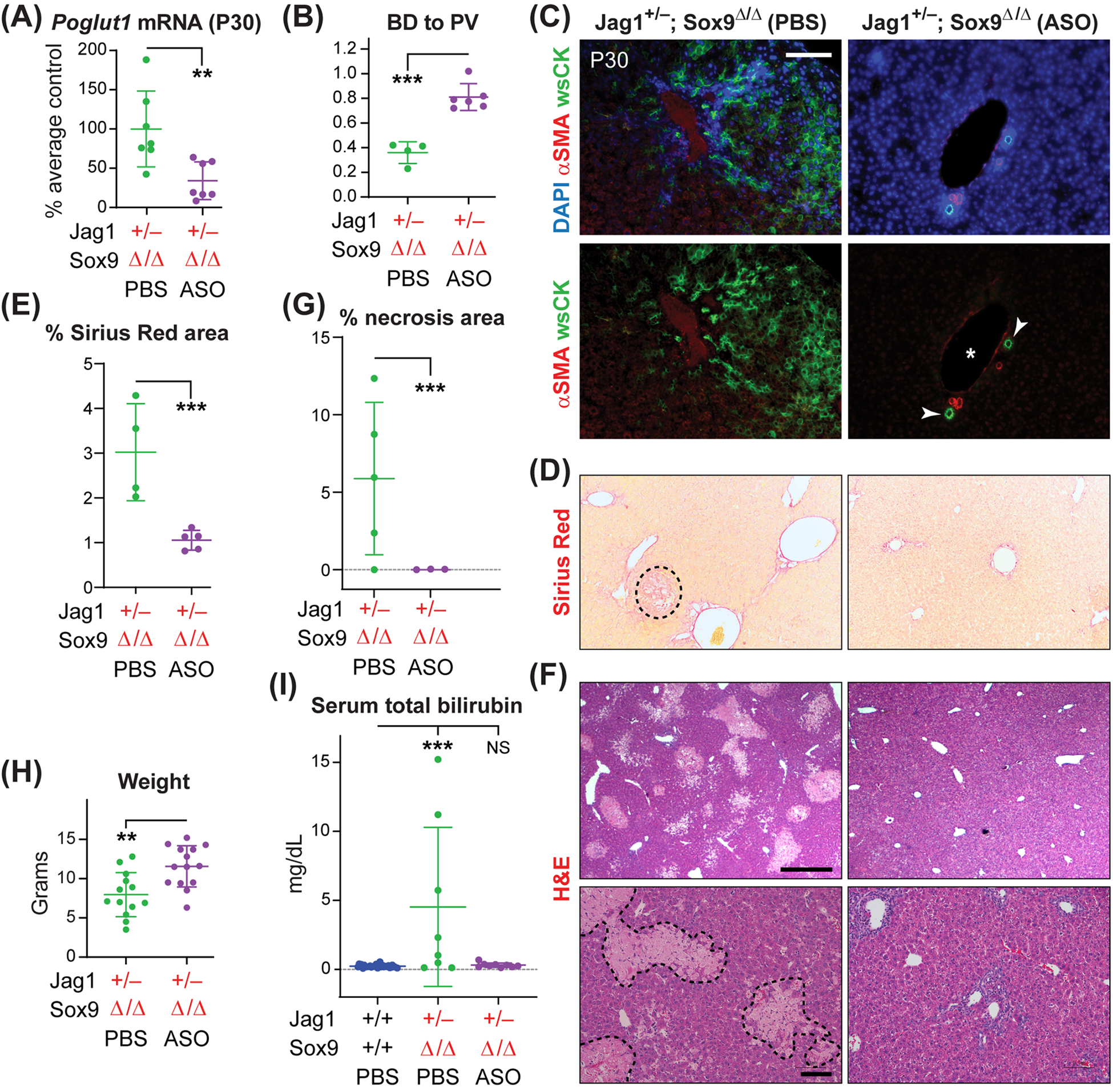
(A) qRT-PCR assays for Poglut1 mRNA levels on Jag1+/−;Sox9Δ/Δ P30 livers injected with ASO or PBS. (B) Average BD to PV ratio in Jag1+/−;Sox9Δ/Δ mice injected with PBS or ASO. (C) P30 liver sections of each genotype stained with αSMA, wide-spectrum cytokeratin (wsCK) and DAPI. Asterisks mark PVs, arrowheads mark patent BDs. Scale bar is 100 μm. (D) Sirius Red staining of P30 livers from Jag1+/−;Sox9Δ/Δ animals injected with ASO or PBS. The dashed shape marks a necrotic region. (E) The degrees of liver fibrosis in the indicated genotypes are measured as the percent area of each liver stained with Sirius Red. (F) H&E staining of P30 liver sections from Jag1+/−;Sox9Δ/Δ animals injected with ASO or PBS. Dashed shapes in the lower-left panel mark areas of necrosis. Scale bars are 500 μm (top panels) and 100 μm (bottom panels). Note that some degree of periportal inflammation persists in ASO-injected livers. (G) Percent area of necrotic regions in H&E staining of liver sections form the indicated genotypes. (H) Weights for P30 Jag1+/−;Sox9Δ/Δ animals injected with ASO or PBS. (I) Serum total bilirubin for the indicated genotypes. In all graphs, each circle represents an animal and horizontal lines show mean ± SD. NS: not significant, **P<0.01, ***P<0.001, using one-way ANOVA with Tukey’s multiple comparisons test (I) or unpaired t-test (A, B, E, G, H).
At P30, Jag1+/−;Sox9+/Δ and Jag1+/−;Sox9Δ/Δ animals show rather severe ductular reactions, accompanied by hypercellularity in the liver parenchyma, fibrosis and extensive necrosis (Figs. 3 and 4).[15] ASO injections dramatically improved these phenotypes, including a full rescue of necrosis (Fig. 3, C–G and Fig. 4, C–G). These phenotypic improvements were accompanied by a significant increase in the average body weight and a significant reduction in the serum total bilirubin of both genotypes (Fig. 3, H and I, and Fig. 4, H and I). We conclude that two postnatal injections of an anti-Poglut1 ASO can significantly improve the severe liver phenotypes observed in these two ALGS models.
ASO-1 dramatically improves the biliary tree structure in ALGS models
Using retrograde ink injections into the common BD, we have previously reported that the biliary tree fails to extend to the liver periphery in P30 Jag1+/− animals and that this phenotype is worsened upon removing one copy of Sox9 in the liver.[15] In agreement with this report, P30 PBS-injected Jag1+/− and Jag1+/−;Sox9+/Δ animals showed a stepwise reduction in the degree of biliary tree extension towards liver periphery compared to PBS-injected WT animals (Fig. 5A). Furthermore, ink injections showed a rudimentary biliary tree in PBS-injected Jag1+/−;Sox9Δ/Δ animals, mostly limited to the hilar region of the liver (Fig. 5A). Similarly, quantification of the area of the liver covered by the biliary tree indicated a stepwise reduction in biliary density in Jag1+/− and the other two models (Fig. 5B). Our two-dose ASO regimen did not cause any biliary tree abnormalities in WT animals (Fig. 5, A and B). However, the ASO led to biliary tree augmentation in all three models (Fig. 5, A and B). In the two severe models, ASO injections led to a remarkable improvement in the extension of the biliary tree towards the liver lobe periphery and the density of biliary tree coverage in the liver (Fig. 5, A and B). These data reveal the beneficial effects of Poglut1 KD in promoting biliary tree development in ALGS mouse models.
Fig. 5. ASO-1 injections result in a dramatic improvement in the biliary tree structure of ALGS models.
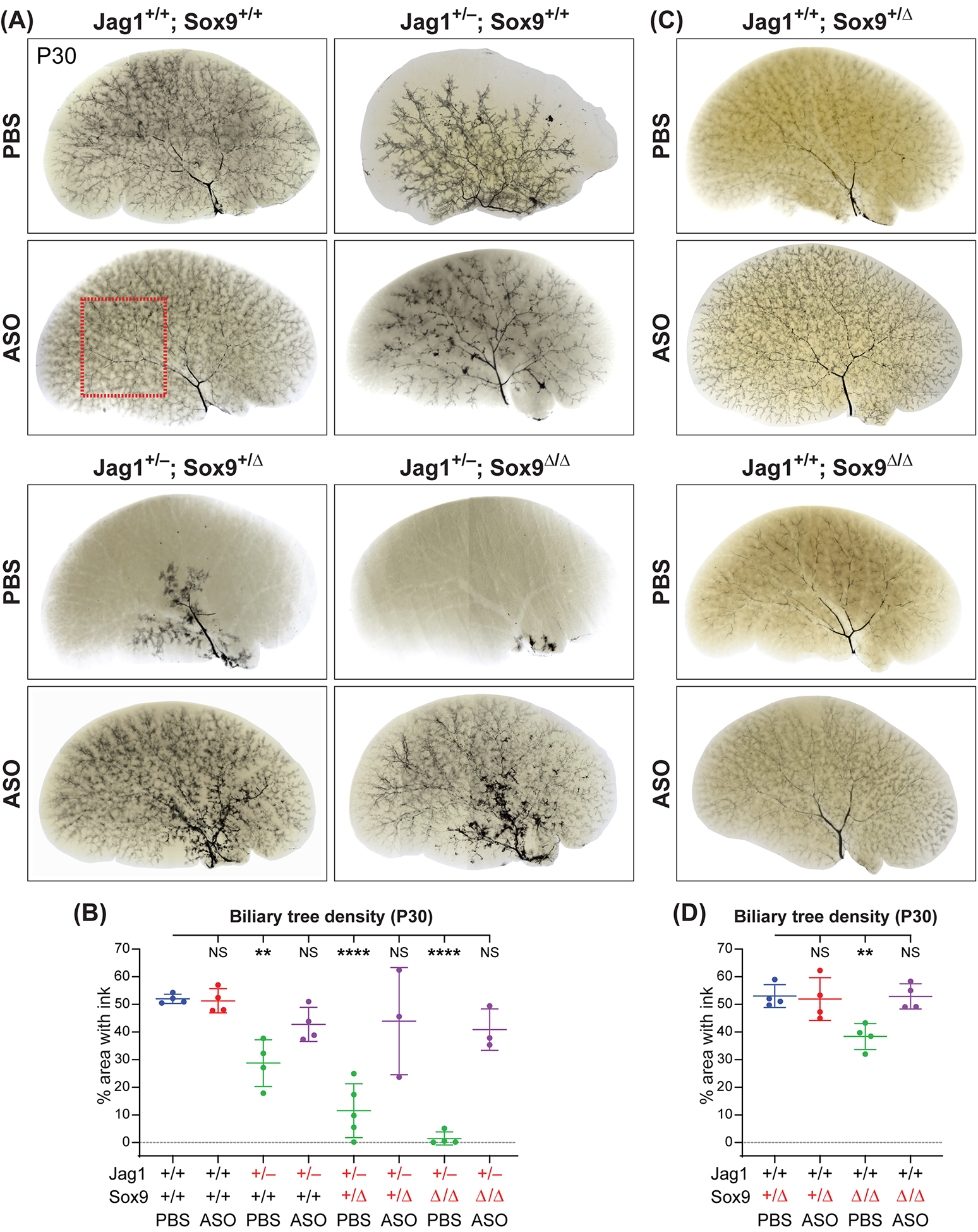
(A, C) Ink injection images of the left liver lobes of P30 animals with indicated genotypes injected with two doses of ASO-1 or PBS. Each image is representative of 3–5 animals for each condition. The pale structures in the liver parenchyma mark the outline of portal veins and are easier to see in the absence of the biliary tree. (B, D) Quantification of the percent area covered by the ink-filled biliary tree in a flattened image of each left liver lobe. The red dashed box in A indicates the area used for quantification of the biliary tree density except in PBS-injected Jag1+/−;Sox9+/Δ and Jag1+/−;Sox9Δ/Δ livers, where the red dashed box was placed over the hilar region to maximize the ink-filled area used in quantification. Each circle represents an animal and horizontal lines show mean ± SD. NS: not significant, **P<0.01, ****P<0.0001, using one-way ANOVA with Dunnett’s multiple comparisons test.
Analysis of liver sections from mice with conditional loss of Sox9 in the liver with albumin/α-fetoprotein-Cre (Alfp-Cre) was reported to cause a delay in BD morphogenesis, which fully resolved by five weeks of age.[24] In the course of our ink injection studies, we noticed a mild but statistically significant reduction in the biliary tree density of Albumin-Cre;Sox9flox/flox animals (Fig. 5, C and D; Jag1+/+;Sox9Δ/Δ). Notably, the anti-Poglut1 ASO rescued this phenotype as well, suggesting that the biliary tree enhancement caused by Poglut1 KD is not limited to animals lacking one copy of Jag1.
The ASO induces significant Poglut1 KD in the muscle but does not reduce the number of muscle stem cells
We have reported that recessive missense mutations in POGLUT1 cause a form of limb-girdle muscular dystrophy in human patients called LGMD R21 (OMIM #617232; also called LGMD 2Z).[25,26] Biochemical experiments indicate that these mutations generally result in an 80–85% decrease in POGLUT1’s enzymatic activity.[25,26] Analysis of patient muscle samples showed reduced NOTCH1 signaling and a severe decrease in the number of PAX7+ muscle stem cells (satellite cells).[25,26] Poglut1+/− mice do not exhibit any gross morphological defects and growth abnormalities,[14,18] suggesting that removing one copy of Poglut1 does not affect muscle development and function. Nevertheless, to establish ASO-mediated Poglut1 KD as a therapeutic strategy in ALGS, it is important to demonstrate that the rescue of ALGS liver phenotypes in Jag1-haploinsufficient animals upon ASO-mediated Poglut1 KD is not accompanied by a reduction in the number of PAX7+ satellite cells. As shown in Supporting Fig. S2A, two injections of ASO-1 resulted in a significant decrease in Poglut1 mRNA levels in quadriceps muscles. However, staining of quadriceps muscle sections of P30 animals indicated that a comparable number of PAX7+ cells are present in ASO-injected versus PBS-injected WT and Jag1+/− animals (Supporting Fig. S2, B and C). These data suggest that Poglut1 KD by systemic injection of a cEt ASO does not reduce the number of satellite cells in WT or Jag1+/− mice.
POGLUT1 negatively regulates JAG1 protein level and JAG1-mediated signaling
To elucidate the effects of reducing Poglut1 levels on JAG1, we generated WT and Poglut1+/− mouse embryonic fibroblasts (MEFs) and stably infected them with a tetracycline-inducible human JAG1 transgene (tet-JAG1). Treating WT-tet-JAG1 MEFs with doxycycline led to accumulation of JAG1 protein and a robust induction of endogenous NOTCH1 signaling in these cells (Fig. 6A). Treating Poglut1+/−-tet-JAG1 MEFs with doxycycline led to the induction human JAG1 mRNA at levels comparable to those seen in WT-tet-JAG1 MEFs (Fig. 6B). However, the levels of human JAG1 protein and endogenous N1-ICD were higher in Poglut1+/− MEFs compared to WT MEFs (Fig. 6A), indicating that loss of one copy of Poglut1 in MEFs increases JAG1 protein level and NOTCH1 activation.
Fig. 6. POGLUT1 negatively regulates JAG1 protein level and JAG1-mediated signaling.
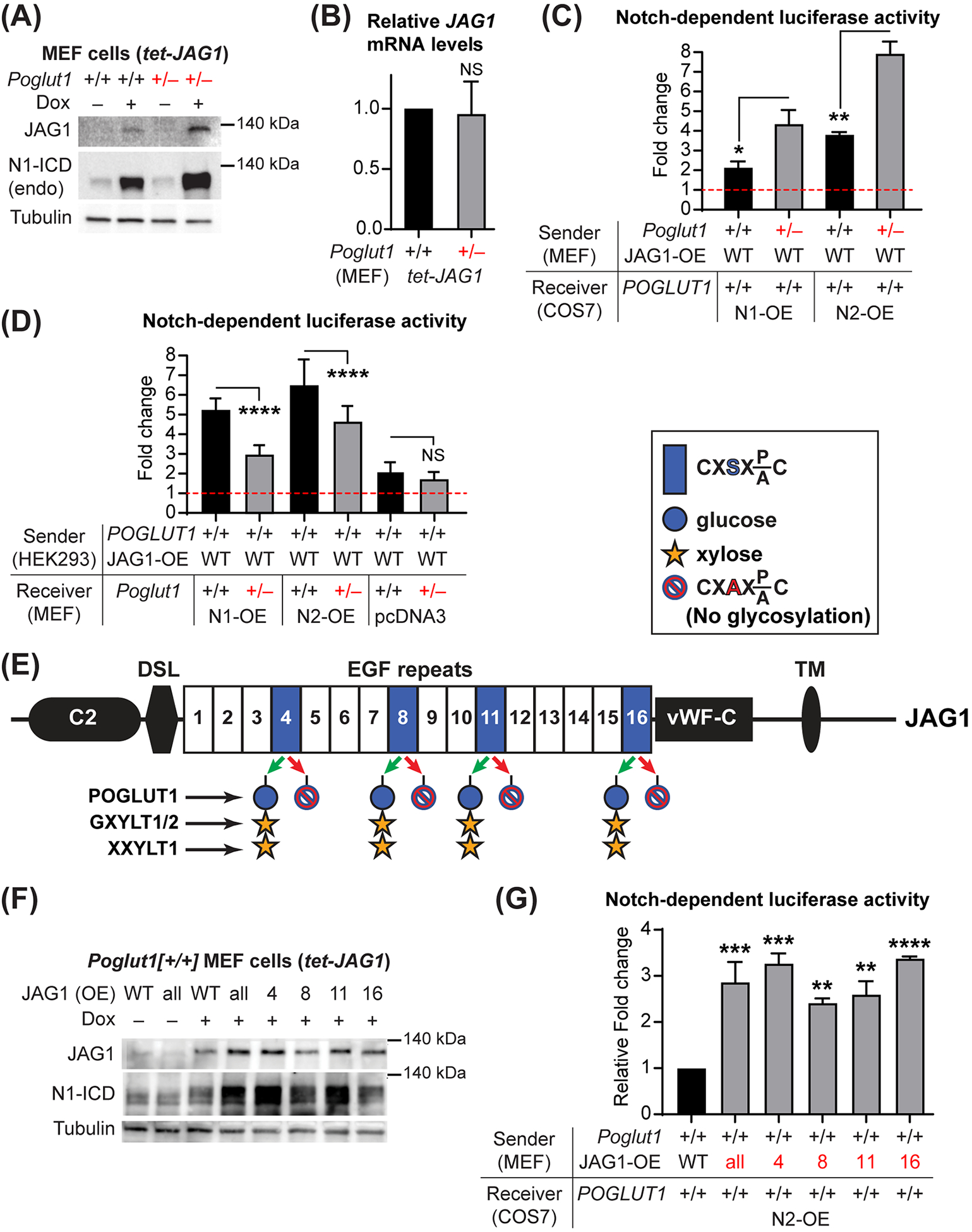
(A) Immunoblots with anti-JAG1, anti-cleaved NOTCH1 intracellular domain (N1-ICD) and anti-Tubulin (loading control) antibodies are shown for WT and Poglut1+/− MEFs with or without doxycycline (Dox) induction of human JAG1 expression. Data are representative of 3 independent experiments. endo: endogenous. (B) qRT-PCR assays for human JAG1 mRNA in WT and Poglut1+/− MEFs treated with Dox to induce human JAG1 expression. (C) Co-culture signaling assays between WT or Poglut1+/− MEFs with or without human JAG1 overexpression and POGLUT1+/+ COS7 cells overexpressing NOTCH1 (N1-OE) or NOTCH2 (N2-OE) along with the TP1–1-luciferase construct. For each co-culture, the fold change of luciferase induction upon Dox-induced expression of human JAG1 compared to the same co-culture without human JAG1 induction (red line set at 1) is shown. (D) Fold change of Notch-dependent luciferase induction upon human JAG1 expression is shown for the indicated co-culture conditions. (E) Schematic of the human JAG1 protein. Blue boxes show EGF repeats with a POGLUT1 target site, along with the trisaccharide and the enzymes responsible for its formation. C2, phospholipid recognition; DSL, Delta-Serrate-Lag2; vWF-C, von Willebrand factor type C; TM, transmembrane. S-to-A mutations prevent O-glucosylation by POGLUT1. (F) Immunoblots for WT cells with or without Dox induction of human JAG1 (WT or harboring S-to-A mutations in individual or all POGLUT1 target sites). Data are representative of 4 independent experiments. (G) Co-culture signaling assays to assess the impact of the POGLUT1 target site mutations on the ability of JAG1 to induce NOTCH2 signaling in neighboring cells. Signaling induced by WT JAG1 was set to 1; signaling by mutant JAG1 proteins are shown as relative fold change compared to that induced by WT JAG1. In C, D and G, n=9 for each column. Each graph shows the results from a representative experiment from 3–4 independent experiments. In B, n=4 for each column. In all graphs, mean + SD is shown. NS: not significant, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, using one-way ANOVA with Tukey’s multiple comparisons test (D, G) or unpaired t-test (B, C).
Since both JAG1 and NOTCH receptors are glycosylated by POGLUT1,[14,18,27] the increased NOTCH1 activation observed in Poglut1+/− cells can theoretically result from the effects of Poglut1 heterozygosity in signal-receiving or signal-sending cells (or both). To address this issue, we performed co-culture assays between MEFs (WT or Poglut1+/–) and another cell type (COS7 or HEK293T, both POGLUT1+/+) and used luciferase activity driven by a Notch-responsive reporter (TP1–1-luc)[28] as the readout for Notch signaling. Induction of JAG1 expression in WT-tet-JAG1 MEFs led to ~2-fold increase in NOTCH1-mediated signaling and ~4-fold increase in NOTCH2-mediated signaling in COS7 cells compared to parallel co-cultures without JAG1 induction (Fig. 6C; black bars). Poglut1+/−-tet-JAG1 MEFs were able to induce significantly more NOTCH1 and NOTCH2-mediated signaling compared to WT-tet-JAG1 MEFs (Fig. 6C; gray bars), indicating that reducing Poglut1 levels in signal-sending cells can increase the ability of JAG1 to activate NOTCH receptors in neighboring cells.
In reciprocal experiments, we used MEFs as signal-receiving cells and cultured them with POGLUT1+/+-tet-JAG1 HEK293T cells. Induction of JAG1 in HEK293T cells induced a 5–6-fold increase in NOTCH1 and NOTCH2 signaling in WT MEFs compared to parallel co-cultures without JAG1 induction (Fig. 6D). Removing one copy of Poglut1 in signal-receiving MEFs led to a statistically significant decrease in JAG1-mediated NOTCH1 and NOTCH2 signaling (Fig. 6D). These experiments indicate that decreasing Poglut1 in signal-receiving cells leads to a reduction in NOTCH1 and NOTCH2 signaling upon receptor overexpression. We also co-cultured POGLUT1+/+-tet-JAG1 HEK293T cells with MEFs transfected with empty pcDNA3 vector to assess the impact of decreasing Poglut1 expression level on signaling by endogenous NOTCH receptors. JAG1 from HEK293T cells led to a 2-fold increase in signaling mediated by endogenous NOTCH receptors in WT MEFs (Fig. 6D). Reducing one copy of Poglut1 in signal-receiving MEFs did not lead to a significant reduction in JAG1-mediated Notch signaling in these cells (Fig. 6D). Together, these experiments indicate that Poglut1 heterozygosity in signal-receiving and signal-sending cells affects JAG1-Notch signaling in opposite directions.
JAG1 has four POGLUT1 modifications sites (Fig. 6E), all of which are efficiently O-glucosylated by POGLUT1.[14] To examine the role of JAG1 O-glucosylation in Notch signaling, we generated Poglut1+/+ MEFs stably infected with tetracycline-inducible human JAG1 transgenes harboring point mutations that abolish O-glucosylation in individual POGLUT1 modification sites or in all four. We found that loss of O-glucosylation sites on JAG1 recapitulates the effects of Poglut1 heterozygosity on JAG1 protein level and on JAG1’s ability to induce NOTCH1 and NOTCH2 signaling (Fig. 6, F and G). These experiments strongly suggest that reducing the POGLUT1-mediated glycosylation of JAG1 increases the JAG1 protein level and the ability of JAG1 to induce Notch signaling in neighboring cells.
Discussion
Intrahepatic BD paucity can result from various etiologies, including genetic, infectious, nutritional and endocrine.[29] BD paucity commonly leads to cholestatic liver disease and often impacts the liver function severely enough to require liver transplantation.[29] Other than liver transplant, the standard of care for BD paucity is mostly geared toward symptomatic relief, although the recent approval of ileal bile acid transporter (IBAT) inhibitors for clinical use in ALGS and progressive familial intrahepatic cholestasis (PFIC) has been a major advance in improving the course of disease and quality of life in patients.[10,30,31] Nevertheless, no approved mechanism-based strategy exists to enhance the intrahepatic biliary tree formation. Here, we report that two injections of an anti-Poglut1 ASO during the early postnatal period lead to a statistically significant improvement in the BD to PV ratio and the assessed clinical and pathologic criteria in all three models of the ALGS liver disease examined in this study. Jag1+/− animals already show a dramatic reduction in the number of SOX9+ biliary cells at embryonic day 18, a lack of patent BDs in the peripheral areas of the liver at P0, and a significant decrease in BD to PV ratio as early as P3.[14,15] Therefore, our ASO injection regimen starts when the animals already show BD paucity. The BD to PV ratio is not fully rescued by the ASO. Nevertheless, ASO treatment results in a strong reduction in the degree of ductular reactions and periportal inflammation, dramatically improves the biliary tree structure—both the degree of extension toward liver periphery and the density of the tree—and almost completely prevents the appearance of the liver damage response in ALGS models, as evidenced by the rescue of fibrosis, necrosis and bilirubin levels. These observations suggest that ASO-mediated reduction of POGLUT1 in the liver of ALGS patients might have the potential to improve their BD paucity enough to avert the need for liver transplantation.
As shown here and previously,[14,15] there is a negative correlation between the level of SOX9 in hepatobiliary cells and the severity of ALGS liver phenotypes in our mouse models and in human patients. In addition, a recent study reported that a small molecule with putative Notch activating properties can induce the expression of Sox9b and rescue the liver phenotypes in a zebrafish model of ALGS.[32] Lastly, Sox9 has been shown to be a direct target of JAG1-Notch signaling in hepatoblasts.[33]Accordingly, we expected that ASO-mediated Poglut1 KD will improve the Jag1+/− liver phenotypes in a SOX9-dependent manner. Rather surprisingly, not only wasn’t this the case, but ASO injections led to a stronger rescue of the liver phenotypes in Jag1+/−;Sox9+/Δ and especially Jag1+/−;Sox9Δ/Δ animals compared to the original Jag1+/− model. These data indicate that expression of SOX9 in biliary cells is not essential for the improvement of Jag1-haploinsufficient liver phenotypes by anti-Poglut1 ASO. This conclusion is in agreement with the observation that ASO injections also rescue the mild biliary tree defects caused by conditional loss of Sox9 in the liver. Given the transient and redundant role played by SOX9 in BD development,[24,34] a likely explanation is that upon enhanced JAG1-mediated signaling caused by Poglut1 KD, Notch pathway targets other than SOX9 are induced in biliary cells and lead to SOX9-independent rescue of the phenotypes. Regardless of the mechanism, our data suggest that the beneficial effects of Poglut1 KD on postnatal biliary development are not limited to a Jag1-haploinsufficient context. How can reducing SOX9 make the Jag1+/− phenotype worse but paradoxically improve the therapeutic response to Poglut1 KD? It has recently been reported that biliary cells express SOX9 at variable levels and that the transcriptomes of SOX9low and SOX9high biliary cells are quite distinct from each other,[35] providing a potential mechanism for our observation which awaits experimental verification.
Experiments on Poglut1-mutant Drosophila (official gene name: rumi), Poglut1 KD Drosophila S2 cells, Poglut1–/– mice, several Poglut1 KD mammalian cells, and muscle samples from LGMD R21 patients have indicated that POGLUT1 promotes the activation of Drosophila Notch and mammalian NOTCH1.[18,25,26,36,37] Moreover, the current work indicates that removing one copy of Poglut1 in signal-receiving MEFs reduces the activation of overexpressed NOTCH1 and NOTCH2 by JAG1 from neighboring cells. Given our previous report that mutating the O-glucosylation sites of Notch in transgenic Drosophila phenocopies the effects of loss of Poglut1on Notch signaling,[37] these observations suggest that reducing Notch O-glucosylation leads to reduced Notch activation. However, the improvements in Jag1+/− liver phenotypes upon genetic [14] and ASO-mediated (this study) reduction in Poglut1 suggest that POGLUT1 reduces JAG1-mediated Notch signaling. Moreover, similar to Poglut1 heterozygosity, mutating the POGLUT1 targets sites on JAG1 is associated with an increase in JAG1 protein level and signaling ability, strongly suggesting that reducing JAG1 O-glucosylation leads to increased JAG1-mediated Notch activation. Accordingly, we suggest that the opposite effects of reducing Poglut1 in signal-receiving versus signal-sending cells on Notch pathway activation can be explained by differential roles played by O-glucose residues on NOTCH1 and NOTCH2 versus JAG1, although we cannot exclude that an increase in the level of endogenous JAG1 in Poglut1+/− signal-receiving cells reduces NOTCH1/2 signaling via cis-inhibition.[38] Importantly, when JAG1 was overexpressed in a monoculture of Poglut1+/− MEFs, endogenous NOTCH1 activation was increased, not decreased (Fig. 6A). Altogether, our data suggest that the effect Poglut1 reduction on JAG1-mediated NOTCH1/2 signaling depends on the relative expression levels of JAG1 versus Notch receptors, and that in a Jag1+/− context, decreasing Poglut1 leads to a net increase in JAG1-mediated signaling, thereby improving the Jag1+/− haploinsufficient liver phenotypes.
ASO-mediated KD in the liver is already employed in clinic to treat several metabolic disorders.[39,40] However, to our knowledge, no ASO-based therapy has been approved for clinical use in a genetic childhood disease affecting liver development. Our cell-based signaling assays provide strong evidence that the JAG1-expressing, signal-sending cell is the cell type in which Poglut1 KD promotes biliary development. Previous work has established that the periportal mesenchymal cells are the major source of JAG1 during biliary development.[41] Moreover, our RNAscope analysis indicates that ASO-1 injections lead to efficient KD of Poglut1 in Jag1-expressing periportal mesenchymal cells. Together, these observations strongly suggest that ASO-mediated Poglut1 KD in periportal mesenchymal cells mediates the observed rescue of the liver phenotypes in our ALGS models. Of note, we previously reported that genetic removal of one copy of Poglut1 in hepatobiliary cells (Albumin-Cre) and periportal mesenchymal cells (SM22α-Cre) can both improve the Jag1+/− liver phenotypes,[14] suggesting that the beneficial effects of reducing Poglut1 in ALGS models are not limited to one cell type. It will remain to be seen whether reducing Poglut1 specifically in postnatal hepatocytes also contributes to the observed improvement of the ALGS biliary phenotypes upon ASO injections.
Our data provide proof of concept that postnatal targeting of a genetic modifier can improve the liver phenotypes in a pediatric liver disease and establish a framework for developing a therapeutic strategy for ALGS liver disease. However, a number of key issues related to the therapeutic potential of Poglut1 KD in ALGS remain to be addressed in preclinical models. It is important to examine whether the BDs formed upon ASO injections remain stable beyond P30, and to determine whether ASO injections after the appearance of fibrosis and necrosis are still able to improve the ALGS liver phenotypes. Since ALGS is a multi-system disorder, it is also important to assess whether ASO-mediated Poglut1 KD can improve other ALGS phenotypes. Finally, given the observed reduction in NOTCH1 and NOTCH2 signaling in Poglut1+/− signal-receiving cells overexpressing these receptors (Fig. 6D), it is critical to ensure that systemic anti-Poglut1 ASO injections do not negatively affect the cardiovascular system and other organs that depend on NOTCH1 and NOTCH2 signaling.[42]
Supplementary Material
Acknowledgments:
We thank Shakeel Thakurdas for initiating the ASO studies in the Jafar-Nejad laboratory; Joshua Adams for discussions; Bob Haltiwanger for constructs; Shinako Kakuda for advice on Notch signaling assays; Natacha Mitchell, Patricia Castro and Maciej Zelazowski for assistance with the staining and scanning of the H&E slides; and Tom Gridley for the Jag1+/− strain. Image collection for ink injections was performed in the Optical Imaging & Vital Microscopy Core Facility at Baylor College of Medicine. Imaging for liver fibrosis was performed at the Neurovisualization Core Facility at Baylor College of Medicine Intellectual and Developmental Disabilities Research Center. A special thank you to J. Matthew Kofron, CCHMC Confocal Imaging Core Director, for guidance of the RNAscope image analyses. The Graphical Abstract was created with BioRender.com.
Financial support:
Supported by the National Institutes of Health (R01 DK109982, R35 GM130317, and R01 AR076770 to H.J.N.; R01 DK120765 and R01 DK107553 to S.S.H.; R01 DK132751 jointly to H.J.N. and S.S.H.); a Pilot/Feasibility Award from the Texas Medical Center Digestive Disease Center to H.J.N. (NIH P30 DK56338); two Alagille Syndrome Accelerator Awards from the Medical Foundation established by generous funding from an anonymous private donor to H.J.N; Funds from the Department of Molecular & Human Genetics, Baylor College of Medicine to H.J.N.; and T32 GM08307 training grant to D.F; Neurovisualization Core Facility at Baylor College of Medicine Intellectual and Developmental Disabilities Research Center (U54 HD083092); Integrative Morphology Core – Pathology Research Service and Confocal Imaging of the Digestive Diseases Research Core Center in Cincinnati (P30 DK078392). All costs involved in the design, screening and production of ASO-1 was covered by Ionis Pharmaceuticals.
List of Abbreviations:
- ANOVA
analysis of variance
- αSMA
alpha smooth muscle actin
- ASO
antisense oligonucleotide
- ALGS
Alagille syndrome
- BD
bile duct
- cEt
constrained ethyl
- CK
cytokeratin
- H&E
hematoxylin and eosin
- JAG1
jagged 1
- KD
knockdown
- LGMD
limb-girdle muscular dystrophy
- MEF
mouse embryonic fibroblast
- N1-ICD
NOTCH1 intracellular domain
- OMIM
Online Mendelian Inheritance in Man
- OPN
osteopontin
- PAX7
paired box 7
- POGLUT1
protein O-glucosyltransferase 1
- PV
portal vein
- qRT-PCR
quantitative reverse-transcriptase polymerase chain reaction
- SOX9
sex determining region Y-box 9
- SD
standard deviation
- wsCK
wide-spectrum cytokeratin
- WT
wild-type
Footnotes
Conflicts of interest: Armand Soriano owns stock in and is employed by Ionis Pharmaceuticals. Frank Rigo owns stock in and is employed by and owns intellectual property rights in Ionis Pharmaceuticals. Paymaan Jafar-Nejad owns stock in and is employed by Ionis Pharmaceuticals. The other authors declare no conflicts.
Supporting Information
References
- 1.Alagille D, Odievre M, Gautier M, Dommergues JP. Hepatic ductular hypoplasia associated with characteristic facies, vertebral malformations, retarded physical, mental, and sexual development, and cardiac murmur. The Journal of pediatrics 1975;86:63–71. [DOI] [PubMed] [Google Scholar]
- 2.Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology 1999;29:822–829. [DOI] [PubMed] [Google Scholar]
- 3.Li L, Krantz ID, Deng Y, Genin A, Banta AB, Collins CC, et al. Alagille syndrome is caused by mutations in human Jagged1, which encodes a ligand for Notch1. Nature genetics 1997;16:243–251. [DOI] [PubMed] [Google Scholar]
- 4.Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, et al. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nature genetics 1997;16:235–242. [DOI] [PubMed] [Google Scholar]
- 5.Warthen DM, Moore EC, Kamath BM, Morrissette JJ, Sanchez-Lara PA, Piccoli DA, et al. Jagged1 (JAG1) mutations in Alagille syndrome: increasing the mutation detection rate. Human mutation 2006;27:436–443. [DOI] [PubMed] [Google Scholar]
- 6.Gilbert MA, Bauer RC, Rajagopalan R, Grochowski CM, Chao G, McEldrew D, et al. Alagille syndrome mutation update: Comprehensive overview of JAG1 and NOTCH2 mutation frequencies and insight into missense variant classification. Human mutation 2019;40:2197–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kamath BM, Bauer RC, Loomes KM, Chao G, Gerfen J, Hutchinson A, et al. NOTCH2 mutations in Alagille syndrome. Journal of medical genetics 2012;49:138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krantz ID, Piccoli DA, et al. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. American journal of human genetics 2006;79:169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mouzaki M, Bass LM, Sokol RJ, Piccoli DA, Quammie C, Loomes KM, et al. Early life predictive markers of liver disease outcome in an International, Multicentre Cohort of children with Alagille syndrome. Liver international : official journal of the International Association for the Study of the Liver 2016;36:755–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gonzales E, Hardikar W, Stormon M, Baker A, Hierro L, Gliwicz D, et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): a randomised phase 2 study. Lancet 2021;398:1581–1592. [DOI] [PubMed] [Google Scholar]
- 11.Ayoub MD, Kamath BM. Alagille Syndrome: Diagnostic Challenges and Advances in Management. Diagnostics (Basel) 2020;10:907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kamath BM, Ye W, Goodrich NP, Loomes KM, Romero R, Heubi JE, et al. Outcomes of Childhood Cholestasis in Alagille Syndrome: Results of a Multicenter Observational Study. Hepatology communications 2020;4:387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vandriel SM, Li LT, She H, Wang JS, Gilbert MA, Jankowska I, et al. Natural history of liver disease in a large international cohort of children with Alagille syndrome: Results from the GALA study. Hepatology 2023;77:512–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thakurdas SM, Lopez MF, Kakuda S, Fernandez-Valdivia R, Zarrin-Khameh N, Haltiwanger RS, et al. Jagged1 heterozygosity in mice results in a congenital cholangiopathy which is reversed by concomitant deletion of one copy of Poglut1 (Rumi). Hepatology 2016;63:550–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adams JM, Huppert KA, Castro EC, Lopez MF, Niknejad N, Subramanian S, et al. Sox9 is a modifier of the liver disease severity in a mouse model of Alagille syndrome. Hepatology 2020;71:1331–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue Y, Gao X, Lindsell CE, Norton CR, Chang B, Hicks C, et al. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Human molecular genetics 1999;8:723–730. [DOI] [PubMed] [Google Scholar]
- 17.Postic C, Shiota M, Niswender KD, Jetton TL, Chen Y, Moates JM, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. The Journal of biological chemistry 1999;274:305–315. [DOI] [PubMed] [Google Scholar]
- 18.Fernandez-Valdivia R, Takeuchi H, Samarghandi A, Lopez M, Leonardi J, Haltiwanger RS, et al. Regulation of mammalian Notch signaling and embryonic development by the protein O-glucosyltransferase Rumi. Development 2011;138:1925–1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akiyama H, Chaboissier MC, Martin JF, Schedl A, de Crombrugghe B. The transcription factor Sox9 has essential roles in successive steps of the chondrocyte differentiation pathway and is required for expression of Sox5 and Sox6. Genes & development 2002;16:2813–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adams JM, Jafar-Nejad H. Determining Bile Duct Density in the Mouse Liver. Journal of visualized experiments : JoVE 2019;146:e59587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schaub JR, Huppert KA, Kurial SNT, Hsu BY, Cast AE, Donnelly B, et al. De novo formation of the biliary system by TGFbeta-mediated hepatocyte transdifferentiation. Nature 2018;557:247–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, et al. Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proceedings of the National Academy of Sciences of the United States of America 2002;99:7142–7147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang F, Flanagan J, Su N, Wang LC, Bui S, Nielson A, et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. The Journal of molecular diagnostics : JMD 2012;14:22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Antoniou A, Raynaud P, Cordi S, Zong Y, Tronche F, Stanger BZ, et al. Intrahepatic bile ducts develop according to a new mode of tubulogenesis regulated by the transcription factor SOX9. Gastroenterology 2009;136:2325–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Servian-Morilla E, Takeuchi H, Lee TV, Clarimon J, Mavillard F, Area-Gomez E, et al. A POGLUT1 mutation causes a muscular dystrophy with reduced Notch signaling and satellite cell loss. EMBO molecular medicine 2016;8:1289–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Servian-Morilla E, Cabrera-Serrano M, Johnson K, Pandey A, Ito A, Rivas E, et al. POGLUT1 biallelic mutations cause myopathy with reduced satellite cells, alpha-dystroglycan hypoglycosylation and a distinctive radiological pattern. Acta Neuropathol 2020;139:565–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rana NA, Nita-Lazar A, Takeuchi H, Kakuda S, Luther KB, Haltiwanger RS. O-glucose trisaccharide is present at high but variable stoichiometry at multiple sites on mouse Notch1. The Journal of biological chemistry 2011;286:31623–31637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kakuda S, Haltiwanger RS. Deciphering the Fringe-Mediated Notch Code: Identification of Activating and Inhibiting Sites Allowing Discrimination between Ligands. Developmental cell 2017;40:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meena BL, Khanna R, Bihari C, Rastogi A, Rawat D, Alam S. Bile duct paucity in childhood-spectrum, profile, and outcome. Eur J Pediatr 2018;177:1261–1269. [DOI] [PubMed] [Google Scholar]
- 30.Shneider BL, Spino CA, Kamath BM, Magee JC, Ignacio RV, Huang S, et al. Impact of long-term administration of maralixibat on children with cholestasis secondary to Alagille syndrome. Hepatology communications 2022;6:1922–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loomes KM, Squires RH, Kelly D, Rajwal S, Soufi N, Lachaux A, et al. Maralixibat for the treatment of PFIC: Long-term, IBAT inhibition in an open-label, Phase 2 study. Hepatology communications 2022;6:2379–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao C, Matalonga J, Lancman JJ, Liu L, Xiao C, Kumar S, et al. Regenerative failure of intrahepatic biliary cells in Alagille syndrome rescued by elevated Jagged/Notch/Sox9 signaling. Proceedings of the National Academy of Sciences of the United States of America 2022;119:e2201097119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zong Y, Panikkar A, Xu J, Antoniou A, Raynaud P, Lemaigre F, et al. Notch signaling controls liver development by regulating biliary differentiation. Development 2009;136:1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poncy A, Antoniou A, Cordi S, Pierreux CE, Jacquemin P, Lemaigre FP. Transcription factors SOX4 and SOX9 cooperatively control development of bile ducts. Developmental biology 2015;404:136–148. [DOI] [PubMed] [Google Scholar]
- 35.Tulasi DY, Castaneda DM, Wager K, Hogan CB, Alcedo KP, Raab JR, et al. Sox9(EGFP) Defines Biliary Epithelial Heterogeneity Downstream of Yap Activity. Cellular and molecular gastroenterology and hepatology 2021;11:1437–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Acar M, Jafar-Nejad H, Takeuchi H, Rajan A, Ibrani D, Rana NA, et al. Rumi is a CAP10 domain glycosyltransferase that modifies Notch and is required for Notch signaling. Cell 2008;132:247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leonardi J, Fernandez-Valdivia R, Li YD, Simcox AA, Jafar-Nejad H. Multiple O-glucosylation sites on Notch function as a buffer against temperature-dependent loss of signaling. Development 2011;138:3569–3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pandey A, Niknejad N, Jafar-Nejad H. Multifaceted regulation of Notch signaling by glycosylation. Glycobiology 2021;31:8–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raal FJ, Santos RD, Blom DJ, Marais AD, Charng MJ, Cromwell WC, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet 2010;375:998–1006. [DOI] [PubMed] [Google Scholar]
- 40.Witztum JL, Gaudet D, Freedman SD, Alexander VJ, Digenio A, Williams KR, et al. Volanesorsen and Triglyceride Levels in Familial Chylomicronemia Syndrome. The New England journal of medicine 2019;381:531–542. [DOI] [PubMed] [Google Scholar]
- 41.Hofmann JJ, Zovein AC, Koh H, Radtke F, Weinmaster G, Iruela-Arispe ML. Jagged1 in the portal vein mesenchyme regulates intrahepatic bile duct development: insights into Alagille syndrome. Development 2010;137:4061–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Masek J, Andersson ER. The developmental biology of genetic Notch disorders. Development 2017;144:1743–1763. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.