

Summary
Mutations in MPV17 are a major contributor to mitochondrial DNA (mtDNA) depletion syndromes, a group of inherited genetic conditions due to mtDNA instability. To investigate the role of MPV17 in mtDNA maintenance, we generated and characterized a Drosophila melanogaster Mpv17 (dMpv17) KO model showing that the absence of dMpv17 caused profound mtDNA depletion in the fat body but not in other tissues, increased glycolytic flux and reduced lifespan in starvation. Accordingly, the expression of key genes of glycogenolysis and glycolysis was upregulated in dMpv17 KO flies. In addition, we demonstrated that dMpv17 formed a channel in planar lipid bilayers at physiological ionic conditions, and its electrophysiological hallmarks were affected by pathological mutations. Importantly, the reconstituted channel translocated uridine but not orotate across the membrane. Our results indicate that dMpv17 forms a channel involved in translocation of key metabolites and highlight the importance of dMpv17 in energy homeostasis and mitochondrial function.
Subject areas: model organism, molecular biology, physiology
Graphical abstract
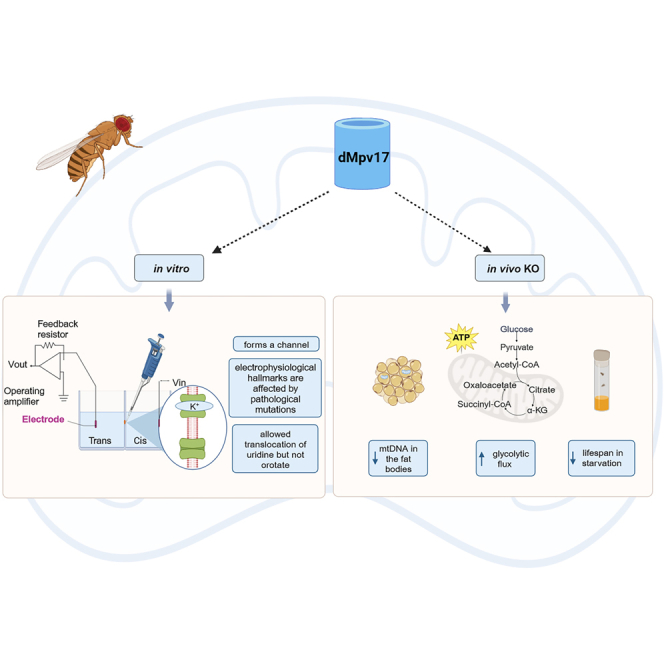
Highlights
-
•
An Mpv17 fly KO recapitulates the main features of human MPV17-related disease
-
•
dMpv17 forms a channel when inserted in lipid bilayers
-
•
dMpv17 electrophysiological properties are affected by pathological mutations
-
•
Uridine but not orotate is transported by dMpv17 across the lipid bilayer
Model organism; Molecular biology; Physiology
Introduction
Mitochondrial DNA (mtDNA) depletion syndromes (MDSs) are autosomal recessive diseases mainly characterized by severe reduction in mtDNA copy number in one or several tissues. MDSs are caused by mutations in nuclear genes encoding proteins involved in the regulation of mitochondrial or cytosolic nucleotide pools, mtDNA replication and repair pathways, and mtDNA nucleoid packaging.1 Genes implicated in MDS-disease include several that are involved in mtDNA replication and nucleotide metabolism. However, some of the genes responsible for MDS have poorly defined functions. Among these, MPV17 encodes a small and highly hydrophobic protein embedded in the inner mitochondrial membrane (IMM), the function of which remains unknown despite many years of intense investigations. In humans, mutations in MPV17 are a prominent cause of infantile-onset hepatocerebral MDS2 (OMIM: 266810256810), including Navajo neurohepatopathy (NHH),3 accounting for about 50% of the cases. A rapid deterioration of hepatic function and severe early-onset hypoglycemia are predominant features of MPV17-dependent MDS, later complicated by severe neurological impairment. In most cases, progressive peripheral neuropathy and cerebellar degeneration appear in the patients who survive early-onset severe metabolic impairment associated with liver failure. Patients with NHH caused by the specific p.R50Q homozygous founder mutation have longer life expectancy than patients with MPV17-related MDS. A later-onset neuromyopathic disease associated with multiple mtDNA deletions in muscle has also been described.4,5 However, the common feature of MPV17-related syndromes is mtDNA instability, indicating a potential role for MPV17 in mtDNA maintenance. In line with this hypothesis, it has been shown that the lack of MPV17 leads to a decrease in deoxyguanosine triphosphate (dGTP) and deoxythymidine triphosphate (dTTP) pools in mouse liver mitochondria, enhancing the incorporation of riboGTP and leading to mtDNA replication arrest.6 More recently, a role for MPV17 in maintaining deoxythymidine monophosphate (dTMP) levels in mitochondria has been suggested.7 In zebrafish, the loss of Mpv17 has been associated to pyrimidine nucleotide metabolism through the impairment of dihydroorotate dehydrogenase (DHODH).8 In addition, Sym1, the yeast homologue of MPV17, has a role in the replenishment of tricarboxylic acid (TCA) cycle intermediates, and its absence causes depletion of glycogen storage.9
However, as a whole, the data summarized previously remain inconclusive as to the precise role of MPV17. There is evidence to indicate that it forms a non-selective ion channel, the gating properties of which can be modified by membrane potential, phosphorylation and redox state.10 The MPV17 channel has been shown to be closed under physiological conditions and to be activated in situations of impaired oxidative phosphorylation (OXPHOS).10 In addition, it has been reported that different mutations can either interfere with the protein amount/localization or with the electrophysological properties of the channel. For instance, the G94R mutation in human MPV17 abolishes protein localization to the IMM, while the MPV17S170F mutation has been suggested to act as a leaky channel which affects the inner membrane potential Δψm.11 Despite intensive research, the ions/metabolites transported through the channel remain unknown. Interestingly, the yeast homolog Sym1 has also been shown to function as an ion channel—at least in vitro—its size being sufficient to allow the transport of large molecules such as metabolites across the IMM.12 Recently, a pan-neuronal knock down of the Drosophila melanogaster Mpv17 (dMpv17) ortholog has been described, which showed larval locomotor and cognitive phenotypes.13 Furthermore, reduced mtDNA copy number and ATP levels, and increased lactate levels in the brain were documented, again in larvae.
The aim of the present study was to define the function of Mpv17 in a knockout D. melanogaster model. We first characterized the phenotype associated with dMpv17 KO. Then we investigated the possibility that dMpv17 may function as a channel also in flies. Finally, we attempted to identify potential transported metabolites, and also to characterize the effect of pathological mutations on channel activity.
Results
CG11077 encodes for the D. melanogaster ortholog of human MPV17
To study MPV17 function in the fly, we first carried out a bioinformatic analysis to identify the putative Drosophila melanogaster ortholog. The integrative orthology prediction tool DIOPT14 assigned the highest homology score to the protein coding gene CG11077 (hereafter named dMpv17 and dMpv17 protein). dMpv17 localizes on the 4th chromosome and encodes a 168 amino acid protein. The overall sequence similarity between the human and fly proteins calculated by Muscle15 was 36% (Figure 1A). Our results are in agreement with those recently reported by Kodani et al. (2022).13
Figure 1.

Characterization of dMpv17
(A) Muscle alignment between human MPV17 and its fly ortholog, displayed by use of the Clc main workbench (Qiagen). The identical amino acidic residues are highlighted in red.
(B) Subcellular localization of dMpv17. Drosophila cells were transiently transfected with dMpv17-HA (green), incubated with MitoTracker dye (red), and analyzed by confocal microscopy. Overlay of images (yellow) confirmed the mitochondrial localization of dMpv17.
(C) dMpv17 3D structure predicted using AlphaFold, which produces a per-residue confidence score (pLDDT) ranging from 0 to 100. Dark blue, very high (pLDDT >90); light blue, good confidence (90>pLDDT>70).
(D) dMpv17 topology predicted using Deep-TMHMM. Upper panel: predicted protein topology; lower panel: associated probability.
As for human MPV17 and yeast Sym1, dMpv17 has no cleavable N-terminal mitochondrial targeting sequence according to the online prediction tools Target P16 and MitoProt II.17 To test whether dMpv17 localizes to mitochondria, we cloned the cDNA into the pAct expression vector with an HA-tag at the C-terminus (dMpv17-HA) and transfected S2R+ Drosophila cells. Confocal imaging of immunostained S2R+ cells using an antibody against HA-tag showed colocalization with MitoTracker dye, confirming that Drosophila ortholog is indeed a mitochondrial protein (Figure 1B).
We then used the 3D structure prediction tool AlphaFold18 and the topology prediction tool DeepTMHMM19 to assess the protein topology. These tools predicted the presence of five transmembrane domains (Figures 1C and 1D), suggesting that, as for hMPV17, dMpv17 is a highly hydrophobic protein, possibly harboring TM domains, compatible with the observed membrane localization.
dMpv17 KO causes mtDNA depletion in the fat body
To investigate the physiological role of dMpv17, we generated a knockout (KO) Drosophila strain by CRISPR/Cas9 genome editing. A stop codon was inserted 45 nucleotides after the ATG start codon, leading to premature termination of translation (Figure S1A). This was performed by inserting four additional nucleotides, three of which (UAA) generating the stop codon and an additional one generating a frameshift immediately after the stop codon (Figure S1B). We investigated the effect of the genetic manipulation by quantifying RNA levels by qPCR. The dMpv17 transcript was reduced by 80% in the fat bodies of KO vs. w1118 (white 1118 strain - Bloomington Drosophila Stock Centre) WT flies, and by 60% in their thoracic muscles (Figures 2A and 2B), suggesting partial decay of dMpv17 transcript.
Figure 2.

Characterization of dmpv17 KO flies
(A) dMpv17 mRNA quantification in fat body. w1118 is the wild-type strain. Error bars represent ±SEM. The asterisks represent the significance levels calculated by unpaired, two-tailed Student’s t test: ∗∗∗p < 0.001.
(B) dMpv17 mRNA quantification in thoracic muscles. w1118 is the wild-type strain. Error bars represent ±SEM. The asterisks represent the significance levels calculated by unpaired, two-tailed Student’s t test: ∗∗p < 0.01.
(C) mtDNA content measured by qPCR in the fat body from dMpv17 KO and wild-type flies. Error bars represent ±SEM. The asterisks represent the significance levels calculated by unpaired, two-tailed Student’s t test: ∗∗p < 0.01.
(D) mtDNA content measured by qPCR in the thoracic muscles from dMpv17 KO and wild-type flies. Error bars represent ±SEM.
Since severe mtDNA depletion in the liver is a hallmark of MPV17 disease, we measured mtDNA copy number in the Drosophila tissues, including the fat body—the analog of vertebrate liver and white adipose tissue—and skeletal muscle. In the fat body, mtDNA copy number was reduced by 50% compared to control flies, while no difference was observed in mtDNA amount in thoracic muscles (Figures 2C and 2D).
Perturbations of deoxynucleoside triphosphate (dNTP) pools are a well-known cause of mitochondrial genomic instability. Therefore, to test if dNTP pool imbalance could be the cause of mtDNA depletion, we downregulated the expression of endogenous dMpv17 in S2R+ cells and tested the effects on cytosolic and mitochondrial dNTP pool levels. In cells incubated for 72 h with dsRNA, real time RT-PCR analysis showed that dMpv17 mRNA was decreased by 80% (Figure S2A), while mtDNA was only slightly reduced (Figure S2B). Considering that the size of the dNTP pools is strongly influenced by the cell cycle phase,20 we quantified the number of cells in the S-phase by FACS analysis in naive and dMpv17 silenced cells (Figure S2C). Since no difference was detected, we were able to directly analyze the concentrations of dNTPs in mitochondria and, again, pools were similar in both cell lines (Figure S2D). These data suggest that dMpv17 is not directly involved in dNTPs metabolism in Drosophila cells.
We then investigated whether the mitochondrial step of pyrimidine de novo synthesis was affected in dMpv17 KO flies, as previously reported in mpv17−/− zebrafish larvae.8 The conversion of dihydroorotic acid (DHOA) into orotic acid (OA) is a metabolic reaction catalyzed by DHODH, the only mitochondrial enzyme involved in pyrimidine de novo synthesis. We measured the activity of this enzyme in mitochondria isolated from whole flies and no differences were observed between dMpv17 KO and control flies (Figure S3).
Taken together, these results indicate that the absence of dMpv17 in flies is associated with tissue-specific mtDNA depletion, which is not related to obvious/detectable alterations in nucleotide pools.
Drosophila Mpv17 is required to maintain mitochondrial ultrastructure
In mammals (human and mouse) and yeast, the absence of MPV17 or its orthologs is associated with profound alterations in mitochondrial cristae morphology, which, in turn, may have an impact on mtDNA stability.9 In addition, it has been recently demonstrated that moderate overexpression of Opa1 in Mpv17−/− mouse preserved mitochondrial network and cristae ultrastructure, leading to increased mtDNA content,21 suggesting that Mpv17 is either directly or indirectly involved in cristae organization.
To characterize mitochondrial morphology in dMpv17 KO and WT flies, we analyzed mitochondrial ultrastructure in the fat body by electron microscopy. Mitochondrial ultrastructure showed swelling of the organelles and disappearance of the cristae in 5-day-old flies, while control flies’ mitochondria displayed densely packed lamellar cristae (Figure 3A). Quantitative analyses documented a reduction in mitochondrial area, length and width (Figure 3B) By contrast, mitochondrial ultrastructure in thoracic muscles was similar in WT and dMpv17 KO flies (Figures 3C and 3D), although KO occasionally presented abnormal cristae structures (swirls) compared to WT flies.
Figure 3.

Electron microscopy analysis
(A) Representative TEM images from Drosophila control (w1118) and dMpv17 KO fat body. Abnormal mitochondria (red arrows) in dMpv17 KO can be easily observed by comparison with wild-type flies. Size bar: 500 nm.
(B) Quantification of the mitochondrial area, length and width (n = 130 mitochondria) in the fat body. LD, lipid droplet. Error bars: SEM. The asterisks represent the significance levels calculated by unpaired, two-tailed Student’s t test: ∗∗∗∗p < 0 0.0001.
(C) Representative TEM images from Drosophila control (w1118) and dMpv17 KO flight muscles. The overall structure of muscle fibers is preserved. Abnormal cristae structures (swirls) can be observed (red arrows) in dMpv17 KO flies. Size bar: 2 μm.
(D) Quantification of mitochondrial area, length and width (n = 100 mitochondria in the flight muscles). Error bars: SEM.
Mitochondrial ultrastructure alterations were also present in dMpv17 KD Drosophila S2R+ cells, which showed a decrease in mitochondrial area and major diameter (Figure S4).
These results suggest that dMpv17 is a major player in the maintenance of mitochondrial ultrastructure, and both its deletion and overexpression alter mitochondrial ultrastructure (Figure S5). Such situation resembles the morphological consequences of abnormalities in the mitochondrial ATP-dependent K+ channel.22
dMpv17 KO leads to alterations in energy metabolism
Specific hMPV17 mutations or deletion of the protein have been linked to altered mitochondrial bioenergetics and metabolism.11 Therefore, we investigated the role of dMpv17 in Drosophila metabolism. Since in humans a hallmark of MPV17-dependent diseases is severe hypoglycaemia, we wondered how dMpv17 KO flies would respond to fasting. We thus examined the survival probability of dMpv17 KO and WT adult flies on normal diet and food deprivation (1% agar to provide a uniform source of water and avoid desiccation).23 No differences were observed in either sex on a standard diet (Figure 4A). By contrast, both male and female dMpv17 KO flies showed a significant reduction of the lifespan in 1% agar compared to WT flies (Figure 4B). Taken together, these results suggest that dMpv17 is dispensable under normal conditions, but has important roles in stress conditions such as starvation.
Figure 4.

Energy metabolism in dMpv17 KO flies
(A) Kaplan-Meier survival analysis in normal diet (females, left; males, right).
(B) Kaplan-Meier survival analysis in food deprivation (females, left; males, right). ∗∗∗∗p < 0.0001 calculated by log rank (Mantel Cox) test.
(C) ATP content in whole 5-day-old flies on normal diet. Error bars represent ±SEM. The asterisks represent the significance levels calculated by unpaired, two-tailed Student’s t test: ∗p < 0.05.
(D) Lactate content in whole 5-day-old flies on normal diet. Error bars represent ±SEM. The asterisks represent the significance levels calculated by unpaired, two-tailed Student’s t test: ∗p < 0.05.
(E) ldh mRNA quantification in 5-day-old flies on normal diet. Error bars represent ±SEM. The asterisks represent the significance levels calculated by unpaired, two-tailed Student’s t test: ∗p < 0.05.
(F) ATP content in flies starved for 18 h. Error bars represent ±SEM. The asterisks represent the significance levels calculated by unpaired, two-tailed Student’s t test: ∗∗∗p < 0.001.
(G) ldh mRNA quantification in starved for 18 h. Error bars represent ±SEM. The asterisks represent the significance levels calculated by unpaired, two-tailed Student’s t test: ∗p < 0.05.
(H) Glycogen content in whole 5-day-old flies on normal diet. Error bars represent ±SEM. The asterisks represent the significance levels calculated by unpaired, two-tailed Student’s t test: ∗p < 0.05.
(I) Glycogen content in flies starved for 18 h. Error bars represent ±SEM.
(J) tobi mRNA quantification in whole 5-day-old flies on normal diet. Error bars represent ±SEM. The asterisks represent the significance levels calculated by unpaired, two-tailed Student’s t test: ∗∗∗p < 0.001.
Since no differences were observed between males and females, we pooled flies from both sexes for subsequent analyses. We analyzed mitochondrial respiratory chain (MRC) in isolated mitochondria from whole dMpv17 KO and WT flies by one-dimensional (1D) blue native gel electrophoresis (BNGE). We found no obvious alterations in MRC complexes amount by Coomassie staining (Figure S6A). In addition, the respiration rate was similar between the two genotypes (Figure S6B). These results indicate no major alterations of the MRC, although we cannot exclude that this is related to the fact that mtDNA depletion is only present in fat bodies. Interestingly, we found increased ATP levels in KO vs. WT flies at day 5 on normal diet (Figure 4C), while they were reduced at day 20 (Figure S6C). In addition, steady state levels of lactic acid, an index of the glycolytic flux, was increased by 2-fold in young flies (day 5) compared to age-matched wild-type controls, suggesting an increase in glycolysis (Figure 4D). This finding was confirmed by a 1.4-fold increase in the transcript level of Impl3, the fly ortholog of mammalian lactate dehydrogenase (Figure 4E).
On starvation, ATP levels were ∼50% lower in dMpv17 KO flies compared to WT controls, suggesting that KO flies utilized their energy reserves faster than controls, thus leading to premature death (Figures 4F–4H). There was a 2-fold increase in Impl3 mRNA in KO compared to WT flies.
These data indicate that, as in cells with hMPV17 deletion, an increase in glycolytic flux occurs in dMpv17 KO compared to WT flies.
Glycogen is the primary form of glucose storage in Drosophila.24 Since fat bodies store large quantities of glycogen, we measured glycogen content in whole dMpv17 KO and WT flies. Glycogen levels were significantly increased in normally fed dMpv17 KO compared to WT flies (Figure 4H). Interestingly, glycogen content was similar in KO and WT flies after 18 h of starvation, suggesting that dMpv17 KO flies have a higher rate of glycogenolysis during starvation (Figure 4I). Accordingly, we also observed a strong up-regulation in the expression levels of the target of brain insulin gene (tobi), encoding an evolutionarily conserved α-glucosidase,25 that breaks down starch and disaccharides into glucose (Figure 4J). These results indicate an alteration of glucose metabolism in Mpv17 KO flies, which is consistent with the severe hypoglycemia that arise 3–4 h after a meal in MPV17 patients.
dMpv17 forms a channel in planar lipid bilayers and its conductance is affected by pathological mutations
Given that both yeast and human proteins have been shown to form an aqueous pore in an artificial lipid bilayer,10,12 we tested whether dMpv17 also forms channels. We thus measured channel activity in a planar lipid bilayer using a recombinant, LDAO-solubilized in vitro-expressed dMpv17 protein with a His-tag at the C-terminus (Figures S7 and S8). In the presence of K+, opening of the channel was detected with a prevalent single channel conductance of 330 picoSiemens (pS) (Figure 5A), which was consistently observed in different preparations and in independent experiments (n = 24). The putative pore size of approximately 1.8 nm supports the hypothesis that dMpv17, possibly in a multimeric structure, possesses the channel-like activity in the IMM that would be required for the transport of relatively large molecules, including metabolic intermediates. As for the human protein,10 dMpv17 was characterized by a flickering behavior and often adopted substates (Figure 5A). The reversal potential of the channel in asymmetric KCl solution (400 mM KCl cis/135 mM KCl trans) was −19.6 mV (Figure 5B), indicating a PK+/PCl− ratio of 4.5. This value is in agreement with that detected for human MPV17, indicating a slight preference for cations over anions. In addition, a linear current-voltage relationship was observed, similar to that of human MPV17.10
Figure 5.

Single channel activity of dMpv17
(A) Representative current traces recorded at the indicated voltages. Electrolyte was symmetrical 150 mM KCl buffered with 10 mM Tris-Cl, pH 7.4 (n=18). Conductance of the fully opened channel was 330 pS. Right: Amplitude histogram derived from the recording of channel activity at −60 mV shown in the upper panel. Current levels for closed, and one and three simultaneously opened channels are labelled as c, o1, o2 and o3 (c=closed, o=opened; s2 and s1 indicate substates 2 and 1).
(B) Representative traces recorded in asymmetrical KCl solutions at the indicated voltages (cis). Single-channel recordings were made in asymmetrical K+ with 400 mM KCl in the cis side and 135 mM in trans side (n=3). The graph to the right represents the current-voltage relationship. Mean current values +/- SEM are shown (from n=3 independent experiments).
As mentioned previously, orotate has been suggested to rescue the phenotype of mpv17-deficient zebrafish. Therefore, by combining electrophysiology and mass spectrometry analysis, it was possible to add small molecules to the cis-side of the membrane and assess whether these molecules were able to pass to the trans-side via an active dMpv17 channel.26 In particular, mass spectrometry analysis was used to determine if the metabolite added to one chamber is eventually found in the other, suggesting translocation (Figure 6A). These experiments indicate that dMpv17 passes uridine but not orotate. A representative experiment (channel shown in the right part of Figure 6B exhibiting persistent activity) documented translocation of uridine added to the cis side of the chamber (10 mM final concentration) to the trans side, yielding 240 nM final concentration. Sampling from the trans side was performed 6 min after adding uridine to the cis side. Similar results were obtained in three further instances, where the detected concentration of uridine on the trans side was 162.1, 35.2, and 29.7 nM, respectively. In three further experiments where channel activity was characterized by brief bursts, the final concentration of uridine was <10 nM. In addition, in 3 control experiments uridine was added to empty membranes and, as expected, did not pass the membrane (data not shown). In 10 independent experiments orotate was added either alone or in combination with uridine to the membrane exhibiting persistent dMpv17 activity, but in none such cases orotate was detected in the trans chamber on subsequent mass spectrometry analysis.
Figure 6.

Electrophysiological features of dMpv17
(A) Representation of the experimental setup.
(B) Representative channel activity in the presence of uridine. Electrolyte was symmetrical 150 mM KCl buffered with 10 mM Tris-Cl, pH 7.4. The channel activity allowed the translocation of uridine added to the cis side of the chamber (at 10 mM final concentration) (representative of n=4 independent experiments, see text for details), yielding 240 nM final concentration in the opposite chamber (trans) in the experiment shown in the figure. The presence of uridine was measured by mass spectrometry.
We next analyzed whether pathogenic mutations significantly affected the slope conductance of the channel. Importantly, many of the sites known to be mutated in MPV17 patients are conserved in the Drosophila protein. We selected as candidate amino acids the conserved Ser-170, one of the three predicted phosphorylation sites the mutation of which causes MDS, and the Arg-50, that has been linked to NHH syndrome. Thus, we introduced the point mutations C488T and G122A in the wild-type dMpv17 sequence resulting in S163F and R41Q changes (corresponding to human hS170F and hR50Q changes) and analyzed their channel properties. The S163F mutation prevented the full opening of the channel in physiological conditions (150 mM KCl, 10 mM HEPES pH 7.4) (Figure 7A). By contrast, the R41Q mutation showed a relevant decrease (down to 55 pS [Figure 7B]), but not the abolition of channel conductance. This is compatible with the less severe phenotype associated with NHH.3
Figure 7.

Channel conductance of dMpv17 is decreased by specific mutations
Representative current traces and corresponding histograms are shown for two mutant forms of dMpv17, i.e. S163F and R41Q. (A) The S163F mutant showed 10pS conductance of the fully opened channel (n=18). Amplitude histograms were obtained from 30-s long current traces. Only in 2 further cases a conductance of 80 pS could also be observed. Amplitude histograms were obtained from 30-s long current traces.
(B) The R41Q mutant showed a conductance of around 40 pS (n=9). Amplitude histograms were obtained from 30-s long current traces.
Finally, to determine whether dMpv17 is part of a complex, as previously reported for Sym1,12 we carried out Western blot analysis from two-dimensional (2D) BNGE analysis using mitochondria isolated from a Drosophila strain expressing an HA-tagged Mpv17 (fused on the C-terminus of the dMpv17 protein sequence). As for the yeast and mouse protein, dMpv17-HA was mainly present in a site corresponding to a complex of 120 kDa, compatible with a hexamer (Figure 8). However, we cannot exclude a more complex stoichiometry due to the presence of other auxiliary subunits in vivo, which could serve, for example, as a “lid” regulating the opening of the pore.
Figure 8.

Blue native gel electrophoresis
(A) Mitochondria from UAS-dMpv17-HA>Act5C-Gal4 and parental (UAS-dMpv17-HA>+ and Act5C-Gal4>+) controls were analyzed by 2D BNGE. The asterisk marks dMpv17-HA specific spot (the two asterisks mark an unspecific spot due to the protein not correctly assembled in the complex).
(B) Molecular weight of dMpv17-HA comprising complex. The protein is present in a spot corresponding to a complex of molecular weight between complex II and IV.
Discussion
Despite clinical heterogeneity, MPV17-related MDS are all characterized by mtDNA instability, indicating a role for MPV17 in mtDNA maintenance. However, the very function of MPV17 remains elusive. Here, we provide robust evidence that dMpv17 forms an ion channel in lipid bilayers, as previously reported,10 and show for the first time that the electrophysiological properties of dMpv17 are modified by pathogenetic mutations, the severity of which roughly correlates with the severity of the human condition. Accordingly, the S163F mutation completely abolished channel opening, while the R41Q mutation, which is typical of NNHP,3 decreased without abolishing channel conductance. It is interesting to note that in HEK293 cells transfected with mutant forms of the protein—in particular the R50W (but to a lesser extent also the R50Q) mutant—enhanced oxidative stress, mitochondrial ROS production and lactate levels, and this was not due to mislocalization of R50Q mutant.11 Interestingly, the S170F mutant did not cause these changes. This mutation in hMpv17 has been hypothesized to lead to a “leaky” channel based on the observation that expression of hMPV17S170F led to loss of Δψm.11 When interpreting these results, one should keep in mind that not only does the S170F mutation change the channel characteristics, but it also leads to dramatically decreased protein levels.11
Recently, biophysical and NMR methods have shown that MPV17 folded into detergent micelles adopts a compact structure consisting of six α-helices. Furthermore, using a nanodisc reconstitution approach, it has been shown that disease-related mutations of MPV17 abolish its oligomerization properties in the membrane.27 By contrast, our in silico analysis identified 5 transmembrane domains, which is in agreement with AlphaFold prediction for both Drosophila and human MPV17. The structural similarity between the fly and human proteins suggests a conserved function. The NMR data suggest that dMpv17 monomers can self-assemble to form oligomers and this process is controlled by ROS levels, which act on specific cysteines (at positions 59, 99, 103, and 160) to trigger the formation of disulfide bonds.27 These are instrumental to stabilize the oligomers and to form a functional ion channel. However this latter assumption has not been directly demonstrated and warrants future investigation. Notably, pathological mutations, especially R50W and Q93P, in MPV17 cause a reduction in oligomerization.27
One of our main findings is that dMpv17 electric activity is coupled with the translocation of uridine across the lipid bilayer, suggesting that the protein may act as a carrier for small metabolites. Future work is warranted to investigate if and which other metabolites are transported. However, our findings on uridine transport are intriguing. Defects in mitochondrial uridine may explain the mtDNA depletion, as uridine is incorporated into the mRNA primers needed for mtDNA replication.28,29 It is well known that the uridine salvage pathway can support pyrimidine synthesis in OXPHOS defects.30 In addition, uridine is linked to several additional metabolic pathways, including lipid and amino acid metabolism, and the TCA cycle.31 Recent evidence suggests that it can also contribute to energy metabolism by conversion of its ribose moiety into fructose-6-P and glyceraldehyde-3-P via the pentose phosphate pathway.32 All these observations position uridine as a central metabolite with multiple roles, and may reconcile previous findings relating MPV17 to hypoglycemias in patients,2 the replenishment of intermediates of the TCA cycle,9 the decrease in dGTP and dTTP levels,6 and the reduced dTMP content.7
Martorano and colleagues have previously reported a drastic reduction of DHODH in zebrafish larvae, pointing to a role for zebrafish Mpv17 in orotate transport.8 We failed to detect a reduction in DHODH activity in our dMpv17 KO model. However, the results reported by Martorano and coworkers do not rule out the possibility that provision of orotate may actually increase uridine availability. Accordingly, dUTP has been reported to robustly rescue the number of iridophores in Mpv17 KO larvae in the same study.8
The critical metabolic role of dMpv17 is supported by our in vivo data. Our dMpv17 KO flies show reduced glycogen content in fat bodies, increased glucose flux through glycolysis and increased lactate levels, accompanied by reduced survival in starvation. A huge increase in tobi encoded beta galactosidase α-glucosidase—which breaks down starch and disaccharides to glucose—confirms the increased use of glycogen stores in dMpv17 KO flies. This is compatible with the hypoglycemias observed in patients. A recent study has characterized an RNAi-based dMpv17 model showing that dMpv17 knockdown induces both locomotor and cognitive defects in larvae.13 However, in our study we generated a knockout fly model, and carried out an extensive characterization of the adult phenotype. Most importantly, we have provided mechanistic insight into MPV17 function by combining in vivo and in vitro techniques.
In conclusion, we have demonstrated that dMpv17 forms a channel of the IMM. Our BNGE data suggest that dMpv17 may form a homohexamer, and possibly interact with other proteins. Proteomic analysis of the native complex will clarify this essential point. High resolution structural analysis of the potential new pore will contribute to elucidate its function, distribution and functional plasticity, and understand the effect of mutant variants as well. The channel is potentially able to translocate metabolites and other relatively big compounds, such as uridine, with a selective capacity. Systematic analysis on the translocation capacity of different compounds will help elucidate the number and features of these structures. Since the pore sits on the IMM, additional structures must be present in a complex, which are able to regulate the opening and closure of the pore, in order to prevent the dissipation of the mitochondrial electrochemical gradient.
Limitations of the study
While we showed that dMpv17 plays an important role in fly energy metabolism and that it works as a channel in an artificial lipid bilayer, whether dMpv17 works as a channel in vivo remains to be established. Similarly, while we proved that dMpv17 transports uridine but not orotate across the lipid bilayer in vitro, whether other metabolites (and which ones) are also transported needs to be investigated.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| monoclonal mouse α-HA antibody | Sigma | Cat# H3663-100UL |
| Alexa Fluor®488 α-mouse IgG | Thermo Fisher | Cat# A32723 |
| CellFectin II Reagent | Invitrogen | Cat# 10362100 |
| MitoTracker® Red CMXROS | Thermo Fisher Scientific | Cat# M46752 |
| SYBR Green | Promega | Cat# A6001 |
| NativePAGE 3–12% Bis-Tris gels | Thermo Fischer Scientific | Cat# BN1001BOX |
| Schneider's Drosophila Medium | Thermo Fisher Scientific | Cat# 21720024 |
| Chemicals, peptides, and recombinant proteins | ||
| LDAO | Sigma | Cat# 40236 |
| L-α-Phosphatidylcholine | Sigma | Cat# P7443 |
| 4ME 16:0 PC 1,2-diphytanoyl-sn-glycero-3-phosphocholine | Avanti Polar Lipids, Inc | Cat# 850356 |
| Decane | Sigma | Cat# 457116 |
| Octane | Sigma | Cat# 74820 |
| L-DHOA | Sigma | Cat# D7128 |
| Alamethicin | Sigma | Cat# A4665 |
| Leflunomide | Sigma | Cat# PHR1378 |
| sodium azide | Sigma | Cat# S2002 |
| antimycin A | Sigma | Cat# A8674 |
| Rotenone | Sigma | Cat# rotenone |
| 3-nitropropionic acid | Sigma | Cat# N22908 |
| Coenzyme Q1 | Sigma | Cat# C7956 |
| 2,6-dichlorophenolindophenol | Sigma | Cat# 36190 |
| Critical commercial assays | ||
| Lactate-Glo Assay | Promega | Cat# J5021 |
| Glycogen Colorimetric/Fluorimetric Assay | Promega | Cat# K646-100 |
| ATPlite luminescence assay system | PerkinElmer | Cat# 6016731 |
| T7 MEGAscript kit | Ambion | Cat# AM1334 |
| RTS100 Wheat Germ CECF Kit (5’-PRIME) | Fisher Scientific | |
| SuperScript II | Life Technologies | Cat# 18064014 |
| DC™ Protein Assay Kit | BioRad | 5000111 |
| Experimental models: Cell lines | ||
| S2R+ | DGRC, Indiana University, Bloomington Campus, USA | Stock number: 150 |
| Experimental models: Organisms/strains | ||
| dMpv17 KO lines | This paper | N/A |
| Oligonucleotides | ||
| Rpl32 Fw (AGGCCCAAGATCGTGAAGAA) | Thermo Fisher | N/A |
| Rpl32 Rv (TGTGCACCAGGAACTTCTTGAA) | Thermo Fisher | N/A |
| 16S Fw (AAAAAGATTGCGACCTCGAT) | Thermo Fisher | N/A |
| 16S Rv (AAACCAACCTGGCTTACACC) | Thermo Fisher | N/A |
| dMpv17 Fw (GTTTCGGCATCGTTGGACTG) | Thermo Fisher | N/A |
| dMpv17 Fw (CAGCATTTTGGTAACGCCCC) | Thermo Fisher | N/A |
| Rp49 Fw (ATCGGTTACGGATCGAACAA) | Thermo Fisher | N/A |
| Rp49 Rv (GACAATCTCCTTGCGCTTCT) | Thermo Fisher | N/A |
| tobi Fw (CCACCAAGCGAGACATTTACC) | Thermo Fisher | N/A |
| tobi Rv (GAGCGGCGTAGTCCATCAC) | Thermo Fisher | N/A |
| Impl3 Fw (CATCATCCCCAAGCTGGTAG) | Thermo Fisher | N/A |
| Impl3 Rv (CCAGGCCACGTAGGTCAT) | Thermo Fisher | N/A |
| Recombinant DNA | ||
| pAc5-STABLE2-neo vector | Addgene | Plasmid #32426 |
| pIVEX 1.3 WG vector | Biotechrabbit | Cat # BR1401301 |
| pACT vector | Promega | Part# TM049 |
| Software and algorithms | ||
| ORIGIN 6.1 | OriginLab | N/A |
| Prism GraphPad | Dotmatics | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Prof. Rodolfo Costa (rodolfo.costa@unipd.it).
Materials availability
The dMpv17 KO fly line generated in this study is available from the lead contact on a collaborative basis.
Experimental model and study participant details
Flies
Drosophila melanogaster dMpv17 KO line was created by CRISPR/Cas9-mediated genome editing through the insertion of a stop codon after the ATG sequence (WellGenetics Inc.). The w1118 Drosophila strain was used as a control because the Mpv17 KO was created on this background. Flies were raised on standard cornmeal medium (1 litre: 50 g inactivated yeast powder, 8.5 g agar, 72 g cornmeal, 79.3 g sucrose, 13.5 ml Nipagin in 75% ethanol) and maintained at 23°C under a 12hr:12hr light:dark cycle. For starvation experiment, 5-day old flies were maintained in 1% agar. An equal number of 5-day-old or 20-day-old male and female flies were used in the different experiments. For lifespan analysis, males and females were assayed separately.
Cells
The Drosophila S2R+ cell line was derived from a primary culture of late stage (20–24 hours old) Drosophila embryos33 and it was purchased from the Drosophila Genomics Resource Center (DGRC, Indiana University, Bloomington Campus, USA). S2R+ cells were maintained at 25°C in Schneider's Drosophila Medium (Thermo Fisher Scientific) supplemented with 10% heat-inactivated Fetal Bovine Serum (Euroclone). We assessed absence of mycoplasma contamination by DAPI staining.
Method details
Fat body and muscles dissection
For thoracic muscle isolation, flies were anesthetized on a CO2 pad, and dissected under a microscope by severing both the head and the abdomen. The wings and legs were also removed. For fat body dissection, the fly head and thorax were removed. The abdominal carcass was dissected in Schneider's Drosophila Medium (Thermo Fisher Scientific), by splitting the ventral tissue longitudinally and removing the gonads, gut, and Malpighian tubules, and then exposing the fat body layer attached to the cuticle.
mtDNA copy number quantification
DNA (genomic and mitochondrial) from 5 adult flies (5 days old unless otherwise indicated) was extracted in triplicate using phenol/chloroform precipitation. The amount of mtDNA was assessed by the ratio of mtDNA with respect to nuclear DNA (nDNA) copy number determined by quantitative real time amplification of the mitochondrial 16S gene and the nuclear Rpl32 gene. We generated two gene-specific calibration curves with six 10-fold serial dilutions (100–10.000.000) of plasmids containing the cloned target sequences. Concentration of plasmid stock solutions was assessed with an ND-1000 spectrophotometer (NanoDrop), and the plasmid copy number of dilutions was calculated using Avogadro's number. Reactions were performed in triplicate using SYBR Green chemistry (Promega) in a 7500 Real Time PCR System instrument (Applied Biosystems, Thermo Fisher).
RNA isolation, retrotranscription and qRT-PCR
Total RNA was extracted in triplicate from 10 adult flies (5-day old unless otherwise indicated) or 2 × 106 cells using TRIzol reagent (Thermo Fisher Scientific) according to the manufacturer's instructions and further purified by precipitation with 8 M LiCl in order to reduce the contamination of carbohydrates. 1 μg of total RNA was used for first strand cDNA synthesis, employing 10 mM deoxynucleotides (dNTPs), 300 ng/μl random examers and SuperScript II (Life Technologies) following manufacturer’s instructions. qRT-PCRs were performed in triplicate in a 7500 Real Time PCR system (Applied Biosystems, Thermo Fisher Scientific) using SYBR Green chemistry (Promega). The 2−ΔΔCt (RQ, relative quantification) method was used to calculate the relative expression ratio.34 Rp49 was used as endogenous control.
All oligonucleotides used were designed using the on-line Primer3 software (http://bioinfo.ut.ee/primer3/) and are listed below.
Lifespan assay
Lifespan was assayed as in Broughton et al. 2005.35 Flies were reared at standard low density, and virgin males and females were collected in a five-hour window. Adults of the same sex were kept at a density of 10 per vial (for a total of 60 individuals). Flies were transferred to fresh medium, without anesthesia, three times per week and deaths were scored.
For starvation experiment, 5-day old flies were maintained in 1% agar. Flies were transferred to fresh medium every day and deaths were scored four times per day.
ATP quantification
ATP content was measured using the ATPlite luminescence assay system (PerkinElmer). Briefly, 10 adult flies per replicate (for a total of 3 replicates) were homogenized in 150 μL of mammalian cell lysis solution on ice and centrifuged at 14000 X g at 4°C for 5 min to remove insoluble material. The supernatant was diluted 1:10 in HBSS (Thermo Fischer Scientific) and quantified according to the manufacturer’s protocol. ATP content was normalized by protein content assessed by Bradford method.
Lactate quantification
For lactate quantification, 30 5-day-old adults per replicate were analyzed using Lactate-Glo Assay (Promega) according to the manufacturer’s protocol. Lactate content was normalized by protein content assessed by Bradford method (BioRad).
Glycogen quantification
Glycogen levels were measured using Glycogen Colorimetric/Fluorimetric Assay (Biovision) according to the manufacturer’s protocol. 6 5-day-old adults per replicate were used and glycogen content was normalized by protein content assessed by Bradford method.
Dihydroorotate dehydrogenase activity quantification
Flies were homogenized in 100 mM HEPES pH 7.5, 150 mM NaCl, 0.01% Triton X-100, with the addition of protease and phosphatase inhibitors. 60 μg of lysate was pre-incubated for 10 min with the substrate L-DHOA (10 mM) and alamethicin (2 mM), and separately with the Dhodh inhibitor leflunomide (200 μM) for 10 min. After the pre-incubation time, sodium azide (250 mM), antimycin A (2 mM), rotenone (2 mM), 3-nitropropionic acid (2 mM), Coenzyme Q1 (10 mM) and 2,6-dichlorophenolindophenol (DCPIP; 5 mM) were added. The activity of the enzyme was assessed at 30°C through spectrophotometric recordings following the reduction of DCPIP at 600 nm. Values were normalized for protein amount.
Mitochondria isolation
Mitochondria were isolated by differential centrifugation from 100 flies.36 Flies (5 days old) were homogenized using a Dounce glass potter and a loose-fitting glass pestle in isotonic isolation buffer (225 mM mannitol, 75 mM sucrose, 5 mM HEPES, 1 mM EGTA, pH 7.4) with 1% BSA. Samples were centrifuged at 1.000xg (Beckman Avanti J-25 Centrifuge, Beckman 2550 rotor) at 4°C for 10 minutes. The supernatant was filtered through a fine mesh, and the supernatant centrifuged at 6.000xg at 4°C for 10 minutes. The pellet was re-suspended in 10 ml of isolation buffer without BSA before being centrifuged at 6.000xg under the same conditions as above and re-suspended in minimal volume of isolation buffer without BSA. Protein concentration was measured by the Bradford assay (Bio-Rad protein assay).
Blue native gel electrophoresis
Isolated mitochondria were resuspended in 1.5 M aminocaproic acid, 50 mM Bis-Tris/HCl pH 7.0. The samples were solubilized with 4 mg digitonin (Merck) per mg of protein. After 5 min. of incubation on ice, samples were centrifuged at 18,000 ×g at 4°C for 30 min. The supernatant was collected and resuspended with Sample Buffer (750 mM aminocaproic acid, 50 mM Bis-Tris/HCl pH 7.0, 0.5 mM EDTA, and 5% Serva Blue G). Native samples were run in NativePAGE 3–12% Bis-Tris gels (Thermo Fischer Scientific) according to the manufacturer’s protocol.
Cell transfection and subcellular localization
CG11077 cDNA was cloned into pACT (Promega) vector, fused with the HA tag. Drosophila S2R+ cells were transfected using CellFectin II Reagent (Invitrogen) following manufacturer’s instruction. After 48 hrc, cells were washed once in 1X PBS and incubated with 10 nM MitoTracker® Red CMXROS (Thermo Fisher Scientific) and 1 μg/ml cyclosporin H (Sigma) in Schneider's Drosophila Medium for 20 minutes. Cells were then washed once in PBS and fixed in 4% paraformaldehyde (PFA) for 20 minutes. For dMpv17-HA, cells were washed again in PBS and permeabilized for 5 minutes with 50 mM NH4Cl in PBS + 0.1% Triton X-100. Cells were then blocked for 1 hr in 3% goat serum in PBS, washed again, and incubated overnight in 1:100 monoclonal mouse α-HA antibody (Sigma) at 4°C. Cells were washed other three times with PBS and then incubated with 1:500 Alexa Fluor®488 α-mouse IgG (Thermo Fisher) with 2% goat serum for 45 minutes at room temperature. The slides were mounted with Vectashield mounting medium (Vector laboratories). Images were taken with a Zeiss LSM700 confocal microscope at 63X magnification.
dsRNA production and RNAi procedures
Double-stranded RNAs (dsRNAs) were prepared employing the T7 MEGAscript kit (Ambion). The oligonucleotide primers used to synthesize dsRNA starting from cDNA were dMpv17_T7 forward (F) and reverse (R) possessing a T7-promoter sequence overhang. These primers give two complementary 600 bp RNA products that anneal each other forming a final 600 bp dsRNA. About 2 × 106 cells were treated with 5 μg of dsRNA in serum-free medium and incubated at room temperature for 1 h. Subsequently, 1 volume of complete medium (2×) was added, and cells were grown in the presence of dsRNA for 3 days at 25°C and then used for subsequent analysis. dsRNA of human insulin like growth factor binding protein 2 gene was used as mock control.
dNTP pools extraction and analysis
About 10 × 106 S2R+ cells were centrifuged in 15-ml tubes for 10 minutes at 400 × g, and the pellet was washed twice with ice-cold PBS. The cells were then re-suspended in 200 μl of extraction buffer (0.21 M mannitol, 0.07 M sucrose, 0.2 M ethyleneglycol-bis(2-aminoethylether)-N,N,N',N'-tetraacetic acid EGTA, 10 mM Tris-HCl, pH 7.5, 0.5% BSA), and a suspension of glass beads (0.1 mm diameter), corresponding to about half of the volume of the cellular pellet, was added. The cell/bead suspension was introduced in a Bullet Blender Storm homogenizer (Next Advance) and shaken for 2 minutes at speed 8, then 400 μl of extraction buffer were added, and the glass beads were removed by a short centrifugation. Mitochondrial nucleotide pools were isolated from the whole cell homogenate by differential centrifugation and methanol extraction.37 The pellet remaining after mitochondrial pool extraction was dissolved in 1 ml of 0.3 M NaOH. The A260 nm of the NaOH fraction was used to normalize the number of cells from which the pools of the different samples were extracted. The sizes of the dNTP pools were determined with a DNA polymerase-based assay.38 The A260 nm of the NaOH fraction was used to normalize the number of cells from which the pools of the different samples were extracted. Two different aliquots of each pool extract were analyzed, and pool sizes were expressed as pmol of dNTPs/million cells or pmol/OD NaOH.
Electron microscopy
S2R+ cells were fixed for 1 hour in 2.5% glutaraldehyde in sodium cacodylate buffer. Samples were washed in 0.1 M cacodylate buffer (pH 7.4), and then post-fixed for 1 hour at room temperature with 1% OsO4 in 0.1 M sodium cacodylate buffer (pH 7.4). Cells were washed once with water and then incubated in 1% uranyl acetate in H2O for 1 hour at room temperature. Subsequently, cells were dehydrated through an ethanol gradient. Finally, the samples were embedded in Epon resin (Sigma) and polymerized at 60°C overnight.
Thoraxes and fat bodies from adult male and female flies were fixed in 2.5% glutaraldehyde overnight. Samples were rinsed in 0.1 M cacodylate buffer with 1% tannic acid and then fixed in 1:1 2% OsO4 and 0.2 M cacodylate buffer for 1 h. Samples were rinsed, dehydrated in ethanol and embedded in Epon resin. Ultrathin sections (400Å) were examined and photographed with a FEI Tecnai G2 electron microscope. Fat body and muscles from adult male flies were fixed in 2.5% glutaraldehyde overnight. Samples were rinsed in 0.1 M cacodylate buffer with 1% tannic acid and then fixed in 1:1 2% OsO4 and 0.2 M cacodylate buffer for 1 h. Samples were rinsed, dehydrated in ethanol, and embedded in Epon. Ultrathin sections (400 Å) were examined and photographed with a FEI Tecnai G2 electron microscope.
Correlative microscopy
To overexpress dMpv17, S2R+ cells were transfected with pAc5-STABLE2-neo vector expressing dMpv17-GFP and then transferred to glass bottom dishes provided with gridded coverslips (MatTek Corporation). After three days of transfection, cells were fixed in 2.5% glutaraldehyde in sodium cacodylate buffer for one hour. Cells were imaged with a fluorescent microscope and the fields containing GFP positive cells were acquired. EM analysis was performed in GFP positive cells to determine mitochondria morphology. Control cells were transfected with pAc5-STABLE2-neo vector expressing GFP.
Electrophysiology experiments in lipid bilayer
dMpv17 coding sequence was cloned into pIVEX 1.3 WG vector (Roche) using In-Fusion® HD Cloning kit (Clontech) following the manufacturer’s instructions. Primer used were Fw CCACAACAGCTTGTCGA ACCATGAAGAGACTTAAAGCGTA and Rv TGATGATGAGAACCCCCCCCGCTATTAAGTATCATGGAGAG for the D. melanogaster sequence and Fw CCACAACAGCTTGTCGAACCATGGCACTCTGGCGGGCA and Rv TGATGATGAGAACCCCCCCCGAGCCGATGTGCCTTCC for the human CDS. In vitro expression was performed using a RTS100 Wheat Germ CECF Kit (5’-PRIME, Fisher Scientific). After expression, the reaction mix was solubilized with 2% LDAO for 90 min at 30°C under 1400 rpm shaking and solubilization was verified by SDS-PAGE and western blot. The preparation was incorporated into planar bilayers for determination of channel activity. Electrophysiology experiments were carried out using a Warner Instruments Incorporated electrophysiological planar bilayer apparatus. Bilayers were prepared using L-α-Phosphatidylcholine in decane (Sigma-Aldrich) containing 1% chloroform (Sigma-Aldrich) or 4ME 16:0 PC 1,2-diphytanoyl-sn-glycero-3-phosphocholine (Avanti Polar Lipids, Inc) in octane (Sigma-Aldrich) across a 250 μM hole in a polystyrene cuvette (Warner Instruments). L-α-Phosphatidylcholine was partially purified by precipitation with cold acetone from a chloroform solution. The bilayer membrane was considered to be satisfactory for further experiment if it exhibited an approximately 150–200 pF capacity. The lipid membrane divided the trans and the cis compartment. All reported voltages refer to the cis chamber, zero being assigned to the trans (grounded) side. The bilayer set up was connected to the external circuit through salt bridges (1 M KCl) with Ag/AgCl electrodes. Experiments were performed in 150 mM KCl concentration media and the protein was added to the cis compartment. All measurements were made at room temperature. Unitary currents were recorded using an Axon BC-525C patch clamp amplifier (Warner Instruments Incorporated). Recordings were filtered at 1 kHz, digitized at 300 KHz and analyzed with pClamp 8.1 and ORIGIN 6.1 software.
Quantification and statistical analysis
Data are presented as mean ± standard error. Error bars in the graphs represent the standard error of the mean. Pairwise comparisons were carried out by unpaired, two-tailed Student’s t-test. Data analyses were performed with Prism v6 (GraphPad Software). Statistical significance: ∗p<0.05, ∗∗p<0.01, ∗∗∗p<0.001, ∗∗∗∗p<0.0001.
Acknowledgments
Our work is supported by: Telethon Foundation (GGP11011 and GGP15041 to R.C., GGP19118 to I.S., GGP19007 and GMR 23T1065 to M.Z. and GGP20013 to C.V.); Comparative Insect Chronobiology (CINCHRON), EU Horizon 2020, Marie Sklodowska-Curie Initial Training Network grant (agreement N°765937) to R.C.; Associazione Luigi Comini Onlus (MitoFight2) to C.V., AFM-Telethon 23706 to C.V.; University of Padova – PRAT 2014 N. CPDA142980 to C.D.; Cariparo Foundation Excellence Project 2021 and PRIN 2022 (grant number 2022ZY7ATN) to V.C.; University of Padova – Department of Biology BIRD 182052 to S.C.; University of Padova – Department of Biomedical Sciences SID2022 and BIRD2022 to C.V.; UMDF grant to M.Z.
Author contributions
Conceptualization: R.C., I.S., C.V., and M.Z.; Methodology: S.C., V.C., M.B., C.R., E.B., M.C.G., K.C., M.R., D.D.S., and S.B.; Data Curation: S.C., V.C., M.B., C.D., C.V., I.S., R.C., and D.D.S.; Investigation: S.C., V.C., M.B., C.R., E.B., M.C.G., K.C., M.R., D.D.S., and S.B.; Writing – Original Draft: S.C., V.C., M.B., and C.V.; Writing – Review and Editing: I.S., R.C., C.V., and M.Z.; Supervision: R.C., I.S., C.V., and M.Z.; Funding Acquisition: R.C., I.S., M.Z., C.V., V.C., C.D., and S.C.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: September 16, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2023.107955.
Contributor Information
Ildiko Szabò, Email: ildiko.szabo@unipd.it.
Rodolfo Costa, Email: rodolfo.costa@unipd.it.
Supplemental information
Data and code availability
-
•
Data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Viscomi C., Zeviani M. MtDNA-maintenance defects: syndromes and genes. J. Inherit. Metab. Dis. 2017;40:587–599. doi: 10.1007/s10545-017-0027-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Spinazzola A., Viscomi C., Fernandez-Vizarra E., Carrara F., D’Adamo P., Calvo S., Marsano R.M., Donnini C., Weiher H., Strisciuglio P., et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat. Genet. 2006;38:570–575. doi: 10.1038/ng1765. [DOI] [PubMed] [Google Scholar]
- 3.Karadimas C.L., Vu T.H., Holve S.A., Chronopoulou P., Quinzii C., Johnsen S.D., Kurth J., Eggers E., Palenzuela L., Tanji K., et al. Navajo Neurohepatopathy Is Caused by a Mutation in the MPV17 Gene. Am. J. Hum. Genet. 2006;79:544–548. doi: 10.1086/506913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blakely E.L., Butterworth A., Hadden R.D.M., Bodi I., He L., McFarland R., Taylor R.W. MPV17 mutation causes neuropathy and leukoencephalopathy with multiple mtDNA deletions in muscle. Neuromuscul. Disord. 2012;22:587–591. doi: 10.1016/j.nmd.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garone C., Rubio J.C., Calvo S.E., Naini A., Tanji K., Dimauro S., Mootha V.K., Hirano M. MPV17 Mutations Causing Adult-Onset Multisystemic Disorder With Multiple Mitochondrial DNA Deletions. Arch. Neurol. 2012;69:1648–1651. doi: 10.1001/archneurol.2012.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalla Rosa I., Cámara Y., Durigon R., Moss C.F., Vidoni S., Akman G., Hunt L., Johnson M.A., Grocott S., Wang L., et al. MPV17 Loss Causes Deoxynucleotide Insufficiency and Slow DNA Replication in Mitochondria. PLoS Genet. 2016;12:e1005779. doi: 10.1371/journal.pgen.1005779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alonzo J.R., Venkataraman C., Field M.S., Stover P.J. The mitochondrial inner membrane protein MPV17 prevents uracil accumulation in mitochondrial DNA. J. Biol. Chem. 2018;293:20285–20294. doi: 10.1074/jbc.RA118.004788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Martorano L., Peron M., Laquatra C., Lidron E., Facchinello N., Meneghetti G., Tiso N., Rasola A., Ghezzi D., Argenton F. The zebrafish orthologue of the human hepatocerebral disease gene MPV17 plays pleiotropic roles in mitochondria. Dis. Model. Mech. 2019;12:dmm037226. doi: 10.1242/dmm.037226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dallabona C., Marsano R.M., Arzuffi P., Ghezzi D., Mancini P., Zeviani M., Ferrero I., Donnini C. Sym1, the yeast ortholog of the MPV17 human disease protein, is a stress-induced bioenergetic and morphogenetic mitochondrial modulator. Hum. Mol. Genet. 2010;19:1098–1107. doi: 10.1093/hmg/ddp581. [DOI] [PubMed] [Google Scholar]
- 10.Antonenkov V.D., Isomursu A., Mennerich D., Vapola M.H., Weiher H., Kietzmann T., Hiltunen J.K. The Human Mitochondrial DNA Depletion Syndrome Gene MPV17 Encodes a Non-selective Channel That Modulates Membrane Potential. J. Biol. Chem. 2015;290:13840–13861. doi: 10.1074/jbc.M114.608083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacinto S., Guerreiro P., de Oliveira R.M., Cunha-Oliveira T., Santos M.J., Grazina M., Rego A.C., Outeiro T.F. MPV17 Mutations Are Associated With a Quiescent Energetic Metabolic Profile. Front. Cell. Neurosci. 2021;15:641264. doi: 10.3389/fncel.2021.641264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reinhold R., Krüger V., Meinecke M., Schulz C., Schmidt B., Grunau S.D., Guiard B., Wiedemann N., van der Laan M., Wagner R., et al. The channel-forming Sym1 protein is transported by the TIM23 complex in a presequence-independent manner. Mol. Cell Biol. 2012;32:5009–5021. doi: 10.1128/MCB.00843-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kodani A., Yamaguchi M., Itoh R., Huynh M.A., Yoshida H. A Drosophila model of the neurological symptoms in Mpv17-related diseases. Sci. Rep. 2022;12:22632. doi: 10.1038/s41598-022-27329-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu Y., Flockhart I., Vinayagam A., Bergwitz C., Berger B., Perrimon N., Mohr S.E. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinf. 2011;12:357. doi: 10.1186/1471-2105-12-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Emanuelsson O., Nielsen H., Brunak S., von Heijne G. Predicting subcellular localization of proteins based on their N-terminal amino acid sequence. J. Mol. Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 17.Claros M.G., Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- 18.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hallgren J., Tsirigos K.D., Pedersen M.D., Almagro Armenteros J.J., Marcatili P., Nielsen H., Krogh A., Winther O. DeepTMHMM predicts alpha and beta transmembrane proteins using deep neural networks. Bioinformatics. 2022 doi: 10.1101/2022.04.08.487609. [DOI] [Google Scholar]
- 20.Ferraro P., Pontarin G., Crocco L., Fabris S., Reichard P., Bianchi V. Mitochondrial deoxynucleotide pools in quiescent fibroblasts: a possible model for mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) J. Biol. Chem. 2005;280:24472–24480. doi: 10.1074/jbc.M502869200. [DOI] [PubMed] [Google Scholar]
- 21.Luna-Sanchez M., Benincá C., Cerutti R., Brea-Calvo G., Yeates A., Scorrano L., Zeviani M., Viscomi C. Opa1 Overexpression Protects from Early-Onset Mpv17-/--Related Mouse Kidney Disease. Mol. Ther. 2020;28:1918–1930. doi: 10.1016/j.ymthe.2020.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paggio A., Checchetto V., Campo A., Menabò R., Di Marco G., Di Lisa F., Szabo I., Rizzuto R., De Stefani D. Identification of an ATP-sensitive potassium channel in mitochondria. Nature. 2019;572:609–613. doi: 10.1038/s41586-019-1498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tennessen J.M., Barry W.E., Cox J., Thummel C.S. Methods for studying metabolism in Drosophila. Methods. 2014;68:105–115. doi: 10.1016/j.ymeth.2014.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arrese E.L., Soulages J.L. Insect fat body: energy, metabolism, and regulation. Annu. Rev. Entomol. 2010;55:207–225. doi: 10.1146/annurev-ento-112408-085356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Buch S., Melcher C., Bauer M., Katzenberger J., Pankratz M.J. Opposing Effects of Dietary Protein and Sugar Regulate a Transcriptional Target of Drosophila Insulin-like Peptide Signaling. Cell Metab. 2008;7:321–332. doi: 10.1016/j.cmet.2008.02.012. [DOI] [PubMed] [Google Scholar]
- 26.Szabò I., Bàthori G., Tombola F., Coppola A., Schmehl I., Brini M., Ghazi A., De Pinto V., Zoratti M. Double-stranded DNA can be translocated across a planar membrane containing purified mitochondrial porin. FASEB J. 1998;12:495–502. doi: 10.1096/fasebj.12.6.495. [DOI] [PubMed] [Google Scholar]
- 27.Sperl L.E., Hagn F. NMR Structural and Biophysical Analysis of the Disease-Linked Inner Mitochondrial Membrane Protein MPV17. J. Mol. Biol. 2021;433:167098. doi: 10.1016/j.jmb.2021.167098. [DOI] [PubMed] [Google Scholar]
- 28.Fusté J.M., Wanrooij S., Jemt E., Granycome C.E., Cluett T.J., Shi Y., Atanassova N., Holt I.J., Gustafsson C.M., Falkenberg M. Mitochondrial RNA Polymerase Is Needed for Activation of the Origin of Light-Strand DNA Replication. Mol. Cell. 2010;37:67–78. doi: 10.1016/j.molcel.2009.12.021. [DOI] [PubMed] [Google Scholar]
- 29.Posse V., Al-Behadili A., Uhler J.P., Clausen A.R., Reyes A., Zeviani M., Falkenberg M., Gustafsson C.M. RNase H1 directs origin-specific initiation of DNA replication in human mitochondria. PLoS Genet. 2019;15:e1007781. doi: 10.1371/journal.pgen.1007781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.King M.P., Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y., Guo S., Xie C., Fang J. Uridine Metabolism and Its Role in Glucose, Lipid, and Amino Acid Homeostasis. BioMed Res. Int. 2020;2020:7091718. doi: 10.1155/2020/7091718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Skinner O.S., Blanco-Fernández J., Goodman R.P., Kawakami A., Shen H., Kemény L.V., Joesch-Cohen L., Rees M.G., Roth J.A., Fisher D.E., et al. Salvage of ribose from uridine or RNA supports glycolysis in nutrient-limited conditions. Nat. Metab. 2023;5:765–776. doi: 10.1038/s42255-023-00774-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schneider I. Cell lines derived from late embryonic stages of Drosophila melanogaster. J. Embryol. Exp. Morphol. 1972;27:353–365. [PubMed] [Google Scholar]
- 34.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 35.Broughton S.J., Piper M.D.W., Ikeya T., Bass T.M., Jacobson J., Driege Y., Martinez P., Hafen E., Withers D.J., Leevers S.J., Partridge L. Longer lifespan, altered metabolism, and stress resistance in Drosophila from ablation of cells making insulin-like ligands. Proc. Natl. Acad. Sci. USA. 2005;102:3105–3110. doi: 10.1073/pnas.0405775102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brischigliaro M., Frigo E., Fernandez-Vizarra E., Bernardi P., Viscomi C. Measurement of mitochondrial respiratory chain enzymatic activities in Drosophila melanogaster samples. STAR Protoc. 2022;3:101322. doi: 10.1016/j.xpro.2022.101322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pontarin G., Gallinaro L., Ferraro P., Reichard P., Bianchi V. Origins of mitochondrial thymidine triphosphate: dynamic relations to cytosolic pools. Proc. Natl. Acad. Sci. USA. 2003;100:12159–12164. doi: 10.1073/pnas.1635259100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferraro P., Franzolin E., Pontarin G., Reichard P., Bianchi V. Quantitation of cellular deoxynucleoside triphosphates. Nucleic Acids Res. 2010;38:e85. doi: 10.1093/nar/gkp1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.