

Abstract
A method for the photoinduced evolution of atomic oxygen from pyridazine N-oxides was developed. This underexplored oxygen allotrope mediates arene C─H oxidation within complex, polyfunctional molecules. A water-soluble pyridazine N-oxide was also developed and shown to promote photoinduced DNA cleavage in aqueous solution. Taken together, these studies highlight the utility of pyridazine N-oxides as photoactivatable O(3P) precursors for applications in organic synthesis and chemical biology.
Graphical Abstract
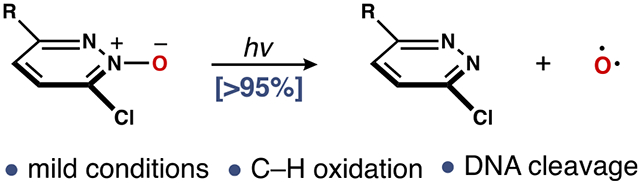
Reactive allotropes of oxygen play a central role in chemistry and biology. Ozone (O3) is the most familiar, given its importance in atmospheric chemistry and as a reagent for chemical synthesis (Figure 1).1 Similarly, singlet oxygen (1O2) and superoxide (O2•−) serve as redox equivalents in vivo.2,3 Compared to these well-studied oxidants, the chemistry of atomic oxygen [O(3P)] remains underexplored, because there are few practical ways to generate this reactive species.4 Certain heterocyclic N-oxides release O(3P) under photochemical conditions, albeit in low yield.5-8 In contrast, the photoinduced deoxygenation of dibenzothiophene S-oxide (DBTO, 1) produces O(3P) via cleavage of the S─O bond.9,10 This photochemistry has been used to interrogate the reactivity of O(3P) with small organic molecules9,11 and biomolecules in solution.12,13 Nevertheless, the utility of 1 as a O(3P) surrogate is hampered by a requirement for short-wavelength light (~300 nm) to promote deoxygenation and the poor solubility profile of 1 in aqueous media. Efforts to address these limitations have relied exclusively on tailored variants of 1 designed to be compatible with lower-energy light and/or more soluble in water (e.g., 2–4).13,14 In this Communication, we describe the photodeoxygenation of 3,6-dichloropyridazine N-oxide (DPNO, 5a), a commercially available heterocycle that can scaffold a new class of O(3P) precursors with improved properties.
Figure 1.

Reactive allotropes of oxygen involved in chemistry and biology.
In their seminal work on the photochemistry of pyridazine N-oxides (6), Buchardt,15 Ogata,16 and Igeta17 identified two primary photoreactions (Scheme 1). The predominant pathway involved photoisomerization of 6 to diazo intermediate 7 (path I). This rearrangement channels though oxazridine A, a transient species that connects to 7 via diazoxepine intermediate B.18 With this mechanism in mind, we previously established that the addition of an electron-donating group at C6 of 6 significantly accelerated photoisomerizaiton to 7. This modification facilitated direct access to functionalized pyrazoles, furans, or indoles from pyridazine N-oxides via controlled secondary reactions of isomer 7.19
Scheme 1. Photochemistry of Pyridazine N-Oxides: Mechanistic Hypotheses and Structure–Function Relationships.

a Photochemistry was carried out at 35 °C in CH2Cl2 (0.1 M) in a Rayonet photoreactor using broadly emitting 24 W 350 nm lamps. b Conversions were determined by 1H NMR spectroscopy using Me2SO2 as an internal standard. cThe remaining yield consisted of products derived from 7. dThe remaining yield was unreacted N-oxide 6. eNonspecific decomposition of 6g was observed.
Photodeoxygenation of 6 to liberate pyridazine 8 with concomitant generation of O(3P) is a competing, albeit inefficient, pathway.17,20 With the goal of developing 6 into a useful O(3P) surrogate, we focused on fate of oxazridine A. Given that photoisomerization is accelerated when a nucleophilic loan pair on X can weaken the C6─N σ-bond,19a we reasoned that changing X to an electron-withdrawing group might prolong the lifetime of A, thereby allowing entry to C via homolysis of the N─O bond (path II).21 While other mechanistic pathways for deoxygenation were considered, this thinking provided a framework to explore structure–function relationships.22 To this end, we prepared a series of pyridazine N-oxides with modifications at C3 and C6 and determined the extent of their photodeoxygentaion in CH2Cl2 using 350 nm UV-A light.23 As predicted, the nature of X proved to be critical for partitioning between the isomerization and deoxygenation pathways. Thus, whereas 6a (X = NHBn) and 6b (X = OBn) rearranged to 7, we observed partial deoxygenation of 6c (X = Ph).15 In contrast, deoxygenation was the only photoreaction when a chlorine atom was added at C6 (X = Cl). As noted by comparing 6e to 6f, the rate of this process was modified by the substituent at C3. However, the optimal result was obtained using 5a (R, X = Cl), which cleanly deoxygenated to give 3,6-dichloropyridazine (8a) in 98% yield after 3.5 h.
The deoxygenation of 5a was subsequently examined using benzene as an oxygen-atom acceptor, a reaction mediated by O(3P) in the gas phase or generated in solution using S-oxide 1 (Table 1).9,24 Following an optimization,25 we found that the photolysis of 5a (hv = 350 nm) in the mixture of C6H6/CH2Cl2 (1:1 v/v) provided 9 in 47% yield (entry 1). In general, reactions were carried out under air-free conditions in anhydrous solvent to establish 5a as the only oxygen-atom source. Nevertheless, we obtained similar results when no precautions were taken to exclude air (entry 2). In contrast, the addition of 9 (1 equiv) to the reaction reduced the efficiency of oxygen-atom transfer to 28% (entry 3). This observation suggests that the O─H bond in 9 (BDE = 87 kcal/mol)26 inhibits oxygen-atom transfer, possibly by reacting with the oxidant released from 5a.27 Importantly, N-oxide 5a was compatible with lamps operating in the range of 254–430 nm. As such, 5a underwent full deoxygenation (>95%) within 3.5 h using broadly emitting 254, 300, and 350 nm UV lamps; however, the best yield of 9 was achieved with the 350 nm light source (entries 1, 4, and 5).23 Notably, we obtained a comparable result with visible light (hv = 430 nm, blue LED) after 24 h of irradiation (entries 6 and 7). Modifications to 5a provided no significant improvement. For example, bromine derivative 5b gave 9 in 50% yield (entry 8), a result analogous to 5a when considering standard experimental error. In contrast, substituted N-oxides 5c and 5d were comparatively inferior to 5a (entries 9 and 10).28 The same was true for S-oxide 1, which was incompatible with our 350 nm lamps (entry 11). In this case, a 300 nm light source was required. Under these modified conditions, 1 provided 9 in 12% yield after 24 h of irradiation (entry 12).
Table 1.

| |||
|---|---|---|---|
| entry | deviation from optimal conditions | 8 (%) | 9 (%) |
| 1 | none | >95 | 47 |
| 2 | under ambient atmospherec | >95 | 44 |
| 3 | 1 equiv of 9 as additive | >95 | 28 |
| 4 | hv = 254 nm lamp | >95 | 18 |
| 5 | hv = 300 nm lamp | >95 | 36 |
| 6 | hv = 430 nm (blue LED) | 38 | 19 |
| 7 | hv = 430 nm (blue LED), 24 h | 92 | 39 |
| 8 | 5b in place of 5a | >95 | 50 |
| 9 | 5c in place of 5a | 70 | 22 |
| 10 | 5d in place of 5a | 39 | 0 |
| 11 | 1 in place of 5a | n/ad | 0 |
| 12 | 1 in place of 5a, hv = 300 nm, 24 h | n/ae | 12 |
Yields of 8 and 9 were determined by 1H NMR spectroscopy using Me2SO2 as an internal standard and were calculated with respect to N-oxide 5. Reported values are the average of three independent experiments (error = ±5%).
Reactions were performed under air-free conditions.
No precautions were taken to exclude air from the reaction.
~2% deoxygenation of 1 was observed.
>95% deoxygenation of 1 was observed.
The ability of 5a to mediate alkane oxidation was examined by replacing benzene with cyclooctane (Scheme 2). Consistent with a C(sp3)─H abstraction mechanism, these conditions generated cyclooctanol (11) in 12% yield alongside 6% yield of alkylated pyridazine 12. However, a competition experiment using a 1:1 mixture of benzene (BDE = 113 kcal/mol) and cyclooctane (BDE = 96 kcal/mol) indicated selective oxidation of benzene (9:11 = ~2:1). This result mirrors the reactivity of O(3P), which has been shown to oxidize benzene and cycloalkanes with similar rates in solution, albeit via different mechanisms.7a,9,24 It should be noted that the short lifetime of O(3P) complicates detection of this species and may contribute to the low yields observed here.29 To resolve this issue, we carried out common intermediate experiments to compare 5a with known O(3P) surrogate 1. We initially examined the oxidation of 4-chlorotoluene (13) as a test reaction, which was expected to generate a mixture of regioisomers 14a and 14b. To ensure a direct comparison was made, we utilized a 300 nm light source shown to be compatible with both heterocycles (Table 1). Following this modification, deoxygenation of 1 and 5a afforded comparable mixtures of 14 (14a/14b ≈ 1.1:1) in 4 and 13% yields, respectively.30 Inspired by the reported selectivity of O(3P) for thiols, we also examined the oxidation of cysteine analogue 15 to disulfide 16.11 As expected, 1 and 5a gave similar yields of 16 (43% for 1; 38% for 5a) after 24 h of irradiation in the presence of 15.31 Notably, 16 was not observed in the absence of 1 or 5a. Taken together, these results indicate that the same reactive oxygen species is released from 1 and 5a.
Scheme 2. Control Experimentsa,b.

a Yields were determined by 1H NMR spectroscopy using Me2SO2 as an internal standard and were calculated with respect to oxidant (i.e., 1 or 5a). bReactions were carried out under air-free conditions. cYield achieved under optimal conditions for 5a (hv = 350 nm, 3.5 h).
Oxygen-atom transfer from 5a to a series of other acceptors (17, 5 equiv) was explored using 350 nm UV-light in degassed CH2Cl2 (Scheme 3). In each case, O(3P) evolved from 5a was captured with modest efficiency to give oxidized products 18. Electron-rich arenes (17a–d) were better acceptors than deactivated arenes (17e,f), an outcome consistent with the electrophilic nature of O(3P).9 More intriguingly, we observed complete selectivity for the oxidation aryl C─H bonds. For example, the aryl ring of esterone analogue 17g was oxidized to 18g (C2:C4 = 1.3:1) in 19% yield, and tyrosine derivative 17h was converted to l-DOPA analog 18h in 34% yield. In addition, 5a mediated the oxidation of N-Boc-Met-OMe (17i) to sulfoxide 18i in 36% yield. This reactivity draws analogy to the direct epoxidation of arenes by cytochrome P450 monooxygenases.32,33 In support of this view, photolysis of 5a in the presence of N-Boc-Trp-OMe (17j) gave oxindolylalinine analogue 18j (25% yield) alongside traces of kynurenine derivative 19 (1–2% yield). Notably, the same products are involved in tryptophan metabolism by P450 enzymes.34 Thus, in this setting, 5a behaves as a monooxygenase equivalent.
Scheme 3. Survey of Small Molecule Oxidation Mediated by the Photolysis of 5aa-c.

a Yields were calculated with respect to 5a and represent isolated yield after purification by silica gel chromatography. b Product ratios were established by 1H NMR spectroscopy of unpurified reaction mixtures. c Except where noted, reactions were carried out under air-free conditions. d Yield of an identical reaction carried out under aerobic conditions.
The ability of 5a to recapitulate oxidative post-translational modifications of amino acid derivatives inspired us to remodel this chemotype for use in aqueous media (Scheme 4).35 Having established that a chlorine atom at C6 was needed for effective deoxygenation, we carried out a regioselective cross-coupling of 5a with potassium vinyltetrafluoroborate. In line with our previous investigation, 20 was formed as a single isomer in 81% yield when dppf was used as a ligand.19a Oxidative cleavage of the pendent alkene followed by reductive amination of the resultant aldehyde gave morpholine adduct 21 in 46% yield. Exposure of 21 to aq. HCl subsequently provided salt 22 as a water-soluble (solubility = 430 mg/mL, 23 °C) solid in excellent yield.
Scheme 4. Design and Synthesis of Water-Soluble Pyridazine N-Oxide 22.

As depicted in Scheme 4, we compared the photochemical properties of 22 with those of 1 and 5a. The UV–vis spectra of these heterocycles were collected in a mixture of MeCN/H2O (6:1). Analysis of these data revealed absorption in the range of 315–360 nm for 5a and 22 that was significant relative to S-oxide 1. This feature corresponds to the n → π* transition for pyridazine N-oxides36 and accounts for the improved compatibility of these N-heterocycles with lower-energy light relative to 1. Likewise, the efficiency of photodeoxygenation for each structure was evaluated by measuring the quantum yield of oxide disappearance (ϕdeox). In our hands, the quantum yield for 1 was 2.4%, which is consistent with reported values.37 In comparison, the ϕdeox values for N-oxides 5a and 22 were found to be 22.1 and 15.2%, respectively. These values are superior to 1, with 5a performing at ~ 10-fold the efficiency of the S-oxide.
To illustrate the potential utility of pyridazine N-oxide photodeoxygentaion in biological settings, we explored the ability of 22 to cleave plasmid DNA under aerobic conditions in water.12a As outlined in Figure 2, photoinduced DNA cleavage by 22 was monitored using gel electrophoresis by measuring the conversion of supercoiled pcDNA 3.1 plasmid DNA (form I) to linear DNA (form II) arising from double-strand breaks or nicked circular DNA (form III) generated from a single-strand break. In these experiments, aqueous solutions of plasmid (40 ng·μL−1) and 22 (1.0 mM) were irradiated at 35 °C with fluorescent bulbs centered at 350 nm in sealed Pyrex vials. Under conditions of ambient oxygenation, significant DNA cleavage was observed after 4 min (lane 4) to generate predominately nicked DNA (form III). The reaction was essentially complete after 32 min (lane 7) with both nicked (85%) and linear DNA (form II, 12%) present. This time-dependent increase in strand scission is typical for DNA cleavage agents.39 Importantly, in all of the reported experiments, approximately 25% of form III DNA was present prior to photolysis (lane 2). Additionally, a small, time-dependent increase of nicked DNA was observed in control samples irradiated in the absence of 22 (≤10%, lanes 8 and 9). Nevertheless, these data clearly show that photolysis of 22 mediates DNA strand cleavage. We observed no significant difference in replicate experiments carried out under anaerobic conditions. Similarly, no change in plasmid DNA was noted in control reactions with 22 protected from light. The rate of photoinduced DNA cleavage was also dependent on the concentration 22. As such, results similar to those in Figure 2 were obtained using 0.50 or 0.25 mM solutions of 22, albeit with extended reaction times required for complete consumption of plasmid DNA.25
Figure 2.

Photocleavage of plasmid DNA by 22 (1.0 mM) in H2O under aerobic conditions: DNA cleavage reactions used 40.0 ng of supercoiled plasmid DNA (pcDNA 3.1) in H2O (50 μL). Photolysis was carried out in Pyrex vials at 35 °C using 350 nm light. aReported values reflect the average of three independent experiments (standard error was ±4%). bLane 1 and lane 10 show the DNA ladder. cSupercoiled plasmid DNA. dLinearized DNA, confirmed by comparison to EcoRI digestion of plasmid DNA shown in lane 3. eNicked DNA. fS = mean number of strand breaks per plasmid molecule calculated using the equation: S = −ln(% form I DNA).38
In conclusion, we have shown that pyrdaizne N-oxides can serve as useful photoactivated oxidants. The incorporation of a chlorine atom at C6 of the pyridazine nucleus was found to be essential to the efficiency of this photochemistry. In analogy to S-oxide 1, the reactive oxygen species evolved from 5a is likely to be O(3P) or an excited-state species that mimics the reactivity profile of O(3P) in solution. Additional mechanistic work is needed to confirm freely diffusing O(3P) as the terminal oxidant. However, in contrast to S-oxide 1, N-oxide 5a allows for efficient generation of this oxidant with standard light sources operating in the range of 350–430 nm. As exemplified by hydrochloride salt 22, the properties of 5a can be tailored for applications with biomolecules in solution. These features provide enthusiasm for the continued development of heterocyclic N-oxides as O(3P) precursors for applications in organic synthesis and chemical biology.
Supplementary Material
ACKNOWLEDGMENTS
This work was support by Florida State University (FSU), and in part, by the National Institutes of Health (R01-GM125926). We thank Dr. Jack Saltiel (FSU) and Dr. Brian Miller (FSU) for their assistance with quantum yield measurements and DNA cleavage experiments, respectively. We also thank Dr. Xinsong Lin (FSU) for his assistance with X-ray crystallography.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.2c00227.
Reaction optimization, experimental procedures, and characterization data for all new compounds (PDF)
Accession Codes
CCDC 2112757 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Bailey PS The reactions of ozone with organic compounds. Chem. Rev 1958, 58, 925–1010. [Google Scholar]; (b) Lerner RA; Eschenmoser A Ozone in biology. Proc. Natl. Acad. Sci, USA 2003, 100, 3013–3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ogilby PR Singlet oxygen: there is indeed something new under the sun. Chem. Soc. Rev 2010, 39, 3181. [DOI] [PubMed] [Google Scholar]
- (3).Dickinson BC; Chang CJ Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Bio 2011, 7, 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Thomas KB; Greer A Gauging the significance of atomic oxygen [O(3P)] in sulfoxide photochemistry. A method for hydrocarbon oxidation. J. Org. Chem 2003, 68, 1886–1891. [DOI] [PubMed] [Google Scholar]
- (5).(a) Albini A; Alpegiani M Photochemistry of the N-oxide function. Chem. Rev 1984, 84, 43–71. [Google Scholar]; (b) Petrosyan A; Hauptmann R; Pospech J Heteroarene N-oxides as oxygen source in organic reactions. Eur. J. Org. Chem 2018, 2018, 5237–5252. [Google Scholar]
- (6).(a) For select examples, see: Hata N; Ono I; Kawasaki M The photoinduced deoxygenation reaction of heterocyclic N-oxides. Chem. Lett 1975, 4, 25–28. [Google Scholar]; (b) Albini A; Fasani E; Frattini V Photochemistry of 2- and 4-benzoylpyridine N-oxides. J. Photochem 1987, 37, 355–361. [Google Scholar]; (c) Xu HJ; Lin SQ; Shen SY; Li L; Chen DW; Xu GZ Radical mechanism in the photoinduced oxygen transfer from acridine N-oxides to cyclohexene. J. Photochem. Photobiol A 1989, 48, 53–59. [Google Scholar]; (d) Solntsev KM; Clower CE; Tolbert LM; Huppert D 6-Hydroxyquinoline-N-oxides: A new class of “super” photoacids. J. Am. Chem. Soc 2005, 127, 8534–8544. [DOI] [PubMed] [Google Scholar]
- (7).(a) The photodeoxygenation of pyridine N-oxides has been described: Bucher G; Scaiano JC Laser flash photolysis of pyridine N-oxide: Kinetic studies of atomic oxygen [O(3P)] in solution. J. Phys. Chem 1994, 98, 12471–12473. [Google Scholar]; (b) Carraher JM; Bakac A Generation of free oxygen atoms O(3P) in solution by photolysis of 4-benzoylpyridine N-oxide. Phys. Chem. Chem. Phys 2014, 16, 19429–19436. [DOI] [PubMed] [Google Scholar]
- (8).(a) In contrast, the photodeoxygention of pyrimidopteridine N-oxides proceeds via a charge-transfer complex and does not involve O(3P): Sako M; Shimada K; Hirota K; Maki Y Photochemical oxygen-atom transfer reaction by heterocycle N-oxides involving single-electron transfer process: Oxidative demethylation of N. N-dimethylaniline. J. Am. Chem. Soc 1986, 108, 6039–6041. [DOI] [PubMed] [Google Scholar]; (b) Sako M; Ohara S; Shimada K; Hirota K; Maki Y Photo-oxygenation of benzene derivatives by a novel derivative of the heterocyclic N-oxide, pyrimido[5,4g]pteridine 5-oxide, involving a single-electron-transfer process. J. Chem. Soc., Perkin Trans 1 1990, 863. [Google Scholar]
- (9).(a) Wan Z; Jenks WS Oxenoid reactivity observed on the photolysis of certain aromatic sulfoxides. J. Am. Chem. Soc 1995, 117, 2667–2668. [Google Scholar]; (b) Gregory DD; Wan Z; Jenks WS Photodeoxygenation of dibenzothiophene sulfoxide: Evidence for a unimolecular S─O cleavage mechanism. J. Am. Chem. Soc 1997, 119, 94–102. [Google Scholar]
- (10).Omlid SM; Dergunov SA; Isor A; Sulkowski KL; Petroff JT II; Pinkhassik E; McCulla RD Evidence for diffusing atomic oxygen uncovered by separating reactants with semi-permeable nanocapsule barrier. Chem. Commun 2019, 55, 1706–1709. [DOI] [PubMed] [Google Scholar]
- (11).Omlid SM; Zhang M; Isor A; McCulla RD Thiol reactivity toward atomic oxygen generated during the photodeoxygentaion of dibenzothiophene S-oxide. J. Org. Chem 2017, 82, 13333–13341. [DOI] [PubMed] [Google Scholar]
- (12).(a) Wauchope OR; Shakya S; Sawwan N; Liebman JF; Greer A Photocleavage of plasmid DNA by dibenzothiophene S-oxide under anaerobic conditions. J. Sulfur Chem 2007, 28, 11–16. [Google Scholar]; (b) Zhang M; Ravilious GE; Hicks LM; Jez JM; McCulla RD Redox switching of adenosine-5′-phosphosulfate kinase with photoactivatable atomic oxygen precursors. J. Am. Chem. Soc 2012, 134, 16979–16982. [DOI] [PubMed] [Google Scholar]; (c) Bourdillon MT; Ford BA; Knulty AT; Gray CN; Zhang M; Ford DA; McCulla RD In vitro oxidations of plasmalogen, low-density lipoprotein and RAW 264.7 cells by photoactivatable atomic oxygen precursors. Photochem. Photobiol 2014, 90, 386–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Isor A; O’Dea AT; Grady SF; Petroff JT II; Skubic KN; Aziz B; Arnatt CK; McCulla RD Effects of photodeoxygentaion on cell biology using dibenzothiophene S-oxide derivatives as O(3P)-precursors. Photochem. Photobiol. Sci 2021, 20, 1621–1633. [DOI] [PubMed] [Google Scholar]
- (14).(a) For select examples, see: Lucien E; Greer A Electrophilic oxidant produced in the photodeoxygenation of 1,2-benzodiphenylene sulfoxide. J. Org. Chem 2001, 66, 4576–4579. [DOI] [PubMed] [Google Scholar]; (b) McCulla RD; Jenks WS Deoxygenation and other photochemical reactions of aromatic selenoxides. J. Am. Chem. Soc 2004, 126, 16058–16065. [DOI] [PubMed] [Google Scholar]; (c) Rockafellow EM; McCulla RD; Jenks WS Deoxygenation of dibenzothiophene-S-oxide and dibenzoselenophene-Se-oxide: A comparison of direct and sensitized photolysis. J. Photochem. Photobiolol., A 2008, 198, 45–51. [Google Scholar]; (d) Korang J; Grither WR; McCulla RD Photodeoxygenation of dibenzothiophene S-oxide derivatives in aqueous media. J. Am. Chem. Soc 2010, 132, 4466–4476. [DOI] [PubMed] [Google Scholar]; (e) Isor A; Chartier BV; Abo M; Currens ER; Weerapana E; McCulla RD Identifying cysteine residues susceptible to oxidation by photoactivatable atomic oxygen precursors using a proteome-wide analysis. RSC Chem. Biol 2021, 2, 577–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Kumler PL; Buchardt O Photochemical studies. XIV. The photolysis of 3,6-diphenylpyridazine N-oxide. Detection of a transient diazo compound. J. Am. Chem. Soc 1968, 90, 5640–5641. [Google Scholar]
- (16).Ogata M; Kano K A photochemical reaction of pyridazine N-oxides. Chem. Commun 1967, 1176–1177. [Google Scholar]
- (17).Igeta H; Tsuchiya T; Yamada M; Arai H Photoinduced oxygenation of hydrocarbons by pyridazine N-oxides. Chem. Pharm. Bull 1968, 16, 767–769. [Google Scholar]
- (18).Ma J; Wagner BD; Li M-D; Lei Y; Phillips DL; Bucher G Detection and identification of reaction intermediates in the photorearrangement of pyridazine N-oxide: Discrepancies between experiment and theory. J. Org. Chem 2019, 84, 10032–10039. [DOI] [PubMed] [Google Scholar]
- (19).(a) Portillo M; Maxwell MA; Frederich JH Synthesis of nitrogen heterocycles via photochemical ring opening of pyridazine N-oxides. Org. Lett 2016, 18, 5142–5145. [DOI] [PubMed] [Google Scholar]; (b) Borger M; Frederich JH Pyridazine N-oxides as precursors to 2-aminofurans: Scope and limitations in complexity building cascade reactions. Org. Lett 2019, 21, 2397–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Borger M; Callen DP; Frederich JH A photoinduced cascade for the synthesis of 1H-indole-2-acetamide derivatives. Asian J. Org. Chem 2019, 8, 1111–1114. [Google Scholar]
- (20).(a) Tsuchiya T; Arai H; Igeta H Photoinduced oxygenation by pyridazine N-oxides. II. Formation of epoxides from ethylenic compounds. Tetrahedron Lett. 1969, 10, 2747–2750. [DOI] [PubMed] [Google Scholar]; (b) Tsuchiya T; Arai H; Igeta H Photoinduced oxygenation by pyridazine N-oxides. III. Oxygenation of polymethylbenzenes. Tetrahedron Lett. 1970, 11, 2213–2216. [Google Scholar]
- (21).N─O bond homolysis has been proposed for oxazridines analogous to A generated from the photolysis of phenanthridine N-oxides: Taylor ED; Spence GG Further studies on the photochemical reactions of 6-substituted phenanthridine 5-oxides. Chem. Commun 1968, 1037–1038. [Google Scholar]
- (22).For example, direct cleavage of the N─O bond from an excited-state species cannot be ruled out.
- (23).For most of the reactions described herein, we used broadly emitting 24 W UV lamps (325–375 nm), where ~90% of emitted light was 350 nm (RPR-3500A, hv = 350 nm). More details regarding the light sources used in this study are available in Table S1 of the Supporting Information.
- (24).Taatjes CA; Osborn DL; Selby TM; Meloni G; Trevitt AJ; Epifanovsky E; Krylov AI; Sirjean B; Dames E; Wang H Products of the benzene + O(3P) reaction. J. Phys. Chem. A 2010, 114, 3355–3370. [DOI] [PubMed] [Google Scholar]
- (25).See the Supporting Information for details.
- (26).Mulder P; Korth H-G; Pratt DA; DiLabio GA; Valgimigli L; Pedulli GF; Ingold KU Critical re-evaluation of the O─H bond dissociation enthalpy in phenol. J. Phys. Chem. A 2005, 109, 2647–2655. [DOI] [PubMed] [Google Scholar]
- (27).There was no evidence of phenylhydroperoxide in the unpurified reaction mixture; however, it is reasonable to expect that this species would be labile. We also did not observe formation of dihydroxybenzene isomers (e.g., catechol).
- (28).UV–vis absorption spectra for 5b–5d were collected and compared with 5a. These data can be found in the Supporting Information.
- (29).The lifetime of O(3P) in the gas phase has been estimated at ~30 ns. See reference 10 for a discussion of the challenges of surrounding direct detection of O(3P) in solution.
- (30).Traces of 4-chlorobenzaldehyde were also detected using both 1 and 5a.
- (31).Photodeoxygenation of 5a was complete after 3.5 h under these modified conditions. Notably, disulfide 16 was produced in 51% yield when 5a was irradiated using 350 nm lamps for 3.5 h.
- (32).For a review on this topic, see: Goodwin BL In Handbook of biotransformations of aromatic compounds; CRC Press: Boca Raton, FL, 2004; pp. 109–124. [Google Scholar]
- (33).As discussed in reference 22, benzene oxide is an intermediate in the production of phenol from benzene and O(3P) in the gas phase.
- (34).We suspect that 18j arises from rearrangement of the corresponding 2,3-indole oxide intermediate. This same species is implicated in the biocatalytic tryptophan oxidation: Mondal P; Wijeratne GB Modeling tryptophan/indoleamine 2,3-dioxygenase with heme superoxide mimetics: Is ferryl the key intermediate? J. Am. Chem. Soc 2020, 142, 1846–1856. [DOI] [PubMed] [Google Scholar]
- (35).For an overview of this topic, see: Ryan BJ; Nissim A; Winyard PG Oxidative post-translational modifications and their involvement in the pathogenesis of autoimmune diseases. Redox Biol. 2014, 2, 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Tomer KB; Harrit N; Rosenthal I; Buchardt O; Kumler P; Creed D Photochemical behavior of aromatic 1,2-diazine N-oxides. J. Am. Chem. Soc 1973, 95, 7402–7406. [Google Scholar]
- (37).Zheng X; Baumann SM; Chintala SM; Galloway KD; Slaughter JB; McCulla RD Photodeoxygenation of dinaphthothiophene, benzophenanthrothiophene, and benzonaphthothiophene S-oxides. Photochem. Photobiol. Sci 2016, 15, 791–800. [DOI] [PubMed] [Google Scholar]
- (38).Hertzberg RP; Dervan PB Cleavge of DNA with methidiumpropyl-EDTA-iron(II): reaction conditions and product analyses. Biochemistry 1984, 23, 3934–3945. [DOI] [PubMed] [Google Scholar]
- (39).(a) Grant KB; Terry CA; Gude L; Fernandez M-J; Lorente A Synthesis and DNA photoclevage by a pyridine-linked chromophore in the presence of copper(II): Ionic strength effects. Bioorg. Med. Chem. Lett 2011, 21, 1047–1051. [DOI] [PubMed] [Google Scholar]; (b) Gonzalez MM; Vignoni M; Pellon-Maison M; Ales-Gandolfo M; Gonzalez-Baro MR; Erra-Balsells R; Epe B; Cabrerizo FM DNA damage photo-induced by chloroharmine isomers: hydrolysis versus oxidation of nucelobases. Org. Biomol. Chem 2012, 10, 1807–1819.22249177 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.