

Abstract
The quinoline-3-carboxamide, Tasquinimod, is orally active as a maintenance therapy with an on-target mechanism-of-action via allosteric binding to HDAC4. This prevents formation of the HDAC4/NCoR1/HDAC3 complex, disrupting HIF-1α transcriptional activation and repressing MEF-2 target genes needed for adaptive survival signaling in the compromised tumor microenvironment. In phase 3 clinical testing against metastatic castration-resistant prostate cancer (mCRPC), Tasquinimod (1 mg/day) increased time-to-progression, but not overall survival. Here, we document that, on this regimen, blood levels are 10-fold lower than the optimal concentration (≥2 μM) needed for anti-cancer activity, suggesting higher daily doses are needed. Unfortunately, we also demonstrate that Tasquinimod is an aryl hydrocarbon receptor (AHR) agonist, which binds with an EC50 of 1 μM to produce unwanted off-target side effects. Therefore, we screened a library of Tasquinimod analogs to maximize on-target vs. off-target activity. Using this approach, we identified ESATA-20, which has ~10-fold lower AHR agonism and >5-fold greater potency against prostate cancer patient-derived xenografts. This increased therapeutic index nominates ESATA-20 as a lead candidate for clinical development as an orally active 3rd generation quinoline-3-carboxamide analog that retains its on-target ability to disrupt HDAC4/HIF-1α/MEF-2-dependent adaptive survival signaling in the compromised TME found in mCRPC.
Introduction
Linomide (a.k.a., Roquinimex), Figure 1A, is a first generation oral quinoline-3-carboxamide with robust documented efficacy against solid malignances, particularly metastatic castration-resistant prostate cancer (mCRPC), as a maintenance therapy in preclinical models via its anti-angiogenic, immunomodulatory, and anti-metastatic properties1–7. Linomide’s clinical development was halted after phase III trials using chronic doses of ≤0.1 mg/kg resulted in several cases of pericarditis and neuropathy8. This side effect profile is consistent with a Linomide-induced systemic pro-inflammatory response9. To overcome this dose-limiting side effect profile, the therapeutic index of additional quinoline-3-carboxamides were evaluated9.
Figure 1: Tasquinimod (TasQ) is an Aryl Hydrocarbon Receptor (AHR) agonist.

Chemical structure of A, Linomide [Roquinimex (INN); 4-hydroxy-N,1-dimethyl-2-oxo-N-phenyl-1,2-dihydroquinoline-3-carboxamide], and B, Tasquinimod [a.k.a., ABR-215050 (INN); 4-hydroxy-5-methoxy-N,1-dimethyl-2-oxo-N-[(4-trifluoromethyl) phenyl]-1,2-dihydroquinoline-3-caboxamide]. C, Left side Y-axis: Dose-response inhibition of in vivo growth of CWR22-RH human prostate cancer PDXs in castrate male NSG mice (n = 5/group) over a 3-week treatment period with the indicated oral daily dose of TasQ initiated when xenografts were ~0.2 cc. Right side Y-axis: TasQ serum Cmax level at indicated dose. D, Summary of % PAL AHR binding inhibited by indicated concentration of TasQ. E, Real Time (RT)-PCR analysis of CYP1A1 mRNA level in LNCaP cells (n = 3) treated for 12 hrs with increasing concentrations of TasQ. F, Western immunoblot (IB) analysis of the cytoplasmic (C) vs. nuclear (N) level of AHR protein in LNCaP human prostate cancer cells treated for 30 minutes with increasing TasQ concentrations. 100,000 cells loaded/lane. G, Time course of change in the cytoplasmic (C) vs. nuclear (N) level of AHR protein in LNCaP cells treated with TasQ (10 μM). H, Relative expression of mouse Liver Cyp1A1 (mCYP1A1) in animals (n = 3) 24 hrs after receiving indicated oral dose of TasQ normalized to animals receiving only vehicle. 100,000 cells loaded/lane.
From these studies, Tasquinimod (TasQ), Figure 1B, was identified as a second-generation oral quinoline-3-carboxamide with increased potency as a maintenance therapy in anti-angiogenic, tumor growth, and metastasis assays9–18. PK analysis in these preclinical studies determined that maximal therapeutic activity requires daily oral dosing of ≥5 mg/kg to maintain serum TasQ concentrations at >2 μM, which is consistent with TasQ being ≥100-fold more potent based on serum drug level compared to Linomide16. In phase I clinical development, oral dose escalation over 4–6 weeks improved tolerability so that patients could be maintained on TasQ at a maximum tolerated dose (MTD) of 1 mg/day [0.014 mg/kg/d], which maintains a steady-state plasma level of 0.42 +/− 0.05 μM19. The dose-limiting toxicity was sinus tachycardia and asymptomatic elevation in amylase19.
Following this dose-finding study, a randomized phase II trial in mCRPC patients progressing on Androgen Deprivation Therapy (ADT) demonstrated that TasQ improved the median progression-free survival (PFS) by 4.3 months (p < 0.01)20,21. In an independent randomized, double-blind, placebo-controlled phase II trial of mCRPC patients progressing on maintenance ADT but responsive to or stabilized during first-line docetaxel chemotherapy, the median radiologic PFS (rPFS) increased from 22.7 weeks to 31.7 weeks in the oral 1 mg/day TasQ maintenance arm [p = 0.0162]22. In a phase III trial to confirm these findings, daily oral TasQ significantly reduced the risk of radiographic progression or death vs. placebo by 36% in mCRPC patients progressing on maintenance ADT. However, at this 1 mg/day dose, overall survival (OS) as the secondary endpoint was not significantly enhanced23.
In preclinical xenograft studies, TasQ synergizes with both ADT and docetaxel11. Therefore, mCRPC patients progressing despite initial docetaxel therapy were treated in a phase 1B trial with ADT maintenance plus a combination of TasQ and cabazitaxel24. Due to side effects, when combined with cabazitaxel and prednisone, the MTD of TasQ had to be lowered to 0.5 mg/d24, a regimen that maintains a plasma concentration of only 0.23 +/− 0.03 μM19. Based upon RECIST criteria, 3 patients (12%) had a complete or partial response (CR/PR) and 12 (48%) had stable disease24. While encouraging, the combined human trial results using an oral daily maintenance dose of 0.5–1.0 mg of TasQ were not of a sufficient therapeutic magnitude for clinical approval in mCRPC patients24. An explanation for this limitation is that in preclinical xenograft studies, a plasma Cmax of >10 μM is required for optimal anti-cancer activity10–16. In mice, this translates into a requirement for daily oral TasQ dosing of >5 mg/kg/d10,16. Unfortunately, in clinical trials increasing oral daily dosing above 0.1 mg/kg as needed to raise the blood Cmax >1 uM produces unacceptable host toxicity25.
Linomide and its quinoline-3-carboxamide analog, Laquinimod, Supplemental Figure 1A, are Aryl Hydrocarbon Receptor (AHR) agonists, which induce downstream target gene effects [i.e., increased expression of phase I activating enzymes (e.g., CYP1A1), thymic regression, and immune suppression via upregulation of regulatory T-cells (T-regs)]3,26–28. This raised the issue of whether TasQ is also an agonistic AHR ligand. AHR agonists, even those that are non-mutagenic like β-naphthoflavone (BNF), are known to cause cardiomyopathies29. Such cardiomyopathies are mediated via mitochondrial dysfunction, including reactive oxygen species (ROS) production, in a Cyp1a1/1a2-dependent manner29–31. If TasQ is also an AHR agonist, this provides a mechanism for the observed cardiotoxicity observed during clinical development that limited dose escalation into optimal therapeutic ranges19. TasQ also binds to S100A9 produced by tumor-infiltrating neutrophils and macrophages, inhibiting its interaction with pro-inflammatory receptors, including Toll-like receptor 4 (TLR4) and receptor of advanced glycation end products (RAGE)17,32. In addition, TasQ is a low nM allosteric inhibitor of HDAC4, an interaction that inhibits endothelial cell sprouting and thus tumor angiogenesis, enhancing hypoxia while inhibiting transcriptional adaptive survival signaling within the compromised tumor microenvironment (TME)10,13,15,16,18. This begs the question of whether TasQ’s anti-cancer on-target mechanism of action (MoA) requires binding to either AHR or S100A9, or whether this therapeutic effect is solely due to HDAC4 inhibition. If the latter is correct, then a 3rd generation quinoline-3-carboxamide analog that retains on-target HDAC4 inhibition while decreasing off-target AHR binding and side effects is possible. Thus, allowing higher daily oral maintenance dosing to enhance therapeutic efficacy in humans.
Methods
Reagents
Linomide, Tasquinimod [TasQ] and ESATA-4 and −5 were provided by Active Biotech Research AB (Lund, Sweden); Trichostatin-A (Sigma-Aldrich); trifluoromethy-acetylysine7-amino-methylcoumarin (aka Lys(Tfa)-AMC) [i.e., HDAC4 specific substrate; Bachem Inc.] AHR: rabbit polyclonal Ab (Cell Signaling, Cat# 13790); HDAC4: rabbit polyclonal Ab (Active Motif, Cat # 40969) Flag: clone M2: mouse monoclonal Ab (Sigma-Aldrich, Cat # F3165); HIF-1α(H-206): rabbit polyclonal Ab [Santa Cruz, Cat # sc-10790 (IP)]; HIF-1α: mouse monoclonal Ab [BD Transduction Laboratories, Cat # 610958 (IB)]; HIF-1α(H-206): rabbit polyclonal [Bethyl Laboratories, Cat # A300–286A (IB)]; NCoR1: rabbit polyclonal Ab (Bethyl Laboratories, Cat # A301–146A); MEF-2: mouse monoclonal Ab (Santa Cruz Cat # SC-17785). Full-length (fl) [A.A. 1–1082] recombinant human histone deacetylase 4 (rhHDAC4) protein as either: wild-type or C669/H675-double mutant, or R681A/R798A double mutant protein with or without an N-terminal GST-tag was produce in HEK-293-T cells based upon a pcDNA vector containing human N-terminal flag-tag fl wild-type histone deacetylase 4 (HDAC4) obtained from Addgene (Cat # 13821) as described previously (18). N-terminal GST-tagged truncated recombinant HDAC4 protein (A.A. 614–1084) was obtained from Abcam, Cambridge, UK (cat number ab104029). Recombinant fl-human nuclear receptor co-repressor-1(NCoR1) protein (N-terminal FLAG-tagged; a.a. 1–2440) was purchased from Abcam, Cambridge, UK (cat. no. ab82239).
Chemical Synthesis
All solvents and reagents used were bought from commercial sources and used without further purification. The 1H and 13C NMR spectra were obtained on a Bruker Avance III 500 MHz NMR spectrometer at 500 MHz and 125 MHz, respectively in deuterated chloroform (CDCl3), deuterated methanol (CD3OD) or deuterated dimethylsulfoxide (C2D6OS). MALDI-Mass spectra were obtained on Voyager DE-STR MALDI-TOF. Analytical thin-layer chromatography was performed using 0.25 mm precoated silica gel 60 F254 plates (Anal- tech Uniplates). Flash column chromatography was performed using silica gel 60 (200–400 mesh, Sorbent Technologies). As needed, further purification of synthetic compounds, including all tasquinimod analogs was done with preparative HPLC using Waters Delta 600 Controller equip with a variable wavelength UV-VIS detector. Purity of the compounds was determined using reverse phase-HPLC.
Synthesis of TasQ Analogs
The synthesis of the carboxamide analogs of TasQ is divided into three parts. Part A involves the synthesis of the intermediate ESATA-2. Part B is the synthesis of the amines that are not commercially available and part C involves the reaction of the intermediate ESATA-2 with the amine shown.
PART A: Synthesis of compound 4 (aka ESATA-2)

Compound 4 was synthesized as reported in literature33 with some modifications. 1, 4-dioxane was placed in a round bottomed flasked in ice bath. To this was added 1.5 molar equivalent of triphosgene. Then 1 molar equiv of 2-amino-6-methoxybenzoic acid slurry in DCM was added to the triphosgene solution gradually. The ice bath was then removed and the slurry (light creamy brown solution) was left to stir at room temperature for 2 hours. Solvent was then evaporated completely and the residue was triturated in diethyl ether (because triphosgene is soluble in ether). It was then filtered and washed with diethyl ether and then air dried to give compound 2 in 99% yield. It was analyzed and characterized with H1 NMR and C13 NMR.
Compouund 2 was dissolved in DMF. It was then placed in ice bath and cooled to 0°C. Then 1.5 molar equivalent of NaH in 60% mineral oil was added gradually. Then MeI was added dropwisely and rinsed down with DMF. Ice bath was removed and the heterogenous solution (slurry) obtained was stirred at room temperature for 3 hours. Solvent and all volatiles were then evaporated completely under reduced pressure to produce compound 3. To this was added DMF and 2.0 molar equivalent of NaH protionwise. This was followed by additon of 2 molar equivalent of dimethyl malonaate dropwisely. The mixture was then placed in a oil bath, heated to 130 °C and stirred at this temperature with a reflux condenser connected for 1 hour. It was then cooled to room temperature, and 400 mL of 1.0 M HCl was added. The slurry formed was filtered and then left to dry on suction overnight to give compound 4 in 93.7% yield. It was analyzed and characterized with H-1 NMR, C-13 NMR and HPLC. Its purity is 98% as measured by the HPLC.
Part B: Synthesis of amines
Structure of amines used for the synthesis of TasQ carboxamide 5a to 5j

The amines 5c and 5d were synthesized according to the scheme shown below.

4-(Trifluoromethyl)aniline (1 molar equiv) was dissolved in dichloromethane and to this was added the appropriate acyl chloride (3 molar equivalent) followed by DIPEA (3 molar equiv). The reaction mixture was stirred at room temperature overnight. Solvent was then evaporated and the crude was dissolved in dichloromethane followed by washing twice with water and brine. The organic layer was then dried with anhydrous sodim sulfate. Solvent was evaporated under reduced pressure to give solid residue which were purified either by trituration or by flash column chromatography using silica gel to give pure amide. Each of the amides obtained were then reduced to amine by reaction one amide equivalent with 10 molar equivalent of LiAlH4 using established method. The LiAlH4 was placed in ice-bath and cold tetrahydrofuran (THF) was added. This solution was stirred gently and 1 molar equivalence of the amide dissolved in THF was added dropwisely. The ice bath was removed and the reaction mixture was stirred overnight at room temperature (12–24 h). Water (3 times LiAlH4 molar equivalence) was added dropwisely followed by 20% (w/v) of NaOH solution (3 times LiAlH4 equiv) and then water (5 times LiAlH4 equiv). The solution formed a huge gelatinous precipitates and was filtered. The organic volatile part of the filtrate was evaporated, and the aqueous mixture left over was washed with dichloromethane (twice) to remove organic compounds. The organic layer was dried over anhydrous sodium sulfate and solvent was evaporated. The crude was purified to obtain the amines 5c and 5d using flash column chromatography with gradient elution, using hexane and ethyl acetate mixtures. All compounds were completely analyzed and characterized with H-1 NMR and C-13 NMR.
Compound 5e was synthesized starting from benzyl alcohol as shown in the synthetic scheme 3 below.
![]()
Benzyl alcohol in a round bottomed flask was dissolved in THF and the mixture was placed in ice bath. To this was added NaH portionwise. The mixture was stirred for 5 minutes and then a solution of 2-bromoacetic acid (1 molar equiv) in THF was added dropwisely. The ice bath was removed and the mixture stirred overnight at room temperature. All solvent was evaporated under reduced pressure, water was added and concentraed HCl was added to lower the pH to 2–4. Aqueous layer was washed three times with diethyl ether to extract the desired product. The organic phase was dried with anhydrous magnesium sulfate and solvent was evaporated to give a brown oil which is the desired product. The yeild is 85%.
This product was dissolved in DCM and DIPEA followed by a solution of HATU in DMF. This mixture was stirred for 5 minutes at room temperature. Then 4-(Trifluoromethyl) aniline was added. The mixture was then stirred overnight at room temperature. Reaction was stopped and solvent was evaporated under reduced pressure. The crude product was purified using preparative HPLC to give compound 7.
Reduction of compound 7 to 5e using LiAlH4 was done as described above for 5c and 5d. It was completely analyzed and characterized with H-1 NMR and C-13 NMR.The amines 5f to 5h were synthesized by reductive amination as shown below.
Synthesis of 5f-5h
The reaction was done as reported in literature34 with some modifications. To a stirred solution of SnCl2.2H2O (0.2 equivalent) in MeOH was added appropriate ketone compound (1.2 mol equiv), 4-(Trifluoromethyl)aniline (1 mol equv) and 2.5 mol equiv of PMHS at room temperature. The mixture was placed in oil bath at 70°C and then refluxed for 4 hours. Heat was removed and the reaction was left to cool down to room temperature. It was filtered under suction and the filtrate was dried over anhydrous sodium sulfate. Solvent was evaporated under reduced pressure to give a crude product which was purified using flash column chromatography with 100% hexanes. All compounds were completely analyzed and characterized with H-1NMR and C-13 NMR.
PART C: Synthesis of carboxamide analogs of TasQ by reaction of ESATA-2 with amines.

ESATA-2 was disolved in toluene to obtain a clear solution and then a heptane solution of appropriate amine in excess molar equivalence was added. The mixture was heated under reflux. Solvent and volatiles were evaporated by distillation to about 15% solution after 1 hour to remove all the methanol that is formed during this reaction. Then more solvent was added and the same process repeated after another hour. This was done repeatedly every hour over a 5 hour reaction time. The heat was removed after 6 hours and the reaction was stirred at room temperature overnight. Precipitates were formed and this was filtered off. The residue is the unreacted ESATA-2. The desired product is in the filtrate. Solvent was evaporated from the filtrate and it was purified by preparative HPLC to obtain each of the derivatives in pure form as described below. Each was fully characterized by H1-NMR, C-13 NMR, MALDI-MS, and HPLC. Yield and percent purity are shown below.
ESATA-11 Purity 95%, yield 63%
ESATA-14 Purity 92%, yield 66%
ESATA-18 Purity 96%, yield 68%
ESATA-20 Purity 95%, yield 63%
ESATA-22 Purity 97%, yield 72%
ESATA-27 Purity 98%, yield 77%
ESATA-28 Purity 96%, yield 76%
ESATA-30 Purity 95%, yield 79%
ESATA-24 Purity 90%, yield 50%.
Both compound ESATA-25 and ESATA-26 were synthesized from ESATA-20 as shown in synthetic below.

Both ESATA-25 and ESATA-26 were synthesized from ESATA-20 by Mitsunobu reaction in separate reactions. To a solution of ESATA-20 in THF was added 2 molar equivalents of DIAD and PPh3. The reaction was stirred at room temperature for 10 minutes and the respective alcohol was added. The reaction mixture was then stirred at room temperature for 24 hours. Solvent was evaporated and the crude product was purified first with flash column chromatography and then with preparative HPLC to obtained the desired product. Each was fully analyzed and characterized using H-1 NMR, C-13 NMR, MALDI-MS and HPLC. ESATA-25 Purity 95%, yield 58%, ESATA-26 Purity 90%, yield 54%.
Prostate Cancer Models
The source, history, and characteristics of the human prostate cancer cell lines and PDXs used, as well as cell culture conditions for their in vitro maintenance and the in vivo protocol for xenograft growth in triple immune‐deficient NSG (i.e. NOD.Cg‐PrkdcScidIl2rgtm1Wji/Szj), adult (>8 week old) male mice obtained from the Sidney Kimmel Comprehensive Cancer Center (SKCCC) Animal Core Facility are described previously16,35,36. Similar information concerning the TRAMP-C2 and B6CaP mouse prostate cancers in syngeneic C57BL/6J intact male mice and Myc-Cap-CR mouse prostate cancer in syngeneic FVB/NJ castrate male mice is described previously37–39.
All lines were mycoplasma negative using the MycoSensor PCR Assay kit (Agilent Technologies) and genetically authenticated within the last 6 months using short tandem repeat profiling conducted by the Johns Hopkins Genetic Resource Core Facility. In vitro growth curves were determined as described15. In vivo growth response to oral daily TasQ (10 mg/kg/d) of TRAMP-C2 mouse prostate cancers was evaluated in syngeneic C57BL/6J wild type vs. NOX-2 vs. iNOS knockout male mice obtained from Jackson Laboratory (Bar Harbor, ME). Growth response to oral daily TasQ (10 mg/kg/d) of TRAMP-C2 and B6CaP mouse prostate cancers was also evaluated in S100A9 knockout C57BL/6J male mice obtained from Dmitry Gabrilovich (Wistar Institute). Growth response to daily oral TasQ vs. ESATA-20 (both at 10 mg/kg/d) of Myc-CaP-CR mouse prostate cancer was evaluated in syngeneic FVB/NJ castrate male mice from Jackson Laboratory. Tumor volume measurements were as described previously15. In vivo experiments were repeated multiple independent times for each model used with 3–5 mice per treatment grouping.
To evaluate AHR agonist induced thymus regression, groups of five 4-week old FVB male mice from Jackson Laboratory were treated with either vehicle, Linomide, TasQ, or its analogs daily by orally gavage for 2 weeks and then the thymus removed and weighted.
Generation of stable shRNA Knockdown (KD) Variants
Stable shRNA HDAC4, and HIF-1α knockdown (KD) were as described previously15. Stable shRNA AHR KD as described previously15 using ATCCACAGTCAGCCATAATAA as the targeting sequence.
Western Blot Analysis
Analysis and quantification of protein expression via immunoblot was performed as previously described40. All assays were performed with total cell lysates from an equal number (i.e., 100,000) of cells loaded per lane for accurate comparisons across cell lines and conditions.
HDAC4 Enzymatic Assay
HDAC4 activity of the various recombinant forms used through these studies was assayed using the small non-protein Lys(Tfa)-AMC as substate as described previously41. The specificity of this assay is documented via its inhibition by a small molecule trifluoroacetylthiophene-trifluoromethylketone derivative [aka TFT-PIP] described previously41.
Real-time quantitative PCR (RT-qPCR) analysis
Total RNA was extracted by using RNeasy plus mini kit from Qiagen Cat # 74134. One microgram of total RNA was reverse transcribed using iScript cDNA synthesis kit from Bio Rad cat # 1708891. One fifth of the first strand cDNA reaction was used for Q-PCR amplification. Q-PCR was performed in an iCYCLER real-time PCR machine (BioRad) using SYBR-Green chemistry (BioRad). Test gene Ct values were normalized to Ct values of the house keeper gene GAPDH and fold differences, as compared to untreated controls, were calculated. For semi quantitative PCR one fifth of the first strand cDNA reaction was used for PCR amplification and amplified samples run on a 2% agarose gel stained with ethidium bromide. For mouse CYP1A1 the primer sets were FP: 5’ ATT CCT GTC CTC CGT TAC CTG 3’ and RP: 5’ GTG GCC CTT CTC AAA TGT CC 3’. For human CYP1A1 primer sets were FP: 5’ CCC AAC CCT TCC CTG AAT G 3’ and RP: 5’ TTC TCC TGA CAG TGC TCA ATC 3’ of the functional HDAC4-NCoR1 complex was tested by pre-incubation with NCoR1 or HDAC4.
Determination of IC50, EC50, and IC90 values
Dose response data were graphically analyzed to determine concentrations for the indicated parameter.
Oral Drug treatment
TasQ at the indicated dose is initially dissolved with equal molar 1 N sodium hydroxide. This solution is diluted with sterile water and the mixture stirred for 1 hr. The pH is then adjusted with 1 N HCl to 8.9 and 200 μl given daily via oral gavage. Benzo[a]pyrene (Sigma-Aldrich) and each of the TasQ analogs was dissolved in DMSO and diluted to 10% DMSO with 20% 2-hydoxypropyl-β-cyclodextrin (HPCD) in saline and 200 μl given orally daily by gavage at the indicated dose.
Pharmacokinetic Analysis
At various times post oral gavage, blood was collected and 50 μL of serum transferred into a 1.7 mL microfuge tube. Three volumes of Acetonitrile containing 0.1% Trifluoroacetic acid (TFA) is added and the sample vortexed for 1 to 3 min, then centrifuged at 13,000 RPM in a micro centrifuge. One hundred and twenty microliters of the supernatant loaded into a sample loop with a cut off volume of 100uL was injected onto a C18 reverse phase HPLC column. The HPLC system is a WATERS 600 series quaternary system with a 2487 dual wavelength detector set at 215 nm and 332 nm. Mobile phase A is 5% Acetonitrile/water, 0.1% TFA Mobile phase B is 95% Acetonitrile/water, 0.1% TFA. 1.5mL/min 20%B to 98% B in 10 minutes, hold for 5 minutes return to initial condition for 4 minutes. Retention time is 9.4 min for Linomide vs 11.3 min for TasQ and 13.5 min for ESATA-20 with no observable interference from endogenous mouse peaks.
AHR Binding Assay
Photo-affinity competitive ligand binding to the human AHR was performed as described previously40.
AHR Agonist Assay
Human Aryl Hydrocarbon Receptor (AHR) Reporter Assay System [Indigo Biosciences (State College, PA) Cat #IB06001] was used as per manufacture’s protocol.
Surface Plasmon Resonance (SPR) Binding Analysis
SPR analysis was performed on the Biacore 3000™ system using CM5 sensor chips, certified buffers (HBS-P −10 mM Hepes, 0.15 M NaCl, pH 7.4, containing 0.005% v/v Surfactant P; HBS-EP-HBS-P with 3 mM EDTA) and reagents for immobilization and surface regeneration from Biacore, GE Healthcare, Uppsala Sweden. Evaluation of sensorgrams was performed using the BIA-evaluation Software version 4.1 and GraphPad Prism 4. Buffer of protein reagents were changed to HBS-P on Zeba spin desalting columns (Thermo Scientific) prior to being used in SPR analysis.
A. Binding of wild-type and mutant forms of HDAC4 to immobilized TasQ
TasQ (ABR-215050; mol wt 406.4) was dissolved as a 40 mM solution in water with 1.5 equivalents of NaOH. TasQ with an amino-linker [i.e. -(CH2)2O(CH2)2-NH2] (ABR-225180) in amide nitrogen position was used for amine coupling on to a Biocore CM5 sensor chip. A 1 mg/mL solution of ABR-225180 in HBS-P buffer was prepared and immobilized on a CM5 chip by injection at a flow rate of 5 or 10 μL/min until a stable response level was reached. Interaction of various HDAC4 forms with immobilized TasQ was studied by injection over this surface in HBS-P buffer containing 10 or 20 μM ZnCl2. Bound HDAC4 was removed by a pulse of 3 mM EDTA in HBS-P or 0.5 to 6.25 mM NaOH in water. In a second series of experiments, the ability of TasQ in solution to displace this binding was studied by pre-incubation of HDAC4 with TasQ as competitor.
B. Binding of TasQ to immobilized HDAC4 and NCoR1
Different variants of HDAC4 (full-length wild-type and mutant forms and the truncated protein) and full-length NCoR1 were immobilized on a CM5 chip using random amine coupling. Briefly, proteins were dissolved in 10 mM sodium acetate buffer at pH 5.0 or 5.5 and immobilized to the aimed level using the standard protocol from GE Healthcare. Serially diluted TasQ in HBS-P buffer with 10 or 20 μM Zn was injected (for 2 or 3 min at a flow rate of 30 μL/min) either directly or after a 1st injection of 1 mM DTT to reduce cystines in the zinc conformation site in HDAC4. Regeneration was made with a short pulse of HBS-EP.
C. Inhibition of HDAC4-NCoR1 interaction by TasQ
The interaction between HDAC4 and NCoR1 was studied either by injecting NCoR over immobilized HDAC4 or with an alternative orientation where HDAC4 was passed over a surface with covalently coupled NCoR1. Whether TasQ can inhibit the formation
Computer docking modeling
This is conducted using Schrödinger’s molecular modeling environment Maestro (www.schrodinger.com) as described previously15.
RNA Sequencing Analysis of In vivo Growing Xenografts
This is performed as described previously36,40 as expressed as fragments normalized per kilo base-pair gene length per million reads library size (FPKM). These data are deposited in NCBI’s Gene Expression Omnibus and are accessible through accession number GSE211375. for the raw and mouse-gene subtracted xenograft data.
Three Dimensional (3D) Endothelial Sprouting Assay
The assay was performed using a minimum of 3 replicate wells of a 48 well tissue culture plate per drug dose per experiment, repeated 3 independent times in which human umbilical vein endothelial cells (HUVECs) obtained from Lonza Walkersville Inc (Walkersville, MD) attached to gelatin-coated dextran microcarrier bead are embedded in a fibrin matrix as described previously18.
Targeted kinase screen
Inhibition of enzymatic activity of the panel of kinases presented in Supplemental Table 1 in the presence of 10 μM ESATA-20 is analyzed by the Kinase Profiling Service at Thermo Fisher Scientific.
Study Approval.
All animal procedures were approved by the Johns Hopkins University School of Medicine Institutional Animal Care and Use Committee.
Statistical Analysis
Results are representative and expressed as mean +/− SEM. A p-value < 0.05 (T-test or ANOVA test when appropriate) are stated as significant.
Results
TasQ is an agonistic AHR ligand
In vitro growth of human prostate cancer cell lines is only modestly inhibited by TasQ (i.e., IC50 >50 μM) under optimal normoxic, nutrient, and pH culture conditions independent of their androgen responsiveness10,15. In contrast to such optimized in vitro conditions, cancers outgrow their blood supply in vivo, producing a compromised TME that is hypoxic and nutrient-limited with an acidic pH. Under these compromised TME conditions, growth of prostate cancer xenografts is profoundly inhibited by oral daily dosing with TasQ10–17. This is due to TasQ inhibiting the downregulation of global histone acetylation via inhibition of HDAC4, which decreases overall transcription needed for cancer cell survival and growth in the compromised TME while also inhibiting the transcription of a select group of survival genes15. In addition to its direct effects on cancer cells, TasQ’s anti-cancer efficacy also involves dose-dependent anti-angiogenic activity on endothelial cells leading to decreased tumor blood vessel density10,15,16,18, Supplemental Figure 1B.
Due to these combined effects, maximal significant growth inhibition of CWR22-RH, a castration-resistant prostate cancer patient-derived xenograft (PDX), is obtained as maintenance therapy with oral daily dosing of ≥5 mg/kg/d, Figure 1C. PK analysis documents that oral dosing with 5 mg/kg/d of TasQ produces a serum Cmax of 20 μM at 1 hr post-dosing, Figure 1C, and a nadir of 1.08 ± 0.21 μM at 24 hrs16. In previous studies, we documented that in vitro treatment of human prostate cancer cells (LNCaP) with 1–10 μM TasQ rapidly (i.e., ≤1 day) induces the transcription of a series of AHR target genes (e.g., Cyp1a1, Cyp1b1, AHRR, TiPARP, GDF15)12. Likewise, upregulation of AHR target genes is also induced in LNCaP xenografts within one day of oral dosing with TasQ at 10 mg/kg12.
TasQ is metabolized via microsomal cytochrome P450 (i.e., CYP) enzymes, but inhibition of such metabolism does not affect TasQ’s ability to inhibit endothelial cell sprouting in vitro or in vivo growth of human prostate cancer xenografts16. This raises the issue of whether the induction of AHR target genes is a result of TasQ or one of its metabolites binds to AHR as a ligand. To test whether TasQ is an AHR ligand, liver cytosol lacking microsomal CYPs was isolated from liver-specific human AHR (hAHR)-expressing transgenic mice. This cytosol was subjected to a rapid (i.e., 20 min) competitive ligand binding assay using a fixed saturating dose of the AHR-specific photo-affinity label (PAL) 2-azido-3-[125I]iodo-7,8-dibromodibenzo-p-dioxin with increasing amounts of TasQ. These results document that TasQ is a competitive ligand for human AHR with an EC50 of 1 μM, Figure 1D and Supplemental Figure 1C. As a reference for comparison, the classic polycyclic aromatic hydrocarbon (PAH) AHR agonist, benzo[a]pyrene (B[a]P), has an EC50 value of 1 μM for binding to hAHR in this PAL assay42.
To determine whether such ligand binding is agonistic vs. antagonistic, a human cell-based luciferase reporter gene assay functionally driven by an AHR-responsive promoter was utilized. These studies document that TasQ is a dose-dependent AHR agonist with an EC50 value of 1.0 +/− 0.2 μM. AHR potency of TasQ in this reporter assay is consistent with the significant in vitro dose-response induction of Cyp1a1 transcription in LNCaP cells, Figure 1E. Further confirmation that TasQ is an AHR agonist is provided by its dose-response ability to induce rapid (i.e., <30 min) translocation of the AHR from the cytosol to the nucleus of LNCaP cells, Figure 1F. Also characteristic of agonistic binding by TasQ is the subsequent rapid (i.e., ~2–4 hrs) downregulation of the AHR protein, which is stereotypically induced by agonist binding43, Figure 1G. The systemic in vivo AHR agonist ability of TasQ is confirmed by the fact that oral dosing of 4-week old FVB male mice with 10 mg/kg/d over 14 days maintains the serum drug level at >1 μM, which induces significant Cyp1a1 transcription in the liver, Figure 1H, and significantly decreases thymus weight (72 +/− 3 vs. 41.6 +/− 6 mg in vehicle- and TasQ-treated animals, respectively).
TasQ’s therapeutic efficacy against prostate cancer growth is independent of the AHR
To evaluate whether this AHR agonist ability is required for the MoA responsible for prostate cancer in vivo growth inhibition, a newly established prostate cancer PDX, LvCaP-3, growing in castrate hosts was tested for its response to daily oral dosing with 10 mg/kg TasQ vs. B(a)P alone and in combination, Figure 2A. This PDX is an example of a double-negative prostate cancer (DNPC) xenograft, which lacks both androgen receptor (AR) and neuroendocrine (NE) marker expression, and grows equally well in an intact vs. castrate hosts40. At this daily dose, B(a)P is a systemic AHR agonist based upon its ability to induce liver Cyp1a144. Like B(a)P, TasQ alone and in combination induces a >50-fold induction of liver Cyp1a1 in treated animals. In contrast, only TasQ alone or in combination with B(a)P significantly inhibits the growth of LvCaP-3 xenografts by ~2/3 at the end of study, while B(a)P completely lacks therapeutic efficacy, Figure 2A. This is despite oral daily dosing of B(a)P (10 mg/kg/d) for 14 days producing a robust systemic AHR agonist response documented by its ability to significantly decrease thymus weight (>70 vs. 27 +/− 8 mg in vehicle- and B(a)P-treated animals, respectively).
Figure 2: TasQ anti-tumor activity is independent of AHR, NOX1, iNOS, and S100A9.

A, Growth response of LvCaP-3 human prostate cancer PDXs in castrate mice to daily oral dosing with TasQ (10 mg/kg/d) or Benzo[a]pyrene [B(a)P] alone or in combination initiated when xenografts were ~0.2 cc. (n = 5/group). B, Initial growth response of LNCaP parental control vs. AHR KD cells in castrate mice (n = 5/group) and their subsequent response to oral dosing with TasQ (10 mg/kg/d). Insert: IB of AHR protein level in control vs. AHR KD LNCaP cells. Arrow indicates time of initiation of TasQ treatment. C, Growth response of TRAMP-C2 mouse prostate cancer in C57BL/6 wt vs. NADPH oxidase Knockout (KO), iNOS Knockout (KO), or S100A9 Knockout (KO) intact male mice to daily oral dosing with TasQ [10 mg/kg/d, (n=5/group)]. D, Computer-based docking of TasQ to inactive (non-NCoR binding) conformation of regulatory zinc-binding domain (ZRD) within the catalytic domain (amino acids 648–1051) of human HDAC4. Magenta colored ball is Zn2+ in the ZRD. In the right panel is an enlargement to indicate the position of R681 and R798. E, Inhibition of wildtype (wt) vs. R681A/R798A mutant HDAC4 binding to immobilized TasQ after pre-incubation with 0.1–500 μM TasQ in solution. F, SPR response detected binding of 50 nM wt fl-HDAC4 (upper sensorgram) vs. R681A/R798A mutant fl-HDAC4 (lower sensorgram) to immobilized TasQ.
To further evaluate the role of the AHR in TasQ’s MoA, the in vivo growth response to TasQ was compared between castration-sensitive LNCaP vs. LNCaP-AhR shRNA knockdown (LNCaP/AHR-KD) xenografts growing in intact male hosts. This documents that a reduction in AHR protein expression by >90%, Figure 2B insert, results in a significant decrease in the growth rate of LNCaP/AHR-KD xenografts compared to the parental LNCaP xenografts in untreated intact male hosts, Figure 2B. Despite this difference, both types of xenografts are equally inhibited when daily oral dosing with TasQ (10 mg/kg/d) is initiated at day 53 post-inoculation, Figure 2B. These combined data document that AHR agonist binding by host cells infiltrating the TME alone (i.e., AHR-KD results) or in combination with prostate cancer cells (i.e., B(a)P results) is not required for TasQ’s efficacy.
TasQ’s MoA does not require NADPH Oxidase, iNOS, or S100A9 expression by host cells
Earlier studies documented that TasQ binds with nanomolar (nM) affinity to S100A932. S100A9 is a Ca2+-binding pro-inflammatory protein produced by tumor-infiltrating monocytes and macrophages that binds TLR4 and RAGE on tumor-infiltrating myeloid-derived suppressor cells (MDSCs). This binding stimulates reactive oxygen species (ROS) production by NADPH Oxidase-2 (NOX2) and reactive nitrogen species (RNS) produced by inducible Nitric Oxide Synthase (iNOS)17. This stimulation is inhibited by the binding of TasQ to S100A917,32. To evaluate the importance of S100A9 inhibition in tumor-infiltrating host cells, the growth response of TRAMP-C2 mouse prostate cancer allografts to daily oral dosing with TasQ (10 mg/kg/d) was compared in syngeneic C57BL/6J wild type hosts vs. NOX2-, iNOS-, or S100A9-knockout (KO) hosts was evaluated, Figure 2C. These results document that expression of neither NOX2, iNOS, nor S100A9 by host cells is required for TasQ’s >70% inhibition of tumor growth over a 3-week period. This lack of a requirement for S100A9 in host cells for TasQ’s efficacy is reproduced in an additional, newly established mouse prostate cancer model (i.e., B6CaP38) also grown in syngeneic C57BL/6J S100A9-KO mice, Supplemental Figure 1D.
TasQ binding requires full-length HDAC4 protein in its open, not closed, conformation
TasQ is an allosteric inhibitor of HDAC4 function15. HDAC4 has an N-terminal association domain at amino acid (AA) 68–208, which contains a self-dimerization subdomain at AA 68–155 and a repressive Myocyte Enhancer Factor-2 (MEF-2) binding subdomain at AA 168–18435,45. Additionally, there is a nuclear localization signal (NLS) at AA 244–279, as well as serine residues at AA 246, 467, and 632, whose phosphorylation is required for binding 14–3-3, which restricts HDAC4 localization to the cytoplasm; in addition to a sumoylation site at Lysine 55935,45. Furthermore, there is an HDAC domain at AA 648–1051, which includes an activating HIF-1α binding domain at AA 822–1051; and a nuclear export signal (NES) at AA 1051–108435,45. While HDAC4 lacks intrinsic DNA-binding activity, it selectively binds a subset of client transcription factors as part of either repressive (e.g., MEF2) or stimulatory (e.g., HIF-1) complexes at specific promoters and enhancers46,47.
Within its C-terminal HDAC domain (AA 648–1051), there is a zinc-bound catalytic domain (ZCD) involving AA 802–950; however, HDAC4 is enzymatically inactive against classic acetylated protein substrates41,48. Within the HDAC domain there is a zinc-bound regulatory domain [ZRD, (AA 648–751], which has two alternative conformations41,48. When Zn2+ is coordinated by C667, C669, H675, and C751, HDAC4 is in an active “closed” conformation, which allows binding of the transcriptional co-repressor NCoR1/HDAC3 complex via the RD3 domain of NCoR1 to D759-H842-F871 at the rim of the ZCD entry site of HDAC441,48. In this HDAC4/NCoR1/HDAC3 complex, HDAC3 is active and deacetylates client proteins tethered to the complex via binding to HDAC445. Alternatively, when Zn2+ is coordinated by H665, C667, H678, and C751, the ZRD is shifted to an inactive “open” conformation unable to bind NCoR1/HDAC3, and thus not able to deacetylate HDAC4-bound client proteins41,48.
To evaluate the conformational requirements for TasQ’s binding to HDAC4, surface plasmon resonance (SPR) analysis was utilized. To maximize conformational flexibility, TasQ is immobilized onto the surface of a Biocore CM5 sensor chip via an amino-linker [i.e.-(CH2)2-O-(CH2)2-NH2] in its amide nitrogen position. Using such amine coupling for SPR-binding analysis documented that immobilized TasQ binds in a Zn+2-dependent saturable manner in a 1:1 stoichiometry with full-length (fl)-HDAC4 protein in solution with an affinity in the nanomolar range (KD ~ 20 nM calculated from the on- and off-rates, Supplemental Figure 1E, or KD ~130 nM calculated from the steady-state dose-response curve, Supplemental Figure 1F). To confirm these results, SPR-binding analysis was repeated, but with the fl-HDAC4 immobilized using random amine coupling onto the surface of a CM5 chip. TasQ in solution demonstrates Zn2+-dependent saturable 1:1 binding to immobilized fl-HDAC4 with a KD of 4.9 +/− 1.4 μM (n = 4), Supplemental Figure 1G. By comparison, Linomide, even at >30 μM in solution, binds only 10–15% of the immobilized fl-HDAC4 protein despite the presence of Zn2+, Supplemental Figure 1H. TasQ’s enhanced affinity for HDAC4 binding compared to Linomide is consistent with its >25-fold increase in anti-tumor efficacy8,9. The fact that the KD for binding of soluble TasQ to immobilized fl-HDAC4 is higher than the KD for binding of soluble fl-HDAC4 with immobilized TasQ is predictable. This is because the conformational dynamics of fl-HDAC4 immobilized on the chip are more restrictive due to the multiple random amine coupling than the conformational dynamics of TasQ immobilized via a single amino-linker.
Previously, we documented that Zn2+-dependent TasQ binding allosterically inhibits formation of HDAC4/NCoR115. SPR analysis determined that TasQ in solution has no detectable binding to immobilized fl-NCoR1 protein. Thus, TasQ’s inhibition of HDAC4/NCoR1 formation involves only its binding to HDAC4. To resolve whether such TasQ binding involves the ZCD vs. ZRD in HDAC4, we took advantage of the fact that while HDAC4 has only marginal deacetylase activity against acetylated lysine-containing proteins, it can deacetylate a small non-protein trifluoroacetamide substrate (a.k.a., Lys(Tfa)-AMC). This reaction is inhibited by a small molecule trifluoroacetylthiophene-trifluoromethylketone derivative [a.k.a., TFT-PIP, (Supplemental Figure 2A-B)] due to its binding to the catalytic Zn2+ in the ZCD, which locks the ZRD in the open conformation41. In contrast, TasQ did not inhibit the enzymatic activity of fl-HDAC4 using this Lys(TFA)-AMC substrate, Supplemental Figure 2B. Importantly, while TFT-PIP locks the ZRD in the open conformation, it does not prevent TasQ binding of fl-HDAC4, Supplemental Figure 2C. These results document that TasQ binds the open conformation of fl-HDAC4 at a site other than the ZCD. This is consistent with our previous demonstration that TasQ binds the fl-C669A/H675A double mutant HDAC4 in which the ZRD is also locked in an open conformation15.
Unlike the situation for fl-wild type or the fl-C669A/H675A double mutant, the truncated C-terminal HDAC domain (AA 614–1084) containing both the ZCD and ZRD has low and non-saturable binding to immobilized TasQ, and there is no binding to an AA 551–648 HDAC4 fragment, Supplemental Figure 1H. These results are significant since energy-minimization modeling predicts that TasQ binds to a specific allosteric site only present within the open, not the closed, conformation of the ZRD between AA612–80019. Modeling predicts that R681 and R798 within the open conformation of the ZRD region are critically important for TasQ binding, Figure 2D15. This suggests that acquisition of the open conformation needed for TasQ binding to R681 and R798 requires cooperativity with N-terminal sequences (i.e. AA1–612) present in the full length, but not in the truncated C-terminal HDAC domain (AA614–1084). Such cooperativity is consistent with the presence of a self-dimerization domain in the N-terminal of fl-HDAC4 located between AA 99–179, and thus missing in the truncated C-terminal HDAC domain (AA 614–1084)35.
To address this possibility, a competitive binding assay was developed based upon the ability of TasQ in solution to compete with immobilized TasQ on the SPR-chip for binding to fl-HDAC4. These results document that the concentration of TasQ in solution needed to reduce binding of fl-wild-type HDAC4 to the immobilized TasQ by 50% (i.e., IC50) is 1.3 μM, Figure 2E. In contrast, fl-R681A/R798A double mutant HDAC4 protein has a >66% reduction in binding immobilized TasQ compared to fl-wild type protein and binding of this fl-R681A/R798A double mutant HDAC4 protein to immobilized TasQ is not competitively displaced by TasQ in solution, Figure 2E.
TasQ inhibits binding between HDAC4 and NCoR1
To confirm that TasQ inhibits formation of the functional complex between HDAC4 and NCoR1, fl-NCoR1 in solution was injected over immobilized fl-HDAC4 after pre-incubation with TasQ in solution at concentrations ranging from 1 to 100 μM, Supplemental Figure 2D. Kinetic analysis of the SPR-sensorgram without competitor determined a high affinity binding (i.e., KD ~0.6 nM) of fl-NCoR1 to immobilized fl-HDAC4, mainly due to a slow off-rate. TasQ in solution inhibits this strong interaction by 50% at 10 μM, Supplemental Figure 2E.
To confirm and extend these results, SPR-analysis was performed with the alternative orientation in which NCoR1 is immobilized on the surface with fl-wild type vs. fl-R681A/R798A double mutant HDAC4 injected in the presence of TasQ in solution as a competitor. These additional analyses again documented high-affinity binding of the wild type fl-HDAC4 protein to fl-NCoR1 with such binding being competitively inhibited by pre-incubation with TasQ in solution, Figure 2F. When the inhibition curve is fit to a sigmoidal dose-response model, the IC50 for TasQ competitive inhibition of fl-HDAC4 binding to immobilized NCoR1 is 1.1 μM, which matches the IC50 of 1.3 μM for TasQ binding to immobilized fl-HDAC4, Figure 2E. In contrast, binding of the fl-R681A/R798A double mutant HDAC4 protein to fl-NCoR1 is reduced by 50% with such reduced binding only slightly inhibited by TasQ in solution (i.e., <35% inhibition reaching a plateau at 2 μM TasQ), Figure 2F.
HDAC4 disruption inhibits tumor growth under hypoxic conditions
Under optimal normoxic (i.e., 21% O2), nutrient, and pH conditions, in vitro growth of human prostate cancer cell lines is only modestly inhibited by TasQ (i.e., IC50 >50 μM)15. Similarly, under such optimal conditions, knocking down HDAC4 expression by 50% with shRNA only decreases the growth of LNCaP cells by <25%, which is equivalent to the inhibition by TasQ at 10μM, Figure 3A. Even under such optimal normoxic conditions, there is a significant enhancement of growth inhibition (i.e., >50%) when HDAC4 KD cells are treated with 10 μM TasQ, Figure 3A. In contrast to normoxic in vitro conditions, cancers outgrow their blood supply in vivo, producing a TME that is hypoxic, nutrient-limited, and acidic (low pH). For example, when prostate cancer xenografts growing subcutaneously reach a size of ~0.2–0.3 cc, the TME is already hypoxic with a pO2 of 3.6 +/− 1.5 mmHg compared to 37.5 +/− 8.6 mmHg in normal tissue13. When LNCaP cells are switched from 21% O2 (i.e., 160 mmHg) to a hypoxic condition of 1% O2 (i.e., 7.6 mmHg), their growth decreases by 50%, Figure 3A. This hypoxia-induced growth inhibition is enhanced by either knocking down HDAC4 or by treatment with TasQ. Figure 3A. Most significantly, when HDAC4 KD cells are exposed to hypoxia and treated with 10μM TasQ, the cells die, Figure 3A. These results document that HDAC4 function is critically required for the survival of prostate cancer cells under hypoxic stress.
Figure 3: HDAC4 disruption inhibits tumor growth under hypoxic conditions.
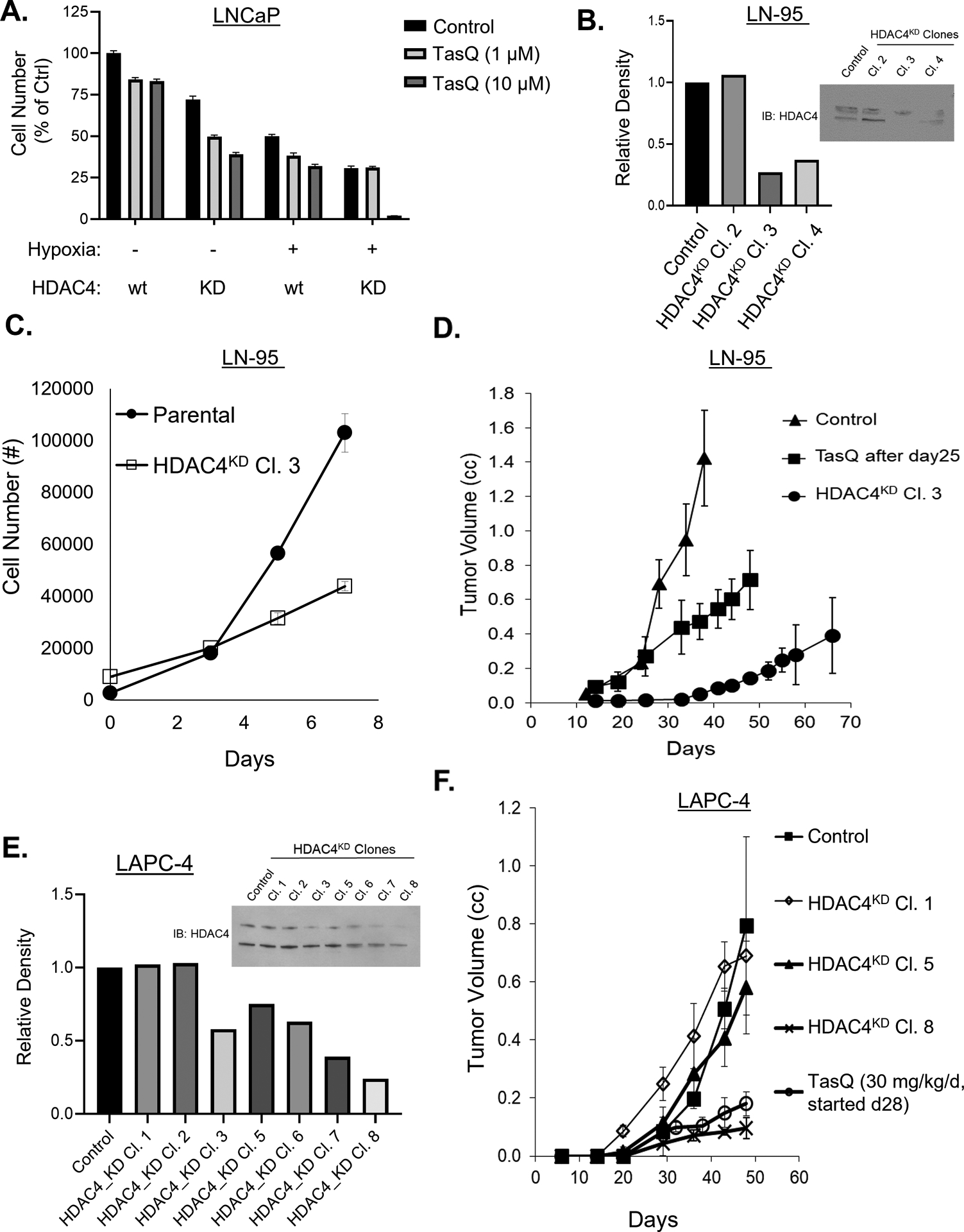
A, Dose response growth inhibition by TasQ of wt parental vs. HDAC4 KD LNCaP cells in normoxia vs. hypoxia. B, IB of HDAC4 protein in LN-95 parental control cells vs. HDAC4 KD clones and densitometry. C, In vitro growth of LN-95 parental cells (vs. HDAC4 KD clone 3 (cl. 3) cells (n = 8/group). D, In vivo growth response (n = 5/group) of LN-95 parental xenografts in untreated castrate mice vs. xenografts in castrated mice treated daily with TasQ (10 mg/kg/d) starting on day 25 post-inoculation vs. HDAC4 KD cl. 3 xenograft in untreated castrated mice. E, IB of HDAC4 protein level in control vs. HDAC4 KD clones of LAPC-4 cells (note upper band is the sumoylated form, lower band is unsumoylated ~140 kDa size) and densitometry. 100,000 cells loaded/lane. F, In vivo growth response (n = 5/group) of LAPC-4 parental vs. HDAC4 KD clones xenografted in untreated intact mice vs. parental LAPC4 xenografts in intact mice treated daily with TasQ (30 mg/kg/d) starting on day 28 post-inoculation.
To extend these results, additional prostate cancers lines were evaluated for their growth response to HDAC4 KD alone and in combination with TasQ treatment. In LN-95 cells, a castration-resistant variant of LNCaP, when HDAC4 is KD by >70% [i.e., clone 3, (Figure 3B)], growth is significantly inhibited under the stressful condition of in vitro growth in androgen-depleted media, Figure 3C and in vivo growth in castrated hosts, Figure 3D. These in vivo results are consistent with the fact that in prostate cancer xenografts, hypoxia is enhanced >2-fold by daily oral TasQ (10 mg/kg/d) lowering pO2 in the TME from >3 mmHg to 1.4 +/− 1.0 mmHg16. Similarly, when HDAC4 is KD by >75% (i.e., only in clone 8 and not in clones 1 and 5), Figure 3E, the in vivo growth of castration-sensitive LAPC-4 xenografts in intact male hosts is significantly inhibited, Figure 3F. The growth inhibition of both LN-95 HDAC4 KD xenografts in castrates and LAPC-4 KD xenografts in intact hosts is essentially identical to that of the parental wild type xenografts once daily oral TasQ is initiated, Figure 3D–F. These combined results document that HDAC4 disruption characteristically enhances hypoxia-induced tumor growth inhibition in vivo in both castration-sensitive and -resistant prostate cancer xenografts.
To test if this hypoxia-dependent growth inhibition is due to the loss of HDAC4/NCoR1 formation, LNCaP cells were transfected with a dominant-negative (DN) H863L mutant HDAC449, Supplemental Figure 2F. This mutant protein, like the wild type, lacks protein deacetylase activity, but also like the wild type protein has enzymatic activity when assayed with the small non-protein Lys(TFA)-AMC substrate, Supplemental Figure 2G. This mutation, however, is DN because it competitively binds to the same subset of transcription factors (e.g., HIF-1α and MEF-2) as wild type HDAC445. Unlike wild type, however, the H863L mutant HDAC4 protein does not co-bind NCoR1, Figure 4A, thus preventing deacetylation by the NCoR1/HDAC3 complex of HDAC4-bound client proteins15. This defect results in profound inhibition of in vivo growth, resulting in a significant 3-fold increase in the time for the xenografts to reach 0.5 cc, which is nearly identical to the growth inhibition by HIF-1α kd, Figure 4B.
Figure 4: TasQ decreases HIF-1α protein by preventing its N-terminal lysine deacetylation.

A, IB of NCoR1 protein in parental LNCaP cells transfected with flag-tagged fl-wt vs. H863L dominant-negative (DN) mutant fl-HDAC4 immunoprecipitated (IP) with Flag. 100,000 cells loaded/lane. B, Inhibition in in vivo growth of LNCaP xenografts expressing H863L-mutant HDAC4 or HIF-1α KD in intact mice expressed as the increase in time post-inoculation to reach 500 mm3 (n = 5/group). C, IB of HIF-1α protein in LNCaP cells in normoxic vs. hypoxic (i.e., 1% pO2) in vitro conditions with and without the addition of indicated TasQ concentration. 100,000 cells loaded/lane. D, Nuclear HIF-1α protein level under hypoxia in untreated wt LNCaP cells vs. treated with TasQ vs. HIF-1α KD or HDAC4 KD cells. E, IB of HIF-1α protein in control LNCaP cells in normoxic (N) vs. hypoxic (H) in vitro conditions and in HIF-1α KD clones (cl) under these same conditions. 100,000 cells loaded/lane. F, IB of HIF-1α protein in parental CWR-22Rv1 cells in normoxia (N) vs. hypoxia (H) in vitro conditions and in control shRNA vs. HIF-1α shRNA-KD under these same conditions. 100,000 cells loaded/lane. G, In vivo inhibition of growth of parental control vs. HIF-1α shRNA KD xenografts in untreated castrate mice or mice given a daily oral dose of TasQ [10 mg/kg/d, (n = 5/group)]. H, In vivo growth response of CWR-22Rv1 parental control cells vs. cells expressing the dominant-positive mutant fl-HIF-1α in untreated castrate mice vs. mice with the parental vs. mutant fl-HIF-1α treated daily with TasQ (10 mg/kg/d) starting when the xenografts were 0.05–0.1 cc in size. Insert, IB of flag-tagged HIF-1α protein in parental wt CWR-22Rv1 vs. cells transfected with flag-tagged wt fl-HIF-1α or fl-HIF-1α in which the N-terminal lysine 10, 11, 12, 19, and 21 are mutated to arginine control (i.e. dominant-positive mutant) under normoxia (N) or hypoxia (H) conditions. 100,000 cells loaded/lane.
TasQ decreases HIF-1α protein by preventing its N-terminal lysine deacetylation
TME hypoxia decreases hydroxylation and acetylation of HIF-1α protein, increasing its cellular level to a point where it activates a transcriptional “pro-angiogenic switch” enhancing tumor angiogenesis and adaptive metabolic survival signaling50. This hypoxia-induced upregulation of HIF-1α protein is an early event in human prostatic carcinogenesis associated with poor clinical outcome51 and involves a decrease in HIF-1α acetylation15,47. Under normoxic conditions, HIF-1α is acetylated on its first five N-terminal lysine residues (lysine 10, 11, 12, 19, and 21), which destabilizes HIF-1α and enhances its proteosomal dependent degradation47. In addition, these N-terminal acetylated lysines are within HIF-1α’s DNA binding domain, and while not preventing HIF-1α/HIF-1β (a.k.a., ARNT; aryl hydrocarbon receptor nuclear translocator) heterodimerization52, they disrupt DNA binding of this heterodimer to hypoxia response elements53.
HIF-1α is an HDAC4 client15,54,55 binding between AA 603–788 in its inhibitory domain to HDAC455. As a client, the HDAC4/NCoR1/HDAC3 complex deacetylates the N-terminal lysines, which stabilizes and thus activates HIF-1α as a transcription factor15,47. In LNCaP cells under normoxic conditions, HIF-1α protein is low, cytoplasmic, and mostly unphosphorylated; while under hypoxic conditions, it is increased, nuclear, and phosphorylated (i.e., slower migrating band on western blot)15, Figure 4C. HDAC4 KD in LNCaP prostate cancer cells prevents deacetylation-dependent stabilization, resulting in a significant reduction of nuclear HIF-1α protein, Figure 4D. As predicted based upon its ability to inhibit formation of the HDAC4/NCoR1/HDAC3 complex, TasQ treatment, like HDAC4 KD, decreases nuclear HIF-1α protein levels in LNCaP cells under both normoxic and hypoxic conditions in vitro with an IC50 of 2.5 μM, Figure 4C–D. The causal importance of decreased HIF-1α protein for in vivo growth was evaluated by knocking down HIF-1α protein expression in LNCaP cells, Figure 4D, and assessing xenograft growth. Such HIF-1α KD results in significant inhibition of LNCaP growth in vivo, resulting in a 2-fold increase in the time for the xenografts to reach 0.5 cc, which is ~2/3 of the growth inhibition produced by the DN H863L mutant HDAC4, Figure 4B.
To confirm the generalizability of this finding, HIF-1α expression was KD in CWR-22Rv1 cells, Figure 4F. When these HIF-1α KD cells are xenografted, their growth over a 3 week period is significantly inhibited compared to controls, Figure 4G, to a point equal to the growth inhibition of parental wild type xenografts in which their HIF-1α protein level has been decreased by daily oral TasQ (10 mg/kg/d) for 3 weeks, Figure 4G. The suppressed growth of these HIF-1α KD xenografts in castrate hosts is only modestly enhanced by the addition of daily treatment with TasQ (10 mg/kg/d), Figure 4G, consistent with the lowering of HIF-1α being a significant part of the MoA of TasQ.
To further query whether decreasing HDAC4-dependent HIF-1α N-terminal deacetylation is a significant component of TasQ’s MoA, CWR-22Rv1 cells were transfected with an expression vector encoding flag-tagged HIF-1α in which the N-terminal lysines (K10, 11, 12, 19, and 21) are mutated to arginines, Figure 4H insert. These mutations prevent acetylation resulting in a gain-of-function (i.e., dominant-positive transcriptional effect) due to stabilization of the protein’s half-life47. When xenografted, these dominant-positive mutant HIF-1α cells grow at a rate similar to the untransfected wild type parental cells, Figure 4H. Unlike the wild type cells whose growth is significantly inhibited by >80% when the host is given daily oral TasQ (10 mg/kg/d), growth of these dominant-positive mutant HIF-1α tumors is only inhibited by <25% in TasQ-treated hosts, Figure 4H.
TasQ suppresses HIF-1α target genes and inhibits repression of MEF-2 target genes needed for survival in the compromised hypoxic TME
RNAseq analysis was performed on CWR22-RH PDX tissue growing in the compromised hypoxic TME of castrated hosts with or without daily oral TasQ (10 mg/kg/d). Such dosing produces optimal CWR22-RH growth inhibition, Figure 1C, consistent with downregulation of the expression of proliferation-associated target genes in addition to the AR and its target genes, Table 1A. In addition, such daily TasQ treatment lowers HIF-1α protein in tumors by >50%12 suppressing HIF1-dependent cell survival target genes, Table 1B. Significantly, this daily oral dose of TasQ (10 mg/kg/d) maintains blood concentrations of >1 μM16. As predicted at this drug level, elevated expression of AHR and a series of its target genes is observed, Table 1C.
Table 1.
TasQ phenocopies HDAC4 knockdown suppression of HIF-1α target gene transcription.
| Proliferation Target Genes (Fold Decrease by TasQ) | AR Target Genes (Fold Decrease by TasQ) | |||
| MKI67 | −1.7 | AR | −2.0 | |
| E2F1 | −2.0 | TMPRSS2 | −1.9 | |
| CDC6 | −1.5 | NKX3.1 | −2.3 | |
| CDT1 | −2.2 | KLKP1 | −2.3 | |
| MCM2 | −1.9 | SLC45A3 | −3.7 | |
| MCM3 | −1.6 | |||
| MCM5 | −2.0 | |||
| DTYMK | −1.6 | |||
| HIF-1a Target Genes (Fold Decrease by TasQ) | MEF-2 Target Genes (Fold Decrease by TasQ) | |||
| IGFBP5 | −18.0 | EDN1 | 15.0 | |
| IGF1 | −11.0 | THB5 | 8.0 | |
| PTN | −6.1 | S100P | 5.7 | |
| LOX | −4.0 | BHLHE41 | 2.9 | |
| TTR | −3.8 | CA12 | 2.7 | |
| TNC | −2.5 | CEACAM1 | 1.8 | |
| BNIP3 | −1.9 | NR4A2 | 1.8 | |
| IGFBP2 | −1.8 | CAPN2 | 1.6 | |
| P4HA1 | −1.8 | NR4A1 | 1.4 | |
| HILPDA | −1.7 | |||
| PDK1 | −1.7 | |||
| LDHA | −1.5 | |||
| VEGFB | −1.5 | |||
| ALDOC | −1.4 | |||
| AHR Target Genes (Fold Increases by TasQ) | ||||
| CYP1B1 | 10.0 | |||
| AGR2 | 5.5 | |||
| TIPARP | 3.4 | |||
| AHR | 3.0 | |||
| NQOI | 2.9 | |||
| GDF15 | 2.3 | |||
While HIF-1α is required for the upregulation of hypoxia survival genes, it is not required for the hypoxia-induced widespread transcriptional repression, which is also needed for enhanced survival in the compromised TME56. MEF-2 is also a HDAC4 client57. MEF-2 dimers bind to target gene promoters, forming a hydrophobic groove composed of AA 1–78 of each monomer to which HDAC4 binds via its amphipathic helix at AA 168–18458. When MEF-2 binds, HDAC4 forms a NCoR1/HDAC3 complex, which deacetylates MEF2 and histones within its proximity; thereby, repressing expression of its target genes57. When CWR-22Rv1 cells are switched from a 21% to a 1% O2 environment in vitro, there is a 3-fold increase in NCoR1 immunoprecipitated with MEF-2 from nuclear extracts, Figure 5A. Co-treatment with TasQ inhibits this hypoxia-induced increase in nuclear MEF2/HDAC4/NCoR1 formation with an IC50 of 5 μM, Figure 5A. These in vitro results are consistent with gene expression analysis of CWR22-RH PDX tissue from castrate hosts in vivo, which document that daily oral TasQ (10 mg/kg/d) inhibits the binding of NCoR1, and thus HDAC3 to the MEF-2/HDAC4 complex, relieving repression of MEF-2 target genes needed for survival under hypoxic conditions, Table1B.
Figure 5: Mechanism of TasQ’s anti-angiogenic activity involves repression of MEF-2 target genes.
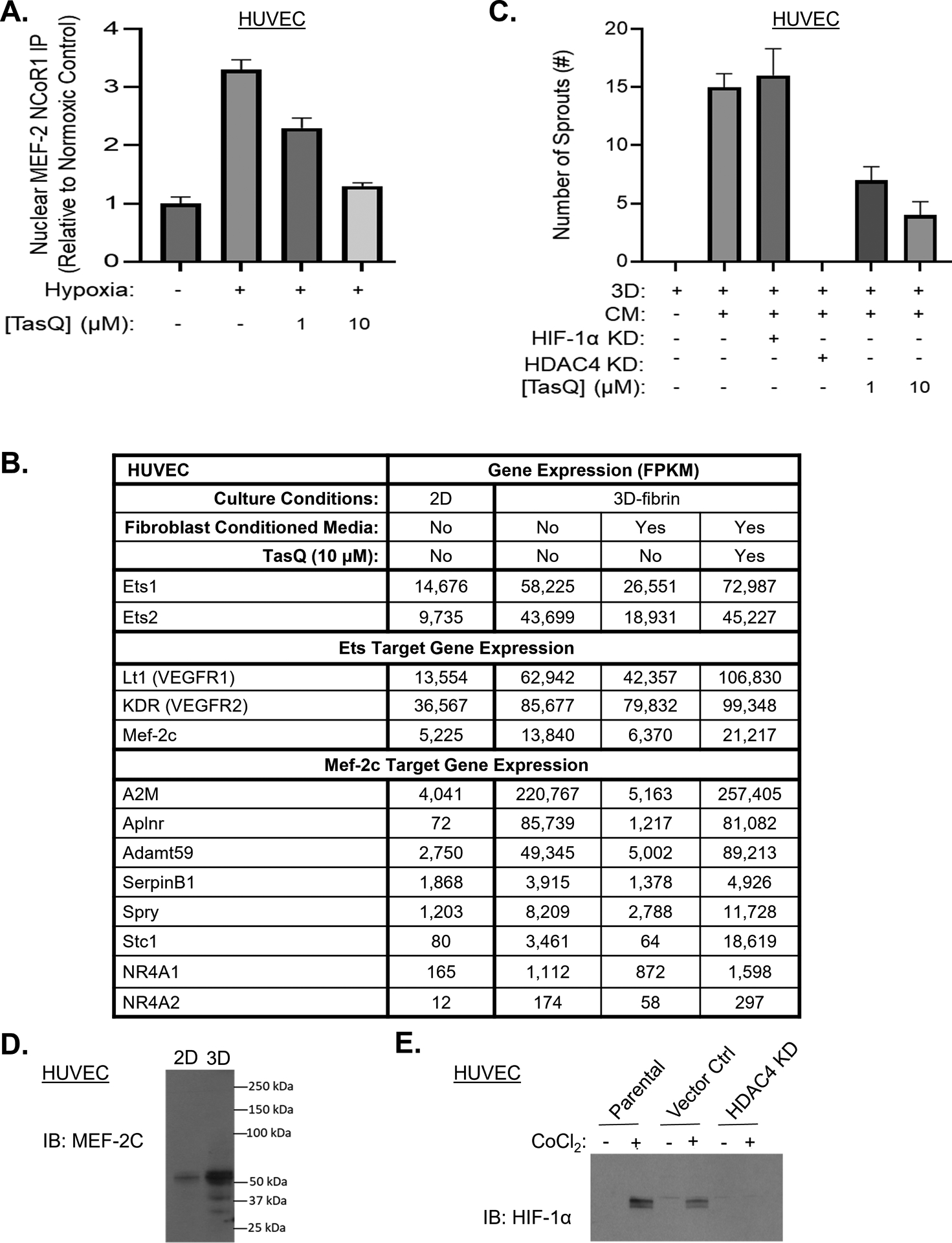
A, Level of nuclear NCoR1 immunoprecipitated with MEF-2 antibody in CWR-22Rv1 cells under normoxia vs. hypoxia with or without treatment with TasQ normalized to normoxic controls. B, mRNA expression normalized as FKPM of HUVEC in 2D vs. 3D-fibrin gels with or without addition of human dermal fibroblast (HDF)-conditioned media (CM) with or without TasQ (10 μM). C, Sprouting in 3D-fibrin gels with and without HDF-CM by wt vs. HIF-1α KD or HDAC4 KD HUVEC vs. wt cells treated with TasQ. Results are expressed as number of sprouts per HUVEC-coated microcarrier bead. D, IB of MEF-2c in HUVEC in 2D vs. 3D-fibrin culture. 100,000 cells loaded/lane. E, IB of HIF-1α in wt vs. control shRNA vs. HIF-1α KD HUVEC with or without exposure to cobalt chloride (CoCl2) to mimic hypoxia. 100,000 cells loaded/lane.
TasQ’s anti-angiogenic MoA involves inhibiting repression of MEF-2 target genes
Besides its therapeutic effects on cancer cells, TasQ’s anti-cancer efficacy also involves its dose-dependent anti-angiogenic activity10–16,18. To clarify the MoA for this anti-angiogenic activity, a 3-dimensional (3D) endothelial cell sprouting assay was used. In this assay, human umbilical vein endothelial cells (HUVECs) in basal media supplemented with VEGF, EGF, FGF, IGF1 and 2% FBS in standard 2D cultures are attached to collagen-coated beads embedded in a 3D-fibrin gel in the same growth factor-supplemented media. Under these 3D-basal conditions, the embedded endothelial cells undergo a major transcriptional reprogramming, but despite a 4–5 fold increase in ETS Proto-Oncogenes 1 and 2 Transcription Factors (Ets-1 and −2) coupled with a 2–3 fold increase in Ets target genes like Flt1 (VEGFR1) and KDR (VEGFR2), Figure 5B, they do not sprout, Figure 5C. This lack of sprouting is associated with an upregulation in Mef-2c mRNA, Figure 5B, and protein, Figure 5D, and its target genes, Figure 5B, like Alpha 2 Macroglobulin (A2M). These results are consistent with earlier studies documenting that sprouting is inhibited in this 3D-basal media due to enhanced expression of Mef-2c coordinately upregulating expression of A2M59. When the 3D-fibrin gels are further supplemented with conditioned media (CM) from human dermal fibroblasts (HDF) to provide additional angiogenic factors typically present within the TME60, expression of the MEF-2c target genes, including A2M, is blocked, Figure 5B; enabling endothelial cell proliferation and invasion into the gels (i.e., “sprouting”); eventually forming canalized and anastomosing neovascular tubes within 7 days, Figure 5C. Importantly, 3D-sprouting occurs, Figure 5C, even when HIF-1α expression is KD in the HUVEC cells, Figure 5E. In contrast, as little as a 60% KD in HDAC4 completely inhibits HUVEC sprouting in this 3D-assay despite the addition of HDF-CM, Figure 5C. Similarly, when these CM-supplemented 3D cultures are treated with TasQ (10 μM), the downregulation of Mef-2c target genes like A2M in the embedded endothelial cells is inhibited, Figure 5B, and sprouting prevented, Figure 5C.
Third-Generation Quinoline-3-Carboxamide Analogs with Reduced AHR Agonism
Combined xenograft and PK results document that the plasma concentration of TasQ required to inhibit HDAC4 binding to NCoR1 sufficiently to suppress HIF-1α transcriptional hypoxic survival signaling and thus maximally inhibit in vivo prostate cancer xenograft growth is >2 μM. Unfortunately, this is higher than the EC50 value of 1 μM for TasQ as an AHR ligand, Figure 1E, as an agonist in the in vitro cell-based ARE-driven luciferase reporter gene assay, Figure 6A, and as a CYP1A inducer in LNCaP cells, Figure 1H. This means that in the clinical setting, dose-escalation above 1 mg/d of TasQ as needed to reach this >2 μM blood level for optimal anti-cancer efficacy will also enhance unwanted off-target AHR induction. Therefore, a medicinal chemical approach was undertaken to identify third generation quinoline-3-carboxamide analogs with increased HDAC4 inhibition, but with reduced AHR agonism. The approach taken is based upon the earlier SPR analysis documenting that TasQ linked to the surface chip via its aniline-N is a potent binder of HDAC4. Thus, a series of quinoline-3-carboxamide analogs were synthesized focusing upon adduct changes at the aniline-N, Figure 6B. These analogs were initially screened for their AHR agonist activity using the in vitro human cell-based ARE-driven luciferase reporter gene assay, Figure 6A. Based upon this primary screen, a subset of analogs was evaluated for their percent inhibition of the growth of CWR22-RH xenografts in castrate NSG male mice vs. the percent thymus regression in intact FVB male mice following daily oral dosing at 10 mg/kg/d over a 3-week period, Figure 6A.
Figure 6. Third generation qunoline-3-carboxamide analogs with reduced AHR antagonism and identification of lead compound, ESATA-20.

A, AHR agonist activity of analogs expressed as EC50 values in an in vitro cell-based ARE-driven luciferase reporter gene assay (n = 4 independent assays/analog) vs. induction of thymus regression expressed as percent (%) decrease in organ weight in mice (n = 5/group) treated orally for 2 weeks with the respective compound (10 mg/kg/d) vs. inhibition of in vivo growth of CWR22-RH xenografts in castrate mice treated orally over a 3 week period with the respective compound [10 mg/kg/d, (n = 5/group)]. ND = not determined. B, Chemical structures of third-generation Quinoline-3-Carboxamide analogs. C, Summary of % PAL AHR binding inhibited by indicated concentration of TasQ vs. ESATA-20. D, Nuclear HIF-1α protein level under hypoxia in untreated wt LNCaP cells vs. treated with TasQ or ESATA-20 vs. HIF-1α KD or HDAC4 KD cells. E, Level of nuclear NCoR1 immunoprecipitated with MEF-2 antibody in CWR-22Rv1 cells under normoxia vs. hypoxia with or without treatment with TasQ or ESATA-20 normalized to normoxic controls. F, Sprouting in 3D-fibrin gels with and without HDF-CM by wt vs. HIF-1α KD or HDAC4 KD HUVEC vs. wt cells treated with TasQ or ESATA-20. Results are expressed as number of sprouts per HUVEC-coated microcarrier bead. G, Dose-response inhibition of in vivo growth of CWR22-RH human prostate cancer PDXs in castrate male NSG mice over a 3-week treatment period with oral daily TasQ or ESATA-20 initiated when xenografts were ~200 mm3. H, Relative expression of mouse Liver Cyp1A1 (mCYP1A1) in animals (n = 3) 24 hrs after receiving indicated oral dose of TasQ vs. ESATA-20 normalized to animals receiving only vehicle.
Structure-activity relationship (SAR) analysis documents that for a TasQ analog to retain potency as a growth inhibitor (GI) of CWR22-RH xenografts but decreased potency as an AHR agonist requires that: 1) the quinoline-3-carboxamide linked aniline moiety be present (e.g., ESATA-2, −3: no GI activity); 2) the aniline-N adduct is not too long (e.g., ESATA-14, −18, −22: no GI activity) or too short (e.g., ESATA-4: high GI activity, but also much more potent AHR agonism; and ESATA-27, −30: no GI activity); 3) the 4-position adduct is not too long (e.g., ESATA-25, −26: no GI activity); and 4) a para-trifluoromethyl group be present in the aniline moiety (e.g., ESATA-28: no GI activity).
ESATA-20 is a more selective/more potent lead third generation quinolone-3-carboxamide analog
Based upon these SAR requirements, the ESATA-20 analog in which the aniline-N has a cyclo-hexane ring adduct is identified as the lead 3rd-generation quinolone-3-carboxamide. This is based upon the fact that ESATA-20 has a 10-fold lower binding affinity than TasQ in both the competitive PAL AHR binding assay, Figure 6C and Supplemental Figure 1C, and the agonist response in the cell-based AHR-promoter assay, Figure 6A (i.e., EC50 of 10 vs. 1 μM for TasQ). In contrast, ESATA-20 is statistically more potent than TasQ at decreasing nuclear HIF-1α protein (i.e., IC50 of 1 vs. 2.5 μM for TasQ), Figure 6D, nuclear MEF-2 and NCoR1 IP (i.e. IC50 <1 vs. 5 μM for TasQ), Figure 6E, and inhibiting endothelial cell sprouting (i.e., the same degree of inhibition by ~1 μM ESATA-20 is equivalent to that produced by ~10 μM TasQ), Figure 6F.
ESATA-20 is less water soluble than TasQ requiring the use of DMSO in a 1:9 ratio with 20% 2-hydoxypropyl-β-cyclodextrin (HPCD) in saline as its vehicle for oral dosing. In this vehicle, ESATA-20 is less bioavailable than TasQ as documented by the fact that a 10 mg/kg oral dose of ESATA-20 produces a serum Cmax of 7.91 +/− 18 μM vs. a Cmax of 44 +/− 6 μM following an equivalent oral dose (10 mg/kg) of TasQ. Despite this 5.5-fold lower bioavailability, PK analysis documented ESATA-20 has an alpha half-life for tissue distribution of 3 hrs, while the beta half-life for serum elimination is 10–12 hrs with >98% of the drug bound to serum proteins, which is very similar to these serum parameters for TasQ16. ESATA-20 (10 mg/kg/d) produces the same significant maximal anti-cancer efficacy as TasQ at the same 10 mg/kg/d dose, Figure 6G. Thus, on a molar basis, ESATA-20 is at least 5-fold more potent than TasQ in inhibiting CWR22-RH xenograft growth. It is also more selective than TasQ as documented by its lack of induction of both liver Cyp1a1, Figure 6H, and thymus regression, Figure 6A, even when ESATA-20 is given orally at a 3-fold higher dose (i.e., 30 mg/kg/d).
Discussion
Solid malignancies outgrow their blood supply producing hypoxia and nutrient limitations within their TME. This enhances glycolysis and amino acid catabolism, in addition to increasing lactate secretion, resulting in an acidic extracellular TME50. Previously, we have documented that such a hypoxic TME occurs early within growing prostate cancer xenografts even when they are less than 0.3 cc13. In this compromised TME, enhanced stability of HIF-1α protein has a major role in promoting cancer growth, progression, and chemotherapy resistance through metabolic reprogramming by upregulating the expression of angiogenic and cell stress survival genes50. Such hypoxia-induced enhancement of HIF-1α protein is an early event in human prostate carcinogenesis51. We and others have documented that compounds that decrease HIF-1α protein are therapeutic in vivo against a variety of preclinical solid malignancy models, including prostate cancer13,15,54,61,62. The present studies document that TasQ reversibly binds with a 1 μM EC50 to HDAC4 producing a negative allosteric effect that disrupts HDAC4/NCoR1/HDAC3 complex formation. This prevents deacetylation-dependent stability of HIF-1α protein, and thus upregulation of its target genes coupled with inhibiting the downregulation of MEF-2 target genes; both responses needed for lethal cancer growth in the compromised TME. Therefore, for optimal therapeutic efficacy, TasQ must be maintained at a blood concentration of >2 μM.
Unfortunately, in addition to this on-target HDAC4-dependent MoA, TasQ also is an agonistic ligand for the AHR, which is associated with significant cardiomyopathies29–31. This is consistent with TasQ dose limiting toxic effects on the heart as detected in clinical Phase I testing19. The present studies document that such AHR agonist binding is an “off-target” effect with an EC50 of 1 μM, which is equivalent to the EC50 for binding to HDAC4. Thus, clinical dose escalation of TasQ above 1 mg/d as needed to maximize its HDAC4-dependent therapeutic activity will also enhance its unwanted systemic side effects (e.g., thymus regression and liver Cyp1A1 induction). Fortunately, TasQ’s therapeutic MoA against prostate tumor models is independent of this off-target AHR agonism.
These results validate the development of a 3rd-generation quinoline-3-carboxamide analog that retains on-target HDAC4/HIF-1α/MEF-2 disruption, while decreasing off-target AHR side effects, and thus allowing higher daily oral dosing to optimize its anti-prostate cancer efficacy in humans. ESATA-20 is such a 3rd-generation quinoline-3-carboxamide systemic analog, which is a ≥5-fold more potent HDAC4 inhibitor and a 10-fold less potent AHR agonist. This translates into ESATA-20’s maximal anti-prostate cancer efficacy produced at a serum concentration of <2 μM, which is below its EC50 (10 μM) for AHR agonism. Importantly, ESATA-20 at this dose (10 μM) produced <15% change in activity in any of the 68 kinases tested in a targeted kinase screen representing a wide range of kinase families, Supplemental Figure 3 and Supplemental Table 1. Thus, daily oral dosing of ESATA-20 should be titrated to produce a serum drug level of 2–5 μM; thereby, optimizing its therapeutic efficacy as an HDAC4/HIF1α/MEF-2 disruptor when given to mCRPC patients without inducing the unwanted off-target AHR agonist side effects.
Though ESATA-20 as a single oral agent inhibits CRPC growth via its disruption of HIF-1α/MEF-2 transcriptional signaling within the compromised hypoxic TME; it does not produce tumor regression. Thus, optimal use of ESATA-20 even at oral doses that produce an optimal blood level will not be administered as monotherapy. Instead, its decrease in off-target AHR agonism should allow continuous maintenance on oral ESATA-20 as early as possible in the progression of metastatic prostate cancer and combined with other agents given episodically that induce cancer cell death (e.g., taxanes, PARP inhibitors, platins, immunotherapy, etc.). This is presently being tested in preclinical models.
Supplementary Material
Statement of Significance:
Tasquinimod is an orally active quinoline-3-carboxamide with documented clinical activity against metastatic castration-resistant prostate cancer, but failed due to dose-limiting toxicity. Herein, we demonstrate this toxicity is an off-target effect due to AHR agonism and report a 3rd generation analog as a lead clinical candidate with an improved therapeutic index.
Acknowledgements
We wish to thank Per Björk at Active Biotech for his Biocore Surface Plasmon Resonance (SPR) Binding Analysis. We acknowledge the following sources of financial support: Department of Defense Prostate Cancer Research Program Awards W81XWH-16-1-0410 and W81XWH-18-1-0348 (to JTI); W81XWH-18-1-0347 and W81XWH-17-1-0415 (to PSN); W81XWH-17-1-0528 (to WNB); NIH-Prostate SPORE grant P50 CA058236 (to JTI); NIH R35 ES028244 (GHP); NIH R01 CA255259 (WNB), Emerson Collective Cancer Research Fund [643396, (WNB)]; Allegheny Health Network-Johns Hopkins University Cancer Research Fund (WNB); and the NIH-SKCCC Cancer Center Support Grant award P30 CA006973, which provides support for the Oncology Tissue Services, Animal Resources, Cell Imaging, and Genetic Resources. We would also like to thank the Johns Hopkins School of Medicine Mass Spectrometry and Proteomics Core facility supported by P30 CA006973 for the MALDI-MS analysis; and Johns Hopkins School of Medicine Pharmacology Department for the H-1 and C-13 NMR analysis.
Footnotes
Conflict of Interest: The authors have declared that no conflict of interest exists.
References
- 1.Ichikawa T, Lamb JC, Christensson PI, Hartley-Asp B & Isaacs JT The antitumor effects of the quinoline-3-carboxamide linomide on Dunning R-3327 rat prostatic cancers. Cancer Res 52, 3022–3028 (1992). [PubMed] [Google Scholar]
- 2.Vukanovic J et al. Antiangiogenic effects of the quinoline-3-carboxamide linomide. Cancer Res 53, 1833–1837 (1993). [PubMed] [Google Scholar]
- 3.Vukanovic J & Isaacs JT Linomide inhibits angiogenesis, growth, metastasis, and macrophage infiltration within rat prostatic cancers. Cancer Res 55, 1499–1504 (1995). [PubMed] [Google Scholar]
- 4.Vukanovic J, Hartley-Asp B & Isaacs JT Inhibition of tumor angiogenesis and the therapeutic ability of linomide against rat prostatic cancers. Prostate 26, 235–246, doi: 10.1002/pros.2990260503 (1995). [DOI] [PubMed] [Google Scholar]
- 5.Joseph IB, Vukanovic J & Isaacs JT Antiangiogenic treatment with linomide as chemoprevention for prostate, seminal vesicle, and breast carcinogenesis in rodents. Cancer Res 56, 3404–3408 (1996). [PubMed] [Google Scholar]
- 6.Joseph IB & Isaacs JT Macrophage role in the anti-prostate cancer response to one class of antiangiogenic agents. J Natl Cancer Inst 90, 1648–1653, doi: 10.1093/jnci/90.21.1648 (1998). [DOI] [PubMed] [Google Scholar]
- 7.Khan SR, Mhaka A, Pili R & Isaacs JT Modified synthesis and antiangiogenic activity of linomide. Bioorg Med Chem Lett 11, 451–452, doi: 10.1016/s0960-894x(00)00699-5 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Tan IL, Lycklama a Nijeholt GJ, Polman CH, Ader HJ & Barkhof F Linomide in the treatment of multiple sclerosis: MRI results from prematurely terminated phase-III trials. Mult Scler 6, 99–104, doi: 10.1177/135245850000600208 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Isaacs JT The long and winding road for the development of tasquinimod as an oral second-generation quinoline-3-carboxamide antiangiogenic drug for the treatment of prostate cancer. Expert Opin Investig Drugs 19, 1235–1243, doi: 10.1517/13543784.2010.514262 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Isaacs JT et al. Identification of ABR-215050 as lead second generation quinoline-3-carboxamide anti-angiogenic agent for the treatment of prostate cancer. Prostate 66, 1768–1778, doi: 10.1002/pros.20509 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Dalrymple SL, Becker RE & Isaacs JT The quinoline-3-carboxamide anti-angiogenic agent, tasquinimod, enhances the anti-prostate cancer efficacy of androgen ablation and taxotere without effecting serum PSA directly in human xenografts. Prostate 67, 790–797, doi: 10.1002/pros.20573 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Olsson A, Bjork A, Vallon-Christersson J, Isaacs JT & Leanderson T Tasquinimod (ABR-215050), a quinoline-3-carboxamide anti-angiogenic agent, modulates the expression of thrombospondin-1 in human prostate tumors. Mol Cancer 9, 107, doi: 10.1186/1476-4598-9-107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dalrymple SL, Becker RE, Zhou H, DeWeese TL & Isaacs JT Tasquinimod prevents the angiogenic rebound induced by fractionated radiation resulting in an enhanced therapeutic response of prostate cancer xenografts. Prostate 72, 638–648, doi: 10.1002/pros.21467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jennbacken K et al. Inhibition of metastasis in a castration resistant prostate cancer model by the quinoline-3-carboxamide tasquinimod (ABR-215050). Prostate 72, 913–924, doi: 10.1002/pros.21495 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Isaacs JT et al. Tasquinimod Is an Allosteric Modulator of HDAC4 survival signaling within the compromised cancer microenvironment. Cancer Res 73, 1386–1399, doi: 10.1158/0008-5472.CAN-12-2730 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Isaacs JT et al. Anti-cancer potency of tasquinimod is enhanced via albumin-binding facilitating increased uptake in the tumor microenvironment. Oncotarget 5, 8093–8106, doi: 10.18632/oncotarget.2378 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen L et al. Tasquinimod modulates suppressive myeloid cells and enhances cancer immunotherapies in murine models. Cancer Immunol Res 3, 136–148, doi: 10.1158/2326-6066.CIR-14-0036 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brennen WN et al. Assessing angiogenic responses induced by primary human prostate stromal cells in a three-dimensional fibrin matrix assay. Oncotarget 7, 71298–71308, doi: 10.18632/oncotarget.11347 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bratt O et al. Open-label, clinical phase I studies of tasquinimod in patients with castration-resistant prostate cancer. Br J Cancer 101, 1233–1240, doi: 10.1038/sj.bjc.6605322 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pili R et al. Phase II randomized, double-blind, placebo-controlled study of tasquinimod in men with minimally symptomatic metastatic castrate-resistant prostate cancer. J Clin Oncol 29, 4022–4028, doi: 10.1200/JCO.2011.35.6295 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Armstrong AJ et al. Long-term survival and biomarker correlates of tasquinimod efficacy in a multicenter randomized study of men with minimally symptomatic metastatic castration-resistant prostate cancer. Clin Cancer Res 19, 6891–6901, doi: 10.1158/1078-0432.CCR-13-1581 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fizazi K et al. A randomized, double-blind, placebo-controlled phase II study of maintenance therapy with tasquinimod in patients with metastatic castration-resistant prostate cancer responsive to or stabilized during first-line docetaxel chemotherapy. Ann Oncol 28, 2741–2746, doi: 10.1093/annonc/mdx487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sternberg C et al. Randomized, Double-Blind, Placebo-Controlled Phase III Study of Tasquinimod in Men With Metastatic Castration-Resistant Prostate Cancer. J Clin Oncol 34, 2636–2643, doi: 10.1200/JCO.2016.66.9697 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Armstrong AJ et al. Phase Ib Trial of Cabazitaxel and Tasquinimod in Men With Heavily Pretreated Metastatic Castration Resistant Prostate Cancer (mCRPC): The CATCH Trial. Prostate 77, 385–395, doi: 10.1002/pros.23277 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gong P et al. Efficacy of tasquinimod in men with metastatic castration-resistant prostate cancer: A meta-analysis of randomized controlled trials. Medicine (Baltimore) 97, e13204, doi: 10.1097/MD.0000000000013204 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahiout S et al. Toxicological characterisation of two novel selective aryl hydrocarbon receptor modulators in Sprague-Dawley rats. Toxicol Appl Pharmacol 326, 54–65, doi: 10.1016/j.taap.2017.04.020 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Berg J et al. The immunomodulatory effect of laquinimod in CNS autoimmunity is mediated by the aryl hydrocarbon receptor. J Neuroimmunol 298, 9–15, doi: 10.1016/j.jneuroim.2016.06.003 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Kaye J et al. Laquinimod arrests experimental autoimmune encephalomyelitis by activating the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A 113, E6145–E6152, doi: 10.1073/pnas.1607843113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gerger CJ, Thomas JK, Janz DM & Weber LP Acute effects of beta-naphthoflavone on cardiorespiratory function and metabolism in adult zebrafish (Danio rerio). Fish Physiol Biochem 41, 289–298, doi: 10.1007/s10695-014-9982-z (2015). [DOI] [PubMed] [Google Scholar]
- 30.Zhou B et al. Mitochondrial activity and oxidative stress functions are influenced by the activation of AhR-induced CYP1A1 overexpression in cardiomyocytes. Mol Med Rep 16, 174–180, doi: 10.3892/mmr.2017.6580 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anandasadagopan SK et al. beta-Naphthoflavone-Induced Mitochondrial Respiratory Damage in Cyp1 Knockout Mouse and in Cell Culture Systems: Attenuation by Resveratrol Treatment. Oxid Med Cell Longev 2017, 5213186, doi: 10.1155/2017/5213186 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kallberg E et al. S100A9 interaction with TLR4 promotes tumor growth. PLoS One 7, e34207, doi: 10.1371/journal.pone.0034207 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jonsson S et al. Synthesis and biological evaluation of new 1,2-dihydro-4-hydroxy-2-oxo-3-quinolinecarboxamides for treatment of autoimmune disorders: structure-activity relationship. J Med Chem 47, 2075–2088, doi: 10.1021/jm031044w (2004). [DOI] [PubMed] [Google Scholar]
- 34.Nayal OS, Bhatt V, Sharma S & Kumar N Chemoselective Reductive Amination of Carbonyl Compounds for the Synthesis of Tertiary Amines Using SnCl2.2H2O/PMHS/MeOH. J Org Chem 80, 5912–5918, doi: 10.1021/acs.joc.5b00156 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Backs J, Backs T, Bezprozvannaya S, McKinsey TA & Olson EN Histone deacetylase 5 acquires calcium/calmodulin-dependent kinase II responsiveness by oligomerization with histone deacetylase 4. Mol Cell Biol 28, 3437–3445, doi: 10.1128/MCB.01611-07 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu Y et al. Role of androgen receptor splice variant-7 (AR-V7) in prostate cancer resistance to 2nd-generation androgen receptor signaling inhibitors. Oncogene 39, 6935–6949, doi: 10.1038/s41388-020-01479-6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foster BA, Gingrich JR, Kwon ED, Madias C & Greenberg NM Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res 57, 3325–3330 (1997). [PubMed] [Google Scholar]
- 38.Simons BW et al. A mouse model of prostate cancer bone metastasis in a syngeneic immunocompetent host. Oncotarget 10, 6845–6854, doi: 10.18632/oncotarget.27317 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watson PA et al. Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res 65, 11565–11571, doi: 10.1158/0008-5472.CAN-05-3441 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Brennen WN et al. Resistance to androgen receptor signaling inhibition does not necessitate development of neuroendocrine prostate cancer. JCI Insight, doi: 10.1172/jci.insight.146827 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bottomley MJ et al. Structural and functional analysis of the human HDAC4 catalytic domain reveals a regulatory structural zinc-binding domain. J Biol Chem 283, 26694–26704, doi: 10.1074/jbc.M803514200 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Flaveny CA, Murray IA, Chiaro CR & Perdew GH Ligand selectivity and gene regulation by the human aryl hydrocarbon receptor in transgenic mice. Mol Pharmacol 75, 1412–1420, doi: 10.1124/mol.109.054825 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davarinos NA & Pollenz RS Aryl hydrocarbon receptor imported into the nucleus following ligand binding is rapidly degraded via the cytosplasmic proteasome following nuclear export. J Biol Chem 274, 28708–28715, doi: 10.1074/jbc.274.40.28708 (1999). [DOI] [PubMed] [Google Scholar]
- 44.Uno S et al. Oral exposure to benzo[a]pyrene in the mouse: detoxication by inducible cytochrome P450 is more important than metabolic activation. Mol Pharmacol 65, 1225–1237, doi: 10.1124/mol.65.5.1225 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Martin M, Kettmann R & Dequiedt F Class IIa histone deacetylases: regulating the regulators. Oncogene 26, 5450–5467, doi: 10.1038/sj.onc.1210613 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Di Giorgio E et al. MEF2 is a converging hub for histone deacetylase 4 and phosphatidylinositol 3-kinase/Akt-induced transformation. Mol Cell Biol 33, 4473–4491, doi: 10.1128/MCB.01050-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geng H et al. HDAC4 protein regulates HIF1alpha protein lysine acetylation and cancer cell response to hypoxia. J Biol Chem 286, 38095–38102, doi: 10.1074/jbc.M111.257055 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Park SY et al. Structural basis of the specific interaction of SMRT corepressor with histone deacetylase 4. Nucleic Acids Res 46, 11776–11788, doi: 10.1093/nar/gky926 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matsuoka H et al. Disruption of HDAC4/N-CoR complex by histone deacetylase inhibitors leads to inhibition of IL-2 gene expression. Biochem Pharmacol 74, 465–476, doi: 10.1016/j.bcp.2007.05.002 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Samanta D & Semenza GL Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim Biophys Acta Rev Cancer 1870, 15–22, doi: 10.1016/j.bbcan.2018.07.002 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Zhong H, Semenza GL, Simons JW & De Marzo AM Up-regulation of hypoxia-inducible factor 1alpha is an early event in prostate carcinogenesis. Cancer Detect Prev 28, 88–93, doi: 10.1016/j.cdp.2003.12.009 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Jiang BH, Rue E, Wang GL, Roe R & Semenza GL Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem 271, 17771–17778, doi: 10.1074/jbc.271.30.17771 (1996). [DOI] [PubMed] [Google Scholar]
- 53.Michel G, Minet E, Mottet D, Remacle J & Michiels C Site-directed mutagenesis studies of the hypoxia-inducible factor-1alpha DNA-binding domain. Biochim Biophys Acta 1578, 73–83, doi: 10.1016/s0167-4781(02)00484-0 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Qian DZ et al. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res 66, 8814–8821, doi: 10.1158/0008-5472.CAN-05-4598 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Seo HW, Kim EJ, Na H & Lee MO Transcriptional activation of hypoxia-inducible factor-1alpha by HDAC4 and HDAC5 involves differential recruitment of p300 and FIH-1. FEBS Lett 583, 55–60, doi: 10.1016/j.febslet.2008.11.044 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Denko NC Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat Rev Cancer 8, 705–713, doi: 10.1038/nrc2468 (2008). [DOI] [PubMed] [Google Scholar]
- 57.McKinsey TA, Zhang CL & Olson EN MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci 27, 40–47, doi: 10.1016/s0968-0004(01)02031-x (2002). [DOI] [PubMed] [Google Scholar]
- 58.Han A, He J, Wu Y, Liu JO & Chen L Mechanism of recruitment of class II histone deacetylases by myocyte enhancer factor-2. J Mol Biol 345, 91–102, doi: 10.1016/j.jmb.2004.10.033 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Sturtzel C, Testori J, Schweighofer B, Bilban M & Hofer E The transcription factor MEF2C negatively controls angiogenic sprouting of endothelial cells depending on oxygen. PLoS One 9, e101521, doi: 10.1371/journal.pone.0101521 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nakatsu MN et al. Angiogenic sprouting and capillary lumen formation modeled by human umbilical vein endothelial cells (HUVEC) in fibrin gels: the role of fibroblasts and Angiopoietin-1. Microvasc Res 66, 102–112, doi: 10.1016/s0026-2862(03)00045-1 (2003). [DOI] [PubMed] [Google Scholar]
- 61.Zhang H et al. Digoxin and other cardiac glycosides inhibit HIF-1alpha synthesis and block tumor growth. Proc Natl Acad Sci U S A 105, 19579–19586, doi: 10.1073/pnas.0809763105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee K et al. Acriflavine inhibits HIF-1 dimerization, tumor growth, and vascularization. Proc Natl Acad Sci U S A 106, 17910–17915, doi: 10.1073/pnas.0909353106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.