

Abstract
Lung cancer is one of the leading causes of cancer-related death. Most advanced lung adenocarcinoma (LUAD) patients have poor survival because of drug resistance and relapse. Neglecting intratumoral heterogeneity might be one of the reasons for treatment insensitivity, while single-cell RNA sequencing (scRNA-seq) technologies can provide transcriptome information at the single-cell level. Herein, we combined scRNA-seq and bulk RNA-seq data of LUAD and identified a novel cluster of malignant epithelial cells - KRT81+ malignant epithelial cells - associated with worse prognoses. Further analysis revealed that the hypoxia and EMT pathways of these cells were activated to predispose them to differentiate into metastatic lung adenocarcinoma cells. Finally, we also studied the role of these tumor cells in the immune microenvironment and their role in the classification and prognosis prediction of lung adenocarcinoma patients.
Keywords: Lung adenocarcinoma, Single-cell RNA-Seq, Prognosis prediction, Lung cancer
Graphical abstract

Graphical Abstract
1. Introduction
Lung cancer is the leading cause of cancer-related death [1]. About 80–85% of lung cancer cases are non-small cell lung cancer and include two main histological subtypes: lung squamous cell carcinoma and lung adenocarcinoma (LUAD). Besides, LUAD accounts for most new cases. Advanced lung cancer is the leading cause of death in lung cancer cases. Although the overall mortality rate of lung cancer has rapidly decreased with the clinical application of PD-1/PD-L1 immunotherapy and EGFR-TKIs targeted therapy in advanced lung cancer, many cases are still insensitive to these treatments [2]. Therefore, finding new prognostic monitoring biomarkers and therapeutic targets remains necessary.
As RNA sequencing (RNA-seq) matures, its high-throughput and efficient advantages help identify tumor molecular features to mine new therapeutic targets and clinical biomarkers [3]. RNA-seq also has a great role in promoting the current concept of precision treatment. However, bulk RNA-seq is based on whole tissue transcriptomes, ignoring intratumoral heterogeneity. At the same time, although many algorithms [[4], [5], [6]] use bulk RNA-seq data to infer the composition of immune cells in the tumor microenvironment, the inability to obtain transcriptome information of immune cells also weakens the understanding of the tumor microenvironment. Intratumoral heterogeneity often leads to different responsiveness of patients throughout immunotherapy and targeted therapy cycles [7]. Moreover, tumor cells that are insensitive to treatment are one of the causes of drug resistance and relapse after treatment [8,9]. Therefore, understanding the transcriptional landscape within tumors in a higher dimension is urgent. Meanwhile, single-cell RNA-seq (scRNA-seq) can solve the lack of understanding of intratumoral heterogeneity and tumor microenvironment in bulk RNA-seq. Additionally, scRNA-seq provides transcriptome sequencing at single-cell resolution, and various associated single-cell algorithms have deepened the understanding of single-cell transcriptional characteristics, cell dynamics, cell-cell interactions, and intratumoral heterogeneity [10,11].
The single-cell transcriptome landscape of LUAD has been reported in several articles [[12], [13], [14], [15], [16], [17], [18], [19]], establishing a preliminary understanding of its intratumoral heterogeneity and providing valuable raw data for subsequent studies. Herein, we combined scRNA-seq and bulk RNA-seq data from LUAD to identify a novel cluster of LUAD tumor cells associated with prognosis. We also profoundly investigated the transcriptome characteristics, the role in the tumor microenvironment, and the unique role of prognostic monitoring. Overall, we revealed an entirely novel cluster of tumor cells that can be used for prognostic prediction and might be potential therapeutic targets for LUAD.
2. Results
2.1. Primary and metastatic LUAD single-cell atlas
The single-cell RNA-seq dataset used in this study contains eight primary cases, five metastatic LUAD, and four normal lung tissues (Table S1). After quality control, dimension reduction, and clustering, we identified 21 unique clusters (Fig. 1, figs1A). According to the marker gene expression in clusters, cells were mainly annotated as epithelial cells (cell marker: EPCAM, CDH1, KRT7, KRT19, KRT8 and KRT18), immune cells (cell marker: PTPRC, CD3E, GZMB, CD68, CD79A, and MS4A2), and stromal cells (cell marker: DCN, COL1A1, COL1A2, THY1, PECAM1, CLDN5, and RAMP2) (Fig. 1B–C). For a more refined annotation of immune cells, we extracted them and re-performed dimensionality reduction and clustering (Fig. 1D). Similarly, we annotated each of the seven major cell types based on their cell-specific gene expression marker (Fig. 1E–F). In the UMAP graph for all cells, cells of the same type were adjacent and showed large genomic differences between different cell types (Fig. 1G). Most immune or stromal cells of the same type were adjacent, while a larger distance was detected between epithelial cell clusters. Since LUAD is derived from epithelial cells [20,21], this result reflected the heterogeneity between LUAD cancer cells. After counting the proportion of cells in each sample, we found that the cell proportion distribution also had a large difference between samples (Fig. 1H). Altogether, these results suggest that there is significant heterogeneity in LUAD cells.
Fig. 1.

Single cell atlas of primary and metastatic lung adenocarcinoma. (A) UMAP plot for all cells clustered into 21 clusters. (B) Expression of cell marker genes in each cell cluster. (C) All cells were annotated as three major cell types including immune cells, epithelial cells, and stromal cells. (D) UMAP plot for immune cells clustered into 22 clusters. (E) Expression of immune cell marker genes in each cell cluster. (F) Cell clusters were annotated as seven different types of immune cells. (G) UMAP Plot for all cells after all cell annotations are integrated. (H) Proportion of different cell types in each sample.
2.2. Copy number analysis of epithelial cells in LUAD
To exclude the incorporation of normal epithelial cells into tumor samples due to sampling, we used a previously published method [16] to infer the exact malignant cells using copy number variation (CNV). According to previously published articles, epithelial cells with high CNV were considered as malignant cells [[22], [23], [24]]. In our result, almost all tumor tissues, most epithelial cells presented higher CNV compared with artificially inserted epithelial cells derived from normal lung tissues. We defined these cells as tumor cells and identified the remaining fraction as normal lung epithelial cells incorporated during tissue sampling (Fig. 2A). Further, we used inferCNV [25] to characterize each chromosome and showed good agreement with the above methods. Notably, we found significant copy number deletions on chromosome 13 in almost all tumor cells (Fig. 2B). Previous studies have reported frequent loss of heterozygosity of alleles in the chromosome 13 region in lung cancer, leading to the inactivation of multiple tumor suppressor genes [[26], [27], [28]]. Therefore, CNV and loss of heterozygosity on chromosome 13 might be associated with lung cancer development. Next, all normal epithelial cells were re-clustered and cell-type annotated. We identified normal epithelial cells as alveolar type I, alveolar type II, secretory club, and airway ciliated cells (Fig. 2C–E). After integrating all epithelial cells and performing re-clustering, AT2 and Club cells were closer to tumor cells, which might be related to previous studies indicating that AT2 and Club cells are potential LUAD progenitors [21,29] (Fig. 2F).
Fig. 2.

Copy Number Analysis for All Epithelial Cells. (A) CNV analysis of all tumor tissue-derived epithelial cells with inserted normal tissue-derived normal cells; the gray dots represent inserted normal tissue-derived normal cells, blue dots are defined as normal epithelial cells in tumor tissue, and red dots are defined as tumor cells. (B) Analysis of copy number loss or amplification of each chromosome in all epithelial cells by InferCNV algorithm.(C) UMAP plot for all normal epithelial cells clustered into four clusters. (D) Expression of cell marker genes in each cell cluster. (E) All clusters were annotated as four major cell types. (F) UMAP plot of all normal and malignant epithelial cells.
2.3. Trajectory analysis of LUAD tumor cells reveals heterogeneity in lung adenocarcinoma
Subsequently, We performed trajectory analysis on all tumor cells using the 'moncel2' R package, an algorithm that can identify cellular transcriptional heterogeneity from single-cell data. We perform trajectory inference on all tumor cells. The two trajectory inflection points identified showed differences in the heterogeneity of lung adenocarcinoma tumor cells. In addition, we also identified developmental pseudotimes and five cell subpopulations on the trajectory (Fig. 3A). In pseudo-time, primary tumor cells showed three different developmental trajectories to metastatic tumor cells, illustrating the heterogeneity presented by the development of primary tumor cells into metastatic LUAD tumor cells (Fig. 3A). In pseudo time, we identified the top 100 hypervariable genes. These genes were clustered into 6 clusters according to their pseudotemporal expression, with a total of 50 upregulated and 50 downregulated (Fig. 3B). After Gene Ontology (GO) functional enrichment analysis, we found that upregulated genes were involved in epithelial cell proliferation, chemotaxis, and migration of various immune cells. However, downregulated genes were enriched in the maintenance and homeostasis of epithelial cells (Fig. 3C–D and Tables S2–S3). These results indicated that uncontrolled cell proliferation and loss of homeostasis maintenance are involved in the heterogeneous metastasis of lung cancer tumor cells and can regulate chemotaxis and migration of various immune cells.
Fig. 3.

Cell trajectory analysis of tumor cells. (A) All tumor cells differentiation trajectories, pseudotime distribution, and cell clusters on pseudotime. (B) Top 50 highly expressed genes and top 50 lowly expressed genes in pseudo-time. (C) Gene Ontology Functional Enrichment Analysis of Top 50 highly expressed genes. (D) Gene Ontology Functional Enrichment Analysis of Top 50 lowly expressed genes. *BP:Biological Process, CC:Cell Component, MF:Molecular Function. And the numbers on the x-axis represent the number of genes enriched in each pathway.
2.4. KRT81+ tumor cells proportion are associated with poor prognosis in LUAD patients
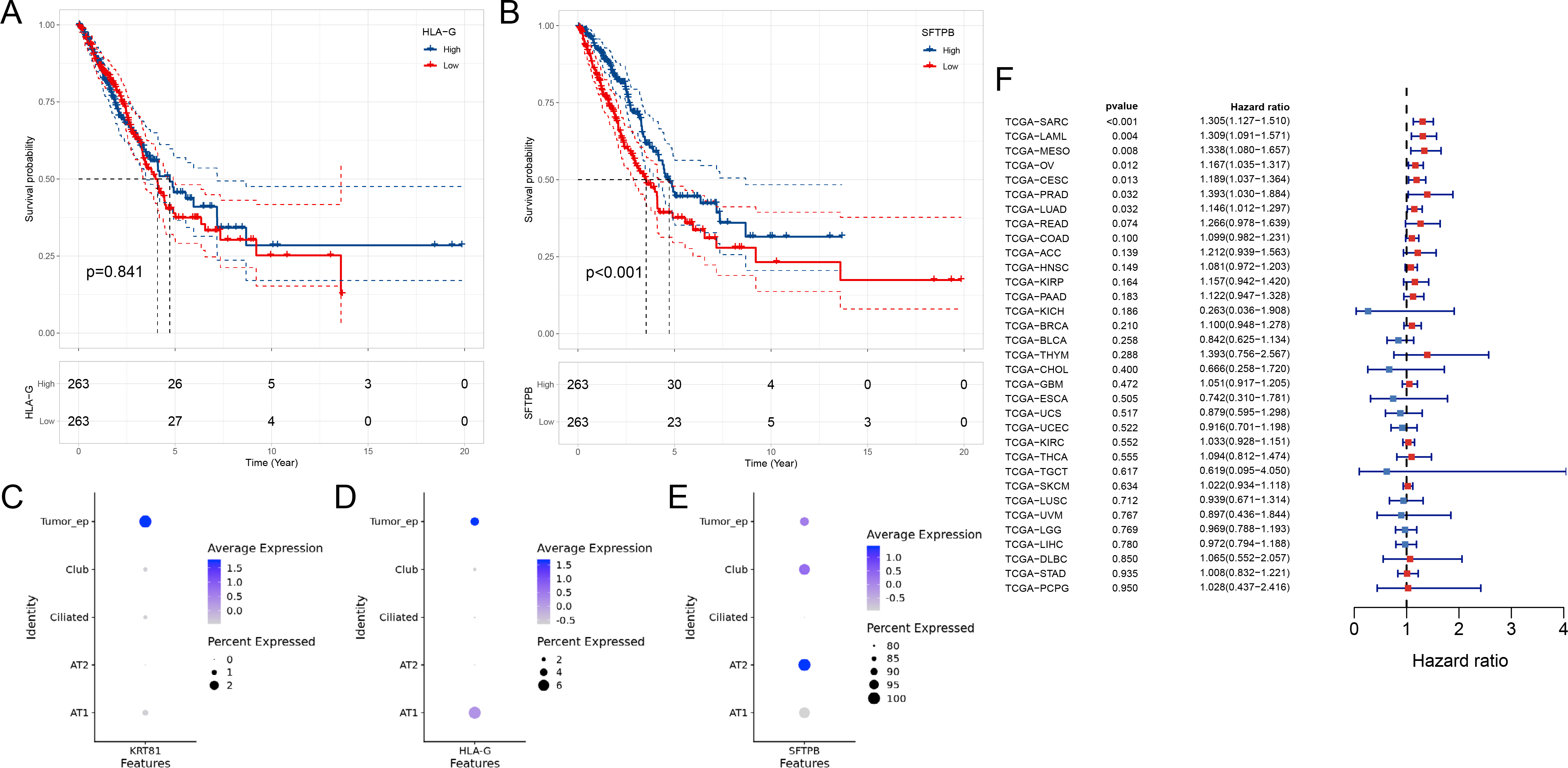
To further study the potential functions of these heterogeneous tumor cells.We further clustered all tumor cells and named them according to the characteristically expressed genes of each cluster (Fig. 4A–B). All 10 clusters emerged as unique clusters in the UMAP plot, demonstrating large differences in their transcriptome status (Fig. 4C). Since single-cell data sets are limited by sample size and prognostic follow-up information, we used the CIBERSORT [5] algorithm to deconvolute the TCGA-LUAD dataset to evaluate the impact of the proportion of these cell clusters on prognosis (Fig. 4D). The clinical information of the TCGA-LUAD dataset has been summarized in Table S4. Then, in the TCGA-LUAD cohort, we performed a univariate Cox analysis of the estimated proportion of each tumor cell cluster. The proportion of KRT81+ tumor cells was a risk factor for the poor prognosis of LUAD patients (Fig. 4E). The survival curves also confirmed this result for KRT81+ tumor cell proportion or KRT81 gene expression grouping (Fig. 4F–G). In addition, we also examined the effect of the expression of another high-expression gene HLA-G and a low-expression gene SFTPB of KRT81+ cell cluster on the prognosis in lung adenocarcinoma. The results showed that the expression level of HLA-G was not related to the prognosis, but the patients with high expression of SFTPB had a better prognosis (Figs. S2A–B). However, it is worth noting that when we examined the expression of these three genes in all epithelial cells in single-cell dataset, only KRT81 was uniquely expressed in tumor cells, and SFTPB was more expressed in normal alveolar epithelium (Figs. S2C–E). Therefore, the association between KRT81+ cell cluster and prognosis may be more driven by the KRT81 gene. We also examined the association of KRT81 gene expression with prognosis in other cancers using univariate COX, and the results showed that it was also associated with poorer prognosis in seven other cancers from the TCGA database. But it is worth noting that KRT81 was only associated with poorer prognosis in lung adenocarcinoma compared with lung squamous cell carcinoma (Fig. S2F). Therefore, the association of KRT81 with poor prognosis is not specific to lung cancer, but its role in lung adenocarcinoma is relatively specific for other lung cancer pathological subtypes. In the TCGA-LUAD cohort, we detected significant activation of hypoxia and epithelial-mesenchymal transition pathways in the high KRT81 expression group (Fig. 4H–I). This result was consistent with the higher scoring of gene sets for epithelial-mesenchymal transition (EMT), hypoxia, metastasis, and invasion phenotypes for the KRT81+ cell cluster in the scRNA-seq data (Fig. 4J-M). Overall, we identified a new cluster of tumor cells associated with poor prognosis in LUAD, KRT81+ tumor cells, that might be associated with the activation of EMT and hypoxia pathways.
Fig. 4.

KRT81+ tumor cells proportion are associated with worse prognosis in lung adenocarcinoma. (A) All tumor cells were divided into 10 clusters. (B) Top 5 highly expressed genes and top 5 low expressed genes in each tumor cell cluster. (C) Ten tumor cell clusters were named according to the highest differentially expressed genes. (D) Deconvolute the TCGA-LUAD cohort using the CibersortX algorithm to infer the proportion of each cell type in each sample. (E) Univariate COX analysis of the proportion of each tumor cell cluster in the TCGA-LUAD cohort, p-value <0.05 was considered to be associated with prognosis. (F) Survival analysis of the TCGA-LUAD cohort grouped according to the median proportion of KRT8+ tumor cells. (G) Survival analysis of the TCGA-LUAD cohort grouped according to the median KRT81 gene expression. (H–I) GSEA functional enrichment analysis of the TCGA-LUAD cohort grouped according to the median KRT81 gene expression. (J–M) Gene set scoring in single-cell datasets using the AddModuleScore function of the "Seurat" R package.
2.5. Experimental validation of the influence of KRT81 on migration and invasion in lung adenocarcinoma cell lines
Through the analysis above, we conclude that KRT81 may be associated with a poorer prognosis in lung adenocarcinoma. Previous research indicates that KRT81 enhances cell migration and invasion in breast cancer [30] and melanoma [31]. Therefore, to investigate its role in lung adenocarcinoma cell lines, we carried out the following experimental validation. We constructed an overexpression plasmid marked with Flag tag for KRT81 and three shRNAs for KRT81 knockdown, and transfected them into A549 and H1299 cell lines, with the transfection effects confirmed by Western blot. The results demonstrated that the overexpression plasmid achieved good overexpression of KRT81 in both cell lines, and among the three shRNAs, the 2nd one yielded good knockdown effect on KRT81 at the protein level (Fig. 5A–D). Hence, sh-KRT81#2 was used for the subsequent experiments. The wound healing experiment was used to measure cell migration ability. The results indicated that compared to the control group, the wound healing ratio in A549 and H1299 cells overexpressing KRT81 significantly increased, while the two cell lines with knockdown of KRT81 demonstrated a decrease in migration ability (Fig. 5E–F). The Transwell assay indicated that cells overexpressing KRT81 invade more into the lower chamber, whereas the number of invasive cells decreased following the knockdown of KRT81 (Fig. 5G). In conclusion, these experimental results indicate that KRT81 promotes the migration and invasion of LUAD cell lines.
Fig. 5.

Experimental validation in lung adenocarcinoma cell lines. (A–D) KRT81 protein expression was detected by Western blot in A549 and H1299 cell lines transfected with overexpression or knockdown plasmids. The relative expression of KRT81 was calculated for statistical analysis. (E) After the overexpression or knockdown of KRT81 in A549 and H1299 cells in the 6-well plate reached 100% growth density, artificial wounds were made and replaced with serum-free medium. Wound pictures were taken at 0h and 6h respectively, and the percentage of wound healing was calculated. Percentage of wound healing = (Area of wound at 0h - Area of wound at 6h)/Area of wound at 0h. (F) Inoculate the same number of control cells and cells with overexpression or knockdown of KRT81 in the upper chamber. After 24 h, stain and photograph the cells in the lower chamber. Count the number of cells that have invaded the lower chamber and perform statistical analysis between groups. In above figures: * represents p-value <0.05, ** represents p-value <0.01, *** represents p-value <0.001, **** represents p-value <0.0001.
2.6. Transcriptional characterization of KRT81+ tumor cell cluster and interactions with immune cells
Based on the association of KRT81+ tumor cells with worse prognosis in LUAD patients, we further analyzed this tumor cell cluster. To study the transcriptional state of KRT81+ tumor cells, we used a python-based "Velocity" algorithm. This algorithm characterizes RNA velocity based on the ratio of spliced and unspliced mRNA and infers developmental direction [32]. In our results, KRT81+ tumor cell cluster (a subset of primary tumor cells) had a tendency to develop into metastatic tumor cells. Thus, such clusters of tumor cells may contribute to disease progression (Fig. 6A–C). Moreover, in the transcriptional factor analysis of various tumor cell clusters, We identified two transcription factors of interest, TFAP2A and WRNIP1, in KRT81+ tumor cells. (Fig. 6D). TFAP2A has been reported to promote LUAD progression by activating the EMT [[33], [34], [35]] and promoting angiogenesis in acquired resistance [36]. WRNIP1 is associated with cell cycle promotion in LUAD [37] and tumor radioresistance [38].
Fig. 6.

Transcriptional characterization of KRT81+ tumor cells and interactions with immune cells. (A) RNA velocity analysis of malignant epithelial cells. (B) UMAP plot of all malignant epithelial cells. (C) All tumor epithelial cells are annotated according to primary or metastatic origin. (D) Transcriptional factor analysis of all malignant epithelial cells. (E) Analysis of ligand-receptor interaction between KRT81+ tumor cells and immune cells using "CellPhoneDB" R package.
Furthermore, we analyzed the interaction of KRT81+ tumor cells with immune cells using the "CellphoneDB" R package [39], an algorithm for analyzing cell and cell-cell interactions in single-cell datasets, to study their role in the tumor microenvironment (Fig. 6E). The NRP1/SEMA3A interacting pair can recruit macrophages into hypoxic tumor areas to promote angiogenesis and suppress antitumor immunity [40]. Additionally, the CXCL9, -10, −11/CXCR3 axis has dual roles. Its autocrine axis can promote tumor cell proliferation and metastasis. In contrast, its paracrine axis can activate the migration and differentiation of various immune cells [41]. Hence, inhibiting its autocrine and activating the paracrine axis might be a potential cancer therapeutic target. This idea was experimentally validated in preclinical models of colon cancer [42], breast cancer [43,44], and osteosarcoma [45] but has not yet been reported in LUAD. Herein, we detected a significant enrichment of the CXCL9, -10, −11/CXCR3 axis in KRT81+ tumor cells, indicating that this strategy might equally possess potential therapeutic value in LUAD. In summary, these results demonstrated the potential of the KRT81+ tumor cell cluster to differentiate into metastatic LUAD cells, which might be mediated by the characteristic transcription factors TFAP2A and WRNIP1. We also revealed the complex regulatory role of the KRT81+ tumor cell cluster in the tumor microenvironment.
2.7. The phenotype of KRT81+ tumor cells can be used as a prognostic biomarker in LUAD patients
To further assess the clinical value of KRT81+ tumor cells, we performed consensus clustering of all TCGA-LUAD cohorts based on the expression of KRT81+ tumor cell markers. We divided them into two clusters according to the expression of KRT81+ tumor cell markers: KRT81+ and KRT81– phenotypes (Fig. 7A and Fig. S2). The KRT81+ phenotype was associated with poor prognosis (Fig. 7B) and was significantly enriched in various hallmark gene sets in the GSVA analysis (Fig. 7C). Besides, We used the ESTIMATE algorithm [4] to infer immune-stromal cell scores in TCGA-LUAD data. The KRT81+ phenotype presented lower immune stromal cell infiltration but higher tumor purity (Fig. 7D–G). The poor prognosis for the KRT81+ phenotype might be associated with antitumor immunosuppression.
Fig. 7.

Identification of KRT81+ Phenotypes. (A) Consensus cluster analysis of the TCGA-LUAD cohort using KRT81+ tumor cell marker gene expression. (B) Survival analysis of two groups after consensus clustering. (C) GSVA analysis of two groups using the HALLMARK gene set. (D–G) ESTIMATE algorithm estimates immune, stromal and tumor purity score.
Next, we performed differential gene expression analysis between the two clusters to investigate the power of the two phenotypes in predicting LUAD prognoses (Fig. 8A). We assessed the predictive effect of all differentially expressed genes on prognosis using univariate Cox (Table S5) and Random Forest (Fig. 8B) algorithms. Finally, we obtained eight robust prognosis-related genes (ASB11, MUCL1, LIPF, SLC6A5, UNC80, RBP3, CDH16, PDX1) based on the intersection of genes with p < 0.05 in the univariate Cox results and top 10 ranked genes with importance in the Random Forest algorithm (Fig. 8C). To prevent model overfitting, we used the least absolute shrinkage and selection operator (LASSO) algorithm to screen the variables, and the smallest λ was detected when the six variables were retained. Therefore, the prognostic model was constructed for these six genes (ASB11, MUCL1, LIPF, SLC6A5, UNC80, and CDH16) (Fig. 8D–E).
Fig. 8.

KRT81+ tumor cells phenotype can be used as a prognostic biomarker in patients with lung adenocarcinoma. (A) Identification of differentially expressed genes in two groups. (B) Random Forest algorithm to evaluate the effect of DEGs on survival. (C) The intersection of univariate COX result and the top ten importance in random forest identified robust prognosis-related genes. (D–E) Further screening of prognosis-related genes by LASSO algorithm. (F) Univariate Cox analysis of Risk Score and clinical index in the TCGA-LUAD cohort. (G) Multivariate Cox analysis of Risk Score and clinical index in the TCGA-LUAD cohort. (H) ROC curves of the predictive ability of the prediction model for the 1, 2, and 3-year prognosis in the training dataset (TCGA-LUAD). (I) ROC curves of the predictive ability of the prediction model for the 1, 2, and 3-year prognosis in the testing dataset (GSE30219 and GSE31210). (J) Constructing a nomogram for prognosis prediction in the TCGA-LUAD dataset. (K) ROC curves of risk score and clinical information on prognosis prediction. (L–N) Calibration curves of the nomogram at 1, 2, and 3 years.
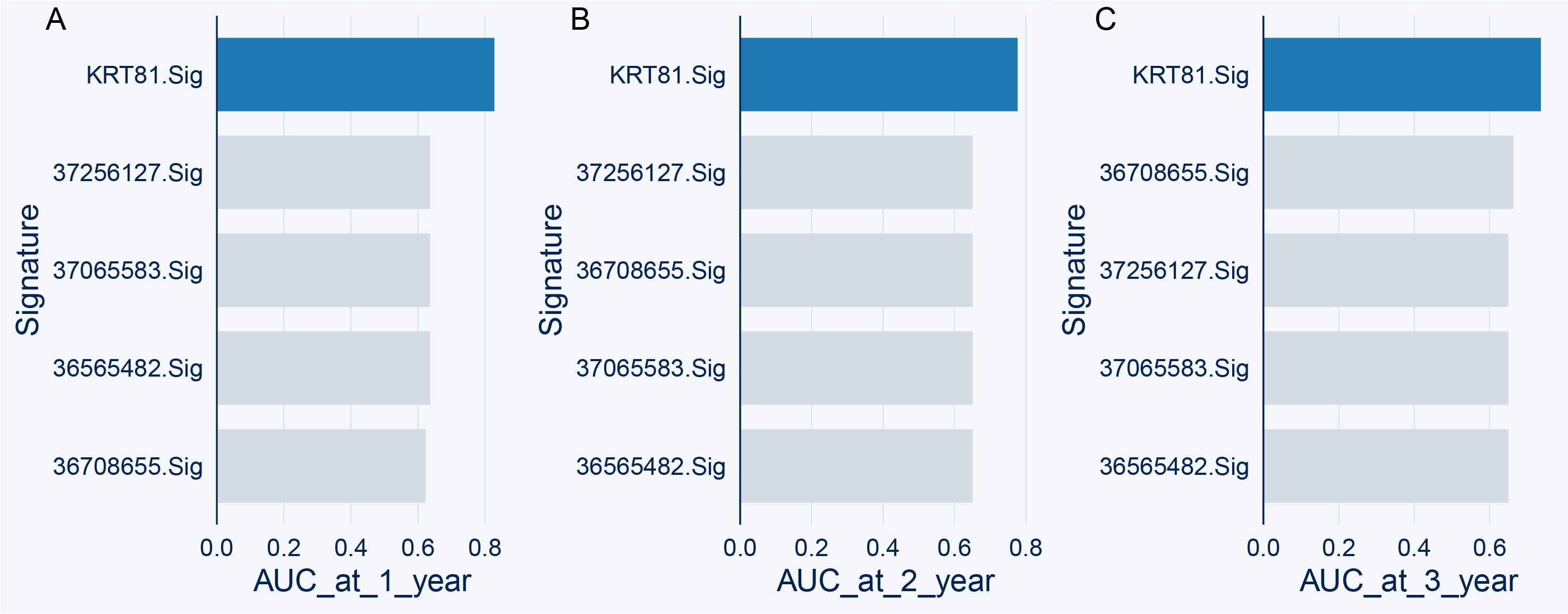
According to our constructed model, we calculated the risk score for each TCGA-LUAD patient. The p-value of the risk score was <0.05 in both univariate and multivariate Cox analyses, indicating that the risk score can be used as an independent prognostic risk factor for LUAD patients (Fig. 8F–G). Next, we used ROC curves to evaluate the ability of the model to predict prognosis. Since the proportion of TCGA-LUAD patients who survived more than 5 years was small, we only assessed the short-term prognosis. The ROC curves at 1, 2, and 3 years demonstrated the model's good predictive ability. Additionally, the AUC values observed at 1, 2, and 3 years were >0.7 in the external GEO cohort (Fig. 8H–I). Additionally, we compared the predictive performance of our model with previously published prognostic models [[46], [47], [48], [49]] constructed using LUAD scRNA-seq data in testing dataset. Our model demonstrated better predictive ability (Figs. S4A–C). These results indicated that our constructed model has a good short-term survival predictive ability for LUAD patients. After contrasting the prognostic predictive ability with other clinical indexes, we found that the risk score had similar AUC values to the clinical stage (Fig. 8K). Therefore, we constructed a nomogram combining the risk score and clinical stage to enhance the current use of the clinical stage alone and to guide patients requiring further intervention (Fig. 8J). The nomogram calibration curves showed good accuracy for short-term prognosis (Fig. 8L-N). Overall, our current findings suggested a novel subtype of LUAD patients with the KRT81+ tumor cell phenotype and proposed a prognostic model associated with this phenotype to assist the currently used clinical stage and to enhance the prediction of short-term prognosis and clinical treatment guidance.
3. Discussion
Despite the widespread use of targeted and immunotherapy for advanced LUAD, relapse and drug resistance remain major concerns [50,51]. Thus, deepening the understanding of LUAD and finding new therapeutic targets and prognostic monitoring biomarkers is urgent. RNA-seq has already provided strong evidence on disease diagnosis, prognostic monitoring, and selection of therapeutic targets. However, this strategy uses whole tissue transcriptome sequencing and ignores intratumoral heterogeneity [3,52]. Notably, intratumoral heterogeneity is one of the main causes of recurrence and drug resistance after treatment [[53], [54], [55], [56], [57]]. The scRNA-seq provides transcriptome data in a single cell dimension to precisely address this problem, allowing a deeper understanding of heterogeneity at the cell level [58]. For LUAD, several articles related to scRNA-seq have delineated the landscape in an atlas approach and provided valuable raw data for subsequent studies [[15], [16], [17],19]. Therefore, searching for new prognostic biomarkers or therapeutic targets in LUAD from the scRNA-seq perspective has advantages over bulk RNA-seq that ignore intratumoral heterogeneity. Herein, we investigated the heterogeneity of malignant epithelial cells in LUAD by combining scRNA-seq and bulk RNA-seq. We found a new malignant epithelial cell cluster associated with prognoses and conducted an in-depth study of its characteristics and clinical significance.
We collected scRNA-seq data from four normal lung tissues, eight primary LUAD, and five distant metastatic LUAD tissues. After dimensionality reduction, clustering, and cell type annotation, heterogeneous tumor purity and immune cell infiltration were reflected in each sample, indicating the importance of individualized treatment for LUAD patients. Regarding the CNV, most tumor cells have higher CNV than normal lung epithelium. Previous studies have found extensive amplification of chromosome 3q in lung squamous cell carcinoma [59]. However, we did not observe it in our LUAD analysis. Additionally, on chromosome 13, LUAD tumor cells presented significant copy number loss. Nevertheless, due to the limited sample size and clinical information investigated, how to correlate this CNV signature with potential clinical predictive value remains to be further determined. In the pseudo-time analysis of all primary and metastatic LUAD tumor cells, we observed a heterogeneous developmental orientation rooted in the primary tumor cells, with malignant epithelial cell proliferation activated and structural homeostasis disrupted during the development of heterogeneous metastatic tumor cells. We also observed the activation of neutrophil chemotaxis and migration pathways. The notion that neutrophils can promote LUAD cell proliferation and metastasis [60] and are associated with worse prognosis [61] has already been validated in previous studies. To further investigate the potential association of these heterogeneous tumor cells with clinical outcomes, we divided all tumor cells into ten clusters and named them by highly expressed genes. Based on highly expressed genes from each cluster to deconvolute bulk RNA-seq data from TCGA-LUAD, we obtained more clinical information and found that KRT81+ tumor cells were associated with a worse LUAD prognosis. Besides, the high KRT81 expression group was enriched for EMT and hypoxia pathways and were validated in the scRNA-seq data. The EMT can lead to tumor cell metastasis by enhancing the migration, invasion, and resistance to apoptotic stimuli of tumor cells. EMT-derived tumor cells can also acquire stemness and significant therapeutic resistance [62,63]. The activation of the hypoxia pathway can promote tumor angiogenesis and cell proliferation [64,65]. Hypoxia can also induce the activation of the EMT pathway [66]. The synergistic effect of hypoxia and EMT might be responsible for the association between KRT81+ tumor cells and poor prognosis. Subsequentially, we revealed that KRT81+ tumor cells tend to differentiate and metastasize, and that their characteristic transcription factor TFAP2A might be responsible for the activation of the EMT pathway. Moreover, activation of the NRP1/SEMA3A ligand-receptor pair by KRT81+ tumor cells in the tumor microenvironment recruits macrophages to hypoxic tumor areas, promoting angiogenesis and suppressing antitumor immunity [40]. Hence, targeting TFAP2A and NRP1 might potentially treat KRT81+ tumor cells. Strategies targeting NRP1 have been reported in previous studies [67], but our findings of TFAP2A might comprehend an entirely novel potential therapeutic target. Previous studies have reported that KRT81 promotes the migration and invasion of breast cancer [30]. Besides, KRT81 knockdown inhibits the proliferation, invasion, and migration of melanoma and promotes apoptosis by downregulating the expression of IL-8 [31], Although previous studies have reported that KRT81 is a risk factor for the prognosis of lung adenocarcinoma [68], no further research has been done on the role of cell cluster with high expression of KRT81. Therefore, this study focused on revealing the prognostic impact and potential functions of KRT81 high-expression tumor cell cluster in single-cell data sets. Further analysis showed that the new LUAD subtypes divided according to the marker genes of KRT81+ tumor cells had significant prognostic differences and had a strong predictive ability for the short-term prognosis of LUAD patients. In contrast to previous studies, our research not only involved the construction of a prognostic prediction model but also delved into in-depth investigations. These investigations encompassed exploring the activation of their transcription factors and their interactions with immune cells. Moreover, we identified several potential therapeutic targets specific to this cell subpopulation.
In conclusion, we combined scRNA-seq and bulk RNA-seq data from LUAD to profile the heterogeneity of LUAD tumor cells. We identified a novel cluster of KRT81+ tumor cells that might tend to metastasize through the activation of the EMT and hypoxia pathways. Furthermore, we showed that the KRT81+ tumor cell phenotype has a good predictive ability for the short-term prognosis of LUAD patients. Our study provides new ideas for predicting the prognosis and treatment strategies of LUAD. But for the current situation, single-cell data sets often lack large sample size and prognosis follow-up data due to high cost. We have to try our best to combine bulk RNA-seq datasets with single-cell datasets to make up for each other's shortcomings, so developing robust integration algorithms is a matter of concern.
4. Materials and methods
4.1. Cell culture
Human A549 and NCI–H1299 cell lines was purchased from Procell Life Science & Technology Co. Ltd., located in Wuhan, China. Both cell lines were cultured using RPMI-1640 medium (Gibco, USA) supplemented with 10% fetal bovine serum (VivaCell Biosciences, Shanghai XP Biomed Ltd.) and incubated at 37 °C with 5% CO2.
4.2. Datasets and processing methods
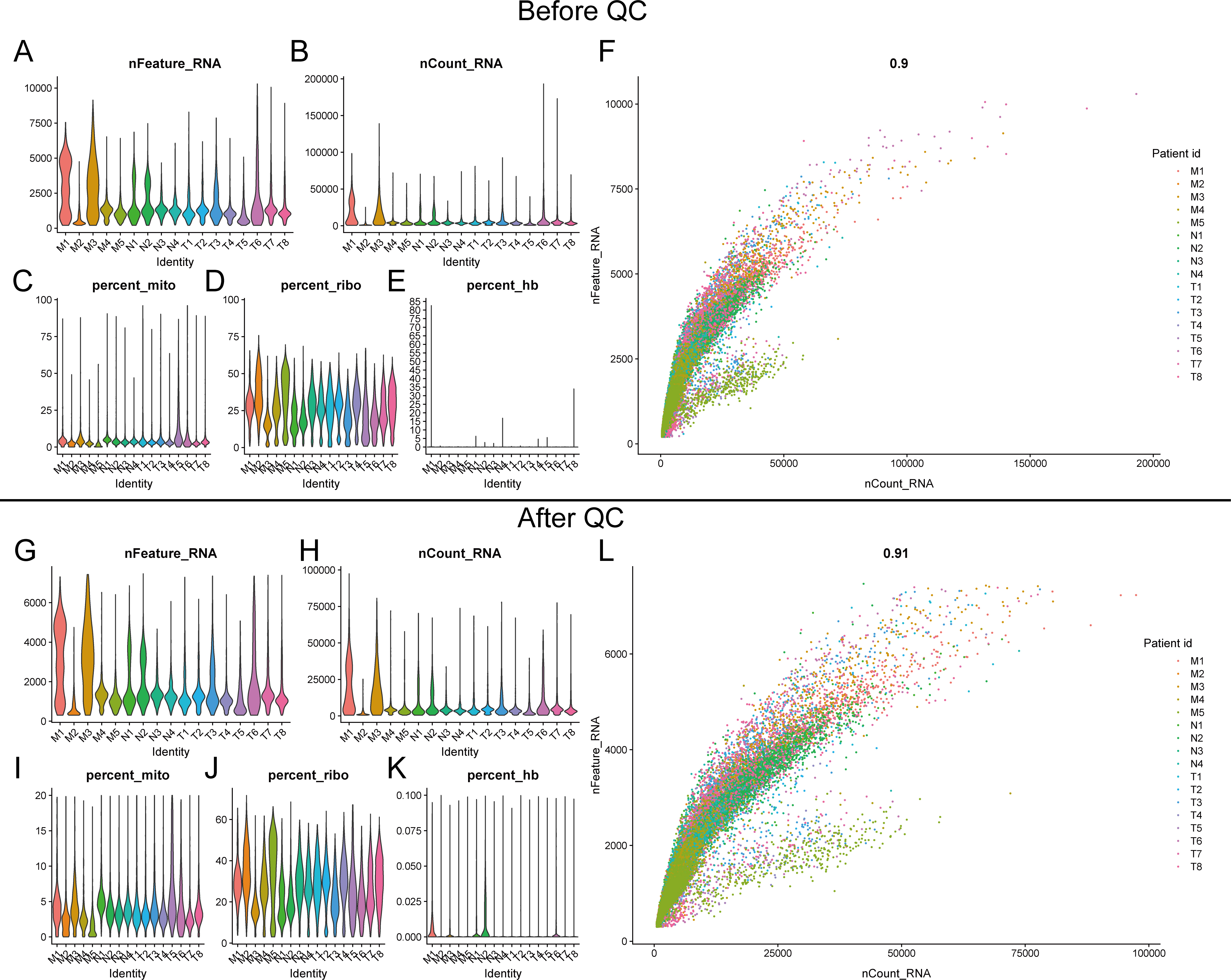
The raw human lung adenocarcinoma single-cell RNA-seq data were obtained from The European Nucleotide Archive (ENA) database (Accession ID: PRJNA510251 [69]). Clinical information for all samples is available from the original publication [69]. Raw sequencing data were matched to the genome and quantified for gene expression following Cell Ranger version.7.0 software Official running pipelines. The R package "Seurat" [70] was used for merge and quality control of multiple sample data. Cells with nFeature between 300 and 7500, nCount between 300 and 100000 and Mitochondrial genes expression less than 20% were retained for subsequent analysis. Data were normalized using the "SCTransform" method followed by dimensionality reduction clustering and visualization. In addition, the annotation of cell types is a characteristic marker of various cells summarized according to previous published articles and CellMarker database (http://xteam.xbio.top/CellMarker/). The Bulk RNA-seq and microarray data used in this study were obtained from the TCGA and GEO databases (TCGA-LUAD, GSE30219 and GSE31210).
4.3. Copy Number Analysis
For inferring normal epithelium mixed in tumor samples, we used methods published in a previous article [16]. Briefly, epithelial cells from each tumor sample were first extracted and incorporated into a certain number of normal epithelial cells from normal samples so that the number of epithelial cells from the tumor samples were below 20%. Genes expressed in fewer cells were eliminated as background genes. Following conversion of expression to z-score, copy number signals were inferred using expression. Two measures were used to determine cell types, which were the estimated mean squared CNV signal and the correlation between each cell and the mean copy number of the top 5% of cells (Code and detailed instructions are available at https://github.com/SGI-LungCancer/SingleCell). We used inferCNV [71] algorithm to revalidate the above results and characterized the copy number landscape of each chromosome in detail. Eventually all normal epithelial cells underwent dimensionality reduction clustering, visualization and cell-type annotation using the Seurat R package.
4.4. Cell trajectory analysis
Cell trajectory analysis was performed using the Monocle [72] R package. The parameters used in the analysis were all the default parameters, and the cell trajectories were visualized using the "DDRTree" method, setting the primary tumor cells as the pseudo-time root, and searching for differential expressed genes in the pseudo-time sequence. Eventually, we extracted the top 50 genes that were up- or down-regulated according to the pseudo-time and performed GO functional enrichment analysis using the clusterProfiler [73,74] R package.
4.5. Identification of tumor cell cluster associated with poor prognosis
All tumor cells were subjected to dimensionality reduction, clustering and visualization using the Seurat R package and identification of cluster signature genes using the FindallMarker function. The clusters were annotation according to highest expressed genes of each cluster. Similarly, we used the FindallMarker function in all other cell types to search for signature genes for each cell type. For all cell clusters, we calculated the average expression profile of the top 50 highly expressed genes in each cell and combined them as a reference data set. Finally, the TCGA-LUAD bulk RNA-seq data was deconvoluted with our constructed reference dataset using the CibersortX [75] algorithm to estimate the proportion of various cell clusters in TCGA lung adenocarcinoma samples. Univariate Cox analysis was used to identify risk factors associated with poor prognosis. Finally, we grouped patients by median cell proportion or median gene expression, and plotted survival curves using the "survival" R package. The EMT and hypoxia pathway gene sets used for functional enrichment are from the "MsigDB" database, and the metastasis and invasion pathway gene sets are from the "cancerSEA" database.
4.6. Plasmid transfection
The KRT81 gene overexpression and shRNA plasmids were purchased from Shanghai GeneChem Co.,Ltd. The sequence for KRT81 shRNA is as follows:
KRT81-shRNA#1: 5′-GATCCCGCGGCCAATTGAACACCACCTCTCGAGAGGTGGTGTTCAATTGGCCGCTTTTTGGAT-3’; KRT81-shRNA#2: 5′-GATCCCCCTCACATTTCTCTGTGTGATCTCGAGATCACACAGAGAAATGTGAGGTTTTTGGAT-3’; KRT81-shRNA#3: 5′-GATCCCAGGCATTGGGGCTGTGAATGTCTCGAGACATTCACAGCCCCAATGCCTTTTTTGGAT-3’.
A549 and H1299 cell lines were seeded in six-well plates and cultured overnight until reaching a density of 50%–80%. Transfection was performed using Lipofectamine 2000 transfection reagent (Invitrogen; CA, USA) according to the manufacturer's instructions. Furthermore, the cells were cultured in the presence of G418 (Beyotime Biotechnology, Shanghai, China) at a concentration of 500 μg/ml for one week to perform resistance selection.
4.7. Western blot
After removing the culture medium, the cells were washed three times with pre-chilled PBS buffer. Total cellular proteins were extracted using RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China) supplemented with a protease inhibitor cocktail (Beyotime Biotechnology, Shanghai, China) and PMSF (Beyotime Biotechnology, Shanghai, China). The protein concentration was determined using the BCA protein assay kit (Thermo Fisher Scientific; CA, USA). Appropriate SDS-PAGE loading buffer was added to prepare protein samples for Western blot analysis. The samples were resolved by 10% SDS-PAGE gel electrophoresis, with a protein ladder (Thermo Fisher Scientific; CA, USA) serving as the molecular weight marker. After electrophoresis, the proteins were transferred onto a PVDF membrane (Millipore, USA) for subsequent antibody incubation. The primary antibody used in this study were anti-KRT81(Proteintech, Wuhan, China, 1:1000) and anti-βactin (Proteintech, Wuhan, China, 1:10000), and the secondary antibody used was Goat anti-Rabbit HRP (Proteintech, Wuhan, China, 1:10000). The visualization of protein bands was achieved using the ECL detection kit (Beyotime Biotechnology, Shanghai, China).
4.8. Wound healing assay
The cells were seeded in six-well plates and allowed to reach 100% confluency. Scratch wounds were created using a 200 μl pipette tip, carefully scraping the cell monolayer. Subsequently, the cells were cultured in serum-free 1640 medium. At designated time intervals, photographs were captured to monitor the healing process of the scratch wounds at the identical locations.
4.9. Transwell assay
The Transwell assay was conducted using 24-well, 8 μm transwell chambers (NEST Biotechnology Co.LTD. Wuxi, China). The cells were dissociated into a single-cell suspension, and a seeding density of 1x105 cells was used for the upper chamber. The upper chamber was supplemented with serum-free medium, while the lower chamber contained medium with 10% FBS. After 24 h of incubation, the cells were fixed with 4% paraformaldehyde (Beyotime Biotechnology, Shanghai, China) and stained using 1% crystal violet (Beyotime Biotechnology, Shanghai, China). After gently removing the cells on the upper surface of the chamber, images were captured using an inverted microscope in at least three random fields of view.
4.10. RNA velocity, transcription factor and cellular interaction analysis
RNA Velocity were analyzed using the python-based "velocity [32]" package. BAM files for all samples were used in this pipeline to assess lineage developmental orientation in an unsupervised manner by the proportion of spliced mRNA and unspliced mRNA and mapped onto UMAP for visualization. Transcription factor analysis was performed using the python-based SCENIC [76] package to mine the characteristic transcription factors of single cell clusters by cis-regulatory analysis to provide biological insights into the heterogeneous mechanisms of cell clusters. Cell-to-cell communication analysis was performed using the python-based CellphoneDB [39] package. In processing of the results, we extracted ligand-receptor pairs between clusters of KRT81+ tumor cells and other cells and screened them using means > 1.
4.11. Construction of prognostic model associated with KRT81+ Phenotype
In the TPM expression data from the TCGA-LUAD cohort, univariate COX analysis of the KRT81+ tumor cell marker genes obtained prognostic KRT81+ tumor cell markers. Expression of these genes in the TCGA-LUAD cohort was then consensus clustered using the "ConsensusClusterPlus [77]" R package. The cluster algorithm was set to k-means, and sampling proportion set to eighty precents. Eventually we identified two clusters and defined them as KRT81+ phenotype cluster and KRT81− phenotype cluster based on the KRT81+ marker genes expression. Survival curves for the two clusters were plotted using the "survival" R package. Next, we performed functional enrichment analysis of the two clusters using the "GSVA" R package, and the HALLMARK gene set was obtained from MSigDB database. Differentially expressed genes from clusters were identified using the "edgeR" R package with |FoldChange| > 1.5 and p-value <0.05 as criterion. To identify robust prognosis related DEGs, we used two algorithms for screening, univariate COX analysis and random forest. By intersection of genes with p-value <0.05 in univariate COX analysis and top 10 genes of importance in random forest algorithm, we obtained eight robust prognosis-related genes. Random forest analysis used the "randomForestSRC" R package with the number of trees set to 1000 and node size set to 15. The LASSO algorithm used the "glmnet" R package, which retains seven genes for subsequent prognostic model construction when λ is a minimum. Prognostic models were constructed using multivariate COX analysis. The "timeROC" R package was used to plot ROC curves for prognostic models to assess the predictive effect of the model. For external validation of the model, we combined two GEO datasets (GSE30219 and GSE31210) that both used the GPL570 platform after removing batch effects using the "ComBat" algorithm and then converted to z-scores. The nomogram is constructed using the "rms" R package and the calibrate curve is plotted using "caret" R package.
4.12. Statistical analysis
The ImageJ software [78] was used for quantitative analysis of Western blot, wound healing, and Transwell assays, with all experiments conducted in triplicate for statistical analysis. The T-test was used for comparisons between two groups, while one-way ANOVA was employed for analysis of three or more groups.
Author contribution statement
Xu Ran: Performed the experiments; Analyzed and interpreted the data; Wrote the paper. </p>
Lu Tong; Wang Chenghao; Li Qi; Peng Bo; Wang Jun: Performed the experiments; Analyzed and interpreted the data. </p>
Zhao Jiaying: Contributed reagents, materials, analysis tools or data. </p>
Zhang Linyou: Conceived and designed the experiments. </p>.
Data availability statement
Data included in article/supplementary material/referenced in article.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We thank Dr.Jianming Zeng (University of Macau), and all the members of his bioinformatics team, biotrainee, for generously sharing their experience and codes.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.heliyon.2023.e20164.
Appendix A. Supplementary data
The following is/are the supplementary data to this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
References
- 1.Siegel R.L., Miller K.D., Fuchs H.E., Jemal A. Cancer statistics, 2022. CA Cancer J Clin. 2022;72(1):7–33. doi: 10.3322/caac.21708. [DOI] [PubMed] [Google Scholar]
- 2.Herbst R.S., Morgensztern D., Boshoff C. The biology and management of non-small cell lung cancer. Nature. 2018;553(7689):446–454. doi: 10.1038/nature25183. [DOI] [PubMed] [Google Scholar]
- 3.Costa C., Gimenez-Capitan A., Karachaliou N., Rosell R. Comprehensive molecular screening: from the RT-PCR to the RNA-seq. Transl. Lung Cancer Res. 2013;2(2):87–91. doi: 10.3978/j.issn.2218-6751.2013.02.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yoshihara K., Shahmoradgoli M., Martinez E., Vegesna R., Kim H., Torres-Garcia W., et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013;4:2612. doi: 10.1038/ncomms3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12(5):453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Becht E., Giraldo N.A., Lacroix L., Buttard B., Elarouci N., Petitprez F., et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17(1):218. doi: 10.1186/s13059-016-1070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dagogo-Jack I., Shaw A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018;15(2):81–94. doi: 10.1038/nrclinonc.2017.166. [DOI] [PubMed] [Google Scholar]
- 8.Vitale I., Shema E., Loi S., Galluzzi L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat Med. 2021;27(2):212–224. doi: 10.1038/s41591-021-01233-9. [DOI] [PubMed] [Google Scholar]
- 9.Prasetyanti P.R., Medema J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer. 2017;16(1):41. doi: 10.1186/s12943-017-0600-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lei Y., Tang R., Xu J., Wang W., Zhang B., Liu J., et al. Applications of single-cell sequencing in cancer research: progress and perspectives. J. Hematol. Oncol. 2021;14(1):91. doi: 10.1186/s13045-021-01105-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gohil S.H., Iorgulescu J.B., Braun D.A., Keskin D.B., Livak K.J. Applying high-dimensional single-cell technologies to the analysis of cancer immunotherapy. Nat. Rev. Clin. Oncol. 2021;18(4):244–256. doi: 10.1038/s41571-020-00449-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zilionis R., Engblom C., Pfirschke C., Savova V., Zemmour D., Saatcioglu H.D., et al. Single-cell Transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity. 2019;50(5):1317–1334 e10. doi: 10.1016/j.immuni.2019.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu F., Fan J., He Y., Xiong A., Yu J., Li Y., et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 2021;12(1):2540. doi: 10.1038/s41467-021-22801-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maynard A., McCoach C.E., Rotow J.K., Harris L., Haderk F., Kerr D.L., et al. Therapy-induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell. 2020;182(5):1232–1251 e22. doi: 10.1016/j.cell.2020.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leader A.M., Grout J.A., Maier B.B., Nabet B.Y., Park M.D., Tabachnikova A., et al. Single-cell analysis of human non-small cell lung cancer lesions refines tumor classification and patient stratification. Cancer Cell. 2021;39(12):1594–1609 e12. doi: 10.1016/j.ccell.2021.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim N., Kim H.K., Lee K., Hong Y., Cho J.H., Choi J.W., et al. Single-cell RNA sequencing demonstrates the molecular and cellular reprogramming of metastatic lung adenocarcinoma. Nat. Commun. 2020;11(1):2285. doi: 10.1038/s41467-020-16164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kashima Y., Shibahara D., Suzuki A., Muto K., Kobayashi I.S., Plotnick D., et al. Single-cell analyses reveal diverse mechanisms of resistance to EGFR Tyrosine kinase inhibitors in lung cancer. Cancer Res. 2021;81(18):4835–4848. doi: 10.1158/0008-5472.CAN-20-2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo X., Zhang Y., Zheng L., Zheng C., Song J., Zhang Q., et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med. 2018;24(7):978–985. doi: 10.1038/s41591-018-0045-3. [DOI] [PubMed] [Google Scholar]
- 19.Chen J., Tan Y., Sun F., Hou L., Zhang C., Ge T., et al. Single-cell transcriptome and antigen-immunoglobin analysis reveals the diversity of B cells in non-small cell lung cancer. Genome Biol. 2020;21(1):152. doi: 10.1186/s13059-020-02064-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rowbotham S.P., Kim C.F. Diverse cells at the origin of lung adenocarcinoma. Proc Natl Acad Sci U S A. 2014;111(13):4745–4746. doi: 10.1073/pnas.1401955111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarode P., Mansouri S., Karger A., Schaefer M.B., Grimminger F., Seeger W., et al. Epithelial cell plasticity defines heterogeneity in lung cancer. Cell. Signal. 2020;65 doi: 10.1016/j.cellsig.2019.109463. [DOI] [PubMed] [Google Scholar]
- 22.Xu J., Qin S., Yi Y., Gao H., Liu X., Ma F., et al. Delving into the heterogeneity of different breast cancer subtypes and the prognostic models utilizing scRNA-seq and bulk RNA-seq. Int. J. Mol. Sci. 2022;23(17) doi: 10.3390/ijms23179936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen K., Wang Y., Hou Y., Wang Q., Long D., Liu X., et al. Single cell RNA-seq reveals the CCL5/SDC1 receptor-ligand interaction between T cells and tumor cells in pancreatic cancer. Cancer Lett. 2022;545 doi: 10.1016/j.canlet.2022.215834. [DOI] [PubMed] [Google Scholar]
- 24.Chen K., Liu X., Liu W., Wang F., Tian X., Yang Y. Development and validation of prognostic and diagnostic model for pancreatic ductal adenocarcinoma based on scRNA-seq and bulk-seq datasets. Hum. Mol. Genet. 2022;31(10):1705–1719. doi: 10.1093/hmg/ddab343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344(6190):1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yokota J., Sugimura T., Terada M. Chromosomal localization of putative tumor-suppressor genes in several human cancers. Environ. Health Perspect. 1991;93:121–123. doi: 10.1289/ehp.9193121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tamura K., Zhang X., Murakami Y., Hirohashi S., Xu H.J., Hu S.X., et al. Deletion of three distinct regions on chromosome 13q in human non-small-cell lung cancer. Int. J. Cancer. 1997;74(1):45–49. doi: 10.1002/(sici)1097-0215(19970220)74:1<45::aid-ijc8>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 28.Kwong F.M., Wong P.S., Lung M.L. Genetic alterations detected on chromosomes 13 and 14 in Chinese non-small cell lung carcinomas. Cancer Lett. 2003;192(2):189–198. doi: 10.1016/s0304-3835(02)00698-5. [DOI] [PubMed] [Google Scholar]
- 29.Cheung W.K., Nguyen D.X. Lineage factors and differentiation states in lung cancer progression. Oncogene. 2015;34(47):5771–5780. doi: 10.1038/onc.2015.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nanashima N., Horie K., Yamada T., Shimizu T., Tsuchida S. Hair keratin KRT81 is expressed in normal and breast cancer cells and contributes to their invasiveness. Oncol. Rep. 2017;37(5):2964–2970. doi: 10.3892/or.2017.5564. [DOI] [PubMed] [Google Scholar]
- 31.Zhang K., Liang Y., Zhang W., Zeng N., Tang S., Tian R. KRT81 knockdown inhibits malignant progression of melanoma through regulating interleukin-8. DNA Cell Biol. 2021;40(10):1290–1297. doi: 10.1089/dna.2021.0317. [DOI] [PubMed] [Google Scholar]
- 32.La Manno G., Soldatov R., Zeisel A., Braun E., Hochgerner H., Petukhov V., et al. RNA velocity of single cells. Nature. 2018;560(7719):494–498. doi: 10.1038/s41586-018-0414-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yuanhua L., Pudong Q., Wei Z., Yuan W., Delin L., Yan Z., et al. TFAP2A induced KRT16 as an oncogene in lung adenocarcinoma via EMT. Int. J. Biol. Sci. 2019;15(7):1419–1428. doi: 10.7150/ijbs.34076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiong Y., Feng Y., Zhao J., Lei J., Qiao T., Zhou Y., et al. TFAP2A potentiates lung adenocarcinoma metastasis by a novel miR-16 family/TFAP2A/PSG9/TGF-beta signaling pathway. Cell Death Dis. 2021;12(4):352. doi: 10.1038/s41419-021-03606-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guoren Z., Zhaohui F., Wei Z., Mei W., Yuan W., Lin S., et al. TFAP2A induced ITPKA serves as an oncogene and interacts with DBN1 in lung adenocarcinoma. Int. J. Biol. Sci. 2020;16(3):504–514. doi: 10.7150/ijbs.40435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang L.L., Lu J., Liu R.Q., Hu M.J., Zhao Y.M., Tan S., et al. Chromatin accessibility analysis reveals that TFAP2A promotes angiogenesis in acquired resistance to anlotinib in lung cancer cells. Acta Pharmacol. Sin. 2020;41(10):1357–1365. doi: 10.1038/s41401-020-0421-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mano Y., Takahashi K., Ishikawa N., Takano A., Yasui W., Inai K., et al. Fibroblast growth factor receptor 1 oncogene partner as a novel prognostic biomarker and therapeutic target for lung cancer. Cancer Sci. 2007;98(12):1902–1913. doi: 10.1111/j.1349-7006.2007.00610.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang W., Han X., Wang J., Wang L., Xu Z., Wei Q., et al. miR-22 enhances the radiosensitivity of small-cell lung cancer by targeting the WRNIP1. J. Cell. Biochem. 2019;120(10):17650–17661. doi: 10.1002/jcb.29032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Efremova M., Vento-Tormo M., Teichmann S.A., Vento-Tormo R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. Nat. Protoc. 2020;15(4):1484–1506. doi: 10.1038/s41596-020-0292-x. [DOI] [PubMed] [Google Scholar]
- 40.Casazza A., Laoui D., Wenes M., Rizzolio S., Bassani N., Mambretti M., et al. Impeding macrophage entry into hypoxic tumor areas by Sema3A/Nrp1 signaling blockade inhibits angiogenesis and restores antitumor immunity. Cancer Cell. 2013;24(6):695–709. doi: 10.1016/j.ccr.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 41.Tokunaga R., Zhang W., Naseem M., Puccini A., Berger M.D., Soni S., et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - a target for novel cancer therapy. Cancer Treat Rev. 2018;63:40–47. doi: 10.1016/j.ctrv.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cambien B., Karimdjee B.F., Richard-Fiardo P., Bziouech H., Barthel R., Millet M.A., et al. Organ-specific inhibition of metastatic colon carcinoma by CXCR3 antagonism. Br. J. Cancer. 2009;100(11):1755–1764. doi: 10.1038/sj.bjc.6605078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu G., Yan H.H., Pang Y., Jian J., Achyut B.R., Liang X., et al. CXCR3 as a molecular target in breast cancer metastasis: inhibition of tumor cell migration and promotion of host anti-tumor immunity. Oncotarget. 2015;6(41):43408–43419. doi: 10.18632/oncotarget.6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walser T.C., Rifat S., Ma X., Kundu N., Ward C., Goloubeva O., et al. Antagonism of CXCR3 inhibits lung metastasis in a murine model of metastatic breast cancer. Cancer Res. 2006;66(15):7701–7707. doi: 10.1158/0008-5472.CAN-06-0709. [DOI] [PubMed] [Google Scholar]
- 45.Pradelli E., Karimdjee-Soilihi B., Michiels J.F., Ricci J.E., Millet M.A., Vandenbos F., et al. Antagonism of chemokine receptor CXCR3 inhibits osteosarcoma metastasis to lungs. Int. J. Cancer. 2009;125(11):2586–2594. doi: 10.1002/ijc.24665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Z., Zhu H., Wang X., Lin S., Ruan C., Wang Q. A novel basement membrane-related gene signature for prognosis of lung adenocarcinomas. Comput. Biol. Med. 2023;154 doi: 10.1016/j.compbiomed.2023.106597. [DOI] [PubMed] [Google Scholar]
- 47.Zhang P., Liu J., Pei S., Wu D., Xie J., Liu J., et al. Mast cell marker gene signature: prognosis and immunotherapy response prediction in lung adenocarcinoma through integrated scRNA-seq and bulk RNA-seq. Front. Immunol. 2023;14 doi: 10.3389/fimmu.2023.1189520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang J., Liu X., Huang Z., Wu C., Zhang F., Han A., et al. T cell-related prognostic risk model and tumor immune environment modulation in lung adenocarcinoma based on single-cell and bulk RNA sequencing. Comput. Biol. Med. 2023;152 doi: 10.1016/j.compbiomed.2022.106460. [DOI] [PubMed] [Google Scholar]
- 49.Huang X., Xiao H., Shi Y., Ben S. Integrating single-cell and bulk RNA sequencing to develop a cancer-associated fibroblast-related signature for immune infiltration prediction and prognosis in lung adenocarcinoma. J. Thorac. Dis. 2023;15(3):1406–1425. doi: 10.21037/jtd-23-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kurahara H., Maemura K., Mataki Y., Tanoue K., Iino S., Kawasaki Y., et al. Lung recurrence and its therapeutic strategy in patients with pancreatic cancer. Pancreatology. 2020;20(1):89–94. doi: 10.1016/j.pan.2019.11.015. [DOI] [PubMed] [Google Scholar]
- 51.Kobayashi S., Boggon T.J., Dayaram T., Janne P.A., Kocher O., Meyerson M., et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005;352(8):786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 52.Shukla S., Evans J.R., Malik R., Feng F.Y., Dhanasekaran S.M., Cao X., et al. Development of a RNA-seq based prognostic signature in lung adenocarcinoma. J Natl Cancer Inst. 2017;109(1) doi: 10.1093/jnci/djw200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marjanovic N.D., Hofree M., Chan J.E., Canner D., Wu K., Trakala M., et al. Emergence of a high-plasticity cell state during lung cancer evolution. Cancer Cell. 2020;38(2):229–246 e13. doi: 10.1016/j.ccell.2020.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lim Z.F., Ma P.C. Emerging insights of tumor heterogeneity and drug resistance mechanisms in lung cancer targeted therapy. J. Hematol. Oncol. 2019;12(1):134. doi: 10.1186/s13045-019-0818-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jamal-Hanjani M., Wilson G.A., McGranahan N., Birkbak N.J., Watkins T.B.K., Veeriah S., et al. Tracking the evolution of non-small-cell lung cancer. N. Engl. J. Med. 2017;376(22):2109–2121. doi: 10.1056/NEJMoa1616288. [DOI] [PubMed] [Google Scholar]
- 56.de Sousa V.M.L., Carvalho L. Heterogeneity in lung cancer. Pathobiology. 2018;85(1–2):96–107. doi: 10.1159/000487440. [DOI] [PubMed] [Google Scholar]
- 57.Fang W., Jin H., Zhou H., Hong S., Ma Y., Zhang Y., et al. Intratumoral heterogeneity as a predictive biomarker in anti-PD-(L)1 therapies for non-small cell lung cancer. Mol. Cancer. 2021;20(1):37. doi: 10.1186/s12943-021-01331-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chong Z.X., Ho W.Y., Yeap S.K., Wang M.L., Chien Y., Verusingam N.D., et al. Single-cell RNA sequencing in human lung cancer: applications, challenges, and pathway towards personalized therapy. J. Chin. Med. Assoc. 2021;84(6):563–576. doi: 10.1097/JCMA.0000000000000535. [DOI] [PubMed] [Google Scholar]
- 59.Mendez P., Ramirez J.L. Copy number gains of FGFR1 and 3q chromosome in squamous cell carcinoma of the lung. Transl. Lung Cancer Res. 2013;2(2):101–111. doi: 10.3978/j.issn.2218-6751.2013.03.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sangaletti S., Ferrara R., Tripodo C., Garassino M.C., Colombo M.P. Myeloid cell heterogeneity in lung cancer: implication for immunotherapy. Cancer Immunol. Immunother. 2021;70(9):2429–2438. doi: 10.1007/s00262-021-02916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Azevedo P.O., Paiva A.E., Santos G.S.P., Lousado L., Andreotti J.P., Sena I.F.G., et al. Cross-talk between lung cancer and bones results in neutrophils that promote tumor progression. Cancer Metastasis Rev. 2018;37(4):779–790. doi: 10.1007/s10555-018-9759-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pan G., Liu Y., Shang L., Zhou F., Yang S. EMT-associated microRNAs and their roles in cancer stemness and drug resistance. Cancer Commun. 2021;41(3):199–217. doi: 10.1002/cac2.12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mittal V. Epithelial mesenchymal transition in tumor metastasis. Annu. Rev. Pathol. 2018;13:395–412. doi: 10.1146/annurev-pathol-020117-043854. [DOI] [PubMed] [Google Scholar]
- 64.Wang W.J., Ouyang C., Yu B., Chen C., Xu X.F., Ye X.Q. Role of hypoxiainducible factor2alpha in lung cancer (Review). Oncol Rep. 2021;45(5) doi: 10.3892/or.2021.8008. [DOI] [PubMed] [Google Scholar]
- 65.Ancel J., Perotin J.M., Dewolf M., Launois C., Mulette P., Nawrocki-Raby B., et al. Hypoxia in lung cancer management: a Translational approach. Cancers. 2021;13(14) doi: 10.3390/cancers13143421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tirpe A.A., Gulei D., Ciortea S.M., Crivii C., Berindan-Neagoe I. Hypoxia: overview on hypoxia-mediated mechanisms with a focus on the role of HIF genes. Int. J. Mol. Sci. 2019;20(24) doi: 10.3390/ijms20246140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dumond A., Pages G. Neuropilins, as relevant oncology target: their role in the tumoral microenvironment. Front. Cell Dev. Biol. 2020;8:662. doi: 10.3389/fcell.2020.00662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu C., Li X., Shao H., Li D. Identification and validation of two lung adenocarcinoma-development characteristic gene sets for diagnosing lung adenocarcinoma and predicting prognosis. Front. Genet. 2020;11 doi: 10.3389/fgene.2020.565206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Laughney A.M., Hu J., Campbell N.R., Bakhoum S.F., Setty M., Lavallee V.P., et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat Med. 2020;26(2):259–269. doi: 10.1038/s41591-019-0750-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184(13):3573–3587 e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Puram S.V., Tirosh I., Parikh A.S., Patel A.P., Yizhak K., Gillespie S., et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck cancer. Cell. 2017;171(7):1611–1624 e24. doi: 10.1016/j.cell.2017.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Trapnell C., Cacchiarelli D., Grimsby J., Pokharel P., Li S., Morse M., et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 2014;32(4):381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16(5):284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., et al. clusterProfiler 4.0: a universal enrichment tool for interpreting omics data. Innovation. 2021;2(3) doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Newman A.M., Steen C.B., Liu C.L., Gentles A.J., Chaudhuri A.A., Scherer F., et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019;37(7):773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aibar S., Gonzalez-Blas C.B., Moerman T., Huynh-Thu V.A., Imrichova H., Hulselmans G., et al. SCENIC: single-cell regulatory network inference and clustering. Nat. Methods. 2017;14(11):1083–1086. doi: 10.1038/nmeth.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wilkerson M.D., Hayes D.N. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics. 2010;26(12):1572–1573. doi: 10.1093/bioinformatics/btq170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9(7):671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data included in article/supplementary material/referenced in article.