

Abstract
BACKGROUND
Trials of monoclonal antibodies that target various forms of amyloid at different stages of Alzheimer’s disease have had mixed results.
METHODS
We tested solanezumab, which targets monomeric amyloid, in a phase 3 trial involving persons with preclinical Alzheimer’s disease. Persons 65 to 85 years of age with a global Clinical Dementia Rating score of 0 (range, 0 to 3, with 0 indicating no cognitive impairment and 3 severe dementia), a score on the Mini–Mental State Examination of 25 or more (range, 0 to 30, with lower scores indicating poorer cognition), and elevated brain amyloid levels on 18F-florbetapir positron-emission tomography (PET) were enrolled. Participants were randomly assigned in a 1:1 ratio to receive solanezumab at a dose of up to 1600 mg intravenously every 4 weeks or placebo. The primary end point was the change in the Preclinical Alzheimer Cognitive Composite (PACC) score (calculated as the sum of four z scores, with higher scores indicating better cognitive performance) over a period of 240 weeks.
RESULTS
A total of 1169 persons underwent randomization: 578 were assigned to the solanezumab group and 591 to the placebo group. The mean age of the participants was 72 years, approximately 60% were women, and 75% had a family history of dementia. At 240 weeks, the mean change in PACC score was −1.43 in the solanezumab group and −1.13 in the placebo group (difference, −0.30; 95% confidence interval, −0.82 to 0.22; P = 0.26). Amyloid levels on brain PET increased by a mean of 11.6 centiloids in the solanezumab group and 19.3 centiloids in the placebo group. Amyloidrelated imaging abnormalities (ARIA) with edema occurred in less than 1% of the participants in each group. ARIA with microhemorrhage or hemosiderosis occurred in 29.2% of the participants in the solanezumab group and 32.8% of those in the placebo group.
CONCLUSIONS
Solanezumab, which targets monomeric amyloid in persons with elevated brain amyloid levels, did not slow cognitive decline as compared with placebo over a period of 240 weeks in persons with preclinical Alzheimer’s disease. (Funded by the National Institute on Aging and others; A4 ClinicalTrials.gov number, NCT02008357.)
The pathological processes that are associated with Alzheimer’s disease — accumulation of amyloid-beta (Aβ) into fibrillar plaques and hyperphosphorylated tau into paired helical filament neurofibrillary tangles — begin more than a decade before clinically evident cognitive impairment. Approximately 20 to 40% of cognitively unimpaired older persons show evidence of elevated amyloid accumulation.1–5 Some evidence suggests that these “amyloid-positive” but cognitively unimpaired older persons represent a preclinical, or asymptomatic, stage of Alzheimer’s disease6,7 and may be at high risk for cognitive decline.1,4
Anti-Amyloid Treatment in Asymptomatic Alzheimer’s Disease (A4) was an early-intervention trial aiming to slow cognitive decline at the stage of preclinical Alzheimer’s disease in older persons who were not cognitively impaired at baseline but had elevated amyloid levels on screening positron-emission tomography (PET).8 We tested solanezumab,9 an immunoglobulin G1 monoclonal antibody that binds to the mid-domain of the Aβ monomer, as compared with placebo for 4.5 years in a phase 3 trial. For the purposes of this trial, new outcome measures were developed to track early stages of cognitive and functional decline.10,11
METHODS
TRIAL OVERSIGHT
We conducted the trial at 67 sites in Australia, Canada, Japan, and the United States; sites were selected on the basis of experience in performing Alzheimer’s disease–related clinical trials. Approval from an institutional review board was obtained at each of the trial sites, and all the participants provided written informed consent. The trial was conducted in accordance with International Council for Harmonisation Good Clinical Practice guidelines.
The trial was conducted as a public–private partnership, with funding from the National Institutes of Health, Eli Lilly (holder of the investigational new drug application), and philanthropic donors. The trial was designed by five academic authors in collaboration with three authors employed by Eli Lilly and other colleagues. Site principal investigators (see the Supplementary Appendix, available with full text of this article at NEJM.org) acted as the trial steering committee. The trial was coordinated (including data collection and management, site monitoring, safety oversight and regulatory oversight) by the academic team with assistance from Eli Lilly. Data were analyzed by two academic authors; parallel analyses were conducted by an author employed by Eli Lilly. An independent data and safety monitoring board reviewed masked data on a quarterly basis. The double-blind phase of the trial was conducted from 2014 through 2022, and database lock of this phase occurred in early 2023.
Eli Lilly provided the trial drug and matching placebo. There were confidentiality agreements between Eli Lilly and the academic authors; however, the authors were not restricted with regard to access to data, analysis, interpretation, presentation, and publication of results. The initial draft of the manuscript was written by four academic authors and was revised and approved by all the authors. The authors vouch for the completeness and accuracy of the data, the fidelity of the trial to the protocol (available at NEJM.org), and complete and up-to-date reporting of adverse events.
TRIAL DESIGN
The screening process for the trial has been published previously12 and is briefly described here and in the protocol. Candidates for the trial referred themselves. Centralized and local recruitment strategies (performed by the academic coordinating center and trial sites, respectively) were implemented in several ways, including community outreach, national and local advertising, paid and unpaid media coverage, and interrogation of national and local registries.
Participants were eligible for screening if they were 65 to 85 years of age and were living independently without a diagnosis of mild cognitive impairment or dementia; the absence of cognitive impairment was confirmed by a global Clinical Dementia Rating (CDR) score of 0 (on an ordinal scale of 0 to 3, with 0 indicating no cognitive impairment and 3 indicating severe dementia), by a Mini–Mental State Examination (MMSE) score of 25 to 30 (range, 0 to 30, with lower scores indicating poorer cognitive performance), and by a Wechsler Memory Scale Logical Memory Delayed Recall (LMDR) score of 6 to 18 (range, 0 to 25, with lower scores indicating fewer details recalled). Persons with LMDR scores greater than 18 (>1.5 SD above normal values for this age range) were excluded to enhance the likelihood of enrolling persons with elevated brain amyloid levels. Persons with unstable medical conditions were excluded, although participants with stable hypertension, diabetes, hypercholesterolemia, mild-to-moderate small-vessel ischemic disease, and other medical conditions were eligible. Each participant was required to have a partner who provided information about the participant’s functioning.
Brain amyloid levels were assessed by means of 18F-florbetapir PET imaging with the use of a mean cortical standardized uptake value ratio (SUVR) with a whole cerebellar reference region. Amyloid status was assessed with an algorithm that combined quantitative SUVR methods and qualitative visual reading performed at a central laboratory. A quantitative SUVR threshold of 1.15 or more was used as the primary criterion to define elevated amyloid levels13 in the identification of persons with early amyloid accumulation.14 An SUVR of 1.10 to less than 1.15 was considered to be elevated only when a visual reading of images was also considered to be positive by a two-reader consensus determination.
Eligible participants were randomly assigned in a 1:1 ratio to receive intravenous solanezumab or placebo. Randomization was stratified according to apolipoprotein E (ApoE) ε4 carrier status, years of education (≤12 or >12), and trial site. The dose was initially 400 mg intravenously every 4 weeks, but the results of a completed phase 3 trial involving persons with mild dementia due to Alzheimer’s disease suggested that this dose may be inadequate,15 and the protocol was changed to adjust the dose to 1600 mg intravenously every 4 weeks in 2017, when approximately 970 participants had already been enrolled (Fig. S1 in the Supplementary Appendix). At the same time, the double-blind phase of the trial was extended to 240 weeks to ensure substantial exposure to the higher dose and sufficient time to evaluate efficacy.
END POINTS
The primary efficacy end point was the change in the Preclinical Alzheimer Cognitive Composite (PACC) score11 at 4.5 years. The PACC scale is a validated1,16–18 scale that was devised for the measurement of amyloid-related cognitive decline in unimpaired populations in clinical trials. The PACC is composed of four components: the total score on the Free and Cued Selective Reminding Test (FCSRT)19, the delayed paragraph recall on the Logical Memory IIa test from the Wechsler Memory Scale,20,21 the Digit Symbol Substitution Test from the Wechsler Adult Intelligence Scale–Revised,22 and the MMSE23 total score. For the FCSRT, the total was the sum of the free and cued scores plus the free-recall score, resulting in a total ranging from 0 to 96, with lower scores indicating greater memory impairment. We used alternate versions of the component subtests to minimize practice effects.21 Each component score was converted to a z score by subtracting the baseline mean for that component and dividing by the baseline standard deviation for that component. The PACC is the sum of the four resulting z scores, with negative scores indicating worsening of cognitive performance. The testing was conducted by certified site psychometrists who were unaware of the trial-group assignments and adverse events.
The first secondary end point in the graphical testing scheme described below was the change in the Cognitive Function Index (CFI) score,10,24 which is based on a set of 15 questions that capture subjective concerns related to cognitive function. The response to each question is scored 1 or 0 (or 0.5 in some cases), indicating the presence or absence of concerns as compared with 1 year earlier (range, 0 to 15, with higher scores indicating greater concern). Responses by the participant and trial partner were added together (range, 0 to 30).
Additional secondary end points include the score on the Alzheimer’s Disease Cooperative Study Activities of Daily Living (ADL) Prevention Questionnaire,25 which was assessed by the partner (range, 0 to 45, with higher scores indicating more difficulty); the CDR–Sum of Boxes (CDR-SB) score26 (range, 0 to 18, with higher scores indicating greater impairment in global functioning); and progression of the global CDR score to a nonzero score indicating mild cognitive impairment or dementia. The PACC was scheduled to be administered at baseline and every 24 weeks thereafter. Assessments of the CFI, ADL, and CDR scores were scheduled to be performed at baseline and at weeks 48, 108, 168, 204, and 240. The visit timing was disrupted by the coronavirus disease 2019 (Covid-19) pandemic, as noted below.
Imaging end points included the change in findings on 18F-florbetapir amyloid PET and volumetric magnetic resonance imaging (MRI). 18F-flortaucipir tau PET was performed in a subgroup of participants, with the primary imaging end point measured from a composite of neocortical regions and a secondary imaging end point in medial temporal lobe regions at baseline and 240 weeks.
STATISTICAL ANALYSIS
Sample size and power were estimated with the use of data from observational studies involving cognitively unimpaired, amyloid-positive persons. The mean (±SD) differences in the change in the PACC score between participants without elevated brain amyloid levels and those with elevated brain amyloid levels at 240 weeks in these studies ranged from 2.13±2.85 to 2.66±3.08. Given these estimates and assuming 30% attrition, we calculated that 1150 participants would provide the trial with 80% power (at a 5% two-sided alpha level) to detect a between-group difference of 0.53 to 0.57 points in the PACC score. This effect size was chosen for power analyses because this change corresponds to a difference of approximately 30% in cognitive decline between amyloid-negative and amyloid-positive persons, a slowing of cognitive decline that was judged to be clinically meaningful over time.
The primary objective was to test the hypothesis that solanezumab, administered for 240 weeks, slows cognitive decline as compared with placebo in participants with preclinical Alzheimer’s disease. The originally planned analysis used a mixed-model, repeated-measures approach, which treats time as categorical and flexibly models the mean at each time category in each group as free parameters. As originally planned, trial visits would occur at fixed intervals (24 weeks [±10 days]) so that assessments could be grouped according to visit number, and this grouping would also reflect the time during which the trial agent was received. The hiatus in dose administration and testing in the trial due to social-distancing measures during the Covid-19 pandemic resulted in many visits being conducted later than the original target date. Therefore, the statistical analysis plan (available with the protocol) was amended to use days since baseline derived from examination dates as the time variable, rather than categorical visit. This change was made before results were unblinded to the trial team and investigators. The amended primary analysis is a constrained longitudinal data analysis that uses natural cubic splines of the primary end-point measure, the PACC score.27 Secondary end points were assessed with a similar approach. The spline model was fit by restricted maximum likelihood with the use of all available follow-up observations without multiple imputation or carrying observations backward or forward for missing data. Sensitivity analyses included a mixed-model, repeated-measures analysis, which required observations to be carried backward owing to Covid-19 disruptions. The variables in the spline models are provided in the statistical analysis plan.
The primary analysis was performed in the modified intention-to-treat population of persons who had a baseline score and at least one post-baseline assessment. If the unstructured covariance approach resulted in a lack of model convergence, simpler prespecified alternatives would be attempted. Secondary end points were analyzed with a similar approach, except the effect for test version was excluded when not applicable.
The time to progression of the global CDR score (defined as an increase in the score from 0 to 0.5, 1, or 2, indicating progression to mild cognitive impairment or mild dementia) was assessed in a Kaplan–Meier analysis. Time to progression of the global CDR score was defined as the time from randomization to the first of two consecutive visits with a nonzero score or to a final visit with a nonzero score. Data from participants who did not have progression were censored at the time of the last assessment of the global CDR score.
A graphical testing scheme to account for multiple comparisons of the primary and secondary end points (see the statistical analysis plan) was prespecified in February 2023, before unblinding, with the change in the PACC score as the primary end point followed by the secondary end points. Confidence intervals for cognitive and functional outcomes are reported with adjustment according to this graphical testing scheme.
A post hoc analysis was conducted to characterize the effect of the baseline amyloid level on florbetapir PET, expressed in centiloids, in the modified intention-to-treat population (Fig. S4). This analysis grouped participants according to tertiles of baseline florbetapir centiloids. The spline model for this analysis included two spline basis expansion terms for each of three tertile groups and did not constrain the baseline means to be the same. Other terms in the model were similar to those in the primary model, except that baseline florbetapir SUVR was excluded. There was no prespecified plan for adjustment of the widths of confidence intervals for multiple comparisons, and no conclusions can be drawn from this analysis.
Imaging biomarkers, including florbetapir amyloid PET composite, flortaucipir tau PET neocortical composite, and MRI hippocampal and total gray-matter volume, were analyzed by means of analysis of covariance. Change from baseline was the response variable, and age, ApoE ε4 carrier status, trial group, and baseline biomarker value were covariates. Only multiplicity-unadjusted 95% confidence intervals are provided for the imaging end-point analyses, and these cannot be used for statistical inferences. All analyses were conducted with the use of R software, version 4.2.1.28–31
RESULTS
PARTICIPANTS
A total of 6763 persons underwent cognitive and medical screening, and 4485 were eligible to proceed to amyloid PET (Fig. 1). Of these 4485 persons, 1322 (29.5%) had elevated brain amyloid levels and were eligible to continue in screening for the trial. Apart from nonelevated amyloid levels, the main reasons for exclusion were cognitive test scores outside the specified range and active medical issues. Ultimately, 1169 participants were randomly assigned to a trial group, 578 to receive solanezumab and 591 to receive placebo; the modified intention-to-treat population included 564 participants who received solanezumab and 583 who received placebo. A total of 379 participants who received solanezumab and 396 who received placebo continued to an open-label extension phase. Final-visit PACC observations were missing for 30.6% of the participants in the solanezumab group and 28.4% of those in the placebo group.
Figure 1. Screening, Randomization, and Follow-up.
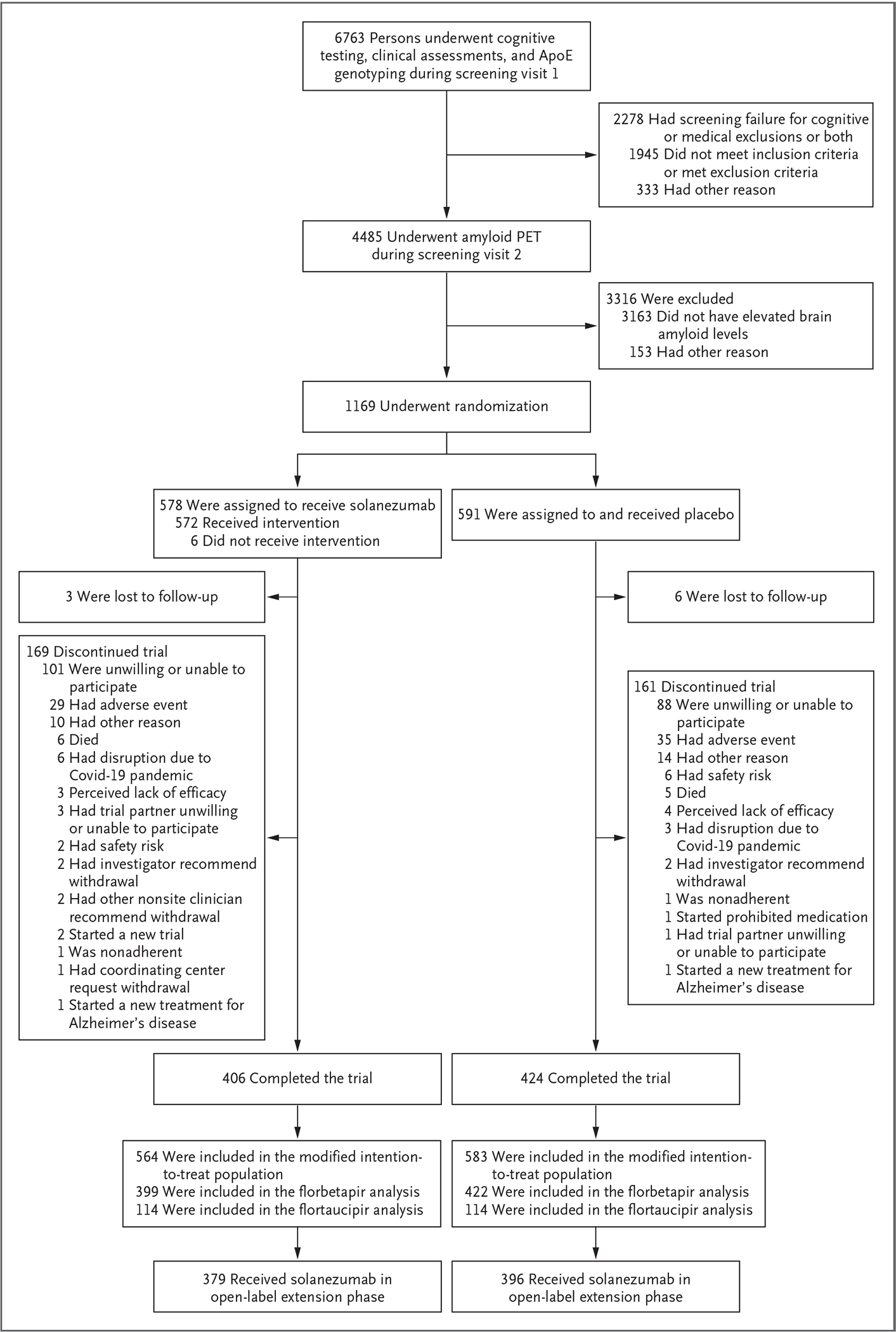
The modified intention-to-treat population included randomly assigned participants who received at least one dose of solanezumab or placebo and underwent assessment for the primary end point. Participants who completed visit 66 (target, 240 weeks) were considered to have completed the double-blind phase. ApoE denotes apolipoprotein E, Covid-19 coronavirus disease 2019, and PET positron-emission tomography.
The two groups had similar characteristics at baseline, including mean (±SD) PACC scores (0.0±2.8 in the solanezumab group and 0.0±2.6 in the placebo group), ApoE ε4 carrier status (59.0% and 58.7%, respectively), and amyloid burden on PET (66.2 centiloids and 65.9 centiloids, respectively) (Table 1). Participants who identified as Black or as Hispanic or Latino were underrepresented as compared with the general population (see Table S1 on the representativeness of the trial population).
Table 1.
Characteristics of the Participants at Baseline (Modified Intention-to-Treat Population).*
| Characteristic | Solanezumab (N = 564) | Placebo (N = 583) |
|---|---|---|
|
| ||
| Age — yr | 72.0±4.7 | 71.9±5.0 |
| Female sex — no. (%) | 329 (58.3) | 352 (60.4) |
| Education — yr | 16.6±2.7 | 16.6±2.9 |
| Race — no. (%)† | ||
| White | 531 (94.1) | 549 (94.2) |
| Black | 12 (2.1) | 15 (2.6) |
| Asian | 11 (2.0) | 13 (2.2) |
| Other or missing | 10 (1.8) | 6 (1.0) |
| Ethnic group — no. (%)† | ||
| Not Hispanic or Latino | 542 (96.1) | 560 (96.1) |
| Hispanic or Latino | 16 (2.8) | 18 (3.1) |
| Unknown or not reported | 6 (1.1) | 5 (0.9) |
| Family history of dementia: parent or sibling — no. (%) | 411 (72.9) | 449 (77.0) |
| ApoE genotype — no. (%) | ||
| ε2/ε2 | 1 (02) | 0 |
| ε2/ε3 | 28 (5.0) | 33 (5.7) |
| ε2/ε4 | 13 (2.3) | 22 (3.8) |
| ε3/ε3 | 202 (35.8) | 208 (35.7) |
| ε3/ε4 | 273 (48.4) | 273 (46.8) |
| ε4/ε4 | 47 (8.3) | 47 (8.1) |
| ApoE ε4 carrier | 333 (59.0) | 342 (58.7) |
| Amyloid burden on 18F-florbetapir PET | ||
| SUVR | 1.3±0.2 | 1.3±0.2 |
| Centiloids | 66.2±33.5 | 65.9±32.1 |
| PACC score‡ | 0.0±2.8 | 0.0±2.6 |
| Wechsler Memory Scale LMDR score§ | 12.6±3.8 | 12.7±3.5 |
| MMSE score¶ | 28.8±1.3 | 28.8±1.2 |
| CFI combined score‖ | 4.0±3.6 | 3.6±3.3 |
| ADL partner score** | 43.4±2.7 | 43.5±2.6 |
| CDR-SB score†† | 0.1±0.2 | 0.0±0.2 |
Plus–minus values are means ±SD. The modified intention-to-treat population included randomly assigned participants who received at least one dose of solanezumab or placebo and underwent assessment for the primary end point. Percentages may not total 100 because of rounding. More details on baseline characteristics are provided in Table S1. ApoE denotes apolipoprotein E, PET positron-emission tomography, and SUVR standardized uptake value ratio.
Race and ethnic group were reported by the participant.
The Preclinical Alzheimer Cognitive Composite (PACC) score is the sum of four z scores, with higher scores indicating better cognitive performance.
Wechsler Memory Scale Logical Memory Delayed Recall (LMDR) scores range from 0 to 25, with lower scores indicating fewer details recalled.
Scores on the Mini–Mental State Examination (MMSE) range from 0 to 30, with lower scores indicating poorer cognition.
Scores on the Cognitive Function Index (CFI) range from 0 to 15, with higher scores indicating greater subjective concerns about cognitive function. Responses by the participant and participant’s partner were added together (range, 0 to 30).
Scores on the Alzheimer’s Disease Cooperative Study Activities of Daily Living (ADL) Prevention Questionnaire, as assessed by the participant’s partner, range from 0 to 45, with higher scores indicating more difficulty in performing activities of daily living.
Scores on the Clinical Dementia Rating (CDR)–Sum of Boxes (CDR-SB) range from 0 to 18, with higher scores indicating greater impairment in global functioning.
TRIAL END POINTS
The mean change from baseline in the PACC score was −1.43 (95% confidence interval [CI], −1.83 to −1.03) in the solanezumab group and −1.13 (95% CI, −1.45 to −0.81) in the placebo group (difference, −0.30; 95% CI, −0.82 to 0.22; P = 0.26) (Table 2 and Fig. 2A), which indicated no significant between-group difference and a numerically greater decline in the solanezumab group. Prespecified subgroup analyses showed similar results (see Fig. S2). Because the between-group difference in the primary end point failed to reach significance, the graphical testing scheme did not allow claims of significance for subsequent end points (Table 2 and Fig. 2B through 2E). Across both groups, 33.4% of the participants had progression of the global CDR score at 240 weeks.
Table 2.
Primary and Secondary End Points (Modified Intention-to-Treat Population).*
| End Point | Solanezumab | Placebo |
|---|---|---|
| Primary end point: PACC score | ||
| No. of participants evaluated† | 401 | 423 |
| Adjusted mean change | −1.43 | −1.13 |
| Adjusted mean difference vs. placebo (95% CI)‡ | −0.30 (−0.82 to 0.22) | |
| P value | 0.26 | |
| CFI combined score | ||
| No. of participants evaluated† | 403 | 418 |
| Adjusted mean change | 2.09 | 1.51 |
| Adjusted mean difference vs. placebo (95% CI)‡ | 0.58 (−0.18 to 1.34) | |
| ADL partner score | ||
| No. of participants evaluated† | 403 | 419 |
| Adjusted mean change | −2.24 | −1.63 |
| Adjusted mean difference vs. placebo (95% CI)‡ | −0.61 (−1.44 to 0.23) | |
| CDR-SB score | ||
| No. of participants evaluated† | 405 | 421 |
| Adjusted mean change | 0.71 | 0.60 |
| Adjusted mean difference vs. placebo (95% CI)‡ | 0.12 (−0.06 to 0.29) | |
| Progression of the global CDR score§ | ||
| No. of participants evaluated† | 406 | 424 |
| Probability of progression at 240 weeks | 0.35 | 0.32 |
Results were analyzed with the use of a spline model, except for progression of the global CDR score, which was assessed in a Kaplan–Meier analysis.
Counts include all the participants who had a final visit, which was initially targeted for 240 weeks after randomization. Some participants had evaluations delayed beyond 240 weeks owing to the coronavirus disease 2019 pandemic.
95% confidence intervals were adjusted for multiple testing according to a prespecified graphical testing scheme.
Progression of the global CDR score was defined as two consecutive scores greater than zero or a final-visit score greater than zero. Global CDR scores range from 0 to 3, with 0 indicating no cognitive impairment and 3 indicating severe dementia.
Figure 2. Primary and Secondary End Points over the Course of the Trial.
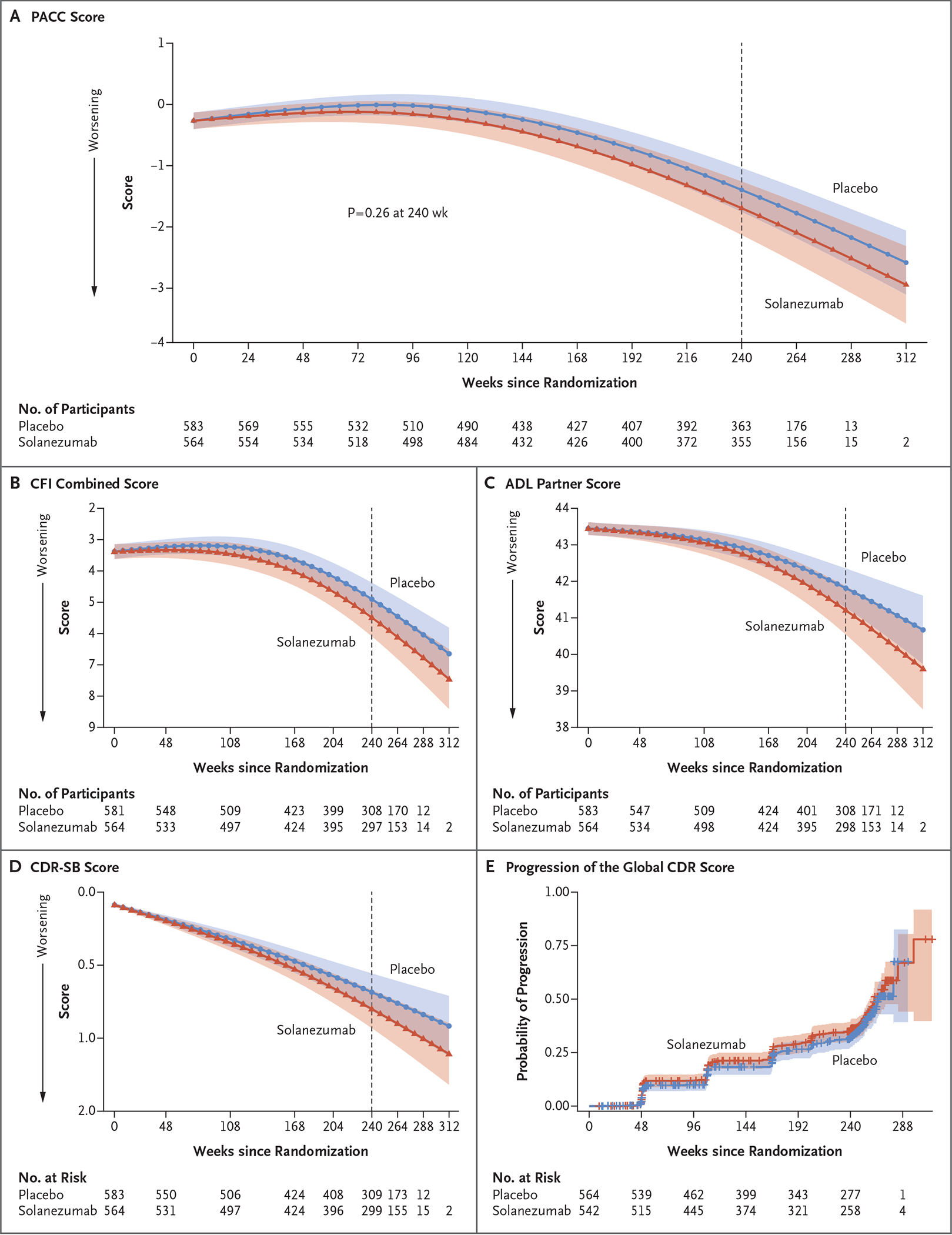
All panels show results for the modified intention-to-treat population. The 95% confidence intervals (indicated by shaded regions) were adjusted for multiplicity according to the prespecified graphical testing scheme. Panel A shows the primary end point, the Preclinical Alzheimer Cognitive Composite (PACC) score. The PACC is the sum of four z scores, with higher scores indicating better cognitive performance. The adjusted means, 95% confidence intervals, and P value were estimated with the use of a natural cubic spline model. Panel B shows the spline model–adjusted mean Cognitive Function Index (CFI) combined score (participant and partner). Scores for participant or partner range from 0 to 15, with higher scores indicating greater subjective concerns about cognitive function (range for combined score, 0 to 30). Panel C shows the spline model–adjusted mean score on the Alzheimer’s Disease Cooperative Study Activities of Daily Living (ADL) Prevention Questionnaire, as assessed by the participant’s partner. Scores range from 0 to 45, with higher scores indicating more difficulty in performing activities of daily living. Panel D shows the spline model–adjusted mean Clinical Dementia Rating (CDR)–Sum of Boxes (CDR-SB) score. Scores range from 0 to 18, with higher scores indicating greater impairment in global functioning. Panel E shows Kaplan–Meier estimates of progression of the global CDR score, defined as two consecutive scores greater than zero or a final-visit score greater than zero. Global CDR scores range from 0 to 3, with 0 indicating no cognitive impairment and 3 indicating severe dementia.
Amyloid PET showed that amyloid continued to accumulate above baseline levels in both trial groups. The increase was numerically larger in the placebo group than in the solanezumab group (19.3 centiloids vs. 11.6 centiloids; mean difference in change, 7.7 centiloids; 95% CI, 5.1 to 10.4), but statistical inferences from these results have limitations (Fig. S3A).
On tau PET (performed in 114 participants in each group), increases in neocortical and medial temporal tau were similar in the two groups (Table S3 and Fig. S3B and S3C). Changes in volumetric MRI measures in the hippocampus and total gray matter were similar in the two groups (Fig. S3D and S3E). Baseline amyloid levels and their association with cognitive and functional decline and the risk of progression to symptomatic Alzheimer’s disease are shown in Figure S4. In both groups combined, higher baseline amyloid levels were associated with faster cognitive and functional decline and a greater risk of progression to symptomatic Alzheimer’s disease, but no formal statistical conclusions can be drawn from those analyses.
SAFETY
Overall and serious adverse events were similar in the two groups in both type and incidence (Table 3 and Table S4). One case of amyloidrelated imaging abnormalities (ARIA) with edema was reported in the solanezumab group, and two cases were reported in the placebo group. ARIA with microhemorrhage or hemosiderosis occurred in 29.2% of the participants in the solanezumab group and 32.8% of those in the placebo group.
Table 3.
Adverse Events.*
| Event | Solanezumab (N = 572) | Placebo (N = 591) |
|---|---|---|
| number (percent) | ||
| Any adverse event | 560 (97.9) | 577 (97.6) |
| Adverse event definitely related to solanezumab or placebo† | 6 (1.0) | 8 (14) |
| Serious adverse event | 172 (30.1) | 158 (26.7) |
| Death | 6 (1.0) | 7 (12) |
| Adverse event leading to discontinuation of solanezumab or placebo | 28 (4.9) | 34 (5.8) |
| ARIA | 167 (29.2) | 194 (32.8) |
| ARIA with edema | 1 (02) | 2 (0.3) |
| ARIA with microhemorrhage or hemosiderosis | 167 (29.2) | 194 (32.8) |
| Microhemorrhage | 158 (27.6) | 189 (32.0) |
| Superficial siderosis | 19 (3.3) | 19 (3.2) |
Shown are adverse events that emerged or worsened after the first dose of solanezumab or placebo. ARIA denotes amyloid-related imaging abnormalities.
The relatedness of the adverse event to solanezumab or placebo was determined by the site investigator.
DISCUSSION
The A4 trial results indicate that the anti–monomeric amyloid antibody solanezumab did not slow the progression of preclinical Alzheimer’s disease as compared with placebo on the basis of the primary or secondary cognitive and functional end points over a period of 4.5 years. Solanezumab was selected for this trial on the basis of its safety profile and cognitive benefits in a meta-analysis involving persons with mild Alzheimer’s disease.32 Because solanezumab does not bind to fibrillar deposits, the imaging results accorded with the expectation that it would not reduce amyloid plaque load on follow-up PET imaging below baseline levels. Solanezumab appeared to slow amyloid accumulation over the course of the trial, a finding consistent with target engagement. In the tau PET sub-study, there were similar increases in neocortical and medial temporal tau in the two trial groups.
Candidates for participation in the trial were selected for screening primarily on the basis of age and unimpaired clinical cognitive status. The screening process showed that close to 30% of these persons had abnormal amyloid PET scans, a finding consistent with those of previous studies.1–5 Cognitive and clinical decline, with progression of the global CDR score to more than 0 in 36% of the participants, in conjunction with accumulation of tau pathologic features shown on flortaucipir PET, is consistent with the view that the accumulation of brain amyloid in an unimpaired population may represent a marker for early-stage Alzheimer’s disease.
Numerical worsening with respect to the primary and secondary end points in the solanezumab group as compared with the placebo group, although not significantly different between groups, was unexpected in view of the results of a meta-analysis of the EXPEDITION trials, in which treatment with solanezumab at a dose of 400 mg intravenously every 4 weeks was associated with modest cognitive benefit in participants with mild dementia due to Alzheimer’s disease, although none of the constituent trials gave positive results.33 One conjecture is that the increase in the dose by a factor of four (to 1600 mg every 4 weeks) during our trial may have severely depleted monomeric Aβ and yielded cognitive worsening, similar to that seen with inhibition of γ- or β-secretase.34 Further analyses will explore the effect of the dose increase on cognitive and clinical outcomes.
This trial has several important limitations. Participants were not representative of older persons at risk for cognitive decline, and there were few Black participants. Our trial was complicated by a midtrial dose increase. The trial had widespread disruption of activities at the sites due to the Covid-19 pandemic.
Several trials involving persons with early symptomatic Alzheimer’s disease have shown that substantial reduction in brain fibrillar amyloid toward normal levels, as measured by amyloid PET, is associated with slowing of clinical and cognitive progression.35–37 In contrast, lesser reductions in amyloid levels have yielded disappointing results in symptomatic Alzheimer’s disease.35,38 The A4 results, in a cognitively unimpaired population, are generally consistent with these results; slowing amyloid accumulation without reduction of fibrillar amyloid levels below baseline did not slow clinical progression.
In a phase 3 trial, solanezumab did not slow the progression of cognitive and functional decline in persons with preclinical Alzheimer’s disease as compared with placebo over a period of 4.5 years.
Supplementary Material
Acknowledgments
Supported by a public–private–philanthropic partnership, including funding from the National Institute on Aging of the National Institutes of Health (R01AG063689, U19AG010483, and U24AG057437), Eli Lilly (also the supplier of active medication and placebo), the Alzheimer’s Association, the Accelerating Medicines Partnership through the Foundation for the National Institutes of Health, the GHR Foundation, the Davis Alzheimer Prevention Program, the Yugilbar Foundation, an anonymous foundation, and additional private donors to Brigham and Women’s Hospital, with in-kind support from Avid Radiopharmaceuticals, Cogstate, Albert Einstein College of Medicine, and the Foundation for Neurologic Diseases.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
A data sharing statement provided by the authors is available with the full text of this article at NEJM.org.
The members of the A4 Study Team are listed in the Supplementary Appendix, available at NEJM.org
Contributor Information
Reisa A. Sperling, Center for Alzheimer Research and Treatment, Brigham and Women’s Hospital, Massachusetts General Hospital, Harvard Medical School, Boston
Michael C. Donohue, Alzheimer’s Therapeutic Research Institute, Keck School of Medicine, University of Southern California, San Diego
Rema Raman, Alzheimer’s Therapeutic Research Institute, Keck School of Medicine, University of Southern California, San Diego
Michael S. Rafii, Alzheimer’s Therapeutic Research Institute, Keck School of Medicine, University of Southern California, San Diego
Keith Johnson, Departments of Neurology and Radiology, Massachusetts General Hospital, Harvard Medical School, Boston
Colin L. Masters, Florey Institute, University of Melbourne, Melbourne, VIC, Australia
Christopher H. van Dyck, Departments of Psychiatry, Neurology, and Neuroscience, Yale School of Medicine, New Haven, CT
Takeshi Iwatsubo, Department of Neuropathology, Graduate School of Medicine, University of Tokyo, Tokyo
Gad A. Marshall, Center for Alzheimer Research and Treatment, Brigham and Women’s Hospital, Massachusetts General Hospital, Harvard Medical School, Boston
Roy Yaari, Eli Lilly, Indianapolis
Michele Mancini, Eli Lilly, Indianapolis
Karen C. Holdridge, Eli Lilly, Indianapolis
Michael Case, Eli Lilly, Indianapolis
John R. Sims, Eli Lilly, Indianapolis
Paul S. Aisen, Alzheimer’s Therapeutic Research Institute, Keck School of Medicine, University of Southern California, San Diego
References
- 1.Donohue MC, Sperling RA, Petersen R, Sun C-K, Weiner MW, Aisen PS. Association between elevated brain amyloid and subsequent cognitive decline among cognitively normal persons. JAMA 2017;317:2305–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jack CR Jr, Wiste HJ, Weigand SD, et al. Age-specific and sex-specific prevalence of cerebral β-amyloidosis, tauopathy, and neurodegeneration in cognitively unimpaired individuals aged 50–95 years: a cross-sectional study. Lancet Neurol 2017;16:435–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jansen WJ, Ossenkoppele R, Knol DL, et al. Prevalence of cerebral amyloid pathology in persons without dementia: a meta-analysis. JAMA 2015;313:1924–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roberts RO, Aakre JA, Kremers WK, et al. Prevalence and outcomes of amyloid positivity among persons without dementia in a longitudinal, population-based setting. JAMA Neurol 2018;75:970–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 2010;31:1275–83. [DOI] [PubMed] [Google Scholar]
- 6.Jack CR Jr, Bennett DA, Blennow K, et al. NIA-AA research framework: toward a biological definition of Alzheimer’s disease. Alzheimers Dement 2018;14:535–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sperling RA, Aisen PS, Beckett LA, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7:280–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sperling RA, Rentz DM, Johnson KA, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med 2014;6:228fs13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Imbimbo BP, Ottonello S, Frisardi V, et al. Solanezumab for the treatment of mild-to-moderate Alzheimer’s disease. Expert Rev Clin Immunol 2012;8:135–49. [DOI] [PubMed] [Google Scholar]
- 10.Amariglio RE, Donohue MC, Marshall GA, et al. Tracking early decline in cognitive function in older individuals at risk for Alzheimer disease dementia: the Alzheimer’s Disease Cooperative Study Cognitive Function Instrument. JAMA Neurol 2015;72:446–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Donohue MC, Sperling RA, Salmon DP, et al. The preclinical Alzheimer cognitive composite: measuring amyloidrelated decline. JAMA Neurol 2014;71:961–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sperling RA, Donohue MC, Raman R, et al. Association of factors with elevated amyloid burden in clinically normal older individuals. JAMA Neurol 2020;77:735–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Royse SK, Minhas DS, Lopresti BJ, et al. Validation of amyloid PET positivity thresholds in centiloids: a multisite PET study approach. Alzheimers Res Ther 2021;13:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Johnson KA, Sperling RA, Gidicsin CM, et al. Florbetapir (F18-AV-45) PET to assess amyloid burden in Alzheimer’s disease dementia, mild cognitive impairment, and normal aging. Alzheimers Dement 2013;9:Suppl:S72–S83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Honig LS, Vellas B, Woodward M, et al. Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med 2018;378:321–30. [DOI] [PubMed] [Google Scholar]
- 16.Bransby L, Lim YY, Ames D, et al. Sensitivity of a Preclinical Alzheimer’s Cognitive Composite (PACC) to amyloid β load in preclinical Alzheimer’s disease. J Clin Exp Neuropsychol 2019;41:591–600. [DOI] [PubMed] [Google Scholar]
- 17.Mormino EC, Papp KV, Rentz DM, et al. Early and late change on the preclinical Alzheimer’s cognitive composite in clinically normal older individuals with elevated amyloid β. Alzheimers Dement 2017;13:1004–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papp KV, Rofael H, Veroff AE, et al. Sensitivity of the Preclinical Alzheimer’s Cognitive Composite (PACC), PACC5, and Repeatable Battery for Neuropsychological Status (RBANS) to amyloid status in preclinical Alzheimer’s disease — atabecestat phase 2b/3 EARLY clinical trial. J Prev Alzheimers Dis 2022;9:255–61. [DOI] [PubMed] [Google Scholar]
- 19.Grober E, Hall CB, Lipton RB, Zon-derman AB, Resnick SM, Kawas C. Memory impairment, executive dysfunction, and intellectual decline in preclinical Alzheimer’s disease. J Int Neuropsychol Soc 2008;14:266–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wechsler D WMS-R: Wechsler Memory Scale–Revised: manual. San Antonio, TX: Psychological Corporation, 1987. [Google Scholar]
- 21.Morris J, Swier-Vosnos A, Woodworth C, Umfleet LG, Czipri S, Kopald B. Development of alternate paragraphs for the Logical Memory subtest of the Wechsler Memory Scale-IV. Appl Neuropsychol Adult 2014;21:143–7. [DOI] [PubMed] [Google Scholar]
- 22.Wechsler D Wechsler Adult Intelligence Scale–Revised. San Antonio, TX: Psychological Corporation, 1981. [Google Scholar]
- 23.Folstein MF, Folstein SE, McHugh PR. “Mini-Mental State”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–98. [DOI] [PubMed] [Google Scholar]
- 24.Walsh SP, Raman R, Jones KB, Aisen PS. ADCS Prevention Instrument Project: the Mail-in Cognitive Function Screening Instrument (MCFSI). Alzheimer Dis Assoc Disord 2006;20:Suppl 3:S170–S178. [DOI] [PubMed] [Google Scholar]
- 25.Galasko D, Bennett DA, Sano M, Marson D, Kaye J, Edland SD. ADCS Prevention Instrument Project: assessment of instrumental activities of daily living for community-dwelling elderly individuals in dementia prevention clinical trials. Alzh eimer Dis Assoc Disord 2006;20:Suppl 3:S152–S169. [DOI] [PubMed] [Google Scholar]
- 26.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–4. [DOI] [PubMed] [Google Scholar]
- 27.Donohue MC, Langford O, Insel PS, et al. Natural cubic splines for the analysis of Alzheimer’s clinical trials. Pharm Stat 2023;22:508–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.R: a language and environment for statistical computing. R Foundation for Statistical Computing (https://www.R-project.org/).
- 29.Bates D, Machler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J Stat Softw 2015;67:1–48. [Google Scholar]
- 30.Lenth RV. R package emmeans: estimated marginal means (aka least-squares means). GitHub. 2023. (https://github.com/rvlenth/emmeans). [Google Scholar]
- 31.Pinheiro J, Bates D, DebRoy S, et al. nlme: Linear and nonlinear mixed effects models. R package, version 3.1–162. 2023. (https://CRAN.R-project.org/package=nlme). [Google Scholar]
- 32.Siemers ER, Sundell KL, Carlson C, et al. Phase 3 solanezumab trials: secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement 2016;12:110–20. [DOI] [PubMed] [Google Scholar]
- 33.Holdridge KC, Yaari R, Hoban DB, Andersen S, Sims JR. Targeting amyloid β in Alzheimer’s disease: meta-analysis of low-dose solanezumab in Alzheimer’s disease with mild dementia studies. Alzheimers Dement 2023. March 22 (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- 34.Imbimbo BP, Ippati S, Watling M, Imbimbo C. Role of monomeric amyloid-β in cognitive performance in Alzheimer’s disease: insights from clinical trials with secretase inhibitors and monoclonal antibodies. Pharmacol Res 2023;187:106631. [DOI] [PubMed] [Google Scholar]
- 35.Budd Haeberlein S, Aisen PS, Barkhof F, et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J Prev Alzheimers Dis 2022;9:197–210. [DOI] [PubMed] [Google Scholar]
- 36.Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in early Alzheimer’s disease. N Engl J Med 2021;384:1691–704. [DOI] [PubMed] [Google Scholar]
- 37.van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in early Alzheimer’s disease. N Engl J Med 2023;388:9–21. [DOI] [PubMed] [Google Scholar]
- 38.Genentech. Genentech provides update on phase III GRADUATE program evaluating gantenerumab in early Alzheimer’s disease. November 13, 2022. (https://www.gene.com/media/press-releases/14974/2022-11-13/genentech-provides-update-on-phase-iii-g).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.