

Abstract
DNA methyltransferase 3A (DNMT3A) is a de novo cytosine methyltransferase responsible for establishing proper DNA methylation during mammalian development. Loss-of-function (LOF) mutations to DNMT3A, including the hotspot mutation R882H, frequently occur in developmental growth disorders and hematological diseases, including clonal hematopoiesis and acute myeloid leukemia (AML). Accordingly, identifying mechanisms that activate DNMT3A is of both fundamental and therapeutic interest. Here, we applied a base editor mutational scanning strategy with an improved DNA methylation reporter to systematically identify DNMT3A activating mutations in cells. By integrating an optimized cellular recruitment strategy with paired isogenic cell lines with or without the LOF hotspot R882H mutation, we identify and validate three distinct hyperactivating mutations within or interacting with the regulatory ADD domain of DNMT3A, nominating these regions as potential functional target sites for pharmacological intervention. Notably, these mutations are still activating in the context of a heterozygous R882H mutation. Altogether, we showcase the utility of base editor scanning for discovering functional regions of target proteins.
Graphical Abstract
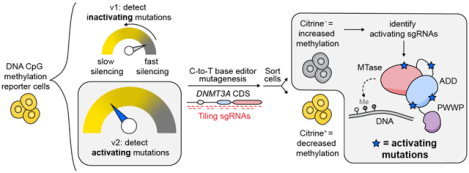
Introduction:
DNMT3A is one of the most highly mutated proteins in hematopoietic disorders, with more than 20% of patients with acute myeloid leukemia (AML) displaying an alteration in DNMT3A.1 About 65% of these occur at the hotspot R882 codon, which is most frequently mutated to histidine (R882H).1 The R882H mutation has been shown to have a dominant-negative effect on DNMT3A activity.1–4 Of the non-R882H mutations, a majority have also been shown to be LOF.5 DNMT3A LOF mutations also occur in developmental disorders, such as Tatton-Brown–Rahman Syndrome.6 Mutations to DNMT3A in hematopoietic disorders are typically heterozygous and associated with a worse prognosis for AML patients.1,7 In addition, DNMT3A mutations are frequently observed in pre-leukemic hematopoietic stem cell (HSC) populations, in which they promote self-renewal.8 Taken together, these observations indicate that DNMT3A serves as a tumor suppressor, where mutations drive clonal expansion of HSCs that can eventually lead to AML. Because these disorders all involve LOF mutations in DNMT3A, determining mechanisms that may activate DNMT3A or restore its activity could inspire strategies to therapeutically correct disease-associated loss of DNMT3A activity.
DNMT3A has three protein domains: the Pro-Trp-Trp-Pro (PWWP) domain, the ATRX-DNMT3-DNMT3L (ADD) domain, and the catalytic methyltransferase (MTase) domain, along with a disordered N-terminus.9 Crystallographic and biochemical data10,11 support an allosteric mechanism by which the ADD domain regulates enzyme activity; in the absence of unmodified histone 3 lysine 4 (H3K4me0), DNMT3A adopts an autoinhibited conformation where the ADD domain blocks the binding of the DNA substrate to the catalytic domain. When the ADD domain binds to H3K4me0, it undergoes a conformational change, revealing the DNA binding site to relieve autoinhibition and increase the activity of DNMT3A.10 Profiling mutations5 that can perturb these regulatory mechanisms, beyond those found clinically, may provide further insight into strategies to activate DNMT3A.
Motivated by these prospects, we conducted in situ mutational scanning on DNMT3A using base editing.12 Base editing is a form of precision genome editing that has enabled the direct mutational scanning of a variety of endogenous proteins.13–16 Base editing has the advantage of targeting the native gene, avoiding the confounding effects of overexpressing mutants of a protein of interest. By coupling a fluorescence-based DNA methylation reporter system with base editing to generate a library of mutants, we previously identified a wide variety of LOF mutations in DNMT3A, and highlighted the importance of the PWWP domain for enzyme activity, particularly in its ability to bind to DNA.12 However, this study only identified a single activating mutation to DNMT3A, which disrupts the known autoinhibitory ADD–MTase interaction.10 Whether additional activating mechanisms exist remains an open question.
To explore potential novel activating mutations, in this study we employ base editor scanning coupled to an optimized DNA methylation reporter assay specifically tailored to discover activating DNMT3A mutations. By performing the screen in isogenic cell lines with or without the hotspot LOF DNMT3AR882H mutation, we identify mutations that both boost the activity of DNMT3AWT and restore activity in the context of DNMT3AR882H. By further validating these mutations biochemically, we nominate the ADD domain and the ADD–MTase interface of DNMT3A as promising functional sites for future pharmacologic modulation to potentiate DNMT3A activity.
Results and Discussion:
Development of a screening approach for discovery of activating mutations
To identify activating DNMT3A mutations, we sought to adapt our previously reported base editor scanning approach coupled to a fluorescence-based reporter of DNA methylation.12,17 In this system, we employed a reporter construct that contains TetO sites upstream of a promoter controlling the expression of citrine (Figure 1a). In our original system (v1), we fused DNMT3L, a catalytically inactive accessory protein that binds to DNMT3A18,19 to rTetR, such that adding doxycycline (dox) to the cells recruited rTetR-DNMT3L to the citrine promoter. Methylation of the citrine promoter by endogenous DNMT3A then silenced citrine expression, which could be used as a proxy for DNMT3A activity. After introducing a library of single-guide RNAs (sgRNAs) to the cells containing this reporter system, differential silencing upon addition of dox was used to sort cells based on the activity of the DNMT3A mutants resulting from each sgRNA (Figure 1b).
Figure 1: Strategy for and validation of DNMT3A activating screen.
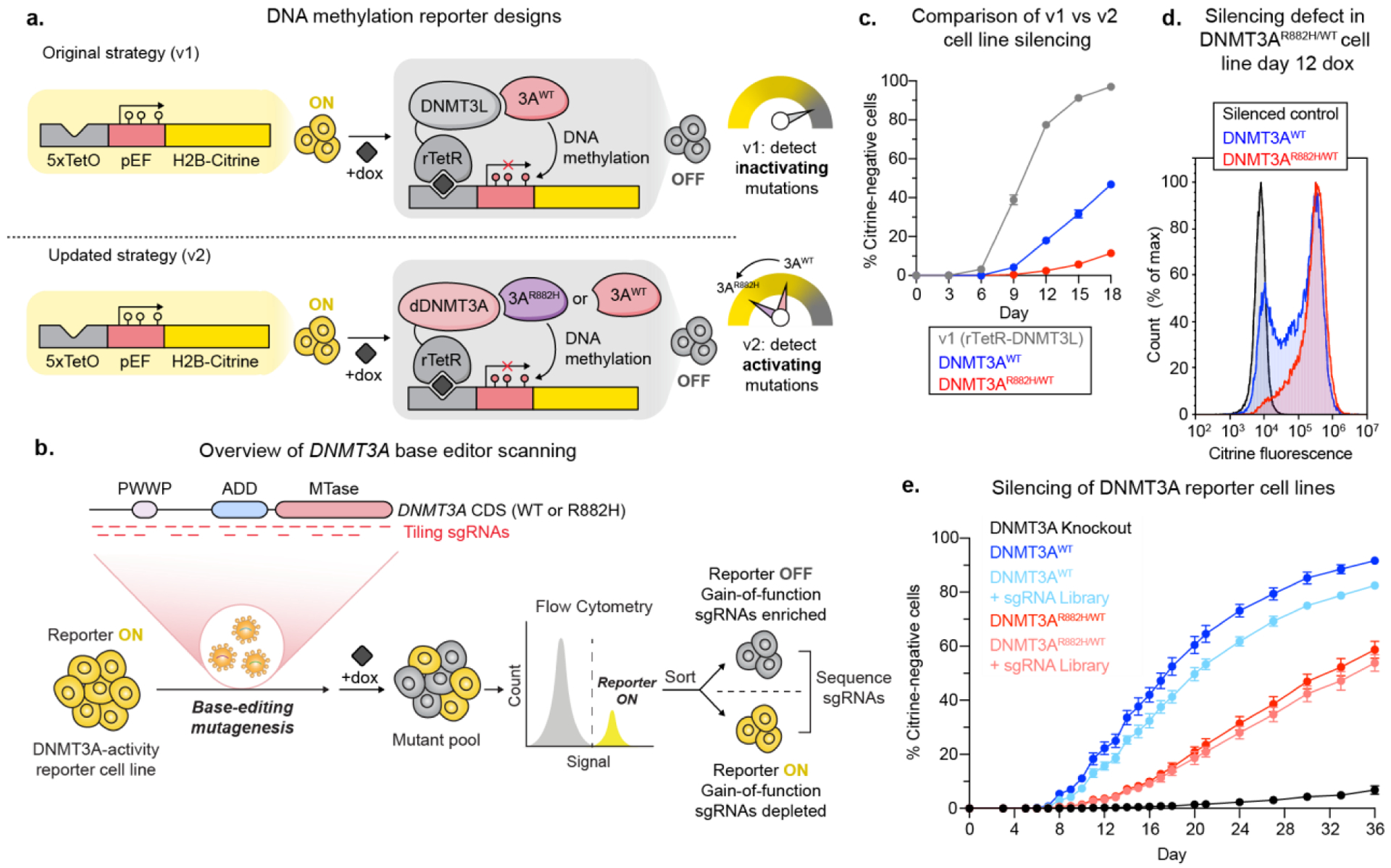
a) Cartoon showing the original base editor scanning12 and updated strategy for identifying DNMT3A activating mutations. By lowering the background activity of our reporter, we decreased the probability that activating mutations would be hidden by the limited dynamic range between baseline and maximum DNMT3A activity. dDNMT3A = codon-switched DNMT3A2E756A.
b) Cartoon showing our strategy for base editor scanning and separating cells based on DNMT3A activity.
c) Line plot showing silencing over time for rTetR-DNMT3L (v1), DNMT3AWT or DNMT3AWT/R882H (v2) cells. See Figure S2c. for significance testing.
d) Histogram showing silencing of citrine reporter in cells with DNMT3AWT or DNMT3AWT/R882H after 12 days of dox treatment. See Figure S2d. for significance testing.
e) Line plot showing silencing over time of DNMT3AWT or DNMT3AWT/R882H cells with and without the base editing library. Data are mean ± SD of n = 3 replicates. See Figure S2e. for significance testing.
While this reporter setup successfully identified inactivating DNMT3A mutations, it was not optimized to identify activating mutations since DNMT3L itself activates DNMT3A,18 resulting in a much higher baseline level of silencing (Figure 1a). Therefore, we modified the reporter system to address these limitations. First, we swapped DNMT3L for a catalytically dead version of DNMT3A (dDNMT3A) harboring a mutation to the catalytic glutamate (E756A),20 which can also recruit endogenous DNMT3A due to DNMT3A’s active state being a tetramer.2 To prevent base editing of dDNMT3A, we adjusted the codons encoding dDNMT3A by introducing synonymous mutations that disrupt the PAM sequence and hence sgRNA targeting (see Methods). To further decrease the background silencing rate of our reporter system and also identify base edits that could activate the most common clinical DNMT3A mutant, we also knocked in a heterozygous DNMT3AR882H mutation into our K562-based reporter cell line, resulting in isogenic DNMT3AWT/WT/WT and DNMT3AWT/R882H/R882H cell lines (Figure S1). The new cell lines were observed to silence more slowly than our previous v1 rTetR DNMT3L cell line (Figure 1c). We then performed parallel screens in these two cell lines (termed DNMT3AWT and DNMT3AWT/R882H, respectively). Altogether, by using dDNMT3A as a recruitment bait and introducing the R882H mutation, we aimed to create a larger dynamic range between the reporter silencing rate in cells that receive an activating mutation versus a neutral mutation (Figure 1a).
As expected, the DNMT3AWT/R882H cell line had fewer silenced cells after 12 days of dox treatment than DNMT3AWT cells (Figure 1d) and silenced the citrine reporter more slowly (Figure 1c,e). In order to verify that citrine silencing reported on DNMT3A activity, we knocked out either DNMT3A or the other human de novo DNA methyltransferase DNMT3B in our reporter cell lines, treated the cells with dox, and tracked the silencing of the reporter over time, confirming that reporter silencing is DNMT3A-dependent (Figure S2a). A silencing time course in each of our cell lines with or without the sgRNA library demonstrated that the DNMT3AWT/R882H cell line silences more slowly than the DNMT3AWT cell line across the entire time course of the experiment (Figure 1e).
Paired base editor screens identify activating mutations
To conduct DNMT3A base editor scanning, we next transduced a lentiviral library containing our previously employed DNMT3A sgRNA library, which contains 730 sgRNAs and the C-to-T base editor BE3.9max15, into the reporter cell lines. sgRNAs were annotated to edit within a given window from the +4 to +8 positions of the sgRNA with the protospacer adjacent motif (PAM) defined as positions +21–23. We treated transduced cells with dox to recruit rTetR-dDNMT3A and subsequently endogenous DNMT3A to the reporter and then sorted for cells that had silenced their citrine reporter genes, using fully silenced cells as a gating control (Figure S2b). Sorted cells were collected when the cells were ~10–15% silenced (see Methods), corresponding to day 10 for DNMT3AWT or day 17 for DNMT3AWT/R882H. By comparing the abundance of cells harboring a particular sgRNA between the citrine negative (dark) and unsorted cells, we identified sgRNAs that accelerate citrine reporter silencing by base editing DNMT3A (Figure 2a,b).
Figure 2: Results of base editor screens in paired isogenic cell lines.
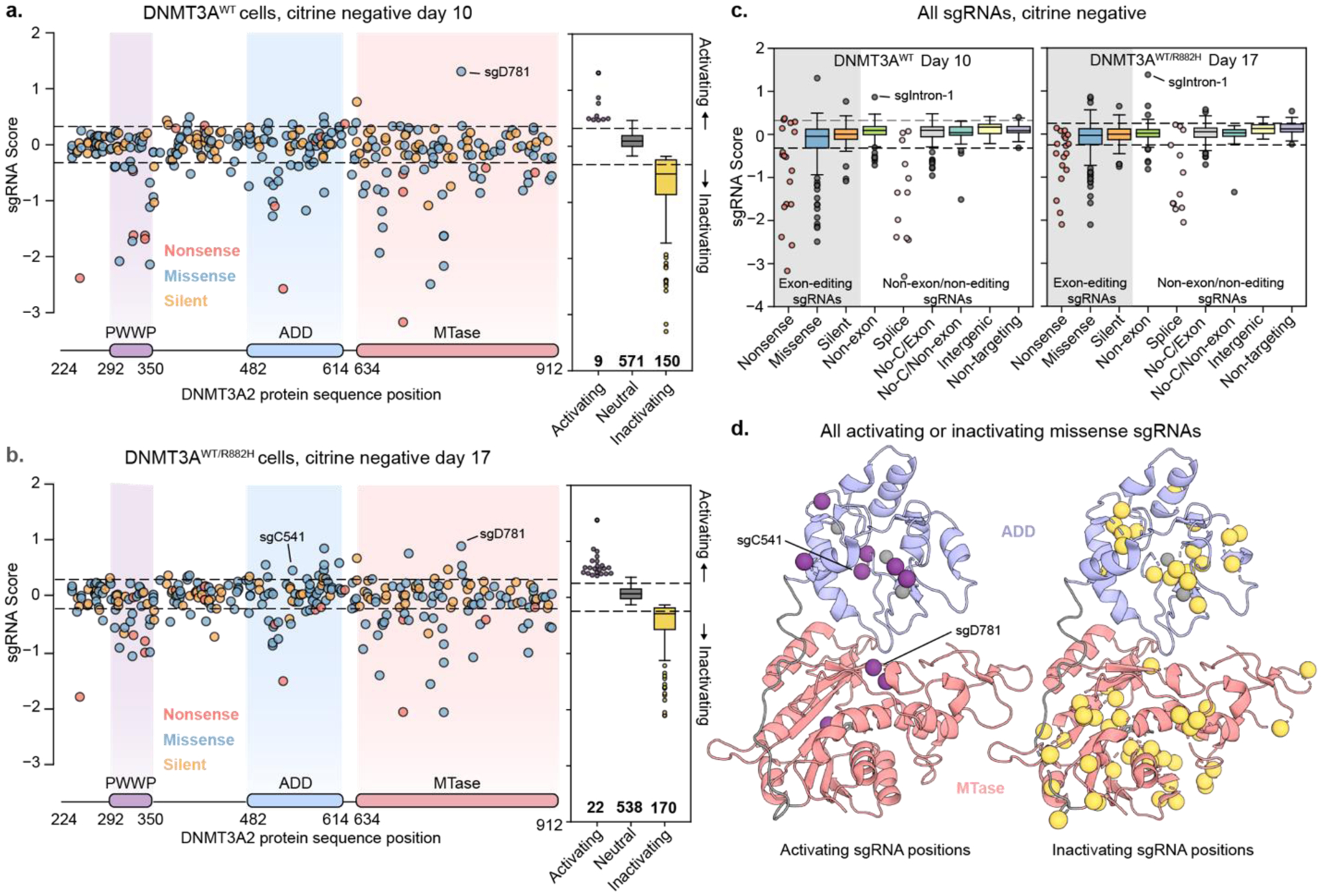
a) Scatter plot of sgRNA enrichment across the coding sequence of DNMT3A in DNMT3AWT cells. sgRNA score corresponds to log2 fold-change of citrine-negative cells/unsorted cells. Enrichment scores for all guides can be found in Table S4.
b) Same as a) but in DNMT3AWT/R882H cells. Enrichment scores can be found in Table S5.
c) Box plot showing enrichment of sgRNAs sorted by category in both screens. sgRNAs in the gray box are predicted to edit within a DNMT3A exon, while those in other categories either edit outside of DNMT3A exons or are not predicted to edit.
d) Pymol cartoon model showing predicted mutated residues from missense activating (left) and inactivating (right) sgRNAs mapped to the crystal structure of the DNMT3A ADD and MTase domains (PDB: 4u7p).10
Comparison of the parallel screens revealed that many more sgRNAs scored as activating in the DNMT3AWT/R882H cell line than in the DNMT3AWT cell line (Figure 2a,b boxplots), supporting the notion that lowering DNMT3A background activity increases the dynamic range for identifying activating mutants. Despite these differences, sgRNAs predicted to introduce nonsense mutations were depleted in both cell lines and control sgRNAs were largely unchanged between sorted and unsorted cells (Figure 2c), indicating that the base editor scans faithfully reported on DNMT3A activity. In both cell lines, activating sgRNAs enriched in the ADD and MTase domains, suggesting that they may disrupt autoinhibitory functions of the ADD domain (Figure 2d). Conversely, inactivating sgRNAs generally spanned the entire protein, as we observed previously12 (Figure 2d).
Interestingly, sgRNAs predicted to introduce nonsense and splice site disruption mutations exhibited a greater negative effect in the DNMT3AWT cell line than in the DNMT3AWT/R882H cell line (paired two-sided student’s t-tests p=0.007 for splice guides and p=0.0230 for nonsense guides, Figure 2c), perhaps due to knockout of the dominant negative R882H copy being advantageous for activity. Of note, one sgRNA annotated as intronic (sgIntron-1, editing window chr2:25243984–25243988) was enriched in both cell lines. Out-of-window editing by sgIntron-1 could potentially edit the exonic region of DNMT3A, so we highlighted it for future investigation along with the enriched sgRNAs predicted to introduce missense mutations.
Cellular and in vitro characterization of activating mutations
To directly compare the base editor results, we plotted the enrichment score of each sgRNA in each cell line against each other (Figure 3a). While more activating mutations were identified in our DNMT3AWT/R882H screen than in the DNMT3AWT cell line (See Table S6 for details on enriched guides), the sgRNA scores were still correlated across each cell line. Next, we sought to validate top activating sgRNAs by introducing them into reporter cells individually. First, we confirmed that DNMT3A base editing did not significantly destabilize the protein (Figure S3). Next, we tested the silencing ability of the base-edited cells compared to cells containing a control nontargeting (Luc) sgRNA (Figure 3b). In this format, five of the seven tested sgRNAs were validated in the DNMT3AWT/R882H, while three out of seven validated in the DNMT3AWT cells. Of note, while sgR631 was not validated, low levels of activation were observed in one replicate of this experiment (Figure S4b), suggesting that variability in editing efficiency might sometimes obscure our ability to detect DNMT3A activation. In our single sgRNA experiments as well as in the screen itself, we also observed that sgG532, our activating hit from our previous study21, was only modestly activating (Figure 3a,b, Figure S4), possibly indicating mechanistic differences in how DNMT3L vs dDNMT3A recruit DNMT3A.
Figure 3: Validation of sgRNA hits.
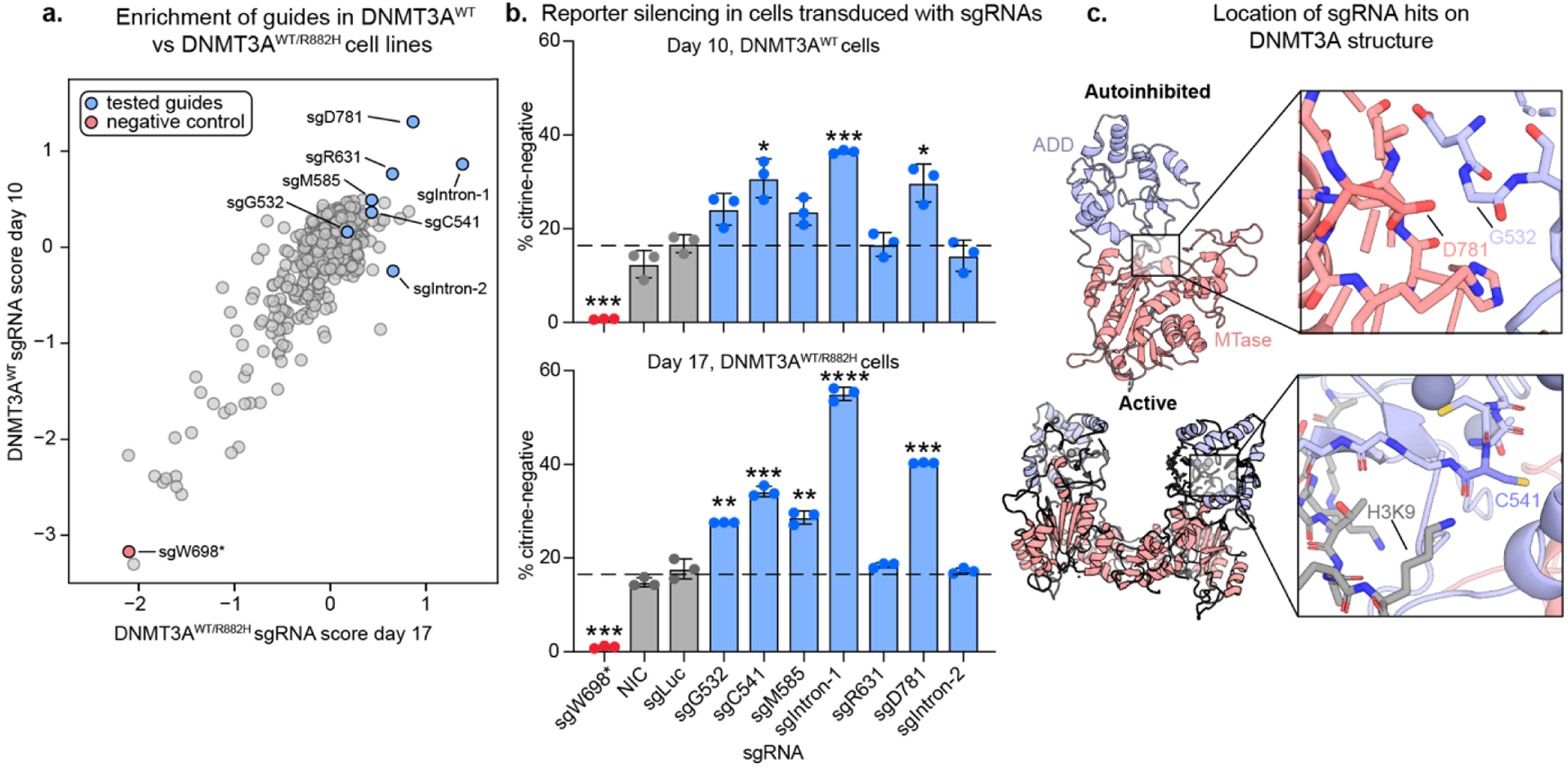
a) Scatter plot showing sgRNA scores in DNMT3AWT vs DNMT3AWT/R882H reporter cells. sgRNAs tested in b) and c) are highlighted. Spearman R = 0.51, p=8.1×10−44
b) Bar plots showing % silenced cells treated with single sgRNAs after 10 (DNMT3AWT) or 17 (DNMT3AWT/R882H) days of dox treatment. sgLuc = Luciferase-targeting sgRNA, NIC = non-infection control. Data are mean ± SD of n = 3 replicates. * Correspond to significance testing using two-sided students t-tests comparing each guide to sgLuc with Bonferroni multiple hypothesis correction. *p ≤ 0.0167, **p ≤ 0.0033, ***p ≤ 0.00033, ****p ≤ 3.33 ×10−5 See Figure S4 for additional sgRNAs and replicates and table S6 for a summary of all sgRNAs tested.
c) PyMOL models showing the structural location of key activating mutants. PDB IDs: 4u7t (active) and 4u7p (autoinhibited).10
After performing silencing assays on cells transduced with individual sgRNAs, we next genotyped the edited cells and compared the observed mutations with the predicted edits. Of the validated sgRNAs, sgD781 introduced the predicted D781N mutation (Figure 4a), which is positioned directly across the autoinhibitory interface from the activating mutation found in our previous work, G532N12 (Figure 3c). sgC541 introduced the predicted C541Y mutation in the ADD domain, which lies close to the interaction site for K9 of the activating H3K4me0 peptide. Interestingly, this mutation is distal to the autoinhibitory interface that is perturbed by the G532N or D781N mutants.
Figure 4: Genotyping of sgRNA hits.
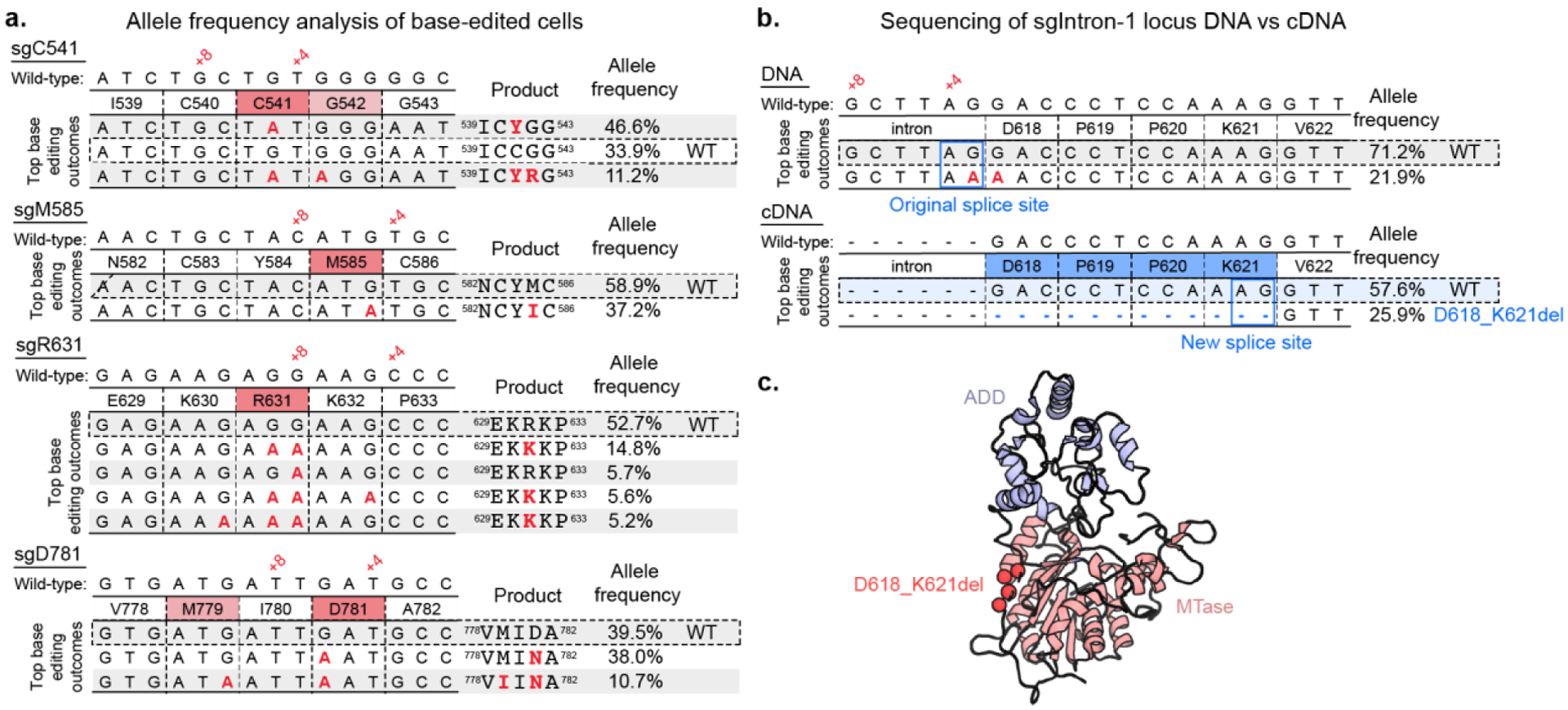
a) Tables showing base editing outcomes with >5% allele frequency from treatment of DNMT3AWT reporter cells with single missense sgRNAs (See Figure S5 for DNMT3AWT/R882H cell line). Analysis performed with CRISPResso222.
b) Scheme showing genomic DNA and cDNA sequencing of DNMT3AWT reporter cells treated with sgIntron-1. (See Figure S5 for DNMT3AWT/R882H cell line).
c) PyMOL model showing location of D618_K621del (PDB ID 4u7p)10.
sgIntron1 was strongly activating as well despite its editing window being in an intron (Figure 3b). Genotyping of the targeted genomic locus revealed editing at the +3 position of the sgRNA, one base pair outside of the canonical editing window (Figure 4b). Notably, the edited C complements the G of a splice acceptor site. Therefore, we hypothesized that the editing of this residue could induce alternative splicing that utilizes an “AG” sequence motif 12 base pairs into the next exon, causing a four-amino acid deletion in the resulting translated protein (D618_K621del). To test this hypothesis, we sequenced the DNMT3A cDNA from cells transduced with sgIntron-1 and observed that 29% and 38% of transcripts from DNMT3AWT and DNMT3AWT/R882H cells, respectively, were alternatively spliced as predicted (Figure 4b). The resulting translation contains a deletion of residues 618–621 in the disordered linker region between the ADD and MTase domains (Figure 4c). In addition, the cryptic splice site leading to the D618_K621del mutation is also found in several species of colobinae monkeys, indicating that this mutation has been sampled in the course of DNMT3A evolution (Figure S6). This demonstrates an unexpected mechanism for generating a deletion mutation using a base editor. Overall, we identified the activating mutants D781N, C541Y, and D618_K621del as strongly activating in our reporter assay and selected them to study in vitro.
Having validated a subset of activating sgRNAs in the cellular reporter system, we next evaluated the activity of the corresponding purified DNMT3A mutants compared to WT DNMT3A and the single activating hit from our previous work21, G532N. In a biochemical methyltransferase assay, G532N demonstrated ~9-fold higher activity than WT DNMT3A, while the mutants identified in the updated screen were ~4 fold more active than WT (Figure 5a). We also confirmed that our mutants are stable by differential scanning fluorimetry (Table S3). We next considered whether these activating mutants are stimulated by H3K4me0. Interestingly, all new mutants showed increased enzymatic activity in the presence of 5 μM H3K4me0 peptide (KD for H3K4me0 binding to DNMT3A ADD domain is 1.75 μM ± 0.1110) (Figure 5b). These results, along with the positions of the D618_K621del and C541Y mutations in the protein structure, suggest that the mutations do not activate the protein by fully relieving the known autoinhibition mechanism, although they do suggest that these mutants could still be perturbing the equilibrium between the autoinhibited and active conformations. Overall, we identified three mutations to DNMT3A through base editor screens that are in and around the ADD domain and are activating both in our cellular reporter system and in vitro.
Figure 5: Biochemical validation of activating mutants.
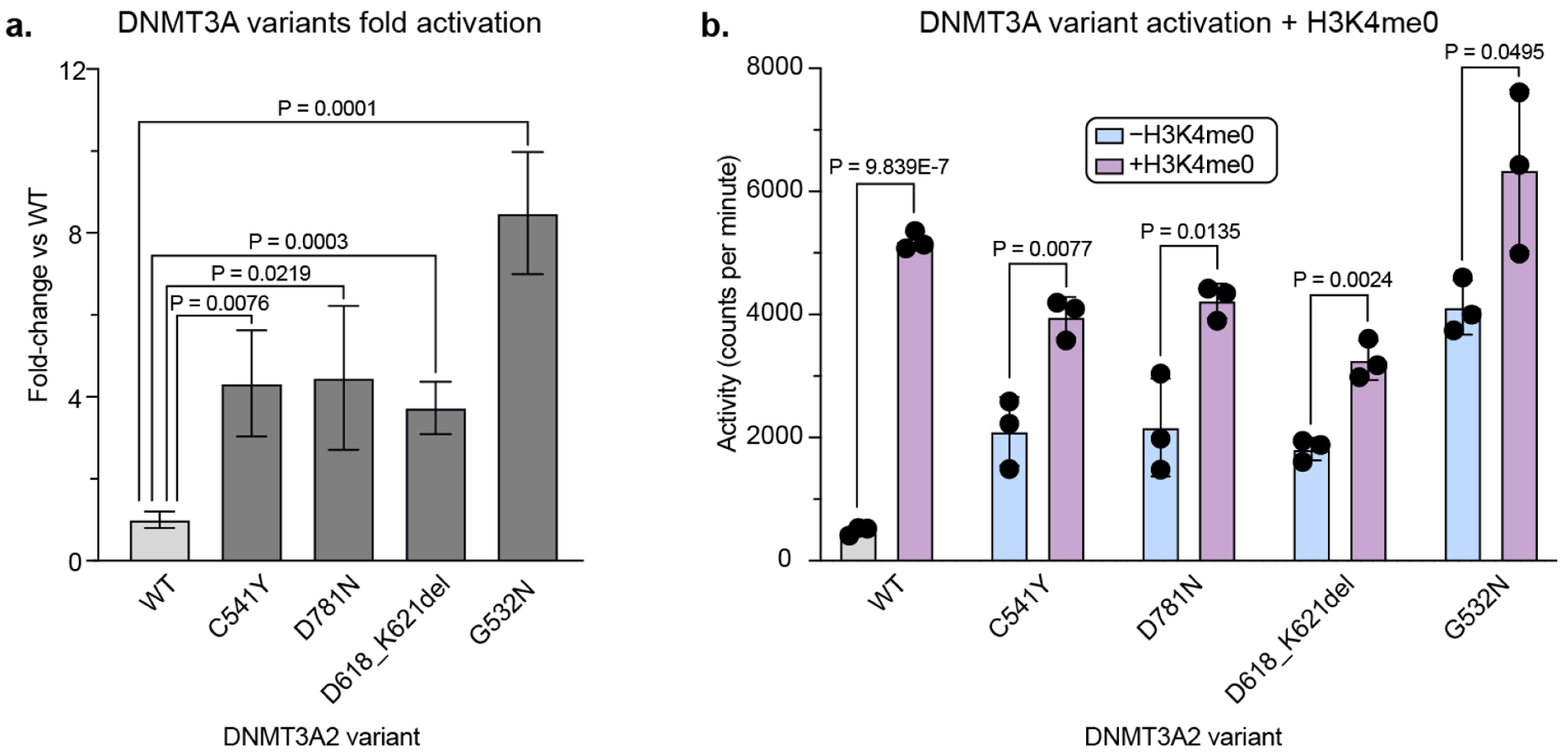
a) Bar plot showing fold-change in DNMT3A methyltransferase activity of mutants vs WT, based on data in b). P-values were calculated by unpaired two-tailed students’ t-tests. Fold-change errors were propagated from the individual standard deviations.
b) Bar plot showing methyltransferase activity of DNMT3A mutants ± 5 μM H3K4me0. Data are mean ± SD of n = 3 replicates. P-values were calculated by unpaired two-tailed students’ t-tests. See Figure S7 for SDS-PAGE gel of purified proteins and replicate 2.
By innovating new DNMT3A reporter strategies combining enzyme hypomorphs with base editing technology, we expanded our ability to discover activating mutations to DNMT3A. Our approach demonstrates the ability of the adapted DNMT3A reporter strategy to identify new activating mutants with more intermediate effects, which was not possible with our initial strategy. These results highlight the importance of considering the baseline activity of the protein of interest and selection in designing an appropriate screening strategy. Furthermore, we provide a blueprint for how parallel base editor screens can be performed in isogenic cell lines. While the mutant DNMT3AWT/R882H cell line provided additional dynamic range in which to detect activating mutations, ultimately most activating mutations identified in the mutant cell line were also activating in the WT cell line. The ability of these mutants to increase DNMT3A activity in both the WT and the most common clinical mutant contexts further highlights these areas of DNMT3A as valuable and broadly applicable pharmaceutical targets. We anticipate that base editing approaches in isogenic cell lines could be a valuable strategy to investigate other proteins (i.e., oncogenes, tumor suppressors) and their corresponding mutants of interest. We also highlight how well-designed selection strategies can enable the identification of hypermorphic variants of a protein of interest with base editor scanning.
One limitation of this study was the use of a cytosine base editor with an “NGG” protospacer-adjacent motif requirement, which reduced the number of accessible mutations. We chose to use a cytidine versus an adenosine base editor due to the availability of efficient C vs A editors at the conception of this study, but A editors such as ABE8e23 would likely be successful in a similar study. Despite these limitations, our optimized screening strategy successfully identified several new activating mutations, suggesting that future studies employing next-generation expanded PAM SpG and SpRY cytidine and adenosine base editors24,25 could uncover even more activating mutations. Furthermore, the ability of base editing to target splice sites—which in the past has been used to disrupt genes,26 induce exon skipping,27 or as a control for LOF mutations—could serve as an interesting tool to probe splice sites in clinically-relevant proteins for potential gain-of-function (GOF) mutations caused by alternative splicing. Altogether, our results suggest that base editing of splice sites might be a good strategy to explore cryptic splicing variants of different proteins of interest.
One additional limitation of this study is that the nature of base editing makes determining whether our mutations in the DNMT3AWT/R882H cell line are in cis or in trans with the R882H mutation very challenging. Innovations in long read sequencing technology could make this type of analysis more routine and feasible for future studies. Additionally, biochemical purification and evaluation of double mutants comprising R882H and our activating mutants would help determine if the activating mutations can directly rescue the effect of R882H. Moreover, these experiments could inform whether these activating mutations directly influence the catalytic activity of DNMT3A or indirectly influence activity by altering oligomerization3, sequence specificity, and/or DNA binding ability28,29 of the catalytically relevant multimeric species.
In this study, we identified and validated three new mutations that demonstrate novel ways to activate DNMT3A, which point to the ADD domain as a master regulator of DNMT3A function and a possible target site to potentiate DNMT3A activity. The D781N mutation likely demonstrates another example of disrupting the ADD–MTase autoinhibitory interface. The D618_K621del activating mutation alters a disordered linker region of DNMT3A, nominating this unstructured region as potentially important for DNMT3A regulation. The location of C541Y on the structure of the protein indicates that areas of the ADD domain beyond the known autoinhibitory interface can also be involved in DNMT3A activation. Notably, the C541Y mutation involves a cysteine to tyrosine substitution, suggesting that covalent modification of the cysteine by a small molecule electrophile might phenocopy the effect.30 Given the large body of evidence indicating that DNMT3A LOF promotes developmental disorders and leukemogenesis in hematopoietic disorders, development of small molecules that could activate DNMT3A activity would be of significant interest. Exploiting these activating mutations could be critically instructive in identifying functional sites for small molecule agonists of DNMT3A. We anticipate that combining innovative selection strategies with base editing mutagenesis could eventually help inform drug discovery efforts to recover activity in other tumor suppressor targets.
Methods:
Safety statement
No unexpected or unusually high safety hazards were encountered.
General plasmid construction
sgRNA plasmids were cloned as described in ref12. Gibson Assembly with NEBuilder HiFi (New England Biolabs) was used to clone additional plasmids. NEB Stable (lentiviral vectors) and NEB 5-alpha (other plasmids) (New England Biolabs) were used as cloning strains. Final constructs were validated by Sanger (Quintara Biosciences) or Oxford Nanopore (Plasmidsaurus) sequencing. Plasmids and oligonucleotides are provided in Tables S1 and S2, respectively.
Construction of rTetR-dDNMT3A reporter
To recruit endogenous DNMT3A to the reporter promoter, a catalytically dead DNMT3A2 mutant (DNMT3A2E756A) was fused to rTetR, in an adaptation of our previously reported strategy12. The gene encoding the recruiting DNMT3A2E756A was engineered to prevent its recognition and mutagenesis by the base editor scanning DNMT3A2 sgRNA library. A custom python script was used to insert synonymous substitutions in silico throughout the DNMT3A2 coding sequence to block sgRNA recognition. Briefly, for each library sgRNA recognizing an exon or exon boundary, the ‘NGG’ protospacer-adjacent motif (PAM) was disrupted where possible; otherwise, protospacer positions were mutated to create at least 2 mismatches, beginning at the PAM-proximal and moving toward the PAM-distal end. After considering all sgRNAs, protospacer mismatches were enumerated. For any sgRNA with 3 or fewer protospacer mutations, none of which were in the 6 most PAM-proximal positions, an additional mismatch was introduced if possible. This resulted in a gene encoding wild-type DNMT3A2 but containing mismatches with each library sgRNA. Finally, the E756A substitution was introduced. The DNMT3A2E756A gene was synthesized by IDT and cloned into pRRL-pEF-H2B-mCherry-T2A-rTetR-Dest-SV40 (Addgene #186968).
Cell culture
K562 (ATCC), 293FT (Thermo Fisher Scientific), and HEK293T (a gift from B. E. Bernstein, Massachusetts General Hospital) cell lines were used in this study. All cell lines were cultured at 37 °C and 5% CO2 in a humidified incubator and were tested for mycoplasma. All media were supplemented with 100 U ml−1 penicillin, 100 μg ml−1 streptomycin (Gibco), and 10% FBS (Peak Serum). K562s were cultured in RPMI-1640 (Gibco), HEK293Ts were cultured in DMEM (Gibco), and 293FTs were cultured in DMEM with 2 mM GlutaMAX (Gibco) and 1× MEM nonessential amino acids (Gibco).
Lentiviral transduction
To produce lentivirus, GAG/POL and VSVG plasmids were co-transfected with transfer plasmid into HEK293Ts or HEK293FTs using FuGENE HD (Promega, 3.33:1 FuGENE:DNA), or lipofectamine 3000 Reagent (Thermo Fisher Scientific). Medium was then replaced after 6–8 hours. 48–60 hours later, the medium was passed through a 0.45-μm filter and either used immediately or snap-frozen for storage at −80 °C. To generate reporter cell lines and introduce sgRNAs, K562s were transduced with the appropriate lentivirus by spinfection (1,800g, 90 min, 37 °C) with 12 μg ml−1 polybrene (Santa Cruz Biotechnology). 2 μg ml−1 puromycin (Gibco) or fluorescence-activated cell sorting (FACS) were used to select for transduced cells. A clonal methylation reporter cell line (DNMT3AWT) was first derived and used to generate the DNMT3AWT/R882H cell line. This clonal cell line or the DNMT3AWT/R882H clonal cell line was used for all individual sgRNA transductions.
Generation of DNMT3AWT/R882H reporter cell line
All CRISPR reagents were obtained from Integrated DNA Technologies (IDT) unless otherwise specified. In order to knock in the R882H mutation to DNMT3A in our reporter cell line, CRISPOR31 was used to design an appropriate guide RNA targeting the DNMT3A R882 locus (see guide sequences in Table S2). The guide RNA was ordered as an Alt-R CRISPR-Cas9 crRNA. The Alt-R HDR Donor Oligo was designed using the IDT web tool. To prepare the gRNA complex, crRNA and Alt-R CRISPR-Cas9 tracrRNA were mixed in equimolar ratio, heated to 95C for 5 min, then allowed to cool to room temperature. To prepare the RNP complex, the gRNA complex was combined with Alt-R Cas9 enzyme and incubated at RT for 20 min. The Neon™ Transfection System was used to electroporate 2 ×105 DNMT3AWT reporter cells with the assembled RNP complex and HDR Donor. Electroporated cells were recovered in antibiotic-free media for 72 hours. Single cell clones were sorted by FACS and screened by amplicon sanger sequencing for the presence of the R882H mutation. The final clone was confirmed to have two copies or the DNMT3AR882H allele and one copy of the DNMT3AWT allele by Illumina amplicon sequencing (Figure S1).
Reporter silencing assays
Reporter silencing assays were conducted as in ref12 with the following changes: cells were treated with 1μg ml−1 dox (Sigma-Aldrich) except for v1 cells which were treated with 0.1 μg/mL dox, and gates were set based on reference reporter cells cultured in parallel without dox and reference fully silenced reporter cells (See gating scheme in Figure S2).
Genotyping
The QIAamp DNA Blood Mini kit (Qiagen) was used to purify genomic DNA. 100 ng of DNA was inputted into a first round of PCR (27 cycles, Q5 hot start high-fidelity DNA polymerase (New England Biolabs)) to attach common overhangs and amplify the target locus. To attach barcoded adapters, 1 μL of each PCR1 reaction was amplified in a second round of PCR (8 cycles). Primer sequences are provided in Table S2. Gel extraction (Zymo) was used to purify final amplicons, which were then sequenced on an Illumina MiSeq. Data were processed using CRISPResso222.
Reverse transcription and sequencing of sgIntron_1 locus
The RNeasy Mini Kit (Qiagen) was used to isolate RNA. Then, MultiScribe Reverse Transcriptase (Invitrogen) was used with 10x RT Random Primers (Invitrogen) to amplify 1.8 ug of RNA for each sample. ~50 ng of the resulting cDNA was then used as template for amplification with pEG1.136_1R and pEG1.236_1F for 40 cycles of PCR. The resulting products were then barcoded and sequenced in the same manner as our DNA genotyping.
Immunoblotting
Immunoblotting was performed as in ref12. The following antibodies and dilutions were used: Primary antibodies: DNMT3A (Cell Signaling Technology, catalog no. 32578, D2H4B, 1:5,000), GAPDH (Santa Cruz Biotechnology, catalog no. sc-47724, 0411, 1:8,000). Secondary antibodies: anti-rabbit IgG HRP conjugate (Promega, catalog no. W4011, 1:100,000 for DNMT3A), anti-mouse IgG HRP conjugate (Promega, catalog no. W4021, 1:400,000 for GAPDH). Immunoblots were visualized using SuperSignal West Femto (DNMT3A) or SuperSignal West Pico PLUS (GAPDH) chemiluminescent substrates (Thermo Fisher Scientific).
Base editor scanning
This study employs the sgRNA library described in ref12. Titering of library lentivirus was performed by measuring cell counts of transduced cells after puromycin selection. 30 × 106 reporter cells per cell line were transduced with library lentivirus at a multiplicity of infection <0.3 and selected with puromycin for 7 days. Following selection and expansion cells were split into three replicates and treated with 1 ug/mL dox. Timing of sorting was chosen to match when cells were ~10–15% silenced. This timepoint balances our ability to enrich activating guides with our ability to sort an adequate number of cells for library coverage. DNMT3AWT reporter cells were passaged on days 3, 6, and 7 of dox treatment, and cells were sorted on day 10 for sequencing analysis. DNMT3AWT/R882H reporter cells were passaged on days 3, 6, 9, 11, 14, and 17 of dox treatment, and cells were sorted on day 17 for sequencing analysis. Cells were sorted on a FACSAria II (BD), collecting citrine+, citrine− and unsorted (all mCherry+) cells. Fully silenced control cells were used for citrine gating (see Figure S2). Library preparation for Illumina sequencing and data analysis was performed as in ref12. Library sgRNA annotations and base editor scanning data are provided in Tables S4 and S5.
Protein expression and purification
Full-length DNMT3A2 was recombinantly expressed and purified according to ref.12 with the following modifications: Base buffer consisted of 50 mM Tris-HCl, pH 8.0 cold, 500 mM NaCl, 1 mM TCEP, and lysis buffer was supplemented with 10 mM imidazole and 0.2% Triton X-100. DNMT3A storage buffer consisted of Base buffer but with 300mM NaCl.
Activity assays
DNMT3A activity was measured using the protocol described in ref12. For histone stimulation experiments, 5 μM peptide was added prior to addition of enzyme. The following peptides were used: H3K4me0 (ARTKQTARKSTG-NH2, Biomatik).
Differential scanning fluorimetry
First, 2 μM purified DNMT3A2 was incubated in triplicate with 5× SYPRO Orange (Thermo Fisher Scientific) in assay buffer (50 mM Tris-HCl, pH 8, 300 mM NaCl, 1mM TCEP). Samples were heated from 10°C to 95°C (10 s at each 0.5°C step) using a CFX Connect qPCR with CFX Manager software (Bio-Rad). Melting temperatures were calculated with DSFWorld32 (by model 2).
Colobinae monkey sequence analysis
The jackhmmer webserver33 (accessed April 2020) was used with human DNMT3A (UniProt: Q9Y6K1) as the query sequence and an E-value cutoff of 0.01 to generate a multiple sequence alignment. This alignment was then manually searched for sequences containing the D618_K621del mutation generated by sgIntron-1, and the resulting sequences were realigned using Geneious. Sequence identities were also calculated using Geneious.
Statistics
All statistical tests are described in figure legends and were performed with SciPy, GraphPad Prism, or Pandas.
Supplementary Material
Table S6: Additional data on activating guides.
Table S2: Primers and oligos used in this study.
Table S1: Plasmids constructed in this study.
Table S3: Melting temperatures of recombinant DNMT3A2 variants by DSF.
Figure S1: Pie chart and allele table showing targeted deep sequencing of R882 codon of DNMT3A in DNMT3AWT/R882H cells.
Figure S2: Silencing dynamics of dDNMT3A reporter system, representative gating scheme, and additional plots associated with Figure 1.
Figure S3: Western Blot of reporter cells transduced with base editing sgRNAs.
Figure S4: Additional sgRNAs tested.
Figure S5: Genotyping of DNMT3AWT/R882H cells treated with sgRNAs.
Figure S6: D618_K621del mutation is found in colobinae monkeys.
Figure S7: Additional in vitro validation of DNMT3A variants.
Table S5: WT-R882H cell line screen results.
Table S4: WT cell line screen results.
Acknowledgements:
We would like to thank members of the Liau Lab and others for insightful comments and discussion on the manuscript, in particular J. Morris, H. Kwok, and S. Berry. We additionally thank Z. Niziolek and C. Maesner at the Bauer Core Facility at Harvard University for help with FACS, and D. Bolduc for biochemistry advice.
E.M.G. and N.Z.L. were supported by National Science Foundation Graduate Research Fellowships (grant no. DGE1745303). E.M.G. was also supported by a Landry Cancer Biology Fellowship. B.B.L. is a Damon Runyon-Rachleff Innovator supported in part by the Damon Runyon Cancer Research Foundation (grant no. DDR 60S-20). This research was additionally supported by award no. 1DP2GM137494 from the National Institute of General Medical Sciences and startup funds from Harvard University.
Conflict of Interest Statement:
B.B.L. holds sponsored research projects with AstraZeneca and Eisai, is a scientific consultant for Imago BioSciences and Exo Therapeutics and is a shareholder and member of the scientific advisory board of Light Horse Therapeutics.
References:
- (1).Brunetti L; Gundry MC; Goodell MA DNMT3A in Leukemia. Cold Spring Harb. Perspect. Med 2017, 7 (2), a030320. 10.1101/cshperspect.a030320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Russler-Germain DA; Spencer DH; Young MA; Lamprecht TL; Miller CA; Fulton R; Meyer MR; Erdmann-Gilmore P; Townsend RR; Wilson RK; Ley TJ The R882H DNMT3A Mutation Associated with AML Dominantly Inhibits Wild-Type DNMT3A by Blocking Its Ability to Form Active Tetramers. Cancer Cell 2014, 25 (4), 442–454. 10.1016/j.ccr.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Nguyen T-V; Yao S; Wang Y; Rolfe A; Selvaraj A; Darman R; Ke J; Warmuth M; Smith PG; Larsen NA; Yu L; Zhu P; Fekkes P; Vaillancourt FH; Bolduc DM The R882H DNMT3A Hot Spot Mutation Stabilizes the Formation of Large DNMT3A Oligomers with Low DNA Methyltransferase Activity. J. Biol. Chem 2019, 294 (45), 16966–16977. 10.1074/jbc.RA119.010126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kim SJ; Zhao H; Hardikar S; Singh AK; Goodell MA; Chen T A DNMT3A Mutation Common in AML Exhibits Dominant-Negative Effects in Murine ES Cells. Blood 2013, 122 (25), 4086–4089. 10.1182/blood-2013-02-483487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Huang Y-H; Chen C-W; Sundaramurthy V; Słabicki M; Hao D; Watson CJ; Tovy A; Reyes JM; Dakhova O; Crovetti BR; Galonska C; Lee M; Brunetti L; Zhou Y; Tatton-Brown K; Huang Y; Cheng X; Meissner A; Valk PJM; Van Maldergem L; Sanders MA; Blundell JR; Li W; Ebert BL; Goodell MA Systematic Profiling of DNMT3A Variants Reveals Protein Instability Mediated by the DCAF8 E3 Ubiquitin Ligase Adaptor. Cancer Discov. 2022, 12 (1), 220–235. 10.1158/2159-8290.CD-21-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Tatton-Brown K; Seal S; Ruark E; Harmer J; Ramsay E; del Vecchio Duarte S; Zachariou A; Hanks S; O’Brien E; Aksglaede L; Baralle D; Dabir T; Gener B; Goudie D; Homfray T; Kumar A; Pilz DT; Selicorni A; Temple IK; Van Maldergem L; Yachelevich N; van Montfort R; Rahman N Mutations in the DNA Methyltransferase Gene DNMT3A Cause an Overgrowth Syndrome with Intellectual Disability. Nat. Genet 2014, 46 (4), 385–388. 10.1038/ng.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Ley TJ; Ding L; Walter MJ; McLellan MD; Lamprecht T; Larson DE; Kandoth C; Payton JE; Baty J; Welch J; Harris CC; Lichti CF; Townsend RR; Fulton RS; Dooling DJ; Koboldt DC; Schmidt H; Zhang Q; Osborne JR; Lin L; O’Laughlin M; McMichael JF; Delehaunty KD; McGrath SD; Fulton LA; Magrini VJ; Vickery TL; Hundal J; Cook LL; Conyers JJ; Swift GW; Reed JP; Alldredge PA; Wylie T; Walker J; Kalicki J; Watson MA; Heath S; Shannon WD; Varghese N; Nagarajan R; Westervelt P; Tomasson MH; Link DC; Graubert TA; DiPersio JF; Mardis ER; Wilson RK DNMT3A Mutations in Acute Myeloid Leukemia. N. Engl. J. Med 2010, 363 (25), 2424–2433. 10.1056/NEJMoa1005143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Shlush LI; Zandi S; Mitchell A; Chen WC; Brandwein JM; Gupta V; Kennedy JA; Schimmer AD; Schuh AC; Yee KW; McLeod JL; Doedens M; Medeiros JJF; Marke R; Kim HJ; Lee K; McPherson JD; Hudson TJ; Pan-Leukemia Gene Panel Consortium, T. H.; Brown AMK; Yousif F; Trinh QM; Stein LD; Minden MD; Wang JCY; Dick JE Identification of Pre-Leukaemic Haematopoietic Stem Cells in Acute Leukaemia. Nature 2014, 506 (7488), 328–333. 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gowher H; Jeltsch A Mammalian DNA Methyltransferases: New Discoveries and Open Questions. Biochem. Soc. Trans 2018, 46 (5), 1191–1202. 10.1042/BST20170574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Guo X; Wang L; Li J; Ding Z; Xiao J; Yin X; He S; Shi P; Dong L; Li G; Tian C; Wang J; Cong Y; Xu Y Structural Insight into Autoinhibition and Histone H3-Induced Activation of DNMT3A. Nature 2015, 517 (7536), 640–644. 10.1038/nature13899. [DOI] [PubMed] [Google Scholar]
- (11).Zhang Y; Jurkowska R; Soeroes S; Rajavelu A; Dhayalan A; Bock I; Rathert P; Brandt O; Reinhardt R; Fischle W; Jeltsch A Chromatin Methylation Activity of Dnmt3a and Dnmt3a/3L Is Guided by Interaction of the ADD Domain with the Histone H3 Tail. Nucleic Acids Res. 2010, 38 (13), 4246–4253. 10.1093/nar/gkq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lue NZ; Garcia EM; Ngan KC; Lee C; Doench JG; Liau BB Base Editor Scanning Charts the DNMT3A Activity Landscape. Nat. Chem. Biol 2023, 19 (2), 176–186. 10.1038/s41589-022-01167-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Sangree AK; Griffith AL; Szegletes ZM; Roy P; DeWeirdt PC; Hegde M; McGee AV; Hanna RE; Doench JG Benchmarking of SpCas9 Variants Enables Deeper Base Editor Screens of BRCA1 and BCL2. Nat. Commun 2022, 13 (1), 1318. 10.1038/s41467-022-28884-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cuella-Martin R; Hayward SB; Fan X; Chen X; Huang J-W; Taglialatela A; Leuzzi G; Zhao J; Rabadan R; Lu C; Shen Y; Ciccia A Functional Interrogation of DNA Damage Response Variants with Base Editing Screens. Cell 2021, 184 (4), 1081–1097.e19. 10.1016/j.cell.2021.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hanna RE; Hegde M; Fagre CR; DeWeirdt PC; Sangree AK; Szegletes Z; Griffith A; Feeley MN; Sanson KR; Baidi Y; Koblan LW; Liu DR; Neal JT; Doench JG Massively Parallel Assessment of Human Variants with Base Editor Screens. Cell 2021, 184 (4), 1064–1080.e20. 10.1016/j.cell.2021.01.012. [DOI] [PubMed] [Google Scholar]
- (16).Coelho MA; Cooper S; Strauss ME; Karakoc E; Bhosle S; Gonçalves E; Picco G; Burgold T; Cattaneo CM; Veninga V; Consonni S; Dinçer C; Vieira SF; Gibson F; Barthorpe S; Hardy C; Rein J; Thomas M; Marioni J; Voest EE; Bassett A; Garnett MJ Base Editing Screens Map Mutations Affecting Interferon-γ Signaling in Cancer. Cancer Cell 2023. 10.1016/j.ccell.2022.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bintu L; Yong J; Antebi YE; McCue K; Kazuki Y; Uno N; Oshimura M; Elowitz MB Dynamics of Epigenetic Regulation at the Single-Cell Level. Science 2016, 351 (6274), 720–724. 10.1126/science.aab2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Chédin F; Lieber MR; Hsieh C-L The DNA Methyltransferase-like Protein DNMT3L Stimulates de Novo Methylation by Dnmt3a. Proc. Natl. Acad. Sci 2002, 99 (26), 16916–16921. 10.1073/pnas.262443999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Jia D; Jurkowska RZ; Zhang X; Jeltsch A; Cheng X Structure of Dnmt3a Bound to Dnmt3L Suggests a Model for de Novo DNA Methylation. Nature 2007, 449 (7159), 248–251. 10.1038/nature06146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Reither S; Li F; Gowher H; Jeltsch A Catalytic Mechanism of DNA-(Cytosine-C5)-Methyltransferases Revisited: Covalent Intermediate Formation Is Not Essential for Methyl Group Transfer by the Murine Dnmt3a Enzyme. J. Mol. Biol 2003, 329 (4), 675–684. 10.1016/S0022-2836(03)00509-6. [DOI] [PubMed] [Google Scholar]
- (21).Lue NZ; Garcia EM; Ngan KC; Lee C; Doench JG; Liau BB Base Editor Scanning Charts the DNMT3A Activity Landscape. Nat. Chem. Biol 2022, 1–11. 10.1038/s41589-022-01167-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).CRISPResso2 provides accurate and rapid genome editing sequence analysis | Nature Biotechnology. https://www.nature.com/articles/s41587-019-0032-3 (accessed 2023-03-24). [DOI] [PMC free article] [PubMed]
- (23).Richter MF; Zhao KT; Eton E; Lapinaite A; Newby GA; Thuronyi BW; Wilson C; Koblan LW; Zeng J; Bauer DE; Doudna JA; Liu DR Phage-Assisted Evolution of an Adenine Base Editor with Improved Cas Domain Compatibility and Activity. Nat. Biotechnol 2020, 38 (7), 883–891. 10.1038/s41587-020-0453-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Lue NZ; Liau BB Base Editor Screens for in Situ Mutational Scanning at Scale. Mol. Cell 2023, 83 (13), 2167–2187. 10.1016/j.molcel.2023.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Walton RT; Christie KA; Whittaker MN; Kleinstiver BP Unconstrained Genome Targeting with Near-PAMless Engineered CRISPR-Cas9 Variants. Science 2020, 368 (6488), 290–296. 10.1126/science.aba8853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Kluesner MG; Lahr WS; Lonetree C; Smeester BA; Qiu X; Slipek NJ; Claudio Vázquez PN; Pitzen SP; Pomeroy EJ; Vignes MJ; Lee SC; Bingea SP; Andrew AA; Webber BR; Moriarity BS CRISPR-Cas9 Cytidine and Adenosine Base Editing of Splice-Sites Mediates Highly-Efficient Disruption of Proteins in Primary and Immortalized Cells. Nat. Commun 2021, 12 (1), 2437. 10.1038/s41467-021-22009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Gapinske M; Luu A; Winter J; Woods WS; Kostan KA; Shiva N; Song JS; Perez-Pinera P CRISPR-SKIP: Programmable Gene Splicing with Single Base Editors. Genome Biol. 2018, 19 (1), 107. 10.1186/s13059-018-1482-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Emperle M; Rajavelu A; Kunert S; Arimondo PB; Reinhardt R; Jurkowska RZ; Jeltsch A The DNMT3A R882H Mutant Displays Altered Flanking Sequence Preferences. Nucleic Acids Res. 2018, 46 (6), 3130–3139. 10.1093/nar/gky168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Anteneh H; Fang J; Song J Structural Basis for Impairment of DNA Methylation by the DNMT3A R882H Mutation. Nat. Commun 2020, 11 (1), 2294. 10.1038/s41467-020-16213-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Boike L; Henning NJ; Nomura DK Advances in Covalent Drug Discovery. Nat. Rev. Drug Discov 2022, 21 (12), 881–898. 10.1038/s41573-022-00542-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Concordet J-P; Haeussler M CRISPOR: Intuitive Guide Selection for CRISPR/Cas9 Genome Editing Experiments and Screens. Nucleic Acids Res. 2018, 46 (Web Server issue), W242–W245. 10.1093/nar/gky354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wu T; Yu J; Gale-Day Z; Woo A; Suresh A; Hornsby M; Gestwicki JE Three Essential Resources to Improve Differential Scanning Fluorimetry (DSF) Experiments. bioRxiv March 25, 2020, p 2020.03.22.002543. 10.1101/2020.03.22.002543. [DOI] [Google Scholar]
- (33).Potter SC; Luciani A; Eddy SR; Park Y; Lopez R; Finn RD HMMER Web Server: 2018 Update. Nucleic Acids Res. 2018, 46 (W1), W200–W204. 10.1093/nar/gky448. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S6: Additional data on activating guides.
Table S2: Primers and oligos used in this study.
Table S1: Plasmids constructed in this study.
Table S3: Melting temperatures of recombinant DNMT3A2 variants by DSF.
Figure S1: Pie chart and allele table showing targeted deep sequencing of R882 codon of DNMT3A in DNMT3AWT/R882H cells.
Figure S2: Silencing dynamics of dDNMT3A reporter system, representative gating scheme, and additional plots associated with Figure 1.
Figure S3: Western Blot of reporter cells transduced with base editing sgRNAs.
Figure S4: Additional sgRNAs tested.
Figure S5: Genotyping of DNMT3AWT/R882H cells treated with sgRNAs.
Figure S6: D618_K621del mutation is found in colobinae monkeys.
Figure S7: Additional in vitro validation of DNMT3A variants.
Table S5: WT-R882H cell line screen results.
Table S4: WT cell line screen results.