

Abstract
This review article discusses the importance and oncogenic signaling pathways of tumor-initiating cells (TICs) in several etiologies of hepatocellular carcinomas (HCCs) induced by hepatitis C virus (HCV), alcohol, obesity and/or chemicals. Stem cells may be present in cancer tissue, and a hierarchy of cells is formed, as is the case for normal tissue. Tumor formation, growth and propagation are maintained by a small proportion of cells with stem cell-like properties. TICs are present in alcoholfed HCV transgenic mice, diethylnitrosamine/phenobarbital-treated mice (chemical carcinogenesis) and Spnb2 +/− mice (defective TGF-β signal). Alcohol/obesity-associated endotoxemia induces the stem cell marker Nanog through TLR4 signaling to generate TICs and liver tumors in several HCC models. The oncogenic pathway (such as the STAT3 and TLR4-NANOG pathway) and mechanism of generation of TICs of HCCs associated with HCV, alcohol and obesity are discussed. Understanding the molecular stemness signaling and cellular hierarchy and defining key TIC-specific genes will accelerate the development of novel biomarkers and treatment strategies. This review highlights recent advances in understanding the pathogenesis of liver TICs and discusses unanswered questions about the concept of liver TICs. (This project was supported by NIH grants 1R01AA018857 and P50AA11999).
Keywords: Hepatocellular carcinoma, Tumor-initiating cells, Hepatitis C virus, Alcohol, TLR4, Nanog
Synergistic increase in the HCC odds ratio according to environmental factors (alcoholic liver diseases and obesity) with virus infection (HBV and HCV)
Alcoholic liver disease (ALD) is a major health problem affecting 15 million people [1] in the USA. The obese and NAFLD populations are also increasing alarmingly in the USA. Both alcohol and obesity increase the gut permeability and lead to increased blood levels of endotoxin [2], which in turn activates TLR4 [3] and induces the production of cytokines and the inflammatory response, leading to liver injury and the development of ALD and obesity [1]. Chronic liver damage caused by viral infection and alcohol can result in an increased risk of HCC. In particular, chronic infection with HBV or HCV represents a major risk factor for HCC [4], which is the third most deadly cancer in the world [4–6]. HCC is highly prevalent in Africa and Asia (which have a higher prevalence of HBV and HCV than other areas) and does not respond well to conventional therapy [4]. HCV affects more than 170 million people worldwide [4, 7, 8]. Ample epidemiological evidence demonstrates that HCV and alcohol consumption or obesity synergistically accelerate the development of liver damage [9]. The risk of HCC, as assessed by the odds ratio, increases from 8–12 to 48–54 if HCV patients have concomitant alcoholism or obesity [10–12]. Therefore, understanding the molecular mechanisms of the HCV-induced hepatocarcinogenesis of alcoholic or obese patients is required for the eventual development of improved therapeutic modalities [13].
Stem-cell-like signature, chemoresistance and poor prognosis markers in tumor-initiating stem-like cells (TICs) of HCCs
Forty percent of HCC is clonal and originates from progenitor/stem cells [14–17] with three characteristics, including a self-renewal ability, asymmetric/multiple cell division (clonality) and plasticity. Liver TICs are responsible for tumor relapse, metastasis and chemoresistance. Liver TICs dictate a hierarchical organization that is shared in both organogenesis and tumorigenesis. Liver TICs in tumors give rise to different types of tumor cells (HCC and cholangiocarcinomas) and express “sternness” genes, including Oct3/4/Bmi and morphogene-induced signaling pathways (Wnt/p-β-catenin, Notch and Hedgehog/SMO) [18–20]. The CD133 + cells isolated from the HCC cell lines and human HCCs with higher expression of CD44/CD34 have a self-renewal ability and repeatedly give rise to tumors when injected into immunodeficient mice [21].
The percent of TICs ranges from less than 1 % to 5–8 % in tumor tissues induced by alcohol, HCV and HCV/alcoholic animal models or patients. CD133+ tumor cells isolated from HCC cells possess tumor-initiating properties and have characteristics similar to those of LPCs with the expression of “sternness” genes, the ability to self-renew and the ability to differentiate into non-parenchymal cell lineages [21]. These CD133 + TICs confer resistance to chemotherapy, and this presents a major obstacle to the treatment of HCC (Fig. 1). One potential reason for this chemoresistance may lie in the plasticity of TICs with dysregulated signaling and gene expression. Among many stemness factors, Nanog is one of the master transcription factors in pluripotent embryonic stem cells (ESCs) [22] required for maintaining the self-renewal ability and pluripotency of both human and mouse embryonic stem cells [23–26]. Overexpression of Nanaog induces and maintains the self-renewal characteristics and pluripotency of ESCs [27]. Nanog expression is elevated in human neoplasms, including HCC [28], breast carcinomas [29], osteosarcoma [30] and certainly germ cell tumors [29, 31–33]. Furthermore, ectopic Nanog expression induces an oncogenic potential in NIH3T3 [34]. Therefore, stemness/morphogen genes play a pivotal role in TIC oncogenesis and self-renewal of ESC.
Fig. 1.
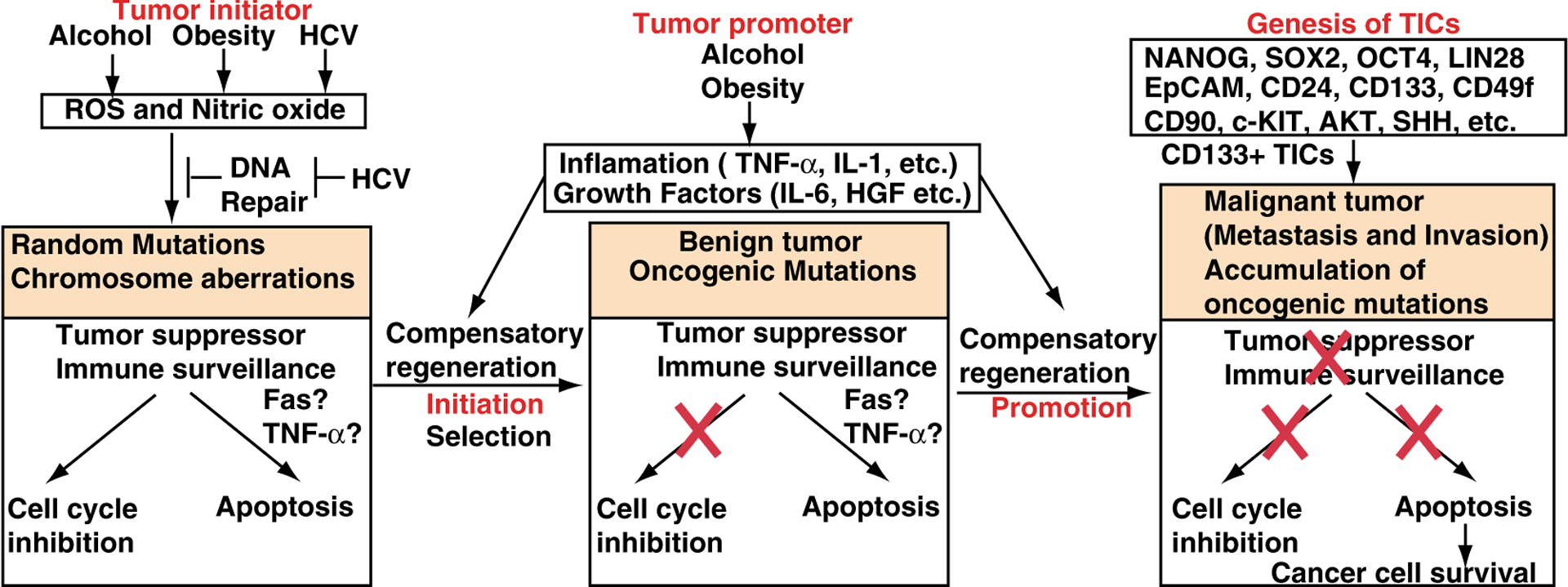
Hypothetical models of the development of HCCs and TICs. Left In the tumor initiation step, alcohol, obesity or HCV induces DNA mutations. Middle In the tumor promotion step, DNA mutations are fixed by inflammatory responses and growth factors. Right TICs are generated, leading to metastatic tumor development
Higher levels of CD133 and EpCAM+ (pancarcinoma antigen epithelial cell adhesion molecule) of HCC cell expression are correlated with increased tumor grade, advanced disease stage, shorter overall survival, tumor progression, invasiveness and higher recurrence rates compared to patients with low CD133 expression [35] (Fig. 1). EpCAM is an early biomarker of HCC [36, 37], which is a direct transcriptional target of the Wnt-β-catenin canonical signaling pathway. EpCAM+ α-fetoprotein (AFP)+ HCC subtype has features of LPCs. Furthermore, CD24(+) HCC cells are critical for the maintenance, self-renewal, differentiation and metastasis of tumors and significantly impact patient prognosis [38]. CD24 is induced in chemoresistant (cisplatin) tumors compared to bulk tumors [38]. The upregulation of Nanog, together with p53 depletion, is significantly associated with clinically late stage HCC. HCC patients who express biliary cell markers (CK7 and CK10) have poor prognoses and higher recurrence rates post surgical resection/transplantation [15].
Rapamycin and sorafenib are respectively used as single chemotherapy agents for HCC, and a recent HCC xenograft study suggests a better effect when given they are together [39]. Clinical evidence, however, reveals eventual chemoresistance to these drugs in HCC patients [40, 41]. We believe this chemoresistance is caused by TICs. Indeed, Nanog antagonism targeting TICs enhances the efficacy of these two drugs in tumor-bearing mice and achieves ~90 % tumor growth suppression [39].
Origin, target cell type and genesis of TICs
Different cell types in the hepatic lineage are considered the origin of HCC, including mature hepatocytes, cholangiocytes, ductular bipotential progenitor cells and periductular stem cells. C-kit inhibition by the tyrosine kinase inhibitor imatinib mesylate attenuates c-KIT-expressing LPC expansion and inhibits liver tumor formation in mice via antiproliferative effects [42]. Further, carcinogenic insult may begin with mature hepatocytes but alter their phenotype toward bipotential progenitor cells before causing transformation in Notch-intracellular-domain (NICD)/AKT-mediated cholangiocarcinoma [43]. Human liver progenitor cells (LPCs) may give rise to HCC and ICC [44–47] since many tumors contain a mixture of mature hepatocytes and LPCs. LPCs have also been noted in hepatoblastoma, the most common liver tumors in children, which are widely believed to be stem cell derived given there can be both epithelial and mesenchymal tissue components. These tumors can even have structures mimicking intrahepatic bile ducts and form ductal plate-like structures [48]. These stemness genes are highly expressed in LPCs, bile duct epithelium and premalignant hepatic tissues, but not in adult hepatocytes [37, 49].
Before onset of HCC nodules, HcPCs are generated in dysplastic areas. HCC progenitor cells (HcPCs), isolated and characterized from different mouse HCC models, are malignantly transformed in a manner depending on autocrine IL-6-LIN28 signaling from inflammatory cells [50]. Both liver undergoing chronic damage and compensatory proliferation are stimulated in vivo and transform HcPCs into cancer. Interestingly, bipotential hepatobiliary progenitors do not give rise to tumors [50].
Our study so far supports the notion that liver progenitor cells (LPCs) are the potential source of transformed cells in HCC [51]. In fact, rat oval cells transfected with c-RasH give rise to HCC in immunocompromized mice [52], and oval cells chemically transformed in vitro produce cholangiocarcinomas and HCC when transplanted in vivo [53]. While mature hepatocytes proliferate to maintain the liver homeostasis after partial hepatectomy [54], differentiation from other sources such as liver progenitor cells (LPCs) may occur after injury, particularly when hepatocyte proliferation is impaired [55]. Indeed, cells whose lineage is traced to LPCs are increased after partial hepatectomy plus 2-AAF or feeding a methionine/choline-deficient diet supplemented with 0.15 % ethionine (MCDE) [56, 57]. These LPCs with bipotential activities are restricted to a subset of biliary duct cells antigenically defined as CD45−/CD11b−/CD31−/MIC1–1C3+/CD133+/CD26− [55].
Maelstrom promotes HCC metastasis by inducing epithelial-mesenchymal transition by way of Akt/GSK-3β/Snail signaling [58]. A novel oncogene, Maelstrom (MAEL), at 1q24, enhanced AKT activity with subsequent GSK-3β phosphorylation and Snail stabilization, finally inducing epithelial-mesenchymal transition (EMT) and promoting tumor invasion and metastasis [58].
Markers and isolation of TICs from HCCs
TICs in HCC are identified by cell surface antigens including CD133, CD49f, CD24, CD90, CD44, OV6, c-KIT and CD326 (EpCAM), or by selecting the side population (SP) cells by Hoechest dye staining. Therefore, several LPC markers, such as CD133, CD49f, EpCAM, CD24 and CD90, are used to isolate liver tumor-initiating cells (TICs) with stem cell features. The TIC marker CD133 is a predictor of the effectiveness of pegylated interferon α−2b therapy against advanced HCCs [59]. CD24 is a liver TIC marker that drives TIC genesis through STAT3-mediated Nanog regulation [38]. Furthermore, ICAM-1 is a marker of HCC stem cells in humans and mice; ICAM-1 inhibitors slow tumor formation and metastasis in mice. ICAM-1 expression is regulated by the stem cell transcription factor Nanog [60]. Insulin-like growth factor (IGF)2 and IGF receptor (IGF1R) are upregulated in Nanog + TICs [61]. The Nanog gene is also induced in metastatic human liver cancer cells and human HCC tissues (120).
Stem cells have three major characteristics, self-renewal, asymmetric and multiple cell division (clonality), and plasticity. The liver has a high regenerative potential, and hepatic small oval progenitor cells around the peripheral branches of the bile ducts, the canals of Hering, can differentiate into biliary epithelial cells and hepatocytes [62]. These oval liver progenitor cells share molecular markers with adult hepatocytes [albumin, cytokeratin 7 (CK7), CK19, oval cell markers (OV-6, A6, and OV-1), chromogranin-A, NCAM (neural cell adhesion molecule)] and fetal hepatocytes (α-fetoprotein) (Table 1) [15, 62]. Indeed, a quarter to half of HCCs (28–50 %) express LPC markers (CK7 and CK19) identified by immunophenotyping of HCCs [63]. They are also positive for more common stem cell markers such as CD34+, Thy-1+, c-Kit+ and Flt-3+ (FMS-like tyrosine kinase 3) [64]. Thus, it currently remains unclear whether these stem cells are derived from the bone marrow and just migrate to this niche or represent true resident liver stem/progenitor cells. Binding of stroma-derived factor-1α (SDF-1α) to its surface receptor CXCR4 activates oval hepatic cells [65]. Forty percent of HCCs have clonality and thus are considered to originate from progenitor/stem cells [14–17]. Recent studies of HCC have centered on TICs, including detection of TICs in cancer, identification of TIC markers and isolation of TICs from human HCC cell lines. TICs were identified as a CD117+/CD133+ LPC in regenerating liver tissue [66] and a CD45−/CD90+ subpopulation of tumor cells in HCC [67]. The CD90+ cells are not present in the normal liver and, when injected into immunodeficient mice, create tumors repeatedly. In human HCC and HCC cell lines, specifically CD133+ cells, not CD133− cells, had the ability to self-renew, create differentiated progenies and form tumors [21].
Table 1.
Markers for liver tumor-initiating cells
| Gene name | Other name | Function | Species | Organ | References |
|---|---|---|---|---|---|
| CD133 | Prominin 1 (PROM1) | Glycoprotein, membrane protrusions | Human, mouse | Liver, brain | [21, 28, 73–76] |
| CD49f | Integrina chain a6 (ITGA6) | Cell adhesion, cell signaling | Mouse | Liver | [21, 76] |
| CD24 | CD24 | Mouse | Liver, lung | [38] | |
| CK19 | Cytokeratin 19 | Biliary lineage marker | Mouse | Liver | [77, 78] |
| OV-6 | Oval cell marker | Early progenitor cells | Human | Liver | [78] |
| CD34 | Glycoprotein | Cell-cell adhesion factor | Mouse | Liver, leukemia | [79] |
| AFP | α-Fetoprotein | Fetal counterpart of serum albumin | Mouse | Liver | [72] |
| CD90 | Thy-1 | Glycophosphatidylinositol (GPI) anchor | Mouse | Liver | [21] |
| CD44 | Hyaluronic acid receptor | Cell adhesion and migration, metastasis | Mouse | Liver, breast | [21, 68] |
| CD117 | KIT | C-kit receptor, cytokine receptor | Mouse | Liver | [21] |
When compared to CD133− cells, the CD133+ cells isolated from the HCC cell lines showed higher expression of CD44 (cell adhesion molecule) and CD34, but both CD133 subpopulations displayed similar expression for CD29 (integrin β1), CD49f (integrin α6), CD90 and CD117 (c-kit: gastrointestinal stroma tumor), indicating these makers are still not definitive TIC markers [21]. Nevertheless, these makers are differentially expressed in other cancers, such as CD44+/CD24−/Iow in breast cancer [68], CD34+/CD38− in acute myeloid leukemia [69] and CD44+/α2b1hi/CD133+ in prostate cancer [70]; for this reason, identification of other surface markers, expressed along with CD133, as a goal to better characterize liver TICs is worthwhile.
A minority of the CD133+ tumor cell population isolated from HCC cells possesses tumor-initiating properties and has characteristics similar to those of progenitor cells including the expression of “sternness” genes, the ability to self-renew and the ability to differentiate into nonhepatocyte-like lineages [21]. Markers of stemness genes, including CD133, CD90, CD24 and CD49f, are used for isolation of TICs. In addition to the marker-based isolation of TICs, TICs are isolated by sorting the side population (SP) from HCC cells, which have high oncogenic potential and anti-apoptotic properties compared with those of non-SP cells [71, 72].
TICs induced by interactions between HCV and alcohol
Compelling evidence identifies obesity and hepatitis C virus (HCV) as comorbidity risk factors for hepatocellular carcinoma (HCC), which contains TICs. Chronic liver damage caused by viral infection and environmental factors (such as alcohol or metabolic syndrome) can result in increased risk for HCC. Clearly, understanding the molecular mechanisms of HCV-induced hepatocarcinogenesis is required for the eventual development of improved therapeutic modalities for this disease [13]. In particular, chronic infection with HBV or HCV represents a major risk factor for HCC [4]. HCV affects more than 170 million people worldwide [4, 7, 8]. HCV may induce cellular transformation by the induction of chronic liver inflammation mediated by the immune cells [80]. This chronic liver inflammation leads to cell deaths, hepatocellular regeneration and the emergence of mutated cells that may be oncogenic. Interactions between alcohol and HCV synergize immune dysfunctions and liver damage [80].
Hepatitis C and alcohol exacerbate liver injury by suppression of FOXO3 [81]. FOXO3 functions as a protective factor preventing alcoholic liver injury. The combination of HCV and alcohol, but not either condition alone, inactivates FOXO3, causing a decrease in expression of its target genes and an increase in liver injury. Modulation of the FOXO3 pathway is a potential therapeutic approach for HCV-alcohol-induced liver injury [81], with regulation of FOXO3 by phosphorylation and methylation in HCV infection and alcohol exposure [82]. The development of this novel capillary isoelectric focusing (IEF) method for the simultaneous quantification of differently modified FOXO3 species allowed us to demonstrate how HCV and alcohol combine to modify a complex pattern of FOXO3 posttranslational modifications (PTMs) that contribute to the pathogenesis [81, 82].
The high rate of chronic HCV infection in alcoholics is due to ethanol’s effects on antiviral immune responses since ethanol inhibits the humoral and cellular immune responses to genetic immunization of HCV protein (antibody levels and the CD4 + proliferative immune response to this HCV nonstructural protein) [83, 84]. Chronic ethanol consumption inhibits cellular immune responses to HCV core proteins that are restored by genetic immunizations via DNA-based immunization approach using cytokine-expressing plasmids [83, 84]. Chronic ethanol feeding inhibits Th cells and CTL activities with reduced cytokine secretion (a switch from Th1 to Th0 subtype) in proliferating CD4 + T cells [83, 84].
Several possible mechanisms may explain the high prevalence rate of HCV among alcoholics and the increased severity of liver diseases in these patients. First, alcohol may enhance the replication of HCV and thus increase the expression of viral RNA and proteins, resulting in more severe HCV-induced liver injury, independent of the damage induced by alcohol alone. Indeed, the HCV titer has been shown to exhibit a positive correlation with the amount of alcohol consumption [85]. This enhanced effect on HCV replication could be caused directly by the metabolites of ethanol, such as acetaldehyde and free radicals, which may stimulate HCV replication and gene expression. It could also be caused indirectly through alcohol-induced inhibition of the antiviral immune response. Indeed, HCV replication is more active in immunodeficient patients, such as HIV-infected patients [86], and ethanol consumption can cause immunosuppression [87].
Our recent studies using HCV transgenic mice as a model have revealed pivotal insights into the mechanisms underlying synergism between ALD and HCV. By using mice with liver-specific expression of the HCV NS5A protein, we found that mice fed an alcohol or high-cholesterol high-fat diet for 12 months developed liver tumors in a manner dependent on TLR4, which was induced by NS5A (Fig. 2, top). Our further studies indicated that the NS5A-induced TLR4 was activated by endotoxemia associated with obesity/alcoholism, leading to accentuated TLR4 signaling, which in turn upregulated the stem cell marker Nanog to accelerate liver oncogenesis [88] (Fig. 2, middle). HCV NS5A transgenic mice have liver-specific expression of NS5A, not in monocytes or macrophages. Therefore, NS5A transgenic mice do not have enhanced expression of TLR4 in monocytes or macrophages, but in hepatocytes. Therefore, this HCV NS5A-TLR4-NANOG axis may contribute to the mechanisms of the synergism between HCV and alcohol/obesity in liver pathogenesis and carcinogenesis (Fig. 2, bottom). Mechanistic understanding of the role of TLR4-mediated TICs in HCC associated with ALD/obesity and HCV addresses an important public health problem.
Fig. 2.
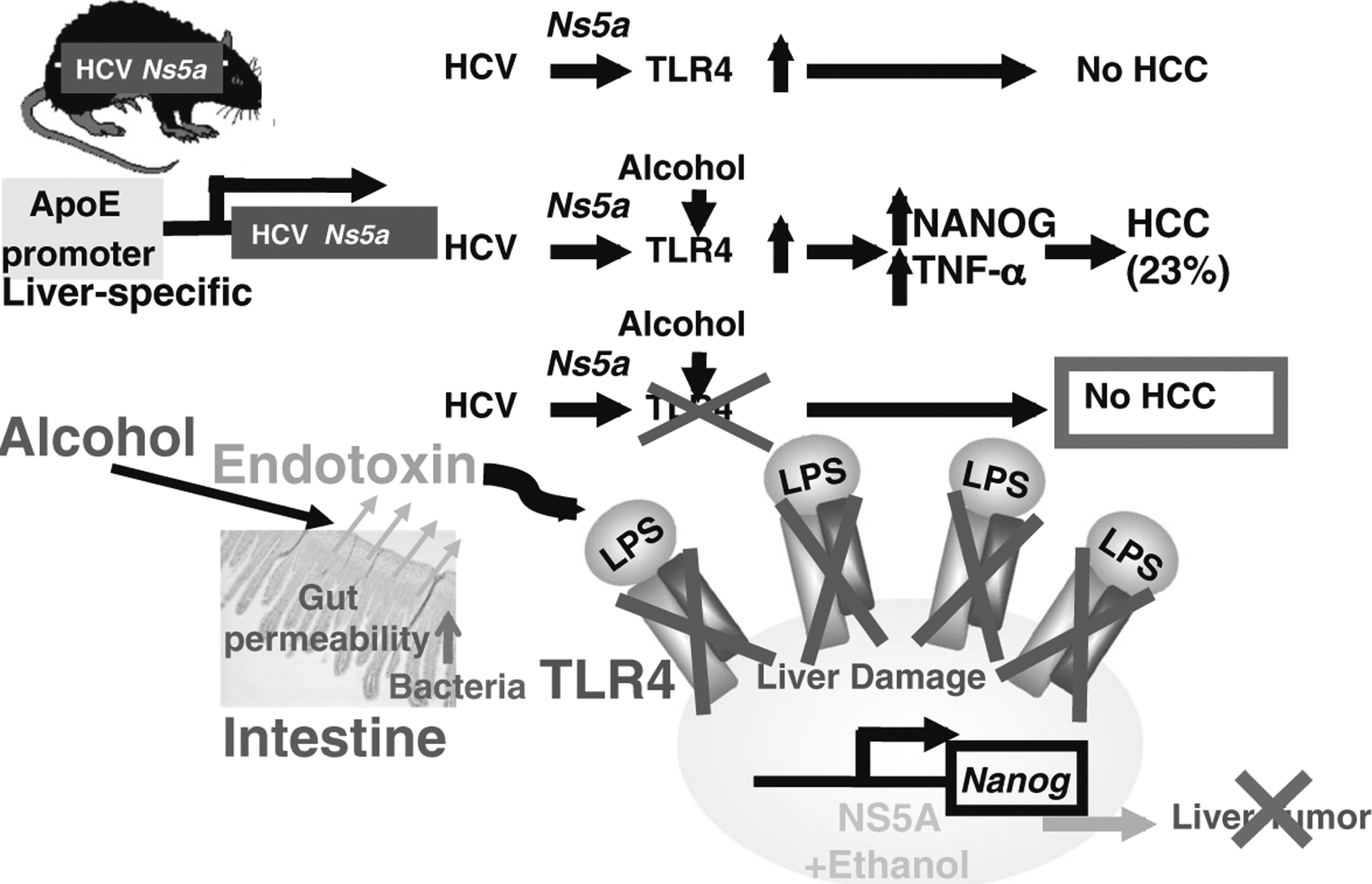
Hypothetical network of HCV-TLR4-Nanog signaling. Top HCV Ns5a or alcohol feeding induces TLR4 gene expression in transgenic (Tg) mouse models (HCV Ns5a Tg mice) that do not develop liver tumors without alcohol feeding. Middle Alcohol-associated endotoxemia then activates TLR4 signaling, resulting in the induction of the stem cell marker Nanog expression and liver tumors. TLR4 is induced by viral proteins such as NS5A, but also by endotoxin, a condition common in alcoholic and non-alcoholic liver diseases [17, 98, 107], and TICs isolated from the models are indeed TLR4-dependent; TICs are isolated from the liver tumors of these mice and patients. Bottom We have also discovered that TLR4 deficiency attenuates the incidence of HCC in these models
Obesity and TICs
The interaction between obesity and liver cancer is particularly strong, and obesity promotes liver diseases such as non-alcoholic fatty liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH). Obesity promotes the HCC burden compared to infection with hepatitis viruses [89]. Both dietary and genetic obesity promotes liver tumors by accentuated expression of IL-6 and TNF-a, leading to liver inflammation and tumorigenesis through activation of the oncogenic transcription factor STAT3 [90]. Activation of JNK induces both the activation of transcription factor activator protein-1 and transcription-independent induction of effector molecules, leading to regulation of cell death and survival, differentiation, proliferation, insulin signaling, ROS and metabolism in the liver [91]. Both loss and hyperactivation of the JNK pathway promote inflammation, leading to metabolic diseases (obesity, steatosis, and insulin resistance), ultimately leading to fibrosis and cancer development [91].
Misregulation of a pluripotency-associated transcription factor network in adult tissues is associated with the expansion of rare, highly malignant tumor-initiating stem cells (TICs) through poorly understood mechanisms. Leptin is the most important adipose-derived hormone, and its circulating concentration is proportional to the total amount of body fat. We demonstrate that robust and selective expression of the receptor for the adipocyte-derived peptide hormone leptin (OB-R) is a characteristic feature of TICs and of a broad array of embryonic and induced pluripotent stem cells and is mediated directly by the core pluripotency-associated transcription factors OCT4 and SOX2 [92]. The induction of OCT4 and SOX2 was observed in leptin-stimulated cells. TICs exhibit sensitized responses to leptin, including the phosphorylation and activation of the pluripotency-associated oncogene STAT3 and induction of Oct4 and Sox2, thereby establishing a self-reinforcing signaling module. The induction of OB-R is also observed in human HCC. We also found that TICs were highly sensitized to leptin exposure in vitro, as judged by the phosphorylation of Stat3-Y705 [92]. Further, leptin promotes the growth of tumors from TICs implanted sub-cutaneously into immune-compromised NOG mice (NOD/Shi-scid/IL-2Rγnull). Exposure of cultured mouse embryonic stem cells to leptin sustains pluripotency in the absence of leukemia inhibitory factor. By implanting TICs into leptin-deficient ob/ob mice or into comparably over-weight Leprdb/db mice that produce leptin, we have provided evidence of a central role for the leptin-TIC–signaling axis in promoting obesity-induced tumor growth [92]. Differential responses to extrinsic, adipocyte-derived cues may promote the expansion of tumor cell subpopulations and contribute to oncogenesis [92]. These findings provide a direct link between the adipose-derived hormone leptin (obesity) and TICs.
The mechanisms of TIC generation
Poorly differentiated and aggressive human tumors have the gene expression signature of embryonic stem cells. Several oncogenic signaling pathways in cancer stem cells of HCC have been described, including activated PI3 K/AKT [28], signal transducer and activator of transcription 3 (STAT3) [93, 94], Notch [95], hedgehog [96, 97] and transforming growth factor-beta (TGF-β) (Fig. 1) [98, 99]. One potential reason for this chemoresistance may lie in the plasticity of cancer stem cells with dysregulated signaling and gene expression. The initiation of hepatocarcinogenesis is linked to chronic inflammation, which promotes a transformed phenotype and stem cell population through a positive circuit loop involving NF-кB, Lin28B, let-7 and IL-6 [100]. The mammalian homologs of lin-28 of C. elegance, LIN28 and LIN28B are overex-pressed in primary human tumors and human cancer cell lines (overall frequency 15 %) [101]. Transduction of Lin28, OCT4, Nanog and reprogram human somatic fibroblasts to pluripotency [102]. Overexpression of Lin28, OCT4 and Nanog drives self-renewal and proliferation, leading to oncogenesis [102]. Lin28B is an oncofetal circulating TIC marker associated with recurrence, advanced disease and poor clinical outcome in HCCs [101, 103–105]. Lin28 and Lin28B bind to the terminal loop of the precursors of let-7 family miRNAs and block their processing into mature miRNAs [101, 106].
Conclusions
Taken together, a mechanistic understanding of the role of TICs in HCC associated with ALD, obesity and HCV addresses an important public health problem, including the excessive morbidity, mortality and economic burden due to uncontrolled HCC. Therefore, antagonism of oncogenic signaling pathways may become a new therapeutic modality for HCC caused by HCV, obesity and alcohol and also opens a door for the potential application of a similar therapeutic approach for HCCs.
Acknowledgements
We thank Akiko Ueno and Raul Lazaro, the Animal Core personnel, for performing mouse experiments and Ratna Ray (Saint Louis University) for providing HCV Ns5a Tg mice. This project was supported by NIH grant 1R01AA018857-01, pilot project funding (5P30DK048522-13), P50AA11999 (Animal Core, Morphology Core, and Pilot Project Program), R24AA012885 (Non-Parenchymal Liver Cell Core) and RC2AA019392-01. This research was also supported by Research Scholar Grant RSG-12-177-01-MPC and pilot funding (IRG-58-007-48) from the American Cancer Society. Tissue pathological slide preparation was performed by Ms. Moli Chen, Translational Pathology Core of Norris Comprehensive Cancer Center.
Footnotes
Compliance with ethical requirements and Conflict of interest
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (USC, USA) and with the Helsinki Declaration of 1975, as revised in 2008 [5]. Informed consent was obtained from all patients for inclusion in the study. All institutional and national guidelines for the care and use of laboratory animals were followed. Keigo Machida, Chia-Lin Chen and Hidekazu Tsukamoto declare that they have no conflict of interest.
Contributor Information
Chia-Lin Chen, Department of Molecular Microbiology and Immunology, University of Southern California, Los Angeles, CA 90033, USA.
Hidekazu Tsukamoto, Southern California Research Center for ALPD and Cirrhosis, Los Angeles, CA, USA; Department of Pathology, University of Southern California, Los Angeles, CA 90033, USA.
Keigo Machida, Department of Molecular Microbiology and Immunology, University of Southern California, Los Angeles, CA 90033, USA; Southern California Research Center for ALPD and Cirrhosis, Los Angeles, CA, USA.
References
- 1.Fleming S, Toratani S, Shea-Donohue T, Kashiwabara Y, Vogel SN, Metcalf ES. Pro- and anti-inflammatory gene expression in the murine small intestine and liver after chronic exposure to alcohol. Alcohol Clin Exp Res 2001;25:579–589 [PubMed] [Google Scholar]
- 2.Bode C, Bode JC. Activation of the innate immune system and alcoholic liver disease: effects of ethanol per se or enhanced intestinal translocation of bacterial toxins induced by ethanol? Alcohol Clin Exp Res 2005;29:166S–1671S [DOI] [PubMed] [Google Scholar]
- 3.Hritz I, Mandrekar P, Velayudham A, Catalano D, Dolganiuc A, Kodys K, et al. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 2008;48:1224–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Okuda K Hepatocellular carcinoma. J Hepatol 2000;32:225–237 [DOI] [PubMed] [Google Scholar]
- 5.Sanyal AJ, Yoon SK, Lencioni R. The etiology of hepatocellular carcinoma and consequences for treatment. Oncologist 2010;15(Suppl 4):14–22 [DOI] [PubMed] [Google Scholar]
- 6.Sanyal AJ, Banas C, Sargeant C, Luketic VA, Sterling RK, Stravitz RT, et al. Similarities and differences in outcomes of cirrhosis due to nonalcoholic steatohepatitis and hepatitis C. Hepatology 2006;43:682–689 [DOI] [PubMed] [Google Scholar]
- 7.Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM, Weinman SA. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 2002;122:366–753. [DOI] [PubMed] [Google Scholar]
- 8.Yao F, Terrault N. Hepatitis C and hepatocellular carcinoma. Curr Treat Options Oncol 2001;2:473–483. [DOI] [PubMed] [Google Scholar]
- 9.Brechot C, Nalpas B, Feitelson MA. Interactions between alcohol andhepatitis viruses intheliver. ClinLabMed 1996;16:273–287 [PubMed] [Google Scholar]
- 10.Yuan JM, Govindarajan S, Arakawa K, Yu MC. Synergism of alcohol, diabetes, and viral hepatitis on the risk of hepatocellular carcinoma in blacks and whites in the U.S. Cancer 2004; 101:1009–1017 [DOI] [PubMed] [Google Scholar]
- 11.Hassan MM, Hwang LY, Hatten CJ, Swaim M, Li D, Ab-bruzzese JL, et al. Risk factors for hepatocellular carcinoma: synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology 2002;36:1206–1213 [DOI] [PubMed] [Google Scholar]
- 12.Artinyan A, Mailey B, Sanchez-Luege N, Khalili J, Sun CL, Bhatia S, et al. Race, ethnicity, and socioeconomic status influence the survival of patients with hepatocellular carcinoma in the United States. Cancer 2010;116:1367–1377 [DOI] [PubMed] [Google Scholar]
- 13.Crippin JS, McCashland T, Terrault N, Sheiner P, Charlton MR. A pilot study of the tolerability and efficacy of antiviral therapy in hepatitis C virus-infected patients awaiting liver transplantation. Liver Transpl 2002;8:350–355 [DOI] [PubMed] [Google Scholar]
- 14.Alison MR. Liver stem cells: implications for hepatocarcinogenesis. Stem Cell Rev 2005;1:253–1260 [DOI] [PubMed] [Google Scholar]
- 15.Roskams T Liver stem cells and their implication in hepatocellular and cholangiocarcinoma. Oncogene 2006;25:3818–3822 [DOI] [PubMed] [Google Scholar]
- 16.Zender L, Spector MS, Xue W, Flemming P, Cordon-Cardo C, Silke J, et al. Identification and validation of oncogenes in liver cancer using an integrative oncogenomic approach. Cell 2006;125:1253–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang Y, Kitisin K, Jogunoori W, Li C, Deng CX, Mueller SC, et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-beta and IL-6 signaling. Proc Natl Acad Sci USA 2008;105:2445–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Valk-Lingbeek ME, Bruggeman SW, van Lohuizen M. Stem cells and cancer; the polycomb connection. Cell 2004;118:409–418 [DOI] [PubMed] [Google Scholar]
- 19.Chambers I, Smith A. Self-renewal of teratocarcinoma and embryonic stem cells. Oncogene 2004;23:7150–7160 [DOI] [PubMed] [Google Scholar]
- 20.Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature 2004;432:324–331 [DOI] [PubMed] [Google Scholar]
- 21.Ma S, Chan KW, Hu L, Lee TK, Wo JY, Ng IO, et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 2007;132:2542–2556 [DOI] [PubMed] [Google Scholar]
- 22.Martin GR. Isolation of a pluripotent cell line from early mouse embryos cultured in medium conditioned by teratocarcinoma stem cells. Proc Natl Acad Sci USA 1981;78:7634–7638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet 2006;38:431–440 [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Rao S, Chu J, Shen X, Levasseur DN, Theunissen TW, et al. A protein interaction network for pluripotency of embryonic stem cells. Nature 2006;444:364–368 [DOI] [PubMed] [Google Scholar]
- 25.Rao S, Orkin SH. Unraveling the transcriptional network controlling ES cell pluripotency. Genome Biol 2006;7:230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pan G, Thomson JA. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res 2007;17:42–49 [DOI] [PubMed] [Google Scholar]
- 27.Chambers I, Colby D, Robertson M, Nichols J, Lee S, Tweedie S, et al. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 2003;113:643–655 [DOI] [PubMed] [Google Scholar]
- 28.Ma S, Lee TK, Zheng BJ, Chan KW, Guan XY. CD133 ? HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008;27:1749–1758 [DOI] [PubMed] [Google Scholar]
- 29.Ezeh UI, Turek PJ, Reijo RA, Clark AT. Human embryonic stem cell genes OCT4, NANOG, STELLAR, and GDF3 are expressed in both seminoma and breast carcinoma. Cancer 2005;104:2255–2265 [DOI] [PubMed] [Google Scholar]
- 30.Gibbs CP, Kukekov VG, Reith JD, Tchigrinova O, Suslov ON, Scott EW, et al. Stem-like cells in bone sarcomas: implications for tumorigenesis. Neoplasia 2005;7:967–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoei-Hansen CE, Almstrup K, Nielsen JE, Brask Sonne S, Graem N, Skakkebaek NE, et al. Stem cell pluripotency factorNANOGis expressed in human fetal gonocytes, testicular carcinoma in situ and germ cell tumours. Histopathology 2005;47:48–56 [DOI] [PubMed] [Google Scholar]
- 32.Hart AH, Hartley L, Parker K, Ibrahim M, Looijenga LH, Pauchnik M, et al. The pluripotency homeobox gene NANOG is expressed in human germ cell tumors. Cancer2005;104:2092–2098 [DOI] [PubMed] [Google Scholar]
- 33.Santagata S, Ligon KL, Hornick JL. Embryonic stem cell transcription factor signatures in the diagnosis of primary and metastatic germ cell tumors. Am J Surg Pathol 2007;31:836–845 [DOI] [PubMed] [Google Scholar]
- 34.Zhang J, Wang X, Chen B, Suo G, Zhao Y, Duan Z, Dai J. Expression of Nanog gene promotes NIH3T3 cell proliferation. Biochem Biophys Res Commun 2005;338:1098–1102 [DOI] [PubMed] [Google Scholar]
- 35.Song W, Li H, Tao K, Li R, Song Z, Zhao Q, et al. Expression and clinical significance of the stem cell marker CD133 in hepatocellular carcinoma. Int J Clin Pract 2008;62:1212–1218 [DOI] [PubMed] [Google Scholar]
- 36.Kim JW, Ye Q, Forgues M, Chen Y, Budhu A, Sime J, et al. Cancer-associated molecular signature in the tissue samples of patients with cirrhosis. Hepatology 2004;39:518–527 [DOI] [PubMed] [Google Scholar]
- 37.Yamashita T, Budhu A, Forgues M, Wang XW. Activation of hepatic stem cell marker EpCAM by Wnt-beta-catenin signaling in hepatocellular carcinoma. Cancer Res 2007;67:10831–10839. [DOI] [PubMed] [Google Scholar]
- 38.Lee TK, Castilho A, Cheung VC, Tang KH, Ma S, Ng IO. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell 2011;9:50–63 [DOI] [PubMed] [Google Scholar]
- 39.Huynh H, Ngo VC, Koong HN, Poon D, Choo SP, Thng CH, et al. Sorafenib and rapamycin induce growth suppression in mouse models of hepatocellular carcinoma. J Cell Mol Med 2009;13:2673–2683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Villanueva A, Chiang DY, Newell P, Peix J, Thung S, Alsinet C, et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology 2008;135:1972–1983, 1983 e1971–1911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen YC, Hsu C, Cheng AL. Molecular targeted therapy for advanced hepatocellular carcinoma: current status and future perspectives. J Gastroenterol 2010;45:794–807 [DOI] [PubMed] [Google Scholar]
- 42.Knight B, Tirnitz-Parker JE, Olynyk JK. C-kit inhibition by imatinib mesylate attenuates progenitor cell expansion and inhibits liver tumor formation in mice. Gastroenterology 2008;135:969-979, 979 e961 [DOI] [PubMed] [Google Scholar]
- 43.Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest 2012;122:2911–2915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michalopoulos GK, DeFrances MC. Liver regeneration. Science 1997;276:60–66 [DOI] [PubMed] [Google Scholar]
- 45.Shafritz DA, Oertel M, Menthena A, Nierhoff D, Dabeva MD. Liver stem cells and prospects for liver reconstitution by transplanted cells. Hepatology 2006;43:S89–S98 [DOI] [PubMed] [Google Scholar]
- 46.Theise ND, Yao JL, Harada K, Hytiroglou P, Portmann B, Thung SN, et al. Hepatic ‘stem cell’ malignancies in adults: four cases. Histopathology 2003;43:263–271 [DOI] [PubMed] [Google Scholar]
- 47.Yao Z, Mishra L. Cancer stem cells and hepatocellular carcinoma. Cancer Biol Ther 2009;8:1691–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zimmermann A Hepatoblastoma with cholangioblastic features (‘cholangioblastic hepatoblastoma’) and other liver tumors with bimodal differentiation in young patients. Med Pediatr Oncol 2002;39:487–491 [DOI] [PubMed] [Google Scholar]
- 49.Yamashita T, Forgues M, Wang W, Kim JW, Ye Q, Jia H, et al. EpCAM and alpha-fetoprotein expression defines novel prognostic subtypes of hepatocellular carcinoma. Cancer Res 2008;68:1451–1461 [DOI] [PubMed] [Google Scholar]
- 50.He G, Dhar D, Nakagawa H, Font-Burgada J, Ogata H, Jiang Y, et al. Identification of liver cancer progenitors whose malignant progression depends on autocrine IL-6 signaling. Cell 2013;155:384–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sell S, Dunsford HA. Evidence for the stem cell origin of hepatocellular carcinoma and cholangiocarcinoma. Am J Pathol 1989;134:1347–1363 [PMC free article] [PubMed] [Google Scholar]
- 52.Braun L, Goyette M, Yaswen P, Thompson NL, Fausto N. Growth in culture and tumorigenicity after transfection with the ras oncogene of liver epithelial cells from carcinogen-treated rats. Cancer Res 1987;47:4116–41124 [PubMed] [Google Scholar]
- 53.Tsao MS, Grisham JW. Hepatocarcinomas, cholangiocarcinomas, and hepatoblastomas produced by chemically transformed cultured rat liver epithelial cells. A light- and electron-microscopic analysis. Am J Pathol 1987;127:168–181 [PMC free article] [PubMed] [Google Scholar]
- 54.Sell S Alpha-fetoprotein, stem cells and cancer: how study of the production of alpha-fetoprotein during chemical hepatocarcinogenesis led to reaffirmation of the stem cell theory of cancer. Tumour Biol 2008;29:161–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dorrell C, Erker L, Schug J, Kopp JL, Canaday PS, Fox AJ, et al. Prospective isolation of a bipotential clonogenic liver progenitor cell in adult mice. Genes Dev 2011;25:1193–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jensen CH, Jauho EI, Santoni-Rugiu E, Holmskov U, Teisner B, Tygstrup N, et al. Transit-amplifying ductular (oval) cells and their hepatocytic progeny are characterized by a novel and distinctive expression of delta-like protein/preadipocyte factor 1/fetal antigen 1. Am J Pathol 2004;164:1347–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Furuyama K, Kawaguchi Y, Akiyama H, Horiguchi M, Kodama S, Kuhara T, et al. Continuous cell supply from a Sox9-expressing progenitor zone in adult liver, exocrine pancreas and intestine. Nat Genet 2011;43:34–41 [DOI] [PubMed] [Google Scholar]
- 58.Liu L, Dai Y, Chen J, Zeng T, Li Y, Chen L, et al. Maelstrom promotes hepatocellular carcinoma metastasis by inducing epithelial-mesenchymal transition by way of Akt/GSK-3beta/Snail signaling. Hepatology 2014;59:531–543 [DOI] [PubMed] [Google Scholar]
- 59.Hagiwara S, Kudo M, Ueshima K, Chung H, Yamaguchi M, Takita M, et al. The cancer stem cell marker CD133 is a predictor of the effectiveness of S1 + pegylated interferon alpha-2b therapy against advanced hepatocellular carcinoma. J Gastroenterol 2011;46:212–221 [DOI] [PubMed] [Google Scholar]
- 60.Liu S, Li N, Yu X, Xiao X, Cheng K, Hu J, et al. Expression of intercellular adhesion molecule 1 by hepatocellular carcinoma stem cells and circulating tumor cells. Gastroenterology 2013;144:1031–1041, e1010 [DOI] [PubMed] [Google Scholar]
- 61.Shan J, Shen J, Liu L, Xia F, Xu C, Duan G, et al. Nanog regulates self-renewal of cancer stem cells through the insulinlike growth factor pathway in human hepatocellular carcinoma. Hepatology 2012;56:1004–1014 [DOI] [PubMed] [Google Scholar]
- 62.Roskams TA, Theise ND, Balabaud C, Bhagat G, Bhathal PS, Bioulac-Sage P, et al. Nomenclature of the finer branches of the biliary tree: canals, ductules, and ductular reactions in human livers. Hepatology 2004;39:1739–1745 [DOI] [PubMed] [Google Scholar]
- 63.Durnez A, Verslype C, Nevens F, Fevery J, Aerts R, Pirenne J, et al. The clinicopathological and prognostic relevance of cytokeratin 7 and 19 expression in hepatocellular carcinoma. A possible progenitor cell origin. Histopathology 2006;49:138–151 [DOI] [PubMed] [Google Scholar]
- 64.Burke ZD, Thowfeequ S, Peran M, Tosh D. Stem cells in the adult pancreas and liver. Biochem J 2007;404:169–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hatch HM, Zheng D, Jorgensen ML, Petersen BE. SDF-1alpha/CXCR4: a mechanism for hepatic oval cell activation and bone marrow stem cell recruitment to the injured liver of rats. Cloning Stem Cells 2002;4:339–351 [DOI] [PubMed] [Google Scholar]
- 66.Craig CE, Quaglia A, Selden C, Lowdell M, Hodgson H, Dhillon AP. The histopathology of regeneration in massive hepatic necrosis. Semin Liver Dis 2004;24:49–64 [DOI] [PubMed] [Google Scholar]
- 67.Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, et al. Significance of CD90 + cancer stem cells in human liver cancer. Cancer Cell 2008;13:153–166 [DOI] [PubMed] [Google Scholar]
- 68.Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003;100:3983–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 1997;3:730–737 [DOI] [PubMed] [Google Scholar]
- 70.Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res 2005;65:10946–10951 [DOI] [PubMed] [Google Scholar]
- 71.Forbes SJ, Alison MR. Side population (SP) cells: taking center stage in regeneration and liver cancer? Hepatology 2006;44:23–26 [DOI] [PubMed] [Google Scholar]
- 72.Chiba T, Kita K, Zheng YW, Yokosuka O, Saisho H, Iwama A, et al. Side population purified from hepatocellular carcinoma cells harbors cancer stem cell-like properties. Hepatology 2006;44:240–251 [DOI] [PubMed] [Google Scholar]
- 73.Ho JW, Pang RW, Lau C, Sun CK, Yu WC, Fan ST, et al. Significance of circulating endothelial progenitor cells in hepatocellular carcinoma. Hepatology 2006;44:836–843 [DOI] [PubMed] [Google Scholar]
- 74.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, et al. Identification of human brain tumour initiating cells. Nature 2004;432:396–401 [DOI] [PubMed] [Google Scholar]
- 75.Shmelkov SV, St Clair R, Lyden D, Rafii S. AC133/CD133/Prominin-1. Int J Biochem Cell Biol 2005;37:715–719 [DOI] [PubMed] [Google Scholar]
- 76.Rountree CB, Senadheera S, Mato JM, Crooks GM, Lu SC. Expansion of liver cancer stem cells during aging in methionine adenosyltransferase 1A-deficient mice. Hepatology 2008;47: 1288–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tirnitz-Parker JE, Tonkin JN, Knight B, Olynyk JK, Yeoh GC. Isolation, culture and immortalisation of hepatic oval cells from adult mice fed a choline-deficient, ethionine-supplemented diet. Int J Biochem Cell Biol 2007;39:2226–2239 [DOI] [PubMed] [Google Scholar]
- 78.Libbrecht L, De Vos R, Cassiman D, Desmet V, Aerts R, Roskams T. Hepatic progenitor cells in hepatocellular adenomas. Am J Surg Pathol 2001;25:1388–1396 [DOI] [PubMed] [Google Scholar]
- 79.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994;367:645–648 [DOI] [PubMed] [Google Scholar]
- 80.Szabo G, Wands JR, Eken A, Osna NA, Weinman SA, Machida K, et al. Alcohol and hepatitis C virus-interactions in immune dysfunctions and liver damage. Alcohol Clin Exp Res 2010;34:1675–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tumurbaatar B, Tikhanovich I, Li Z, Ren J, Ralston R, Kuravi S, et al. Hepatitis C and alcohol exacerbate liver injury by suppression of FOXO3. Am J Pathol 2013;183:1803–1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tikhanovich I, Kuravi S, Campbell RV, Kharbanda KK, Artigues A, Villar MT, et al. Regulation of FOXO3 by phosphorylation and methylation in hepatitis C virus infection and alcohol exposure. Hepatology 2014;59:58–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Geissler M, Gesien A, Wands JR. Inhibitory effects of chronic ethanol consumption on cellular immune responses to hepatitis C virus core protein are reversed by genetic immunizations augmented with cytokine-expressing plasmids. J Immunol 1997;159:5107–5113 [PubMed] [Google Scholar]
- 84.Encke J, Wands JR. Ethanol inhibition: the humoral and cellular immune response to hepatitis C virus NS5 protein after genetic immunization. Alcohol Clin Exp Res 2000;24:1063–1069 [PubMed] [Google Scholar]
- 85.Oshita M, Hayashi N, Kasahara A, Hagiwara H, Mita E, Naito M, et al. Increased serum hepatitis C virus RNA levels among alcoholic patients with chronic hepatitis C. Hepatology. 1994;20:1115–20 [PubMed] [Google Scholar]
- 86.Sherman KE, O’Brien J, Gutierrez AG, Harrison S, Urdea M, Neuwald P, et al. Quantitative evaluation of hepatitis C virus RNA in patients with concurrent human immunodeficiency virus infections. J Clin Microbiol 1993;31:2679–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Paronetto F Immunologic reactions in alcoholic liver disease. SeminLiverDis 1993;13:183–195 [DOI] [PubMed] [Google Scholar]
- 88.Machida K, Tsukamoto H, Mkrtchyan H, Duan L, Dynnyk A, Liu HM, et al. Toll-like receptor 4 mediates synergism between alcohol and HCV in hepatic oncogenesis involving stem cell marker Nanog. Proc Natl Acad Sci USA 2009;106:1548–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sun B, Karin M. Obesity, inflammation, and liver cancer. J Hepatol 2012;56:704–713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, et al. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010;140:197–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Seki E, Brenner DA, Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 2012;143:307–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Feldman DE, Chen C, Punj V, Tsukamoto H, Machida K. Pluripotency factor-mediated expression of the leptin receptor (OB-R) links obesity to oncogenesis through tumor-initiating stem cells. Proc Natl Acad Sci USA 2012;109:829–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wurmbach E, Chen YB, Khitrov G, Zhang W, Roayaie S, Schwartz M, et al. Genome-wide molecular profiles of HCV-induced dysplasia and hepatocellular carcinoma. Hepatology 2007;45:938–947 [DOI] [PubMed] [Google Scholar]
- 94.Yeoh GC, Ernst M, Rose-John S, Akhurst B, Payne C, Long S, et al. Opposing roles of gp130-mediated STAT-3 and ERK-1/2 signaling in liver progenitor cell migration and proliferation. Hepatology 2007;45:486–494 [DOI] [PubMed] [Google Scholar]
- 95.Dando JS, Tavian M, Catelain C, Poirault S, Bennaceur-Gris-celli A, Sainteny F, et al. Notch/Delta4 interaction in human embryonic liver CD34 + CD38- cells: positive influence on BFU-E production and LTC-IC potential maintenance. Stem Cells 2005;23:550–560 [DOI] [PubMed] [Google Scholar]
- 96.Sicklick JK, Li YX, Jayaraman A, Kannangai R, Qi Y, Vive-kanandan P, et al. Dysregulation of the Hedgehog pathway in human hepatocarcinogenesis. Carcinogenesis 2006;27:748–757 [DOI] [PubMed] [Google Scholar]
- 97.Sicklick JK, Li YX, Melhem A, Schmelzer E, Zdanowicz M, Huang J, et al. Hedgehog signaling maintains resident hepatic progenitors throughout life. Am J Physiol Gastrointest Liver Physiol 2006;290:G859–G870 [DOI] [PubMed] [Google Scholar]
- 98.Kitisin K, Ganesan N, Tang Y, Jogunoori W, Volpe EA, Kim SS, et al. Disruption of transforming growth factor-beta signaling through beta-spectrin ELF leads to hepatocellular cancer through cyclin D1 activation. Oncogene 2007;26:7103–7110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nguyen LN, Furuya MH, Wolfraim LA, Nguyen AP, Holdren MS, Campbell JS, et al. Transforming growth factor-beta differentially regulates oval cell and hepatocyte proliferation. Hepatology 2007;45:31–41 [DOI] [PubMed] [Google Scholar]
- 100.Iliopoulos D, Hirsch HA, Struhl K. An epigenetic switch involving NF-kappaB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 2009;139:693–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Viswanathan SR, Powers JT, Einhorn W, Hoshida Y, Ng TL, Toffanin S, et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat Genet 2009;41:843–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yamanaka S Pluripotency and nuclear reprogramming. Philos Trans R Soc Lond B Biol Sci 2008;363:2079–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cheng SW, Tsai HW, Lin YJ, Cheng PN, Chang YC, Yen CJ, et al. Lin28B is an oncofetal circulating cancer stem cell-like marker associated with recurrence of hepatocellular carcinoma. PLoS ONE 2013;8:e80053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang YC, Chen YL, Yuan RH, Pan HW, Yang WC, Hsu HC, et al. Lin-28B expression promotes transformation and invasion in human hepatocellular carcinoma. Carcinogenesis 2010;31:1516–1522 [DOI] [PubMed] [Google Scholar]
- 105.Guo Y, Chen Y, Ito H, Watanabe A, Ge X, Kodama T, et al. Identification and characterization oflin-28 homolog B (LIN28B) in human hepatocellular carcinoma. Gene 2006;384:51–61 [DOI] [PubMed] [Google Scholar]
- 106.Chang TC, Zeitels LR, Hwang HW, Chivukula RR, Wentzel EA, Dews M, et al. Lin-28B transactivation is necessary for Myc-mediated let-7 repression and proliferation. Proc Natl Acad Sci USA 2009;106:3384–3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science 2003;299:574–577 [DOI] [PubMed] [Google Scholar]