

Abstract
Hepatocellular carcinoma (HCC) is a malignant tumor with high mortality. This study aimed to build a prognostic signature for HCC patients based on immune-related genes (IRGs) and epigenetics-related genes (EPGs). RNA-seq data from Gene Expression Omnibus were used for dynamic network biomarker (DNB) analysis to identify 56 candidate IRG–EPG–DNBs and their first-neighbor genes. These genes were screened using LASSO-Cox regression analysis to finally obtain five candidate genes—RNF2, YBX1, EZH2, CAD, and PSMD1—which constituted the prognostic signature panel. According to this panel, patients in The Cancer Genome Atlas and International Cancer Genome Consortium were divided into high- and low-risk groups. The prognosis, clinicopathological features, and immune cell infiltration significantly differed between the two risk groups. The prognostic ability of the signature panel and expression profiling were further validated using online databases. We used an independent cohort of patients to validate the expression profiles of the five genes using reverse transcription–PCR. CMap and CellMiner predicted four small molecule drug–protein pairs based on the five prognostic genes. Of them, two market drugs approved by the Food and Drug Administration (AT-13387 and KU-55933) have emerged as candidates for HCC study. This new signature panel may serve as a potential prognostic marker, engendering the possibility of novel personalized therapy with classification of HCC patients.
Keywords: Hepatocellular carcinoma, Immune-related genes, Epigenetics-related genes, Dynamic network biomarker analysis, Prognostic signature, Drugs
Graphical Abstract

1. Background
Globally, hepatocellular carcinoma (HCC) is the most common type of primary liver cancer, with the fourth highest cancer-related mortality rate [1]. Surgical resection of the tumor is the most common treatment method, but due to the lack of obvious early symptoms, most patients are diagnosed in the middle or late phase of the disease, thus missing the best therapeutic window for surgical resection [2], [3]. Besides, the diagnosis and treatment of HCC is challenging, mainly due to its high metastatic ability and recurrence rate, and the 5-year survival rate of patients is below 20% [4].
During the development of HCC, highly activated inflammatory signaling and complex mutations are observed in hepatocytes from chronic inflamed liver tissues, which is conducive for their transformation into cancerous cells [5]. The liver is a central immune modulator that maintains the balance of immune tolerance in the tumor microenvironment. The development of immune checkpoint inhibition has greatly changed the clinical management of HCC [6]. The mechanism of immune escape-assisted tumor progression has been extensively studied, with a focus on two immune checkpoint proteins: programmed cell death protein 1 (PD-1) and programmed death-ligand 1 (PD-L1). However, the remission rate after immune checkpoint inhibition monotherapy is only 20% [7]. The PD-1-blocking antibodies nivolumab and pembrolizumab elicited responses in less than 20% of HCC patients. Sorafenib is the first systemic drug for HCC approved by the Food and Drug Administration (FDA), but the individual patient responses to sorafenib vary greatly. Sorafenib combined with PD-1 or PD-L1 demonstrated a good synergistic therapeutic effect in the treatment of HCC. In particular, the combination of atezolizumab (anti-PD-L1) and bevacizumab (anti-vascular endothelial growth factor monoclonal antibody) achieved higher survival rates than sorafenib alone, thus emerging as a new standard in the first-line treatment of advanced HCC [8]. These studies indicate that HCC interacts with the immune system in a complex way. Biomarkers for clinical prediction are also urgently needed to accurately classify patients, so that their response to immune checkpoint inhibitors is enhanced.
Epigenetic elements are potentially heritable elements in the genome that are not encoded in the nucleotide sequence [9]. Abnormal DNA methylation is one of the most common epigenetic modifications in HCC. Immune cells also show the ability to modulate DNA methylation. Over the past 35 years, epigenetic modifications have emerged as major drivers of cancer initiation and progression. Previous research on cancer epigenetics focused on the abnormal expression of certain chromatin-modifying enzymes and their regulatory roles in tumor cells. Nowadays, the relationship between epigenetic mechanisms and the tumor microenvironment is being investigated [10]. For example, tumor-associated macrophages were found to upregulate DNMT1 in gastric cancer, which silences the tumor suppressor gene G SN [11]. Through genome-wide DNA methylation analysis and other methods, many studies have identified different methylation patterns in HCC tissues compared with normal tissues. Genes with significant hypermethylation, such as EZH2 [12], CDKN2A [13], RASSF1 [14], and APC [15], have been studied.
In this study, we used multiple statistical methods to construct diagnostic and prognostic models of HCC based on key immune-related genes (IRGs), epigenetics-related genes (EPGs), and their closely related neighbor genes identified via dynamic network biomarker (DNB) analysis. The prognostic model, consisting of five key genes, divided HCC patients into high- and low-risk groups, and was statistically correlated with the immune status as well as the prognosis of patients. Furthermore, we performed molecular docking, CellMiner analysis, and drug–gene profiling using CMap to explore drug repositioning for HCC.
2. Material and methods
2.1. Data source and clinical information
For this study, we selected a dataset from Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo) that satisfied the following criteria. Inclusion criteria: data on HCC associated with chronic liver disease, which evolved from precancerous lesions and early HCC (eHCC) into advanced cancer; larger sample size; Homo sapiens; and expression profiling. Exclusion criteria: cell line data and missing adjacent normal samples. The dataset GSE114564 was selected, and the data matrix was preprocessed using the “GEO2R” package in R. This dataset contains next-generation sequencing RNA-seq data on 118 cases, including healthy (n = 15), chronic hepatitis (n = 20), cirrhosis (n = 10), dysplastic nodules (n = 10), eHCC (n = 18), and advanced HCC (n = 45) [16], [17]. The causes of these patients include HBV, HCV, alcohol and others [17]. Next, we mapped the full list of 2490 IRGs and EPGs to the data matrix to obtain their expression profiles in healthy to advanced HCC cases for DNB analysis. Besides, we obtained transcriptome profiling data on 424 samples (normal and tumor specimens) and the corresponding clinical information on HCC patients from the Cancer Genome Atlas (TCGA; https://portal.gdc.cancer.gov/), and data on another independent 231 tumor specimens from the International Cancer Genome Consortium (ICGC; https://dcc.icgc.org/projects/LIRI-JP).
2.2. IRGs and EPGs
The full list of IRGs was downloaded from ImmPort (https://www.immport.org/home) on Feb 2, 2022. We extracted 720 EPGs from the EpiFactors database (https://epifactors.autosome.ru/). The full list of IRGs and EPGs is provided in Supplementary Table S6.
2.3. DNB analysis
The DNB method, widely employed to identify the tipping point or critical transition in disease progression, has proven effective in establishing disease markers. The DNB theory is based on the fact that strong fluctuations in several molecules (DNB members) cause dramatic changes in biological systems.
CVI is the average coefficient of variance (CV) of DNBs, PCCin is the average Pearson correlation coefficient (PCC) of molecules in the candidate cluster, and PCCout is the average PCC of molecules inside as well as outside the cluster [18].
IRGs and EPGs extracted from GSE114564 were subjected to DNB analysis to obtain IRG–EPG–DNBs (IEDs), which were individually mapped to the integrated functional linkage networks to obtain their respective first-neighbor genes. The network was constructed using various databases. The available functional linkage information for Homo sapiens was downloaded and combined from the following databases: KEGG (www.genome.jp/kegg), TRED (www.rulai.cshl.edu/cgi-bin/TRED/), BioGrid (https://thebiogrid.org), IntAct (https://www.ebi.ac.uk/intact/), MINT (https://mint.bio.uniroma2.it/), UniProt (https://www.uniprot.org/), PINA (https://omics.bjcancer.org/pina/home.action), and STRING (https://cn.string-db.org/). This integrated functional linkage protein–protein interaction (PPI) network was built using Cytoscape (version 3.8.2) [19].
2.4. Construction and validation of a prognostic EPG signature using univariate, multivariate, and least absolute shrinkage and selection operator (LASSO)-Cox analysis
To evaluate the prognostic value of candidate genes, their relationship with overall survival (OS) in TCGA data was explored using Cox regression analysis. The LASSO-Cox regression model ("glmnet" package, version 4.1) was used to screen diagnostic markers. The penalty parameter (λ) was determined by the lowest standard. Logistic regression was used to build a prognostic signature panel model. A risk score was calculated for each patient in TCGA and ICGC, and they were segregated into high- or low-risk groups based on the median risk score. The OS of patients in the high- and low-risk groups was analyzed using the “survival” (version 3.3)and “survminer” (version 0.4.7) packages in R (version 4.2.1). P < 0.05 was considered statistically significant.
2.5. Predictive nomogram for TCGA and ICGC data
A nomogram is a clinically applicable tool for predicting the 1-year, 2-year, and 3-year survival rates of HCC patients. Three clinical features each were selected from TCGA and ICGC: age, gender, and stage, and gender, tumor grade, and existence of prior malignancy, respectively. The “RMS” (version 6.7) package from R was applied to establish nomograms using the Cox method, which could evaluate the prognostic significance of these features with clinical data, such as survival time and survival status.
2.6. Tumor-infiltrating immune cell analysis
We assessed the immune microenvironment of both the risk groups. The CIBERSORT algorithm identified 22 types of tumor-infiltrating immune cells. The composition of the immune infiltrates was calculated for both the groups. The ESTIMATE algorithm was used to further analyze the tumor microenvironment of HCC patients from both the groups. Finally, we calculated the stromal, immune, and ESTIMATE scores for both the groups.
2.7. Changes in genetic expression, co-expression, and neighbor gene network analyses of the five candidate genes
Gene expression from the TCGA and Genotype-Tissue Expression data was validated using GEPIA (http://gepia.cancer-pku.cn/index.html). Gene expression profiles from the Human Protein Atlas (HPA) dataset (https://www.proteinatlas.org/) were explored using immunohistochemistry. Gene mutation analysis in HCC samples from TCGA was performed using cBioPortal (www.cbioportal.org/), an interactive platform to visualize and analyze genetic data from cancer studies. Calculating the mutation landscape of 5 genes in high and low risk group samples in the TCGA was performed in “Maftools” (version 2.16.0) package in R.
The five candidate genes were mapped to the integrated functional linkage PPI network previously constructed during DNB analysis to identify genes closely related to them. These genes were strictly screened by edge selection criteria with physical association, or experimentally validated, as known as high confidence scores.
The core genes from the PPI network were identified using CytoHubba and CytoNCA, both of which are Cytoscape applications. CytoHubba can predict and explore important nodes and subnetworks in a given network using several topological algorithms, such as Maximal Clique Centrality. We selected the top 20 nodes ranked by Maximal Clique Centrality. Similarly, CytoNCA evaluates the central nodes of networks. We selected the top 20 nodes ranked by degree value. The overlapping genes in these two algorithms were identified as core genes for drug repositioning.
2.8. RNA extraction and reverse transcription–quantitative PCR (RT–qPCR) validation
Total RNA was extracted using TRIzol reagent (Invitrogen, CA, USA) according to the manufacturer’s protocols. cDNA was synthesized using reverse transcriptase (TOYOBO, Osaka, Japan). RT–qPCR was performed using the Power SYBR Green PCR Master Mix (TOYOBO, Osaka, Japan) on the Applied Biosystems Step-one PCR system (Thermo Fisher Scientific, CA, USA). Briefly, multiplex one-step RT-PCR was carried out in a final reaction volume of 25 μL containing 2 × SYBR Green Realtime RT-PCR Master Mix, 0.4 μM primers, and 0.5 μL of template cDNA. The primers used are listed in Table S7. Gene expression levels were quantified and normalized to the expression of the reference gene, ACTB, using an optimized comparative Ct (ΔCt) method.
2.9. Drug repositioning and molecular docking
CMap (https://clue.io/; accessed on 20 June 2022) is an integrative platform that contains data on drugs or drug candidates from large perturbational datasets in the clinical experimental, investigational, and approved-for-treatment stages. Drugs likely to interact with the core genes were retrieved from CMap [44]. The correlation between hub genes and the drug response was predicted using the CellMiner database (https://discover.nci.nih.gov/cellminer/).
Molecular docking was carried out to further verify the effective binding of the selected compounds to the core target. The structure energy was minimized using the ChemBioDraw 3D module. The crystal structure of the candidate target was obtained from the Protein Data Bank (https://www.rcsb.org/). The PDB IDs of EZH2 and YBX1 are 4w2r and 1h95, respectively. The 3D structures of small molecule compounds were downloaded from PubChem (https://pubchem.ncbi.nlm.nih.gov/) in Structure-Data File format.
The receptor structure was modified using AutoDockTools 1.5.621 (dehydration and hydrogenation) and exported in the PDBQT format. After defining the grid on the active site of the receptor protein, docking was performed using Autodock Vina 1.1.2, and the output score was displayed in kcal / mol. PyMOL 2.3.0 and BIOVIA Discovery Studio 2016 were used for result processing and visualization.
2.10. Data processing
A series of R packages, including "survminer"(version 0.4.7), "survival"(version 3.3), and "time-ROC"(version 0.4), were used to perform time-dependent receiver operating characteristic (ROC) curve analyses. A P-value ≤ 0.05 was considered to be statistically significant.
3. Results
3.1. Dynamic changes in IRGs and EPGs during hepatocarcinogenesis
The DNB algorithm could effectively identify the critical transition of IRGs and EPGs in HCC by calculating the dramatic changes of CI value in their expression correlated with disease progression (Fig. 1a). Three criteria were levied for selecting DNBs (Fig. 1b): (1) high fluctuation in candidate IED expression with larger CVs; (2) high correlation between candidate IEDs (high PCCin); and (iii) weak association between IEDs and non-IEDs (low PCCout). eHCC was defined as the critical stage with the maximum confidence interval (CI) value. Then, we individually mapped the IEDs to the whole-gene regulation networks to construct a series of networks whose structure illustrated the dramatic changes and expression fluctuations in DNBs (Fig. 1c). We identified 55 IEDs that had high CVs, fluctuated strongly, and shared close relationships with each other during eHCC (Table S1). Co-expression relationships between IEDs dramatically changed when the biological system approached the critical stage. We mapped individual IEDs to the integrated functional linkage network to obtain their first-neighbor genes. A total of 529 genes were identified (Table S2), which, along with the 55 IEDs, were used as seed genes for subsequent studies.
Fig. 1.

Dynamic network biomarker (DNB) identification in the GSE114564 dataset. (a) Brief description of the DNB analysis. (b) Four diagrams for criteria results of DNBs over six disease stages in GSE114564. The confidence interval was calculated based on the three criteria to identify the critical transition phase. (c) Series of immune-related genes–epigenetics-related genes–DNBs (IEDs) mapped to gene regulation networks. The color shade of the node represents the magnitude of the change in coefficient of variation of the molecule. DNBs strongly fluctuate and closely correlate with each other during early HCC (eHCC). Healthy (n = 15), Chronic hepatitis (n = 20), Cirrhosis (n = 10), Dysplastic nodules (n = 10), eHCC (n = 18), and Advanced HCC (n = 45).
3.2. Construction of prognostic models based on IEDs and closely related neighbor-genes
The set of 529 DNBs and their neighbor-genes were subjected to LASSO-Cox regression analysis using 10-time cross-validation for tuning the parameter selection model. As a result, five genes were identified: RNF2, YBX1, EZH2, CAD, and PSMD1 (Fig. 2a,b; Table 1). RNF2, YBX1, and EZH2 are EPGs, while PSMD1 and CAD are IRGs. In the regression equation, the coefficients of signature variable (genes) were calculated using LASSO algorithms, and each significant gene has its own coefficient value. The coefficient values represent the degree of influence of the independent variable (genes) on the dependent variable (risk score). Thus, the risk score was calculated using the gene signature as follows: (5.204e-05 × RNF2) + (0.0018 × YBX1) + (0.0168 × PSMD1) + (0.023 × EZH2) + (0.002 × CAD). In this case, EZH2 has the most impact on the risk score, having the highest coefficient (0.023) among the five genes. We analyzed the relationships between different risk scores and survival times, survival events, and expression changes in the five genes based on TCGA data (n = 357). Low- and high-risk groups were formed based on the median risk score. We found that when the risk score increased (x-axis from left to right, Fig. 2c, top), patient mortality increased and the survival time of patients decreased significantly (Fig. 2c, middle). The expression levels of the five genes were positively correlated with the risk score (Fig. 2c).
Fig. 2.

Construction of prognostic models based on genes selected from independent cross-link analysis of IEDs and their first-neighbor genes. (a) The vertical line indicates the minimum partial likelihood deviation of the least absolute shrinkage and selection operator (LASSO) coefficient distribution. The two vertical dashed lines are λ.min and λ.1se. (b) LASSO coefficient profiles of these candidate genes with non-zero coefficients determined by the optimal λ. (c) Risk score and survival status of patients in the Cancer Genome Atlas (TCGA) cohort determined using the five genes.
Table 1.
Univariate, multivariate, and LASSO-Cox analysis of the five candidate genes.
| Gene | Cox regression analysis |
LASSO-Cox regression |
||
|---|---|---|---|---|
| Coefficients | p-value | HR (95% CI) | LASSO Coefficient | |
| YBX1 | 0.0120 | 0.0910 | 1.01 (0.998–1.03) | 0.0024 |
| PSMD1 | 0.5200 | 0.0000 | 1.68 (1.37–2.06) | 0.0204 |
| CAD | 1.8000 | 0.0000 | 6.06 (3.97–9.25) | 0.0048 |
| EZH2 | 0.4400 | 0.0000 | 1.56 (1.34–1.81) | 0.0286 |
| RNF2 | 0.0780 | 0.0000 | 1.08 (1.05–1.11) | 0.0005 |
LASSO, least absolute shrinkage and selection operator; HR, hazards ratio; CI, confidence interval
3.3. Independent prognostic ability of the risk score
Univariate Cox regression analysis revealed that the risk score was significantly associated with poor survival in both the TCGA and ICGC datasets (hazards ratio [HR]: 6.060, 95% CI: 3.970 −9.250, and HR: 2.770, 95% CI: 1.600 −4.780, respectively; Fig. 3a, c). Clinical feature of age and gender except tumor stage was significantly correlated with OS (Fig. 3a).
Fig. 3.

Risk score panel derived from the five genes. Risk scores were calculated for the TCGA and the International Cancer Genome Consortium (ICGC) datasets. Risk scores were combined with clinical information, survival time, and survival status for regression analysis in the TCGA and ICGC datasets. (a,b) Univariate and multivariate Cox regression in TCGA; (c,d) Univariate and multivariate Cox regression in ICGC.
Multivariate analysis also revealed that the risk score was an independent indicator of OS in the TCGA and ICGC datasets (HR: 5.410, 95% CI: 3.40 −8.61, and HR: 2.960, 95% CI: 1.710 −5.130, respectively; Fig. 3b, d). These results suggest that the risk score generated from the five-gene signature panel could be used as an independent prognostic indicator of OS in HCC patients.
In both TCGA and ICGC datasets, Kaplan–Meier curves showed that patients in the low-risk group had markedly longer OS than those in the high-risk group (P < 0.001; Fig. 4a,c). Area under the curve (AUC) of time-dependent ROC curves was used to further assess the prognostic ability of the risk score. The AUC for the TCGA dataset was 0.79 at 1 year, 0.72 at 3 years, and 0.74 at 5 years (Fig. 4a). For the ICGC dataset, the AUC was 0.70 at 100 days, 0.71 at 300 days, and 0.60 at 600 days (Fig. 4b,d). These results suggest that the predictive ability of the five-gene signature-based risk score was favorable.
Fig. 4.

Verification of the five-gene signature in survival analysis and its predictive capacity. (a,b) Survival analysis using the five-gene-based risk score panel and its capacity to predict the 1-, 3-, and 5-year survival rates of patients in the TCGA cohort (n = 357). (c,d) Survival analysis using the five-gene-based risk score panel and its capacity to predict the 1-, 3-, and 5-year survival rates of patients in the ICGC cohort (n = 231).
3.4. Nomogram analysis
We constructed a nomogram that could estimate the 1-year, 2-year, and 3-year survival rates to build a clinically applicable method of predicting the survival probability of HCC patients. The nomogram included three features from TCGA—age, gender, and stage—and three features from ICGC—gender, tumor grade, and the existence of prior malignancy (Fig. 5a). TNM stage is recognized as a traditional prognostic indicator. The TNM staging system for cancers is an internationally accepted system used to determine the disease stage based on the pathological stage. T refers to the size and depth of tumor infiltration, N refers to the presence and number of lymph node metastases, and M refers to the presence of distant organ metastases. Based on this system, clinicians divide patients into stages I, II, III, and IV. In TCGA, TNM stage exhibited good predictive potential (C-index = 0.630, 95% CI: 0.551–0.710; P = 0.001; Fig. 5a). The five-gene signature-derived risk score and stage showed better prognostic ability (C-index = 0.751, 95% CI: 0.700–0.802; P = 2.354e-22) than TNM alone. The “RMS” package from R was used to integrate the data on survival time, survival status, and characteristics to establish nomograms using the Cox method that could evaluate the prognostic significance of these characteristics, thereby making the clinical management of HCC patients more practical. The nomogram predictions for the 1-year, 3-year, and 5-year OS rate closely matched the best prediction performance (Fig. 5b). TNM stage is one of the most widely used and effective prognostic indicators for HCC (C-index = 0.630) [20]. It showed a better prognostic improvement ability than previously developed genetic markers, with an AUC of 0.79 at 1 year, 0.72 at 3 years, and 0.74 at 5 years. An eight-gene signature for classifying patients has been previously reported (AUC = 0.667 at 1 year, 0.630 at 3 years, 0.618 at 5 years), which includes CA9, CCL20, CORO1C, CTSC, LDHA, NDRG1, PTP4A3, and TUBA1B. This signature can assist clinical decision-making by combining information on hypoxia, thereby useful for stratifying patients by risk [21]. Another seven-gene signature [22] (AUC = 0.686 at 1 year, 0.644 at 3 years, and 0.615 at 5 years), including KIF18B, CEP55, CITs, MCM7, CDC45, EZH2, and MCM5, was reported to effectively predict OS in Asian HCC patients.
Fig. 5.

Construction and verification of a nomogram for survival prediction in TCGA. (a) A nomogram constructed by combining gender, tumor grade, existence of prior malignancy, and the risk score. (b) The actual and predictive probability of 1-year, 3-year, and 5-year survival calculated by the nomogram.
3.5. Difference in immune infiltration between high and low-risk groups
CIBERSORT characterizes the cell composition of complex tissues from their gene expression profiles. Applying this method to TCGA and ICGC datasets, we assessed the cell composition of the immune microenvironment in the two risk groups by analyzing the abundance of tumor-infiltrating immune cells. We also assessed the stromal, immune, and ESTIMATE scores for the two risk groups using the ESTIMATE algorithm.
In the TCGA dataset, the high-risk group was significantly more infiltrated by activated memory CD4 T cells (P < 0.05), T follicular helper cells (P < 0.05), M0 macrophages (P < 0.001), resting dendritic cells (P < 0.05), and neutrophils (P < 0.001) than the low-risk group, but notably lesser infiltrated by resting memory CD4 T cells (P < 0.001), monocytes (P < 0.05), and resting mast cells (P < 0.01) (Fig. 6a). In the ICGC dataset, the immune infiltration composition was like that in the TCGA dataset. T follicular helper cells (P < 0.05), regulatory T cells (P < 0.001), and M0 macrophages (P < 0.001) infiltrated the high-risk group significantly more than the low-risk group, whereas M2 macrophages (P < 0.05), activated mast cells (P < 0.01), and eosinophils (P < 0.05) infiltrated the high-risk group significantly lesser (Fig. 6d).
Fig. 6.

Immune infiltration levels in the high- and low-risk groups in the TCGA and ICGC datasets. (a,d) The differences in immune infiltrates between the two groups in the TCGA and ICGC datasets, respectively. (b,c)and (e,f) The infiltration proportion of 22 immune cells in the high- and low-risk groups in the TCGA and ICGC datasets, respectively. ns, P > 0.05; * P < 0.05; * * P < 0.01; * ** P < 0.001.
In the TCGA dataset, the proportion of M2 macrophages was significantly higher than that of any other immune cell subtype in both the groups (Fig. 6b,c). In the low-risk group, two types of T cells (resting memory CD4 T cells [11.21%] and CD8 T cells [12.22%]), macrophages (M2 macrophages [25.96%], M1 macrophages [5.18%], and M0 macrophages [8.38%]), and activated natural killer cells (5.52%) accounted for more than half (62.95%) of the total immune cell infiltrates (Fig. 6b). Meanwhile, the proportions of resting memory CD4 T cells (12.27%), CD8 T cells (12.44%), resting mast cells (7.12%), and M0 macrophages (6.32%) differed from those in the high-risk group (Fig. 6b,c).
In the ICGC dataset, the proportions of M2 macrophages and resting memory CD4 T cells in the tumor microenvironment were significantly higher than those of any other immune cell subtype in both the risk groups (Fig. 6e,f). M2 macrophages (24.62%), resting memory CD4 T cells (17.75%), M0 macrophages (9.47%), M1 macrophages (7.93%), and CD8 T cells (6.55%) constituted more than half of the tumor immune infiltrates in the low-risk group. M2 macrophages infiltrated the high-risk group more than the low-risk group. In the high-risk group, the proportions of M1 macrophages (6.79%), CD8 T cells (5.87%), and M0 macrophages (4.63%) differed from those in the low-risk group (Fig. 6e,f).
3.6. Differences in the immune state of high- and low-risk groups
We compared the stromal, immune, and ESTIMATE scores of the two groups to assess the presence of different infiltrating stromal/immune cells. High ESTIMATE scores and/or immune scores were associated with a higher survival rate. The stromal score significantly differed between the high- and low-risk groups in both the TCGA (P = 0.024) and ICGC (P = 0.001) datasets (Fig. 7a,d). The results slightly differed between the TCGA and ICGC datasets, possibly because of the different sample sources. The immune and ESTIMATE scores did not differ significantly between the two groups in the TCGA dataset, but they could visibly separate the two risk groups in the ICGC dataset. Immunotherapy has emerged as a new promising strategy to treat advanced HCC. Therefore, to explore the relationship between the five-gene signature panel and the response to immunotherapy, we compared the expression levels of common immune checkpoint genes and genes associated with T cell exhaustion between the two risk groups. Checkpoint genes, such as PD1, PD-L1, and CTLA-4, were notably upregulated in the high-risk group compared with the low-risk group (Fig. 8a–c). A similar trend was observed in the expression of genes associated with T cell exhaustion (TIM3 and ICOS; Fig. 8d–f). Logged fold change (logFC) of expression of check point genes between high risk score group vs. low risk score group are as follows: PD-1 (logFC = 1.56), CD274 (logFC = 0.95),CTLA4 (logFC = 1.15), HAVCR2 (logFC = 1.10), TIGIT (logFC = 1.13), and ICOS (logFC = 1.2). The check point genes all elevated in high risk score groups compared to low risk score group.
Fig. 7.

The assessment of stromal, immune, and ESTIMATE scores by the ESTIMATE method. (a–c) The stromal, immune, and ESTIMATE scores of the two risk groups in the TCGA dataset comprising hisk score samples (n = 178) and low risk samples (n = 179). (d–f) The stromal, immune, and ESTIMATE scores of the two risk groups in the ICGC dataset comprising high score samples (n = 111) and low risk samples (n = 112). Mann–Whitney U test was used to ascertain significant expression.
Fig. 8.

Comparative expression of immune checkpoints genes between different clusters. (a–f) Box plots visualizing the differential expression of immune checkpoints between the high- and low-risk groups. PDCD1 (PD-1): programmed cell death 1; CD274 (PD-L1): programmed death-ligand 1; CTLA4: cytotoxic T-lymphocyte-associated protein 4; TIM-3 (HAVCR2): T cell immunoglobulin mucin receptor 3; TIGIT: T cell immunoreceptor with Ig and ITIM domains; ICOS, inducible co-stimulatory molecule.
3.7. Expression of the five candidate genes and their individual prognostic ability
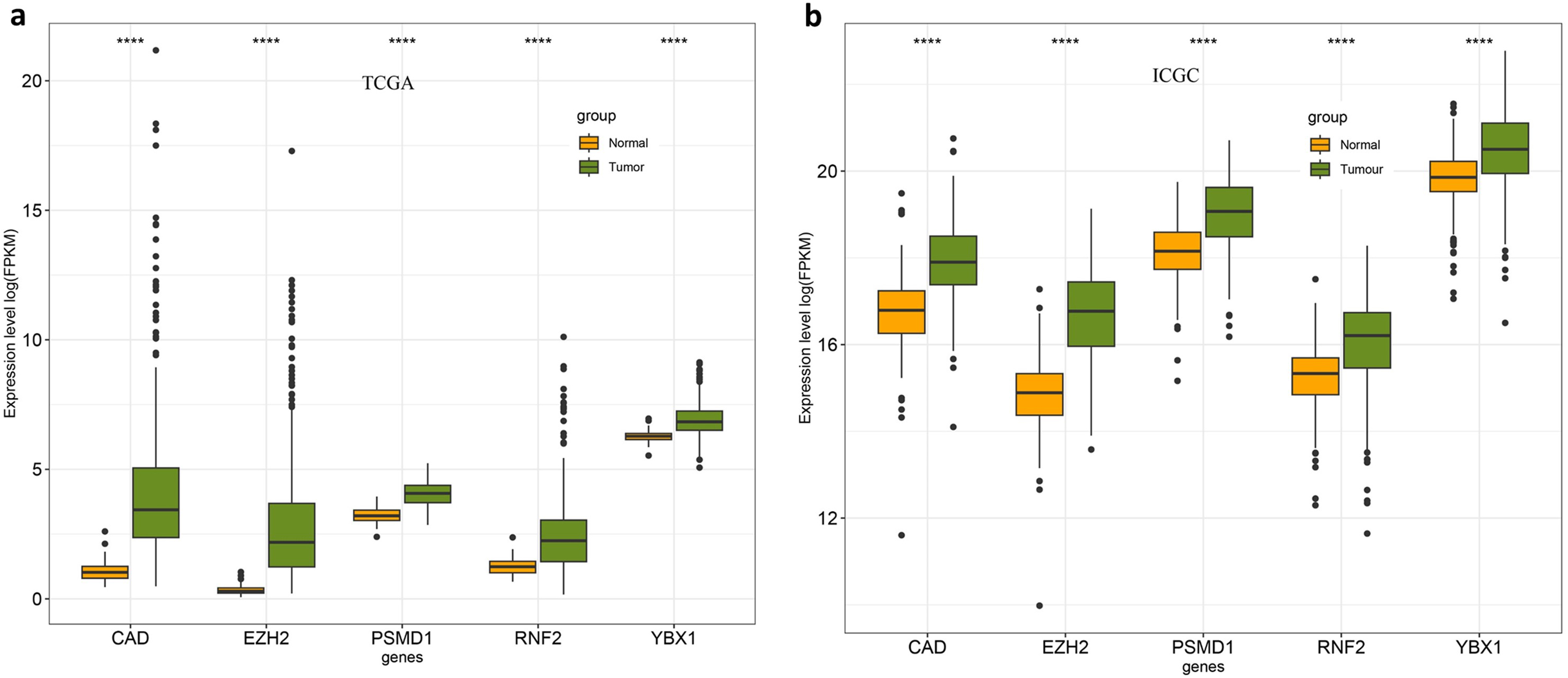
To obtain new insights into expression patterns, prognostic values, and mutations in the five-gene signature panel, we further used various online databases to explore the expression of RNF2, YBX1, EZH2, CAD, and PSMD1 based on RNA sequencing data from HCC patients. Firstly, we compared the transcriptional levels of the five genes in liver cancers using information from the Gene Expression Profiling Interactive Analysis datasets. The mRNA levels of the five genes were significantly higher in HCC than in normal samples (Fig. 9a). We also checked the expression of five signature genes in ICGC datasets. The result is consisting with the expression in TCGA (Fig. S3). Then, we assessed the prognostic values of the five genes in the context of HCC using Kaplan–Meier plots. OS was calculated as an outcome measure for cancer prognosis. We discovered that the increased mRNA levels of the five genes were correlated with lower survival rates (Fig. 9b–f). Both in TCGA and ICGC databases, the expression of five genes elevated in HCC samples. We added the results in Fig. S4.
Fig. 9.

The expression of YBX1, PSMD1, CAD, EZH2, and RNF2 and survival analysis in the TCGA dataset. (a) Expression of the five genes in the TCGA dataset comprising high risk score samples (n = 178) and low risk score samples (n = 179). Mann–Whitney U test was used to detect significant expression. * ** *, P < 0.0001; * ** , P < 0.001; * *, P < 0.01; * , P < 0.05. (b–f) Kaplan–Meier curves based on the five genes to determine the overall survival (OS) of HCC patients in the TCGA dataset.
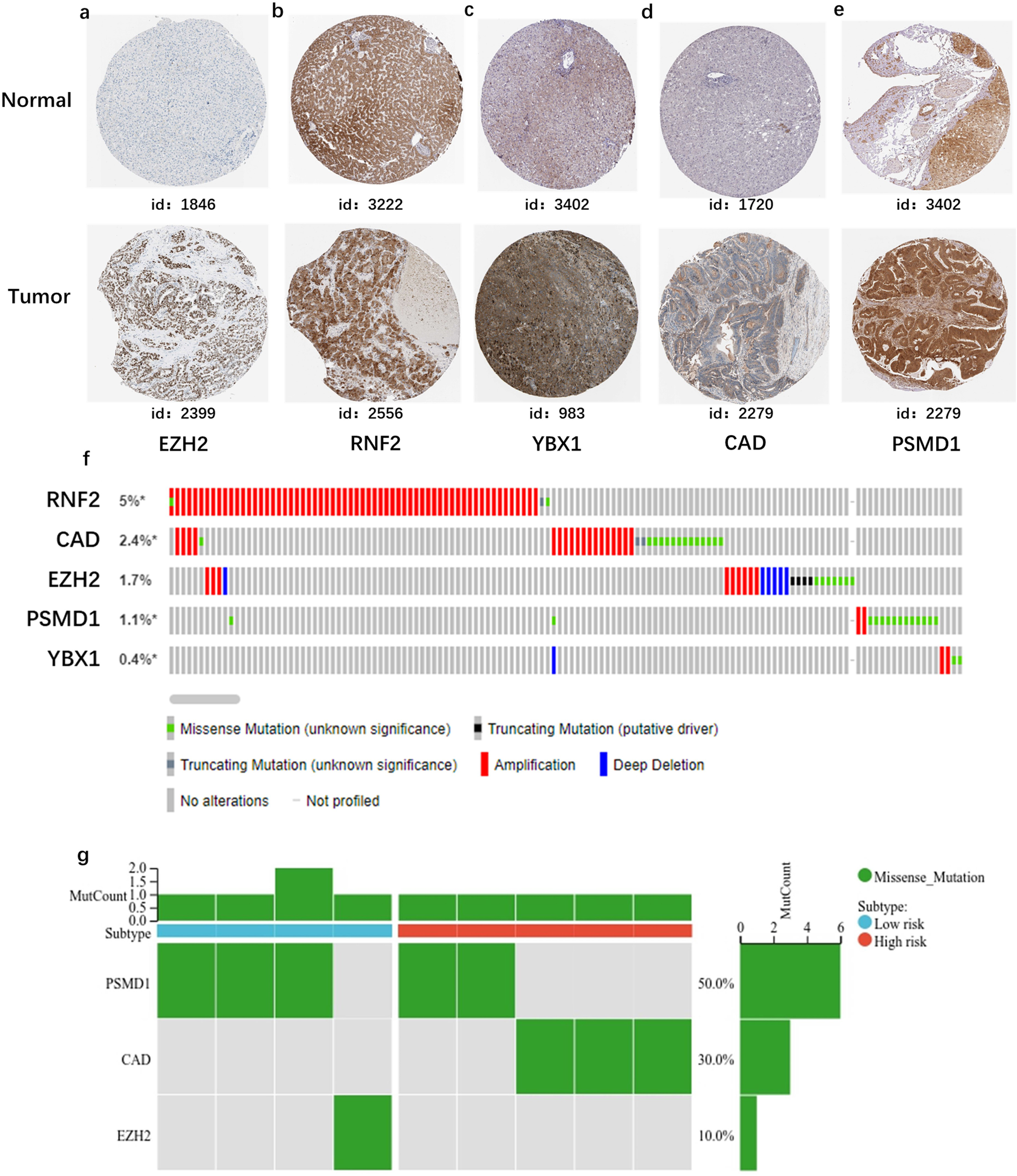
Furthermore, to confirm whether these five genes played an important role in liver carcinogenesis, we used data from two independent databases: immunohistochemical results from clinical samples in the HPA database and gene mutation analysis from the cBioPortal database. From the HPA data, we found that immunohistochemical sections showed that the proteins encoded by these genes were highly expressed in HCC patients (Fig. S1a, c–e). However, the expression of RNF2 only slightly elevated in HCC. (Fig. S1b). Data from cBioPortal also indicated that RNF2 and CAD exhibited the most common genetic variations (5% and 2.4%, respectively), and the most pronounced change was amplification of the mutations (Fig. S1f). Moreover, we compared the genetic variations in signature genes between the high- and low-risk groups. The mutation rates of these five genes did not differ between the two groups (Fig. S1g).
3.8. RT–PCR validation of hub genes
To verify the expression of the candidate genes, we collected and validated their expression profiles in 40 samples from an independent clinical cohort comprising tumor tissue samples and corresponding non-tumor tissue samples (Fig. 10, Table S3). All five genes were upregulated in tumor tissues compared with non-tumor tissues, and the difference was significant for EZH2 (P = 0.046), YBX1 (P = 0.038), and PSMD1 (P = 0.017).
Fig. 10.

Reverse transcription–PCR (RT–PCR) validation of the five genes in samples from other independent participants in our cohort. The vertical axis represents relative expression values normalized to the expression of actin beta (ACTB). The significance of differential expression between Normal (n = 40) and HCC (n = 40) samples were measured using Mann–Whitney U tests. (a) EZH2, (b) RNF2, (c) YBX1, (d) CAD, (e) PSMD1.
3.9. Potential drug candidates for the five hub genes
Co-expression analysis showed that RNF2, YBX1, EZH2, CAD, and PSMD1 strongly correlated with each other in both the TCGA and ICGC datasets (Fig. 11a,b). Furthermore, we used several gene–gene regulation databases, including STRING, IntAct, MINT, and PINA 3.0, to build the regulation network of the five genes and analyze their interactions with other closely related proteins. These genes were strictly screened by edge selection criteria with physical association or experimentally validated to only include those with high confidence scores. The constructed regulation network contained 315 nodes and 359 edges (Fig. 11c). Using CytoHubba and CytoNCA, we obtained a core subnetwork composed of the five candidate genes and their first-neighbor genes. Since the two different algorithms yielded different predictions, we considered the intersection between the two subnetworks. Finally, the five candidate genes along with 15 closely related hub genes were obtained.
Fig. 11.

Correlation within the five-gene signature panel. (a) Correlation among the five genes in the ICGC dataset. (b) Correlation among the five genes in the TCGA dataset. Size of the bubbles: correlation coefficients. The bigger size of the bubble, the higher value of the correlation coefficients. * ** *, P < 0.0001.(c) Regulation network analysis of the five genes. Red dots: RNF2, YBX1, EZH2, CAD, and PSMD1; blue dots: genes from the regulation network that were strictly screened by edge selection criteria from multiple databases. (d) Core subnetwork composed of the five candidate genes and 15 first-neighbor genes obtained using CytoHubba and CytoNCA, separately. (e) Venn analysis of genes intersecting between the two subnetworks.
3.10. Interactions between drug candidates and the gene signature
The CMap database suggested drug candidate molecules for the submitted hub proteins. From CMap, we identified 647 repurposed drugs with the highest negative scores and a cutoff raw score of 0.6 (Table S4). The candidate drugs were further screened by assessing the correlation between the expression of the five genes and the drug response predicted using CellMiner. Resultantly, 40 FDA-approved drugs were obtained (Table S5). The results from CMap and CellMiner were subjected to Venn analysis to identify four potential FDA-approved drugs for the gene signature. Finally, the four gene–drug pairs were identified: EZH2–AT-13387 (Fig. 12a), EZH2–fenretinide, YBX1–carmustine, and YBX1–KU-55933 (Fig. 12d).
Fig. 12.

The correlation between four drug–protein pairs and molecular docking of the repurposed drugs onto the potential target proteins. (a,d): Correlation between protein expression and predicted drug response. The figures show the best docking conformation between the protein and the drug (b,e): The three-dimensional interaction diagrams of the best docking conformation for EZH2–AT-13387 and YBX1–KU-55933, respectively. (c,f): Two-dimensional molecular structure diagram of EZH2–AT-13387 and YBX1–KU-55933, respectively.
The possible interactions between the candidate drugs and the proteins encoded by these genes were studied using molecular docking. EZH2–AT-13387 (onalespib) and YBX1–KU-55933 were most likely to interact through protein–molecule interactions (Fig. 12). EZH2 (4w2r) combines well with AT-13387 (−10.5 kcal/mol), forming binding pockets. The residues TRPE:89 and GLNE:86 of EZH2 (4w2r) form pi–pi stacking bonds and hydrogen bonds, respectively, with AT-13387, which stabilize the interaction. Pi–alkyl bonds, alkyl bonds, hydrocarbon bonds, and extensive van der Waals interactions are also involved. YBX1 (1h95) also interacts well with KU-55933 (−8.2 kcal/mol), and the residue PHEA:16 of KU-55933 forms pi–pi stacking bonds with YBX1, which stabilizes the interaction. Pi–anion bonds, pi–alkyl bonds, alkyl bonds, and van der Waals forces also play a role in their binding. (Fig. 12 b,c and d–f).
EZH2 and fenretinide display unfavorable bonding effects (Fig. S2a). Based on the docking score, the interaction between YBX1 and carmustine is weak (−3.8 kcal/mol), with only van der Waals forces and alkyl bonds existing between the two moieties (Fig. S2b).
4. Discussion
The management of HCC is formidably challenged by the poor prognosis and high mortality of the disease, resulting from insensitive diagnosis and limited treatment strategies [18]. New prognostic biomarkers for precise personalized treatment are urgently needed. The liver is the largest digestive gland of the human body. It secretes bile, performs phagocytosis, and contributes to the immune response. Recent studies have shown that an imbalance in the immune microenvironment and epigenetic disorders can both intimately influence the development of HCC. Therefore, we aimed to develop a prognostic signature based on IRGs and EPGs that could predict OS in HCC patients to provide personalized diagnosis and treatment in the future.
Previous studies on this topic have lacked systematic and dynamic exploration. They mainly focused on single IRGs or EPGs, which were identified based on their differential expression in tumor tissues compared with healthy samples. In this study, we applied the DNB algorithm to identify DNBs and the critical state of HCC progression based on the correlation between IRGs and EPGs and the changes in their expression. IRGs and EPGs are related to each other in terms of gene function, and both mostly perform upstream regulatory roles. In this study, we first collated a list of IRGs and EPGs from typical immune and epigenetics databases. The selected dataset GSE114564 contains transcriptome data of samples ranging from normal to advanced HCC with continuously dynamic characteristics. We subjected this dataset to DNB analysis—an advanced bioinformatic algorithm based on disease–gene specificity that can capture the underlying key regulatory biomolecules—to identify potential IEDs. LASSO-Cox analysis, Kaplan–Meier survival plots, and ROC curves were used to successfully construct a prognostic signature comprising five genes (EZH2, RNF2, YBX1, CAD, and PSMD1). Risk scores based on the prognostic signature were combined with TNM staging to establish nomograms for predicting patient survival. The prognostic signature and the nomograms accurately identified high-risk patients and predicted their prognosis. Therefore, we propose that this novel IED-related signature can serve as a potential prognostic biomarker for HCC.
Recently, some other HCC biomarkers with good prognostic ability have emerged. Yang et al. [23] classified high-risk patients and predicted OS (AUC = 0.798 for 1 year, 0.748 for 3 years, and 0.721 for 5 years). Tang et al. developed a nine-gene signature with AUC values of 0.79, 0.71, and 0.71 for 1-, 3-, and 5-year OS, respectively. These results align with those reported in this study. However, they focus on different mechanisms involved in HCC pathogenesis, such as iron death [24], cellular senescence [25], and angiogenesis [26]. The liver is the largest immune organ in the human body. Immune regulation and epigenetic changes play a crucial role in the progression of HCC. DNB analysis is an effective method to identify the genes regulating the critical transition in the progression of a disease, helping us identify the signature genes in the development of that disease. In addition, unlike the previous studies, we have focused on potential FDA-approved drug candidates that target these signature genes, proposing their potential applications in HCC.
To explore the complexity of the HCC microenvironment, we used the risk score to divide cases from the TCGA and ICGC datasets into high/low-risk groups, and evaluated the differences in immune cell infiltrates between them. We observed highly abundant and abnormal infiltration of the whole macrophage population, resting memory CD4 T cells, and activated natural killer cells. M2 macrophages, which are activated by interleukin-4 (IL-4) and IL-13, often accelerate cancer progression [27]. A large proportion of tumor-infiltrated M2 macrophages were found in both the groups, which further increased (25.96–26.15% in TCGA and 24.62–28.02% in ICGC) with an increase in the risk grade of patients. During cancer cell invasion, M0 macrophages are easily transformed in the tumor microenvironment into the M2 subtype [28], which secretes the inhibitory cytokines IL-10 and transforming growth factor-β and promotes inflammation [29]. In the tumor immune microenvironment, the increased proportion of macrophages may be a significant driver of liver carcinogenesis.
Higher expression of PD-L1 significantly and independently correlated with poor survival in HCC patients [30]. In this study, the high-risk group had low stromal scores (Fig. 7a, d) in both TCGA and ICGC dataset, suggesting a higher tumor purity and a dismal prognosis, which is consistent with a previous study [31]. However, in ICGC, the immune score and Estimate score did not shown significantly difference, which may due to the different patients source of two datasets. Patients with higher expression of immune checkpoints (PD-1, PD-L1, and CTLA-4) may be suitable for treatment with anti-immune checkpoint antibodies, such as nivolumab (anti-PD1) and ipilimumab (anti-CTLA-4) [32]. TIM3 is expressed in various types of T cells and is a candidate target for tumor immunotherapy. High expression of TIM3 and PD-1 in T cells and the enrichment of regulatory T cells suggest that TIM3 may be one of the signs of PD-1 blocking antibody resistance [33]. In preclinical studies, TIM3 and PD-1 inhibitors elicited similar effects. In this study, TIM3 was found to be highly expressed; therefore, the high-risk group may benefit from TIM3-targeted anti-tumor immunity (Fig. 8).
We noticed that the data analysis results slightly differed between the TCGA and ICGC datasets, possibly because of the different sample sources. The ICGC database is an international oncogene collaboration, housing data from dozens of cancer samples from different countries and regions. The TCGA database, on the other hand, is affiliated to the National Cancer Institute in the United States, and contains data on samples only from the United States. In bioinformatic analysis, ICGC datasets are often used for validation. To address this issue, experimental validation is necessary. We validated the mRNA expression of the five signature genes in an independent cohort of HCC patients using RT–PCR. Four out of five genes (except RFN2) were found to be significantly upregulated, corroborating the previous result from TCGA, and indicating that these genes may be dominantly expressed in tumor cells (Figure S1). However, the expression of RNF2 did not show statistical difference, which requires larger sample size for validation in the future. The expression of several signature genes fluctuated not only in tumor cells but also in immune cells. EZH2 was reported to be highly overexpressed in the subset of tumor-associated macrophages (macrophage.3) in glioblastoma [33]. Single-cell sequencing revealed that EZH2 also regulated antitumoral responses in natural killer cells [34]. Takeuchi et al. found that YBX-1 was poorly expressed in T lymphocytes compared with cancer tissues, indicating that the suppression of YBX1 induces the proteolysis of signal transducer and activator of transcription 3 and sensitizes renal cancer to interferon-α [35]. Notably, the prognostic signature genes were significantly related to the abnormal infiltration of macrophages. Among them, EZH2-mediated H3K27me3 was shown to induce the transformation of hepatic macrophages from an M1 to an M2 phenotype [34]. High expression of YBX1 was related to M2 macrophage infiltration and T cell failure in the tumor microenvironment of breast cancer [35]. RNF2 induced persistent tumor rejection and established immune memory by enhancing the infiltration and activation of natural killer and CD4 T cells [36].
EZH2 (enhancer of zeste homolog 2) is a polycomb gene and an epigenetic regulator that inhibits transcription. EZH2 plays an important role in humoral and cell-mediated immunity [34], [37]. In the immune microenvironment of liver fibrosis, EZH2 regulates the enhancer-mediated transcription factor KLF14 of H3K27me3 to aggravate liver fibrosis [38]. Inhibition of EZH2 disrupts myofibroblasts and causes remission of liver fibrosis. It was also demonstrated to alleviate atrial fibrosis caused by EZH2-activating mutations in an experimental as well as clinical setting [39]. EZH2 promotes anticancer immune responses by regulating Th1 chemokine expression, myeloid-derived suppressor cells, or CD8+ T cell infiltration [40], [41]. Multiple studies have proposed EZH2 to be a potential target in HCC [12], [42], which aligns with the results from this study.
YBX1 is a transcription factor that belongs to the cold-shock protein superfamily and is associated with various cancers, such as breast cancer, renal cell carcinoma, and HCC [43], [44]. YBX1 may importantly activate mammalian target of rapamycin signaling by mediating an autoactivation pathway that impairs proteostasis in glioblastoma [45]. Studies have shown that long noncoding RNAs can regulate HCC progression by targeting YBX1 [46].
CAD (carbamoyl phosphate synthase 2, aspartate transcarbamylase, and dihydroorotase) is a multifunctional protein. Dysregulation of CAD-related pathways or CAD mutations can lead to cancer, neurological diseases, and inherited metabolic diseases [47]. Using online cancer datasets, Wang et al. [48] found that pyrimidine metabolic signaling was disrupted in various cancers, and CAD was enriched in cancer types with poor clinical outcomes, such as HCC, breast cancer, and colon cancer. Fu et al. [49] found that CAD-mediated pyrimidine biosynthesis was inhibited by arginine anabolic enzymes via VIP/VIPR1 signaling, suggesting that CAD was involved in the metabolic regulation of HCC. Besides, increased CAD phosphorylation, which stimulates tumorigenesis, has been observed in several types of cancers [50].
PSMD1 encodes a subunit of the 19 S regulatory complex of the 26 S proteasome, making it a component of the ubiquitin–proteasome system. It plays an important role in neutralizing damaged and misfolded proteins. PSMD1 was found upregulated in anaplastic thyroid carcinoma and breast cancer tissues, and it may serve as a novel potential therapeutic target [51], [52]. Rubio et al. [53] found significant differences in the mRNA and protein expression of PSMD1 across various cancers when they analyzed data from TCGA and Clinical Proteomic Tumor Analysis Consortium in UALCAN. They also established that PSMD1 and PSMD3 play oncogenic roles in chronic myeloid leukemia by stabilizing nuclear factor-kappa B [54].
RNF2 (ring finger protein 2) is a well-known E3 ligase that plays oncogenic roles in various cancers, including colon cancer [45], breast cancer [55], and gastric cancer [47]. It is one of the core components of polycomb repressive complex 1, and may play an important role in multiple stages of carcinogenesis, such as normal epithelial cell immortalization, early malignant transformation, and stem cell self‐renewal. Qu et al. [56] reported RNF2 mRNA amplification in 19.7% of the HCC samples in their study.
Finally, we combined the results from CMap and CellMiner to select specific candidate drugs. We validated two FDA-approved market drugs (AT-13387 and KU-55933) for HCC treatment using target protein–drug molecular docking simulations. Drug target binding affinity scores (less than −8.0 kcal/mol) indicated the potential of the two drugs in HCC treatment. These results may be a useful resource for the prevention and early detection of HCC.
The advantage of this study lies in the fact that we used the DNB algorithm to build a prognostic gene signature that divides patients into two risk groups with different immune microenvironment signatures. An accurate measure of the degree and composition of immune infiltrates can guide future clinical immunotherapy. Although the five-gene signature achieved independent prognosis, this study has certain limitations as well. First, it mainly focused on bioinformatic analysis. Although the expression level of signature genes in HCC was verified by immunohistochemistry in HPA and RT–PCR, a mechanistic verification is lacking. Comparing PCR results between patients with better or worse clinical outcomes will make more sense. This requires a multicenter study designed like a prospective clinical trial. Years of follow-up data and patient samples are also needed to verify the gene expression in HCC patients with better or worse clinical outcomes. In the future, we will investigate the specific mechanisms of these genes and design prospective clinical trials to accomplish the task of sample collection.
5. Conclusion
In the current study, we applied the DNB algorithm to evaluate IRGs, EPGs, and their closely related neighbor genes in HCC samples. Using multiple statistical methods, five candidate genes (RNF2, YBX1, EZH2, CAD, and PSMD1) were identified and built into a signature panel with great prognostic ability. The expression of these genes was also validated in an independent patient cohort. This panel was used to establish two risk groups, which confirmed its prognostic ability. Immune checkpoint genes were upregulated in the high-risk group compared with the low-risk group, which indicates possible association of the panel and the response to HCC immunotherapy. It provided a new strategy for classifying HCC patients. These findings may guide the development of precise targeted chemotherapeutic drugs and personalized therapeutic strategies.
CRediT authorship contribution statement
Yuting Hu: Methodology, Software, Resources, Data curation, Writing – original draft. Xingli Zhang: Methodology, Software. Qingya Li: Resources. Qianmei Zhou: Validation. Dongdong Fang: Conceptualization, Resources. Yiyu Lu: Conceptualization, Methodology, Resources, Data curation, Writing – original draft. All authors have read and agreed to the published version of the manuscript.
Declaration of Competing Interest
The authors declared that they have no conflicts of interest to this work. We declare that we do not have any commercial or associative interest that represents a conflict of interest in connection with the work submitted.
Acknowledgements
This study was funded by the National Natural Science Foundation of China (82274183, 82274157, 81503478; 81603607); the Shanghai Science and Technology Committee (STCSM) Science and Technology Innovation Program (No.20ZR1453700); Shanghai Municipal Health Commission's special clinical research project in the health industry (202240243); Henan Natural Science Foundation of China (162300410189). We also thank Fan Jian and Zhu Jian (Qidong Hospital of Nantong University) to provide clinical samples.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.csbj.2023.09.030.
Contributor Information
Dongdong Fang, Email: fangdd1200@shutcm.edu.cn.
Yiyu Lu, Email: yiyulu@shutcm.edu.cn.
Appendix A. Supplementary material
Supplementary material.
.
Supplementary material.
.
Supplementary material.
.
Supplementary material.
.
Supplementary material.
.
Supplementary material.
.
Supplementary material.
.
Figure S1. Protein levels of five genes in HCC and normal tissues by the Human Protein Atlas (HPA) database and mutation analysis. (a) EZH2 (patient id:1846 and 2399), (b) RNF2 (patient id:3222 and 2556), (c) YBX1 (patient id:3402 and 983), (d) CAD (patient id:1720 and 2279), (e) PSMD1 (patient id:3402 and 2279) (f) Mutation frequency of candidate genes based on data from the cBioPortal database. (g) Somatic mutations of signature genes in high and low risk score groups.
{kind=link}
.
Figure S2. Molecular docking results of YBX1- Carmustine and EZH2- Fenretinide. (a) YBX1 (1h95) -Carmustine: weak interaction (−3.8 kcal/mol). (b) EZH2 (4w2r) and Fenretinide has unfavorable bonding effect (red bond).
{kind=link}
.
Figure S3. The expression of YBX1, PSMD1, CAD, EZH2, and RNF2 and survival analysis in the ICGC dataset. (a) Expression of the five genes in the TCGA dataset comprising high risk score samples (n = 111) and low risk score samples (n = 112). Mann–Whitney U test was used to detect significant expression. * ** *, P < 0.0001; * ** , P < 0.001; * *, P < 0.01; * , P < 0.05.
{kind=link}
.
Figure S4. The expression of YBX1, PSMD1, CAD, EZH2, and RNF2 and survival analysis in the TCGA and ICGC dataset. (a) Expression of the five genes in the TCGA dataset comprising Normal samples (n = 50) and Tumor samples (n = 357). (b) Expression of the five genes in the ICGC dataset comprising Normal samples (n = 223) and Tumor samples (n = 223). Mann–Whitney U test was used to detect significant expression. * ** *, P < 0.0001; * ** , P < 0.001; * *, P < 0.01; * , P < 0.05.
{kind=link}
.
References
- 1.Sung H., Ferlay J., Siegel R.L., et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin. 2021;71(3):209–249. doi: 10.3322/caac.21660. [DOI] [PubMed] [Google Scholar]
- 2.Wang J., Ha J., Lopez A., et al. Medicaid and uninsured hepatocellular carcinoma patients have more advanced tumor stage and are less likely to receive treatment. J Clin Gastroenterol. 2018;52(5):437–443. doi: 10.1097/MCG.0000000000000859. [DOI] [PubMed] [Google Scholar]
- 3.Gunasekaran G., Bekki Y., Lourdusamy V., et al. Surgical treatments of hepatobiliary cancers. Hepatology. 2021;73(1):128–136. doi: 10.1002/hep.31325. [DOI] [PubMed] [Google Scholar]
- 4.Zhang X., Wang Z., Tang W., et al. Ultrasensitive and affordable assay for early detection of primary liver cancer using plasma cell-free DNA fragmentomics. Hepatology. 2022;76(2):317–329. doi: 10.1002/hep.32308. [DOI] [PubMed] [Google Scholar]
- 5.Zanotti S., Boot G.F., Coto-Llerena M., et al. The role of chronic liver diseases in the emergence and recurrence of hepatocellular carcinoma: an omics perspective. Front Med. 2022;9 doi: 10.3389/fmed.2022.888850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muhammed A., Fulgenzi C.A.M., Dharmapuri S., et al. The systemic inflammatory response identifies patients with adverse clinical outcome from immunotherapy in hepatocellular carcinoma. Cancers. 2021;14(1) doi: 10.3390/cancers14010186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yau T., Kang Y.K., Kim T.Y., et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: the CheckMate 040 randomized clinical trial. JAMA Oncol. 2020;6(11) doi: 10.1001/jamaoncol.2020.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J., Chen X., Li T., et al. Identification of chromatin organization-related gene signature for hepatocellular carcinoma prognosis and predicting immunotherapy response. Int Immunopharmacol. 2022;109 doi: 10.1016/j.intimp.2022.108866. [DOI] [PubMed] [Google Scholar]
- 9.Domovitz T., Gal-Tanamy M. Tracking down the epigenetic footprint of HCV-induced hepatocarcinogenesis. J Clin Med. 2021;10(3) doi: 10.3390/jcm10030551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee J.E., Kim M.Y. Cancer epigenetics: Past, present and future. Semin Cancer Biol. 2022;83:4–14. doi: 10.1016/j.semcancer.2021.03.025. [DOI] [PubMed] [Google Scholar]
- 11.Wang H.C., Chen C.W., Yang C.L., et al. Tumor-associated macrophages promote epigenetic silencing of gelsolin through DNA methyltransferase 1 in gastric cancer cells. Cancer Immunol Res. 2017;5(10):885–897. doi: 10.1158/2326-6066.CIR-16-0295. [DOI] [PubMed] [Google Scholar]
- 12.Salani F., Latarani M., Casadei-Gardini A., et al. Predictive significance of circulating histones in hepatocellular carcinoma patients treated with sorafenib. Epigenomics. 2022;14(9):507–517. doi: 10.2217/epi-2021-0383. [DOI] [PubMed] [Google Scholar]
- 13.Pal D., Sur S., Roy R., et al. Hypomethylation of LIMD1 and P16 by downregulation of DNMT1 results in restriction of liver carcinogenesis by amarogentin treatment. J Biosci. 2021;46 [PubMed] [Google Scholar]
- 14.Liu Z.J., Huang Y., Wei L., et al. Combination of LINE-1 hypomethylation and RASSF1A promoter hypermethylation in serum DNA is a non-invasion prognostic biomarker for early recurrence of hepatocellular carcinoma after curative resection. Neoplasma. 2017;64(5):795–802. doi: 10.4149/neo_2017_519. [DOI] [PubMed] [Google Scholar]
- 15.Guo Y., Zhao Y.R., Liu H., et al. EHMT2 promotes the pathogenesis of hepatocellular carcinoma by epigenetically silencing APC expression. Cell Biosci. 2021;11(1):152. doi: 10.1186/s13578-021-00663-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eun J.W., Jang J.W., Yang H.D., et al. Serum Proteins, HMMR, NXPH4, PITX1 and THBS4; a panel of biomarkers for early diagnosis of hepatocellular carcinoma. J Clin Med. 2022;11(8):11. doi: 10.3390/jcm11082128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Son J.A., Ahn H.R., You D., et al. Novel gene signatures as prognostic biomarkers for predicting the recurrence of hepatocellular carcinoma. Cancers. 2022;14(4) doi: 10.3390/cancers14040865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Y., Liu C., Qi L., et al. Diagnosis of Pre-HCC disease by hepatobiliary-specific contrast-enhanced magnetic resonance imaging: a review. Dig Dis Sci. 2020;65(9):2492–2502. doi: 10.1007/s10620-019-05981-0. [DOI] [PubMed] [Google Scholar]
- 19.Shannon P., Markiel A., Ozier O., et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abdel-Rahman O. Assessment of the discriminating value of the 8th AJCC stage grouping for hepatocellular carcinoma. HPB: J Int Hepato Pancreato Biliary Assoc. 2018;20(1):41–48. doi: 10.1016/j.hpb.2017.08.017. [DOI] [PubMed] [Google Scholar]
- 21.Chang W.H., Forde D., Lai A.G. A novel signature derived from immunoregulatory and hypoxia genes predicts prognosis in liver and five other cancers. J Transl Med. 2019;17(1) doi: 10.1186/s12967-019-1775-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xiang X.H., Yang L., Zhang X., et al. Seven-senescence-associated gene signature predicts overall survival for Asian patients with hepatocellular carcinoma. World J Gastroenterol. 2019;25(14):1715–1728. doi: 10.3748/wjg.v25.i14.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang Z., Zi Q., Xu K., et al. Development of a macrophages-related 4-gene signature and nomogram for the overall survival prediction of hepatocellular carcinoma based on WGCNA and LASSO algorithm. Int Immunopharmacol. 2021;90 doi: 10.1016/j.intimp.2020.107238. [DOI] [PubMed] [Google Scholar]
- 24.Han F., Cao D., Zhu X., et al. Construction and validation of a prognostic model for hepatocellular carcinoma: Inflammatory ferroptosis and mitochondrial metabolism indicate a poor prognosis. Front Oncol. 2022;12 doi: 10.3389/fonc.2022.972434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang Y., Guo C., Chen C., et al. Characterization of cellular senescence patterns predicts the prognosis and therapeutic response of hepatocellular carcinoma. Front Mol Biosci. 2022;9 doi: 10.3389/fmolb.2022.1100285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang B., Zhang X., Yang X., et al. Construction and validation of an angiogenesis-related scoring model to predict prognosis, tumor immune microenvironment and therapeutic response in hepatocellular carcinoma. Front Immunol. 2022;13 doi: 10.3389/fimmu.2022.1013248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schmieder A., Michel J., Schönhaar K., et al. Differentiation and gene expression profile of tumor-associated macrophages. Semin Cancer Biol. 2012;22(4):289–297. doi: 10.1016/j.semcancer.2012.02.002. [DOI] [PubMed] [Google Scholar]
- 28.Zhang L., Wang Z., Li M., et al. HCG18 participates in vascular invasion of hepatocellular carcinoma by regulating macrophages and tumor stem cells. Front Cell Dev Biol. 2021;9 doi: 10.3389/fcell.2021.707073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang S., Liu G., Li Y., et al. Metabolic reprogramming induces macrophage polarization in the tumor microenvironment. Front Immunol. 2022;13 doi: 10.3389/fimmu.2022.840029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma L.J., Feng F.L., Dong L.Q., et al. Clinical significance of PD-1/PD-Ls gene amplification and overexpression in patients with hepatocellular carcinoma. Theranostics. 2018;8(20):5690–5702. doi: 10.7150/thno.28742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurebayashi Y., Ojima H., Tsujikawa H., et al. Landscape of immune microenvironment in hepatocellular carcinoma and its additional impact on histological and molecular classification. Hepatology. 2018;68(3):1025–1041. doi: 10.1002/hep.29904. [DOI] [PubMed] [Google Scholar]
- 32.Hu B., Yang X.B., Sang X.T. Liver graft rejection following immune checkpoint inhibitors treatment: a review. Med Oncol. 2019;36(11) doi: 10.1007/s12032-019-1316-7. [DOI] [PubMed] [Google Scholar]
- 33.Yang R., Sun L., Li C.F., et al. Galectin-9 interacts with PD-1 and TIM-3 to regulate T cell death and is a target for cancer immunotherapy. Nat Commun. 2021;12(1) doi: 10.1038/s41467-021-21099-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chi G., Pei J.H., Li X.Q. EZH2-mediated H3K27me3 promotes autoimmune hepatitis progression by regulating macrophage polarization. Int Immunopharmacol. 2022;106 doi: 10.1016/j.intimp.2022.108612. [DOI] [PubMed] [Google Scholar]
- 35.Lv Z., Xue C., Zhang L., et al. Elevated mRNA level of Y-box binding protein 1 indicates unfavorable prognosis correlated with macrophage infiltration and T cell exhaustion in luminal breast cancer. Cancer Manag Res. 2021;13:6411–6428. doi: 10.2147/CMAR.S311650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang Z., Luo L., Xing C., et al. RNF2 ablation reprograms the tumor-immune microenvironment and stimulates durable NK and CD4(+) T-cell-dependent antitumor immunity. Nat Cancer. 2021;2(10):1018–1038. doi: 10.1038/s43018-021-00263-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sun S., Yu F., Xu D., et al. EZH2, a prominent orchestrator of genetic and epigenetic regulation of solid tumor microenvironment and immunotherapy. Biochim Et Biophys Acta Rev Cancer. 2022;1877(2) doi: 10.1016/j.bbcan.2022.188700. [DOI] [PubMed] [Google Scholar]
- 38.Du Z., Liu M., Wang Z., et al. EZH2-mediated inhibition of KLF14 expression promotes HSCs activation and liver fibrosis by downregulating PPARγ. Cell Prolif. 2021;54(7) doi: 10.1111/cpr.13072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Song S., Zhang R., Mo B., et al. EZH2 as a novel therapeutic target for atrial fibrosis and atrial fibrillation. J Mol Cell Cardiol. 2019;135:119–133. doi: 10.1016/j.yjmcc.2019.08.003. [DOI] [PubMed] [Google Scholar]
- 40.Peng D., Kryczek I., Nagarsheth N., et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature. 2015;527(7577):249–253. doi: 10.1038/nature15520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ennishi D., Takata K., Béguelin W., et al. Molecular and genetic characterization of MHC deficiency identifies EZH2 as therapeutic target for enhancing immune recognition. Cancer Discov. 2019;9(4):546–563. doi: 10.1158/2159-8290.CD-18-1090. [DOI] [PubMed] [Google Scholar]
- 42.Yao F., Zhan Y., Li C., et al. Single-cell RNA sequencing reveals the role of phosphorylation-related genes in hepatocellular carcinoma stem cells. Front Cell Dev Biol. 2021;9 doi: 10.3389/fcell.2021.734287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y., Su J., Wang Y., et al. The interaction of YBX1 with G3BP1 promotes renal cell carcinoma cell metastasis via YBX1/G3BP1-SPP1- NF-κB signaling axis. J Exp Clin Cancer Res: CR. 2019;38(1):386. doi: 10.1186/s13046-019-1347-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiao L., Zhou Z., Li W., et al. Chromobox homolog 8 (CBX8) interacts with Y-Box binding protein 1 (YBX1) to promote cellular proliferation in hepatocellular carcinoma cells. Aging. 2019;11(17):7123–7149. doi: 10.18632/aging.102241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J., Ouyang X., Zhou Z., et al. RNF2 promotes the progression of colon cancer by regulating ubiquitination and degradation of IRF4. Biochim Et Biophys Acta Mol Cell Res. 2022;1869(1) doi: 10.1016/j.bbamcr.2021.119162. [DOI] [PubMed] [Google Scholar]
- 46.Zhao X., Liu Y., Yu S. Long noncoding RNA AWPPH promotes hepatocellular carcinoma progression through YBX1 and serves as a prognostic biomarker. Biochim Et Biophys Acta Mol basis Dis. 2017;1863(7):1805–1816. doi: 10.1016/j.bbadis.2017.04.014. [DOI] [PubMed] [Google Scholar]
- 47.Lakshmi Ch N.P., Sivagnanam A., Raja S., et al. Molecular basis for RASSF10/NPM/RNF2 feedback cascade-mediated regulation of gastric cancer cell proliferation. J Biol Chem. 2021;297(2) doi: 10.1016/j.jbc.2021.100935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang H., Wang X., Xu L., et al. High expression levels of pyrimidine metabolic rate-limiting enzymes are adverse prognostic factors in lung adenocarcinoma: a study based on The Cancer Genome Atlas and Gene Expression Omnibus datasets. Purinergic Signal. 2020;16(3):347–366. doi: 10.1007/s11302-020-09711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fu Y., Liu S., Rodrigues R.M., et al. Activation of VIPR1 suppresses hepatocellular carcinoma progression by regulating arginine and pyrimidine metabolism. Int J Biol Sci. 2022;18(11):4341–4356. doi: 10.7150/ijbs.71134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee J.S., Adler L., Karathia H., et al. Urea cycle dysregulation generates clinically relevant genomic and biochemical signatures. Cell. 2018;174(6):1559–1570. doi: 10.1016/j.cell.2018.07.019. e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jonker P.K., van Dam G.M., Oosting S.F., et al. Identification of novel therapeutic targets in anaplastic thyroid carcinoma using functional genomic mRNA-profiling: Paving the way for new avenues? Surgery. 2017;161(1):202–211. doi: 10.1016/j.surg.2016.06.064. [DOI] [PubMed] [Google Scholar]
- 52.Xiong W., Wang W., Huang H., et al. Prognostic significance of PSMD1 expression in patients with gastric cancer. J Cancer. 2019;10(18):4357–4367. doi: 10.7150/jca.31543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rubio A.J., Bencomo-Alvarez A.E., Young J.E., et al. 26S proteasome Non-ATPase regulatory subunits 1 (PSMD1) and 3 (PSMD3) as putative targets for cancer prognosis and therapy. Cells. 2021;10(9) doi: 10.3390/cells10092390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bencomo-Alvarez A.E., Rubio A.J., Olivas I.M., et al. Proteasome 26S subunit, non-ATPases 1 (PSMD1) and 3 (PSMD3), play an oncogenic role in chronic myeloid leukemia by stabilizing nuclear factor-kappa B. Oncogene. 2021;40(15):2697–2710. doi: 10.1038/s41388-021-01732-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Penugurti V., Khumukcham S.S., Padala C., et al. HPIP protooncogene differentially regulates metabolic adaptation and cell fate in breast cancer cells under glucose stress via AMPK and RNF2 dependent pathways. Cancer Lett. 2021;518:243–255. doi: 10.1016/j.canlet.2021.07.027. [DOI] [PubMed] [Google Scholar]
- 56.Qu C., Qu Y. Down-regulation of salt-inducible kinase 1 (SIK1) is mediated by RNF2 in hepatocarcinogenesis. Oncotarget. 2017;8(2):3144–3155. doi: 10.18632/oncotarget.13673. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material.
Supplementary material.
Supplementary material.
Supplementary material.
Supplementary material.
Supplementary material.
Supplementary material.
Figure S1. Protein levels of five genes in HCC and normal tissues by the Human Protein Atlas (HPA) database and mutation analysis. (a) EZH2 (patient id:1846 and 2399), (b) RNF2 (patient id:3222 and 2556), (c) YBX1 (patient id:3402 and 983), (d) CAD (patient id:1720 and 2279), (e) PSMD1 (patient id:3402 and 2279) (f) Mutation frequency of candidate genes based on data from the cBioPortal database. (g) Somatic mutations of signature genes in high and low risk score groups.
Figure S2. Molecular docking results of YBX1- Carmustine and EZH2- Fenretinide. (a) YBX1 (1h95) -Carmustine: weak interaction (−3.8 kcal/mol). (b) EZH2 (4w2r) and Fenretinide has unfavorable bonding effect (red bond).
Figure S3. The expression of YBX1, PSMD1, CAD, EZH2, and RNF2 and survival analysis in the ICGC dataset. (a) Expression of the five genes in the TCGA dataset comprising high risk score samples (n = 111) and low risk score samples (n = 112). Mann–Whitney U test was used to detect significant expression. * ** *, P < 0.0001; * ** , P < 0.001; * *, P < 0.01; * , P < 0.05.
Figure S4. The expression of YBX1, PSMD1, CAD, EZH2, and RNF2 and survival analysis in the TCGA and ICGC dataset. (a) Expression of the five genes in the TCGA dataset comprising Normal samples (n = 50) and Tumor samples (n = 357). (b) Expression of the five genes in the ICGC dataset comprising Normal samples (n = 223) and Tumor samples (n = 223). Mann–Whitney U test was used to detect significant expression. * ** *, P < 0.0001; * ** , P < 0.001; * *, P < 0.01; * , P < 0.05.