

Abstract
Furins are serine endoproteases that process precursor proteins into their biologically active forms, and they play essential roles in normal metabolism and disease presentation, including promoting expression of bacterial virulence factors and viral pathogenesis. Thus, furins represent vital targets for development of antimicrobial and antiviral therapeutics. Recent experimental evidence indicated that dichlorophenyl (DCP)-pyridine “BOS” drugs (e.g., BOS-318) competitively inhibit human furin by an induced-fit mechanism in which tryptophan W254 in the furin catalytic cleft (FCC) functions as a molecular gate, rotating nearly 180o through a steep energy barrier about its chi-1 dihedral to an “open” orientation, exposing a buried (i.e., cryptic) hydrophobic pocket 1. Once exposed, the non-polar DCP group of BOS-318, and similar halo-phenyl groups of analogs, enter the cryptic pocket, stabilizing drug binding. Here, we demonstrate flexible-receptor docking of BOS-318 (and various analogs) was unable to emulate the induced-fit motif, even when tryptophan was replaced with less bulky phenylalanine or glycine. While either substitution allowed access to the hydrophobic pocket for most ligands tested, optimal binding was observed only for W254, inferring a stabilizing effect of the indole sidechain. Furthermore, non-equilibrium steered molecular dynamics (sMD) in which the bound drugs (or their fragments) were extracted from the FCC did not cause closure of the open W254 gate, consistent with the thermodynamic stability of the open or closed W254 orientations. Finally, interactive molecular dynamics (iMD) revealed two putative conduits of drug entry and binding into the FCC, each coupled with W254 dihedral rotation and opening of the cryptic pocket. The iMD simulations further revealed ligand entry and binding in the FCC is likely driven in part by energy fluxes stemming from disruption and re-formation of ligand and protein solvation shells during drug migration from the solution phase into the FCC.
Keywords: Furin, Induced-fit drug docking, Interactive molecular dynamics, SARS-CoV-2, Antiviral, Therapeutics
Graphical abstract
1. Introduction
Furin is predominantly a Golgi membrane-localized calcium-dependent serine endopeptidase and member of the broad subtilisin-like proprotein convertase family [2], [3]. Furins proteolytically modify and activate many critical protein precursors (i.e., proprotein substrates), ranging from pathogenic agents to growth factors, receptors, and extracellular matrix proteins [4]. Furin’s mechanism of action involves hydrolytic cleavage of oligopeptides and protein substrates at partially conserved paired (polybasic) recognition sequences (RNRR), e.g., -Lys-Arg- or -Arg-Arg-.
Furins are ubiquitously expressed in human and other mammalian tissues and play central roles in numerous disease processes [4], including diabetes [5], obesity [6], atherosclerosis [7], cancer [4], [8], Alzheimer’s disease [9], [10], and viral infections [4], including COVID-19 [11], [12], [13], [14]. They have been shown to promote the expression of virulence factors (including biotoxins) and enhance the infectivity and pathogenesis of many viral and bacterial species, including Herpes viruses, Corona viruses, Retroviruses, Anthrax toxin, Diphtheria toxin, Pseudomonas exotoxins, Shigella toxins, and more [2]. Furin activates the SARS-CoV-2 spike glycoprotein at characteristic multi-basic recognition sequences, in particular at the primary S1/S2 cleavage site; and at the S2’ site by the trypsin-like protease TMPRSS2 [10], [14], [15], [16], [17], [18], [19], [20], [21], [22] (see Fig. 1). The cleavage of these viral glycoproteins at the arginine rich multi-basic site S1/S2 (680-SPRRARS-686) and multi-basic site S2’ (810-SKPSKRS-816) occurs between the same R-S residues in both sites (685R-686S and 815R-816S, respectively). Because of this, “arginine blockers” (ARBs; e.g., Sartans) containing anionic tetrazole and/or carboxyl functional groups, and in particular Bisartans containing two tetrazole moieties with increased acidity, represent promising repurposed antiviral drugs [23]. Thus, drugs in the Sartan family can block furin activity (and subsequent infection) by obstructing basic amino acids of cleavage sites [24], [25], [26]. Clinical studies have shown the beneficial effect of ARBs Telmisartan, Candesartan, Losartan in hypertension patients infected by SARs-CoV-2 compared to patients not taking ARBs [27], [28]. The interaction of Sartans with the ACE2-RBD angiotensin converting enzyme 2-receptor binding domain (ACE2-RBD) complex by computational and enzyme studies and the role of the warhead tetrazole functionality has been recently reported [29].
Fig. 1.

Homotrimeric SARS-CoV-2 spike glycoprotein (S-protein) showing locations of the S1/S2 and S2' furin cleavage sites. Swiss Model 05, full sequence homology model.
Due to their involvement in the earliest stages of cellular infection, furins represent vital targets for the discovery and design of novel therapeutic antiviral drugs [13], [14], [21]. However, furins are challenging drug targets since this protease family is also broadly involved in many beneficial metabolic processes [30]. Thus, drugs identified or developed to suppress furin activity must also avoid deleterious side effects on beneficial biochemical pathways while targeting only those furin mutational variants that are involved in the disease process.
Dahms and colleagues [1] recently reported several small-molecule 3,5-di-chlorophenyl-pyridine-derived ligands (i.e., “BOS” inhibitors such as BOS-318) that exhibit strong binding to, and competitive inhibition of, human furin. Moreover, selected BOS inhibitors have been shown to block endogenous S-protein cleavage at the S1/S2 and S2’ sites in HeLa cells; and in combination with Camostat, a transmembrane serine protease 2 (TMPRSS2) inhibitor, SARS-CoV-2 infection of lung-derived Calu-3 cells was prevented [31].
X-ray crystallographic evidence demonstrated that when BOS-318 binds to the furin catalytic cleft (FCC), it induced conformational rearrangement of the active-site coupled with dihedral rotation of a central tryptophan residue (W254). Concomitant with drug binding, W254 was pivoted nearly 180 degrees around its chi-1 (N, CA, CB, CG) dihedral bond, thereby exposing a cryptic hydrophobic pocket into which the nonpolar 3,5-dichlorophenyl (DCP) moiety of the inhibitor(s) was stably inserted. Importantly, the authors further demonstrated that the hydrophobic DCP-binding pocket is not well conserved across the human furin family [1]. Thus, it was reasonably speculated that DCP-pyridine-derived inhibitors (or structural analogs thereof) could be tailored to inhibit furin variants more selectively and improve the therapeutic index of such compounds.
Cryptic pockets are defined as hidden potential binding sites that become observable only when drugs bind, or when localized protein conformations deviate sufficiently from their ground-state (normal-mode) configurations through thermal perturbations or other kinetic inputs to reveal a new spatial domain that temporarily opens up for acceptance of a drug or substrate [32]. As such, cryptic pockets provide propitious alternatives to orthodox binding sites for drug discovery and design. For example, Knoverek et al. [33] recently reported that opening of a cryptic pocket under the Ω-loop of excited-state β-lactamase increases penicillinase activity. Identification of cryptic pockets is challenging since at typical physiological temperatures they do not emerge as local minima in the energy landscape of unliganded receptors, and therefore remain obfuscated in conventional molecular dynamics (MD) simulations. Oleinikovas et al. [32] used a novel Hamiltonian Replica Exchange-based method to enhance sampling of protein phase-space to reveal cryptic pockets in MD simulations. More recently, the “PocketMiner”, was developed, a rapid graph neural network trained to predict where druggable cryptic domains are likely to open. Subsequent MD simulations can then be employed to sample structural configurations with the pocket(s) open, which enables structure-based drug design and targeting of pharmacologically relevant proteins that otherwise lack druggable pockets in their ground-state structures [34]. Whereas Dahms and co-workers [1] clearly demonstrated an induced-fit drug binding motif predicated on a major torsional shift of W254 was responsible for optimal receptor binding and ligand specificity, they did not comment on the dynamics of potential binding mechanism(s) by which such a large, and energetically unfavorable, dihedral rotation might occur. Nor were potential drug entry trajectories reported.
Herein, we describe two potential routes of drug entry and binding into the furin catalytic pocket by DCP-pyridine-derived compounds, as well as by several modified anionic tetrazole-bearing homologs of the parent (BOS-318) drug. To achieve this objective, we employed non-equilibrium interactive molecular dynamics (iMD) which, unlike conventional equilibrium MD approaches [35], is able to access the very large temporal and spatial domains required to identify and explore drug entry and binding trajectories [36], [37], [38], [39].
2. Methods
2.1. Equilibrium molecular dynamics
Conventional (NPT ensemble) MD simulations were performed with the Yasara suite [40] using imposed periodic boundaries, a pKa prediction algorithm to adjust protonation states of protein residues at the chosen pH of 7.4 [41] and optimization of the hydrogen bonding network [42] to increase receptor and/or receptor-ligand complex stability. Sodium (Na+) and chloride (Cl-) ions were introduced to physiological concentrations (approximately 0.9 wt%), with an excess of either ion to neutralize the cell. Following steepest descent and simulated annealing minimizations to remove clashes, simulations were typically run for 40–120 ns using the AMBER14 force field [43] for the solute, GAFF2 [44] and AM1BCC [45] for ligands, and TIP3P for water [46], [47]. The cutoff was 8.0 Å for Van der Waals forces (the default used by AMBER [48], no cutoff was applied to electrostatic forces (using the Particle Mesh Ewald algorithm [49]). Equations of motion were integrated with a multiple timestep of 2.5 fs for bonded intramolecular interactions and 5.0 fs for non-bonded interactions at a temperature of 311OK and a pressure of 1 atm (NPT ensemble) using algorithms described previously [50]. Using a separate script, the “free energy” of ligand binding across MD trajectories (including iMD and sMD runs) were calculated by the “Te = Ae + Be + Ce + Ee” method, where Te = total energy, Ae and Be = ligand (A) and receptor (B) energies, Ce = the A-B interaction potential (binding energy), and Ee = estimated entropic cost of exposing one Å^2 of the ligand to the aqueous solvent (cf., Yasara2020/yasara/doc/BindEnergyObj.html).
2.2. Interactive molecular dynamics (iMD)
Interactive MD (iMD) simulations (in 3D Cartesian space) were performed within the YASARA dynamics suite suite using a scripted algorithm for this purpose [40]. The process was initiated by setting up the fully solvated molecular system with periodic boundaries, which included: (1) Visual inspection of the furin receptor to identify potential ligand entry points and relatively unobstructed (i.e., low-resistance) channels or pathways that lead into the catalytic cleft; (2) Positioning the ligand in the TIP3P solution phase outside the entry point of the chosen channel; (3) Invoking a normal NPT eqMD simulation, as described above (see 3.1); (4) Marking a “pulling (lead) atom”, typically at or near the leading terminus of the ligand; (5) Selection of a starting pulling acceleration (1−9) to be applied to the pulling atom; (6) Using the mouse pointer to then guide the ligand into and along the entry channel; (7) Cancellation of the pulling force once the ligand entered the furin catalytic pocket; and (8) Continued NPT equilibration for an additional time period, typically about 100 ps or less. Note that extended NPT eqMD was also performed for selected iMD ligand-furin complexes for 50–100 ns to ascertain complex stability. The pulling strength was computed as “KeyScale x DisScale x TypeScale”, where 'KeyScale' depends on the key pressed to set the initial relative strength (1x for key 1, 2x for key 2, 4x for key 3, 8x for key 4, 16x for key 5, etc., and 256x for key 9); 'DisScale' is the distance between the lead atom and mouse pointer in pixels divided by the height of the application window in pixels (so pulling gets stronger with increasing distance between the marked atom and mouse pointer; and 'TypeScale' is 1e-9 Newton applied as a velocity vector to the marked atom during a steepest descent minimization: 500 m/s if a single hydrogen atom is pulled, or 50 m/s otherwise. In this study, a carbon atom was always designated as a lead atom. Lead atom velocities (V = ɗr/ɗt) and accelerations (ɗV/ɗt) were computed from the raw coordinate and time data associated with the designated atom. It was sometimes useful to monitor lead atom “net velocities” and “net accelerations”, which were defined as the sum of the absolute values of either metric (e.g., net acceleration = |ɗVx/ɗt| + |ɗVy/ɗt| + |ɗVz/ɗt|) and reflected the total forces on the ligand regardless of direction.
2.3. Steered molecular dynamics (sMD)
Steered MD (sMD) simulations were performed using a modified script (see Krieger and Vriend, 2014). The script was applied to an elongated (cuboid) periodic system (x = 175 Å x y = 100 Å x z = 100 Å) in which a ligand-furin complex from a previous iMD docking exercise, and fully solvated in physiological saline (0.9 wt% NaCl), was first energy-minimized using an NPT ensemble by a series of annealing and optimization steps using the AMBER14 force field [43] for the protein, GAFF2 [44] and AM1BCC [45] for ligands, and TIP3P for water [46], [47]. The cutoff was 8.0 Å for Van der Waals forces (the default used by AMBER [48], no cutoff was applied to electrostatic forces (using the Particle Mesh Ewald algorithm [49]). A sMD pulling angle/direction was manually assigned to the ligand. In this study, two directions were employed: (1) one perpendicular to the major axis of the FCC; and (2) the other oriented parallel with the long axis of the FCC “side channel” (see Results, section 4.4) such that the iMD-docked ligand was extracted more-or-less in a reversal of its iMD docking pathway. Following optimizations and setting the pulling direction, an acceleration of 250 pm/ps [2] was applied to the ligand; and this value was increased by 100 pm/ps [2] increments until the ligand began to be extracted from the pocket. The acceleration bias was decreased incrementally while the ligand continued on its trajectory or increased if the ligand slowed due to resistance from obstructing furin side chains. The sMD simulation was halted when the center-of-mass (COM) separation between the ligand and furin reached 30 Å. Results were plotted as force (F) versus distance (D) curves, from which the amount of extraction work (A) at each sMD time point was calculated by FxD. The cumulative work performed to fully separate the ligand (designated ‘Asep’) was estimated by integrating ‘A’ over the trajectory path until all ligand atoms were > 4–6 Å from furin, i.e., the drug was fully solvated. Steered MD simulations were carried out not only on docked intact ligands, but also on ligand fragments (e.g., the DCP group of BOS-318). The fragments were prepared in situ (i.e., inside the FCC) by deleting the extraneous atoms of the bound ligand leaving only the fragment of interest. Resulting unbonded atoms were capped with hydrogen.
2.4. Calculation of dihedral energy barriers
Trp254 dihedral (chi-1 and chi-2) barriers were computed over 80 steps at 4.5-degree increments in a fully (TIP3P) solvated periodic system using the AMBER14 force field with long-range electrostatics (see MD methods). At each rotational increment, residues surrounding W254 within a spherical radius of 10 Å were subjected to steepest-descent minimization prior to resolution of the dihedral strain energy. Energies were compensated for solvation effects based on W254 exposed surface area.
2.5. Flexible-receptor (Induced-Fit) docking
Docking was performed using AutoDock-VINA [51] with default parameters. Partial atomic charges were assigned according to the AMBER03 or AMBER14 force fields [52], and then dampened to mimic the less polar Gasteiger charges used to optimize the AutoDock scoring function [53]. The initial setup was done with the YASARA molecular modeling program [54] and the “free-energy of binding” (accounting for ligand desolvation effects) results from 500 to 900 runs were reported in kcal/mol. Note that in this report, more positive binding energies correspond to stronger ligand-receptor interactions. Receptor flexibility was invoked in the docking routines using the approach described by Morris et al. [55]. Ligand-receptor dissociation constants (Kd) were computed within the automated docking scripts according to the following approach:
The Kd is defined by the molar concentrations of the receptor R, ligand L and complex C in solution:
| (1) |
The molar concentration is given by the number of molecules in solution, divided by the volume of the solution and normalized by Avogadro’s Constant, shown here for the receptor:
| (2) |
The concentration of the receptor is the same as that of the ligand since each cell contains either one unbound receptor-ligand pair or one complex. Thus:
| (3) |
The number of complexes is the total number of cells (i.e., the number of simulation snapshots) minus the number of cells with unbound receptor and ligand:
| (4) |
The volume V is the number of simulation snapshots times the cell volume:
| (5) |
Re-writing the equation, we calculate Kd using [R]= [L], [R], nC and V from above:
| (6) |
3. Results
3.1. Analysis of furin structure and W254 dihedral energy barriers
The structure of unliganded human furin (PDB 5JXG) showing the major catalytic cleft and the location and orientation of Trp254 (W254), which has been shown to undergo a large drug-induced chi-1 dihedral rotation associated with binding of the DCP-pyridine drug, BOS-318, is depicted in Fig. 2 [1]. Fig. 3 A presents chi-1 and chi-2 dihedral energy barrier scans for furin residue W254 calculated at 4.5-degree increments over 80 steps. Each rotational step was accompanied by local energy minimization within a 10-Å sphere surrounding the W254 residue prior to resolution of the strain potential (see Methods). The scan results shown in Fig. 3 A indicate that rotation of W254 about its chi-2 dihedral resulted in an insurmountable torsion barrier (>400,000 kcal/mol) stemming from a direct sidechain clash of W254 with W291 situated at about 3.57 Å from W254. No such clash was observed for the W254 chi-1 dihedral bond rotation when it was evaluated in the presence and absence of furin-associated (chelated) Ca++ ions (Fig. 3 C). Calcium has been shown to be essential for maximal furin activity and is involved in stabilizing the N-terminus and the unique S1 specificity pocket [3].
Fig. 2.

[A] View down the major axis of the FCC of PDB 5JXG. [B] View perpendicular to the catalytic cleft. [C] Magnified view of the 5JXG catalytic cleft showing the location and orientation of W254 in the unliganded “closed” state. The chi-1 dihedral angle is marked with the curved blue arrow. [D] Similar view as in [C], but also showing the location and orientation of W291 (cyan tubes) lying proximal (within 3.57 Å) from W254. Also indicated is the chi-2 torsion angle of W254 (blue curved arrow). Rotation about the chi-2 dihedral (but not the chi-1 dihedral) resulted in a clash with W291. [E] Overview of superimposed human furin x-ray crystallographic structures: BOS-318 liganded form (PDB 7LCU; maroon carbon atoms) and unliganded form (PDB 5JXG; light blue carbon atoms). [F] Magnified view of FCC domain showing bound inhibitor BOS-318 (PDB 7LCU]. W254 is depicted in the “open” fully rotated state in the liganded 7LCU structure; and in the “closed” state in the superimposed unliganded 5JXG structure. Van der Waals shaded atoms (translucent brown spheres) indicate approximate extent of the cryptic hydrophobic pocket.
Fig. 3.

[A]: Comparison of chi-1 (N,CA,CB,CG; blue line) and chi-2 (CA,CB.CG,CD1; orange line) dihedral barrier scans for W254 in PDB 5JXG. The 360-degree scans were produced over 80 steps at 4.5-degrees per step (see Methods). Rotation about the W254 chi-2 dihedral bond resulted in an insurmountable clash with W291 located about 3.57 Å from W254. The energy barrier for this clash exceeded 416,000 kcal/mol). No serious sidechain clashes were observed for chi-1 rotation. [B] Details of W254 rotation about its chi-1 torsion bond. Rotation of this bond in the intMD docking simulations using 5JXG was from right to left (large blue arrow) starting at + 34 degrees (W254 in “closed” orientation) through 180-degrees and terminating at approximately − 145 degrees (W254 in “open” orientation). [C] Comparison of W254 chi-1 dihedral barrier scans for 5JXG with (orange line) and without (blue line) the four chelated calcium ions. The chi-1 peak energy barrier was approximately 158 kcal/mol for the Ca-free receptor and 230 kcal/mol for the receptor with Ca. The presence of Ca ions resulted in an overall (global) reduction in the chi-1 energy landscape by about 50–60 kcal/mol.
The presence of the four coordinated Ca++ atoms in the 5JXG furin structure caused an overall lowering (i.e., greater stability) of the chi-1 rotational barrier of W254 by about 50 kcal/mol compared to the calcium-free receptor. Three main W254 chi-1 dihedral peak locations and energies were observed for the calcium-free scan at − 40 degrees (−13891 kcal/mol), − 55 degrees (−13900 kcal/mol) and − 130 degrees (−13936 kcal/mol). These energy barriers were mainly attributed to repulsive interactions of W254 with Leu152, Leu227, and Asp153. The largest W254 peak dihedral barriers were approximately 158 and 225 kcal/mol.
above their “ground-state” energies, respectively, for the calcium-free and calcium-associated systems under the fully solvated conditions used. Allowing the chi-2 dihedral to remain flexible at each chi-1 rotational step resulted in a moderate reduction in the W254 torsion by about 60 kcal/mol (data not shown), however, the initial starting energy was also elevated by about the same amount.
It should be emphasized that the native resting (i.e., ground-state) orientation of the W254 chi-1 torsion angle in the unliganded 5JXG x-ray structure is about + 34 degrees (“closed” position), whereas that of the BOS-318 liganded 7LCU (or 7QY0) x-ray structure is about − 146 degrees (“open” position). As seen in Fig. 3 A and 3 C, both rotamer orientations fall within local minimum zones and thus can be expected to be thermodynamically stable furin configurations. The dihedral energy landscapes of the calcium-free and calcium-associated scans were quite similar (R2 =0.76), with the exception of the minor barrier at − 130 degrees. For this reason, and the fact that the peak torsion barrier in the calcium-free receptor offered a lower energy pathway for ligand entry and binding, the iMD and sMD studies described in subsequent sections of this report were carried out in the absence of calcium ions.
3.2. Flexible-receptor docking of BOS-318 and selected structural analogs to the furin catalytic cleft
Flexible-receptor virtual ligand screening (fsVLS) using AutoDock VINA (900 runs per ligand; see Methods for details) was carried out to determine whether a fully flexible W254 residue could emulate the induced-fit docking responses reported by Dahms and colleagues [1] in which residue W254 undergoes a major dihedral shift of nearly − 180 degrees from its original “closed” orientation at − 34 degrees (as in 5JXG). In addition to W254, Tyr308 and His194 were also kept fully flexible during the docking runs. The flexible-receptor (i.e., induced-fit) docking was performed against two furin receptor models: (1) unliganded PDB 7QY0 with W254 in the “open” orientation; and (2) unliganded PDB 5JXG with W254 in the “closed” orientation.
In both scenarios, the chi-1 and chi-2 dihedral angles of W254, Y308 and H194 were maintained as fully flexible; and the docking domain dimensions and AMBER14 force field grid matrices (delineated by non-periodic walled boundaries) were precisely matched for the two receptor models (see below Fig. 6, upper panel).
Fig. 6.

Docking of ligands to the FCC domain of receptors 7QY0 (open orientation) and 5JXG (closed orientation), as well as additional furin targets harboring point mutations at the W254 locus. [A] Depiction of the FCC docking domain used in this study showing walled boundaries. [B] Enlarged view of the FCC in the vicinity of the W254 of 7QY0 in the open orientation. The chi-1 and chi-2 dihedral angles of W254 are labeled. [C] VINA flexible-receptor docking results for 15 selected ligands (structures given in Fig. 4) docked to the furin catalytic domain of unliganded 7QY0 (“open” W254; dark blue bars) or 5JXG (“closed” W254; gray bars). Results are also given for five W254x point mutations, including: 5JXG-W254G (green bars), 5JXG-W254F/closed (yellow bars), 7QY0-W254G (magenta bars), 7QY0-W254F/open (light blue bars), and 5JXG-W254A (brown bars). Compounds beginning with the prefix “Mod-” are modifications of the known furin inhibitor BOS-318 [1]. In this series, Mod23 exhibited the best docking score of 12.36 kcal/mol to the unliganded 7QY0 receptor with W254 in the open orientation, whereas BOS-318 and its ethoxylated homolog “7QY0 ligand” scored lower in comparison (11.14 and 10.59 kcal/mol, respectively).
A total of 15 ligands were examined which included: (i) the proven 3,5-dichlorophenyl-pyridine furin inhibitor, BOS-318 reported by Dahms et al. [1]; (ii) a close structural analog of BOS-318 also known to competitively block furin activity, referred to herein as the “7QYO ligand”; and (iii) 13 structural analogs of BOS-318, each bearing a modified non-polar halophenyl group plus one or two terminal anionic alkyl-tetrazole groups. Structures for the modified and unmodified drugs are given in Fig. 4.
Fig. 4.

Structures of BOS-318 and 14 modified forms (“Mod”- prefixes). Included in the group is the bisartan ACC519TT, an anionic benzimidazole-N,N bis biphenyl-tetrazole known to exhibit anti-viral activity. Also included is the ethoxylated BOS-318 homolog (“7QY0-Ligand”) extracted form PDB 7QY0.
It should be noted that AutoDock VINA (as implemented in the Yasara suite; see Methods) was able to accurately reproduce the correct x-ray pose of BOS-318 in the furin 7QY0 catalytic cleft (Fig. 5). Note that W254 is already in the open orientation in the unliganded PDB 7QY0 model used for docking purposes. The overall (per-object) RMSD of the superimposed 7LCU x-ray structure with bound BOS-318 against the 7QY0 structure with the VINA-docked BOS-318 ligand was 1.0597 Å. The per-atom RMSD for only the superimposed ligands was 0.3132 Å. These results provide a modicum of confidence in the use of VINA algorithms as applied in this study, particularly since all the ligands evaluated exhibited structural similarities.
Fig. 5.

AutoDock VINA prediction of correct x-ray crystallographic pose for BOS-318. [A] View of PDB 7QY0 x-ray crystallographic model (dusty-blue carbon atoms) with bound BOS-318 superimposed onto PDB 7QY0 model (maroon carbon atoms) with docked BOS-318. The overall per-object RMSD for the superimposed models was 1.0597 Å. [B] Superimposition of the BOS-318 x-ray crystallographic pose from 7LCU (dusty blue carbons) against the docked pose for 7QY0 (maroon carbons). The per-atom RMSD for the superimposed ligand couple was 0.3231 Å.
Results of the flexible-receptor docking are presented in Fig. 6. Inspection of the data indicate that drug-furin interactions were uniformly stronger for the 7QY0 receptor with W254 in the “open” state and weaker for the 5JXG receptor with W254 in its “closed” ground-state orientation, which is the same trend as reported by Dahms and co-workers [1]. Note that more positive docking scores shown in Fig. 6 correlate with stronger ligand binding free energies.
Although an induced-fit docking methodology was employed throughout in which W254, His194 and Tyr308 remained fully flexible, no drug, including BOS-318 and the ethoxylated “7QY0 ligand”, prompted any large or otherwise significant shift in the W254 dihedrals of the 5JXG furin receptor, which disallowed penetration of the BOS-318 non-polar DCP or halo-phenyl groups of other ligands into the buried hydrophobic pocket. Thus, the VINA docking algorithm was unable to reproduce the induced-fit binding motif that has been experimentally demonstrated by Dahms et al. [1].
The same negative result was obtained (i.e., no substantive change in the W254 dihedral angles of 5JXG) when ensemble docking, or flexible-receptor docking was repeated (using VINA) for the isolated case of BOS-318 docked against the 5JXG receptor with as many as 13 simultaneously flexible pocket residues (data not shown). Similarly, the internal coordinate mechanics (ICM) flexible-receptor docking algorithms from Molsoft, LLC (San Diego, CA, USA) likewise did not reproduce the induced-fit mechanism documented in the experimental studies reported by Dahms and co-workers [1].
The modified ligands (i.e, BOS-318 structural analogs) exhibited the strongest binding free energies to 7QY0 (e.g., Mod21, Mod22 and Mod23, Fig. 6), suggesting it is feasible to incrementally improve drug binding affinity and stability for this receptor. All of the modifications included one or two terminal anionic alkyl-tetrazole groups that were able to extend further along the major axis of the FCC. The ethyl-tetrazolate extensions allowed torsional flexibility for the anionic group and enabled formation of multiple salt bridges (also referred to herein as cation-π interactions) with two basic arginine residues, R193 and R197, located at the “mouth” of the FCC. The VINA docking motifs of the BOS-318 parent compund and the ethyl-monotetrazole Mod23 ligand are compared in Fig. 7. The additional tetrazole-mediated interactions with R193 and R197 strengthened the overall docking scores for Mod21–23 and stabilized their binding in the pocket (see MD simulation results below, Fig. 8). It was also noted that converting the two neighboring pyridine rings of BOS-318 to phenyl groups did not adversely affect (weaken) VINA docking scores. This change could potentially simplify and faciliate synthetic pathways for this category of putative furin inhibitors. Replacement of the chlorine atoms of BOS-318 with paired bromine and fluorine atoms resulted in a slight but significant boost in VINA docking scores for Mod23.
Fig. 7.

Comparison of drug binding motifs for [A] BOS-318 bound to the FCC of PDB 7LCU (original x-ray structure); and [B] Mod23 docked in PDB 7YQ0 using AutoDock VINA. In the latter case, the anionic ethyl-tetrazole group (red circle) extended farther along the FCC to form stabilizing cation-π (salt-bridge) interactions with the basic residues Arg193 and Arg197. Note that residues shown in the magnified views (on the right) are colored by amino acid type: gray = hydrophobic; blue = cationic; red = acidic; cyan = neutral. The hydrophobic pocket is approximately indicated by the purple shading. Hydrophobic interactions tended to dominate the central region of each ligand.
Fig. 8.

[A] Results of a 59.1 ns equilibrium MD simulation (NPT ensemble; see Methods) of Mod23 docked into the furin 7QY0 catalytic domain. Heavy-atom RMSD scores and total system potential energy are plotted for 7QY0 and Mod23 over the 59.1-ns trajectory. [B] Image frame captures at 0, 29.6 and 59.1 ns indicating the Mod23 ligand (rendered as yellow spheres) was stably bound in the catalytic cleft over the 59.1-ns MD trajectory. [C] Ligand free energy of binding (blue line) and heavy-atom RMSD values calculated for the 7QY0 receptor (orange line) and Mod23 (gray line). Note that in this calculation, more positive binding energies correspond to stronger binding.
Mod 21, Mod22 and Mod23 also differed from BOS-318 by substitution of the BOS-318 terminal carboxylate group with a tetrazolate and the incorporation of an amino (-NH2) group in the pyridine ring. This change resulted in formation of two hydrogen bonds between the new amino group and a proximal tyrosine residue (Tyr308). Results presented in Fig. 6 also summarize the effects of various point mutations introduced at the W254 (W254x) site on ligand docking energies (i.e., free energy of binding). Five mutations were examined, including: 5JXG-W254G (green bars), 5JXG-W254F/closed (yellow bars), 7QY0-W254G (magenta bars), 7QY0-W254F/open (light blue bars), and 5JXG-W254A (brown bars). The W254A mutation in 5JXG (brown bars, Fig. 6) resulted in the greatest overall suppression of ligand binding, which was surprising given the relatively small footprint of the alanine methyl sidechain. The W254A mutation completely blocked penetration of the non-polar halo-phenyl group of ligands into the cryptic FCC hydrophobic pocket. This observation suggests that mutations at the W254 locus can have a significanmt influence on the druggability of this site. W254G was the only mutation in 5JXG (green bars, Fig. 6) that allowed the non-polar halo-phenyl group of most ligands tested to occupy the FCC hydrophobic pocket (exceptions included Mod15 and Mod17). However, in spite of having access to the furin hydrophobic pocket, binding energies were consistently lower (weaker) for all ligands compared to the case of 7QY0 (with W254 in the open state; dark blue bars, Fig. 6).
Introduction of phenylalanine in the open orientation at the W254 locus of 7QY0 (light blue bars, Fig. 6) also permitted occupancy of the FCC cryptic pocket by the halo-phenyl group of ligands. However, as was the case for the W254G mutation in the 5JXG receptor, the free energy of binding was lower (weaker) for nearly all drugs tested compared to the open-state W254 of 7QY0 (dark blue bars, Fig. 6). These observations suggest the W254 indole sidechain in the open orientation of 7QY0 provides additional attractive interactions with the ligands that are not available with glycine or phenylalanine at this position. Moreover, it seems likely that the tryptophan residue in the open conformation helps to properly orient ligands with respect to binding of the halo-phenyl group in the hydrophobic pocket.
When docking to the W254G mutation in 5JXG (green bars, Fig. 6) was compared to docking scores for the W254G mutation in 7QY0 (magenta bars, Fig. 6), ligand binding stength in the former case was substantially reduced. This result was somewhat surprising since In both situations access to the hydrophobic pocket was permitted, suggesting the docking scores should have been similar. A possible explanation for the observation is that subtle orientations of the glycine hydrogen atoms can significantly influence ligand binding. Measurement of the dihedral angle of the glycine #2 hydrogen atom of W254G in the 5JXG receptor formed by CA(252)-CA(253)-CA(254)-HA2(254) yielded a value of − 33.815 degrees, wheras the same angle for the W254G mutation in 7QY0 was − 91.606 degrees. This result is consistent with the notion that subtle alterations of the W254 site, such as the orientation of a sidechain hydrogen, can substantially influence drug behavior in a manner similar to the W254A mutation described above.
Equilibrium molecular dynamics (eqMD) was performed (NPT ensemble, in 0.9 wt% physiological saline, constant temperature @ 311oK; see Methods) to determine if the docked Mod23 ligand remained stably bound over ns time scales in the FCC. As shown in Fig. 8, Mod23 was not ejected from the cleft and remained stably bound over the 59.1-ns MD trajectory. Heavy-atom RMSD values for both the 7QY0 furin receptor (<2.5 Å) and the Mod23 ligand (<1.5 Å) indicated relatively minor atomic displacements and were consonant with relatively stable ligand binding. The free enregy of binding (FEB) of Mod23 (i.e., the ligand-furin interaction potential) was also monitored over the eqMD trajectory. The FEB values gradually increased (corresponding to stronger binding) over the course of the trajectory, suggesting the ligand-furin complex became progressively more stable with time. The above findings suggest it may be possible to design next-generation anionic tetrazole-bearing di-halo-phenyl drugs with enhanced furin affinity and specificity compared to the parent BOS-318 ligand.
3.3. Steered MD Simulations of Ligand Extractions
Non-equilibrium sMD simulations were perfomed to determine: (1) if extraction of the iMD-docked ligands, or ligand fragments, would induce closure of the W254 gate of liganded 5JXG; (2) if ligand extraction energies would reflect trends in the docking energies described above; and (3) if the hydrophobic halo-phenyl groups (HPGs) of BOS-318 and Mod23, or their respective fragments lacking the HPG, differed in their binding energies. In the sMD simulations, the iMD-docked drugs (and their fragments) were dynamically dragged from the furin FCC pocket. For each ligand, two different pre-determined extraction directions (angles) were examined, including: (1) Pulling the ligand upward at an approximately perpendicular angle to the long axis of the FCC, referred to as “top” (tpex) extraction; and (2) Pulling the ligand in reverse direction from its iMD-docked position in the FCC, referred to as “reverse side-channel” (rsc) extraction (see iMD results below, section 4.4). The former (tpex) extraction trajectory offered the least obstruction by furin sidechains to drug extraction. The sMD simulations were carried out under NPT periodic conditions with explicit solvation by physiological saline (0.9 wt% NaCl, 311OK, TIP3P water). The main features of the automated sMD script included introduction of an initial pulling force (acceleration vector) applied to the ligand (250 pm/ps2) in the prescribed direction indicated by the red arrows in Fig. 9 C and D. The acceleration was increased in 100-pm/ps2 increments until the ligand began to move on the designated trajectory (see Methods for details). The acceleration was incrementally adjusted upward or downward depending on the rate of forward progress of the ligand. The sMD simulations were stopped when the furin-ligand center-of-mass (COM) separation distance reached 30 Å.
Fig. 9.

[A] Kinetics of steered-MD extraction of iMD-docked BOS-318 and Mod23 intact ligands and ligand fragments from the 5JXG furin FCC. The ordinate is the applied force in pN and the abscissa denotes the number of measurement points to reach a ligand-furin center-of-mass (COM) separation distance of 30 Å. Ligand Suffix Designations: “-int” = native intact ligand; “noHPG” = ligand fragment lacking the hydrophobic group (HPG); “HPGonly” = only the isolated HPG fragment. Ligand Prefixes: “rsc” = reverse extraction of ligand from the side channel; “tpex” = ligand extraction from the “top” of the FCC. Ligand fragments were prepared in situ by deleting the appropriate atoms and capping groups with a hydrogen atom (see Methods). Ligand extraction values (pN-Å) on the ordinate are expressed as the cumulative work (∑force x distance = Asep) required to move (separate) the ligand from its docked state into the TIP3P solvent phase. Separation was completed when all ligand-furin atom distances exceeded about 4–6 Å, which corresponded to 16–22 Å of COM separation, depending on the ligand and fragment makeup. Blue and green bars = BOS-318 data; orange and gray bars = Mod23 data. [B] and [C] Examples of initial (t = 0 ps) setup conditions for the docked BOS-318-furin complex for carrying out sMD simulations. Shown are the “rsc” [B] and “tpex” [C] arrangements with the docked intact BOS-318 ligand. Corresponding arrangements were used for sMD extractions of the iMD-docked Mod23 ligand (not shown). BOS-318 atoms are rendered as spheres: carbon=cyan; nitrogen=blue; oxygen=red; hydrogen=gray; chlorine=green. Direction of the sMD bias acceleration is indicated by the red arrow. The FCC is approximately indicated by yellow-colored residues (stick renditions) and atom-centered Van der Waals surface renditions (light brown spheres).
The principal results of the sMD simulations are summarized in Fig. 9. The kinetics of the ligand or fragment sMD extractions followed a similar motif, whereby higher accelerations (i.e., applied pulling forces) were required to cause initial dislodgement of the bound ligand, followed by a gradual reduction of the applied force as the ligand transitioned from the FCC into the aqueous phase (Fig. 9 A).
It should be noted that even after the ligand entered completely into the solution phase, it encountered sporadic resistance variations resulting from conformational fluctuations (and associated changing hydrogen-bonding interactions with water molecules) and/or random interactions with dissolved ions.
The trend of higher initial force application was especially apparent for the reverse side-channel (rsc) extraction of the intact Mod23 ligand (rsc-Mod23-int) and the Mod23 fragment lacking the hydrophobic HPG (rsc-Mod23-noHPG; yellow and light blues lines, respectively in Fig. 9 A). Both trajectiories were characterized by an abrupt, but brief, increase in the force needed to initiate ligand extraction (approximately 3500 pN). The abrupt early increase in the Mod23 pulling force can be attributed to scission of the relatively strong salt-bridge (i.e., cation-π) interaction of the anionic tetrazole group of Mod23 with furin residues R193 and R197 (see Fig. 7, above), and not the HPG moiety per se. Accordingly, the lowest sMD extraction energies were exhibitied by the isolated HPG fragments, which generally required applied forces falling well below 1000 pN for the major portion of their extraction trajectories (Fig. 9 A and B). This suggests the non-polar ligand HPGs provided a comparatively small contribution to the binding energy of the intact ligands. Instead, the much larger HPG-free ligand fragments contributed diproportionately more to the docking energies. Notwithstanding its minimal energy contribution to overall ligand binding energy in the sMD simulations, the halo-phenyl groups may be mostly involved in helping to properly orient the drug in its approach to the pocket and subsequently help stabilize binding by optimizing drug conformation in the FCC.
Based on the sMD results presented in Fig. 9B, expressed as the cumulative work required to fully extract and separate the ligand from the FCC pocket [∑FxD = Asep], the overall trend in sMD extraction energies was (strongest to weakest): rsc-BOS-318-int > rsc-BOS-318-noHPG > rsc-Mod23-int > tpex-BOS-318-noHPG > tpex-BOS-318-int > tpex-Mod23-int > rsc-HPGonly > tpex-Mod23-HPGonly > tpex-Mod23-noHPG > rsc-Mod23-noHPG > rsc-Mod23-HPG only. Moreover, as expected, the reverse side-channel (rsc) sMD extractions for both ligands required greater work compared to the top (tpex) extractions. For example, the totalized work of extraction of intact BOS-318 via the reverse side-channel pathway (Asep = 7.62 ×107 pN-Å) was > 3-fold more than the amount of work compared to BOS-318 extraction from the “top” of the FCC (2.48 ×107 pN-Å). A similar relationship existed between rsc-Mod23-int extraction (3.71 ×107 pN-Å) and the tpex-Mod23-int extraction (1.41 ×107 pN-Å).
For most of the sMD runs, summation of the separation work (Asep) for the isolated HPG fragment + that of the larger HPG-free fragment was approximately equivalent to the extraction work for the respective intact ligand. The main exception to this observation was the case for the rsc-Mod23 ligand in which the sum of the Asep work of its HPG fragment + the HPG-free fragment was well below that of the work needed to extract the intact Mod23 ligand. This result, as well as the lesser fragment “work-summation” variations observed for the other cases, is believed to be related in part to the series of initial system geometry optimization steps (e.g., annealing and energy minimizations) prior to invocation of the sMD pulling force. Such minimizations could be expected to affect the starting conformations of the HPG-free ligand fragments to a greater extent than the ioslated non-polar HPG fragments, since the former possess comparatively more internal torsional degrees of motion. Thus, some minimizations could cause the larger HPG-free fragments to be more or less stable at the begiining of the sMD simualtions.
Finally, it is noteworthy that W254 was not observed to rotate to the closed state in any of the sMD ligand extraction simulations. This result is consistent with the dihedral bond energetics outlined in Fig. 3, which indicates both the closed and open states of W254 were comparitively stable orientations.
3.4. Interactive MD simulations of ligand docking
3.4.1. Identification of putative ligand entry channels
Failure of flexible-receptor docking approaches to emulate an induced-fit response in 5JXG likely resulted from the very large W254 chi-1 (∼158 kcal/mol) and chi-2 (>400,000 kcal/mol) dihedral barriers described above (see Fig. 3). Most docking algorithms are appropriately designed to handle much smaller torsional energy barriers. Because of this problem, a non-equilibrium interactive MD (iMD) approach was employed to help elucidate and characterize how selected ligands might gain entrance to and navigate their way into the FCC via relatively low-resistance trajectories, while at the same time ensuring the large W254 dihedral shift reported by Dahms and coworkers [1].
IMD methods involve real-time user input to individual or groups of ligand atoms during MD simulations (i.e., introduction of non-equilibrium acceleration biases). Thus, the ligand may be maneuvered by a user along a vector that largely avoids obstructing residue sidechains to gain access to a chosen destination, such as a potential binding pocket. Because of increased ligand velocities, iMD techniques permit exploration of vastly larger spatio-temporal domains compared to docking approaches or conventional eqMD methods [36]. The biased input is incrementally calibrated and adjustable on-the-fly and generally adequate to rotate or otherwise deform side-chain obstacles, e.g., an alkyl group, but insufficient to break chemical bonds at physiologic temperatures [39].
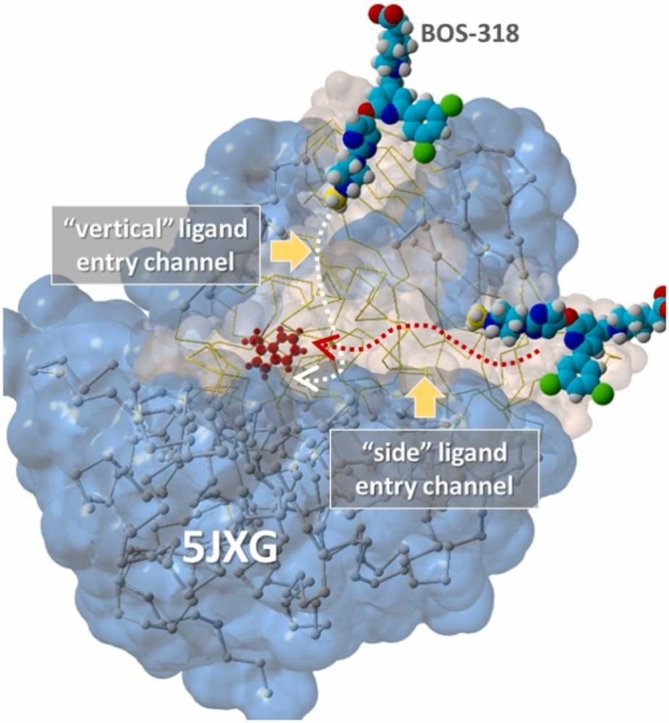
Visual inspection of the furin model PDB 5JXG (closed W254) revealed at least two relatively unobstructed (i.e., minimal sidechain blockage) and potentially low-resistance pathways (“channels”) for ligands to access the main FCC and simultaneously force the W254 chi-1 dihedral to rotate into the “open” orientation. As shown in Fig. 10, a “vertical” channel was oriented approximately perpendicular to the longitudinal major axis of the FCC. Although ligand access to the vertical channel (entry point bracketed by furin residues Asp233, Asp265, Pro256 and Ala267) was relatively unobstructed by residue sidechains, it required that the ligand must undergo an abrupt 90-degree change in direction at one point to become aligned with the W254 residue in order to assure a successful collision that could result in the large shift in its dihedral bond.
Fig. 10.

[A] Periodic boundaries (10-Å cube) showing orientation of the 5JXG furin receptor and BOS-318 ligand(s) positioned at the “vertical” and “side” channel entry points. The side-channel was oriented approximately parallel with the FCC, whereas the vertical channel was roughly perpendicular to the FCC. [B] Close-up view of the 5JXG furin model showing the vertical and side entry channels as Van der Waals surfaces (light brown shading) and the remainder of the protein as the water-accessible surface (1.4-Å probe radius; translucent blue shading). Dotted lines with arrows indicate approximate iMD trajectories for the respective entry channels.
A second “horizontal” or “side” channel was oriented more-or-less parallel with the major (long) axis of the FCC. The ligand entry point for this channel was bracketed (approximately) between furin residues Ser279, Pro446, Gln447, Arg448, Lys449 and Phe275. For both the side and vertical entry channels, the terminal methyl group of BOS-318, or the tetrazole moiety of Mod23, were first to access the channel. The BOS-318 ligand extracted from the 7LCU x-ray structure is shown positioned at the opening of the vertical and side channels in Fig. 10. These ligand arrangements correspond to the iMD starting configurations used in the iMD docking simulations described in the sections that follow.
BOS-318 and the other ligands evaluated in the iMD docking simulations traveled from a bulk aqueous phase into a largely de-solvated furin interior. Thus, it was essential to launch the simulations using a reasonable solution-phase ligand conformation. A 170-ns eqMD simulation of BOS-318 in physiological saline (0.9 wt% NaCl) at a constant temperature of 311oK was performed to compare the solution-phase drug conformation (averaged over 17.0 ns in 170 steps) against that of BOS-318 extracted from the 7LCU x-ray crystallographic structure. Superimposition of the two structures indicated an all-atom RMSD = 2.197 Å (data not shown). Because the difference in the two conformations was relatively small, the BOS-318 x-ray structure was employed in the iMD studies described in this report. While guided ligand entry into either channel could be expected to result in collision with W254 and, with sufficient applied acceleration, induce the chi-1 dihedral bond of W254 to correctly rotate into the desired (open) orientation, no significant change in ligand direction was needed to ensure an effective W254 collision for a side-channel trajectory.
Numerous iMD runs were carried out for this study using either the vertical or side-channel pathways. In both instances, the appropriate W254 torsional shift was observed with concomitant stable binding of the DCP (or halo-phenyl) group of drugs into the newly exposed hydrophobic pocket. However, because the side channel offered a more direct route to W254 and did not require any abrupt direction changes, only results from two exemplary iMD runs using the side channel will be described the sections below.
3.4.2. IMD Simulation of Side-Channel Entry for Ligand BOS-318
The initial setup configuration at t = 0 ps for the side-channel entry iMD docking is shown in Fig. 10; and the simulation kinetics, including the net ligand velocity and acceleration, are summarized in Fig. 11. The total duration of the iMD docking simulation was about 53 ps. Acceleration bias (i.e., iMD) was discontinued at approximately 19–20 ps into the simulation. The side-channel route of entry (depicted in Fig. 10) formed a relatively open pathway to the W254 residue and, unlike the vertical-channel iMD simulation, only required minimal alteration of the direction of the lead methyl carbon atom to remain on an effective collision trajectory with W254.
Fig. 11.

Results of an iMD docking simulation for entry of BOS-318 through the furin side channel. The duration of the simulation was 52 ps. The velocity (blue bars) and acceleration (orange bars) applied to the leading methyl carbon atom (yellow sphere) are plotted as a function of simulation time (ps) in the center panel. Gray line: ligand binding free energy (kcal/mol; higher values = stronger binding). Velocity and acceleration remained relatively consistent for the initial 15–16 ps of the iMD simulation (gray shaded region). As the ligand approached the “closed” W254 residue (green shaded zone), additional acceleration was applied (about 0.5–1.8 Å/ps^2; green arrow) to force the chi-1 dihedral of W254 through a nearly 180-degree rotation, thereby flipping the residue into the “open” orientation. See text for additional details.
As indicated by the kinetic data in Fig. 11, the net velocity and acceleration remained relatively consistent for the initial 15 ps of the iMD simulation (gray shaded region) which corresponded to entry and initial transport of the drug into the channel. However, as the ligand approached the “closed” W254 residue, additional acceleration was required (about 5 Å/ps^2) to overcome (primarily Coulomb) repulsion and force the chi-1 dihedral of W254 through a nearly − 180-degree rotation, thereby flipping the residue into the “open” orientation enabling stable drug binding.
Immediately following the W254 dihedral shift (at around 15–24 ps), the DCP moiety of BOS-318 settled abruptly into the newly exposed hydrophobic pocket (blue shaded region of Fig. 11), an event that marked completion of ligand docking and cessation of all iMD user input bias. The series of image frame captures acquired along the iMD trajectory (Fig. 11, upper and lower panels) reveal the progression of the BOS-318 ligand as it approached and collided with W254. Visual inspection of the dynamics of the sudden entry and binding of the DCP moiety into the cryptic pocket appeared to help orient BOS-318 for optimal interaction with the FCC. Binding of the non-polar DCP group was associated with modest fluctuations of both the net velocity and acceleration of the lead methyl carbon atom. Subsequent to binding of the DCP group at about 25 ps (and discontinuance of iMD input at about 20 ps), the simulation entered a brief 22-ps equilibrium MD phase (orange shaded region, Fig. 11, orange shaded area). During this period the ligand underwent additional energy minimization, and the free energy of ligand binding grew stronger (Note: more positive values in the figure = stronger binding).
Oscillations in the ligand and furin receptor conformations expressed in terms of all-atom RMSD metrics are summarized in Fig. 12. Ligand docking at about 17–20 ps coincided with an approximately 2.0-Å increase in the BOS-318 RMSD value. The steepest rate of ligand conformational change (0.524 Å/ps^2 at 17 ps, calculated as ɗRMSD) coincided with stable docking of BOS-318 into the catalytic cleft.
Fig. 12.

Total system energy (orange line, kcal/mol), and all-atom RMSD values for furin 5JXG (blue line) and BOS-318 (gray line) over the course of the 53-ps intMD docking of ligand through the side-channel entry. Ligand docking was completed by about 19–20 ps. As reflected by an approximately 2.5-Å increase in the ligand RMSD score (gray line), completion of docking at about 15–20 ps was associated with moderate divergence from the initial (solution-phase) ligand conformation. Green shading and discs indicate region where images frames shown in upper panel were captured.
The embedded images in Fig. 12 show that this was the time (about 17 ps) when ligand-W254 collision occurred. An additional 4 ps passed until the W254 dihedral rotation was completed at about 24 ps. This is also the same period when the total system energy increased, suggesting an overall modest (but temporary) instability of the complex. The surge in the system energy was accompanied by a corresponding increase in the furin RMSD value by approximately 1.0 Å, presumably resulting from kinetic (thermal) input into the system by the acceleration bias applied to the ligand. The enhanced thermal motion of the 5JXG receptor may in part explain the observed increase in furin-solvent hydrogen bonding as discussed below (see Fig. 14).
Fig. 14.

Ligand-solvent and receptor(5JXG)-solvent hydrogen bond counts over the 53-ps BOS-318 iMD trajectory demonstrating partial dehydration of the ligand as it entered the side-channel of 5JXG. Primarily solution-phase water molecules located within 5.0 Å of the ligand at t = 0 ps are rendered as yellow spheres. Primarily protein-associated water molecules located within 5.0 Å of the ligand at t = 53 ps are rendered as magenta-colored spheres. All other water molecules and ions have been hidden for clarity. The majority of the solution-phase water associated with the ligand at t = 0 ps were stripped away from the ligand during the intMD trajectory. The ligand was partially rehydrated following docking into the catalytic cleft. Moreover, two trends were apparent: (1) Water molecules (yellow) that dissociated from the ligand during side-channel entry became dispersed as they diffused into the bulk solvent phase, suggesting an increase in system entropy; and (2) Water molecules that ultimately became associated with the bound ligand (magenta) gradually increased in density (around the bound ligand) compared to their initial t = 0 state, presumably due to attractive interactions with the ligand. Re-organization of water molecules around the ligand suggests a possible decrease in system entropy. These observations are consonant with the hydrogen-bonding trends shown in the graph (lower panel), which is color-coded according to the sequence of intMD docking events, including: (1) Initial ligand de-solvation phase (green shading); (2) Ligand binding in the cleft (yellow shading); (3) Post-iMD equilibration phase (gray shading); and (4) Ligand re-hydration (blue shading).
Analysis of BOS-318 motion on a per-atom RMSF basis revealed that the chlorine #3 atom (Cl3) displayed the most positional delocalization, mainly resulting from deformation of the bulky non-polar DCP group as it transited the side channel and docked into the hydrophobic pocket (see Fig. 13B). As shown in the series of associated frame captures in Fig. 13 A, the non-polar group was free to undergo dihedral rotation during the iMD trajectory resulting in large spatial displacements for the two halogen atoms.
Fig. 13.

[A] Image frame captures from the 53-ps iMD docking of BOS-318 through the 5JXG side channel. The 3,5-di-chlorophenyl group underwent significant dihedral rotation and partial deformation (via angle bending) as it traversed the side channel resulting in elevated per-atom RMSF oscillations for this non-polar group. [B] Per-atom RMSF metrics for the BOS-318 ligand tabulated over the 53-ps iMD trajectory. The chlorine atoms exhibited comparatively high RMSF fluctuations.
The behavior of ligand solvation shells was monitored and documented for all of the iMD simulations performed in this investigantion, ostensibly for the reason that entropy effects during ligand solvation/desolvation can strongly influence drug migration and binding [61], [62]. Ligand solvation behaviorial patterns were nearly identical for both the side-channel and vertcial-channel iMD simulations. Ligand-solvent hydrogen bonding analyses presented in Fig. 14 demonstrated that BOS-318 underwent nearly complete dehydration as it traversed from the bulk solution (TIP3P water) phase into the FCC. While the ligand relinquished approximately four (4) solvent hydrogen bonds, the 5JXG receptor gained nearly eighty (80), suggesting ligand transit and docking somehow facilitated solvent interactions and enhanced hydrogen bonding with the protein (Fig. 14, graph in lower panel).
The images shown in Fig. 14 indicate that the bulk of the water molecules (marked as yellow spheres) surrounding the ligand within a 5.0-Å solvation shell at the start of the iMD docking run did not co-migrate with the ligand into the side channel. Thus, the notion that significant numbers of water molecules were drawn into the channel by the ligand and subsequently underwent hydrogen bonding to the protein is unsupported. Moreover, most water molecules that re-hydrated the bound ligand by the end of the iMD simulation (shown as magenta spheres) originated from the bulk solution phase (plus a few from the catalytic cleft domain) near the mouth of the channel. Whereas, the precise mechanism of increased water-furin hydrogen bonding remains unclear, the above observations are perhaps most consistent with a process in which iMD-induced conformational perturbation of the furin receptor led to enhanced water-furin hydrogen bonding, presumably by exposing partially buried hydrophilic sidechains or backbone amine and carbonyl groups.
As indicated by the residue RMSD and RMSF metrics presented in Fig. 15, the majority of furin residues displayed RMSD values less than about 1.5 Å over the course of the iMD docking, with only about 10 residues exceeding 2.0 Å. Not surprisingly, the greatest residue motion displacement was exhibited by W254 with an RMSD value of 5.315 Å (similar to that for the vertical channel simulations; data not shown). This displcement was attributable to the large dihedral shift invoked by ligand collision. The RMSD and RMSF oscillations in atom positions was reflected in the residue B-factor color-coding depicted in the furin image inset in Fig. 15, with yellow being indicative of maximal residue delocalization (relative to mean rotamer displacements) across the iMD trajectory.
Fig. 15.

RMSD and RMSF metrics for furin residues computed over the 53-ps iMD simulation for the BOS-318 side-entry channel. All residues exhibiting RMSD values > 2.0 Å are labeled. Image inset shows the 5JXG furin protease color-coded according to residue B-factor: yellow > >> > mean residue motion (MRM); orange > > MRM; magenta > MRM; blue = MRM.
To evaluate the stability of the iMD-docked BOS-318 ligand, equilibrium MD simulation of the ligand-5JXG complex was carried out in physiological saline (0.9 wt% NaCl) at constant temperature of 311oK for a period of about 102 ns (see Fig. 16). The starting structure for the MD simulation was the last frame of the 53-ps iMD side-channel docking run. The RMSD metrics indicate the iMD-docked ligand remained stably bound in the FCC for the duration of the eqMD simulation.
Fig. 16.

Equilibrium MD simulation of the 53-ps iMD docked BOS-318-furin complex. [A] Ligand (rendered as yellow spheres; gray line data) and 5JXG receptor (orange line) RMSD metrics and total system total energy (blue line) over the 102-ns MD trajectory. W254 is rendered as red ball-and-stick. The iMD-docked ligand remained stably bound in the FCC over the course of the equilibrium MD trajectory, although some minor conformational oscillations were observed between 23 and 35 ns. [B] Ligand free energy of binding (orange line) and ligand RMSD (blue line) as a function of eqMD time. Note: more positive ligand binding energies correspond to stronger binding in the receptor pocket. The aforementioned ligand instability was correlated with diminished ligand binding strength.
There were slightly increased ligand oscillations observed between 23 and 35 ns as indicated by a rise in ligand RMSD values from about 1.0–1.5 Å (Fig. 16B). However, even during this period of minor instability the ligand did not exit the pocket. The ligand free energy of binding was only moderately weaker during this period (Fig. 16C).
Superimposition of the BOS-318-liganded 7LCU x-ray structure against the final BOS-318 liganded iMD 5JXG structure averaged over the 100-ns follow-up eqMD run to dampen thermal noise is shown in Fig. 17. The overall RMSD displacement of the superimposed protein structures (i.e., 100-ns averaged 5JXG versus PDB 7LCU) was 1.859 Å, whereas the RMSD for the superimposed ligands was 3.346 Å. These results are consistent with the view that the iMD approach employed in this study can successfully approximate initial ligand binding motifs and free energy of binding over follow-up eqMD simulations.
Fig. 17.

[A] Superimposition of liganded 7LCU x-ray structure against the last frame (53 ps) for side-entry of iMD simulation. [B] Magnified view showing only the ligands and W254 residues.
3.4.3. IMD Simulation of Side-Channel Entry for Ligand Mod23
As described earlier in this report, it was possible to incrementally improve the docking strength of BOS-318 by means of a series of modifications to its structure. The BOS-318 analog Mod23 bearing a terminal anionic ethyl-tetrazole group exhibited a significant increase in the free energy of binding compared to the parent BOS-318 ligand (see Fig. 6, above). Therefore, it was of interest to: (1) Determine how Mod23 would behave during an iMD docking simulation; and (2) Test if an iMD approach would effectively emulate the VINA docking motif for this ligand. For this evaluation, the more direct and less obstructed furin side channel route was utilized, and the initial iMD setup is shown in Fig. 18. The iMD acceleration bias was applied to the ethyl-carbon atom #7061 near the terminal tetrazole group (see Fig. 18F). This atom was selected as the “lead” atom since it permitted full flexibility of the anionic ethyl-tetrazole group, which was important for the ability of the functional group to negotiate any sidechain obstacles.
Fig. 18.

Interactive MD furin side-channel docking of the Mod23 ligand variant of BOS-318. [A] Starting configuration of the Mod23–5JXG furin complex at t = 0 ps. The Mod23 ligand is viewed down the X-axis (toward the origin where x = 0). [B] Mod23 viewed down the Z-axis (“side” view of complex). [C] Mod23 viewed down the Y-axis (“top-down” view of the complex). Red arrow indicates the approximate intMD trajectory of the Mod23 ligand. [D] Same perspective as in [A] showing the residues (yellow ball-and-cylinder rendering) outlining of the relatively open channel leading to W254 and the binding pocket in the furin catalytic cleft. [E] Magnified view of [D] depicting the relatively open pathway to residue W254 (red colored stick rendition). [F] Structure of Mod23 indicating terminal anionic ethyl-tetrazolate group and carbon atom 7061, to which the acceleration bias was applied.
The iMD docking simulation spanned 132 ps and the kinetic results are summarized in Fig. 19. As indicated by the frame captures shown at 0 ps, 50 ps and 65 ps iMD docking was effectively complete by 65 ps, after which the ligand-furin complex was permitted to equilibrate for an additional 67 ps. Cartesian space acceleration vectors (dVx, dVy, dVz) acting on carbon atom 7061 of the Mod23 ligand over the 132 ps iMD simulation trajectory are plotted in Fig. 19B. Also, the trajectory has been divided by color-coding into key docking events, such as “ligand approach”, “docking in the cleft domain”, etc.
Fig. 19.

Interactive MD trajectory of ligand Mod23. [A] Depicts iMD frame captures at 0 ps, 50 ps and 65 ps showing progress of Mod23 docking into the furin catalytic cleft. Docking was effectively complete by about 65 ps, after which the ligand-furin complex continued to equilibrate. [B] Plotted over the 132 ps iMD simulation are net acceleration (orange bars) and velocity (blue bars) for atom 7061 of the ethyl-tetrazole group of Mod23 (see molecular structure inset). Gray line: Ligand binding energy (kcal/mol); higher values indicate stronger binding. It is noteworthy that the strongest ligand binding was associated with entry of Mod23 into the furin catalytic cleft. The trajectory is color-coded to reflect notable events in the trajectory: Gray shading = period of ligand approach to the furin entry channel; Green shading = ligand entry into and partial binding within the furin catalytic cleft. This is the period in which the chi-1 dihedral of W254 undergoes rotation approximately 180 degrees to better accommodate Mod23; Blue shading = period in which the 3-chloro-5-bromo-phenyl group enters and rapidly transitions into the newly exposed hydrophobic (cryptic) pocket. Yellow shading = post-binding equilibration phase.
The free energy of ligand binding is shown as the blue line in Fig. 19B (note that higher values indicate stronger binding). It is noteworthy that the strongest ligand binding was associated with entry of Mod23 into the furin catalytic cleft at approximately 55–60 ps. The non-polar 3-chloro-5-bromo-phenyl group of Mod23 docked abruptly into the newly exposed cryptic hydrophobic pocket following W254 rotation at about 60–70 ps. Docking of the non-polar group was associated with a significant but transient increase in the dVx acceleration vector of atom 7061. Ligand docking was effectively completed by about 65 ps, at which time iMD acceleration bias was terminated. Inspection of image frame captures suggested that the non-polar halo-phenyl group behaved in a manner to suggest it played a role in influencing the proper approach and orientation of Mod23 to optimize its interaction with the FCC pocket.
Furin residue heavy-atom RMSD and RMSF values are presented in Fig. 20 A, which demonstrates that W254 underwent the greatest conformational change (RMSD > 6.0 Å) over the 132-ps iMD trajectory. A few other residues proximally located along the side-channel pathway (e.g., Val231, Leu227, Gln188) also exhibited moderate to strong perturbations as the ligand traversed the channel. The RMSD and RMSF metrics are reflected in the furin model in Fig. 20B whose residues are color-coded according to their respective B-factors, with yellow coloring indicative of the greatest atomic motion or displacement.
Fig. 20.

[A]: Root mean square fluctuation (RMSF) values as a function of furin residue number over the course of the 132-ps iMD trajectory. RMSF indicates positional differences between entire structures over time and calculates individual residue flexibility. The large fluctuation of W254 is attributed to its forced (ligand induced) rotation about its chi-1 dihedral angle. It is noteworthy that 4–5 additional residues also exhibited some motions in excess of about 1.5 Å RMSF relative to their t = 0 conformations. Such motion was attributed to interaction of Mod23 with residue side chains comprising the entry channel to the furin binding pocket. [B]: B-factor coloration of residues computed over the 132-ps intMD trajectory. B-factor (also referred to as the Debye-Waller factor, temperature factor, or atomic displacement parameter) is used in protein crystallography to quantify attenuation of X-ray scattering by thermal motion and reflects residue flexibility. Furin is depicted as a residue trace with small spheres marking residue positions along the protein backbone. The magnitude of residue motion is color-keyed as follows: Yellow > > Orange > > Magenta > > Blue (frozen).
As was the case for BOS-318 (Fig. 14; also see Supplemental Data), the Mod23 ligand similarly experienced partial dehydration (indicated by a reduction in ligand-solvent hydrogen bonding) as it passed into and transited the length of the furin side channel. As indicated by the color-coded water molecules in Fig. 21, nearly all of the initial 5-Å Mod23 solvation shell was shed (yellow water molecules) as the ligand was guided into and through the channel. Upon docking into the FCC, Mod23 underwent partial re-hydration by water molecules primarily originating in the bulk solvent phase at the mouth of the channel (magenta-colored water).
Fig. 21.

[A] Reduction of ligand-solvent hydrogen bonding and increase in furin-solvent hydrogen bonding over the course of the 132-ps Mod23 iMD side-channel trajectory. The loss of ligand-solvent hydrogen bonds indicates that Mod23 became partially de-solvated as it entered the side channel and docked in the furin catalytic cleft. [B] Image frame captures from the intMD docking simulation. Water molecules located within 5.0 Å from the ligand at t = 0 ps are rendered as yellow ball-and-cylinders. Water molecules located within 5.0 Å from the ligand at t = 132 ps are rendered as magenta-colored ball-and-cylinders. Two trends are evident: (1) Dehydration of the ligand, i.e., loss of solvent-phase water molecules from the original 5.0-Å water shell (yellow) as the ligand entered into and transited the furin side channel; and (2) Ligand re-hydration and densification primarily by solution-phase water molecules (magenta) as docking proceeded. Note that water molecules and ions outside of the 5.0-Å drug solvent shells have been hidden for clarity.
Also similar to the BOS-318 iMD docking runs described above, the initial water shell rapidly dispersed back into the bulk solution around the channel entrance as the ligand approached and negotiated the side channel. Moreover, water molecules that re-hydrated Mod23 progressively increased in density around the docked ligand. Although no quantitative (mass-balance) analysis was performed, the images in Fig. 21 suggest that greater numbers of water molecules underwent dispersion compared to those that eventually re-hydrated the bound ligand. Thus, it may be reasonably speculated that a net gain in entropy could be expected for the system, which in turn might contribute to the thermodynamic stability of the docked ligand.
The iMD-docked Mod23 ligand-furin complex (taken at 132 ps) was used as the starting structure for a 90-ns eqMD simulation run at 311oK in physiological saline (see Methods), the results of which are presented in Fig. 22. As shown in Fig. 22A, the total system energy was relatively constant over the trajectory, although the RMSD values of the ligand and furin each showed modest increases that tended to level off after about 60 ns. Despite the modest RMSD increases, Mod23 remained stably bound in the FCC for the duration of the 90-ns simulation (see images in Fig. 22B).
Fig. 22.

Results from a 90-ns eqMD simulation of the iMD-docked Mod23 ligand (constant-temperature NPT ensemble at 311oK, physiological saline; 0.9 wt%NaCl). The starting complex was the last frame from the 132-ps iMD docking run (see text for details). [A] All-atom RMSD metrics for the Mod23 ligand (gray line) and the 5JXG furin receptor (orange line), and the total system energy as a function of MD simulation time. Although the Mod23 ligand exhibited some conformational fluctuation over the MD trajectory, it remained stably bound in the furin catalytic cleft. [B] Series of image frame captured over the course of the MD trajectory. Furin is shown as the water-accessible surface using a 1.4-Å probe radius.
4. Discussion
Computational results presented in this communication indicate that docking of known inhibitors of human furin (e.g., the dichlorophenyl-pyridine, BOS-318) to the catalytic domain using modern flexible-receptor methods, including AutoDock VINA and ICM from Molsoft, failed to emulate the induced-fit mechanism experimentally demonstrated by Dahms and coworkers [1]. Flexible-receptor docking also failed for a series of BOS-318 analogs (e.g., Mod23 harboring a terminal ethyl-tetrazole group) that exhibited moderately improved docking energies relative to the parent drug.
In the induced-fit mechanism described by Dahms et al. [1], the furin W254 residue acts as a molecular gateway that pivots from its closed ground-state position to an open orientation around its chi-1 dihedral angle nearly 180 degrees in response to binding of BOS-318 (or structurally-similar ligands). When W254 is in the open state, the DCP group (or analogs thereof) of drugs can undergo binding to a newly exposed deep hydrophobic cryptic pocket situated beneath the FCC.
Mutational analyses presented in this report demonstrated that W254G of 7QY0 (open gate) with a hydrogen atom “sidechain”, or W254F (rotated to the open position), permitted ligand access to the cryptic pocket. Interestingly, the order of ligand binding free energies from stronger to weaker was: W254 > W254F > W254G. Thus, despite the much smaller sidechain footprint of glycine, drug binding in the case of W254G was noticeably weaker compared to the open-gate W254 or W254F, suggesting the indole group of the former and the phenyl group of the later enhanced and strengthened ligand-receptor interaction. Inspection of docked poses indicated that the indole and phenyl sidechains of W254 and W254F entered into hydrophobic bonds, π-π resonance and ionic (salt-bridge) interactions with most ligands.
Steered MD simulations in which the iMD-docked BOS-318 and Mod23 ligands were dynamically extracted from the FCC demonstrated the hydrophobic DCP group of BOS-318 and the halo-phenyl group of Mod23 made relatively minor contributions to the overall binding energies of the intact ligands. The hydrophobic groups were likely instrumental in stabilizing drug binding by assisting in guiding ligand approach and final drug conformation in the FCC. It was also observed that none of the sMD extractions caused the open W254 gate to re-close, which was consonant with the comparative stability of the W254 dihedral angles in the fully open and closed orientations. Because flexible-receptor docking could not mimic the induced-fit mechanism of BOS-318 and its analogs, an iMD approach was employed to delineate potential ligand trajectories leading to optimal drug binding in the main catalytic cleft of the human furin endopeptidase 5JXG with the W254 gate in the closed (ground-state) orientation. The iMD docking simulations performed in this study illustrated potential modes of entry of BOS-318, as well as various tetrazole-bearing structural analogs with similar binding strengths, into the FCC coupled with collision-induced chi-1 dihedral rotation of W254 into the open orientation.
Forces applied to guide ligand movements were calculated from the raw coordinates and elapsed-time data for the lead atom considering the mass of the ligand. Ligand forces (for Mod23 with a mass of ∼573 g/mol) ranged from about 3–20 pN as the ligand navigated from the bulk solution phase into the side-channel entry point at velocities ranging from about 3–5 Å/ps. Similar forces were applied during transport of the ligand through the channel interior. However, to achieve complete W254 rotation into the open orientation during ligand collision, applied forces were briefly spiked to ∼160 pN. The calculated forces fall within the range reported for many non-bond type molecular interactions that have been studied, e.g., attractive Van der Waals, hydrophobic and electrostatic interactions that mediate protein interactions, nucleic acid conformations [56], [57], [58], as well as covalent bond breakage (e.g., carbon-iron single bond [59]).
Calculations of the force needed for the chi-1 rotation of W254 through − 180 degrees (from +34 degrees to −146 degrees) based on the observed torsional barrier of ∼158 kcal/mol (see Fig. 3 C) indicated 10,421 pN would be required. This force is approximately 68-fold greater than the computed force applied to Mod23 (about ± 160 pN) in the iMD side-channel entry simulation in which W254 was successfully rotated. Given such a large discrepancy between the applied force and required force, it remains unclear how Mod23 (or BOS-318) could overcome the large 158-kcal/mol dihedral barrier of W254. One possible explanation is that the vectored acceleration applied to the ligands during iMD docking may transfer kinetic energy to furin residues comprising the FCC domain of a sufficient magnitude to lower the W254 torsional barrier below that of the applied force on the ligand. Such a scenario would be equivalent to injecting additional thermal energy into the furin receptor, thereby elevating its overall (or localized) energy landscape to a more excited state where larger torsion barriers and displacements could be sampled. The B-factor calculations (and corresponding colored-keyed images) of 5JXG shown in Fig. 15 and 20 suggest some level of kinetic perturbation of residues in the vicinity of the FCC and entry channels, especially for the case of iMD docking of the Mod23 ligand (Fig. 20). It seems also plausible that increased kinetic input to the protein could lead to a reduction of the dihedral energy barrier of W254, as well as other furin residues, thus allowing the ligand-induced rotations.
The question of whether forces of this magnitude are thermodynamically accessible at ambient and physiological temperatures was beyond the scope of the present investigation. The crucial role of solvation shells in the bioenergetics of drug-receptor interactions is well documented [35], [60], [61]. However, water-solute interactions in the systems modeled herein are complicated by re-solvation of the ligand upon binding in the FCC coupled with expanded solvation of the furin receptor, with the later far exceeding the former in terms of the number of water molecules involved. Theoretical and experimental estimates of the entropic penalty associated with the transfer of a single water molecule to a drug binding cavity range from about + 0.46 kcal/mol for mostly hydrophobic pockets to + 2.67 kcal/mol for more hydrophilic pockets, with an average value of about + 2.0 kcal/mol [62]. Thus, loss of 3 water molecules from the ligand solvation shell during transport into the FCC (see Fig. 13, Fig. 20) could result in an entropy reward of approximately − 6.0 kcal/mol for ligand binding, which translates into more than one order of magnitude increase in ligand binding strength [63]. Furthermore, Huggins [62] argues that the entropic penalty is often offset by larger (favorable) gains in system enthalpy due to formation of stable solvent-solute hydrogen bonding and attractive polar interactions. Lacking a rigorous quantitative analysis, given the extent of ligand dehydration (followed by re-solvation) concomitant with enhanced protein solvation observed in our models during drug entry and binding, entropic and enthalpic fluctuations associated with solvation interactions are likely to play influential roles in docking by mechanisms outlined in this investigation [63], [64], [65]. Additional physico-chemical analytical techniques will be needed to verify and extend the computational findings presented in this paper. For example, high-resolution nuclear magnetic resonance (NMR) (specifically 2D NMR methods) could help resolve and confirm predicted interactions between ligands and proteases, as well between ligands and specific interacting residues [65].
Declaration and Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
J.M.M. and K.K. would like to express their gratitude to start up company New Drug /NeoFar located at Patras Science Park, Greece, for providing the personnel and the infrastructure to develop bisartans the new generation of antivirals. They also thank the Region of Western Greece (Research and Technology) for funding (NeoFar financial support through MIS 5092131-action 110051339 grant) and their invaluable support throughout all the necessary steps in the implementation of the current research. C.T.C. would like to thank the National Research Foundation (NHRF) for supporting the research work by providing a Research Seed Grant. V.A., H.R., J.O. and J.M.M would like to thank the Institute for Health and Sport (IHES), and the Institute for Sustainable Industries and Liveable Cities (ISILC) at Victoria University, Melbourne Australia, for their support. V.A. was also supported through the Vice-Chancellors Distinguished Fellows scheme, VU Research, Victoria University.V.A. and H.R. were supported by a Planetary Health Grant PH098 from Victoria University. V.A. would like to thank the Greek Orthodox Archdiocese of Australia Funds, and the VU Vaccine appeal Funds, whose generous support made possible the research of this paper. G.J.M. would like to thank PepMetics for its support. V.G.G. received support from the National Public Investment Program of the Ministry of Development and Investment/General Secretariat for Research and Technology, in the framework of the Flagship Initiative to address SARS-CoV-2 (2020ΣΕ01300001); the Welfare Foundation for Social & Cultural Sciences (KIKPE), Athens, Greece; H. Pappas donation; grants no. 775 (Hippo) and 3782 (PACOREL) from the Hellenic Foundation for Research and Innovation (HFRI); and NKUA-SARG grant 70/3/8916. V.G.G. is thankful to Bodosakis Foundation for funding the work in Biosafety level 3 facility. We thank Nikolaos Maniotis for editing and proofreading this manuscript.
Author contributions
Conceptualization, H.R, J.M.M and C.T.C; Experimental Work and Methodology, H.R, J.M.M and C.T.C.; Writing—original draft preparation, H.R, J.O, M.T.M, K.K, G.J.M, S.T, J.M.M and C.T.C.; Writing—review and editing, H.R, J.O, M.T.M, K.K, G.J.M, S.T, V.A., V.G.G., J.M.M and C.T.C.;. All authors have read and agreed to the published version of the manuscript.
Contributor Information
Harry Ridgway, Email: ridgway@vtc.net.
Christos T. Chasapis, Email: cchasapis@eie.gr.
Vasso Apostolopoulos, Email: Vasso.Apostolopoulos@vu.edu.au.
John M. Matsoukas, Email: imats1953@gmail.com.
References
- 1.Dahms S.O., et al. Dichlorophenylpyridine-based molecules inhibit furin through an induced-fit mechanism. ACS Chem Biol. 2022;17:816–821. doi: 10.1021/acschembio.2c00103. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol. 2002;3:753–766. doi: 10.1038/nrm934. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Than M.E., et al. The endoproteinase furin contains two essential Ca2+ions stabilizing its N-terminus and the unique S1 specificity pocket. Acta Crystallogr Sect D Biol Crystallogr. 2005;61:505–512. doi: 10.1107/s0907444905002556. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 4.Braun E., Sauter D. Furin‐mediated protein processing in infectious diseases and cancer. Clin Transl Immunol. 2019;8 doi: 10.1002/cti2.1073. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez C., et al. Plasma levels of the proprotein convertase furin and incidence of diabetes and mortality. J Intern Med. 2018;284:377–387. doi: 10.1111/joim.12783. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swärd P., Rosengren B.E., Jehpsson L., Karlsson M.K. Association between circulating furin levels, obesity and pro‐inflammatory markers in children. Acta Paediatr. 2021;110:1863–1868. doi: 10.1111/apa.15774. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 7.Ren K., Jiang T., Zheng X.-L., Zhao G.-J. Proprotein convertase furin/PCSK3 and atherosclerosis: new insights and potential therapeutic targets. Atherosclerosis. 2017;262:163–170. doi: 10.1016/j.atherosclerosis.2017.04.005. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 8.Fitzgerald K. Furin protease: from SARS CoV-2 to anthrax, diabetes, and hypertension. Perm J. 2020;24 doi: 10.7812/tpp/20.187. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bennett B.D., et al. A furin-like convertase mediates propeptide cleavage of BACE, the Alzheimer's β-secretase. J Biol Chem. 2000;275:37712–37717. doi: 10.1074/jbc.M005339200. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y., et al. A second functional furin site in the SARS-CoV-2 spike protein. Emerg Microbes Infect. 2022;11:182–194. doi: 10.1080/22221751.2021.2014284. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jamison D.A., et al. A comprehensive SARS-CoV-2 and COVID-19 review, Part 1: intracellular overdrive for SARS-CoV-2 infection. Eur J Hum Genet. 2022;30:889–898. doi: 10.1038/s41431-022-01108-8. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lamers M.M., Haagmans B.L. SARS-CoV-2 pathogenesis. Nat Rev Microbiol. 2022;20:270–284. doi: 10.1038/s41579-022-00713-0. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 13.Zhang Q., et al. Molecular mechanism of interaction between SARS-CoV-2 and host cells and interventional therapy. Signal Transduct Target Ther. 2021;6 doi: 10.1038/s41392-021-00653-w. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Villoutreix B.O., Badiola I., Khatib A.-M. Furin and COVID-19: structure, function and chemoinformatic analysis of representative active site inhibitors. Front Drug Discov. 2022;2 doi: 10.3389/fddsv.2022.899239. (https://doi.org:) [DOI] [Google Scholar]
- 15.Osadchuk T.V., Shybyryn O.V., Kibirev V.K. Chemical structure and properties of low-molecular furin inhibitors. Ukr Biochem J. 2016;88:5–25. doi: 10.15407/ubj88.06.005. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 16.Wu C., et al. Furin: a potential therapeutic target for COVID-19. iScience. 2020;23 doi: 10.1016/j.isci.2020.101642. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cheng Y.-W., et al. Furin inhibitors block SARS-CoV-2 spike protein cleavage to suppress virus production and cytopathic effects. Cell Rep. 2020;33 doi: 10.1016/j.celrep.2020.108254. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kacprzak M.M., et al. Inhibition of furin by polyarginine-containing peptides. J Biol Chem. 2004;279:36788–36794. doi: 10.1074/jbc.M400484200. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 19.Örd M., Faustova I., Loog M. The sequence at Spike S1/S2 site enables cleavage by furin and phospho-regulation in SARS-CoV2 but not in SARS-CoV1 or MERS-CoV. Sci Rep. 2020;10 doi: 10.1038/s41598-020-74101-0. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sielaff F., Than M.E., Bevec D., Lindberg I., Steinmetzer T. New furin inhibitors based on weakly basic amidinohydrazones. Bioorg Med Chem Lett. 2011;21:836–840. doi: 10.1016/j.bmcl.2010.11.092. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Osman E.E.A., Rehemtulla A., Neamati N. Why all the fury over furin. J Med Chem. 2021;65:2747–2784. doi: 10.1021/acs.jmedchem.1c00518. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 22.Dahms S.O., et al. X-ray structures of human furin in complex with competitive inhibitors. ACS Chem Biol. 2014;9:1113–1118. doi: 10.1021/cb500087x. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swiderski J., et al. Role of angiotensin ii in cardiovascular diseases: introducing bisartans as a novel therapy for coronavirus 2019. Biomolecules. 2023;13:787. doi: 10.3390/biom13050787. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ridgway H., et al. Discovery of a new generation of angiotensin receptor blocking drugs: receptor mechanisms and in silico binding to enzymes relevant to SARS-CoV-2. Comput Struct Biotechnol J. 2022;20:2091–2111. doi: 10.1016/j.csbj.2022.04.010. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ridgway H., et al. Understanding the driving forces that trigger mutations in SARS-CoV-2: MUtational Energetics and the Role of Arginine Blockers in COVID-19 therapy. Viruses. 2022;14:1029. doi: 10.3390/v14051029. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ridgway H., et al. Molecular epidemiology of SARS-CoV-2: the dominant role of arginine in mutations and infectivity. Viruses. 2023;15:309. doi: 10.3390/v15020309. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duarte M., et al. TelmiSartan for treatment of Covid-19 patients: an open multicenter randomized clinical trial. EClinicalMedicine. 2021;37 doi: 10.1016/j.eclinm.2021.100962. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elkahloun A.G., Saavedra J.M. CandeSartan could ameliorate the COVID-19 cytokine storm. Biomed Pharmacother. 2020;131 doi: 10.1016/j.biopha.2020.110653. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kelaidonis K., et al. Computational and enzymatic studies of sartans in SARS-CoV-2 Spike RBD-ACE2 binding: the role of tetrazole and perspectives as antihypertensive and COVID-19 therapeutics. Int J Mol Sci. 2023;24:8454. doi: 10.3390/ijms24098454. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu B., et al. Furin promotes dendritic morphogenesis and learning and memory in transgenic mice. Cell Mol Life Sci. 2018;75:2473–2488. doi: 10.1007/s00018-017-2742-3. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Essalmani R., et al. Distinctive roles of furin and TMPRSS2 in SARS-CoV-2 infectivity. J Virol. 2022;96 doi: 10.1128/jvi.00128-22. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oleinikovas V., Saladino G., Cossins B.P., Gervasio F.L. Understanding cryptic pocket formation in protein targets by enhanced sampling simulations. J Am Chem Soc. 2016;138:14257–14263. doi: 10.1021/jacs.6b05425. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 33.Knoverek C.R., et al. Opening of a cryptic pocket in β-lactamase increases penicillinase activity. Proc Natl Acad Sci. 2021;118 doi: 10.1073/pnas.2106473118. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Meller A., et al. Predicting locations of cryptic pockets from single protein structures using the PocketMiner graph neural network. Nat Commun. 2023;14 doi: 10.1038/s41467-023-36699-3. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shan Y., et al. How does a drug molecule find its target binding site. J Am Chem Soc. 2011;133:9181–9183. doi: 10.1021/ja202726y. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Paci E., et al. Interactive molecular dynamics in virtual reality for accurate flexible protein-ligand docking. Plos One. 2020;15 doi: 10.1371/journal.pone.0228461. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Connor M.B., et al. Interactive molecular dynamics in virtual reality from quantum chemistry to drug binding: an open-source multi-person framework. J Chem Phys. 2019;150 doi: 10.1063/1.5092590. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 38.Dreher M., et al. Interactive molecular dynamics: scaling up to large systems. Procedia Comput Sci. 2013;18:20–29. doi: 10.1016/j.procs.2013.05.165. (https://doi.org:) [DOI] [Google Scholar]
- 39.Grayson P., Tajkhorshid E., Schulten K. Mechanisms of selectivity in channels and enzymes studied with interactive molecular dynamics. Biophys J. 2003;85:36–48. doi: 10.1016/s0006-3495(03)74452-x. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krieger E., Vriend G. YASARA view—molecular graphics for all devices—from smartphones to workstations. Bioinformatics. 2014;30:2981–2982. doi: 10.1093/bioinformatics/btu426. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krieger E., Nielsen J.E., Spronk C.A.E.M., Vriend G. Fast empirical pKa prediction by Ewald summation. J Mol Graph Model. 2006;25:481–486. doi: 10.1016/j.jmgm.2006.02.009. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 42.Krieger E., Dunbrack R.L., Hooft R.W.W., Krieger B. Assignment of protonation states in proteins and ligands: combining pKa prediction with hydrogen bonding network optimization. 2012;819:405–421. doi: 10.1007/978-1-61779-465-0_25. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 43.Maier J.A., et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–3713. doi: 10.1021/acs.jctc.5b00255. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 45.Jakalian A., Jack D.B., Bayly C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 46.Jorgensen W.L. Revised TIPS for simulations of liquid water and aqueous solutions. J Chem Phys. 1982;77:4156–4163. doi: 10.1063/1.444325. (https://doi.org:) [DOI] [Google Scholar]
- 47.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–935. doi: 10.1063/1.445869. (https://doi.org:) [DOI] [Google Scholar]
- 48.Hornak V., et al. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Protein: Struct, Funct, Bioinforma. 2006;65:712–725. doi: 10.1002/prot.21123. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Essmann U., et al. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. doi: 10.1063/1.470117. (https://doi.org:) [DOI] [Google Scholar]
- 50.Krieger E., Vriend G. New ways to boost molecular dynamics simulations. J Comput Chem. 2015;36:996–1007. doi: 10.1002/jcc.23899. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2009 doi: 10.1002/jcc.21334. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Duan Y., et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem. 2003;24:1999–2012. doi: 10.1002/jcc.10349. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 53.Morris G.M., et al. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J Comput Chem. 1998;19:1639–1662. doi: 10.1002/(sici)1096-987x(19981115)19:14<1639::aid-jcc10>3.0.co;2-b. (https://doi.org:) [DOI] [Google Scholar]
- 54.Krieger E., Koraimann G., Vriend G. Increasing the precision of comparative models with YASARA NOVA-a self-parameterizing force field. Protein: Struct, Funct, Bioinforma. 2002;47:393–402. doi: 10.1002/prot.10104. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 55.Morris G.M., et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hummer G., Szabo A. Free energy profiles from single-molecule pulling experiments. Proc Natl Acad Sci. 2010;107:21441–21446. doi: 10.1073/pnas.1015661107. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Souza N. Pulling on single molecules. Nat Methods. 2012;9:873–877. doi: 10.1038/nmeth.2149. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 58.James J.R. Using the force to find the peptides you’re looking for. Proc Natl Acad Sci. 2017;114:10303–10305. doi: 10.1073/pnas.1713970114. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen P., et al. Breaking a dative bond with mechanical forces. Nat Commun. 2021;12 doi: 10.1038/s41467-021-25932-6. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen W., He H., Wang J., Wang J., Chang C.-e A. Uncovering water effects in protein–ligand recognition: importance in the second hydration shell and binding kinetics. Phys Chem Chem Phys. 2023;25:2098–2109. doi: 10.1039/d2cp04584b. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yekta R., et al. The impact of water molecules on binding affinity of the anti-diabetic thiazolidinediones for catalase: kinetic and mechanistic approaches. Arch Biochem Biophys. 2019;664:110–116. doi: 10.1016/j.abb.2019.02.002. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 62.Huggins David J. Quantifying the entropy of binding for water molecules in protein cavities by computing correlations. Biophys J. 2015;108:928–936. doi: 10.1016/j.bpj.2014.12.035. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ladbury J.E. Just add water! The effect of water on the specificity of protein-ligand binding sites and its potential application to drug design. Chem Biol. 1996;3:973–980. doi: 10.1016/s1074-5521(96)90164-7. (https://doi.org:) [DOI] [PubMed] [Google Scholar]
- 64.Young T., Abel R., Kim B., Berne B.J., Friesner R.A. Motifs for molecular recognition exploiting hydrophobic enclosure in protein–ligand binding. Proc Natl Acad Sci. 2007;104:808–813. doi: 10.1073/pnas.0610202104. (https://doi.org:) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Matsoukas J.M., Panagiotopoulos D., Keramida M., Mavromoustakos T., Yamdagni R., Wu Q., et al. Synthesis and contractile activities of cyclic thrombin receptor-derived peptide analogues with a Phe-Leu-Leu-Arg Motif: importance of the phe/arg relative conformation and the primary amino group for activity. J Med Chem. 1996;39(18):3585–3591. doi: 10.1021/jm950690v. [DOI] [PubMed] [Google Scholar]