

Abstract
Subsequent to the discovery of insulin 100 years ago, great strides have been made in understanding its function, especially in the brain. It is now clear that insulin is a critical regulator of the neuronal circuitry controlling energy balance and glucose homeostasis. This review focuses on the effects of insulin and diabetes on the activity and glucose sensitivity of hypothalamic glucose-sensing neurones. We highlight the role of electrophysiological data in understanding how insulin regulates glucose-sensing neurones. A brief introduction describing the benefits and limitations of the major electrophysiological techniques used to investigate glucose-sensing neurones is provided. The mechanisms by which hypothalamic neurones sense glucose are discussed with an emphasis on those glucose-sensing neurones already shown to be modulated by insulin. Next, the literature pertaining to how insulin alters the activity and glucose sensitivity of these hypothalamic glucose-sensing neurones is described. In addition, the effects of impaired insulin signalling during diabetes and the ramifications of insulin-induced hypoglycaemia on hypothalamic glucose-sensing neurones are covered. To the extent that it is known, we present hypotheses concerning the mechanisms underlying the effects of these insulin-related pathologies. To conclude, electrophysiological data from the hippocampus are evaluated aiming to provide clues regarding how insulin might influence neuronal plasticity in glucose-sensing neurones. Although much has been accomplished subsequent to the discovery of insulin, the work described in our review suggests that the regulation of central glucose sensing by this hormone is both important and understudied.
Keywords: diabetes, electrophysiology, hyperglycaemia, hypoglycaemia, oxidative stress
1 |. GLUCOSE-SENSING NEURONES
1.1 |. Electrophysiological methods
This aims to highlight the electrophysiological evidence supporting a critical role of insulin in regulating glucose-sensing neurones. Electrophysiological techniques have been used for over a half of a century to investigate the responses of neurones to extracellular glucose, as well as other nutrients and hormones. These techniques have evolved significantly over time, leading to a variety of different recording configurations. The distinctions between the different configurations are subtle and can be confusing to individuals who are not electrophysiologists. For this reason, we begin the discussion of glucose-sensing and insulin-responsive neurones with a brief introduction into the types of electrophysiological studies encountered most frequently in the literature under review. These include (but are not limited to) single unit extracellular, intracellular (sharp electrode), loose patch and patch clamp recording configurations. Figure 1 provides an illustration of these recording configurations and a common example of the type of data produced.
FIGURE 1.
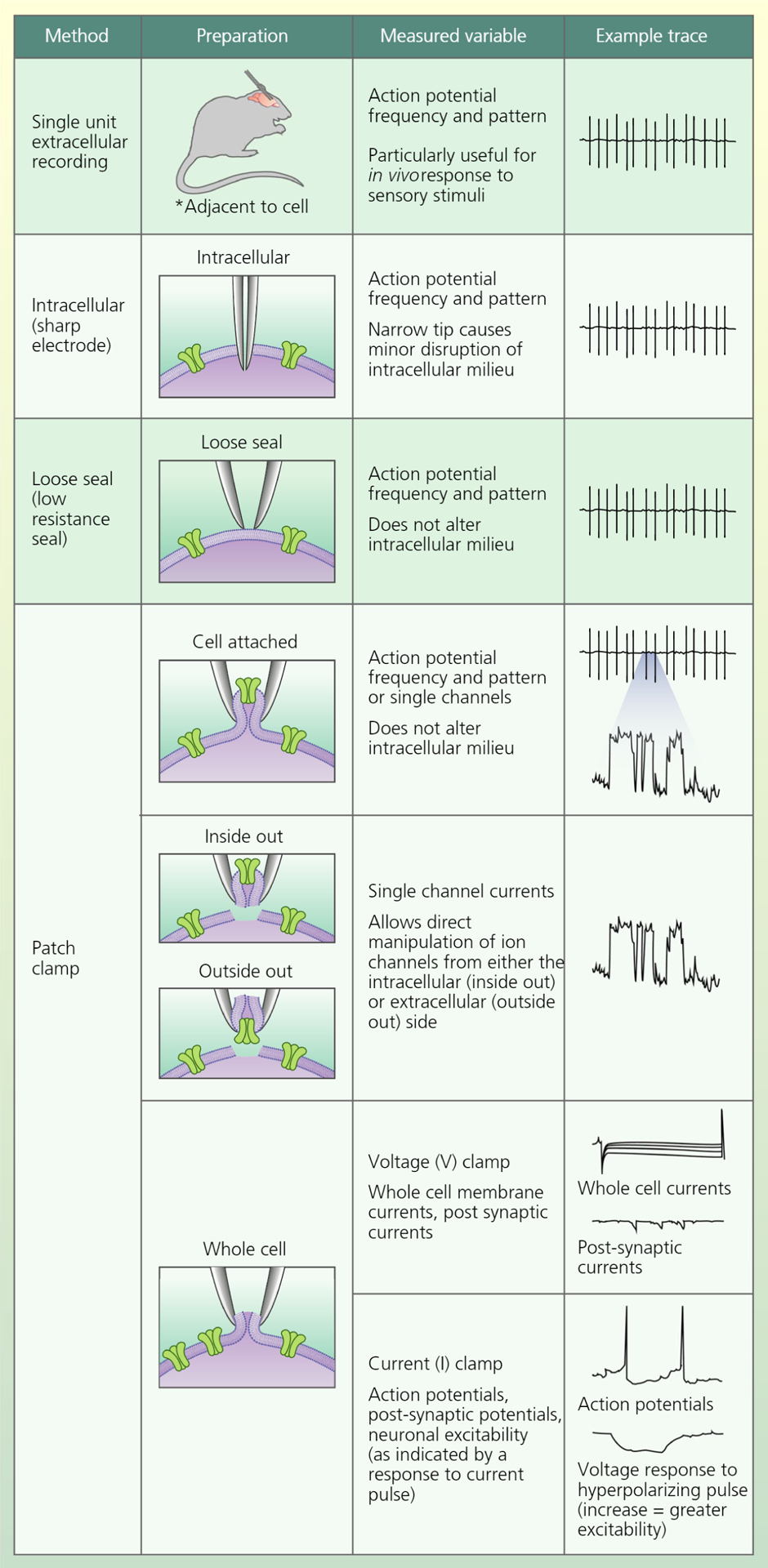
Major electrophysiological recording configurations used to investigate the effects of glucose and insulin on neuronal and ion channel activity
Extracellular, single unit recordings have been used subsequent to the 1950s to investigate neuronal activity (action potentials) in vivo or in brain slices ex vivo.1 This method measures the electrical activity of neurones adjacent to the recording electrode. Often, the goal is to record activity from just one neurone. The nature of the observed wave-form enables the user to identify when that is the case. Extracellular recording is also used to simultaneously measure neuronal activity or the response to excitatory input in a population of neurones. This technique is beneficial for investigating changes in neuronal activity patterns in response to sensory stimuli in alert, freely moving animals. In addition, because the electrode does not contact the cell, this technique is non-invasive with respect to the neurone being recorded. Action potentials can also be measured in vivo or ex vivo using intracellular or sharp electrodes that impale the cell.2 The benefit of this technique is that it is clear that the activity of a single neurone is being recorded when the narrow tip prevents significant disruption of the intracellular milieu. A third electrophysiological technique used to measure action potentials is known as the loose patch technique.3 In loose patch recording, the glass pipette housing the recording electrode is pressed against the cell membrane, which makes a ‘loose’ or low resistance (eg, 500 MΩ) seal against the electrode glass. The advantage of this technique is that it also guarantees that activity of a single neurone is being monitored but it does not impale the neurone and thus does not disrupt the intracellular milieu. This prevents dialysis of cellular contents (eg, ATP, cAMP) or disruption of ionic gradients. Maintaining the intracellular environment is important for investigating hormonal or nutrient regulation that involves intracellular signalling pathways or metabolism. However, one disadvantage is that current leak around the loose seal precludes mechanistic evaluation of ion channel activity.
The patch clamp electrophysiological technique was established by Neher and Sakmann in the 1970s and has become the gold standard for investigating the cellular mechanisms underlying the regulation of action potential frequency and pattern.4,5 In patch clamp recording, a seal is achieved between the pipette glass housing the recording electrode and the cellular membrane. Known as the gigaohm (GΩ) seal, it has very high resistance and allows minimal current leak. As a result, it is possible to measure the currents resulting from flux through single ion channels, as well as whole cell currents in response to changes in the solution bathing the cell or pipette, depending on the recording configuration. Patch clamp recording enables the user to ‘clamp’ either the membrane current or the membrane voltage. In voltage clamp, the membrane voltage is held at a constant value to allow single channel or whole cell ionic currents to be measured with high fidelity. On the other hand, current clamp allows measurement of membrane voltage changes, such as action potentials, in response to experimental manipulation. There are four major patch clamp configurations: cell attached, inside out, outside out and whole cell.4 The cell attached configuration, similar to loose seal, allows for measurement of action potentials without compromising the intracellular milieu. This configuration, which is generally used in the voltage clamp mode, is also able to measure the ionic currents of the ion channels in the piece of membrane directly under the tip of the glass recording pipette. This can be useful for evaluating ion channels that are regulated by cytosolic factors. The inside out and outside out configurations are used in the voltage clamp mode to directly manipulate ion channels. The inside out configuration consists of a piece of cell membrane attached to the pipette with the cytosolic side facing the solution bathing the pipette. By changing the bath solution, it is possible to investigate the regulation of the cytosolic face of the ion channels. This is useful, for example, for investigating the ATP sensitivity of the ATP-sensitive potassium (KATP) channel because it is now simple to manipulate cytosolic ATP concentration.6 The outside out configuration consists of a piece of cell membrane attached to the pipette with the extracellular side facing the bathing solution. This is useful, for example, for investigating the regulation of the KATP channel by neuroactive peptides in the extracellular solution.7 In whole cell recording, the gigaohm seal is formed and then gentle suction is used to rupture the membrane under the tip of the recording pipette. The experimenter is now able to measure changes in whole cell currents (voltage clamp) or membrane voltage (current clamp).
An additional advantage of the whole cell configuration is the ability to dialyze a dye from the pipette solution into the cell. This allows the use of post-recording immunohistochemistry to phenotype the recorded neurone. For example, Wang et al8 used this technique in brain slice preparations to demonstrate that glucose-sensing neurones located in the cell poor region between the arcuate and ventromedial hypothalamus were distinct from the arcuate pro-opiomelanocortin (POMC) neuronal population. The whole cell recording configuration or similar pipette seals can also be used to harvest cellular contents for a single cell reverse transcriptase-polymerase chain reaction. Such studies have revealed an overlap between the insulin receptor and glucose-sensing neurones (as described below).9–11 Measuring action potentials is a common use of the whole cell current clamp mode. However, in this configuration, it is also possible to measure the membrane voltage response to injected current pulses. The relationship between injected current and the cellular voltage response is termed the input resistance and reflects the excitability of the neurone to pre-synaptic input. A larger voltage response to current corresponds to increased cellular electrical resistance, which in turn reflects closure of extra-synaptic ion channels on the cell body. These somatic ion channels are most often permeable to either K+ or Cl− and are responsible for setting the resting membrane potential. Thus, increased resistance would indicate closure of somatic channels and depolarisation and/or increased neuronal excitability in response to input from upstream (pre-synaptic) neurones.12 Whole cell resistance is of particular importance to the topic of this review because these somatic channels are responsive to factors present in the extracellular milieu (eg, glucose, insulin) as opposed to neurotransmitters released at the synapse.
Pre-synaptic input (ie, from upstream neuronal circuits) can be measured in the whole cell configuration as excitatory and inhibitory post-synaptic currents (EPSCs or IPSCs, respectively; voltage clamp) or potentials (E/IPSPs; current clamp). The neurone can be isolated from pre-synaptic input in brain slice preparations with compounds such as tetrodotoxin, which prevents input from pre-synaptic action potentials by blocking sodium channels. This enables the experimenter to measure the direct effects of nutrients or hormones on the neurone that is being recorded. Although astrocytes do not typically produce action potentials, patch clamp recording can also be used to measure astrocytic depolarisation and single channel or whole cell currents produced by these cells. This is of significance because astrocytes not only sense nutrients and hormones, but also are known to regulate action potential generation and pattern in neuronal circuits.13,14
Historically, in vivo studies have predominantly used extracellular, single unit or intracellular sharp electrode recording configurations because movement of the brain in response to blood flow was less likely to disrupt the recording. In comparison, even the slightest movement can disrupt a patch clamp recording pipette seal. However, as the techniques continue to evolve, reports are emerging using patch clamp recording in vivo.15 Finally, the use of intracellular dyes that fluoresce in response to changes in neuronal activity and/or cellular processes (eg, membrane potential, calcium, or nitric oxide [NO] sensitive dyes) are now commonly used to investigate populations of nutrient- and hormone-sensing neurones.9,11,16–20 The benefit of these dyes is the ability to monitor large populations of neurones. The main disadvantages are that these indices are only surrogates for electrical activity and these measurements often must be made in dissociated cells (eg, in the absence of glia and other regulatory input).
1.2 |. Glucose-sensing neurones
Glucose-sensing neurones were first discovered in 1964 by two independent laboratories using in vivo extracellular recordings in cats and dogs.21,22 Together, their findings showed that both food intake and elevated blood glucose increased the activity of approximately half of recorded neurones in the ventromedial hypothalamus (VMH) and decreased the activity of approximately half of the neurones in the lateral hypothalamus (LH). Conversely, fasting resulted in greater neuronal activity within the LH than within the VMH. This suggested that glucose excites a population of VMH neurones and inhibits a population of LH neurones. Interestingly, insulin did the converse of glucose.21 In 1990, Ashford et al23 demonstrated the presence of metabolism dependent glucose-excited (GE) neurones in the VMH using intracellular recording. They then used cell attached patch clamp recording in isolated neurones to show that glucose closed a potassium channel. Subsequent inside out patch clamp recordings revealed this to be the ATP-sensitive K+ (KATP) channel.23 These early studies used a non-physiological range of brain glucose concentrations (0 to 10 mmol L−1); however, GE neurones in both the VMH and arcuate nucleus of the hypothalamus (ARC) were later demonstrated to be sensitive to physiological changes in extracellular glucose (0.1 to 5 mmol L−1) with a K1/2 of approximately 0.5–0.7 mmol L−1.8,24 This is relevant because hypothalamic glucose concentrations are approximately 0.7 mmol L−1 after an overnight fast, 1.5–2 mmol L−1 during the fed state and increase to only 4.5 mmol L−1 when blood glucose is 20 mmol L−1.25−27 VMH GE neurones use the pancreatic form of hexokinase, glucokinase (GK), to sense glucose.9,11 Thus, glucose sensing in VMH and ARC GE neurones is very similar to that of the insulin secreting pancreatic β-cell.28 There is also a subpopulation of ARC GE neurones that are excited only by glucose increases > 5 mmol L−1.29,30 Glucose sensing in this GE population involves the transient receptor potential channel C (TRPC) rather than the KATP channel.31
The early single unit recording and patch clamp studies of the VMH suggested that neurones which are inhibited by glucose (glucose-inhibited [Gl] neurones) were either not present or present in numbers too few to likely be of physiological relevance.21−23,32 However, later studies using patch clamp recording in brain slices or membrane potential and calcium sensitive dyes suggested a significant population of Gl neurones in the VMH. It is now demonstrated that Gl neurones comprise almost 60% of the neurones of the ventrolateral VMH.9,11,19,33,34 Similar to GE neurones, VMH Gl neurones express GK and respond to physiological changes in extracellular glucose.9,11,19,24,34,35 Although they possess functional KATP channels, VMH Gl neurones require phosphorylation and activation of AMP-activated protein kinase (AMPK) and neuronal NO synthase (nNOS) to be activated in low glucose.17,19,36 As noted for ARC GE neurones, there is also a population of ARC Gl neurones that are only inhibited when glucose levels exceed 5 mmol L−1.29 The early single unit recordings also suggested that the LH primarily possessed Gl neurones.21,22 However, similar to the VMH, populations of both LH Gl and GE neurones have been observed using patch clamp recording. LH orexin and GABA neurones are Gl neurones, whereas melanin-concentrating hormone (MCH) neurones are GE neurones.37,38 Additionally, the orexin and GABA Gl neurones are inhibited by the non-metabolisable glucose analogue, 2-deoxyglucose (2DG).38 Thus, rather than responding to changes in glucose metabolism, these Gl neurones sense the glucose molecule itself. Little is known about the mechanism of glucose sensing by MCH neurones; however, whole cell patch clamp recordings indicate that glucose increases cellular resistance consistent with closing an ion channel and increasing excitability. Whether this is the KATP channel remains to be determined.37 One potential explanation for the discrepancy between the extracellular single unit and later patch clamp recording data with regard to VMH Gl neurones is that the former measures the dominant (macro) circuits, whereas the latter enables detection of more discrete (micro) circuitry. Moreover, Gl neurones are concentrated within the ventrolateral VMH and may have been missed when recording elsewhere in the VMH.
In addition to the VMH and LH, glucose-sensing neurones have been found in numerous other hypothalamic and extra-hypothalamic regions. Regions or cell types expressing GE neurones include the pre-autonomic neurones of the hypothalamic paraventricular nucleus (PVN),39 the gonadotrophin-releasing hormone (GnRH) neurones of the anterior hypothalamus,40,41 the ARC POMC neurons,42 the amygdala43 and the brainstem.44 Gl neurones are also found in the PVN,39 the dorsomedial hypothalamus, the ARC neuropeptide Y (NPY) neurones,29 the amygdala43 and brainstem.44 Interestingly mechanisms of glucose sensing differ among distinct subpopulations of glucose-sensing neurones. For example, as noted above, glucose sensing by LH Gl neurones is metabolism independent, whereas VMH Gl neurones require the cellular fuel sensor AMPK to sense glucose. Similarly, VMH GE neurones sense glucose via the KATP channel, whereas GnRH GE neurones do not.41 Moreover, both GnRH GE neurones and the GT1–7 cell lines that are derived from GnRH neurones require AMPK to sense glucose,40,41 but VMH GE neurones do not.45 Finally, single unit recordings in primates demonstrate the presence of glucose-sensing neurones in the orbitofrontal cortex, a region not usually considered in metabolic regulation.46 Thus, it is clear that brain glucose sensing is a diverse and integrated network that likely functions not only to regulate glucose homeostasis, but also to link neuroendocrine and behavioural systems with nutrient availability.
2 |. EFFECTS OF INSULIN ON NEURONAL ACTIVITY AND GLUCOSE SENSING
2.1 |. Effects of insulin on neuronal activity of glucose-sensing neurones
Insulin has widespread effects in the brain and influences both neurones and glial cells.47 A higher percentage of VMH GE and Gl neurones express insulin receptors compared to non-glucose-sensing neurones.11 As expected, insulin regulates both the activity and the glucose sensitivity of glucose-sensing neurones. In supraphysiological glucose concentrations (10 mmol L−1) insulin hyperpolarised, decreased the input resistance (consistent with opening KATP channels) and diminished the action potential frequency of freshly dissociated VMH and ARC GE neurones. This was associated with increased KATP single-channel activity and was mediated by phosphoinositol-3 (PI3) kinase signalling.48 This effect persisted in the absence of pre-synaptic input suggesting that insulin acts directly on GE neurones. Interestingly, the effect of insulin is dependent on glucose concentration. In the presence of 5 mmol L−1 glucose, which might be seen in the brain during diabetic hyperglycaemia,27 insulin had a lesser effect on the input resistance and action potential frequency of VMH GE neurones than in 10 mmol L−1 glucose.45,48 However, although insulin decreased input resistance (eg, opened KATP channels) of VMH GE neurones in 2.5 mmol L−1 glucose, this was not sufficient to reduce action potential frequency, nor did insulin have any effect on ARC GE neurones in this glucose concentration.8,45 This suggests that the ability of insulin to inhibit GE neurones is dependent on the state of the KATP channel. Accordingly, the effect of insulin effect is greatest when the KATP channel is in the closed state in high glucose; however, it appears to have minimal effects on neuronal activity in VMH and arcuate GE neurones at physiological glucose levels. That said, decreased input resistance in 2.5 mmol L−1 glucose would reduce neuronal responses to pre-synaptic input. In 2.5 mmol L−1 glucose, insulin hyperpolarises VMH Gl neurones and increases their NO production. The latter is mediated by PI3 kinase.17 This appears paradoxical because increased NO production is associated with depolarisation of VMH Gl neurones in low glucose.19 However, VMH Gl neurones possess KATP channels that are regulated not by glucose but, instead, by lactate.24 This suggests that the KATP channel- and NO-mediated pathways may have distinct effects in VMH Gl neurones. Moreover, although glucose acts only on the NO pathway, insulin may regulate both NO production and KATP channel activity in Gl neurones. These observations also suggest that the net effect of insulin on metabolically sensitive pathways in VMH GE and Gl neurones is dependent on the status of the neurones in any given nutrient or hormonal milieu.
Insulin effects have also been observed in LH MCH (GE), ARC NPY (Gl) and POMC (GE) neurones. In LH MCH neurones, insulin increases excitability and action potential frequency.49 Insulin effects in POMC and NPY neurones are not unexpected given the well-established role of POMC neurones in promoting satiety and increased energy expenditure, whereas NPY neurones signal for the reverse.50 Interestingly, whether insulin excites or inhibits POMC neurones during whole cell recordings is controversial. A number of studies report that insulin inhibits POMC neurones by activating the KATP channel.51–54 Most of these studies evaluated the effects of the Humulin™ (Eli Lilly, Indianapolis, IN, USA) preparation of insulin in rats or mice.51,53,54 However, Qiu et al55 showed that purified guinea pig or bovine insulin depolarised and increased action potential frequency in the majority (74%) of POMC neurones from guinea pigs, with the remainder being unaffected. Insulin excitation was accompanied by an inward (excitatory) current that reversed at −10 mV, consistent with activation of a cation channel. This insulin effect was mediated by a PI3 kinase-induced activation of the transient receptor potential channel TRPC5.55 Interestingly, the TRPC5 channel may contribute to crosstalk between insulin and glucose because some arcuate GE neurones (those responding to glucose levels above 5 mmol L−1) use the TRPC channel to sense glucose.31 All of these studies used whole cell patch clamp recording to evaluate insulin effects; however, these differences could result from differences in species or insulin preparation. To address this issue, Qiu et al55 evaluated the effects of Humulin™ in their guinea pig preparation and found the majority of POMC neurones were inhibited. Because most Humulin™ preparations contain zinc in a 2:1 insulin:zinc ratio and zinc activates the KATP channel, they hypothesised that hyperpolarisation was a result of zinc activation of KATP channels in the Humulin™ preparation. Consistent with their hypothesis, zinc hyperpolarised POMC neurones from guinea pigs and induced an outward (inhibitory) current, which reversed at the K+ equilibrium potential and was blocked by the KATP channel inhibitor tolbutamide. Their data suggest that the reported inhibitory effect of insulin, or at least the Humulin™ preparation, may actually be a result of zinc-mediated KATP channel activation, masking any insulin-induced depolarisation. However, this explanation does not entirely end the controversy because several groups report that purified insulin inhibits POMC neurones.52,56
A recent study by Dodd et al56 provides an intriguing alternative hypothesis. In their study, also using whole cell patch clamp recording, they found that the majority of POMC neurones from ad lib fed mice were either inhibited or unaffected by reconstituted human insulin (Tocris, Bristol, UK), with only a small subpopulation being excited.56 Whether insulin inhibited or excited POMC neurones in their preparation was dependent on T-cell protein tyrosine phosphatase (TCPTP). That is, the insulin-inhibited POMC neurones expressed TCPTP, whereas the insulin-excited neurones did not. Dodd et al56 then showed that TCPTP is induced in the fasted state when POMC neurones are generally inhibited. When TCPTP was deleted in POMC neurones, the relative proportion of insulin-excited POMC neurones increased with a concomitant decrease in the proportion of those which were inhibited.56 On the other hand, insulin reduced action potential frequency in NPY/agouti-related peptide (AgRP) neurones; an effect which was blocked when TCPTP was deleted in these neurones.57 This suggests an interaction between insulin, TCPTP and energy status. In the fasted state, TCPTP would shift the effects of insulin toward inhibition of POMC GE neurones, at the same time as blocking the inhibitory effect of insulin on the feeding stimulatory NPY/AgRP-GI neurones. Taken together, the above data obtained in the VMH and ARC indicate an interaction between insulin and the steady-state activity of glucose-sensing neurones that is dependent on metabolic status. However, it is important to acknowledge the role of zinc in studies that use Humulin™ in mouse or rat brains. The above data also suggest heterogeneity in the POMC populations. Interestingly, in terms of glucose sensing, it is not un-common for glucose-sensing and non-glucose-sensing neurones to exist within distinct neuropeptide populations (eg, orexin, NPY58,59). Moreover, some studies report POMC neurones to be glucose sensing where others do not.8,29,42 Taken together, these data suggest the likelihood of subpopulation variation in glucose or insulin sensing that could be dependent on different energy states of the cells.
2.2 |. Insulin effects on glucose sensitivity of glucose-sensing neurones
Few studies have evaluated the effects of insulin on glucose sensing. However, as might be expected from the above discussion, insulin affects the glucose sensitivity of GE and Gl neurones. In whole cell patch clamp recordings from VMH GE neurones, insulin (recombinant human) blunts the decreased input resistance and action potential frequency normally seen when glucose levels decrease from 2.5 to 0.1 mmol L−1.45 Thus, in the presence of insulin, a meal-to-meal or fasting-induced decrease in blood glucose would have less impact on VMH GE neurones. Conversely, the increase in input resistance and action potential frequency normally seen in VMH Gl neurones as glucose decreases was significantly blunted in neuronal insulin receptor knockout (NIRKO) mice.60 This was associated with a blunted sympathoadrenal response to hypoglycaemia, suggesting that VMH Gl neurones are necessary to raise blood glucose during insulin hypoglycaemia. However, this may not reflect a role for these Gl neurones in mediating insulin effects during euglycaemia. This latter hypothesis is supported by studies of mice lacking the insulin sensitive glucose transporter 4 (GLUT4). These mice exhibit an impaired response to hypoglycaemia in vivo, and the activation of VMH Gl neurones in low glucose is largely blocked.61
Although this review is focused on glucose-sensing neurones, astrocytes express glucose-sensing machinery including GLUT2, which mediates glucose sensing in liver and pancreatic β-cells.62–65 Astrocytes are important regulators of normal neuronal function and activity.13,66,67 Changes in extracellular glucose alters intracellular calcium levels in tanycytes (astrocytes lining the third ventricle).68 Moreover, tanycytic processes in the VMH change their morphology following repetitive hypoglycaemia, which impairs hypothalamic glucose sensing.69 This suggests that astrocytes may play a role in glucose sensing. Astrocytes also express insulin receptors, suggesting a role for this hormone in astrocytic regulation of neuronal activity.66 For example, dopamine release is impaired in mice lacking the astrocytic insulin receptor. It is tempting to speculate that insulin increases glucose uptake into astrocytes; however, this does not appear to be the case. Rather, insulin increases astrocytic glycogen levels.70 Although the precise function of astrocytes in central insulin and glucose sensing is not known, it is clear that they are poised to play a significant role.
3 |. EFFECTS OF DIABETES MELLITUS ON GLUCOSE-SENSING NEURONES
Type 1 diabetes mellitus (T1DM) is a result of autoimmune destruction of the pancreatic β-cell leading to an absence of insulin secretion as glucose levels increase.71 By contrast, type 2 diabetes mellitus (T2DM) is initially a result of insulin resistance which can be followed by β-cell fatigue and impaired insulin secretion.72 Regardless of the cause of the pathology, both T1DM and T2DM are associated with reduced insulin action. As would be expected, insulin resistance impacts the activity and glucose sensitivity of glucose-sensing neurones. Additionally, hyperglycaemia per se resulting from either a lack of insulin or impaired insulin signalling is associated with impaired glucose sensing.16,20,48,73
3.1 |. Effects of insulin resistance on the activity and glucose sensitivity of glucose-sensing neurones
Surprisingly few studies have evaluated the effects of insulin resistance on glucose-sensing neurones. Most of these studies use either the Zücker fatty rat or the db/db mouse. Zücker fatty rats express a mutated leptin receptor. Heterozygotes for this mutation are phenotypically lean whereas homozygotes are obese. Although VMH and ARC GE neurones from lean Zücker rats are inhibited by insulin, those from obese Zücker rats are insulin insensitive.48 Similarly, diabetic db/db mice are characterised by a leptin receptor mutation and insulin resistance. In these mice, the inhibitory response of VMH GE neurones to low glucose was enhanced and insulin was without effect on glucose sensitivity. The insulin sensitiser, compound 2, normalised the effects of insulin on VMH GE neurones from these mice.73 Additionally, in diabetic db/db mice, spontaneous neuronal firing is higher in PVN pre-autonomic neurones compared to their lean counterparts. These neurones, of which approximately 70% are glucose sensing (both GE and Gl), project to the liver. This increase in neuronal activity is associated with elevated sympathetic activation and may lead to increased hepatic glucose production.74 Moreover, insulin resistance as a result of a high-fat diet blocked the effects of insulin in POMC neurones from male guinea pigs. By contrast, POMC neurones from female guinea pigs in pro-oestrus retained insulin sensitivity, whereas those from ovariectomised guinea pigs did not. The effect of ovariectomy was blocked by oestradiol administration.75 Thus, the ability of insulin to modulate glucose-sensing neurones translates to dysfunction during insulin resistance, the extent of which is further impacted by sex. Although the majority of studies have been performed in males, sex hormones clearly play a role in insulin sensitivity and glucose homeostasis. Female mice lacking oestrogen receptors in either POMC or VMH neurones exhibit impaired glucose homeostasis.76 Furthermore, oestrogen and insulin recruit the PI3 kinase pathway, suggesting overlap in insulin and oestrogen signalling.77 Finally, the glucose sensitivity of VMH Gl neurones is sexually dimorphic and varies across the oestrus cycle.33,78 These data are highly relevant given the dearth of literature in human and non-human females. This is clearly an area that requires further study.
3.2 |. Lack of insulin/diabetic hyperglycaemia
Streptozotocin (STZ) is a toxin that is transported into pancreatic β-cells by the glucose 2 transporter leading to cell death. STZ is often used as a rodent model that mimics diabetic hyperglycaemia/insulin loss in T1DM.16,20,79,80 In this model, it is difficult to distinguish between the effects arising as a result of the lack of insulin and the presence of hyperglycaemia; however, this is the case in human disease as well. In preparations of isolated VMH neurones from STZ rats, decreased glucose did not elicit NO production or neuronal depolarisation as measured by NO or membrane potential sensitive dyes, respectively.16 This indicates an interesting inability to detect VMH Gl neurones in STZ rats. A similar effect was also observed when the neurones were incubated in 5 mmol L−1 glucose in vitro suggesting that this is mediated by hyperglycaemia per se and not the absence of insulin in the STZ model. Both NO production and depolarisation (ie, presence of Gl neurones) were restored by an AMPK activator. VMH GE neurones were not altered in preparations from STZ rats.16 In brain slices of the PVN of STZ mice, the amplitude of IPSCs in whole cell recordings of pre-sympathetic neurones projecting to the kidney were smaller than in non-diabetic controls.81 GABAergic inhibition was also blunted, indicating that a lack of insulin/hyperglycaemia alters the electrophysiological profile of pre-sympathetic PVN neurones. These data further suggest that T1DM leads to remodelling of the sympathetic nervous system.81
3.3 |. Effects of insulin-induced hypoglycaemia on glucose-sensing neurones
Iatrogenic hypoglycaemia is the major limiting factor in treating diabetes with intensive insulin therapy.82 Severe acute hypoglycaemia can lead to seizures, brain damage and death. However, the effects of repeated non-life threatening hypoglycaemia are more insidious. In response to an initial hypoglycaemic episode, the counter-regulatory hormones glucagon, epinephrine, corticosterone and growth hormone are secreted to restore euglycaemia. In addition, sympathetic activation and cognitive arousal alert the individual to falling glucose levels (hypoglycaemia awareness) and elicit behavioural responses such as food intake. However, repeated hypoglycaemia blunts the ability of the brain to detect and correct subsequent hypoglycaemia leading to hypoglycaemia associated autonomic failure (HAAF), which is a result of impaired glucagon and epinephrine release, and hypoglycaemia unawareness, in which cognitive awareness of hypoglycaemia is diminished.83 During HAAF and hypoglycaemia unawareness, blood glucose levels can fall to dangerous, even lethal, levels without detection and correction. There is a significant literature on the effects of HAAF on neuronal activity.20,34,84–89 However, as a result of the lack of an animal model of hypoglycaemia unawareness, until recently,90 less was known about the neural mechanisms involved. When discussing these data, as with those regarding impaired insulin signalling vs hyperglycaemia, it is difficult to differentiate between the effects of hypoglycaemia and the increased insulin level often needed to create it. It is clear, however, that recurrent insulin-induced hypoglycaemia and HAAF impair the ability of VMH GE and Gl neurones to respond to low glucose.
During HAAF, VMH GE neurones experience a shift in glucose sensitivity, requiring a greater decrease in glucose concentration before their activity is inhibited.91,92 Because insulin also blunts inhibition of VMH GE neurones by low glucose,45 it is not clear whether repeated insulin or hypoglycaemia or both contribute to this effect. However, it is clear that VMH GE neurones and KATP channels are important for the response to insulin hypoglycaemia. Mice that lack the inwardly rectifying potassium subunit of the KATP channel (Kir6.2) lack VMH GE neurones and have impaired glucagon production in response to insulin-induced hypoglycaemia.93 Pharmacological closure of VMH KATP channels suppresses, whereas activation enhances, the counter-regulatory hormone response to insulin hypoglycaemia.94–96 Activation of PVN neurones in low glucose is also suppressed during HAAF, suggesting a role for PVN GE neurones as well.97,98
Similar to VMH GE neurones, the threshold for activation of VMH Gl neurones shifts to lower glucose levels during HAAF.34,92 Moreover, ex vivo activation of VMH Gl neurones by low glucose is impaired in animals displaying in vivo dysregulation of hypoglycaemia counterregulation.20,34,60,61,84,92,99 Conversely, when the glucose responsive machinery of VMH Gl neurones (eg, AMPK, nNOS signalling) is impaired in vivo, so also is the counter-regulatory response to hypoglycaemia.36,100–102 These data strongly support a role for these neurones in the counter-regulatory response to hypoglycaemia. Interestingly, although insulin increases NO production in VMH Gl neurones, it also hyperpolarises these neurones.17 Thus, to some extent, it is possible that repeated insulin and repeated hypoglycaemia mediate these effects. In support of a role for insulin in the impaired response to hypoglycaemia in diabetes, VMH insulin infusion normalised the response to hypoglycaemia in STZ treated rats by increasing GLUT4 expression.103
Although the hypothalamus hosts the majority of glucose and feeding responsive neurones, the response to hypoglycaemia may also be modulated by neurones found outside of the hypothalamus. For example, there is very strong evidence for a role of brain-stem glucose sensors in hypoglycaemia counter-regulation.104–109 However, GE and Gl neurones in this region have not been well characterised electrophysiologically, nor has their response to insulin been evaluated. Similarly, in the periphery, glucose-sensing cells within the portal vein of the liver are very important for hypoglycaemia counter-regulation, although the identity of these cells remains unknown.109–115 Furthermore, an early study conducted in 1986 demonstrated that single unit recordings in the amygdala of conscious monkeys showed a decrease in neuronal activity following feeding.116 From these data alone, it was not possible to determine whether this response was a result of the presence of Gl neurones that directly sense glucose, or whether the decreased neuronal activity was a result of pre-synaptic input from glucose-sensing neurones outside the amygdala or even if glucose sensing was involved at all. However, a more recent study was able to confirm the presence of both GE and Gl neurones in the amygdala utilising calcium imaging in isolated cells.43 These glucose-sensing neurones express glucokinase and the urocortin 3 receptor. Amygdala urocortin receptor expressing neurones project to the VMH and approximately 30% of these neurones are active during mild insulin-induced hypoglycaemia. Chemical deletion of these amygdalar neurones with ibotenic acid leads to an impaired counter-regulatory hormone response during a hyperinsulinemic hypoglycaemic clamp. These data suggest a role for amygdalar glucose-sensing neurones in the response to insulin-induced hypoglycaemia and, furthermore, that they may modulate the VMH response. The effects of insulin hypoglycaemia are mimicked by the non-metabolisable glucose analogue, 2DG, which suggests that this effect is mediated by hypoglycaemia per se and not insulin.43
A distinct population of hypothalamic glucose-sensing neurones may play a role in both the counter-regulatory response to insulin-induced hypoglycaemia and hypoglycaemia awareness. Single unit recordings of perifornical neurones in vivo indicated increased activity in response to insulin-induced hypoglycaemia or systemic 2DG, and a number of these neurones were orexin positive.117,118 Perifornical injection of 5-thioglucose, another non-metabolisable glucose analogue, also increased blood glucose and epinephrine secretion.119 The response to non-metabolisable glucose analogues, as well as the observation that neuronal activation coincided with maximal hypoglycaemia, suggests that these effects were likely resulting from a hypoglycaemic state, more so than insulin signalling itself.117–119 These data suggest a role for the LH orexin Gl neurones in sympathetic activation and epinephrine secretion during hypoglycaemia. These neurones also play a role in hypoglycaemia awareness. Otlivanchik et al90 were the first to establish a model of hypoglycaemia awareness that mimics human pathology. These investigators used a conditioned place preference (CPP) paradigm to train rats to prefer one side of a CPP chamber. They then induced insulin-hypoglycaemia on the preferred side and observed that, on the following day, the rats no longer preferred that side. This indicates that the rats were aware of the hypoglycaemia and found it aversive. However, when the rats experienced several hypoglycaemic episodes in their home cages prior to the hypoglycaemia in the CPP chamber, they did not reverse their preference for the trained side. This indicates that they were not aware of the hypoglycaemia and did not develop an aversion. Pretreatment with a selective orexin receptor-1 antagonist, SB-334867A, blocked awareness of hypoglycaemia and mimicked the effects of prior hypoglycaemia, suggesting a role for orexin neurones in hypoglycaemia awareness and unawareness.90,119
It is clear that glucose-sensing neurones both within the hypothalamus and in other regions play a role in the response to insulin-induced hypoglycaemia. What is not clear is whether or how the insulin that results in hypoglycaemia modulates the responses of these neurones. This issue is particularly important when considering repeated hypoglycaemia, which by its nature is also repeated insulin. Further studies to uncover the mechanisms by which insulin-induced hypoglycaemia affects glucose sensing may shed light on this topic.
4 |. MECHANISMS BY WHICH DIABETIC HYPERGLYCAEMIA AND INSULIN-INDUCED HYPOGLYCAEMIA ALTER GLUCOSE SENSING
4.1 |. Glycogen/nutrient compensation
Brain glycogen stores are significant and astrocytic glycogen supports neuronal activity during glucose deprivation.120,121 This suggests the hypothesis that repeated insulin-hypoglycaemia leads to glycogen super compensation, which would then prevent glucose-sensing neurones from sensing glucose deficit. However, brain glycogen levels decreased to a similar degree following either a single episode or three episodes of insulin-hypoglycaemia. Moreover, there was no difference in glycogen levels following recovery from hypoglycaemia in either situation.122 These data suggest that glycogen super compensation does not explain impaired glucose sensing following repeated hypoglycaemia.
An alternative hypothesis is that lactate substitutes for glucose during insulin-induced hypoglycaemia. This hypothesis derives from the now well-known astrocyte-neurone lactate shuttle proposed by Pellerin and Magistretti in 1994.123 The purpose of this shuttle is to allow astrocytes to provide glycolytically derived lactate to neurones as energy to sustain increased neuronal activity.123 Interestingly, insulin and insulin-like growth factor-1 synergistically stimulate glucose uptake into primary cultures of cortical astrocytes.124 Moreover, lactate levels increase following recurrent hypoglycaemia.125 Thus, insulin and the concomitant hypoglycaemia could trigger lactate production in astrocytes to compensate for the lack of glucose. This lactate might prevent glucose-sensing neurones from responding to hypoglycaemia. This hypothesis is consistent with the observation of Borg et al126 that VMH lactate infusion blunts the counter-regulatory response to insulin-induced hypoglycaemia. Moreover, current clamp recordings of VMH GE neurones in brain slices show that lactate closes KATP channels and restores action potential frequency in low glucose.24 However, the story is clearly more complicated. For example, insulin reduces excitability in VMH GE neurones in 2.5 mmol L−1 glucose by activating KATP channels.45,48 However, insulin also blunts the inhibitory response of VMH GE neurones to low glucose.45 Moreover, lactate stimulates VMH Gl neurones by closing the KATP channel, whereas recurrent hypoglycaemia blunts their activation in low glucose.24,34 Additionally, although lactate enhances glucose metabolism and pre-serves neuronal function during hypoglycaemia, glucose is still the major neuronal energy source.125 Taken together, these data suggest that altered brain metabolism as a result of repeated insulin and/or hypoglycaemia may influence central glucose sensing and play a role in HAAF, although we are still far from a clear understanding of the mechanism.
4.2 |. Oxidative stress
Another mechanism underlying impaired glucose sensing during insulin-induced hypoglycaemia and diabetic hyperglycaemia is oxidative stress and the production of reactive oxygen species (ROS). Hypoglycaemia increases mitochondrial ROS (mROS) production.36,84,91,127,128 Central hypoglycaemia is further associated with a corresponding reduction in mitochondrial free radical scavengers, such as glutathione and superoxide-dismutase.128 Taken together, hypoglycaemia represents a significant risk for harmful levels of oxidative stress, to which the brain is particularly susceptible.
First, it should be noted that, although ROS are often cast in a negative light, they do serve a function, in particular, as vital mitochondrial redox signalling molecules.129 ROS help maintain efficient mitochondrial function and additionally serve as important signals in adaptive responses to energy demand.129–132 Indeed, the hypothalamus utilises mROS as a metabolic marker for fluctuating energy status. In the ARC, hypoglycaemia-induced mROS are a necessary component of neuronal glucose sensing, which is blocked upon the addition of antioxidants, and conversely mimicked by mitochondrial destabilisers that generate mROS.132 However, if left unchecked, chronic elevated ROS is implicated in the pathogenesis of several neurological conditions.133
VMH Gl neurones represent one pertinent example of how dysregulated ROS signalling can result in impaired neurological function. As discussed earlier, insulin-induced recurrent hypoglycaemia blunts activation of VMH Gl neurones in low glucose,134,135 partly through the increased NO production that is necessary for activation of VMH Gl neurones in low glucose.36 This is because, in addition to activating VMH Gl neurones in hypoglycaemia, NO also increases oxidative stress. NO inhibits cytochrome c, resulting in temporary impairment of mitochondrial respiration and elevated mROS. Normally, such transient fluxes of mROS are not concerning to long-term neuronal health. However, the combination of elevated NO and ROS, such as O2.−, may result in the formation of ONOO−, a reactive intermediate capable of impairing proteins through post-translational S-nitrosation of sulfur groups.91 Fioramonti et al84 demonstrated that recurrent hypoglycaemia impairs the counter-regulatory response to hypoglycaemia in part through increasing S-nitrosation of VMH proteins, and in particular, of soluble guanylyl cyclase, an endogenous NO receptor. These effects were prevented by the pre-administration of N-acetylcysteine (NAC), a precursor to the antioxidant glutathione. Furthermore, NAC restored typical glucose sensing in VMH Gl neurones from non-diabetic rats following recurrent hypoglycaemia, highlighting the impact of oxidative stress on the neurocircuits involved in hypoglycaemia counter-regulation.84
Oxidative stress is elevated in many brain regions during diabetic hyperglycaemia.89,128,136–142 Moreover, the pathology of diabetes includes impairment of neuronal glucose sensing and is in part a product of neuronal oxidative stress.128 For example, activation of VMH Gl neurones in low glucose and the counter-regulatory response to insulin-induced hypoglycaemia is impaired in STZ diabetes even in the absence of recurrent hypoglycaemia. However, VMH overexpression of the antioxidant enzyme thioredoxin restored normal VMH Gl neuronal glucose sensing and hypoglycaemia counter-regulation in STZ rats under basal conditions.20 Interestingly, transgenic mice deficient in insulin receptor substrate-2 (IRS2−/−) demonstrate elevated hypothalamic oxidative stress, resulting in increased inflammatory markers and eventual cell death.136 Thus, it is possible that both hyperglycaemia per se and impaired insulin signalling increase oxidative stress.
As discussed above, treatment with antioxidants such as NAC have been shown to normalise glucose sensing in VMH Gl neurones following recurrent hypoglycaemia. However, NAC did not normalise baseline glucose sensing in VMH Gl neurones or the counter-regulatory response to hypoglycaemia in STZ rats.20,84 Thus, the glutathione antioxidant system alone is not sufficient to combat the combined oxidative stress of diabetes and insulin-induced recurrent hypoglycaemia. This suggests that perhaps a different antioxidant system, alone or in combination with NAC, might restore normal glucose sensing. In support of this, infusion of the thioredoxin inhibitor auranofin blunted the counter-regulatory response and diminished VMH Gl neuronal activation in low glucose.20 Conversely, VMH thioredoxin-1 overexpression normalised both Gl glucose sensing and the counter-regulatory response in STZ diabetic rats.20 Unfortunately, similar to NAC, VMH thioredoxin overexpression alone was also not able to maintain normal glucose sensing and hypoglycaemia counter-regulation when the STZ rats were subjected to recurrent hypoglycaemia.20 This suggests again, that either a combination of glutathione and thioredoxin or another antioxidant are needed to compensate for the oxidative stress produced when insulin-induced hypoglycaemia occurs on a background of diabetic hyperglycaemia. However, what remains evident is that the counter-regulatory response and VMH glucose sensing are directly impacted by oxidative stress resulting from hyperglycaemia and hypoglycaemia. It is also possible that impaired insulin signalling contributes to this pathology.
4.3 |. Mitochondrial KATP in glucose sensing
Mitochondrial KATP (mKATP) channels located on the inner mitochondrial membrane may provide a link between oxidative stress and glucose sensing. Although the molecular identity of mKATP channels remains controversial, they appear to play several important roles in determining the balance between cell survival and death. These roles include adjusting the K+ balance on either side of the membrane to control mitochondrial swelling, maintaining the mitochondrial membrane potential, sensing mitochondrial ATP to adjust the rate of the electron transport system to ATP requirements, regulating ROS production and affecting the release of cytochrome c. Increased oxidative stress activates mKATP channels, which then reduce ROS levels and confer neuroprotection in cortical cultures following ischaemia or excitotoxicity.143–146
mKATP channels may also play a role in glucose homeostasis. For example, bilateral VMH microinjections of the KATP channel opener diazoxide enhance the release of counter-regulatory hormones during insulin-induced hypoglycaemia in both control rats and rats with HAAF.96 On the other hand, during euglycaemia, diazoxide injected i.c.v. or into the hypothalamus or given orally lowers hepatic glucose output in humans and rodents.147–149 Although diazoxide opens both the mKATP channel and the plasma membrane KATP channel, the diazoxide IC50 for the plasma membrane KATP channel is 2000 times greater than that of the mKATP channel (835 vs 0.4 nmol L−1, respectively).150 The VMH (1 nmol L−1 to 0.1 pmol L−1) and cerebrospinal fluid (3 nmol L−1) concentrations of diazoxide in the above studies are more consistent with an action on the mKATP channel rather than the plasma membrane KATP channel. An action on the mKATP channel makes intuitive sense as well. Opening of the plasma membrane KATP channel causes neuronal hyperpolarisation. Because KATP channels are almost ubiquitous throughout the brain,151 opening the plasma membrane KATP channel would in essence inhibit the entire VMH. Such an action is not consistent with an enhanced counter-regulatory response to hypoglycaemia because anaesthetising or lesioning the VMH blunts the increase in blood glucose following exercise or hypoglycaemia.152,153 On the other hand, during euglycaemia, VMH lesions increase blood glucose. It is interesting that, during euglycaemia, diazoxide lowers hepatic glucose output. This supports the hypothesis that ROS and oxidative stress must remain in balance, with deviations to either side being deleterious to glucose homeostasis. This hypothesis is consistent with the observation that VMH thioredoxin overexpression in STZ rats also lowered basal glycaemia and reduced insulin requirements in addition to restoring the counter-regulatory response to insulin-induced hypoglycaemia and normalising glucose sensing in VMH Gl neurones.135 Interestingly, diabetic humans and rodents are refractory to the glucose lowering effect of diazoxide.154 Consistent with this, mKATP channels are also impaired during diabetes.155,156 Taken together, these observations suggest that the mKATP channel plays a role in glucose homeostasis and is potentially a therapeutic target in diabetes.
5 |. INSULIN REGULATION OF SYNAPTIC PLASTICITY
The effects of insulin on neuronal activity are complex and much work remains to be performed to clearly understand how insulin affects neuronal circuity in homeostatic and glucose-sensing brain regions. Moreover, although insulin effects on activity and glucose sensitivity of glucose-sensing neurones have been studied to some extent, the effects of insulin on synaptic plasticity in homeostatic circuits have not. However, more is known of a mechanistic nature about insulin effects on synaptic activity and plasticity in other brain regions such as the hippocampus. Much of these data derive from studies of insulin effects on cognition, and particularly memory formation.157,158 Although these data may not be directly relevant to glucose sensing, such studies provide clues regarding how to investigate insulin effects on neuronal activity in homeostatic circuitry. Below, the focus is on what is known about the effects of insulin on synaptic plasticity in the hippocampus.
Hippocampal long-term potentiation (LTP) and depression (LTD) are indices of synaptic plasticity and are associated with memory and learning.159,160 Hippocampal glutamate-mediated LTP is experimentally induced by high frequency stimulation of the afferent neurones projecting to the CA1 region of the hippocampus. Conversely, hippocampal glutamate-mediated long-term depression is induced by low frequency stimulation. Often both LTP and LTD can be induced in the same neuronal circuits depending on the frequency of stimulation. LTP and LTD are measured in vivo or ex vivo using extracellular recordings of the field (population) of EPSPs (fEPSPs) which represent the excitatory response to pre-synaptic inputs in the region. LTP is indicated by an increase in slope of the fEPSPs to afferent stimulation, whereas the fEPSP slope is reduced during LTD.161,162 The effects of insulin on LTP and LTD are complex and can depend on a number of factors, including glucose concentration and insulin resistance.
Insulin shifts the frequency for induction of both LTD and LTP in the CA1 region to lower frequencies. This effect is mediated by PI3 kinase signalling.161 Under baseline conditions, low doses of insulin decrease fEPSP slope, whereas higher concentrations can either increase or decrease fEPSP slope. However, when protein kinase C signalling is blocked insulin potentiates fEPSP slope. Furthermore, the effect of insulin on basal fEPSP slope is dependent on glucose concentration. That is, in 5 mmol L−1 glucose, the inhibitory effect of insulin on fEPSP slope is greater than that in 10 mmol L−1 glucose; however, in 20 mmol L−1 glucose, insulin increases the fEPSP slope. By contrast to basal conditions, LTP induction is enhanced when afferent stimulation occurs in the presence of acute insulin application to the hippocampal slice.162 On the other hand, i.c.v. infusion of insulin for 3 months reduced hippocampal LTP induction.163 In addition to modulating hippocampal glutamatergic LTP and LTD, insulin increases hippocampal GABAergic transmission.164 Hippocampal GABA transmission is critical for gating of information into the hippocampus.165 These observations suggest that insulin affects synaptic plasticity and information processing in the hippocampus. Moreover, these effects of insulin are dependent on concentration, time-course and extracellular glucose concentration.
As would be expected from the preceding discussion, the effects of insulin are altered during insulin resistance and diabetes. Hippocampal LTP induction is reduced in obese hyperinsulinemic Zucker rats, as well as in rats treated with STZ or exposed to recurrent hypoglycaemia.166–168 LTP induction is also reduced when insulin receptor expression is reduced in the hippocampus.157,158 Although LTP and LTD are more difficult to investigate in the hypothalamus, glutamate and GABA plasticity have been observed in this brain region.67,88,169–173 Thus, it is not only possible, but also very likely that insulin regulates synaptic plasticity in hypothalamic areas containing glucose-sensing neurones. It is also likely that the pathological conditions discussed in preceding sections (diabetic hyperglycaemia and insulin resistance or insulin-induced hypoglycaemia) may alter the effects of insulin on synaptic plasticity in glucose-sensing neurones.
6 |. CONCLUSIONS
In conclusion, glucose-sensing neurones exist throughout the brain that control glucose homeostasis and are altered by diabetes and its associated factors. Insulin alters both the activity and glucose sensitivity of glucose-sensing neurones. In turn, the effects of insulin are dependent on glucose concentration and metabolic status. Thus, care must be taken when interpreting data to account for experimental conditions. Furthermore, diabetic hyperglycaemia, insulin resistance and repeated episodes of insulin-induced hypoglycaemia alter the function of glucose-sensing neurones. Taken together, these data suggest a critical role for insulin signalling in the regulation of glucose-sensing neurones. The mechanisms by which dysfunctional insulin signalling and other factors associated with diabetes alter central glucose sensing are not clear. However, one attractive option lies in oxidative stress, which appears to be central in innate glucose sensing, as well as symptomatic and/or causative for metabolic conditions such as diabetes and obesity that impair insulin signalling and, consequently, glucose sensing. It will also be important to determine how insulin and altered insulin signalling affect synaptic plasticity in glucose-sensing neurones. This is particularly relevant because the effects of insulin on glucose-sensing neurones are dependent on sustained changes in metabolic status. As a result, insulin has the means to have profound and long-term effects on glucose-sensing neurones and the circuits that control glucose homeostasis. The critical importance of insulin in maintaining metabolic health warrants further investigation of these underlying mechanisms.
ACKNOWLEDGEMENTS
This work was funded in part by the Department of Defense award W81XWH1910015 and National Institute of Heath R01 DK103676 and F31 DK126433.
Funding information
National institute of Health, Grant/Award Number: F31 DK126433 and R01 DK103676; U.S. Department of Defense, Grant/Award Number: W81XWH1910015
Footnotes
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
REFERENCES
- 1.Humphrey DR, Schmidt EM. Extracellular single-unit recording methods. In: Boulton AA, Baker GB, Vanderwolf CH, eds. Neurophysiological Techniques: Applications to Neural Systems. Totowa, NJ: Humana Press; 1990:1–64. [Google Scholar]
- 2.Li WC, Soffe SR, Roberts A. A direct comparison of whole cell patch and sharp electrodes by simultaneous recording from single spinal neurons in frog tadpoles. J Neurophysiol. 2004;92:380–386. [DOI] [PubMed] [Google Scholar]
- 3.Perkins KL. Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J Neurosci Methods. 2006;154:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. [DOI] [PubMed] [Google Scholar]
- 5.Neher E, Sakmann B. Single-channel currents recorded from membrane of denervated frog muscle fibres. Nature. 1976;260:799–802. [DOI] [PubMed] [Google Scholar]
- 6.Routh VH, McArdle JJ, Levin BE. Phosphorylation modulates the activity of the ATP-sensitive K+ channel in the ventromedial hypothalamic nucleus. Brain Res. 1997;778:107–119. [DOI] [PubMed] [Google Scholar]
- 7.Dunne MJ, Bullett MJ, Li GD, Wollheim CB, Petersen OH. Galanin activates nucleotide-dependent K+ channels in insulin-secreting cells via a pertussis toxin-sensitive G-protein. EMBO J. 1989;8:413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang R, Liu X, Hentges ST, et al. The regulation of glucose-excited neurons in the hypothalamic arcuate nucleus by glucose and feeding-relevant peptides. Diabetes. 2004;53:1959–1965. [DOI] [PubMed] [Google Scholar]
- 9.Dunn-Meynell AA, Routh VH, Kang L, Gaspers L, Levin BE. Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes. 2002;51:2056–2065. [DOI] [PubMed] [Google Scholar]
- 10.Kang L, Dunn-Meynell AA, Routh VH. et al. Glucokinase is a critical regulator of ventromedial hypothalamic neuronal glucosensing. Diabetes. 2006;55:412–420. [DOI] [PubMed] [Google Scholar]
- 11.Kang L Routh VH, Kuzhikandathil EV, Gaspers L, Levin BE. Physiological and molecular properties of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes. 2004:53:559. [DOI] [PubMed] [Google Scholar]
- 12.van Welie I, van Hooft JA, Wadman WJ. Homeostatic scaling of neuronal excitability by synaptic modulation of somatic hyperpolarization-activated Ih channels. Proc Natl Acad Sci. 2004;101:5123–5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deemyad T, Luthi J, Spruston N. Astrocytes integrate and drive action potential firing in inhibitory subnetworks. Nat Commun. 2018;9:4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu L, Wang W, Peng Y, Li J, Wang L, Wang X. Electrophysiology and pharmacology of tandem domain potassium channel TREK-1 related BDNF synthesis in rat astrocytes. Naunyn Schmiedebergs Arch Pharmacol. 2014;387:303–312. [DOI] [PubMed] [Google Scholar]
- 15.Wang Y, Liu Y-Z, Wang S-Y, Wang Z. In vivo whole-cell recording with high success rate in anaesthetized and awake mammalian brains. Mol Brain. 2016;9:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Canabal DD, Potian JG, Duran RG, McArdle JJ, Routh VH. Hyperglycemia impairs glucose and insulin regulation of nitric oxide production in glucose-inhibited neurons in the ventromedial hypothalamus. Am J Physiol Regul Integr Comp Physiol. 2007;293:R592–R600. [DOI] [PubMed] [Google Scholar]
- 17.Canabal DD, Song Z, Potian JG, Beuve A, McArdle JJ, Routh VH. Glucose, insulin and leptin signaling pathways modulate nitric oxide (NO) synthesis in glucose-inhibited (Gl) neurons in the ventromedial hypothalamus (VMH). Am J Physiol Regul Integr Comp Physiol. 2007;292:R1418–R1428. [DOI] [PubMed] [Google Scholar]
- 18.Le Foil C, Dunn-Meynell A, Musatov S, Magnan C, Levin BE. FAT/CD36: a major regulator of neuronal fatty acid sensing and energy homeostasis in rats and mice. Diabetes. 2013;62:2709–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murphy BA, Fakira KA, Song Z, Beuve A, Routh VH. AMP-activated protein kinase (AMPK) and nitric oxide (NO) regulate the glucose sensitivity of ventromedial hypothalamic (VMH) glucose-inhibited (Gl) neurons. Am J Physiol Cell Physiol. 2009;297:C750–C758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou C, Routh VH. Thioredoxin-1 overexpression in the ventromedial nucleus of the hypothalamus (VMH) preserves the counterregulatory response to hypoglycemia during type 1 diabetes mellitus in male rats. Diabetes. 2018;67:120–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anand BK, China GS, Sharma KN, Dua S, Singh B. Activity of single neurons in the hypothalamus feeding centers: effect of glucose. Am J Physiol. 1964;2207:1146–1154. [DOI] [PubMed] [Google Scholar]
- 22.Oomura Y, Kimura K, Ooyama H, Maeo T, Iki M, Kuniyoshi N. Reciprocal activities of the ventromedial and lateral hypothalamic area of cats. Science. 1964:143:484–485. [DOI] [PubMed] [Google Scholar]
- 23.Ashford MLJ, Boden PR, Treherne JM. Glucose-induced excitation of hypothalamic neurones is mediated by ATP-sensitive K+ channels. Pfugers Arch. 1990;415:479–483. [DOI] [PubMed] [Google Scholar]
- 24.Song Z, Routh VH. Differential effects of glucose and lactate on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes. 2005;54:15–22. [DOI] [PubMed] [Google Scholar]
- 25.De Vries MG, Arseneau LM, Lawson ME, Beverly JL. Extracellular glucose in rat ventromedial hypothalamus during acute and recurrent hypoglycemia. Diabetes. 2003;52:2767–2773. [DOI] [PubMed] [Google Scholar]
- 26.Silver IA, Erecinska M. Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo-, hypo-, and hyperglycemic animals. J Neurosci. 1994;14:5068–5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silver IA, Erecinska M. Glucose-induced intracellular ion changes in sugar-sensitive hypothalamic neurons. J Neurophysiol. 1998:79:1733–1745. [DOI] [PubMed] [Google Scholar]
- 28.Rorsman P, Ramracheya R, Rorsman NJG, Zhang Q. ATP-regulated potassium channels and voltage-gated calcium channels in pancreatic alpha and beta cells: similar functions but reciprocal effects on secretion. Diabetologia. 2014;57:1749–1761. [DOI] [PubMed] [Google Scholar]
- 29.Fioramonti X, Contie S, Song Z, Routh VH, Lorsignol A, Penicaud L. Characterization of glucosensing neuron subpopulations in the arcuate nucleus: Integration in NPY and POMC networks? Diabetes. 2007;56:1219–1227. [DOI] [PubMed] [Google Scholar]
- 30.Fioramonti X, Lorsignol A, Taupignon A, Penicaud L. A new ATP-sensitive K+ channel-independent mechanism is involved in glucose-excited neurons of mouse arcuate nucleus. Diabetes. 2004;53:2767–2775. [DOI] [PubMed] [Google Scholar]
- 31.Chrétien C, Fenech C, Liénard F, et al. Transient receptor potential canonical 3 (TRPC3) channels are required for hypothalamic glucose detection and energy homeostasis. Diabetes. 2017;66:314–324. [DOI] [PubMed] [Google Scholar]
- 32.Song Z, Levin BE, McArdle JJ, Bakhos N, Routh VH. Convergence of pre- and postsynaptic influences on glucosensing neurons in the ventromedial hypothalamic nucleus. Diabetes. 2001;50:2673–2681. [DOI] [PubMed] [Google Scholar]
- 33.Santiago AM, Clegg DJ, Routh VH. Estrogens modulate ventrolateral ventromedial hypothalamic glucose-inhibited neurons. Mol Metab. 2016;5:823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song Z, Routh VH. Recurrent hypoglycemia reduces the glucose sensitivity of glucose-inhibited neurons in the ventromedial hypothalamus nucleus (VMN). Am J Physiol Regul Integr Comp Physiol. 2006;291:R1283–R1287. [DOI] [PubMed] [Google Scholar]
- 35.Murphy BA, Fioramonti X, Jochnowitz N, et al. Fasting enhances the response of arcuate neuropeptide Y (NPY)-glucose-inhibited (Gl) neurons to decreased extracellular glucose. Am J Physiol Cell Physiol. 2009;296:746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fioramonti X, Marsollier N, Song Z, et al. Ventromedial hypothalamic nitric oxide production is necessary for hypoglycemia detection and counterregulation. Diabetes. 2010;59:519–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burdakov D Gerasimenko O, Verkhratsky A Physiological changes in glucose differentially modulate the excitability of hypothalamic melanin-concentrating hormone and orexin neurons in situ. J Neurosci. 2005;25:2429–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karnani MM, Szabό G, Erdélyi F, Burdakov D. Lateral hypothalamic GAD65 neurons are spontaneously firing and distinct from orexin- and melanin-concentrating hormone neurons. J Physiol. 2013;591:933–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melnick IV, Price CJ, Colmers WF. Glucosensing in parvocellular neurons of the rat hypothalamic paraventricular nucleus. Eur J Neurosci. 2011;34:272–282. [DOI] [PubMed] [Google Scholar]
- 40.Beall C, Hamilton D, Gallagher J, et al. Mouse hypothalamic GT1–7 cells demonstrate AMPK-dependent intrinsic glucose-sensing behaviour. Diabetologia. 2012;55:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roland AV, Moenter SM. Glucosensing by GnRH neurons: inhibition by androgens and involvement of AMP-activated protein kinase. Mol Endocrinol. 2011;25:847–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ibrahim N, Bosch MA, Smart JL, et al. Hypothalamic proopiomela-nocortin neurons are glucose responsive and express K(ATP) channels. Endocrinology. 2003;144:1331–1340. [DOI] [PubMed] [Google Scholar]
- 43.Zhou L, Podolsky N, Sang Z, et al. The medial amygdalar nucleus: a novel glucose-sensing region that modulates the counterregulatory response to hypoglycemia. Diabetes. 2010;59:2646–2652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balfour RH, Hansen AMK, Trapp S. Neuronal responses to transient hypoglycaemia in the dorsal vagal complex of the rat brain-stem. J Physiol. 2006;570:469–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cotero VE, Routh VH. Insulin blunts the response of glucose-excited (GE) neurons in the ventrolateral-ventromedial hypothalamic nucleus (VL-VMN) to decreased glucose. Am J Physiol Endocrinol Metab. 2009;296:E1101–E1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Szabό I, Hormay E, Csetényi B, Nagy B, Lénárd L, Karádi Z. Multiple functional attributes of glucose-monitoring neurons in the medial orbitofrontal (ventrolateral prefrontal) cortex. Neurosci Biobehav Rev. 2018;85:44–53. [DOI] [PubMed] [Google Scholar]
- 47.Ono H Molecular mechanisms of hypothalamic insulin resistance. IntJMol Sci. 2019;20:1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spanswick D, Smith MA, Mirshamsi S, Routh VH, Ashford ML. Insulin activates ATP-sensitive K+ channels in hypothalamic neurons of lean, but not obese rats. Nat Neurosci. 2004;3:757–758. [DOI] [PubMed] [Google Scholar]
- 49.Hausen AC, Ruud J, Jiang H, et al. Insulin-dependent activation of MCH neurons impairs locomotor activity and insulin sensitivity in obesity. Cell Rep. 2016;17:2512–2521. [DOI] [PubMed] [Google Scholar]
- 50.Elmquist JK. Hypothalamic pathways underlying the endocrine, autonomic, and behavioral effects of leptin. Physiol Behav. 2001;74:703–708. [DOI] [PubMed] [Google Scholar]
- 51.Hill JW, Williams KW, Ye C, et al. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Investig. 2008;118:1796–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Könner AC, Janoschek R, Plum L, et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007;5:438–449. [DOI] [PubMed] [Google Scholar]
- 53.Plum L, Ma X, Hampel B, et al. Enhanced PIP3 signaling in POMC neurons causes KATP channel activation and leads to diet-sensitive obesity. J Clin Investig. 2006;116:1886–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Williams KW, Margatho LO, Lee CE, et al. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci. 2010;30:2472–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Qiu J, Zhang C, Borgquist A, et al. Insulin excites anorexigenic proopiomelanocortin neurons via activation of canonical transient receptor potential channels. Cell Metab. 2014:19:682–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dodd GT, Michael NJ, Lee-Young RS, et al. Insulin regulates POMC neuronal plasticity to control glucose metabolism. eLife. 2018;7:e38704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dodd GT, Andrews ZB, Simonds SE, et al. A hypothalamic phosphatase switch coordinates energy expenditure with feeding. Cell Metab. 2017;26:375–393.e7. [DOI] [PubMed] [Google Scholar]
- 58.Hao L, Sheng Z, Potian J, Deak A Rohowsky-Kochan C, Routh VH Lipopolysaccharide (LPS) and tumor necrosis factor alpha (TNFα) blunt the response of neuropeptide Y/agouti-related peptide (NPY/AgRP) glucose inhibited (Gl) neurons to decreased glucose. Brain Res. 2016;1648:181–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sheng Z, Santiago AM, Thomas MP, Routh VH. Metabolic regulation of lateral hypothalamic glucose-inhibited orexin neurons may influence midbrain reward neurocircuitry. Mol Cell Neurosci. 2014;62:30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Diggs-Andrews KA, Zhang X, Song Z, Daphna-lken D, Routh VH, Fisher SJ. Brain insulin action regulates hypothalamic glucose sensing and the counterregulatory response to hypoglycemia. Diabetes. 2010;59:2271–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reno CM, Puente EC, Sheng Z, et al. Brain GLUT4 knockout mice have impaired glucose tolerance, decreased insulin sensitivity, and impaired hypoglycemic counterregulation. Diabetes. 2016;66:587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Elizondo-Vega R, Cortes-Campos C, Barahona MJ, Oyarce KA, Carril CA, Garda-Robles MA. The role of tanycytes in hypothalamic glucosensing. J Cell Mol Med. 2015:19:1471–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leloup C, Arluison M, Lepetit N, et al. Glucose transporter 2 (GLUT 2): expression in specific brain nuclei. Brain Res. 1994:638:221–226. [DOI] [PubMed] [Google Scholar]
- 64.Marty N, Dallaporta M, Foretz M, et al. Regulation of glucagon secretion by glucose transporter type 2 (glut2) and astrocyte-dependent glucose sensors. J Clin Invest. 2005;115:3545–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Penicaud L, Leloup C, Lorsignol A, Alquier T, Guillod E. Brain glucose sensing mechanism and glucose homeostasis. Curr Opin Clin Nutr Metab Care. 2002;5:539–543. [DOI] [PubMed] [Google Scholar]
- 66.Cai W, Xue C, Sakaguchi M, et al. Insulin regulates astrocyte gliotransmission and modulates behavior. J Clin Invest. 2018;128:2914–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gordon GR, Iremonger KJ, Kantevari S, Ellis-Davies GC, MacVicar BA, Bains JS. Astrocyte-mediated distributed plasticity at hypothalamic glutamate synapses. Neuron. 2009;64:391–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Orellana JA, Saez PJ, Cortes-campos C, et al. Glucose increases intracellular free Ca2+ in tanycytes via ATP released through connexin 43 hemichannels. Glia. 2012;60:53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sanders NM, Dunn-Meynell AA, Levin BE. Third ventricular alloxan reversibly impairs glucose counterregulatory responses. Diabetes. 2004;53:1230–1236. [DOI] [PubMed] [Google Scholar]
- 70.Muhič M, Vardjan N, Chowdhury HH, Zorec R, Kreft M. Insulin and insulin-like growth factor 1 (IGF-1) modulate cytoplasmic glucose and glycogen levels but not glucose transport across the membrane in astrocytes. J Biol Chem. 2015;290:11167–11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mordes JP, Bortell R, Blankenhorn EP, Rossini AA, Greiner DL. Rat models of type 1 diabetes: genetics, environment, and autoimmunity. ILAR J. 2004;45:278–291. [DOI] [PubMed] [Google Scholar]
- 72.Boyle PJ, Zrebiec J. Impact of therapeutic advances on hypoglycaemia in type 2 diabetes. Diabetes Metab Res Rev. 2008;24:257–285. [DOI] [PubMed] [Google Scholar]
- 73.Cotero VE, Zhang BB, Routh VH. The response of glucose-excited neurones in the ventromedial hypothalamus to decreased glucose is enhanced in a murine model of type 2 diabetes mellitus. JNeuroendocrinol. 2009;22:65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gao H, Molinas AJR, Miyata K, Qiao X, Zsombok A. Overactivity of liver-related neurons in the paraventricular nucleus of the hypothalamus: electrophysiological findings in db/db mice. J Neurosci. 2017;37:11140–11150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qiu J, Bosch MA, Meza C, et al. Estradiol protects proopiomelanocortin neurons against insulin resistance. Endocrinology. 2017;159:647–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Xu Y, Nedungadi Thekkethil P, Zhu L, et al. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab. 2011;14:453–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhu L, Xu P, Cao X, et al. The ERa-PI3K cascade in proopiomelano-cortin progenitor neurons regulates feeding and glucose balance in female mice. Endocrinology. 2015;156:4474–4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Santiago AM, Clegg DJ, Routh VH. Ventromedial hypothalamic glucose sensing and glucose homeostasis vary throughout the estrous cycle. Physiol Behav. 2016;167:248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Perreault M, Dombrowski L, Marette A. Mechanism of impaired nitric oxide synthase activity in skeletal muscle of streptozotocin-induced diabetic rats. Diabetologia. 2000:43:427–437. [DOI] [PubMed] [Google Scholar]
- 80.Souayah N, Potian JG, Garcia CC, et al. Motor unit number estimate as a predictor of motor dysfunction in an animal model of type 1 diabetes. Am J Physiol Endocrinol Metab. 2009;297:E602–E608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jiang Y, Gao H, Krantz AM, Derbenev AV, Zsombok A. Reduced GABAergic inhibition of kidney-related PVN neurons in strep-tozotocin-treated type 1 diabetic mouse. J Neurophysiol. 2013:110:2192–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Diabetes Control and Complications Trial Research Group; Nathan DM, Genuth S, Lachin J, et al. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. The Diabetes Control and Complications Trial Research Group. N Engl J Med. 1993;329:977–986. [DOI] [PubMed] [Google Scholar]
- 83.Cryer PE. Hypoglycemia-associated autonomic failure in diabetes. Am J Physiol. 2001;281:E1115–E1121. [DOI] [PubMed] [Google Scholar]
- 84.Fioramonti X, Deak A, Deshpande S, et al. Hypothalamic S-nitrosylation contributes to the counter-regulatory response impairment following recurrent hypoglycemia. PLoS One. 2013;8:e68709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Inouye KE, Chan O, Yue JTY, Matthews SG, Vranic M. Effects of diabetes and recurrent hypoglycemia on the regulation of the sympathoadrenal system and hypothalamo-pituitary-adrenal axis. Am J Physiol Endocrinol Metab. 2005;288:E422–E429. [DOI] [PubMed] [Google Scholar]
- 86.Languren G, Montiel T, Julio-Amilpas A, Massieu L. Neuronal damage and cognitive impairment associated with hypoglycemia: an integrated view. Neurochem Int. 2013;63:331–343. [DOI] [PubMed] [Google Scholar]
- 87.Paranjape SA, Briski KP. Recurrent insulin-induced hypoglycemia causes site-specific patterns of habituation or amplification of CNS neuronal genomic activation. Neuroscience. 2005:130:957–970. [DOI] [PubMed] [Google Scholar]
- 88.Szepietowska B, Horvath TL, Sherwin RS. Role of synaptic plasticity and EphA5-EphrinA5 interaction within the ventromedial hypothalamus in response to recurrent hypoglycemia. Diabetes. 2017;63:1140–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Won S, Yoo B, Kauppinen T, et al. Recurrent/moderate hypoglycemia induces hippocampal dendritic injury, microglial activation, and cognitive impairment in diabetic rats. J Neuroinflammation. 2014;9:182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Otlivanchik O, Sanders NM, Dunn-Meynell A, Levin BE. Orexin signaling is necessary for hypoglycemia-induced prevention of conditioned place preference. Am J Physiol Regul Integr Comp Physiol. 2016;310:R66–R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fioramonti X, Song Z, Vazirani RP, Beuve A, Routh VH. Hypothalamic NO in hypoglycemia detection and counter-regulation: a two edged sword. Antioxid Redox Signal. 2010;14:505–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kang L, Sanders NM, Dunn-Meynell AA, et al. Prior hypoglycemia enhances glucose responsiveness in some ventromedial hypothalamic glucosensing neurons. Am J Physiol Regul Integr Comp Physiol. 2008;294:R784–R792. [DOI] [PubMed] [Google Scholar]
- 93.Miki T, Liss B, Minami K, et al. ATP-sensitive potassium channels in hypothalamic neurons play an essential role in the maintenance of glucose homeostasis by controlling glucagon release and food intake. Nat Neurosci. 2001;5:507–512. [DOI] [PubMed] [Google Scholar]
- 94.Evans ML, McCrimmon RJ, Flanagan DE, et al. Hypothalamic ATP-sensitive K+ channels play a key role in sensing hypoglycemia and triggering counterregulatory epinephrine and glucagon responses. Diabetes. 2004;53:2542–2551. [DOI] [PubMed] [Google Scholar]
- 95.Fan X, Ding Y, Brown S, et al. Hypothalamic AMP-activated protein kinase activation with AICAR amplifies counterregulatory responses to hypoglycemia in a rodent model of type 1 diabetes. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1702–R1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McCrimmon RJ, Evans ML, Fan X, et al. Activation of ATP-sensitive K+ channels in the ventromedial hypothalamus amplifies counter-regulatory hormone responses to hypoglycemia in normal and recurrently hypoglycemic rats. Diabetes. 2005:54:3169–3174. [DOI] [PubMed] [Google Scholar]
- 97.Evans SB, Wilkinson CW, Bentson K, Gronbeck P, Zavosh A, Figlewicz DP. PVN activation is suppressed by repeated hypoglycemia but not antecedent corticosterone in the rat. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1426–R1436. [DOI] [PubMed] [Google Scholar]
- 98.Evans SB, Wilkinson CW, Gronbeck P, Bennett JL, Taborsky GJ Jr, Figlewicz DP. Inactivation of the PVN during hypoglycemia partially simulates hypoglycemia-associated autonomic failure. Am J Physiol Regul Integr Comp Physiol. 2003;284:R57–R65. [DOI] [PubMed] [Google Scholar]
- 99.McCrimmon RJ, Song Z, Cheng H, et al. Corticotrophin-releasing factor receptors within the ventromedial hypothalamus regulate hypoglycemia-induced hormonal counterregulation. J Clin Invest. 2006;116:1723–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.McCrimmon RJ, Fan X, Cheng H, et al. Activation of AMP-activated protein kinase within the ventromedial hypothalamus amplifies counterregulatory hormone responses in rats with defective counterregulation. Diabetes. 2006;55:1755–1760. [DOI] [PubMed] [Google Scholar]
- 101.McCrimmon RJ, Fan X, Ding Y, Zhu W, Jacob RJ, Sherwin RS. Potential role for AMP-activated protein kinase in hypoglycemia sensing in the ventromedial hypothalamus. Diabetes. 2004;53:1953–1958. [DOI] [PubMed] [Google Scholar]
- 102.McCrimmon RJ, Shaw M, Fan X, et al. Key role for AMP-activated protein kinase in the ventromedial hypothalamus in regulating counterregulatory hormone responses to acute hypoglycemia. Diabetes. 2008;57:444–450. [DOI] [PubMed] [Google Scholar]
- 103.Agrawal R, Vieira-de-Abreu A, Durupt G, Taylor C, Chan O, Fisher SJ. Insulin regulates GLUT4 in the ventromedial hypothalamus to restore the sympathoadrenal response to hypoglycemia in diabetic rats. Am J Physiol Endocrinol Metab. 2018;315:E1286–E1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Andrew SF, Dinh TT, Ritter S. Localized glucoprivation of hindbrain sites elicits corticosterone and glucagon secretion. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1792–R1798. [DOI] [PubMed] [Google Scholar]
- 105.Li A-J, Wang Q, Elsarelli MM, Brown RL, Ritter S. Hindbrain catecholamine neurons activate orexin neurons during systemic gluco-privation in male rats. Endocrinology. 2015;156:2807–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li A-J, Wang Q, Ritter S. Selective pharmacogenetic activation of catecholamine subgroups in the ventrolateral medulla elicits key glucoregulatory responses. Endocrinology. 2018;159:341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ritter RC, Slusser PG, Stone S. Glucoreceptors controlling feeding and blood glucose: location in the hindbrain. Science. 1981;213:451–452. [DOI] [PubMed] [Google Scholar]
- 108.Ritter S, Dinh TT, Li A-J. Hindbrain catecholamine neurons control multiple glucoregulatory responses. Physiol Behav. 2006;89:490–500. [DOI] [PubMed] [Google Scholar]
- 109.Routh VH, Donovan CM, Ritter S. 2. Hypoglycemia detection. Transl Endocrinol Metab. 2012;3:47–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bohland M, Matveyenko AV, Saberi M, Khan AM, Watts AG, Donovan CM. Activation of hindbrain neurons is mediated by portal-mesenteric vein glucosensors during slow-onset hypoglycemia. Diabetes. 2014;63:2866–2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Donovan CM, Hamilton-Wessler M, Halter JB, Bergman RN. Primacy of liver glucosensors in the sympathetic response to progressive hypoglycemia. Proc Natl Acad Sci. 1994;91:2863–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Donovan CM, Watts AG. Peripheral and central glucose sensing in hypoglycemic detection. Physiology. 2014;29:314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fujita S, Bohland M, Sanchez-Watts G, Watts AG, Donovan CM. Hypoglycemic detection at the portal vein is mediated by capsaicin-sensitive primary sensory neurons. Am J Physiol Endocrinol Metab. 2007;293:E96–E101. [DOI] [PubMed] [Google Scholar]
- 114.Matveyenko AV, Donovan CM. Metabolic sensors mediate hypoglycemic detection at the portal vein. Diabetes. 2006;55:1276–1282. [DOI] [PubMed] [Google Scholar]
- 115.Saberi M, Bohland M, Donovan CM. The locus for hypoglycemic detection shifts with the rate of fall in glycemia: the role of portal-superior mesenteric vein glucose sensing. Diabetes. 2008;57:1380–1386. [DOI] [PubMed] [Google Scholar]
- 116.Nakano Y, Oomura Y, Lénárd L, et al. Feeding-related activity of glucose- and morphine-sensitive neurons in the monkey amygdala. Brain Res. 1986:399:167–172. [DOI] [PubMed] [Google Scholar]
- 117.Korim WS, Bou Farah L, McMullan S, Verberne AJM. Orexinergic activation of medullary premotor neurons modulates the adrenal sympathoexcitation to hypothalamic glucoprivation. Diabetes. 2014;63:1895–1906. [DOI] [PubMed] [Google Scholar]
- 118.Korim WS, Llewellyn-Smith IJ, Verberne AJM. Activation of medulla-projecting perifornical neurons modulates the adrenal sympathetic response to hypoglycemia: involvement of orexin type 2 (OX2-R) receptors. Endocrinology. 2016;157:810–819. [DOI] [PubMed] [Google Scholar]
- 119.Otlivanchik O, Le Foil C, Levin BE. Perifornical hypothalamic orexin and serotonin modulate the counterregulatory response to hypoglycemic and glucoprivic stimuli. Diabetes. 2015;64:226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brown AM, Sickmann HM, Fosgerau K, et al. Astrocyte glycogen metabolism is required for neural activity during aglycemia or intense stimulation in mouse white matter. J Neurosci Res. 2005:79:74–80. [DOI] [PubMed] [Google Scholar]
- 121.Wender R, Brown AM, Fern R, Swanson RA, Farrell K, Ransom BR. Astrocytic glycogen influences axon function and survival during glucose deprivation in central white matter. J Neurosci. 2000;20:6804–6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Herzog Rl, McNay EC, McCrimmon RJ, et al. Effect of acute and recurrent hypoglycemia on regional changes in brain glycogen concentration. Am Diabetes Assoc Sci Sessions. 2006;55:A149. [Google Scholar]
- 123.Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci USA. 1994;91:10625–10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Fernandez AM, Hernandez-Garzon E, Perez-Domper P, et al. Insulin regulates astrocytic glucose handling through cooperation with IGF-I. Diabetes. 2017; 66: 64–74. [DOI] [PubMed] [Google Scholar]
- 125.Herzog Rl, Jiang L, Herman P, et al. Lactate preserves neuronal metabolism and function following antecedent recurrent hypoglycemia. J Clin Investig. 2013;123:1988–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Borg MA, Tamborlane WV, Shulman Gl, Sherwin RS. Local lactate perfusion of the ventromedial hypothalamus suppresses hypoglycemic counterregulation. Diabetes. 2003;52:663–666. [DOI] [PubMed] [Google Scholar]
- 127.McGowan JE, Chen L, Gao D, Trush M, Wei C. Increased mitochondrial reactive oxygen species production in newborn brain during hypoglycemia. Neurosci Lett. 2006;399:111–114. [DOI] [PubMed] [Google Scholar]
- 128.Singh P Jain A, Kaur G Impact of hypoglycemia and diabetes on CNS: correlation of mitochondrial oxidative stress with DNA damage. Mol Cell Biochem. 2004;260:153–159. [DOI] [PubMed] [Google Scholar]
- 129.Cobley JN, Fiorello ML, Bailey DM. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018;15:490–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bailey DM, Bärtsch P, Knauth M, Baumgartner RW. Emerging concepts in acute mountain sickness and high-altitude cerebral edema: from the molecular to the morphological. Cell Mol Life Sci. 2009;66:3583–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jaillard T, Roger M, Galinier A, et al. Hypothalamic reactive oxygen species are required for insulin-induced food intake inhibition. A NADPH oxidase-dependent mechanism. Diabetes. 2009;58:1544–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Leloup C, Magnan C, Benani A, et al. Mitochondrial reactive oxygen species are required for hypothalamic glucose sensing. Diabetes. 2006;55:2084–2090. [DOI] [PubMed] [Google Scholar]
- 133.Wang X, Michaelis EK. Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci. 2010;2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Routh VH, Song Z, Liu X. The role of glucosensing neurons in the detection of hypoglycemia. Diabetes Technol Ther. 2004;6:413–421. [DOI] [PubMed] [Google Scholar]
- 135.Zhou C, Teegala SB, Khan BA, Gonzalez C, Routh VH. Hypoglycemia: role of hypothalamic glucose-inhibited (Gl) neurons in detection and correction. Front Physiol. 2018:9:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Baquedano E, Burgos-Ramos E, Canelles S, et al. Increased oxidative stress and apoptosis in the hypothalamus of diabetic male mice in the insulin receptor substrate-2 knockout model. D/s Model Mech. 2016:9:573–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cardoso S, Santos MS, Seiça R, Moreira PI. Cortical and hippocampal mitochondria bioenergetics and oxidative status during hyperglycemia and/or insulin-induced hypoglycemia. Biochim Biophys Acta. 2010;1802:942–951. [DOI] [PubMed] [Google Scholar]
- 138.Ceriello A, Novials A, Ortega E, et al. Evidence that hyperglycemia after recovery from hypoglycemia worsens endothelial function and increases oxidative stress and inflammation in healthy control subjects and subjects with type 1 diabetes. Diabetes. 2012;61:2993–2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kamboj SS, Sandhir R. Protective effect of N-acetylcysteine supplementation on mitochondrial oxidative stress and mitochondrial enzymes in cerebral cortex of streptozotocin-treated diabetic rats. Mitochondrion. 2011;11:214–222. [DOI] [PubMed] [Google Scholar]
- 140.Lappalainen Z, Lappalainen J, Oksala NKJ, et al. Diabetes impairs exercise training-associated thioredoxin response and glutathione status in rat brain. J Appl Physiol. 2009;106:461–467. [DOI] [PubMed] [Google Scholar]
- 141.Reagan LP, Magarinos A, Yee DK, et al. Oxidative stress and HNE conjugation of GLUT3 are increased in the hippocampus of diabetic rats subjected to stress. Brain Res. 2000;862:292–300. [DOI] [PubMed] [Google Scholar]
- 142.Schulze PC, Yoshioka J, Takahashi T, He Z, King GL, Lee RT. Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin-interacting protein. J Biol Chem. 2004;279:30369–30374. [DOI] [PubMed] [Google Scholar]
- 143.Busija DW, Lacza Z, Rajapakse N, et al. Targeting mitochondrial ATP-sensitive potassium channels—a novel approach to neuroprotection. Brain Res Rev. 2004;46:282–294. [DOI] [PubMed] [Google Scholar]
- 144.Fornazari M, de Paula JG, Castilho RF, Kowaltowski AJ. Redox properties of the adenoside triphosphate-sensitive K+ channel in brain mitochondria. J Neurosci Res. 2008;86:1548–1556. [DOI] [PubMed] [Google Scholar]
- 145.Shi N, He J, Guo Q, Liu T, Han J. Liraglutide protects against diabetes mellitus complicated with focal cerebral ischemic injury by activating mitochondrial ATP-sensitive potassium channels. NeuroReport. 2019;30:479–484. [DOI] [PubMed] [Google Scholar]
- 146.Smith CO, Nehrke K, Brookes PS. The Slo(w) path to identifying the mitochondrial channels responsible for ischemic protection. Biochem J. 2017:474:2067–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Carey M, Lontchi-Yimagou E, Mitchell W, et al. Central KATP channels modulate glucose effectiveness in humans and rodents. Diabetes. 2020;69:1140–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kishore P, Boucai L, Zhang K, et al. Activation of K(ATP) channels suppresses glucose production in humans. J Clin Investig. 2011:121:4916–4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pocai A, Lam TKT, Gutierrez-Juarez R, et al. Hypothalamic KATP channels control hepatic glucose production. Nature. 2005;434:1026–1031. [DOI] [PubMed] [Google Scholar]
- 150.Garlid KD, Paucek P, Yarov-Yarovoy V, Sun X, Schindler PA. The mitochondrial K channel as a receptor for potassium channel openers. J Biol Chem. 1996;271:8796–8799. [DOI] [PubMed] [Google Scholar]
- 151.Dunn-Meynell AA, Rawson NE, Levin BE. Distribution and phenotype of neurons containing the ATP-sensitive K + channel in rat brain. Brain Res. 1998;814:41–54. [DOI] [PubMed] [Google Scholar]
- 152.Borg WP, During MJ,Sherwin RS, Borg MA, Brines ML, Shulman Gl. Ventromedial hypothalamic lesions in rats suppress counterregulatory responses to hypoglycemia. J Clin Invest. 1994;93:1677–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Vissing J Wallace JL, Scheurink AJ, Galbo H, Steffens AB. Ventromedial hypothalamic regulation of hormonal and metabolic responses to exercise. Am J Physiol. 1989;256:R1019–R1026. [DOI] [PubMed] [Google Scholar]
- 154.Esterson YB, Carey M, Boucai L, et al. Central regulation of glucose production may be impaired in type 2 diabetes. Diabetes. 2016;65:2569–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Katakam PVG, Jordan JE, Snipes JA, Tulbert CD, Miller AW, Busija DW. Myocardial preconditioning against ischemia-reperfusion injury is abolished in Zucker obese rats with insulin resistance. Am J Physiol Regul Integr Comp Physiol. 2007;292:R920–R926. [DOI] [PubMed] [Google Scholar]
- 156.Li D, Huang B, Liu J, Li L, Li X. Decreased brain KATP channel contributes to exacerbating ischemic brain injury and the failure of neuroprotection by sevoflurane post-conditioning in diabetic rats. PLoS One. 2013;8(8):e73334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Grillo CA, Piroli GG, Lawrence RC, et al. Hippocampal insulin resistance impairs spatial learning and synaptic plasticity. Diabetes. 2015;64:3927–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Soto M, Cai W, Konishi M, Kahn CR. Insulin signaling in the hippocampus and amygdala regulates metabolism and neurobehavior. Proc Natl Acad Sci. 2019;116:6379–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Malenka RC, Nicoll RA. Long-term potentiation - a decade of progress? Science. 1999;285:1870–1874. [DOI] [PubMed] [Google Scholar]
- 160.Rafalovich IV, Melendez AE, Plotkin JL, Tanimura A, Zhai S, Surmeier DJ. Interneuronal nitric oxide signaling mediates post-synaptic long-term depression of striatal glutamatergic synapses. Cell Rep. 2015;13:1336–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Van Der Heide LP, Kamal A, Artola A, Gispen WH, Ramakers GMJ. Insulin modulates hippocampal activity-dependent synaptic plasticity in a N-methyl-d-aspartate receptor and phosphatidyl-inositol-3-kinase-dependent manner. J Neurochem. 2005;94:1158–1166. [DOI] [PubMed] [Google Scholar]
- 162.Zhao F, Siu JJ, Huang W, Askwith C, Cao L. Insulin modulates excitatory synaptic transmission and synaptic plasticity in the mouse hippocampus. Neuroscience. 2019;411:237–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kamal A, Ramakers GMJ, Gispen WH, Biessels GJ. Effect of chronic intracerebroventricular insulin administration in rats on the peripheral glucose metabolism and synaptic plasticity of CA1 hippocampal neurons. Brain Res. 2012;1435:99–104. [DOI] [PubMed] [Google Scholar]
- 164.Jin Z, Jin Y, Kumar-Mendu S, Degerman E, Groop L, Birnir B. Insulin reduces neuronal excitability by turning on GABAA channels that generate tonic current. PLoS One. 2011;6:el6188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pelkey KA, Chittajallu R, Craig MT, Tricoire L, Wester JC, McBain CJ. Hippocampal GABAergic inhibitory interneurons. Physiol Rev. 2017:97:1619–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Izumi Y, Yamada KA, Matsukawa M, Zorumski CF. Effects of insulin on long-term potentiation in hippocampal slices from diabetic rats. Diabetologia. 2003;46:1007–1012. [DOI] [PubMed] [Google Scholar]
- 167.Kamal A, Ramakers GMJ, Gispen WH, Biessels GJ, Al AA. Hyperinsulinemia in rats causes impairment of spatial memory and learning with defects in hippocampal synaptic plasticity by involvement of postsynaptic mechanisms. Exp Brain Res. 2013;226:45–51. [DOI] [PubMed] [Google Scholar]
- 168.Yamada KA, Rensing NICH, Izumi YUKI, et al. Repetitive hypoglycemia in young rats impairs hippocampal long-term potentiation. Pediatr Res. 2004;55:372–379. [DOI] [PubMed] [Google Scholar]
- 169.Cheng J, Huang X, Liang Y, Xue T, Wang L, Bao J. Plasticity of light-induced concurrent glutamatergic and GABAergic quantal events in the suprachiasmatic nucleus. J Biol Rhythms. 2018;33:65–75. [DOI] [PubMed] [Google Scholar]
- 170.Crosby KM, Murphy-Royal C, Wilson SA, Gordon GR, Bains JS, Pittman QJ. Cholecystokinin switches the plasticity of GABA synapses in the dorsomedial hypothalamus via astrocytic ATP release. J Neurosci. 2018;38:8515–8525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Matikainen-Ankney BA, Kravitz AV. Persistent effects of obesity: a neuroplasticity hypothesis. Ann N Y Acad Sci. 2018;1428:221–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Pak CW, Currás-Collazo MC. Expression and plasticity of glutamate receptors in the supraoptic nucleus of the hypothalamus. Microsc Res Tech. 2002;56:92–100. [DOI] [PubMed] [Google Scholar]
- 173.Tartar JL, Tartar JL, King MA, Devine DP. Glutamate-mediated neuroplasticity in a limbic input to the hypothalamus. Stress. 2006;9:13–19. [DOI] [PubMed] [Google Scholar]